

Abstract
Lysosomes are intracellular acidic organelles with catabolic functions that contribute to the activation of autophagy. Although autophagy abnormality is associated with defects in lysosomal acidification during the progression of nonalcoholic fatty liver disease (NAFLD), the mechanisms of control of lysosomal acidification are not well understood at the molecular level. Thus, we aimed to elucidate the role of the orphan nuclear receptor retinoic acid–related orphan receptor α (RORα) in lysosomal acidification and autophagic flux, particularly in nutrition‐enriched hepatocytes. First, lysosomal acidity was much lower in the hepatocytes obtained from hepatocyte‐specific RORα‐deleted (RORα‐LKO) mice, whereas the infusion of an adenovirus encoding RORα in wild‐type hepatocytes increased lysosomal acidity, as determined by LysoSensor. Second, the lysosomal translocation of the mechanistic target of rapamycin was increased and immature cathepsin D was accumulated in the liver of RORα‐LKO mice. Third, the accumulation of LC3‐II, p62/sequestosome 1 (SQSTM1), and neighbor of BRCA1 gene 1 (NBR1) was increased in the livers of RORα‐LKO mice, indicating an impaired autophagic flux in the livers. Consistently, the number of autolysosomes containing mitochondria and lipid droplets was dramatically reduced in the RORα‐deleted hepatocytes. Finally, we found that RORα induced the transcription of genes involved in lysosomal function, such as Atp6v1g1, a vacuolar H+‐ATPase (v‐ATPase) subunit, which were largely down‐regulated in the livers of mice with high‐fat diet–induced NAFLD and patients with hepatitis. Conclusion: Targeting RORα may be a potential therapeutic strategy to restore lysosomal acidification, which inhibits the progression of NAFLD.
We identified a novel function of RORα that enhanced lysosomal acidification and autophagic flux through the transcriptional activation of Atp6v1g1. Thus, targeting RORα may be a potential therapeutic strategy to restore lysosomal acidification, which inhibits the progression of NAFLD.

Abbreviations
- Ad‐RORα
adenovirus‐encoding RORα
- BSA
bovine serum albumin
- ChIP
chromatin immunoprecipitation
- Ctsd
cathepsin D
- DMEM
Dulbecco’s modified Eagle’s medium
- EM
electron microscopy
- EV
empty vector
- GFP
green fluorescent protein
- GO
Gene Ontology
- HFD
high‐fat diet
- IF
immunofluorescence
- IgG
immunoglobulin G
- IHC
immunohistochemistry
- LFD
low‐fat diet
- LXRα
liver X receptor α
- MAP‐LC3
microtubule‐associated protein light chain 3
- mRNA
messenger RNA
- mTOR
mechanistic target of rapamycin
- NAFLD
nonalcoholic fatty liver disease
- NBR1
neighbor of BRCA1 gene 1
- PD
Parkinson’s disease
- RORα
retinoic acid‐related orphan receptor α
- RORα LKO
hepatocyte‐specific RORα knockout
- SQSTM1
sequestosome 1
- STRING
Search Tool of the Retrieval of Interacting Genes/Proteins
- TFEB
transcription factor EB
- v‐ATPase
vacuolar H+‐ATPase
- WD
Western diet
Nonalcoholic fatty liver disease (NAFLD) is a hepatic metabolic disease with a wide spectrum of abnormalities including triglyceride accumulation, increased oxidative stress, and inflammation. The prevalence of NAFLD has risen recently, with a worldwide increase in the incidence of diabetes and metabolic syndrome.( 1 ) Autophagy is an intracellular degradative process that is essential for cellular quality control and homeostasis. Substances subjected to decomposition are transported to lysosomes during the process of various types of autophagy, such as macroautophagy, chaperone‐mediated autophagy, and microautophagy.( 2 ) Macroautophagy, the most well‐known autophagic pathway, begins with the formation of an autophagosome membrane derived from the endoplasmic reticulum, Golgi apparatus, or mitochondria. Selective sequestration of a specific cargo into autophagosomal membranes is achieved through a physical interaction between the microtubule‐associated protein light chain 3 (MAP‐LC3; also referred to as LC3) and autophagy receptors, such as p62/sequestosome 1 (SQSTM1), neighbor of BRCA1 gene 1 (NBR1), and nuclear dot protein 52 kDa. The targets of selective autophagy include aggregated proteins (aggrephagy), mitochondria (mitophagy), and lipids (lipophagy).( 3 ) Recently, abnormality in autophagic function was indicated as the fundamental cellular defect involved in the pathogenesis of NAFLD. The hepatic expression and serum levels of p62/SQSTM1 were significantly induced in patients with NAFLD.( 4 ) The aggregation of the p62/SQSTM1 protein was significantly correlated with the clinical symptoms of NAFLD.( 5 , 6 ) In experimental animals, the enhancement of the autophagic flux protected against palmitate lipotoxicity in murine hepatocytes.( 7 ) In addition, liver injury and inflammation were increased when hepatic autophagy was reduced by genetic knockout of autophagy‐related 5 in a mouse model.( 8 )
The degradative ability of the autophagic pathway is afforded by lysosomes.( 2 ) The catabolic function of lysosomes is exerted through diverse hydrolytic enzymes, such as proteases, glycosidases and lipases, which are activated in an acidic environment of about pH 4.5 driven by vacuolar H+‐ATPase (v‐ATPase).( 9 ) Therefore, maintaining the acidity of lysosomes is essential for their successful digestive function in the autophagic pathway. Recently, abnormalities in lysosomal acidification accompanied by dysfunction of the autophagic flux were observed in several in vitro and in vivo experiments. Inami et al. showed that impaired autophagosomal acidification was accompanied by a decrease in autophagic proteolysis in hepatocytes from ob/ob mice with hepatic steatosis.( 10 ) Disruption of lysosomal acidification led to protease inactivation in the livers of mice with NAFLD induced by methionine‐choline–deficient diet feeding.( 4 ) Cholesterol loading in Huh7 cells, a human hepatoma cell line, impaired lysosomal acidification and promoted exosome release.( 11 ) Moreover, increasing clinical evidence supported the association between lysosome function and NAFLD. The number of lipid droplet–loaded lysosomes and the expression levels of lysosomal genes were increased with the increase in the NAFLD activity scores in patients with NAFLD.( 6 ) In addition, abnormal expression of cathepsin was observed in hepatocytes from patients with NAFLD.( 5 ) Although these observations indicate the importance of lysosomal acidification in the pathogenesis of NAFLD, the molecular and cellular mechanisms that regulate lysosomal acidity during the progression of the disease are not well understood.
The retinoic acid receptor–related orphan receptor α (RORα) is a transcription factor that plays a protective role against the progression of NAFLD through the modulation of mitochondrial dynamics, lipotoxicity, and liver macrophage polarization, as well as the up‐regulation of microRNA122.( 12 , 13 , 14 ) We observed an accumulation of abnormally swollen mitochondria with structural defects and lipid droplets in the hepatocytes of RORα‐deficient mice with high‐fat diet (HFD)‐induced nonalcoholic steatohepatitis (NASH).( 14 ) Thus, we hypothesized that RORα affects autophagic degradation in hepatocytes. Here, we demonstrated that lysosomal acidification and autophagic activity were defective in the liver of HFD‐fed hepatocyte‐specific RORα knockout (RORα‐LKO) mice. Moreover, we found that RORα regulates the expression of a group of genes encoding proteins that are important for lysosomal function, including the Atp6v1g1 gene (which encodes a v‐ATPase subunit), suggesting that they represent one of the molecular mechanisms underlying the RORα‐induced lysosomal acidity and autophagic activity in hepatocytes.
Materials and Methods
Animal Studies
The RORαfl/fl (fl/fl) and the RORα‐LKO mice were described previously.( 14 ) The RORαfl/fl mutant embryo, which has loxP sites flanking exon 4 of the RORα gene, was provided from the Institut Clinique de la Souris (Illkirch, France). The mutant mouse was generated by in vitro fertilization (Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea). To generate the hepatocyte‐specific RORα knockout mice, RORαfl/fl mice were cross‐bred with AlbCre animals, which highly express Cre recombinase in hepatocytes by the albumin promoter (Jackson Laboratory, Bar Harbor, ME). Six‐week‐old male RORαfl/fl mice or RORα‐LKO mice were fed an HFD (D12492) or low‐fat diet (LFD) (D12450J; Research Diets, New Brunswick, NJ) for 12 weeks, as described previously.( 14 ) To establish diet‐induced NAFLD models, 7‐week‐old wild‐type (WT) littermates were fed Western diet (WD) (D12079B; Research Diets) or LFD (D12450J; Research Diets) for 21 weeks, and 6‐week‐old WT littermates were HFD (D12492; Research Diets) or LFD (D12450J; Research Diets) for 20 weeks. After feeding, liver tissues were excised, and cross sections of the left lobe of the liver were analyzed for protein or fixed in 10% neutral buffered formalin (Sigma‐Aldrich, St. Louis, MO) for immunofluorescence (IF), and immunohistochemistry (IHC). For IF and IHC, 3‐μm sections of paraffin‐embedded tissue or primary hepatocytes were stained with anti–mechanistic target of rapamycin (mTOR) (66888‐1‐Ig; ProteinTech, Rosemont, IL), anti‐Lamp1 (L1418; Sigma‐Aldrich), anti‐SQSTM1/p62 (23214; Cell Signaling Technology, Danvers, MA), and anti‐Atp6v1g1 (sc‐25333; Santa Cruz Biotechnology, Inc., Dallas, TX). All experiments were performed in a blinded and randomized fashion. The experimental protocols were approved by the Seoul National University Institutional Animal Care and Use Committee (SNU‐140424‐2‐10), and all experiments were conducted according to the committee’s guidelines.
