

Abstract
Methanotrophic bacteria utilize the non-heme diiron enzyme soluble methane monooxygenase (sMMO) to convert methane to methanol in the first step of their metabolic cycle under copper-limiting conditions. The structure of the sMMO Fe(IV)2 intermediate Q responsible for activating the inert C-H bond of methane (BDE = 104 kcal/mol) remains controversial, with recent studies suggesting both “open” and “closed” core geometries for its active site. In this study, we employ nuclear resonance vibrational spectroscopy (NRVS) to probe the geometric and electronic structure of intermediate Q at cryogenic temperatures. These data demonstrate that Q decays rapidly during the NRVS experiment. Combining data from several years of measurements, we derive the NRVS vibrational features of intermediate Q as well as its cryoreduced decay product. A library of 90 open and closed core models of intermediate Q is generated using density functional theory (DFT) to analyze the NRVS data of Q and its cryoreduced product, as well as prior spectroscopic data on Q. Our analysis reveals that a subset of closed core models reproduce these newly acquired NRVS data as well as prior data. The reaction coordinate with methane is also evaluated using both closed and open core models of Q. These studies show that the potent reactivity of Q towards methane resides in the “spectator oxo” of its Fe(IV)2O2 core, in contrast to non-heme mononuclear Fe(IV)=O enzyme intermediates that H-atom abstract from weaker C-H bonds.
Graphical Abstract
1. Introduction
The superfamily of binuclear non-heme (NH2Fe) enzymes1 plays major roles in all realms of life including metabolism,2-3 biotechnology,4-5 drug design,6-7 and bioremediation.8 They share in common an active site composed of histidine and carboxylate residues that house the binuclear iron core. The reductive activation of dioxygen by this cofactor initiates a wide range of chemistries with substrates including N-oxygenation,9 electrophilic aromatic substitution (EAS), 10 and hydroxylation.11-12 Spectroscopic studies of O2-activated intermediates in several NH2Fe enzymes have led to an improved understanding of their reaction mechanisms.13-14
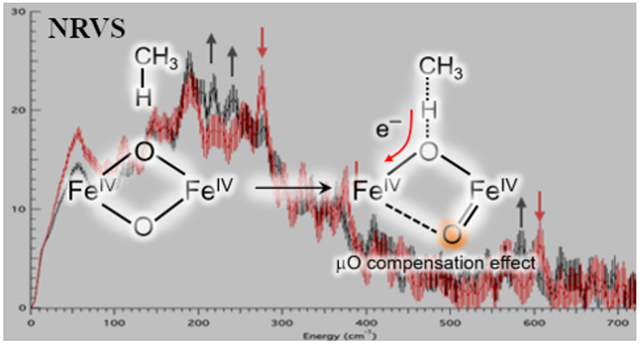
Under low [Cu] conditions, methanotrophic bacteria utilize the NH2Fe enzyme soluble methane monooxygenase (sMMO) to convert methane to methanol in the first step of its metabolic cycle.15,16 The quaternary structure of this enzyme complex consists of three component subunits: a hydroxylase (αβγ)2 (MMOH), which holds the NH2Fe cofactor; an electron transfer reductase (MMOR) that supplies two electrons to the NH2Fe cofactor during catalysis; and a regulatory component (MMOB) that binds to MMOH, greatly enhancing its product yield, O2 reactivity, and turnover frequency.17-20 To produce methanol, this enzyme complex must activate the inert C-H bond of methane (BDE = 104 kcal mol−1) at ambient conditions, a chemical feat otherwise unprecedented in iron enzymology. The catalytic cycle of sMMO has been intensely studied under single-turnover conditions, leading to the identification of several intermediates including the methane-reactive intermediate Q.19-23 Mössbauer spectroscopy has defined intermediate Q as having a high-spin, antiferromagnetically-coupled Fe(IV)2 electronic structure.24 This intermediate stands alone in the NH2Fe superfamily, for which most other substrate-reactive intermediates to date have been characterized as biferric (hydro)peroxides13 (and in some cases ferric superoxide).25,26
The structure of intermediate Q was originally assigned in the late 1990s as a closed “diamond” Fe2(μ-O)2 core from extended X-ray absorption fine structure (EXAFS) data27 indicating a short Fe-Fe distance of 2.46 Å. This assignment was supported by a more recent (2015) continuous flow resonance Raman (rR) study,28 which revealed a 690 cm−1 vibration showing 18O2 and 16O18O isotopic shifts consistent with the vibrational spectra of diamond core model complexes29,30 and O2 loading into the Fe2O2 core. An additional peak at 556 cm−1 observed upon the decay of Q in this study also showed an 18O2 (but no unique 16O18O) shift, and was assigned to an Fe(III)2 mono-μ-O species formed after methanol production (with loss of one labeled oxygen to methanol). Kinetic studies of the reaction of intermediate Q with methane reported a low activation barrier31 (ΔG‡ = 10 kcal mol−1 at 4 °C) and large deuterium kinetic isotope effect32 (KIE = 50), indicating an H-atom abstraction from the strong C-H bond of CH4. These EXAFS and reaction kinetics studies prompted a number of theoretical investigations into the reactivity of methane hydroxylation by closed core Q models.33-39 Various closed core models have been proposed, including a Fe2O2 diamond core with one33 or two μ-1,3-carboxylate bridges,31 and with or without an exogenous water ligand.30 Irrespective of the proposed model, all candidate structures optimized with a high-spin electronic structure resulted in Fe-Fe distances of ≥ 2.65 Å, far longer than measured by EXAFS.
In 2010, a high-spin “open” core O=Fe(IV)─O─Fe(III)─OH model complex was reported that exhibited a second order rate constant for dihydroanthracene oxidation 106 higher than its low-spin diamond core precursor.40,41 This observation fueled speculation into the possibility of an open core assignment for Q. Since this report, two studies using high-energy resolution fluorescence detected (HERFD) X-ray absorption spectroscopy (XAS) on rapid freeze-quenched (RFQ) samples of Q have advocated for the possibility of an open core assignment. First, an analysis of the high intensity of the HERFD-XAS pre-edge region of the Q spectrum showed that the pre-edge was best reproduced by an open core model complex possessing a terminal oxo; this high pre-edge intensity of Q was alternatively not reproduced by a Fe2O2 diamond core model complex.42 Second, remeasurement of the EXAFS region using HERFD-XAS did not reproduce the Fe-Fe 2.46 Å scatterer, which was determined to be a background metallic iron contribution.43 The new RFQ-Q HERFD-EXAFS data were fit with an Fe-Fe distance of 3.30-3.34 Å, longer than the 3.27 Å distance of the ~ 50-60% Fe(II)2 species (MMOHred) also present in the RFQ-Q sample. This finding was interpreted as Q possessing a longer Fe-Fe distance (3.4 Å) than MMOHred, which required an open core structural assignment. Though these studies have cast doubt on the closed core assignment of Q, no general consensus has been reached on its geometric structure and therefore the relevance of past calculations on closed core models.
Defining the geometric structure of Q thus requires further spectroscopic insight. The continuous-flow rR feature at 690 cm−1 informs on the mechanical coupling of O2 incorporated atoms, but this feature alone provides limited structural insight. However, the 57Fe preparations of sMMO used for RFQ-Mössbauer spectroscopy also enable the use of nuclear resonance vibrational spectroscopy (NRVS). This technique probes the vibrational sidebands in the 57Fe nuclear Mössbauer transition and reports on all vibrations with significant 57Fe displacement.44-46 NRVS studies by our group have successfully characterized high-valent oxygen intermediates in binuclear47 and mononuclear enzymes48 and their models,49,50 as well as iron-containing zeolites.51 In the present study we utilize NRVS to study RFQ samples of Q. This hot intermediate is found to rapidly decay in the beam. Scanning multiple samples of Q over several years of experiments has enabled the separation of the Q NRVS spectrum from its decay. Further, the decay of Q in the X-ray beam is found to involve its cryoreduction; thus the NRVS spectrum of Q decay provides a spectral perturbation to assist in the assignment of Q. DFT calculations have been used to generate 90 structural models to correlate to the Q NRVS data as well as to prior spectroscopic data. This study yields deeper insight into the structure of Q as well as its reactivity relative to mononuclear non-heme iron enzymes.
2. Results
2.1. NR VS of Intermediate Q
Intermediate Q is formed by reaction O2 with biferrous MMOH in complex with equimolar (vs MMOH active sites) MMOB. Thus, all samples analyzed here contain a complex of MMOH and MMOB. NRVS data collected on RFQ-Q samples measured at BL09XU exhibit time-dependent changes in the beam at cryogenic temperatures. The NRVS spectrum of a high-purity preparation of Q (50% of total iron, 37% biferrous (MMOHred), 13% biferric (MMOHox); see Figure S1 for quantitation) displays an intense feature at 275 cm−1 in the first 10 scans (1 hour/scan) which disappears after 20 total scans (Figure S2, bottom to top). Concomitant with the decay of the 275 cm−1 feature is the growth of intensity between 200-250 cm−1 during 40 hours of measurement. Other RFQ samples of Q containing a lower quantity of Q (40%) and higher MMOHred (50%) also exhibited weaker intensity at 275 cm−1 that goes away with increasing scans. Therefore, in order to clearly distinguish the NRVS features of Q (early time data) and its decay product (late time points), seven samples of Q were measured up to 80 hours (ranging from 20 to 80 hours per sample). Binning sixty scans from these samples into ten-hour windows demonstrates biphasic, time-dependent NRVS changes (Figure S3, bottom to top): in the first phase, the 275 cm−1 feature and a weaker (but reproducible above noise) feature at 605 cm−1 decay and are no longer present after 20 hours of measurement (bottom to middle, black arrows), while features between 220-240 and at 580 cm−1 grow in (middle, red arrows). In the second decay phase, the 220-240 and 580 features continue to increase in intensity and no longer change after 30 hours of measurement (top, black arrows). The decay of a 375 cm−1 feature is also observed in these phases (top, black line), resulting in a local maximum in the late time point NRVS data at 365 cm−1. NRVS data combined from several samples into sets of 5-hour intervals (Figure S4) corroborate the decay of the 605 and 275 cm−1 features, and the growth of the 220, 240, and 580 cm−1 features within the first 15 hours of measurement (first phase). Final early and late time scan Q NRVS data were generated by compiling over one-hundred scans between 1-15 and 30-80 hours of measurement, and these are shown in Figure 1A top as red and black, respectively. Additionally shown in Figure 1A top (orange) is the NRVS spectrum of a MMOHred sample which was scaled down to reflect the 50% Fe(II)2 present in the “Q” sample. Subtraction of 50% MMOHred and renormalization generates the spectra at the bottom of Figure 1A for the Q (red) and Q decay (black) NRVS data. The overlay of these spectra confirms that the time-dependent changes observed between early and late Q NRVS data (red down arrows and up black arrows, respectively) are outside of noise, as are additional features at 420, 330, and 300 cm−1 which do not change between the early and late Q NRVS data (lighter coincident red and black lines). Thus, NRVS data of Q reveal strong time-dependent changes in the beam at both low (200-400 cm−1) and high (560-610 cm−1) energies. The early time data define the NRVS features of intermediate Q, and the late time data provide the NRVS spectrum of its decayed features in the X-ray beam.
Figure 1.

NRVS spectra of Q samples indicating time-dependent changes in the beam and isotopic perturbations. (A) Top: NRVS spectra (including error) of early Q (hours 1-15; top red), and late decay scans (30+ hours; top black), and MMOHred (multiplied by 0.5; top orange). Late decay scans include 16O2 and 18O2 data in order to maximize S/N. At the top of the figure, thick red lines indicate major features of early Q data. Brown lines indicate features which grow in over two phases. Black thick lines indicate major features of decayed Q data. Thin red and black lines indicate features shared by both early and decayed Q data. Bottom: MMOHred-subtracted and renormalized spectra (Q red and decay black). In the bottom plot, based on poor S/N the intermediate Q and decay spectra past 620 cm−1 are given as Early – Late scan data (no feature above noise is observed). (B) NRVS spectra of 16O2 (71 scans) and 18O2 (110 scans, with data accumulated at ~ 1/2 count rate as 16O2 decay data) labeled Q after 30+ hours of measurement.
In addition, intermediate Q samples were also prepared with 18O2. While the 16O2 Q samples could be generated using chlorite dismutase and were thus concentrated enough to display significant NRVS intensities in the early scans, the 18O2 samples had to be prepared with 18O2-saturated buffer and thus were more dilute (1.00 mM total iron vs. 1.80 mM). The 18O2 generated Q samples required more accumulation time for good S/N and isotope perturbations could only be obtained for the late time (30-80 hour) decay data. Two isotope shifts are observed in the decayed Q NRVS spectrum: a 12-15 cm−1 downshift of the 420 cm−1 feature, and a small downshift of the 580 cm−1 feature (Figure 1B; see Figure S5 for raw data counts in this region). While limited by the S/N of ~ 2, analysis of the 580 cm−1 feature (Figure S6) reveals an isotope shift of 4–7 cm−1.
In summary, Q shows NRVS peaks at 605, 420, 375, 330, 300, and 275 cm−1. The decay of Q results in the loss of the 605, 375, and 275 cm−1 features and produces new peaks at 580, 365, 240, and 220 cm−1. The features at 580 and 420 cm−1 in the decayed Q spectrum show a 16O/18O effect of 4-7 cm−1 and 12-15 cm−1, respectively.
2.2. Mössbauer of Q NRVS Samples and Parallel Data On Cryoreduced Q
It was hypothesized that the decay of Q in the X-ray beam during NRVS data collection was due to its photoreduction. Mössbauer spectra were obtained on RFQ Q samples before and after NRVS experiments to quantify the amount of Q decay and identify any new species formed due to photoreduction. To track the decay of Q during NRVS measurements, parallel experiments were performed on frozen-solution Q samples at BL19LXU at SPring-8. Samples measured at BL19LXU were prepared in 11 x 4 x 1.2 mm rectangular holders; illumination of these samples results in an estimated 25% total volume exposure to the NRVS X-ray beam. The NRVS spectrum of a BL19LXU Q sample after 14 hours overlays well with the NRVS spectrum of BL09XU Q decay (Figure S7; due to the 3 times higher flux at BL19LXU). Importantly, The Mössbauer spectrum of the BL19LXU sample (Figure 2A red, speciation prior to NRVS experiment given in Figure S8) after NRVS measurement at BL19 for 14 hours shows two changes: 1) 20% reduction of the original Q in the full sample volume (Figure 2A, black, arrows pointing up at 0.50 mm/s and −0.14 mm/s where the second component of the Q doublet is located); and 2) concomitant growth of a new species with δ ≈ 0.6 and ΔEQ ≈ 1.60 mm/s (Figure 2A, arrows pointing down at 1.45 mm/s and −0.14 mm/s; note the downward arrow at −0.14 mm/s reflects a compensating increase in signal for the loss of Q at this velocity) indicative of the formation of a ferric species and possibly photoreduction of the NRVS sample.
Figure 2.

Mössbauer spectra collected at 6 K of intermediate Q exposed to the NRVS X-ray beam and to cryoreduction. (A) Mössbauer spectra before (red) and after (black) NRVS measurement of Q. Arrows indicate major changes pre- and post-NRVS measurement. (B) Mössbauer spectra of a Q sample irradiated to 6 Mrad (top to middle). Arrow in the middle spectrum indicates a new feature present after cryoreduction. Bottom spectrum post-cryoreduction in an applied 73 mT parallel field showing the appearance of paramagnetic features (arrows).