Confocal Microscopy
Isolated hepatocytes were plated in a confocal dish (211350; SPL) with Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone Laboratories Inc., Logan, UT) containing 10% fetal bovine serum (FBS). For virus infusion, Ad‐GFP (green fluorescent protein) or Ad‐GFP‐RORα particles (multiplicity of infection = 20) were added 4 hours after seeding and incubated for 18 hours. The production and infusion of Ad‐GFP and Ad‐GFP‐RORα were performed as described previously.( 15 ) For the visualization of lysosomes, cells were stained with 200 nM LysoTracker Red DND‐99 (L7528; Invitrogen, Waltham, MA) or 5 μM LysoSensor Blue DND‐167 (L7533; Invitrogen). To study the proteolytic activity of lysosomes, cells were incubated with 20 μg/ml DQ Red BSA (D12051; Invitrogen) following manufacturer’s instructions.
To visualize LC3 puncta, hepatocytes were treated with an adenovirus carrying mCherry‐GFP‐LC3 or adenovirus expressing enhanced GFP–LC3 (multiplicity of infection = 30), as described previously.( 16 ) For palmitic acid treatment, after hepatocyte seeding, media were exchanged to DMEM without FBS for 12 hours. Subsequently, cells were exposed to DMEM with 0.5 mM palmitic acid conjugated with bovine serum albumin (BSA) for the indicated time. For the visualization of lysosomes, mitochondria, and lipid droplets, hepatocytes were stained with 100 nM LysoTracker Green DND‐26 (L7526; Invitrogen), 150 nM MitoTracker Red CMXRos (M7512; Invitrogen), or LipidTOX Red (H34476; Invitrogen) diluted in media at 1:200. Cells were then examined using a confocal microscope (LSM 700; Carl Zeiss Microscopy, Jena, Germany).
Electron Microscopy
Small tissue blocks were excised from the left lobe of the liver, and primary hepatocytes were isolated from 8‐10‐week‐old male C57BL/6 mice through the perfusion of livers with collagenase type IV (C5138; Sigma‐Aldrich), as described previously.( 17 ) After fixation overnight in 2.5% glutaraldehyde at 4°C and washing in 0.1 M phosphate buffer (pH 7.0), the fixed blocks or primary hepatocytes were postfixed in 1% osmium tetroxide, followed by en bloc staining with 0.5% uranyl acetate. After dehydration with graded ethanol, samples were embedded in Spurr’s resin (14300; Electron Microscopy Sciences, Inc., Hatfield, PA) and polymerized. Ultrathin sections were cut using an EM UC7 ultramicrotome (Leica Biosystems, Wetzlar, Germany) and examined using a Talos L120C transmission electron microscope (120 kV) (FEI, Brno, Czech).
Western Blotting and Quantitative Real‐Time Polymerase Chain Reaction
Western blotting was performed as described previously using specific antibodies against phospho‐mTOR (2971; Cell Signaling Technology), mTOR (2972; Cell Signaling Technology), cathepsin D (Ctsd) (sc‐377299; Santa Cruz Biotechnology), Lamp1 (L1418; Sigma‐Aldrich), LC3 (L8918; Sigma‐Aldrich), NBR1 (9891; Cell Signaling Technology), RORα (sc‐6062; Santa Cruz Biotechnology), Hsp60 (ab45134; Abcam, Cambridge, United Kingdom), and Actin (sc‐1616; Santa Cruz Biotechnology).( 15 ) Relative messenger RNA (mRNA) expression was determined by quantitative real‐time PCR using the ABI StepOnePlus Real‐Time PCR system (Applied Biosystems, Foster City, CA) using specific primers (Supporting Table S2). The mRNA expression of genes was calculated relative to controls using the 2–ΔΔCT method.( 17 )
Human Tissue Array
Liver disease spectrum tissue array (catalog LV20812a) was purchased from US Biomax (Rockville, MD). We analyzed and compared the chronic hepatitis cores with normal tissue cores, which showed no features of inflammation and fatty degeneration. The chronic hepatitis cores, with or without fatty degeneration, showed inflammatory lesions without hepatitis B virus infection. Detailed information on patients can be found on the US Biomax website (biomax.us). The slides were incubated with mouse anti‐Lamp2 (sc‐18822; Santa Cruz Biotechnology) and rabbit anti‐mTOR (2983; Cell Signaling Technology) antibodies. The slides were then incubated with the secondary antibodies anti‐rabbit immunoglobulin G (IgG) Alexa555 (A31572; Invitrogen) and anti‐mouse IgG Alexa488 (A21200; Invitrogen). The stained tissue samples were examined with by confocal microscope (LSM 700; Carl Zeiss Microscopy). For IHC, the liver tissues were incubated with anti‐Atp6v1g1 (sc‐25333; Santa Cruz Biotechnology).
Statistical Analysis
All analyses were performed using the GraphPad Prism software (GraphPad Software, San Diego, CA). Statistical analyses between two groups were conducted using the nonparametric Mann‐Whitney U test (two‐tailed). Data are presented as the mean ± SEM. Statistical significance was set at P < 0.05.
Results
RORα Enhances Lysosomal Acidity in Mouse Hepatocytes
We reported previously that RORα‐LKO mice were more susceptible to HFD‐induced steatohepatitis( 14 ) (Supporting Fig. S1). Excessive fat accumulation and abnormally swollen mitochondria with an irregular membrane structure were observed frequently in the hepatocytes of HFD‐fed RORα‐LKO mice.( 14 ) Thus, we hypothesized that lysosomal dysfunction is involved in the abnormalities detected in RORα‐deleted hepatocytes. To address this issue, first we examined lysosomal acidity in hepatocytes isolated from the livers of RORαfl/fl and RORα‐LKO mice using LysoSensor (pKa 5.1), the fluorescence of which increases in acidic environments. Surprisingly, the intensity of the LysoSensor fluorescence was much lower in the RORα‐deleted compared with RORαfl/fl hepatocytes (Fig. 1A). Furthermore, the infusion of an adenovirus‐encoding RORα (Ad‐RORα) enhanced the fluorescence of LysoSensor in primary hepatocytes (Fig. 1B). The LysoSensor fluorescence in Ad‐RORα‐infused cells was disappeared when the cells were treated with bafilomycin A1, a lysosomal v‐ATPase inhibitor, supporting the specificity of LysoSensor fluorescence as an indicator of lysosomal acidification (Supporting Fig. S2A). When a fluorogenic substrate of lysosomal enzymes, DQ Red BSA, was treated, the red fluorescence was becoming strong following Ad‐RORα infusion, indicating that lysosomal proteolytic activity was enhanced in Ad‐RORα‐infused hepatocytes (Fig. 1C). Furthermore, the decrease in LysoSensor fluorescence in RORα‐LKO hepatocytes was recovered when RORα was restored by infusion of Ad‐RORα (Supporting Fig. S2B). Taken together, these results indicate that RORα enhanced lysosomal acidity in hepatocytes.
FIG. 1.
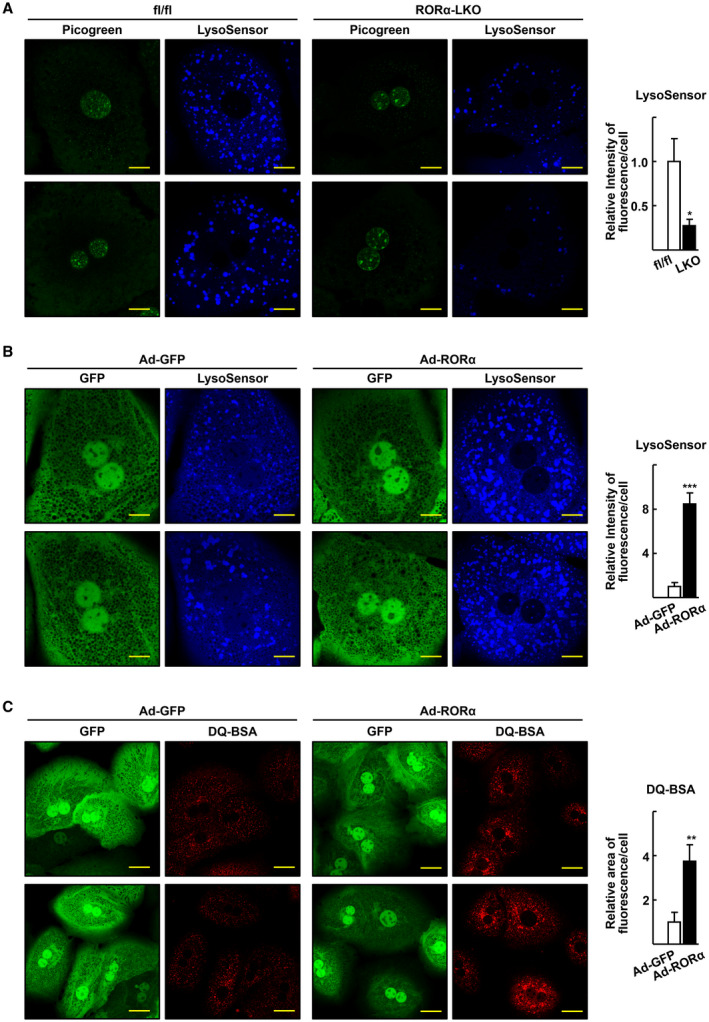
RORα enhances lysosomal acidification in the hepatocytes. (A) Confocal microscopy was performed with primary hepatocytes obtained from the RORαfl/fl (fl/fl) and RORα‐LKO (LKO) and stained using LysoSensor Blue DND‐167. Picogreen was used to stain the nucleus. Representative images of hepatocytes are presented. Fluorescence intensity was quantified in at least 100 cells using ImageJ. Scale bar: 10 μm. *P < 0.05 versus fl/fl. (B) After mouse primary hepatocytes were infected by either Ad‐GFP or Ad‐RORα for 18 hours, cells were stained with LysoSensor Blue DND‐167 and subjected to confocal microscopy. Representative images of hepatocytes are presented. Fluorescence intensity was quantified in at least 100 cells using ImageJ software. Scale bar: 10 μm. ***P < 0.001 versus Ad‐GFP‐infused hepatocytes. (C) The virus‐infused hepatocytes were incubated with DQ Red BSA 20 μg/mL for 16 hours and subjected to confocal microscopy. Representative images are shown. Fluorescent area was quantified in at least 100 cells using ImageJ software. Scale bar: 25 μm. **P < 0.01 versus Ad‐GFP.
Lysosomal Function Is Impaired in the Liver of RORα‐LKO Mice
Next, we examined the lysosomal dysfunction in the livers of RORα‐LKO mice. First, we performed an IF study to examine the expression pattern of the mTOR, because constitutive lysosomal association and aberrant accumulation of mTOR represent a loss of lysosomal function.( 18 , 19 ) We observed that the pattern of mTOR staining in the liver sections of HFD‐fed RORαfl/fl was diffused and that a portion of mTOR staining was co‐localized with Lamp1, a lysosomal surface protein. In contrast, mTOR staining exhibited a punctate pattern, and the areas that co‐localized with Lamp1 were increased in the liver sections of HFD‐fed RORα‐LKO mice (Fig. 2A). The ratio of the immature versus the mature form of the Ctsd protein represents lysosomal acidity, because maturation of Ctsd occurs at the lowest end of the lysosomal pH range.( 20 ) We found that both the mature and immature Ctsd proteins were significantly up‐regulated, and that the ratio of immature versus mature Ctsd was significantly increased in the liver of RORα‐LKO mice (Fig. 2B). Similarly, the mTOR protein was significantly up‐regulated in the lysosomes isolated from the liver of RORα‐LKO mice, whereas the Lamp1 protein was not significantly altered (Fig. 2C). These data indicate that lysosomal dysfunction was apparent in RORα‐deleted livers after HFD feeding. We observed a similar punctate pattern of mTOR expression and accumulation of the mTOR protein in the liver tissues or lysosomes isolated from mice fed with a WD or HFD, indicating that lysosomal dysfunction is present in various mouse models of NAFLD (Supporting Fig. S3).
FIG. 2.
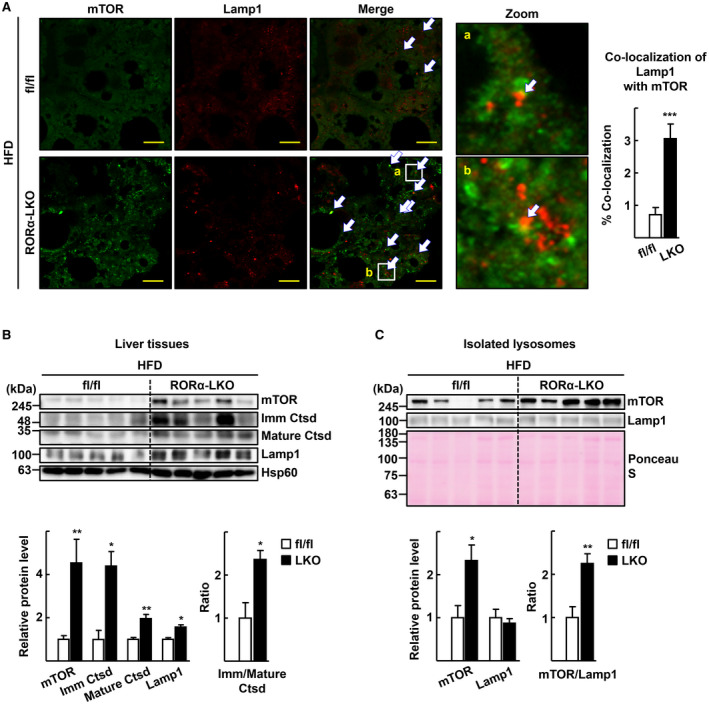
Deletion of RORα results in dysfunction of lysosomes in the liver. Six week‐old RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice were fed with HFD for 12 weeks. (A) Mouse liver tissues were subjected to immunostaining for mTOR (green) and Lamp1 (red) and examined by confocal microscopy. Representative images are shown. Arrows indicate co‐localization of mTOR with Lamp1. Percentage of co‐localization was determined using the JACoP plugin of ImageJ software in three different sections for each liver tissue. Scale bar: 10 μm. ***P < 0.001 versus fl/fl (n = 4). (B) Tissue extracts from mouse liver were subjected to western blotting to analyze expression level of proteins. Densitometry was performed using ImageJ software and normalized to that of Hsp60. The numbers represent the relative protein level of RORα‐LKO livers when the level of fl/fl livers was considered as 1. The ratio of Immature (Imm) Ctsd/Mature Ctsd was determined using the protein levels of two different forms of Ctsd that were normalized with Hsp60, and the ratio of fl/fl livers was considered as 1. *P < 0.05 and **P < 0.01 versus fl/fl (n = 5). (C) Lysosomal fractions were prepared from liver tissues of HFD‐fed RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice. The indicated proteins were analyzed by western blotting. Membrane was stained with Ponceau S as a loading control. Band intensities of each protein were quantified using ImageJ software and normalized to that of Ponceau S. The numbers represent the relative protein levels of RORα‐LKO livers when the level of fl/fl livers was considered as 1. The ratio of mTOR/Lamp1 was determined using the protein level of each protein, and the ratio of fl/fl livers was considered as 1. *P < 0.05 and **P < 0.01 versus fl/fl (n = 5).
Also, we observed that the intensity of LysoSensor staining was lowered significantly in the RORα‐LKO hepatocytes, whereas that of LysoTracker staining remained the same (Supporting Fig. S4A). These data suggested that the lysosome acidification, but not the amounts of lysosomes, was defected in the RORα‐LKO hepatocytes. To support this finding, we evaluated the expression level of transcription factor EB (TFEB) protein, a master regulator of lysosomal biogenesis. The levels of TFEB and its targets, Atp6v1a and Atp6v1d, were not significantly different in the liver tissues from RORαfl/fl and RORα‐LKO mice (Supporting Fig. S4B).( 21 ) Together, these results indicated that lysosomal acidification rather than lysosomal biogenesis was involved in the RORα‐associated enhancement of lysosomal function.