In order to evaluate whether the new ferric species in Figure 2A (downward arrows) is a product of Q photoreduction, samples of Q were independently cryoreduced using 6 Mrad of γ-ray emission from a radioactive 60Co source. Similar experiments were reported by Valentine et al. on MMOH Q isolated from Methylococcus capsulatus (Mc Bath);52 however, Mössbauer data have not yet been reported for cryoreduced Q from Methylosinus trichosporium OB3b (Mt OB3b) MMOH. Mössbauer spectra of Q before (Figure 2B top) and after cryoreduction revealed formation of a ferric species (Figure 2B, middle, arrow) with a feature at 1.45 mm/s, similar to the signal observed in the post-NRVS Mössbauer spectrum (Figure 2A, black), confirming cryoreduction of the NRVS sample. Application of a 73 mT parallel magnetic field resulted in no change of the 1.45 mm/s signal indicating it is a coupled biferric species, and revealed additional features at δ = −3.7, −1.7, and 4.8 mm/s (Figure 2B, bottom, arrows), indicating the presence of a paramagnetic Fe(III)Fe(IV) species, similar to intermediate X52,53 (here labeled Qx) in ribonucleotide reductase. Subtracting the Fe(II)2, an initial Fe(III)2 component present in Q before cryoreduction, and the Fe(III)Fe(IV) species from the bottom spectrum in Figure 2B yields the diamagnetic biferric species (Q3; Figure S9, bottom), consisting of two inequivalent high-spin Fe(III) sites (δ1 = 0.50 mm/s, ΔEQ1 = 1.99 mm/s; δ2 = 0.63, mm/s ΔEQ2 = 1.33 mm/s) of equal intensity. Thus, the loss of the Q Mössbauer signal at 0.50 mm/s (Figure 2A) and the formation of a new ferric signal in the post-NRVS Mössbauer spectrum at −0.14 and 1.45 mm/s can be attributed to photoreduction of Q and formation of a high-spin antiferromagnetically coupled biferric decay product (through the mixed valent Qx). From simulating the BL19 post-NRVS Mössbauer spectrum (Figure S10), all of Q in the BL19 NRVS sample has decayed, leaving a mixture of Qx and Q3 (roughly 40%:60%). While the ratio of these two reduced species would vary with beam exposure, Qx and Q3 are calculated to have (vide infra) and do exhibit similar NRVS features (scan dependence in Figure S3). Thus, this variable is not an issue in the analysis of the NRVS decay spectra in Section 3.4.
3. Analysis
3.1. Structural Models for Intermediate Q
To determine possible structures for Q which best reproduce the NRVS and prior spectroscopic data, a library of 90 DFT models (Table S1) that contain two antiferromagnetically-coupled high-spin Fe(IV) centers was constructed by modifying the X-ray crystal structure of MMOB-associated MMOH from Methylococcus capsulatus (Bath. PDB 4GAM).54 Though these crystals appear to have cryoreduced in the X-ray beam, a crystal structure of oxidized MMOH (without MMOB bound, PDB 1MHY) possesses three oxygen ligands, two O(H)x bridges and one terminal O(H)y moiety.55 Therefore, a model with two (μ-O) bridges and a terminal H2O (bis(μO)-H2O-1) was chosen to preserve charge neutrality of the Q Fe(IV)2 active site and served as a starting structural scaffold for optimization of Q models. This model choice has been justified by the recent report of the higher resolution crystal structure of fully oxidized MMOB-associated MMOH from Methylosinus trichosporium OB3b (see Section 3.4),56 and has been further calibrated with DFT (Figure S11) for determination of the optimal truncation scheme.
The full library of Q models possesses either “closed core” structures with two O(H)x bridges (denoted bis(μO)-L-# in Table S1, where L is a terminal ligand to one Fe(IV)) or “open core” structures with one oxo bridge (denoted OC## in Table S1), with further variations in the protonation level of the carboxylate ligands and oxygenic terminal ligands including oxide, hydroxide, and water, and hydrogen bonding interactions. Closed core Q models with a terminal H2O ligand (bis(μO)-H2O subclass, 16 models) were initially constructed through ligand rearrangements of bis(μO)-H2O-1. These models were further modified by deprotonating or removing the terminal H2O ligand, resulting in bis(μO)-OH and bis(μO) subclasses (17 and 6 models, respectively) containing a variety of Glu coordination, conformation, and protonation states, as well as H-bonding interactions with the bis(μO) core or a non-coordinating H2O molecule. Two other subclasses, (μOμOH) (8 models) and bis(μO)=O (4 models), are included in the SI and will be briefly presented. Representative bis(μO), bis(μO)-OH, and bis(μO)-H2O structures are given at the bottom of Figure 3.
Figure 3.

Structural model subtypes for Q. Includes structural representatives for the OC -OH/-OH (OC10), OC -OH/=O (OC26) subclasses at the top, and bis(μO) (bis(μO-1)), bis(μO)-OH (bis(μO)-OH-16), and bis(μO)-H2O (bis(μO)-H2O-6) subclasses at the bottom.
Open core models for Q were prepared by translocating a proton from the terminal water in bis(μO)-H2O-1 to a μ-O bridge and reoptimizing on the Fe(IV)2 level. Opening of the core in this manner yielded a 5C square pyramidal and 6C site with a terminal OH− trans-axial to a His. Models generated in this way with further variations in the conformations and H-bonds of the 1st-sphere Glu residues form the OH-Fe-O-Fe-OH (OC -OH/-OH) subclass. By either protonating or deprotonating the terminal OH− ligand, 37 total open-core models were generated, including the OH-Fe-O-Fe=O (OC -OH/=O) subclass and three other open core subclasses, O=Fe-O-Fe=O (OC=O/=O), O=Fe-O-Fe-H2O (OC -H2O/=O), and OH-Fe-O-Fe-H2O (OC -OH/-H2O), to be presented in less detail (Table S1). Representative OC -OH/-OH and OC -OH/=O structures are given at the top of Figure 3.
3.2. Vibrational Analysis of Closed and Open Core Models of Q
The 90 possible models for intermediate Q generated in Section 3.1 were initially evaluated by computing their vibrational spectra using DFT methods (with the BP86 functional plus 10% HF) and comparing these results to the prior rR data on Q. This rR study28 revealed that Q exhibits a vibration at 690 cm−1, with a 18O2 isotopic downshift of 36 cm−1 and a 16O18O mixed isotope downshift of 12-15 cm−1 (with no observable 2H perturbation, for an experimental resolution of ~ 3 cm−1). Furthermore, rR data of the methane-reacted product complex, T show a 556 cm−1 vibration with a 18O2 downshift to 533 cm−1, and two 16O18O mixed isotope signals at 556 and 533 cm−1 reflecting the presence a single labeled oxygen atom in T. These rR data on T define it as having a μ-O bridge derived from O2. Therefore, these data together show that Q contains an O2 derived core, with one of these oxygens loaded in a μ-O bridge. To screen candidates for Q from the library of 90 models, vibrational frequencies for 16O2, 18O2, 16O18O, and 2H isotopomers for each of the models were obtained from the DFT calculations and correlated to the experimental rR vibrational data (16O2 spectra for all models given in Table S1, along with tabulated 16O18O, 18O2 and 2H perturbations).
Closed core Q models of the bis(μO), bis(μO)-OH, and bis(μO)-H2O subclasses exhibit three modes above 600 cm−1: a symmetric (Ag) breathing mode at 700 cm−1, as well as two non-symmetric (B3u and B2u) modes (using effective D2h symmetry for a Fe2O2 core) between 600 and 650 cm−1 (ν1, ν2, ν3 in Figure 4 top, respectively). Incorporation of 18O2 and 16O18O into the (μ-O)2 core results in downshifts of the breathing mode by 30-40 cm−1 and 15-20 cm−1, respectively. The absence of proton involvement in the breathing mode results in no predicted 2H effect. The breathing mode of all bis(μO) models, as well as 12 out of 16 bis(μO)-H2O models, reproduces the Q rR 690 cm−1 feature well (highlighted in red in Figure 4 top), and is similar to rR data collected on bis(μO) model complexes.29,30 However, the symmetry of the Fe2O2 core mediates the degree of coupling between the (μ-O)2 units. For example, in bis(μO)-H2O-16, hydrogen bonding (heavy atom distance = 2.5 Å) between E144 and one (μ-O) introduces a C2ν distortion into the core (Fe1-Oa = 1.83, Fe2-Oa = 1.95 Å, Fe1-Ob = 1.74, Fe2-Ob = 1.85). As a result, bis(μO)-H2O-16 possesses two uncoupled ν(Fe1-Oa) and ν(Fe2-Ob) stretches above 600 cm−1 which localize onto the short Fe-O bonds, and prevents the appropriate mixed isotope shift in the 16O18O isotopomer (similar distortions are found in bis(μO)-H2O-5, 7, and 13; see Table S1). In the bis(μO)-OH subclass, the strong ligand field of the terminal OH− produces a large C2h distortion and uncoupling of the core vibrations (models bis(μO)-OH 1-5, 7, 9, 17-18). In contrast to the bis(μO)-H2O subclass, hydrogen bonding to the (μ-O)2 unit in the bis(μO)-OH structures decreases the distortion. This increases the (μ-O)2 mechanical coupling and results in a breathing mode vibration near 690 cm−1 with the appropriate mixed isotope shift. The influence of the OH− ligand is such that, of the remaining 9 bis(μO)-OH models (bis(μO)-OH 6, 8, 10-16) possessing a coupled Ag breathing mode, 7 of them contain a hydrogen bond to the (μ-O)2 unit (models bis(μO)-OH 6, 10-14, 16). For all three bis(μO), bis(μO)-OH, and bis(μO)-H2O subclasses, the 690 cm−1 vibration is well reproduced by a breathing mode in models without strong C2ν or C2h distortions (i.e., in models for which all Fe-O bond lengths in the Fe2O2 core are similar)
Figure 4.

Normal modes for the Q closed core bis(μO), bis(μO)-OH, and bis(μO)-H2O subclasses (top), and Q open core OC -OH/-OH (middle) and OC -OH/=O subclasses (bottom). Closed core modes defined in D2h symmetry. The calculated vibration in each of these models that corresponds to the reported rR 690 cm−1 vibration is highlighted in red. Arrows denote in-plane motion, and “x” and “o” characters denote out-of-plane motions.
The two other closed core Q subclasses, bis(μO)=O and (μOμOH), do not possess a feature above 600 cm−1 with an appropriate mixed isotope downshift. In bis(μO)=O models, the terminal oxo facilitates an opening of the core to a O=Fe-O-Fe=O structure; therefore, only one antisymmetric μ-O stretch (νas(Fe-O-Fe)) is observed between 600-740cm−1. This is true as well for (μOμOH) models which lack a coupling of the μ-OH stretches to the νas(Fe-O-Fe) mode. Thus, these models do not predict the observed average mixed isotope downshift in the 16O18O isotopomer and are excluded from further consideration.
Though previous literature had identified closed core models as structural candidates for Q based on the 690 cm−1 vibration and 16O18O mixed isotope shift, an open question remains as to how an open core model as suggested by recent EXAFS data could reproduce these rR data. This question is now addressed. In Q open core models, the Fe-OH stretch (ν(Fe-OH)) and νas(Fe-O-Fe) appear between 600 and 750 cm−1. If these vibrations occur close in energy, they mix to form symmetric (ν(Fe-OH) + νas(Fe-O-Fe)) and antisymmetric (ν(Fe-OH) − νas(Fe-O-Fe)) combinations (Figure 4, ν1 and ν2 middle row, and ν2 and ν3 bottom row).
For OC -OH/=O models possessing an OH-Fe2-(μ-O)-Fe1=O structure, The Fe1=O bond weakens the Fe1-(μ-O) bond resulting in a shorter Fe2-(μ-O) bond, localizing νas(Fe-O-Fe) on the Fe2 side. This facilitates ν(Fe-OH) + νas(Fe-O-Fe) coupling, resulting in a symmetric mode ν2 which would agree with the 690 cm−1 experimental data (Figure 4, bottom row, red box), as anticipated in a recent computational study of Q.57 Isotope labeling with 18O2 must be incorporated into the μ-O bridge to correlate to the decay product T, as well as either into the -OH or =O terminal ligands. Incorporation of 18O into the =O position does not lead to an observed mixed isotope signal, since the oxo vibration is too high in energy (800 cm−1) to influence ν2 (Figure S12). Therefore, the 18O2 would have to be incorporated into the -OH and μ-O ligands. OC -OH/=O 18O2 and 16O18O isotopomers display a downshift of ν2 by 20-40 and 10-20 cm−1 (respectively), which reproduce the Q rR experimental data. Importantly, the mixed isotope signal strongly depends on the similarity between Fe2-OH and Fe2-(μ-O) bond lengths. Fe2-OH and Fe2-(μ-O) bond lengths differing by more than 0.05 Å results in the uncoupling of the ν(Fe-OH) and νas(Fe-O-Fe) vibrations and the absence of the appropriate mixed isotope shift (OC2, OC22, OC24, OC25). This ν(Fe-OH) + νas(Fe-O-Fe) mode coupling is also facilitated by hydrogen-bond donation to the Fe1=O oxo, which weakens the Fe1=O bond and strengthens the Fe1-(μ-O) bond, resulting in elongation of the Fe2-(μ-O) bond. Of the remaining OC -OH/=O models, OC17 and OC26 exhibit ν2 matching the experimental 690 cm−1 vibration and its isotope shifts, facilitated by hydrogen-bond donation (from a glutamic acid or OH−) to the terminal oxo. Note, however, that two of these models (OC1 and OC3) exhibit ν2 2H downshifts of 8 cm−1 that is too large based on the rR data; an explanation for this effect is provided below.
In OC -OH/-OH models, three vibrations appear between 600 and 750 cm−1: two ν(Fe-OH) (denoted ν(Fe1-OH) and ν(Fe2-OH)) and one νas(Fe-O-Fe) vibration which may mix to give symmetric and antisymmetric combinations (Figure 4, middle). In these OH-Fe1-(μ-O)-Fe2-OH models, the Fe1-OH vector is oriented out-of-plane with respect to the Fe-O-Fe plane and ν(Fe1-OH) couples weakly with the ν(Fe2-OH) and νas(Fe-O-Fe) vibrations. Thus two coupled modes are observed near 700 cm−1: ν(Fe2-OH) + νas(Fe-O-Fe) and ν(Fe2-OH) − νas(Fe-O-Fe) (Figure 4, middle row, ν1 and ν2), as well as one uncoupled ν(Fe1-OH) mode (ν3). Hence, to reproduce the experimental isotope shifts the two atoms of 18O2 must incorporate into the in-plane hydroxide (Fe2-OH) and the μ-O ligands. For OC -OH/-OH models mode ν1 exhibits 18O2 and 16O18O isotopic shifts of 20-40 and 10-20 cm−1 (respectively), agreeing well with the experimental Q rR data. Due to the similarity of the Fe2-OH and Fe2-(μ-O) bond lengths (Δ = 0.02-0.03 Å), 13 out of 14 OC -OH/-OH models display ν1 with a mixed isotope downshift of 10-20 cm−1. However, 9 of these 13 models exhibit a large ν1 downshift upon deuterium labeling (∣Δ2H∣ ≥ 5 cm−1 for OC9, OC11, OC13, OC18, OC19, OC20, OC23, OC33, OC34), as observed in the OC -OH/=O OC1 and OC3 models (vide supra). This is due to significant mixing of the OH torsional twist into the in-plane ν(Fe2-OH) stretch; this torsion mode downshifts by more than 100 cm−1 upon 2H labeling and hence downshifts ν(Fe2-OH) (Figure S13). The remaining models (OC10, OC15, OC16, OC32) possess a ν1 with a mixed isotope signal that reproduces the 690 cm−1 rR vibration well with no significant 2H effect.