Defects in Autophagy Flux in the Liver of RORα‐LKO Mice
We investigated whether the lack of adequate lysosomal acidification led to impaired autophagy in the liver of RORα‐LKO mice. An IHC study showed that the aggregation of p62/SQSTM1 was remarkably increased in the liver of RORα‐LKO mice (Fig. 3A). Moreover, obvious accumulation of LC3‐II and NBR1 was observed in the liver of RORα‐LKO mice, indicating a decrease in the autophagic flux in the liver of RORα‐LKO mice (Fig. 3B). Electron microscopy (EM) revealed differences between the hepatocytes obtained from RORαfl/fl and RORα‐LKO mice after palmitic acid treatment. RORαfl/fl hepatocytes exhibited many autolysosomes in various states of degradation containing partially disrupted membrane‐derived materials. In contrast, the number of autolysosomes was remarkably reduced in RORα‐deleted hepatocytes (Fig. 3C). To support the blockade of the autophagic flux, autolysosomes were visualized using the mCherry‐GFP‐LC3 reporter. In this reporter system, GFP fluorescence was quenched in autolysosomes fused with lysosomes because of a low pH, whereas mCherry fluorescence was affected to a lesser extent.( 22 ) We observed a lower level of formation of both autolysosomes (red in the merged image) and autophagosomes (yellow in the merged image) in the hepatocytes of RORα‐LKO compared with RORαfl/fl mice (Fig. 3D).
FIG. 3.
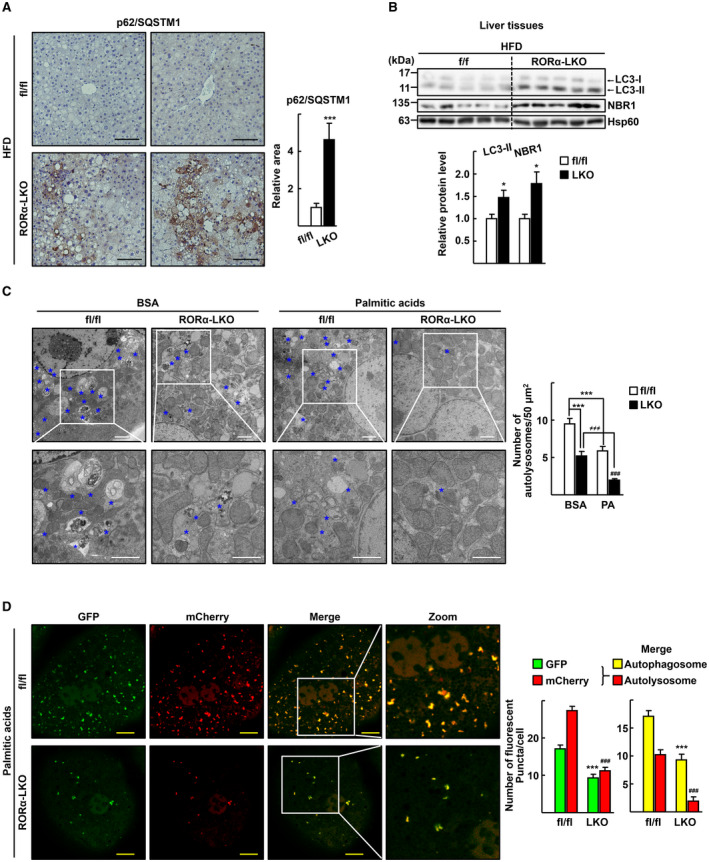
Depletion of RORα impairs autophagic flux in the hepatocytes. (A) Expression of p62/SQSTM1 in the liver sections of the HFD‐fed RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice was visualized by IHC. Representative images are presented. The relative area of p62/SQSTM1 staining was quantified in at least two images from a liver tissue using ImageJ software. Scale bar: 100 μm. ***P < 0.001 versus fl/fl (n = 5). (B) Tissue extracts were prepared from liver of HFD‐fed RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice. Expression of the indicated proteins was analyzed by western blotting. Intensity of each protein band was quantified using ImageJ software and normalized to that of Hsp60. The numbers represent the relative protein level of RORα‐LKO livers when the level of fl/fl livers was considered as 1. *P < 0.05 versus fl/fl (n = 5). (C) Hepatocytes obtained from RORαfl/fl (fl/fl) or RORα‐LKO (LKO) were exposed to 0.5 mM palmitic acid conjugated with BSA or BSA for 1 hour, and then fixed in 2.5% glutaraldehyde. Representative EM images are shown. Blue stars indicate autolysosomes. The number of autolysosomes was counted in at least 20 cells. Scale bar: 2 μm. ***P < 0.001 versus BSA‐treated fl/fl; ### P < 0.001 versus palmitic acid‐BSA treated fl/fl; and ≠≠≠ P < 0.001 versus BSA‐treated LKO. (D) Hepatocytes obtained from RORαfl/fl (fl/fl) or RORα‐LKO (LKO) were transduced by Ad‐mCherry‐GFP‐LC3. After 18 hours, cells were treated with 0.5 mM palmitic acid conjugated with BSA for 1 hour. The autophagosomes (yellow in merge) and autolysosomes (red in merge) in at least 100 cells were counted. Scale bar: 10 μm. ***P < 0.001 versus GFP or autophagosome of fl/fl and ### P < 0.001 versus mCherry or autolysosome of fl/fl.
An electron microscopy study confirmed the decrease in autophagic activity in the liver tissues of RORα‐LKO mice. The liver sections of RORαfl/fl mice contained many autolysosomes surrounded by a single membrane with partially disrupted organelles in various sizes. Conversely, the number of autolysosomes was much lower in the liver sections of RORα‐LKO mice (Fig. 4A,C). In addition, lipophagy, or autophagy of lipid droplets, was observed frequently in the livers of RORαfl/fl, but not in those of RORα‐LKO mice (Fig. 4B). Consistently, confocal microscopy demonstrated a decrease in mitophagy and lipophagy in RORα‐LKO hepatocytes after treatment with palmitic acid. Staining of mitochondria and autolysosomes with MitoTracker and LysoTracker, respectively, revealed the capture of mitochondria by autolysosomes in RORαfl/fl cells, whereas this phenomenon was reduced in RORα‐LKO hepatocytes (Fig. 4D). The pattern of decrease in the number of co‐localized MitoTracker and LysoTracker staining in the RORαfl/fl and RORα‐LKO remained the same in the presence of leupeptin, a lysosomal protease inhibitor, indicating that lysosomal degradation did not affect the pattern of decrease (Supporting Fig. S5). Similarly, the number of autolysosomes capturing lipid droplets was reduced dramatically in RORα‐LKO hepatocytes (Fig. 4E). Moreover, the number of LC3 puncta co‐localized with mitochondria or lipid droplets was reduced in the palmitic acid–challenged RORα‐LKO hepatocytes (Fig. 4F,G). Together, these results indicated that mitophagy and lipophagy are impaired in the RORα‐LKO hepatocytes.
FIG. 4.
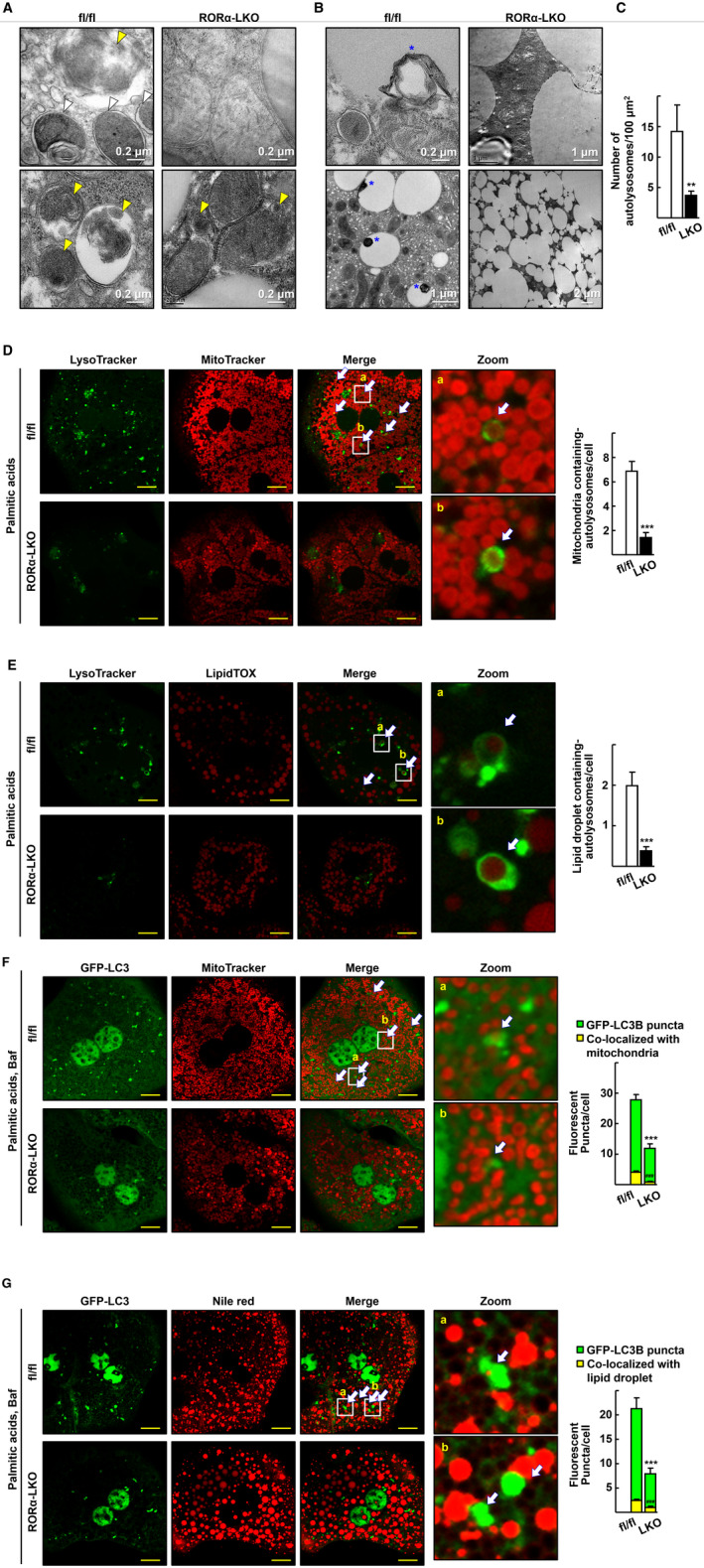
Mitophagy and lipophagy are impaired in the liver of RORα‐LKO mice. (A,B) Representative EM images of the liver sections from the HFD‐fed RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice. (A) White arrowheads indicate autophagosomes that contain clearly discernible mitochondria, and yellow arrowheads indicate autolysosomes that contain partially disrupted materials. (B) Blue stars indicate lipophagy, lipid droplets with multimembrane structures or a dug in lipid droplet. (C) The number of autolysosomes was counted in five different liver sections of each mouse. **P < 0.01 versus fl/fl (n = 4). (D) Hepatocytes obtained from the RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice were treated with 0.5 mM palmitic acids conjugated with BSA for 1 hour, stained with MitoTracker Red CMXRos and LysoTracker Green DND‐26, and subjected to confocal microscopy. The number of autolysosomes containing mitochondria, as indicated by white arrows, was counted in at least 100 cells. Scale bar: 10 μm. ***P < 0.001 versus fl/fl. (E) Hepatocytes obtained from the RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice were treated with 0.5 mM palmitic acids conjugated with BSA for 6 hours and stained with LipidTOX Red and LysoTracker Green DND‐26. The number of autolysosomes containing lipid droplets, as indicated by white arrows, was counted in at least 100 cells. Scale bar: 10 μm. ***P < 0.001 versus fl/fl. (F) Hepatocytes of RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice were transduced by Ad‐GFP‐LC3. After 18 hours, cells were exposed to 0.5 mM palmitic acids conjugated with BSA, and 30 nM bafilomycin A1 (Baf) for 1 hour and then stained using MitoTracker Red CMXRos for confocal microscopy. The number of mitochondria wrapped with LC3 puncta, as indicated by white arrows, was counted in at least 100 cells. Scale bar: 10 μm. ***,### P < 0.001 versus GFP‐LC3 puncta and puncta with mitochondria, respectively, in hepatocytes of fl/fl. (G) The virus‐infused hepatocytes of RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice were treated with 0.5 mM palmitic acid conjugated with BSA for 6 hours, and 30 nM bafilomycin A1 for 1 hour before examination of lipophagosome. Hepatocytes were then stained using Nile red for visualization of lipid droplets. The number of lipids wrapped with LC3 puncta, as indicated by white arrows, was counted in at least 100 cells. Scale bar: 10 μm. ***,### P < 0.001 versus GFP‐LC3 puncta and puncta with lipids, respectively, in hepatocytes of fl/fl.
The function of RORα in autophagic flux was further supported by the infusion of Ad‐RORα. The number of p62‐positive puncta and the level of NBR1 protein were greatly decreased by Ad‐RORα‐infusion in the palmitic acid–challenged hepatocytes (Fig. 5A,B). Consistently, electron microscopy demonstrated that Ad‐RORα infusion increased the number of autolysosomes (Fig. 5C). In addition, more LC3‐II was accumulated in Ad‐RORα‐infused hepatocytes in a classical LC3‐II turnover assay in the Earle’s balanced salt solution medium–mediated starvation system (Fig. 5D). Taken together, these data confirmed that autophagic flux was enhanced by RORα.
FIG. 5.
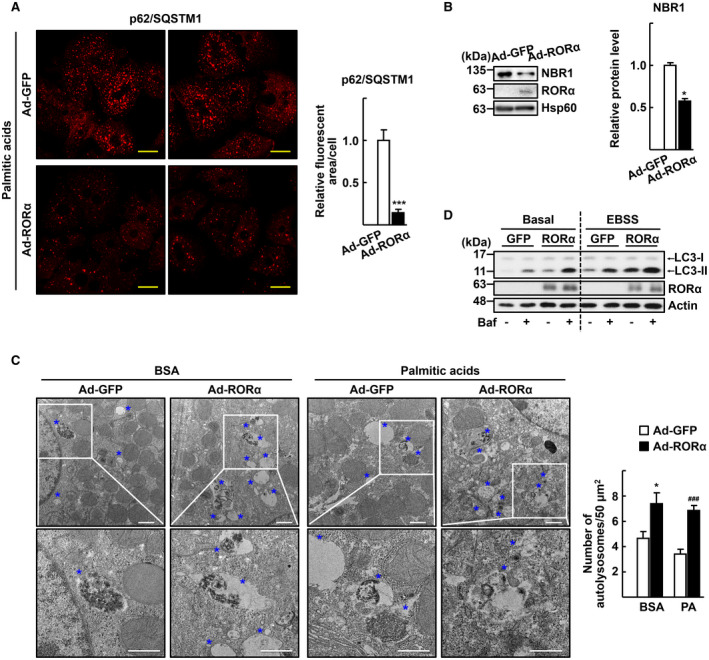
Infusion of Ad‐RORα enhances autophagic flux in the hepatocytes. Primary hepatocytes obtained from C57/BL6 mice were infected by either Ad‐GFP or Ad‐RORα for 18 hours. (A) The virus‐infused hepatocytes were exposed to 0.5 mM palmitic acids conjugated with BSA for 1 hour. Immunostaining was performed for p62/SQSTM1 (red) and examined by confocal microscopy. Representative images are shown. The relative area of fluorescence was quantified using Image J software in at least 100 cells. Scale bar: 25 μm. ***P < 0.001 versus Ad‐GFP. (B) The virus infused hepatocytes were exposed to 0.5 mM palmitic acids conjugated with BSA for 1 hour. Expression of the NBR1 protein was analyzed by western blotting. Band intensity of NBR1 was quantified using ImageJ software and normalized to that of Hsp60. *P < 0.05 versus Ad‐GFP (n = 4). (C) The virus‐infused hepatocytes were exposed to 0.5 mM palmitic acids conjugated with BSA for 1 hour and then subjected to EM. Representative EM images with blue stars indicating autolysosomes are shown. The number of autolysosomes was counted in at least 20 cells. Scale bar: 1 μm. *P < 0.05 versus Ad‐GFP with BSA and ### P < 0.001 versus Ad‐GFP with palmitic acid BSA. (D) The virus‐infused hepatocytes were exposed to Earle’s balanced salt solution for 1 hour with or without 30 nM bafilomycin A1 for examination of autophagy flux. Accumulation of LC3‐II protein was analyzed by western blotting.
RORα Induces the Transcription of Lysosomal Genes Including Atp6v1g1
To identify the mechanism through which RORα induced lysosomal acidification at the molecular level, we deduced a list of 68 lysosomal genes by intersecting the overlapping target genes delineated by our previous RNA‐sequencing analysis (GSE90844), the publicly available chromatin immunoprecipitation (ChIP)–sequencing data set (GSE59486), and the group of genes in the lysosome‐related Gene Ontology (GO) (GO:0005764)( 14 , 23 ) (Fig. 6A). To define the lysosomal core genes that are under RORα regulation, the 68 lysosomal genes were input into the Search Tool of the Retrieval of Interacting Genes/Proteins (STRING) database resource.( 24 ) The STRING protein interaction network showed that the genes related to the biological processes of “vesicle‐mediated transport,” “cellular catabolic process,” “homeostatic process,” and “autophagy” were associated with hepatic RORα deletion (false discovery rate ≤ 0.0001) (Fig. 6B and Supporting Table S1).
FIG. 6.