The remaining OC =O/=O, -H2O/=O and -OH/-H2O subclasses do not exhibit a 690 cm−1 vibration with an appropriate mixed isotope downshift for the 16O18O isotopomer and are excluded from further consideration. Both OC =O/=O and OC -H2O/=O subclasses display a single νas(Fe-O-Fe) vibration between 600-750 cm−1 with no coupling partner. In the OC -OH/-H2O subclass, the Fe-(μ-O) bond on the OH- side is too long to permit ν(Fe-OH) and νas(Fe-O-Fe) coupling.
Thus, both closed core and open core Q models predict vibrations near 690 cm−1 with a 16O18O labeled mixed isotope shift that would correspond to the experimental rR data. The bis(μO), bis(μO)-OH, and bis(μO)-H2O models display the canonical Ag breathing mode, which upon 16O18O labeling into the (μ-O)2 core exhibits an appropriate mixed isotope shift. For OC -OH/=O models, the νas(Fe-O-Fe) mode localizes on the Fe2-OH side and mixes with the ν(Fe2-OH) vibration to produce a symmetric ν(Fe2-OH) + νas(Fe-O-Fe) mode. Labeling of the OH- and μ-O ligands with 16O18O and 18O2 would result in its downshift of 10-20 and 20-40 cm−1 respectively, which would agree well with experiment. The OC -OH/-OH subclass also exhibits a ν(Fe2-OH) + νas(Fe-O-Fe) mode which agrees well with experiment, provided minimal rotation of the Fe2-OH unit (that would result in a large deuterium isotope shift) and selective 18O2 labeling of the in-plane Fe2-OH and μ-O ligands. All calculated isotope perturbations are included in Table S1 for the 90 Q model library.
3.3. TD-DFT Correlations to Absorption and Excited State Distortions
Normal mode vibrations in both Q closed core (ν1, Figure 4 top) and open core models (ν1 and ν2, Figure 4 middle and bottom) have been identified as candidates for the 690 cm−1 feature in its rR spectrum. The UV-vis absorption spectrum for Q exhibits an intense (ε = 10 mM−1cm−1) band at 350 nm (28,600 cm−1; Figure 5A) which, when probed with a 351 nm (28,500 cm−1) laser line, results in the resonance-enhancement of the characteristic 690 cm−1 vibration. The absorption spectrum additionally shows a weak feature at 800 nm (12,500 cm−1; ε = 1.2 mM−1cm−1. Insert Figure 5A), as well as rising intensity between 15,000 and 28,000 cm−1 with a local maximum at 23,000 cm−1 (ε = 8 mM−1cm−1). To correlate the Q models identified in Section 3.2 to the experimental absorption spectrum and its associated rR data, there are two points of focus: 1) prediction of the absorption spectra for structural subtypes of Q and in particular the nature of the transitions in the 350 nm region; and 2) determination of the excited state distortions associated with the 350 nm transitions that would lead to resonance enhancement. Therefore, TD-DFT (using the CAM-B3LYP functional to obtain reasonable carboxylate charge transfer energies)58 was employed to calculate and assign the absorption spectra of the Q models (identified in Section 3.2) and predict the excited state distortions associated with 351 nm excitation.
Figure 5.

Q UV-vis absorption spectrum and TD-DFT calculated spectra and predicted rR enhancement of Q models. (A) Experimental absorption spectrum for intermediate Q. Inset: expanded scale for low energy region. Adapted from reference 16. (B) TD-DFT spectrum (calculated using CAM-B3LYP, downshifted by 3,000 cm−1 based on calibration in Figure S14), left, of bis(μO)-H2O-6 as representative of the bis(μO)-L (L = OH−, H2O, or vacant site) models. A qualitative description of transition 2 is shown on the right, along with the LCT for transition 2 calculated for modes ν1, ν2, and ν3 (Figure 4 top). (C) TD-DFT calculated spectrum (left) of OC10 as a representative spectrum of the OC -OH/-OH models. A qualitative depiction of transition 1 is given on the right, along with the LCTs for transition 1 along modes ν1, ν2, and ν4 (Figure 4, middle). (D) TD-DFT spectrum (left) of OC26 as representative of the OC -OH/=O models. Transition 1 is schematically given on the right, along with LCTs for transition 1 for modes ν2, ν3, and ν4 (Figure 4, bottom).
The TD-DFT calculated spectra for closed core and OC -OH/-OH and OC -OH/=O models are shown in Figure 5. The predicted electronic transitions for the three classes are similar and may be described together (see SI, Figures S14-24 and Tables S2-12 for a full description of the TD-DFT calculations and analysis). The weak feature at 12,500 cm−1 in the experimental data (Figure 5A, inset) is predicted to consist of Fe(IV) d-d transitions (Figure 5B-D, insets). The rising intensity above 15,000 cm−1 in the experimental data (Figure 5A) may be described as LMCT transitions involving μ-O, oxo, and OH− donation into the Fe(IV) dσ* LUMO orbitals (Figure 5B-D; the dσ* LUMOs are given in Figure S17, S20, and S23). Importantly, the TD-DFT calculations predict the third critical spectral region incorporating the 28,600 cm−1 experimental peak (Figure 5A) to contain LMCT transitions involving μ-O and OH− donation into the Fe(IV) dπ* orbitals (Figure 5B-D, numbered transitions; the dπ* orbitals are given in Figure S17, S20, and S23) that are highly localized in nature (Note that for the open core models, the acceptor orbitals reside on Fe2 containing the in-plane OH− ligand in the OC -OH/-OH subclass and the OH− ligand in the OC -OH/=O subclass; for examples, see Tables S8 and S10). In the bis(μO), bis(μO)-H2O, and bis(μO)-OH subclasses, the (μ-O)2 → Fe(IV)2 dπ* LMCT would result in an excited state with an elongation of all four Fe2-(μ-O)2 bonds and resonance enhancement of the Ag breathing mode (Figure 4, top), as observed in M2O2 (M = metal) systems.29,30 This prediction based on past model studies was evaluated by using TD-DFT to calculate the linear coupling terms (LCT) for the different possible normal modes in the 650 – 750 cm−1 region, where a large slope in the LCT (i.e., a large change in excited state transition energy with distortion along a normal mode) for a given vibration indicates a large exited state distortion, and thus significant resonance enhancement in its Raman spectrum. As observed in Figure 5B (right), the LCT for the bis(μO)-L (L = OH−, H2O, or vacant axial position) in the symmetric breathing mode has a larger slope (relative to other modes) and is thus calculated to be the dominant resonance enhanced vibration. From extension to the OC -OH/-OH and OC -OH/=O subclasses, we find that the ν1 mode in the former and the ν2 mode in the latter (both at ~ 690 cm−1) do have large LCT slopes (Figure 5C-D on right; relative to other modes in this region with small slopes) and are also predicted to have significant resonance enhancement. This derives from the assignments of the transitions in this region of the absorption spectrum as μ-O and OH− → Fe2(IV) dπ* LMCT (see Tables S8-9). This would result in significant elongations of both the Fe2-OH and Fe2-(μ-O) bonds that corresponds to a positive distortion along the ν1 (OC -OH/-OH) and ν2 (OC -OH/=O) modes. All three modes identified as candidates for the 690 cm−1 rR feature in the possible Q structural types are thus predicted to be resonance enhanced by excitation at 351 nm (28,500 cm−1). Hence, the TD-DFT calculations herein reproduce findings from previous studies that demonstrate resonance enhancement of the Ag mode (at 690 cm−1) in Fe2-(μ-O)2 closed core systems. Importantly, they also predict resonance enhancement of the symmetric ν1 (OC -OH/OH) and ν2 (OC -OH/=O) modes at 690 cm−1 in Q open core models upon excitation at 351 nm, which results in μ-O and OH− donation into localized Fe2(IV) dπ* orbitals.
3.4. NRVS Structural and Spectral Assignment of Q
In the previous sections, a number of models with open and closed core motifs have been evaluated and a limited number have been identified as structural candidates for Q through a correlation of their predicted absorption and rR spectra to experimental data. The remaining models have OC -OH/=O and OC -OH/-OH cores, as well as bis(μO) cores with only glutamate, or OH− and H2O axial ligands. These models are now further appraised using NRVS. The results from the NRVS experiments in Section 2.1 define unique features for Q in its PVDOS spectrum at 605, 420, 375, 330, 300 and 275 cm−1 (Figure 1A, red spectrum). Decay of these features was observed in two sequential phases (Figure S3), corresponding to the cryoreductive formation of a one-electron reduced high-spin, mixed-valence, antiferromagnetically-coupled Fe(III)Fe(IV) species (Qx), followed by further reduction to an Fe(III)2 species (Q3). In the first decay phase, the 605 cm−1 and 275 cm−1 features are eliminated, with concomitant intensity growing in between 560-580 cm−1 and 220-240 cm−1. In the second decay phase, intensity between 560-580 cm−1 and 220-240 cm−1 continues to grow and reaches a maximum. A feature at 375 cm−1 in the early Q data also decays during the measurement. Hence, the final decay spectrum (Figure 1A, black), containing contributions from a mixture of Fe(III)Fe(IV) and Fe(III)2 species, exhibits a feature between 560-580 cm−1, and at 420, 365, 330, 300, 240, and 220 cm−1. Furthermore, the NRVS spectrum for the 18O2 Q decay sample (Figure 1B) exhibits an observable but modest (4 - 7 cm−1) downshift relative to the 16O2 560 - 580 cm−1 feature, and shows a 12-15 cm−1 downshift of the 16O2 420 cm−1 feature.
In order to screen the possible candidates for Q using NRVS, the DFT-calculated NRVS spectra for all candidate Q models (90 total) were correlated to the NRVS experimental data (Figure 1A, bottom red spectrum). Models with calculated Q NRVS spectra that reproduce the experimental spectrum were then computationally “cryoreduced” for correlation to the cryoreduced-Q NRVS and Mössbauer data as a further screen. From Section 2.2, the NRVS sample at the end of the measurement contains 0% Q, 50% MMOHred, and approximately 20% Fe(III)Fe(IV) (Qx) and 30% Fe(III)2 (Q3) species. Therefore, calculated cryoreduced NRVS spectra were generated using 40% Fe(III)Fe(IV) and 60% Fe(III)2 species (matching the speciation with MMOHred subtracted). Though this chosen component mixture is approximate, the calculations below and the experimental data in Figure S3 show that the NRVS features of Fe(III)Fe(IV) and Fe(III)2 cryoreduced models are similar and therefore deviations from this composition would not significantly impact the calculated NRVS spectra. Two important considerations have been included in the generation of acceptable cryoreduced-Q models. First, in order to maximize the number of models possessing high-spin Fe(III)Fe(IV) and Fe(III)2 electronic structures, cryoreduction-modeling calculations were performed with B3LYP which preferentially stabilizes high-spin states. The Fe(IV)2 Q models obtained with 10% HF included with BP86 were reoptimized with B3LYP, which resulted in only minor changes in structures and vibrational profiles (spectra shown in Figure S25). These re-optimized Q structures were then used for cryoreduction modeling. Second, the cryoreduced-Q models were determined by fully optimizing the B3LYP Fe(IV)2 Q models with high-spin Fe(III)Fe(IV)2 and Fe(III)2 electronic structures. This assumption allowed for bond length changes in the iron-ligand bonds and perturbations of hydrogen-bonding interactions involving the μ-O ligands, which we believe is reasonable at cryogenic temperatures. NRVS spectra for models “cryoreduced” in such a fashion (including spectra for both 16O2 and 18O2 isotopomers) were calculated and compared to the experimental data in Figure 1A (bottom, black spectrum) and Figure 1B (for the isotope perturbation) as an additional screen for the determination of the geometric and electronic structure of Q.
In the following sections, analyses of the NRVS spectra for OC -OH/=O, OC-OH/-OH, and bis(μO)-L models (where L is H2O, OH−, or no water-derived ligand) are presented using the methodology described above.
3.4.1. Open Core Terminal Oxo, Terminal Hydroxide (OC -OH/=O)
Of the eight OC -OH/=O models, only OC1, OC3, OC17, and OC26 possess a ν(Fe-OH) + νas(Fe-O-Fe) mode (ν2 Figure 4, bottom) near 700 cm−1 which would correspond to the resonance Raman feature at 690 cm−1. (Note that the ν2 modes for OC1 and OC3 exhibit a 2H perturbation of > ∣8∣ cm−1, but are included to generally explore the NRVS properties of this structural subclass.) The calculated Q NRVS spectra of these models are provided in Figure 6 (black spectra), and compared to the Early Q – MMOHred data in red (top spectrum; note NRVS calculated spectra for other OC -OH/=O models are provided in Table S1). The experimental data correlate best to the OC17 NRVS spectrum. Below 300 cm−1, the OC17 NRVS spectrum exhibits two peaks at 250 and 281 cm−1 that dominate the spectrum, and may be correlated to the 275 cm−1 and the 300 cm−1 peaks in the experimental data. The two peaks in the calculated spectrum at 329 cm−1 and 356 cm−1 would correlate to the experimental data at 330 and 375 cm−1, respectively, while the calculated less intense 390 and 425 cm−1 peaks would correspond to the weak 420 cm−1 feature in the data. Hence, below 450 cm−1, the calculated OC17 NRVS spectrum reasonably correlates to the experimental Q data.
Figure 6.

Correlation of OC -OH/=O computed NRVS spectra to experimental data. The Q – MMOHred (and renormalized) PVDOS spectrum is shown at top in red (adapted from Figure 1A, bottom). Q data (unsubtracted including error) between 610 and 720 cm−1 are provided in the top right inset. OC -OH/=OH models that possess a ν2 mode (see Figure 4, bottom) with the approximate 690 cm−1 mixed isotope signal are shown below in black (OC1, OC3, OC17, OC26. Their structures are given in Table S1). The blue region indicates low energy bending modes (ν5 & ν6 Figure 4 bottom) and terminal ν(Fe-COO−) stretches. The yellow region also contains ν(Fe-COO−) stretches and the νs(Fe-O-Fe) mode (ν4 Figure 4, bottom). The red region contains ν2 and its antisymmetric pair (ν3 Figure 4, bottom).
Above 450 cm−1, the calculated OC17 NRVS spectrum exhibits one peak at 475 cm−1, followed by a lack of intensity up until ~ 700 cm−1, where two peaks are observed at 691 cm−1 and 722 cm−1. The experimental data above 620 cm−1 show no feature above the increasing high noise within this region (Figure 6, top spectrum, inset on right). However, it is clear that the calculated spectrum does not reproduce the experimental data as there is no calculated peak at 605 cm−1. We thus evaluated whether a vibration of the OC17 model (and OC -OH/=O models more generally) could reproduce the experimental 605 cm−1 peak, with some limited structural perturbation. Vibrations closest to 605 cm−1 in the calculated NRVS spectra are color highlighted in Figure 6 and assessed. The calculated vibrations at 691 and 722 cm−1 correspond to the ν3 (ν(Fe-OH) - νas(Fe-O-Fe); Figure 4 bottom) and ν2 modes, respectively (red region, Figure 6). The ν2 mode corresponds to the resonance Raman 690 cm−1 feature, and is thus required to be near 700 cm−1 based on this assignment. Since ν3 is the antisymmetric version of ν2 and is a result of mixing (between the ν(Fe-OH) and νas(Fe-O-Fe) modes), and since the splitting between ν2 and ν3 is ~ 30-40 cm−1, the ν3 mode cannot be assigned to the 605 cm−1 peak without lowering the energy of ν2 and sacrificing the rR assignment. Note that the 605 cm−1 feature cannot be assigned as the isolated ν(Fe-OH) mode in these models, since ν(Fe-OH) + νas(Fe-O-Fe) mixing is required near 700 cm−1 to reproduce the mixed isotope effect in rR of the ν2 mode. This rules out models such as OC24 (see NRVS spectrum in Table S1), which possesses a particularly short Fe1=O bond that highly localizes the μ-O ligand on the Fe2 side, resulting in strong μ-O donation to Fe2 and an elongated (1.82 Å) Fe2-OH bond. This model exhibits a localized ν(Fe-OH) vibration near 600 cm−1; however, only the νas(Fe-O-Fe) mode is observed near 700 cm−1, which would not produce the mixed isotope effect in the rR spectrum (and also would not show resonance enhancement). Thus, neither the ν2, ν3, nor an isolated ν(Fe-OH) mode can be assigned to the 605 cm−1 peak in the experimental data (indicated by the red X in OC17 Figure 6).
Vibrations below 600 cm−1 were also assessed as possible assignments for the 605 cm−1 feature. The region between 440 and 540 cm−1 (yellow region Figure 6) includes the νs(Fe-O-Fe) mode (ν4 Figure 4, bottom), as well as terminal carboxylate (ν(Fe-COO−)) stretches that further distribute into the 345 and 440 cm−1 region (blue region Figure 6) that mostly contains low energy bending modes (ν5 and ν6, see Figure 4 bottom). The energy of the νs(Fe-O-Fe) mode (and importantly, the higher energy νas(Fe-O-Fe) mode) depends primarily on the Fe-O-Fe angle.59 However, manipulation of the Fe-O-Fe angle to increase the energy of the νs(Fe-O-Fe) mode would decrease the energy of the νas(Fe-O-Fe) mode, which would unacceptably decrease the energy of the ν2 mode that would be assigned to the 690 cm−1 feature in the rR spectrum. Hence, the νs(Fe-O-Fe) mode cannot be assigned to the 605 cm−1 NRVS feature. The ν(Fe-COO−) modes are too low in energy to be reasonably assigned to the 605 cm−1 feature (yellow and blue regions in Figure 6). Their energies are already calculated to be slightly higher than that experimentally observed for a terminal ν(Fe-COO−) mode in a NRVS study48 of a high-spin Fe(IV) oxo intermediate in SyrB2 at 370 cm−1. Therefore, the 605 cm−1 experimental NRVS peak cannot be reasonably correlated to either ν(Fe-COO−) or νs(Fe-O-Fe) modes that must be at energies well below 600 cm−1.
In summary, while OC17 (and OC-OH/=O models in general) reproduces the Q NRVS spectrum reasonably well below 450 cm−1, it fails to capture the 605 cm−1 peak in the experimental data. No vibration in the OC-OH/=O models may be reasonably assigned to the 605 cm−1 NRVS feature; thus, the OC -OH/=O subclass is excluded as a possible candidate for intermediate Q.
3.4.2. Open Core bis-terminal-hydroxide (OC -OH/-OH)
The OC -OH/-OH models (14 total) constitute the remaining open core subclass that are to this point in the analysis possible structures of Q. Of these 14 models, 13 possess a symmetric ν(Fe-OH) + νas(Fe-O-Fe) mode (ν1 Figure 4) near 690 cm−1 with an appropriate 16O18O mixed isotope shift, while only 4 of these models (OC10, OC15, OC16, and OC32) also exhibit a small 2H isotope effect (Δ ≤ 3 cm−1) that would reproduce the rR data. (Models exhibiting a larger 2H isotope effect are OC9, OC11, OC13, OC18, OC19, OC20, OC23, OC33, OC34) The calculated NRVS spectra for OC10, OC15, OC16, and OC32 are provided in Figure 7A, and NRVS spectra for all remaining OC -OH/-OH models are included in Table S1.
Figure 7.

NRVS spectra of OC -OH/-OH models and the cryoreduction decay spectra for OC16. (A) Early Q – MMOHred NRVS data (top), with calculated spectra given below for the four OC -OH/-OH models possessing a ν1 mode (Figure 4 middle) with appropriate 16O18O mixed isotope and deuterium isotope effects (calculated with BP86 and 10% HF). Yellow region contains the ν4 (Figure 4 middle) and terminal ν(Fe-COO−) modes. Red region contains the ν1, ν2, and ν3 modes (Figure 4). (B) Top: Early Q – MMOHred (red spectrum) and Decayed Q – MMOHred (black spectrum) NRVS data, with arrows highlighting changes in the spectra from early to late scans. Middle: NRVS spectrum for the B3LYP DFT-optimized OC16 model (red; see Figure S25 for the close correlation of NRVS calculations between B3LYP vs. 10% HF in BP86), as well the calculated decay spectrum (OC16 Decay) consisting of a 40%/60% mixture of its Fe2(III)Fe1(IV) and Fe(III)2 optimized calculated NRVS spectra, respectively (black). Arrows indicate major changes from OC16 to OC16 Decay. Bottom: Calculated NRVS spectra for the OC16 Decay from Figure 7B middle (black) and its 18O2 labeled isotopomer (18O2 OC16 Decay, blue).
Of the 4 models in Figure 7A, OC16 best reproduces the experimental data. The calculated NRVS spectrum of OC16 (Figure 7) below 450 cm−1 exhibits an intense feature at 278 cm−1, which may be associated with the 275 cm−1 peak in the experimental data (Figure 7A, top red spectrum). The calculated feature at 315 cm−1 would correlate reasonably to the 300 peak in the data, while the broad, intense calculated peak at 357 cm−1 may be associated with the combination of both the 330 and 375 cm−1 features in the data. The 406 cm−1 feature may be correlated to the peaks around 420 cm−1 in the data; hence, the low energy (< 450 cm−1) region of the calculated OC16 spectrum reasonably reproduces the Q NRVS spectrum (with similar results for models OC10, OC15, and OC32).
Above 450 cm−1, both OC16 and OC32 exhibit intensity near 605 cm−1, whereas OC10 and OC15 (as well as the remaining 9 models in the SI) do not. Three vibrations contribute to the calculated OC16 NRVS spectrum in the region between 600 and 750 cm−1: the rR-assigned ν1 mode (the symmetric combination of the in-plane ν(Fe2-OH) stretch and νas(Fe-O-Fe) mode, centered at ~ 712 cm−1); ν2 (Figure 4 middle), the antisymmetric version of ν1 (625 cm−1); and ν3 (Figure 4 middle), the isolated out-of-plane ν(Fe1-OH) mode (666 cm−1). Both the ν2 and ν3 modes are possible assignments for the 605 cm−1 feature in this structure, with the ν2 mode closer to 605 cm−1. Interestingly, the ν1 and ν2 splitting is larger (40-70 cm−1; red region Figure 7A) in OC -OH/-OH vs. OC -OH/=O models (30-40 cm−1). This is due to the higher symmetry of the OC -OH/-OH models (relative to OC -OH/=O); the two similar Fe-(μ-O) bond lengths result in a lowering of the νas(Fe-O-Fe) mode to be closer to ν(Fe2-OH) (with a concomitant increase of νs(Fe-O-Fe)). This increases the νas(Fe-O-Fe) + ν(Fe2-OH) mixing, which results in the larger split ν1 and ν2 modes. For models OC10 and OC15 (and the 9 related models in the SI), the ν2 mode is at ≥ 650 cm−1, which may not be lowered without sacrificing the rR-assigned ν1 mode. However, in OC16, Glu243 is hydrogen-bonded to Fe1-OH as well as Fe2-OH (see structure in Table S1), which weakens its hydrogen-bonding interactions with Fe2-OH (heavy atom distance increases by 0.1 Å, compared to OC10). This decreases the energy of the ν(Fe2-OH) mode, which drives down the energies of both the ν1 and ν2 modes. Hence, up until this point in the analysis of this structural class ν2 is a possible candidate for the 605 cm−1 NRVS assignment.
As an additional screen for evaluating OC16 (and OC32; see Figure S27), this model was cryoreduced in DFT to correlate its simulated decay NRVS spectrum to the experimental data in Figure 7B (top, black spectrum). OC16 was thus optimized with high spin Fe(III)Fe(IV) and Fe(III)2 electronic structures using the methodology outlined above Section 3.4. Optimization of OC16 to a Fe(III)Fe(IV) species afforded two possible models: Fe2(III)Fe1(IV) or Fe2(IV)Fe1(III), with the Fe2(III)Fe1(IV) model lower in energy by ΔE = −3.4 kcal/mol. The NRVS spectra for the cryoreduced OC16 Fe2(III)Fe1(IV) and Fe(III)2 models are provided in Figure S26 (middle and bottom rows) in black (structures for the cryoreduced optimized forms are also provided).
To reproduce the decayed Q NRVS decay spectrum in Figure 7B (top in black) which contains a mixture of both Fe(III)Fe(IV) and Fe(III) species, the cryoreduced mixture spectrum (OC16 Decay) was prepared by summing 40% of the calculated OC16 Fe2(III)Fe1(IV) NRVS spectrum and 60% of the Fe(III)2 spectrum from Figure S26A (black spectra). The changes from OC16 to OC16 Decay (Figure 7B middle, red to black) agree with the data (Figure 7B top, red to black), showing shifts of intensity from 288 to 260 cm−1 (exp 275 to 220-240 cm−1) and 612 to 590 cm−1 (exp 605 to 560-580 cm−1), and decay of the 357 cm−1 feature (exp 375 cm−1). The decay of the calculated 612 cm−1 and formation of the 590 cm−1 features are of particular interest, which in this model would represent the cryoreduced decay of the ν2 mode corresponding to the 605 cm−1 experimental NRVS feature in Q. In the cryoreduced calculation the 590 cm−1 feature corresponds to a localized ν(Fe2-OH) mode (Figure 7B, middle); relative to the ν2 mode in the Fe(IV)2 Q OC16 model (Figure 7B, middle), this vibration appears downshifted and uncoupled from νas(Fe-O-Fe) due elongation of the Fe2(III)-OH bond upon cryoreduction. This lowers the energy of the ν(Fe2-OH) mode and limits its mixing with the νas(Fe-O-Fe) mode. The calculated 590 cm−1 feature in OC16 Decay also contains contributions from νs(Fe-O-Fe) (ν4 Figure 4), which shifted up in energy (~ 40 cm−1) relative to OC16 due to a decrease of the Fe-O-Fe angle upon cryoreduction. However, both of these modes would be expected to exhibit signficant 18O2 perturbations as both oxygen atoms in the modes must be derived from O2 based on the rR assignment of the 690 cm−1 feature in the Fe(IV)2 structure.
The 18O2 spectrum for OC16 Decay (18O2 Q Decay; Figure 7B bottom, blue spectrum) was prepared by summing 40% of the 18O2 OC16 Fe2(III)Fe1(IV) and 60% of the 18O2 Fe(III)2 NRVS spectra (provided in Figure S26B, blue spectra). As anticipated, the NRVS spectrum for 18O2 Q Decay predicts a large (22 cm−1) isotope downshift for the OC16 Decay 590 cm−1 feature, which does not agree with the 4-7 cm−1 shift observed experimentally.
Note that from the analysis above, the ν4 mode would also exhibit a large isotope dependence in the cryoreduced model (Figure S26B, bottom) and can be excluded. Therefore, the remaining possible assignment for the 605 cm−1 feature in the experimental data is the ν3 mode (ν(Fe1-OH)) centered at ~ 690 cm−1 (note this is slightly upshifted relative to the BP86/10% HF calculation value of 666 cm−1). In the OC16 Decay spectrum this mode decreases in energy by 19 cm−1. This mode is not calculated to have an 18O2 isotope dependence in the 18O2 OC16 Decay spectrum (Figure 7B, bottom), since the O2 loading would occur on the Fe2-OH and μ-O oxygen atoms. Weak mixing with the ν4 mode could provide a small ν3 18O2 isotope shift as observed experimentally. However, the energy of the ν3 mode at > 650 cm−1 (Figure 7A, red column) is too high to reproduce the 605 cm−1 NRVS feature and to facilitate mixing with the ν4 mode. In principle, a model possessing a weaker Fe1-OH bond and thus a lower energy ν(Fe1-OH) mode could reproduce the Q 605 cm−1 NRVS feature and the 4-7 cm−1 isotope shift of the Q decay 580 cm−1 feature.
The approximate Fe1-OH bond length required to produce a 605 cm−1 ν(Fe1-OH) mode was calculated using a Badger’s rule analysis (Figure S28).60 The 605 cm−1 feature could be reproduced by a ν(Fe1-OH) mode corresponding to a 1.85 Å Fe-OH bond length. Models with hydrogen-bond donors to the Fe1-OH oxygen (OC9, OC13, OC21, OC33) result in modest elongation of the Fe-OH bond to 1.76 – 1.78 Å, and exhibit NRVS intensity above 660 cm−1 (see Table S1). Indeed, none of the 14 OC -OH/-OH models reproduce this long Fe1-OH bond corresponding to a ν(Fe1-OH) mode near 605 cm−1.
To summarize, the OC -OH/-OH subclass reasonably reproduces the low-energy (< 450 cm−1) region of the Q NRVS spectrum. However, the 18O2 4-7 cm−1 isotope shift of the 560-580 cm−1 feature in the Q decay data eliminates assignment of the Q 605 cm−1 NRVS feature to vibrations strictly containing motion of the in-plane Fe2-OH and μ-O ligands. The one plausible assignment of the 605 cm−1 feature in the OC -OH/-OH structure is the out-of-plane ν3 (ν(Fe1-OH)) mode; however, the calculated Fe1-OH bond length is too short in the OC -OH/-OH subclass (due to the relatively low donor strength of the ligands on Fe1) to produce a ν3 mode near this energy. The relatively high energy of this mode prevents mixing with the ν4 mode that would be required for the 18O2 4-7 cm−1 isotope shift of the Q decay 580 cm−1 feature. Though the assignment of Q to the OC -OH/-OH subclass is not well-supported by our calculations, we leave it as an open possibility pending further experimental evidence.