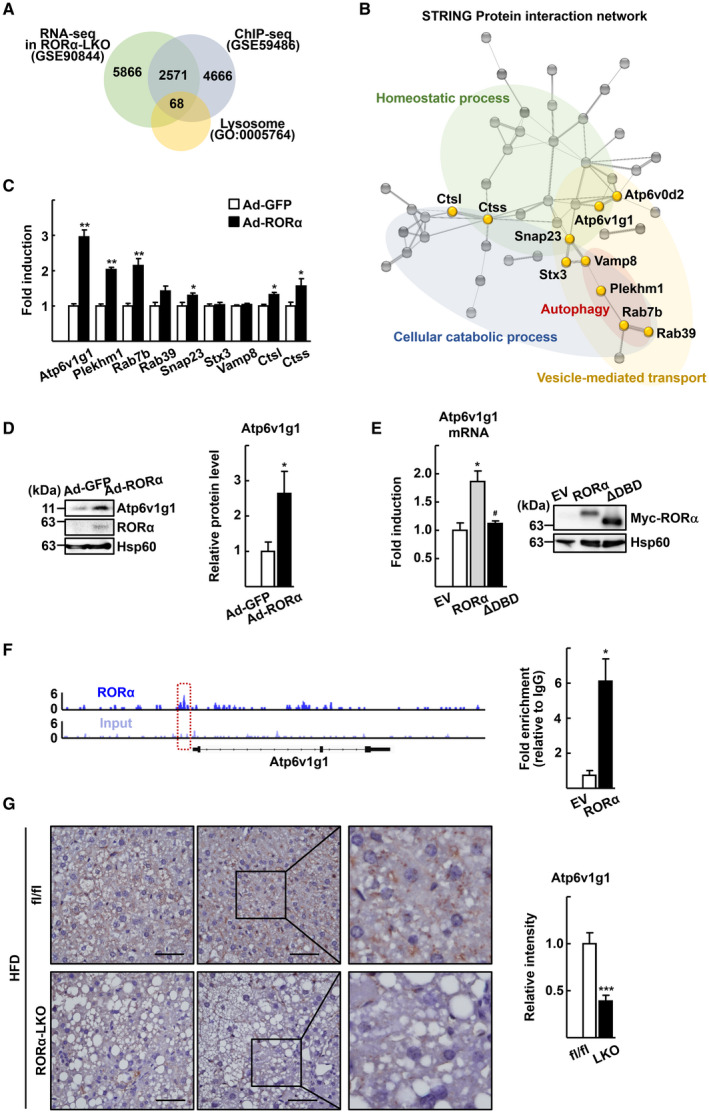
RORα increases expression level of Atp6v1g1. (A) Venn diagram showing the overlapping target genes—those delineated by the RNA‐sequencing (RNA‐seq) analysis (GSE90844), the publicly available ChIP‐sequencing (ChIP‐seq) data set (GSE59486), and lysosomal genes in the GO (GO:0005764). A list of 68 lysosomal genes was deduced. (B) An interaction network was obtained from the list of 68 genes by the STRING database. Genes commonly included in top‐ranked GO biological process are colored in yellow. (C) Primary hepatocytes obtained from C57/BL6 mice were infected by either Ad‐GFP or Ad‐RORα for 18 hours. Total RNA was isolated, and the mRNA level of the indicated genes was measured by quantitative real‐time PCR. *P < 0.05 and **P < 0.01 versus Ad‐GFP (n = 6). (D) Primary hepatocytes obtained from C57/BL6 mice were infected by either Ad‐GFP or Ad‐RORα for 18 hours. Whole cell lysates were prepared, and the expression level of Atp6v1g1 was analyzed by western blotting. Intensity of each protein band was quantified using ImageJ software and normalized to that of Hsp60. *P < 0.05 versus Ad‐GFP (n = 4). (E) Primary hepatocytes were transfected with empty vector (EV) or the expression vector encoding Myc‐RORα (RORα) or Myc‐RORα ΔDBD (ΔDBD) for 18 hours. Total RNA was isolated, and the mRNA level of Atp6v1g1 was measured by quantitative real‐time PCR. Protein level of Myc‐RORα and Myc‐RORα ΔDBD was analyzed by western blotting using anti‐Myc antibody as control. *P < 0.05 versus EV and # P < 0.05 versus Myc‐RORα (n = 4). (F) The binding of RORα to the regulatory region of Atp6v1g1 gene. The ChIP‐sequencing reads of RORα (upper) and liver input control (lower) in the genome loci of Atp6v1g1 are shown in ChIP‐seq tracks. The red box indicates the region where the RORα signal is significantly enriched. A line below the ChIP‐seq tracks represents the transcript of the gene. Primary hepatocytes were transfected with EV or the expression vector encoding Myc‐RORα (RORα). DNA fragments were immunoprecipitated with the anti‐Myc antibodies and then amplified by PCR with specific primers. *P < 0.05 versus EV (n = 4). (G) Expression of Atp6v1g1 in the liver sections of the HFD‐fed RORαfl/fl (fl/fl) and RORα‐LKO (LKO) mice was visualized by IHC. Representative images are presented. The intensity of Atp6v1g1 staining was quantified in at least four images for a liver tissue using ImageJ software. Scale bar: 50 μm. ***P < 0.001 versus fl/fl (n = 4).
We analyzed the transcriptional level of the genes included in the top‐ranked GO biological processes using quantitative real‐time PCR. Among the genes tested, Atp6v1g1, which encodes a component of the peripheral stalk of v‐ATPase, showed the highest induction after the infusion of Ad‐RORα (Fig. 6C). Consistently, RORα overexpression significantly increased the expression of the Atp6v1g1 protein in mouse hepatocytes (Fig. 6D). However, the RORα ΔDBD, which lacked the DNA‐binding domain, did not induce the transcription of Atp6v1g1, indicating that the transcriptional function of RORα was required for the induction of the gene (Fig. 6E). To understand the mechanism how RORα regulates Atp6v1g1 gene expression, we searched RORα‐binding signals in the regulatory region of Atp6v1g1 gene using ChIP‐sequencing data available in public (GSE59486). We found a reads‐enriched signal in the 5’ region of the Atp6v1g1 gene and a ChIP assay confirmed that RORα bound to the region, which indicated that RORα directly regulates transcription of Atp6v1g1 (Fig. 6F). IHC staining showed that the Atp6v1g1 protein was expressed in a punctate pattern in the liver of HFD‐fed RORαfl/fl mice, whereas it was significantly reduced in the liver of RORα‐LKO mice, suggesting that RORα contributes to the enhancement of lysosomal acidity by up‐regulating Atp6v1g1 in the liver (Fig. 6G). Furthermore, the level of the Atp6v1g1 protein was lower in the livers of WD‐fed mice compared with those of control diet–fed mice, suggesting that defects in the maintenance of Atp6v1g1 levels may be associated with the development of NASH in mouse models (Supporting Fig. S6).
Down‐regulation of Atp6v1g1 Accompanies Lysosomal Dysfunction in the Livers of Human Patients With Hepatitis
Finally, we examined the relevance of our findings in human NAFLD. The translocation of mTOR to lysosomes was increased significantly in the human liver specimens studied here, indicating that lysosomal dysfunction was also present in the livers of human patients with hepatitis (Fig. 7A). Moreover, an IHC study showed that the expression level of Atp6v1g1 was reduced significantly in the liver tissues of these patients (Fig. 7B). Taken together, these results suggest that impairment of lysosomal acidification is closely associated with the down‐regulation of Atp6v1g1 in patients with NAFLD.
FIG. 7.