3.4.3. Closed core
The closed core subclasses available as possible assignments for Q are the bis(μO), bis(μO)-H2O, and bis(μO)-OH models. The subset of these models possessing an appropriate 16O18O mixed isotope shift include all 6 bis(μO) models (bis(μO)-1-6), 12 out of 16 of bis(μO)-H2O models (bis(μO)-H2O-1-4, 6, 8-12, 14-15), as well as 9 out of 16 of the bis(μO)-OH models (bis(μO)-OH-6, 8, 10-16). The NRVS spectra for all closed core models are included in Table S1 (along with their structures).
Of these remaining models, four models (bis(μO)-OH-6, bis(μO)-OH-8, bis(μO)-OH-10, bis(μO)-H2O-14) lack in their calculated NRVS spectra the characteristic 275 cm−1 signal present in the early Q data (Figure 8A, red spectrum) and are therefore excluded from further analysis. The calculated NRVS spectrum of bis(μO)-H2O-6 corresponds particularly well to the early Q NRVS data, and is representative of the NRVS spectral behavior of the possible bis(μO) and bis(μO)-H2O closed core models. Below 450 cm−1, the bis(μO)-H2O-6 NRVS spectrum (Figure 8B) exhibits one intense feature at 267 cm−1, which may be associated with the intense 275 cm−1 peak in the early Q data (Figure 8A, red spectrum). At higher energy, four peaks of moderate intensity are calculated at 306, 349, 378 and 413 cm−1, which would correspond to the features in the experimental data at 300, 330, 375, and 420 cm−1. Thus, below 450 cm−1, the calculated NRVS spectrum for bis(μO)-H2O-6 reproduces the early Q NRVS data reasonably well.
Figure 8.

Calculated NRVS spectra of bis(μO)-H2O-6 as representative of bis(μO) and bis(μO)-H2O subclasses. All spectra simulated with B3LYP. (A) Early Q – MMOHred (red) and Decayed Q – MMOHred (black) NRVS data. Arrows indicate major spectral differences. (B) Calculated NRVS spectrum of bis(μO)-H2O-6. (C) Calculated NRVS spectra bis(μO)-H2O-6 (red) and its simulated decay product bis(μO)-H2O-6 Decay (black). Arrows indicate major spectral differences. (D) Calculated NRVS spectra for bis(μO)-H2O-6 Decay (black) and its 18O2 isotopomer (blue).
Above 450 cm−1, two features of similar intensity are calculated at 614 and 631 cm−1. These two features correlate well to the early Q NRVS feature at 605 cm−1; indeed, all 23 possible closed core models possess features between 600 and 640 cm−1 which could correspond to the 605 cm−1 feature in the early Q NRVS data. In bis(μO)-H2O-6, the two features calculated at 614 and 631 cm−1 correspond to the B2u (ν3 Figure 4) and B3u (ν2 Figure 4) non-symmetric modes of the Fe2O2 core. The remaining high-energy Fe2O2 stretching mode, the Ag breathing mode of the core (ν1 Figure 4) that would be assigned to the 690 cm−1 rR vibration, is calculated at 693 cm−1 and is much weaker in NRVS than the non-symmetric modes. Therefore, at this point in the analysis, the high-energy (> 450 cm−1) region of the early Q NRVS data may reasonably be assigned to the non-symmetric Fe2O2 stretches of bis(μO)-H2O-6 (as well as in the other closed core models).
The bis(μO)-H2O-6 and related acceptable models were then further screened through NRVS calculations based on their cryoreducted DFT structures for correlation to the decayed Q data (Figure 8A, black spectrum). For the H2O-Fe2-(μ-O)2-Fe1 structure of bis(μO)-H2O-6, optimization to an Fe2(III)Fe1(IV) electronic structure was lower in energy by 2.8 kcal/mol. The decay spectrum for bis(μO)-H2O-6 (bis(μO)-H2O-6 Decay) is shown in Figure 8C (40% Fe2(III)Fe1(IV) and 60% Fe(III)2, their individual NRVS spectra are shown in Figure S29A (ii) and (iii)). The spectral differences between bis(μO)-H2O-6 and bis(μO)-H2O-6 Decay (Figure 8C red to black) correlate well to the decay of Q features in the beam (Figure 8A, red to black), which include decay of the intense 267 cm−1 feature (exp 275 cm−1) with growth of features between 190 and 215 cm−1 (exp 220-240 cm−1), decay of the 378 cm−1 feature (exp 375 cm−1), and a downshift in energy of the 631 cm−1 feature (exp 605 cm−1) to 560 cm−1 (exp 560 to 580 cm−1). Though the downshift of the calculated 631 cm−1 feature agrees with experiment, this mode corresponds to the ν2 non-symmetric stretch of the Fe2O2 core and is calculated to have an 18O2 isotopic shift (> 20 cm−1; Figure 8D) larger than accommodated by the experimental data (4-7 cm−1 from Figure 1B). However, the related closed core models with the axial H2O replace by an OH− are consistent with experiment.
Unlike the bis(μO) and bis(μO)-H2O subclasses, the bis(μO)-OH models possess an additional mode between 600-700 cm−1 corresponding to the ν(Fe-OH) stretching mode, and are therefore further evaluated. The calculated NRVS spectrum for bis(μO)-OH-12 is given in Figure 9B (and its structure is given in Figure 10). Below 500 cm−1, the calculated NRVS spectrum correlates well to the experimental data (Figure 9A, red spectrum), exhibiting one intense peak at 270 cm−1 (exp 275 cm−1), three additional features at 300 cm−1 (exp 300 cm−1), 336 cm−1 (exp 330 cm−1) and 381 cm−1 (exp 375 cm−1), and intensity at 428 and 467 cm−1 (exp 420 cm−1). Above 450 cm−1, the calculated spectrum shows a shoulder at 589 cm−1, followed by strong intensity between 604 - 621 cm−1 (with a shoulder at ~ 620 cm−1 and a weak feature at 692 cm−1. The intense peak at ~ 604 cm−1 corresponds to the ν(Fe-OH) mode, while the remaining intensity corresponds to the breathing mode (692 cm−1; assigned to the rR feature at 690 cm−1) and two of the non-symmetric core modes (589 and ~ 620 cm−1). Interestingly, this ν(Fe-OH) mode is downshifted by more than 50 cm−1 compared to the out-of-plane ν(Fe-OH) mode in the OC - OH/-OH subclass (Section 3.4.2). This reflects the longer Fe-OH bond length in the bis(μO)-OH (~ 1.83 A) vs. OC -OH/-OH (~ 1.77 A) models, due to the bis(μO) bridges providing a stronger donating equatorial core. From correlation to the experimental Q NRVS data, any one of the calculated three modes in the bis(μO)-OH-12 NRVS spectrum between 580 cm−1 and 630 cm−1 could correspond to the Q NRVS feature at 605 cm−1.
Figure 9.

Calculated NRVS spectra of bis(μO)-OH-12 as representative of the bis(μO)-OH subclass. All spectra simulated with B3LYP. (A) Early Q – MMOHred (red) and Decayed Q – MMOHred (black) NRVS data. Arrows indicate major spectral differences. (B) Calculated NRVS spectrum of bis(μO)-OH-12. (C) Calculated NRVS spectra for bis(μO)-OH-12 (red) and its simulated cryoreduced product (bis(μO)-OH-12 Decay in black; see text for details). Arrows indicate major spectral differences. (D) Calculated NRVS spectra for bis(μO)-OH-12 Decay (black) and its 18O2 isotopomer (blue).
Figure 10.

Structure for bis(μO)-OH-12.
The bis(μO)-OH-12 model was further evaluated by DFT calculated cryoreduction to Fe(III)Fe(IV) and Fe(III)2 electronic structures. Optimization on the Fe(III)Fe(IV) level yielded both Fe2(III)Fe1(IV) and Fe2(IV)Fe1(III) models (hydroxide bonded to Fe2) differing in energy by only 1.5 kcal/mol. Therefore, the NRVS spectra of both models are included in the analysis of the cryoreduction data. Additionally, optimization of bis(μO)-OH-12 to an Fe(III)2 structure resulted in proton transfer from Glu144 to the μ-O bridge it hydrogen-bonds to in the Q structure (Figure 10); structures and bond lengths of these cryoreduced models are found in Figure S30 and Table S13, respectively.
The cryoreduced NRVS spectrum of bis(μO)-OH-12 (bis(μO)-OH-12 Decay) was generated by combining 20%, 20%, and 60% of the bis(μO)-OH-12 Fe2(III)Fe1(IV), Fe2(IV)Fe1(III), and Fe(III)2 NRVS spectra (Figure S30 (ii), (iii), and (iv)). The spectral changes from bis(μO)-OH-12 to bis(μO)-OH-12 Decay (Figure 9C, red to black) correlate reasonably well to the changes in the Q NRVS spectrum observed in the beam (Figure 9A, red to black). Decay of the calculated feature at 270 cm−1 (exp 275 cm−1) is observed with concomitant growth of intensity between 180-215 cm−1 (exp 220-240 cm−1), as well as decay of a feature at 381 cm−1 (exp 375 cm−1). At high energy, a downshift of calculated intensity is observed in the 604-621 cm−1 region (exp 605 cm−1) to 560-583 cm−1 (exp 560-580 cm−1), corresponding to a decrease in the energies of the non-symmetric stretches and ν(Fe-OH) mode. Note that, in the distorted Fe(III)Fe(IV) and (μ-OH)(μ-O) Fe(III)2 structures of the cryoreduced models, the Fe-O core stretches localize (therefore, Fe-(μ-O) stretching modes are indicated as ν(Fe-O) in black in Figure 9C).
The calculated 18O2 NRVS spectrum of bis(μO)-OH-12 Decay (18O2 bis(μO)-OH-12 Decay) reveals a striking difference in the isotope sensitivity of the high energy region ~ 600 cm−1) relative to the other bis(μO) subclasses (Figure 9D black to blue). Vibrational modes with Fe-(μ-O) stretching motion above 500 cm−1 exhibit 18O2 downshifts larger than 20 cm−1, which are not observed in the experimental data (Δ = 4-7 cm−1) and is similar to the findings for the bis(μO) and bis(μO)-H2O subclasses. However, a significant peak at 583 cm−1 in the bis(μO)-OH-12 Decay NRVS spectrum does not exhibit an isotope shift in the calculated 18O2 spectrum (Figure 9D). This corresponds to the ν(Fe2-OH) mode, for which the hydroxide oxygen atom remains unlabeled upon 18O2 loading. Note that the limited 4-7 cm−1 isotope shift observed would reflect some mixing with the non-symmetric Fe2O2 core modes in this energy region (as observed in the calculated bis(μO)-OH-16 NRVS spectra: see Figure S31). Indeed, in comparison to all other subclasses of models analyzed, the bis(μO)-OH subclass best reproduces the two defining characteristics of the Q 605 cm−1 NRVS feature: the small 16O2 → 18O2 4-7 cm−1 isotope shift in the Q decay NRVS spectrum, which reflects the unlabeled Fe-OH bond; and the low energy of this ν(Fe-OH) stretch, reflecting the increased Fe2-OH bond length due to the strong equatorial (μ-O)2 donation in the bis(μO)-OH core. Hence, of the 90 models evaluated in this study, the closed core bis(μO)-OH subclass (Figure 10) best reproduces the Q and Q decay NRVS data in the higher energy region.
With the assignment of this subclass as the best candidate for the structure of Q, assignment of the Q and Q decay NRVS spectra is now provided (Figure 9B) using bis(μO)-OH-12. The most intense mode (Figure 9B) calculated at 270 cm−1 (exp 275 cm−1) corresponds to two ν(Fe-N) stretching modes. The decay of these features with concomitant growth of intensity between 180 - 215 cm−1 (exp 220-240 cm−1) in bis(μO)-OH-12 decay reflects elongation of the Fe-N bonds upon cryroeduction. The two features at 300 cm−1 (exp 300 cm−1) and 336 cm−1 (exp 330 cm−1) are assigned to the Ag accordion (ν5 Figure 4) and Au butterfly (ν6 Figure 4) modes of the closed core, respectively. These vibrations primarily reflect bending motions of the Fe2O2 core, with force constants less dependent on Fe-ligand bond lengths; thus, upon cryoreduction, the energy of these vibrations remains relatively unchanged. The feature at 381 cm−1 (exp 375 cm−1) is assigned to a ν(Fe-COO−) terminal carboxylate stretch, and decay of this feature reflects an elongation of the Fe-COO− bond upon cryoreduction. The two calculated peaks between 428 and 467 cm−1 (exp 420 cm−1) correspond to the Fe2O2 core B1g mode (ν4 Figure 4), mixed into symmetric and antisymmetric combinations with a nearby terminal carboxylate stretching mode. In the cryoreduced models, the structure distorts and the core modes localize (indicated in Figure 9C by ν(Fe-O) in black). The cryoreduced Fe(III)2 (μ-O)(μ-OH) model now has μ-OH antisymmetric νas(Fe-OH-Fe) and symmetric νs(Fe-OH-Fe) modes. The lowest energy ν(Fe-O) and νs(Fe-OH-Fe) modes are calculated to be between 400 and 450 cm−1, in a similar energy regime as the B1g mode in bis(μO)-OH-12, and exhibit an 18O2 downshift in 18O2 bis(μO)-OH-12 Decay (16 cm−1) that corresponds well to the isotope shift of the 420 cm−1 NRVS feature in Q decay (12-15 cm−1). Finally, as presented above, non-symmetric Fe2O2 core stretches as well as the ν(Fe2-OH) mode are calculated to be between 589 – 621 cm−1 in the Q model, which lower in energy to 560-583 cm−1 in bis(μO)-OH-12 Decay. However, from the data the ν(Fe2-OH) mode (calc ~ 604 cm−1) is mostly observed in the NRVS spectrum, based on the limited 16O2 and 18O2 sensitivity of the 560 – 580 cm−1 region in the Q decay data.
In the process of completing this study, a higher resolution XFEL structure was published (Srinivas et al., 2020)56 of MMOB-associated MMOH. Compared to the crystal structure (PDB: 4GAM) of the MMOB/MMOH complex used for the analysis herein,54 the XFEL-derived structure exhibits two oxygenic bridging ligands as well as monodentate ligation of Glu114, Glu243, and Glu209 (in contrast to their bidentate ligation in 4GAM, likely due to cryoreduction).56 Small differences in the Cα constraints are also observed in Glu114 (Δ = 0.80 Å) and Glu243 (Δ = 0.46 Å). The influence of these structural changes on our computational models was determined by generating bis(μO)-H2O-1 and subsequent models of Q from the XFEL-derived structure and resimulating their NRVS spectra (Figure S32). These XFEL-derived structures are very similar and their calculated NRVS spectra show negligible deviations from those used in this analysis.
3.5. Correlation to Reactivity
Intermediate Q in sMMO (Mt OB3b) abstracts an H atom from methane with a kinetic barrier ΔG‡ of 10.3 kcal/mol.31,61 For comparison, the mononuclear non-heme iron enzyme Fe(IV)=O intermediate in SyrB2 has a ΔG‡ ~ 17.7 kcal/mol for H-atom transfer (HAT) from threonine at the same temperature.62 This can be propagated to the stronger C-H bond of CH4 (cf. ΔHBDE(threonine) ~ 94 kcal/mol vs. ΔHBDE(CH4) ~ 104.5 kcal/mol). This 10 kcal/mol worse thermodynamics would raise the HAT reaction barrier between the Fe(IV)=O site in SyrB2 and CH4 to ~ 24 kcal/mol. Thus, the binuclear Fe(IV) intermediate Q is far more reactive than the mononuclear NH Fe(IV)=O intermediate. This substrate-adjusted kinetic barrier for SyrB2 is well reproduced by reaction coordinate calculations, which predict ΔG‡ ~ 22.1 kcal/mol for the CH4 reaction in Figure 11(a) and Table 1.
Figure 11.

Reaction coordinates calculated for hydrogen atom abstraction from methane by (a) the mononuclear Fe(IV)=O site in SyrB2, (b) the open core HO-Fe(IV)-μO-Fe(IV)=O site in OC26, (c) the open-core HO-Fe(IV)-μO-Fe(IV)-OH site in OC15, and (d) the closed core bis(μO)-bridged Fe(IV)2 site in bis(μO)-OH12. The Fe, C, N, O, Cl, and H atoms are displayed in green, gray, blue, red, yellow, and white, respectively.
Table 1.
Potential energy, enthalpy, and free energy changes and intrinsic barriers (in kcal/mol) involved in hydrogen atom abstraction from methane by models in Figure 11
| Systems | ΔE; ΔH; ΔG | ΔE‡; ΔH‡; ΔG‡ | ; ; |
|---|---|---|---|
| Fe(IV)=O | 17.1; 14.7; 14.3 | 25.1; 20.1; 22.1 | 15.3; 11.6; 14.0 |
| HO-Fe(IV)-μO-Fe(IV)=O | 10.8; 7.2; 10.4 | 24.8; 19.7; 25.0 | 19.0; 15.9; 19.4 |
| HO-Fe(IV)-μO-Fe(IV)-OH | 0.7; −0.3; −2.1 | 19.8; 14.8; 15.4 | 19.4; 14.9; 16.4 |
| Fe(IV)-(μO)2-Fe(IV)-OH** | 3.6; 2.1; −1.0 | 15.7; 11.2; 12.4 | 13.9; 10.1; 12.9 |
ΔG values are calculated at 5 °C.
ΔG and
ΔG values for Fe(IV)-(μO)2-Fe(IV)-OH2 are 3.1 and 11.1 kcal/mol.
Three binuclear models, OC26 (-OH/=O), OC15 (-OH/-OH), and bis(μO)-OH12, have been evaluated for their H-atom abstraction ability relative to this mononuclear site. The Fe(IV)=O site of the OC -OH/=O model is predicted to react with CH4 at a similar rate as SyrB2, with the calculated ΔG‡ ~ 25.0 kcal/mol. While this reaction appears more driven by the thermodynamics (ΔΔE ~ 6 kcal/mol and ΔΔG ~ 4 kcal/mol), this difference is due to an H bond that forms to the OH group of OC26 generated upon H-atom abstraction (HAA). Without this H-bond, the HAA product of the OC -OH/=O model is destabilized by 5 and 8 kcal/mol in potential energy and free energy, respectively (Figure S33). These energy profiles indicate that the activity of the Fe(IV)=O site is similar, regardless of whether it is present in a mononuclear or OC μ-O bridged binuclear site.
The H atom abstraction from CH4 by the Fe(IV)-OH site in the open-core -OH/-OH model has a kinetic barrier of ΔG‡ ~ 15.4 kcal/mol and the calculated thermodynamics of ΔG ~ −2.1 kcal/mol. These values are 9.6 and 12.5 kcal/mol lower than those of the Fe(IV)=O site in the OC -OH/=O model, respectively, showing that the Fe(IV)-OH system can be more reactive than the Fe(IV)=O system. When Marcus theory is used to remove the thermodynamic contribution to the barrier, the intrinsic barriers obtained for the Fe(IV)=O and Fe(IV)-OH systems are similar (last column in Table 1; Figure S34). Thus, the lower HAA barrier of the Fe(IV)-OH system is attributed to its greater thermodynamic driving force relative to the Fe(IV)=O system. Indeed, a hypothetical mononuclear Fe(IV)-OH site in SyrB2 was evaluated and gave similar thermodynamics to the -OH/-OH model (Figure S34). Upon HAA, the Mayer bond order between the Fe and the O decreases by 0.883 for the Fe(IV)=O to Fe(III)-OH conversion while it is 0.718 for the Fe(IV)-OH to Fe(III)-OH2 conversion, implying that the latter costs less energy and is thus more favorable. The calculated barrier for the open-core -OH/-OH model of ΔG‡ ~ 15.4 kcal/mol is comparable to the experimental barrier (ΔG‡ ~ 10.3 kcal/mol) measured for intermediate Q. Interestingly, the strong HAA reactivity of the M(IV)-OH unit (M = metal) agrees with findings from a recent study on corrole model complexes, in which the Mn(IV)-OH complex was found to be more reactive than the Mn(V)=O analog.63
The preferred closed core model based on the NRVS data contains the bis(μO) bridges and the terminal OH ligand, which differs from previously suggested models that instead contain a terminal water ligand or two μ-1,3-carboxylato bridges with no water-derived ligands.64-68 This closed core model is calculated to be much more reactive than the mononuclear Fe(IV)=O and OC -OH/=O models, with ΔG‡ ~ 12.4 kcal/mol and ΔG ~ −1.0 kcal/mol (Table 1). This low barrier agrees well with the experimental barrier of intermediate Q. The origin of this high HAA activity of the closed-core relative to the open-core systems reflects their key structural difference. As presented above, the open-core models are very similar to the corresponding mononuclear systems in terms of HAA activity. In contrast, the 2nd μ–O bridge of the closed core systems cooperates with the H atom-accepting μ–O bridge, signifying a key role of the 2nd Fe center in the HAA process. More details will be discussed below.
4. Discussion
In this study, we have systematically evaluated a significant range of open and closed core candidate structures of intermediate Q and assessed their viability in reproducing prior experimental as well as newly acquired NRVS data. Though the original 2.46 Å Fe-Fe distance favoring a closed core structure for Q has been determined to be an artifact,43 still reasonable evidence for this assignment was provided by resonance Raman spectroscopy28 that showed the presence of a 690 cm−1 vibration exhibiting an averaged 16O18O mixed isotope shift. Herein, our analysis of closed core models of Q has shown that a subset of these models also possessing carboxylate (bis(μO)), water (bis(μO)-H2O), and hydroxide (bis(μO)-OH) axial ligands would show a resonance-enhanced Raman vibration near 690 cm−1 with the appropriate isotope perturbations. From TD-DFT calculations of absorption spectra, the transitions associated with the 351 nm line excitation for the closed core structure correspond to charge transfers from both μ-O bridges to both Fe(IV) ions. Such an excited state would result in elongations of all four Fe-(μ-O) bonds, leading to resonance enhancement of the Ag breathing mode characteristic of M2(μ-O)2 (M = metal) cores29,30,69 (ν1 at 690 cm−1 in Fe(IV)2(μ-O)2, Figure 4, top). Incorporation of 16O18O into the (μ-O)2 core would provide the observed mixed isotope shift.
However, the analysis presented here shows that the experimental 690 cm−1 rR feature and its 16O18O shift are not necessarily indicative of a closed core geometry. Two subclasses of open core models have been identified that are predicted to reproduce the rR data for Q: HO-Fe2(IV)-O-Fe1(IV)=O (OC -OH/=O) and HO-Fe2(IV)-O-Fe1(IV)-OH (OC -OH/-OH; Fe1-OH vector oriented out of the Fe-O-Fe plane). Interestingly, the calculated TD-DFT absorption spectra for both subclasses (as well as the closed core subclasses) are similar, predicting a low-energy region (8,000 – 15,000 cm−1) comprised of Fe(IV) d-d transitions, followed by a region (15,000 – 23,000 cm−1) of μ-O and OH− LMCTs to the exchange-stabilized dx2-y2 LUMO. Above 23,000 cm−1 (and relevant to the 351 nm/28,500 cm−1 rR excitation) TD-DFT calculations predict μ-O and OH− charge transfer to localized dπ* orbitals of the Fe(IV)2 centers. Such an excited state would result in a symmetric elongation of the in-plane Fe2-OH and Fe2-(μ-O) bonds. In the OC -OH/=O and -OH/-OH subclasses, this excited state distortion would result in the resonance enhancement of the ν(Fe2-OH) + νas(Fe-O-Fe) symmetric stretching mode (ν2 Figure 4 bottom, and ν1 Figure 4 middle, respectively) near 700 cm−1 that would reproduce the 690 cm−1 experimental feature. Hence, the resonance Raman data for Q do not rule out an open core structural assignment.
Unlike resonance Raman, NRVS observes all vibrations of a 57Fe active site with iron motion and has been used in this study to probe the vibrational spectrum of intermediate Q up to 720 cm−1. The NRVS data of Q (Figure S3) exhibit time-dependent changes in the beam, most dramatically the rapid decay of an intense feature at 275 cm−1 in the early time data with concomitant growth of intensity between 220-240 cm−1 in later scans. Tracking this feature during the NRVS measurement allowed for quantitative chronological binning of the data and elucidation of intermediate Q vs. Q decay NRVS features (Figure 1A), which allowed identification of a key additional feature at 605 cm−1 associated with the Q NRVS spectrum that downshifted to 560 - 580 cm−1 in Q decay. The DFT simulations of the NRVS spectra for the Q models predict an intense low energy (< 300 cm−1) feature for both open and closed core structural subtypes due to Fe(IV)-N(His) vibrations; hence, the 275 cm−1 NRVS feature is a distinguishing feature of Q but is not characteristic of a particular structure. In contrast, these simulations have shown that the energy of the 605 cm−1 NRVS feature, as well as the 16O2 → 18O2 4 – 7 cm−1 isotope shift of the 560 - 580 cm−1 NRVS feature of Q decay (Figure 1B), does enable evaluation of possible structural subtypes for intermediate Q.
Indeed, the presence of this high-energy NRVS feature eliminates the assignment of an OC -OH/=O structure to Q. This subclass had been postulated as a structural candidate for Q based on the high C-H bond reactivity of an OC -OH/=O model complex relative to its closed core precursor,40 as well as the intense pre-edge region of Q that derives from 3d-4p mixing requiring a non-centrosymmetric environment, thought to reflect the terminal Fe(IV)=O unit in a μ-O bridged open core.42 However, the OC -OH/=O model calculations cannot reproduce both the experimental 605 cm−1 NRVS and 690 cm−1 rR features. A strong Fe1=O oxo bond would result in a long Fe1-(μ-O) and short Fe2-(μ-O) bond, resulting in a long Fe2-OH bond that would exhibit a stretching mode near 605 cm−1 (Figure 12); however, this would result in an isolated νas(Fe-O-Fe) mode above 700 cm−1 that would not reproduce the resonance-enhanced mixed isotope shift of the 690 cm−1 rR feature. A weak Fe1=O oxo bond (facilitated through hydrogen-bonding interactions) would reverse this effect, raising the energy of the ν(Fe2-OH) mode to near 690 cm−1 that allows its mixing with νas(Fe-O-Fe) thus reproducing the rR feature and its isotope shifts. However, in this case, no mode near 605 cm−1 would be observed that could be assigned to the NRVS feature in Q. Therefore, due to the donor strength of the terminal oxo and its distortion of the OC -OH/=O core, this subclass cannot accommodate both a 605 cm−1 NRVS and a 690 cm−1 rR feature. A strong oxo can distort the core to provide the NRVS feature, and a weak oxo can distort the core to reproduce the rR feature – but the presence of the oxo prevents both.
Figure 12.

Representation of the ligand donations required to produce a 605 cm−1 NRVS feature in the OC -OH/=O (left), OC -OH/-OH (middle), and bis(μO)-OH (right) subclasses. The direction of donation is symbolized with arrows. In the OC subclasses, the donation required to create a long Fe-OH bond to reproduce the low ν(Fe-OH) 605 cm−1 NRVS feature of Q results in the absence of a ν(Fe-OH) + νas(Fe-O-Fe) symmetric mode that would be assigned to the rR feature at 690 cm−1. In the bis(μO)-OH subclass, the (μ-O)2 core provides strong donation to facilitate a long Fe2-OH bond and thus a ν(Fe2-OH) mode near 605 cm−1, and the Ag breathing mode (ν1 Figure 4) satisfies the 690 cm−1 rR assignment.
The possibility of a new open core structural type, OC -OH/-OH, was further raised in this study and evaluated against the Q and Q decay NRVS data. With two Fe-OH bonds, this model could in principle account for both the 605 cm−1 NRVS feature and the small isotope shift of the Q decay 560 - 580 cm−1 feature (via the out-of-plane Fe1-OH, which has not incorporated O2 but could mix weakly with the μ-18O bridge), and the 690 cm−1 resonance Raman feature and its mixed isotope shifts (due to mixing of the O2-labeled in-plane Fe2-OH and μ-O modes). However, all OC -OH/-OH models evaluated in this study possess ν(Fe1-OH) modes above 650 cm−1 reflecting their relatively short Fe1-OH bonds (1.76 Å). Hydrogen bonding to the Fe1-OH oxygen resulted in a modest elongation of the bond, but did not result in a calculated ν(Fe1-OH) mode near 605 cm−1 that would require a ~ 1.85 Å Fe1-OH bond length. However, even if a long Fe1-OH bond was obtained, this would shorten the Fe1-(μ-O) bond and elongate the Fe2-(μ-O) bond, thus strengthening the Fe2-OH bond (Figure 12). A structure with short Fe2-OH and long Fe2-(μ-O) bonds would likely eliminate the ν(Fe2-OH) + νas(Fe-O-Fe) coupling required to reproduce the mixed isotope shift of the rR 690 cm−1 feature. While our study has not identified a viable OC -OH/-OH model that satisfies the isotope sensitivity of the Q 690 and Q decay 560 - 580 cm−1 features, we leave this model as a possible assignment for Q contingent on further experimental data.
Analysis of the predicted NRVS spectra for closed core models has yielded an assignment for Q that satisfies both the rR and NRVS data. Of all 90 DFT models considered in this report, the bis(μO)-OH subclass best reproduces the low energy of the 605 cm−1 feature in the Q NRVS spectrum and the 4-7 cm−1 18O2 isotope shift in the cryoreduced data (between 560 - 580 cm−1) and is our favored assignment for Q. The closed core bis(μO)-OH subclass possesses an additional mode near 605 cm−1 that corresponds to the ν(Fe2-OH) stretching mode. The low energy of this mode reflects the strongly donating (μ-O)2 equatorial ligand field, which results in an elongated Fe2-OH bond (~ 1.83 Å) compared to the OC -OH/-OH models (Figure 12). The ν(Fe2-OH) mode would show only limited 18O2 isotope dependence in the Q decay NRVS spectrum, since O2 would be loaded into the (μ-O)2 core and thus the isotope shift of this mode would be contingent on mixing with non-symmetric core modes. It should be qualified that this assignment of Q using NRVS assumes the DFT-calculated cryoreduced structures are reasonable allowing use of the isotope perturbations. However, in the DFT analysis of the cryoreduced data the NRVS spectra clearly exclude the OC -OH/=O subclass.