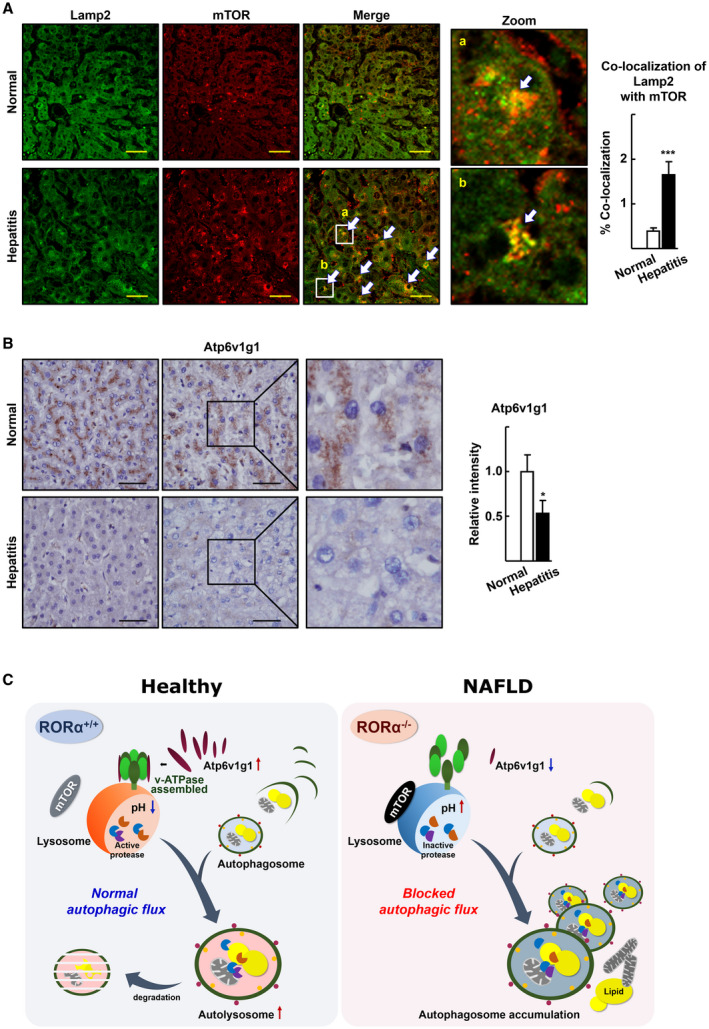
mTOR translocation to lysosomes and decreased expression of Atp6v1g1 in the livers of patients with hepatitis. (A) Liver specimens of patients with hepatitis were examined for the expression pattern of mTOR (red) and Lamp2 (green) using IF and subsequent confocal microscopy. Representative images are shown. Arrows indicate co‐localization of mTOR with Lamp1. Percentage of co‐localization was determined using the JACoP plugin of ImageJ software in two different images for each tissue specimen. Scale bar: 50 μm. ***P < 0.001 versus normal (n = 12 for normal, n = 9 for hepatitis). (B) Expression of Atp6v1g1 in the liver specimens of patients with hepatitis was visualized by IHC. Representative images are presented. The intensity of Atp6v1g1 staining was quantified in two images for a liver specimen using ImageJ software. Scale bar: 50 μm. *P < 0.05 versus normal (n = 12 for normal, n = 9 for hepatitis). (C) Schematic model for gain and loss of function of RORα in lysosomal acidification and autophagic flux.
Discussion
The acidic pH of lysosomes afforded by v‐ATPase leads to the activation of hydrolytic enzymes, thus enabling the successful lysosomal digestion of complex substrates, such as mitochondria and deleterious products.( 9 ) Although the importance of lysosomal acidity in the pathogenesis of NAFLD has been highlighted recently, the regulatory mechanisms of lysosomal acidification in the high fat–loaded liver are not well understood. Here, we discovered that Atp6v1g1, one of v‐ATPase subunits, is associated with defects in lysosomal acidification and autophagic flux in the liver (Fig. 7C). v‐ATPase is a multimeric protein complex consisting of two core subcomplexes: the cytosolic V1 complex, an ATP‐driven rotor, and the membrane‐bounded V0 complex, containing the proton pore.( 25 ) Atp6v1g1 is a V1 subunit that is necessary to structurally connect the V1 sector to the V0 sector, thus forming peripheral stator stalks.( 26 ) A proper level and adequate posttranslational modification of the Atp6v1g1 protein were shown to be essential for the function of the proton pump.( 27 , 28 ) In our study, the protein level of Atp6v1g1 was decreased significantly in the liver of HFD‐fed RORα‐LKO mice and in the liver of human patients with hepatitis (Figs. 6G and 7B). Therefore, we suspect that the down‐regulation of the Atp6v1g1 protein in RORα knockout may lead to the inadequate assembly of v‐ATPases, resulting in poor acidification of lysosomes. Similarly, the reduced level of other subunits of the V1 sector (i.e., Atp6v1a, Atp6v1b, and Atp6v1d) was reported in the liver of obese KKAy mice with steatosis.( 29 ) Interestingly, mutations in the Atp6ap1 and Atp6ap2 genes, the products of which are two accessory proteins of the v‐ATPase complex, were found in the patients with NAFLD.( 30 , 31 ) In addition, genetic mutations were identified in TMEM199, CCDC115, and VMA21, factors required for assembly of v‐ATPase, of the hepatic steatosis patients with increased liver enzymes.( 32 , 33 , 34 ) The molecular details underlying the manner in which these mutations affect the normal function of v‐ATPase and the pathogenesis of NAFLD should be investigated further.
Autophagy is a process that proceeds in the order of phagophore initiation and elongation, autophagosome closure, autophagosome−lysosome fusion, and lysosomal degradation. To date, many studies showed defects in autophagy processes in NAFLD pathogenesis, in particular, in the early and intermediate steps of autophagy (i.e., autophagosome formation and autophagosome−lysosome fusion), which are under the control of mTOR and of TFEB.( 2 , 7 , 8 , 35 , 36 , 37 ) We observed that RORα also functions to regulate autophagosome formation, as the number of autophagosome puncta was decreased in the hepatocytes of RORα‐LKO mice (Fig. 3D). Even though lysosome fusion was blocked using bafilomycin A1, the LC3‐II level was still lower in the RORα‐LKO hepatocytes, indicating decreased autophagosome formation in the absence of RORα (Fig. 4F,G). Liver X receptor α (LXRα) could be a regulator of the early step of the autophagic process in the liver, as it inhibits lipophagy by down‐regulating ATG4B and Rab‐8B, which are involved in autophagosome formation and autophagosome−lysosome fusion, respectively.( 36 ) In a previous study, we showed that RORα inhibits the transcriptional function of LXRα; thus, it is suspected that RORα enhances autophagosome formation by inhibiting LXRα.( 15 ) Taken together, these data indicate that RORα may play a role in multifaceted processes of autophagy (such as autophagosome formation and autophagosome−lysosome fusion), rather than in lysosomal degradation exclusively, thereby providing proper cellular energy homeostasis to prevent the progression of NAFLD in conditions of overnutrition.
mTOR, a serine/threonine kinase, is a master regulator of cellular metabolism, particularly of macroautophagy.( 38 ) In this investigation, we found that the suppression of mTOR signaling was one of the important mechanisms underlying the RORα‐induced autophagy flux in the liver. When nutrient supply is sufficient, mTOR is tethered to the lysosome surface through the lysosomal v‐ATPase−Ragulator complex, to generate active signaling.( 39 ) Thus, the lysosomal accumulation of mTOR proteins in RORα‐deleted hepatocytes may indicate hyperactivation of mTOR because of the impairment of lysosomal degradation in the absence of RORα (Fig. 2).( 19 ) The RORα‐induced acidification of lysosomes may contribute to the degradation of mTOR, which blocks further activation of mTOR signaling. Previously, we reported that RORα suppressed mTOR activity by enhancing the activity of adenosine monophosphate–activated protein kinase in the liver of HFD‐fed mice.( 15 ) Thus, it appears that RORα targets mTOR function through at least two different pathways. Most importantly, mTOR is an emerging pharmacological target in the manipulation of autophagy and related human diseases.( 40 ) Several mTOR inhibitors are currently undergoing preclinical studies to test their usefulness for autophagy induction.( 35 , 40 ) However, significant side effects, such as alteration of insulin signaling and hyperlipidemia, were reported after Sirolimus (Rapamycin) and Everolimus, which are allosteric inhibitors of mTOR, and applied for immune suppression in transplant recipients.( 41 , 42 ) Our results suggest that strategies using RORα and its ligand may provide a potential way to target mTOR for the treatment of autophagy‐related metabolic diseases, including NAFLD.
Lysosomal dysfunction is closely associated not only with NAFLD, but also with other degenerated diseases, such as Alzheimer’s disease, Parkinson’s disease (PD), and amyotrophic lateral sclerosis.( 43 ) Recently, altered acidification of endosomes accompanied by down‐regulation of Atp6v1g1 was reported in primary fibroblasts from a young patient with a juvenile form of PD.( 44 ) In addition, a decrease in the expression of the Atp6v1g1 gene, together with an acidification defect, was observed in the hippocampus and spinal cord of a mouse model of ALS.( 45 ) Interestingly, it has been reported that the abnormal processing of the Rora RNA and Rora loci was associated with the development of ALS and PD.( 46 , 47 ) Thus, the involvement of RORα and Atp6v1g1 in the onset of neurodegenerative diseases, as well as NAFLD, warrants further investigation.
In conclusion, the function of RORα described here (i.e., the enhancement of lysosomal acidification and autophagic flux) may provide a therapeutic strategy to restore lysosomal function, by inhibiting the progression of lysosome‐linked degenerative and metabolic diseases, including NAFLD.