This assignment of Q is also consistent with past spectroscopic results. From Mössbauer spectroscopy, the two iron ions are fairly equivalent but show a measurable difference in the Mc Bath variant. Mössbauer calculations herein (Table S14) on a variety of models demonstrate that the isomer shifts are fairly similar for all subclasses, while the quadrupole splitting shows some variability in the difference (ΔEQ1-ΔEQ2). In the bis(μO)-OH subclass, this difference in quadrupole splitting between the two iron ions is found to primarily depend on the axial carboxylate Glu144-Fe2 interaction (Figure 10), in which a stronger interaction leads to a smaller ΔEQ1-ΔEQ2. From TD-DFT XAS calculations in the SI (Figure S35), the bis(μO)-OH subclass would be consistent with the increased pre-edge intensity of Q as long as at least one Fe(IV) is 5C. Additionally, while an active site with two 6C Fe(IV) would have a K-pre-edge spectrum with a well split dπ-dσpattern with only the dσ being intense, the presence of at least one 5C Fe(IV) site reduces the energy of its dz2 peak and produces additional dipole character in the dπ peak, consistent with the experimental spectrum (Figure S35). The Fe-Fe distance of 3.4 Å from HERFD-EXAFS43 is more of an issue. However, the published data contain 50-60% biferrous with a distance of 3.27 Å, and had been extrapolated from a narrow k range of 2 to 11 Å−1 which affects70 the Fourier transform that had been fit in the analysis. These fits show two Fe-Fe contributions at 3.05 and 3.30 Å. While the longer distance would rule out a closed core species, elongation of the Fe-Fe distance to 3.0 Å in bis(μO)-OH models costs 2-5 kcal/mol and shifts the resonance Raman active Ag breathing mode down by at least 40 cm−1 (with the correct isotope shifts; see Table S15). The NRVS spectra of these closed core Fe-Fe elongated models exhibit only modest changes near 600 cm−1, due to elongation of the Fe-μ-O bonds that shifts core modes to lower energy while shortening the ν(Fe2-OH) bond and increasing its vibrational energy by ~ 10 cm−1 (Figure S36).
The DFT-calculated HAA activity of the closed-core bis(μO)-OH models reproduces the experimental value for intermediate Q. Compared to the mononuclear non-heme Fe(IV)=O site in SyrB2, this closed core model has a lower kinetic barrier by ~10 kcal/mol for HAA from CH4 (Table 1). Given the similarity in intrinsic barriers, the lower barrier of the closed core model can be attributed to the greater thermodynamic driving force for HAA relative to the mononuclear Fe(IV)=O system (ΔΔG ~ −15 kcal/mol). Figure 13 illustrates the origin of this favorable thermodynamics of the binuclear closed-core system based on the concerted motion of the bis(μO) bridges. For the case of the mononuclear system, upon HAA, only the Fe(IV)=O bond significantly changes, while the other ligand-Fe bonds only show minor changes (Figure S37). Alternatively, for the closed-core models, all four Fe(IV)-μO bonds undergo significant variations upon HAA; while Fe2 and μOa in Figure 13 accept the electron and proton of the H atom, respectively, the Fe2-μOb bond is comparably weakened with a MBO decrease of 0.381. Notably, the weakening of the Fe2-μOb bond is compensated by forming a stronger μOb bond to the Fe1 center with an increase in MBO of 0.463 (Figure S38). If the Fe1-μO bonds are kept frozen during HAA, the reaction thermodynamics is predicted to become more endergonic by 12 kcal/mol (Figure 13(b), bottom), equivalent to that of the mononuclear system. Thus, the concerted motion of the bis(μO) bridges coupled to the second Fe center drives the HAA process. After HAA, the rebound of the CH3· radical can occur with a barrier of ΔG‡ ~ 8.4 kcal/mol, lower than the HAA barrier of ΔG‡ ~ 12.4 kcal/mol, via opening of the core with the dissociation of the Fe2(III)-μOa bond (Figure S39).
Figure 13.

H atom abstraction from CH4 by (a) the mononuclear Fe(IV)=O system and (b) the binuclear closed-core Fe2(μO)2 system with (top) and without (core frozen, bottom) the bis(μO) concerted motions. The electron-accepting Fe centers are displayed in green, while the redox-neutral Fe center is colored in orange.
For the case of the OC -OH/-OH model, this compensation effect from the second Fe center is not significant, as the OC -OH/-OH model is calculated to show similar thermodynamics to that of a hypothetical mononuclear Fe(IV)-OH system (Figure S34). Based on its similar calculated low-energy ΔG‡ (15.4 vs. 12.4 kcal/mol for the closed core), the -OH/-OH model could also be a candidate for HAT from CH4. Importantly, the reactivity of the two terminal OH ligands would be constrained by the O2 labeling results. From the rR data, one oxygen atom from O2 remains in the μO bridge after HAT, while the other oxygen atom from O2 is incorporated into the CH3OH product.32 Since the HAT and the hydroxyl rebound cannot occur from the same OH group, the OH group that abstracts the H atom from CH4 must be water-derived. However, unlike the bridging oxygen, the terminal OH ligands can undergo isotope scrambling, which was not observed in the rR data.28 Further, this HAT-active OH group would also have to be out of the Fe-μO-Fe plane, because the in-plane OH group must be derived from O2 to reproduce the 16O18O mixed isotope effect for the rR feature28 at 690 cm−1. Therefore, to match existing data, the out-of-plane OH group of the -OH/-OH model would have to abstract the H atom, while the in-plane OH group would need to react with the CH3 radical to form CH3OH. However, if CH4 can access the in-plane OH group, it is also able to perform HAT with an even lower barrier of ΔG‡ ~ 10.2 kcal/mol (Figure S40). Thus, while the binuclear nature of the site might enable the formation of the Fe(IV)-OH group in the -OH/-OH model, which is capable of HAT and rebound to form CH3OH with a low barrier (Figure S41), there would have to be limited terminal OH− ligand exchange and major positional control of non-polar molecules (CH4 and CH3·) by the active-site pocket.
While finalizing this manuscript, another study was published which computationally evaluates previous data on intermediate Q.71 This study raised the idea that the resonance Raman data of Q can be consistent with an open core structure, which is consistent with our results. However, the authors of this work favor an OC -OH/=O active site geometry, inconsistent with our NRVS data and in particular the 605 cm−1 NRVS feature that excludes this assignment of Q. Additionally, we calculate that this open core structural subclass possesses similar reactivity towards methane as a mononuclear Fe(IV)=O system, inconsistent with the higher reactivity of Q relative to mononuclear non-heme iron enzymes. Alternatively, we do find that closed core models predict larger reactivity due to the compensating effect of the spectator μ-oxo bridge, and additionally find that the OC -OH/-OH subclass may also reproduce the high reactivity of intermediate Q.
5. Conclusion
In this study, NRVS has been used to probe all vibrational modes of intermediate Q and its decay product having Fe motion, thus providing important complimentary information to findings from other spectroscopic techniques. Analyzing these data with DFT calculations provides an experimentally supported geometric/electronic structure for Q that has been combined with kinetic data to obtain insight into its unique reactivity. The analysis herein supports a closed bis(μO) Fe(IV)2 core for Q, where its high reactivity reflects the compensating effect of the μ–O bridge not involved in the H-atom abstraction. This application of NRVS to intermediate Q highlights the power of this technique in characterizing transient intermediates in iron metalloenzymes.
Supplementary Material
Acknowledgements
The authors acknowledge the financial support of this work from U.S. Grants NIH GM 118030 (to J.D.L.) and NIH GM 40392 (to E.I.S.), as well as from the National Research Foundation of Korea (NRF-2018R1C1B6007430 to K.P.). Synchrotron experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposals 2011B1267, 2012A1295, 2013B0105, 2018A0137, 2018B1046, 2018B0137, 2019A1275).
Footnotes
Supporting Information
Materials and Methods. Figures S1-S44 and Tables S1-S15. Additional data and figures including Mössbauer spectra speciation fits, decay of Q NRVS features over time, DFT model used for Q optimizations, DFT vibrational analysis, TD-DFT calibration, TD-DFT natural transition orbital (NTO) analyses, molecular orbital diagrams, additional TD-DFT calculated spectra, TD-DFT calculated linear coupling term (LCT) analyses, DFT correlation of B3LYP and BP86/10% HF functionals, DFT calculated NRVS spectra for cryoreduced Q models, DFT calculated NRVS spectra and structures for 90 Q models including predicted resonance Raman vibrational signals and Fe-Fe distances, reaction coordinate calculations for mononuclear, open core, and closed core Q models, and TD-DFT analysis of Q Fe-K-pre-edge region.
References
- (1).Andrews SC The Ferritin-like Superfamily: Evolution of the Biological Iron Storeman from a Rubrerythrin-like Ancestor. Biochim. Biophys. Acta 2010, 1800 (8), 691–705. 10.1016/j.bbagen.2010.05.010. [DOI] [PubMed] [Google Scholar]
- (2).Xing G; Hoffart LM; Diao Y; Prabhu KS; Arner RJ; Reddy CC; Krebs C; Bollinger JM A Coupled Dinuclear Iron Cluster That Is Perturbed by Substrate Binding in Myo-Inositol Oxygenase. Biochemistry 2006, 45 (17), 5393–5401. 10.1021/bi0519607. [DOI] [PubMed] [Google Scholar]
- (3).Hankes LV; Politzer WM; Touster O; Anderson L Myo-Inositol Catabolism in Human Pentosurics: The Predominant Role of the Glucuronate-Xylulose-Pentose Phosphate Pathway. Ann. N. Y. Acad. Sci 1970, 165 (2), 564–576. 10.1111/j.1749-6632.1970.tb56424.x. [DOI] [PubMed] [Google Scholar]
- (4).Schirmer A; Rude MA; Li X; Popova E; del Cardayre SB Microbial Biosynthesis of Alkanes. Science 2010, 329, 559–562. [DOI] [PubMed] [Google Scholar]
- (5).Fishman A; Tao Y; Bentley WE; Wood TK Protein Engineering of Toluene 4-Monooxygenase of Pseudomonas Mendocina KR1 for Synthesizing 4-Nitrocatechol from Nitrobenzene. Biotechnol. Bioeng 2004, 87 (6), 779–790. 10.1002/bit.20185. [DOI] [PubMed] [Google Scholar]
- (6).Yang B; Hodgkinson A; Millward BA; Demaine AG High Glucose-Induced DNA-Binding Activities of Nuclear Factor of Activated T Cells 5 and Carbohydrate Response Element Binding Protein to the Myo-Inositol Oxygenase Gene Are Inhibited by Sorbinil in Peripheral Blood Mononuclear Cells from Patients with Ty. Int. J. Diabetes Mellit 2010, 2 (3), 169–174. https://doi.org/ 10.1016/j.ijdm.2010.08.005. [DOI] [Google Scholar]
- (7).Shao J; Zhou B; Chu B; Yen Y Ribonucleotide Reductase Inhibitors and Future Drug Design. Curr. Cancer Drug Targets 2006, 6 (5), 409–431. [DOI] [PubMed] [Google Scholar]
- (8).Lee SW; Keeney DR; Lim DH; Dispirito AA; Semrau JD Mixed Pollutant Degradation by Methylosinus Trichosporium OB3b Expressing Either Soluble or Particulate Methane Monooxygenase: Can the Tortoise Beat the Hare. Appl. Environ. Microbiol 2006, 72 (12), 7503–7509. 10.1128/AEM.01604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Choi YS; Zhang H; Brunzelle JS; Nair SK; Zhao H In Vitro Reconstitution and Crystal Structure of P-Aminobenzoate N-Oxygenase (AurF) Involved in Aureothin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (19), 6858–6863. 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sazinsky MH; Bard J; Di Donato A; Lippard SJ Crystal Structure of the Toluene/o-Xylene Monooxygenase Hydroxylase from Pseudomonas Stutzeri OX1: Insight Into The Substrate Specificity, Substrate Channeling, And Active Site Tuning Of Multicomponent Monooxygenases. J. Biol. Chem 2004, 279 (29), 30600–30610. 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- (11).Tinberg CE; Lippard SJ Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res 2011, 44 (4), 280–288. 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Austin RN; Chang H; Zylstra GJ; Groves JT The Non-Heme Diiron Alkane Monooxygenase of Pseudomonas Oleovorans (AlkB) Hydroxylates via a Substrate Radical Intermediate. J. Am. Chem. Soc 2000, 122, 11747–11748. [Google Scholar]
- (13).Solomon EI; Park K Structure/Function Correlations over Binuclear Non-Heme Iron Active Sites. J. Biol. Inorg. Chem 2016, 21, 575–588. 10.1007/s00775-016-1372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jasniewski AJ; Que L Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118 (5), 2554–2592. 10.1021/acs.chemrev.7b00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hanson RS; Hanson TE Methanotrophic Bacteria. Microbiol. Rev 1996, 60 (2), 439–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Banerjee R; Jones JC; Lipscomb JD Soluble Methane Monooxygenase. Annu. Rev. Biochem 2019, 88 (1), 409–431. 10.1146/annurev-biochem-013118-111529. [DOI] [PubMed] [Google Scholar]
- (17).Fox BG; Froland WA; Dege JE; Lipscomb JD Methane Monooxygenase from Methylosinus Trichosporium OB3b. Purification and Properties of a Three-Component System with High Specific Activity from a Type II Methanotroph. J. Biol. Chem 1989, 264 (17), 10023–10033. [PubMed] [Google Scholar]
- (18).Pilkington SJ; Dalton H Soluble Methane Monooxygenase from Methylococcus Capsulatus Bath. Methods Enzymol. 1990, 188, 181–190. 10.1016/0076-6879(90)88032-6. [DOI] [Google Scholar]
- (19).Lee SK; Nesheim JC; Lipscomb JD Transient Intermediates of the Methane Monooxygenase Catalytic Cycle. J. Biol. Chem 1993, 268 (29), 21569–21577. [PubMed] [Google Scholar]
- (20).Liu Y; Nesheim JC; Lee SK; Lipscomb JD Gating Effects of Component B on Oxygen Activation by the Methane Monooxygenase Hydroxylase Component. J. Biol. Chem 1995, 270 (42), 24662–24665. 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- (21).Liu KE; Valentine AM; Wang D; Huynh BH; Edmondson DE; Salifoglou A; Lippard SJ Kinetic and Spectroscopic Characterization of Intermediates and Component Interactions in Reactions of Methane Monooxygenase From Methylococcus-Capsulatus (Bath). J. Amer. Chem. Soc 1995, 117 (7), 10174–10185. 10.1021/ja00146a002. [DOI] [Google Scholar]
- (22).Banerjee R; Meier KK; Münck E; Lipscomb JD Intermediate P* from Soluble Methane Monooxygenase Contains a Diferrous Cluster. Biochemistry 2013, 52 (25), 4331–4342. 10.1021/bi400182y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lee S-K; Lipscomb JD Oxygen Activation Catalyzed by Methane Monooxygenase Hydroxylase Component: Proton Delivery during the O-O Bond Cleavage Steps. Biochemistry 1999, 38 (14), 4423–4432. 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- (24).Lee S-K; Fox BG; Froland WA; Lipscomb JD; Münck E A Transient Intermediate of Methane Monooygenase Catalytic Cycle Containing an FeIVFeIV Cluster. J. Am. Chem. Soc 1993, 115, 6450–6451. 10.1021/ja00067a086. [DOI] [Google Scholar]
- (25).Xing G; Diao Y; Hoffart LM; Barr EW; Prabhu KS; Arner RJ; Reddy CC; Krebs C; Bollinger JM Evidence for C-H Cleavage by an Iron-Superoxide Complex in the Glycol Cleavage Reaction Catalyzed by Myo-Inositol Oxygenase. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (16), 6130–6135. 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Acheson JF; Bailey LJ; Brunold TC; Fox BG In-Crystal Reaction Cycle of a Toluene- Bound Diiron Hydroxylase. Nature 2017, 1–15. 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- (27).Shu L; Nesheim JC; Kauffmann K; Münck E; Lipscomb JD; Que L An Fe2IVO2 Diamond Core Structure for the Key Intermediate Q of Methane Monooxygenase. Science 1997, 275, 515–518. 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- (28).Banerjee R; Proshlyakov Y; Lipscomb JD; Proshlyakov DA Structure of the Key Species in the Enzymatic Oxidation of Methane to Methanol. Nature 2015, 518, 431–434. 10.1038/nature14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xue G; Wang D; De Hont R; Fiedler AT; Shan X; Münck E; Que L A Synthetic Precedent for the [FeIV2(Mu-O)2] Diamond Core Proposed for Methane Monooxygenase Intermediate Q. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (52), 20713–20718. 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wilkinson EC; Dong Y; Zang Y; Fujii H; Fraczkiewicz R; Fraczkiewicz G; Czernuszewicz RS; Que L Raman Signature of the Fe2O2 “diamond” Core. J. Am. Chem. Soc 1998, 120 (5), 955–962. 10.1021/ja973220u. [DOI] [Google Scholar]
- (31).Brazeau BJ; Lipscomb JD Kinetics and Activation Thermodynamics of Methane Monooxygenase Compound Q Formation and Reaction with Substrates. Biochemistry 2000, 39, 13503–13515. 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- (32).Nesheim JC; Lipscomb JD Large Kinetic Isotope Effects in Methane Oxidation Catalyzed by Methane Monooxygenase: Evidence for C-H Bond Cleavage in a Reaction Cycle Intermediate. Biochemistry 1996, 35 (31), 10240–10247. 10.1021/bi960596w. [DOI] [PubMed] [Google Scholar]
- (33).Dunietz BD; Beachy MD; Cao Y; Whittington DA; Lippard SJ; Friesner RA Large Scale Ab Initio Quantum Chemical Calculation of the Intermediates in the Soluble Methane Monooxygenase Catalytic Cycle. J. Am. Chem. Soc 2000, 122 (12), 2828–2839. 10.1021/ja9920967. [DOI] [Google Scholar]
- (34).Basch H; Mogi K; Musaev DG; Morokuma K Mechanism of the Methane → Methanol Conversion Reaction Catalyzed by Methane Monooxygenase: A Density Functional Study. J. Am. Chem. Soc 1999, 121 (31), 7249–7256. 10.1021/ja9906296. [DOI] [Google Scholar]
- (35).Siegbahn PEM; Crabtree RH; Nordlund P Mechanism of Methane Monooxygenase - A Structural and Quantum Chemical Perspective. J. Biol. Inorg. Chem 1998, 3 (3), 314–317. 10.1007/s007750050238. [DOI] [Google Scholar]
- (36).Yoshizawa K Two-Step Concerted Mechanism for Methane Hydroxylation on the Diiron Active Site of Soluble Methane Monooxygenase. J. Inorg. Biochem 2000, 78 (1), 23–34. 10.1016/S0162-0134(99)00201-9. [DOI] [PubMed] [Google Scholar]
- (37).Yoshizawa K; Yumura T A Non-Radical Mechanism for Methane Hydroxylation at the Diiron Active Site of Soluble Methane Monooxygenase. Chem. - A Eur. J 2003, 9 (10), 2347–2358. 10.1002/chem.200204269. [DOI] [PubMed] [Google Scholar]
- (38).Huang S-P; Shiota Y; Yoshizawa K DFT Study of the Mechanism for Methane Hydroxylation by Soluble Methane Monooxygenase (SMMO): Effects of Oxidation State, Spin State, and Coordination Number. Dalt. Trans 2013, 42 (4), 1011–1023. 10.1039/c2dt31304a. [DOI] [PubMed] [Google Scholar]
- (39).Baik MH; Gherman BF; Friesner RA; Lippard SJ Hydroxylation of Methane by Non-Heme Diiron Enzymes: Molecular Orbital Analysis of C-H Bond Activation by Reactive Intermediate Q. J. Am. Chem. Soc 2002, 124 (49), 14608–14615. 10.1021/ja026794u. [DOI] [PubMed] [Google Scholar]
- (40).Xue G; De Hont R; Münck E; Que L Million-Fold Activation of the [Fe(2)(Micro-O)(2)] Diamond Core for C-H Bond Cleavage. Nat. Chem 2010, 2 (5), 400–405. 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).De Hont RF; Xue G; Hendrich MP; Que L; Bominaar EL; Münck E Mössbauer, Electron Paramagnetic Resonance, and Density Functional Theory Studies of Synthetic S = 1/2 Fe(III)-O-Fe(IV)=O Complexes. Superexchange-Mediated Spin Transition at the Fe(IV)=O Site. Inorg. Chem 2010, 49 (18), 8310–8322. 10.1021/ic100870v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Castillo RG; Banerjee R; Allpress CJ; Rohde GT; Bill E; Que L; Lipscomb JD; DeBeer S, High-Energy-Resolution Fluorescence-Detected X-Ray Absorption of the Q Intermediate of Soluble Methane Monooxygenase. J. Am. Chem. Soc 2017, 139 (49), 18024–18033. 10.1021/jacs.7b09560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cutsail GE; Banerjee R; Zhou A; Que L; Lipscomb JD; DeBeer S High-Resolution Extended X-Ray Absorption Fine Structure Analysis Provides Evidence for a Longer Fe· · ·Fe Distance in the Q Intermediate of Methane Monooxygenase. J. Am. Chem. Soc 2018, 140, 16807–16820. 10.1021/jacs.8b10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sturhahn W Nuclear Resonant Spectroscopy. J. Phys. Condens. Matter 2004, 16 (5), S497–S530. 10.1088/0953-8984/16/5/009. [DOI] [Google Scholar]
- (45).Seto M; Yoda Y; Kikuta S; Zhang X; Ando M Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett 1995, 74 (19), 3828–3831. [DOI] [PubMed] [Google Scholar]
- (46).Scheidt WR; Durbin SM; Sage JT Nuclear Resonance Vibrational Spectroscopy--NRVS. J. Inorg. Biochem 2005, 99 (1), 60–71. 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- (47).Park K; Li N; Kwak Y; Srnec M; Bell CB; Liu LV; Wong SD; Yoda Y; Kitao S; Seto M; Hu M; Zhao J; Krebs C; Bollinger JM; Solomon EI Peroxide Activation for Electrophilic Reactivity by the Binuclear Non-Heme Iron Enzyme AurF. J. Am. Chem. Soc 2017, 139 (20), 7062–7070. 10.1021/jacs.7b02997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell CB; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM; Solomon EI Elucidation of the Fe(IV)=O Intermediate in the Catalytic Cycle of the Halogenase SyrB2. Nature 2013, 499 (7458), 320–323. 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Park K; Bell CB; Liu LV; Wang D; Xue G; Kwak Y; Wong SD; Light KM; Zhao J; Alp EE; Yoda Y; Saito M; Kobayashi Y; Ohta T; Seto M; Que L; Solomon EI Nuclear Resonance Vibrational Spectroscopic and Computational Study of High-Valent Diiron Complexes Relevant to Enzyme Intermediates. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (16), 6275–6280. 10.1073/pnas.1304238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bell CB; Wong SD; Xiao Y; Klinker EJ; Tenderholt AL; Smith MC; Rohde JU; Que L; Cramer SP; Solomon EI A Combined NRVS and DFT Study of FeIV=O Model Complexes: A Diagnostic Method for the Elucidation of Non-Heme Iron Enzyme Intermediates. Angew. Chemie - Int. Ed 2008, 47 (47), 9071–9074. 10.1002/anie.200803740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Snyder BER; Böttger LH; Bols ML; Yan JJ; Rhoda HM; Jacobs AB; Hu MY; Zhao J; Ercan Alp E; Hedman B; Hodgson KO; Schoonheydt RA; Sels BF; Solomon EI Structural Characterization of a Non-Heme Iron Active Site in Zeolites That Hydroxylates Methane. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (18), 4565–4570. 10.1073/pnas.1721717115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Valentine AM; Tavares P; Pereira AS; Davydov R; Krebs C; Hoffman BM; Edmondson DE; Boi Hanh Huynh; Lippard SJ Generation of a Mixed-Valent Fe(III)Fe(IV) Form of Intermediate Q in the Reaction Cycle of Soluble Methane Monooxygenase, an Analog of Intermediate X in Ribonucleotide Reductase R2 Assembly. J. Am. Chem. Soc 1998, 120 (9), 2190–2191. 10.1021/ja974169x. [DOI] [Google Scholar]
- (53).Sturgeon BE; Burdi D; Chen S; Huynh BH; Edmondson DE; Stubbe J; Hoffman BM Reconsideration of X, the Diiron Intermediate Formed during Cofactor Assembly in E. Coli Ribonucleotide Reductase. J. Am. Chem. Soc 1996, 118 (32), 7551–7557. 10.1021/ja960399k. [DOI] [Google Scholar]
- (54).Lee SJ; McCormick MS; Lippard SJ; Cho U-S Control of Substrate Access to the Active Site in Methane Monooxygenase. Nature 2013, 494 (7437), 380–384. 10.1038/nature11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Elango NA; Radhakrishnan R; Froland WA; Wallar BJ; Earhart CA; Lipscomb JD; Ohlendorf DH Crystal Structure of the Hydroxylase Component of Methane Monooxygenase from Methylosinus Trichosporium OB3b. Protein Sci. 1997, 6 (3), 556–568. 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Srinivas V; Banerjee R; Lebrette H; Jones JC; Aurelius O; Kim IS; Pham CC; Gul S; Sutherlin KD; Bhowmick A; John J; Bozkurt E; Fransson T; Aller P; Butryn A; Bogacz I; Simon P; Keable S; Britz A; Tono K; Kim KS; Park SY; Lee SJ; Park J; Alonso-Mori R; Fuller FD; Batyuk A; Brewster AS; Bergmann U; Sauter NK; Orville AM; Yachandra VK; Yano J; Lipscomb JD; Kern J; Högbom M High-Resolution XFEL Structure of the Soluble Methane Monooxygenase Hydroxylase Complex with Its Regulatory Component at Ambient Temperature in Two Oxidation States. J. Am. Chem. Soc 2020, 142 (33), 14249–14266. 10.1021/jacs.0c05613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Schulz CE; Castillo RG; Pantazis DA; Debeer S; Neese F Structure-Spectroscopy Correlations for Intermediate Q of Soluble Methane Monooxygenase: Insights from QM/MM Calculations. J. Am. Chem. Soc 2021, 143 (17), 6560–6577. 10.1021/jacs.1c01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Laurent AD; Jacquemin D TD-DFT Benchmarks: A Review. Int. J. Quantum Chem 2013, 113 (17), 2019–2039. 10.1002/qua.24438. [DOI] [Google Scholar]
- (59).Sanders-Loehr J; Wheeler WD; Shiemke AK; Averill BA; Loehr TM Electronic and Raman Spectroscopic Properties of Oxo-Bridged Dinuclear Iron Centers in Proteins and Model Compounds. J. Am. Chem. Soc 1989, 111, 8084–8093. 10.1021/ja00203a003. [DOI] [Google Scholar]
- (60).Gordy W A Relation between Bond Force Constants, Bond Orders, Bond Lengths, and the Electronegativities of the Bonded Atoms. J. Chem. Phys 1946, 14 (5), 305–320. 10.1063/1.1724138. [DOI] [Google Scholar]
- (61).Brazeau BJ; Wallar BJ; Lipscomb JD Unmasking of Deuterium Kinetic Isotope Effects on the Methane Monooxygenase Compound Q Reaction by Site-Directed Mutagenesis of Component B. J. Am. Chem. Soc 2001, 123 (42), 10421–10422. 10.1021/ja016632i. [DOI] [PubMed] [Google Scholar]
- (62).Matthews ML; Krest CM; Barr EW; Vaillancourt H; Walsh CT Substrate-Triggered Formation and Remarkable Stability of the C-H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zaragoza JPT; Siegler MA; Goldberg DP A Reactive Manganese(IV)-Hydroxide Complex: A Missing Intermediate in Hydrogen Atom Transfer by High-Valent Metal-Oxo Porphyrinoid Compounds. J. Am. Chem. Soc 2018, 140 (12), 4380–4390. 10.1021/jacs.8b00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Siegbahn PEM O-O Bond Cleavage and Alkane Hydroxylation in Methane Monooxygenase. J. Biol. Inorg. Chem 2001, 6 (1), 27–45. 10.1007/s007750000184. [DOI] [PubMed] [Google Scholar]
- (65).Baik MH; Newcomb M; Friesner RA; Lippard SJ Mechanistic Studies on the Hydroxylation of Methane by Methane Monooxygenase. Chem. Rev 2003, 103 (6), 2385–2419. 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- (66).Gherman BF; Dunietz BD; Whittington DA; Lippard SJ; Friesner RA Activation of the C-H Bond of Methane by Intermediate Q of Methane Monooxygenase: A Theoretical Study. J. Am. Chem. Soc 2001, 123 (16), 3836–3837. 10.1021/ja0055108. [DOI] [PubMed] [Google Scholar]
- (67).Gherman BF; Lippard SJ; Friesner RA Substrate Hydroxylation in Methane Monooxygenase: Quantitative Modeling via Mixed Quantum Mechanics/Molecular Mechanics Techniques. J. Am. Chem. Soc 2005, 127 (3), 1025–1037. 10.1021/ja049847b. [DOI] [PubMed] [Google Scholar]
- (68).Musaev DG; Basch H; Morokuma K Theoretical Study of the Mechanism of Alkane Hydroxylation and Ethylene Epoxidation Reactions Catalyzed by Diiron Bis-Oxo Complexes. The Effect of Substrate Molecules. J. Am. Chem. Soc 2002, 124 (15), 4135–4148. 10.1021/ja0176393. [DOI] [PubMed] [Google Scholar]
- (69).Baldwin MJ; Roo DE; Pate JE; Fujisawa K; Kitajima N; Solomon EI Spectroscopic Studies of Side-On Peroxide-Bridged Binuclear Copper(II) Model Complexes of Relevance to Oxyhemocyanin and Oxytyrosinase. J. Am. Chem. Soc 1992, 114 (26), 10421–10431. 10.1021/ja00052a043. [DOI] [Google Scholar]
- (70).Dassama LMK; Silakov A; Krest CM; Calixto JC; Krebs C; Bollinger JM; Green MTA 2.8 Å Fe─Fe Separation in the Fe2 III/IV Intermediate, X, from Escherichia Coli Ribonucleotide Reductase. J. Am. Chem. Soc 2013, 135, 16758–16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Schulz CE; Castillo RG; Pantazis DA; Debeer S; Neese F Structure – Spectroscopy Correlations for Intermediate Q of Soluble Methane Monooxygenase : Insights from QM / MM Calculations. 2021. 10.1021/jacs.1c01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.