Supporting information
Supplementary Material
Supported by the National Research Foundation of Korea (2014M3A9D5A01073556, 2017R1A2B3011870, and 2018R1A5A2024425).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yim WW‐Y, Mizushima N. Lysosome biology in autophagy. Cell Discov 2020;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014;16:495‐501. [DOI] [PubMed] [Google Scholar]
- 4. Wang X, Zhang X, Chu ESH, Chen X, Kang W, Wu F, et al. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J 2018;32:37‐51. [DOI] [PubMed] [Google Scholar]
- 5. Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non‐alcoholic fatty liver disease. Hepatol Res 2014;44:1026‐1036. [DOI] [PubMed] [Google Scholar]
- 6. Carotti S, Aquilano K, Zalfa F, Ruggiero S, Valentini F, Zingariello M, et al. Lipophagy impairment is associated with disease progression in NAFLD. Front Physiol 2020;11:850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mwangi SM, Li GE, Ye L, Liu Y, Reichardt F, Yeligar SM, et al. Glial cell line‐derived neurotrophic factor enhances autophagic flux in mouse and rat hepatocytes and protects against palmitate lipotoxicity. Hepatology 2019;69:2455‐2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen Y, Malik SA, Amir M, Kumar P, Cingolani F, Wen J, et al. Decreased hepatocyte autophagy leads to synergistic IL‐1β and TNF mouse liver injury and inflammation. Hepatology 2020;72:595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 2019;21:133‐142. [DOI] [PubMed] [Google Scholar]
- 10. Inami Y, Yamashina S, Izumi K, Ueno T, Tanida I, Ikejima K, et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Bioph Res Co 2011;412:618‐625. [DOI] [PubMed] [Google Scholar]
- 11. Zhao Z, Zhong L, Li P, He K, Qiu C, Zhao L, et al. Cholesterol impairs hepatocyte lysosomal function causing M1 polarization of macrophages via exosomal miR‐122‐5p. Exp Cell Res 2020;387:111738. [DOI] [PubMed] [Google Scholar]
- 12. Chai C, Cox B, Yaish D, Gross D, Rosenberg N, Amblard F, et al. Agonist of RORA attenuates non‐alcoholic fatty liver progression in mice via upregulation of microRNA 122. Gastroenterology 2020;159:999‐1014.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han Y‐H, Kim H‐J, Na H, Nam M‐W, Kim J‐Y, Kim J‐S, et al. RORα induces KLF4‐mediated M2 polarization in the liver macrophages that protect against nonalcoholic steatohepatitis. Cell Rep 2017;20:124‐135. [DOI] [PubMed] [Google Scholar]
- 14. Kim H‐J, Han Y‐H, Na H, Kim J‐Y, Kim T, Kim H‐J, et al. Liver‐specific deletion of RORα aggravates diet‐induced nonalcoholic steatohepatitis by inducing mitochondrial dysfunction. Sci Rep 2017;7:16041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim E‐J, Yoon Y‐S, Hong S, Son H‐Y, Na T‐Y, Lee M‐H, et al. Retinoic acid receptor–related orphan receptor α–induced activation of adenosine monophosphate–activated protein kinase results in attenuation of hepatic steatosis. Hepatology 2012;55:1379‐1388. [DOI] [PubMed] [Google Scholar]
- 16. Lee M‐H, Koh D, Na H, Ka N‐L, Kim S, Kim H‐J, et al. MTA1 is a novel regulator of autophagy that induces tamoxifen resistance in breast cancer cells. Autophagy 2018;14:812‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han Y‐H, Kim H‐J, Kim E‐J, Kim K‐S, Hong S, Park H‐G, et al. RORα decreases oxidative stress through the induction of SOD2 and GPx1 expression and thereby protects against nonalcoholic steatohepatitis in mice. Antioxid Redox Sign 2014;21:2083‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bartolomeo R, Cinque L, De Leonibus C, Forrester A, Salzano AC, Monfregola J, et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J Clin Invest 2017;127:3717‐3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shao Q, Yang M, Liang C, Ma L, Zhang W, Jiang Z, et al. C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy 2019;1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification—the emerging role of v‐ATPase in aging and neurodegenerative disease. Ageing Res Rev 2016;32:75‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, et al. Impaired TFEB‐mediated lysosome biogenesis and autophagy promote chronic ethanol‐induced liver injury and steatosis in mice. Gastroenterology 2018;155:865‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopez A, Fleming A, Rubinsztein DC. Seeing is believing: methods to monitor vertebrate autophagy in vivo. Open Biol 2018;8:180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang B, Everett L, Jager J, Briggs E, Armour S, Feng D, et al. Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell 2014;159:1140‐1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 2019;47:D607‐D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vasanthakumar T, Rubinstein JL. Structure and roles of V‐type ATPases. Trends Biochem Sci 2020;45:295‐307. [DOI] [PubMed] [Google Scholar]
- 26. Ohira M, Smardon AM, Charsky CMH, Liu J, Tarsio M, Kane PM. The E and G Subunits of the yeast V‐ATPase interact tightly and are both present at more than one copy per V1 complex. J Biol Chem 2006;281:22752‐22760. [DOI] [PubMed] [Google Scholar]
- 27. De Luca M, Cogli L, Progida C, Nisi V, Pascolutti R, Sigismund S, et al. RILP regulates vacuolar ATPase through interaction with the V1G1 subunit. J Cell Sci 2014;127:2697. [DOI] [PubMed] [Google Scholar]
- 28. Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol 2017;13:616‐623. [DOI] [PubMed] [Google Scholar]
- 29. Nakadera E, Yamashina S, Izumi K, Inami Y, Sato T, Fukushima H, et al. Inhibition of mTOR improves the impairment of acidification in autophagic vesicles caused by hepatic steatosis. Biochem Bioph Res Co 2016;469:1104‐1110. [DOI] [PubMed] [Google Scholar]
- 30. Jansen EJR, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun 2016;7:11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cannata Serio M, Rujano MA, Simons M. Mutations in ATP6AP2 cause autophagic liver disease in humans. Autophagy 2018;14:1088‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jansen J, Timal S, van Scherpenzeel M, Michelakakis H, Vicogne D, Ashikov A, et al. TMEM199 deficiency is a disorder of golgi homeostasis characterized by elevated aminotransferases, alkaline phosphatase, and cholesterol and abnormal glycosylation. Am J Hum Genet 2016;98:322‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jansen J, Cirak S, van Scherpenzeel M, Timal S, Reunert J, Rust S, et al. CCDC115 deficiency causes a disorder of golgi homeostasis with abnormal protein glycosylation. Am J Hum Genet 2016;98:310‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cannata Serio M, Graham LA, Ashikov A, Larsen LE, Raymond K, Timal S, et al. Mutations in the V‐ATPase assembly factor VMA21 cause a congenital disorder of glycosylation with autophagic liver disease. Hepatology 2020;72:1968‐1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khambu B, Yan S, Huda N, Liu G, Yin X‐M. Autophagy in non‐alcoholic fatty liver disease and alcoholic liver disease. Liver Res 2018;2:112‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim YS, Nam HJ, Han CY, Joo MS, Jang K, Jun DW, et al. LXRα activation inhibits autophagy and lipophagy in hepatocytes by dysregulating ATG4B and Rab‐8B, reducing mitochondrial fuel oxidation. Hepatology 2021;73:1307‐1326. [DOI] [PubMed] [Google Scholar]
- 37. Dossou AS, Basu A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers 2019;11:1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017;168:960‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang C‐S, Jiang B, Li M, Zhu M, Peng Y, Zhang Y‐L, et al. The lysosomal v‐ATPase‐regulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab 2014;20:526‐540. [DOI] [PubMed] [Google Scholar]
- 40. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015;125:25‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morrisett JD, Abdel‐Fattah G, Hoogeveen R, Mitchell E, Ballantyne CM, Pownall HJ, et al. Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J Lipid Res 2002;43:1170‐1180. [PubMed] [Google Scholar]
- 42. Levy G, Schmidli H, Punch J, Tuttle‐Newhall E, Mayer D, Neuhaus P, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12‐ and 36‐month results. Liver transplantation 2006;12:1640‐1648. [DOI] [PubMed] [Google Scholar]
- 43. Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal biology and function: modern view of cellular debris bin. Cells 2020;9:1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guerra F, Girolimetti G, Beli R, Mitruccio M, Pacelli C, Ferretta A, et al. Synergistic effect of mitochondrial and lysosomal dysfunction in Parkinson’s disease. Cells 2019;8:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu JJ, Cai A, Greenslade JE, Higgins NR, Fan C, Le NTT, et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. P Natl Acad Sci USA 2020;117:15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, et al. RNA targets of TDP‐43 identified by UV‐CLIP are deregulated in ALS. Mol Cell Neurosci 2011;47:167‐180. [DOI] [PubMed] [Google Scholar]
- 47. Hook PW, McClymont SA, Cannon GH, Law WD, Goff LA, McCallion AS. Temporal and spatial variation among single dopaminergic neuron transcriptomes informs cellular phenotype diversity and Parkinson’s disease gene prioritization. bioRxiv 2017:148049. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material