

Abstract
Wiedemann-Steiner syndrome (WSS) is an autosomal dominant disorder caused by monoallelic variants in KMT2A and characterized by intellectual disability and hypertrichosis. We performed a retrospective, multicenter, observational study of 104 individuals with WSS from five continents to characterize the clinical and molecular spectrum of WSS in diverse populations, to identify physical features that may be more prevalent in White versus Black Indigenous People of Color individuals, to delineate genotype–phenotype correlations, to define developmental milestones, to describe the syndrome through adulthood, and to examine clinicians' differential diagnoses. Sixty-nine of the 82 variants (84%) observed in the study were not previously reported in the literature. Common clinical features identified in the cohort included: developmental delay or intellectual disability (97%), constipation (63.8%), failure to thrive (67.7%), feeding difficulties (66.3%), hypertrichosis cubiti (57%), short stature (57.8%), and vertebral anomalies (46.9%). The median ages at walking and first words were 20 months and 18 months, respectively. Hypotonia was associated with loss of function (LoF) variants, and seizures were associated with non-LoF variants. This study identifies genotype–phenotype correlations as well as race-facial feature associations in an ethnically diverse cohort, and accurately defines developmental trajectories, medical comorbidities, and long-term outcomes in individuals with WSS.
Keywords: hypertrichosis, KMT2A, MLL1, syndromic intellectual disability, syndromic short stature, Wiedemann-Steiner syndrome
1 ∣. INTRODUCTION
Familial hypertrichosis cubiti or “hairy elbows” syndrome (MIM#605130) was first described in 1970 in a father and two of his five children (Beighton, 1970). Wiedemann subsequently described a Caucasian male with growth deficiency, developmental delay (DD), and physical features including: a round and flat face, short nose, hypertelorism, long philtrum, short palpebral fissures, low-set ears, high-arched palate, short and thick limbs, an alternating convergent strabismus, and dilatation of the renal calyces (Wiedemann et al., 1989). Although similar individuals were reported in the intervening years, it was not until 2000 that Steiner and colleagues reported a female with growth deficiency, DD, distinctive features and hypertrichosis (Steiner & Marques, 2000) and suggested a common diagnosis with Wiedemann's originally reported family. Wiedemann-Steiner syndrome (WSS, MIM#605130), as it is now known, is caused by monoallelic pathogenic variants of KMT2A (previously MLL1, ALL1) and characterized by DD and characteristic facial features (Aggarwal et al., 2017; Baer et al., 2018; Jones et al., 2012; Koenig et al., 2010). The KMT2A gene product is a complex transcriptional regulatory protein that is the mammalian homolog of Drosophila Trithorax, which regulates hox expression. The facial phenotype is characterized by thick eyebrows, long eyelashes, wide nasal bridge, down-slanting palpebral fissures, narrow palpebral fissures, hypertelorism, broad nasal tip, thin vermillion border, and thick hair (Aggarwal et al., 2017; Baer et al., 2018; Jones et al., 2012; Koenig et al., 2010).
Hypertrichosis cubiti was thought to be pathognomonic for this syndrome, but the adoption of genome-scale diagnostics has revealed hypertrichosis in as few as 60% of individuals with WSS (Aggarwal et al., 2017; Baer et al., 2018; Strom et al., 2014). Other clinical features include feeding difficulties, prenatal and postnatal growth retardation, epilepsy including infantile spasms, ophthalmological anomalies, congenital heart disease, hand anomalies such as brachydactyly and clinodactyly, musculoskeletal anomalies such as hypotonia and vertebral anomalies, and renal and uterine anomalies (Aggarwal et al., 2017; Baer et al., 2018; Giangiobbe et al., 2020; Koenig et al., 2010; Li et al., 2018). Immune dysfunction, including early-onset combined variable immune deficiency, recurrent pulmonary infection, and death from sepsis have been reported (Bogaert et al., 2017; Bramswig et al., 2015; Stellacci et al., 2016). Other rare neurological findings, such as syringomyelia (Pavone et al., 2015) periventricular nodular heterotopia (Baer et al., 2018), and cerebral hemisphere asymmetry (Steel et al., 2015) have been noted. Outcomes of two adults have been detailed (Feldman et al., 2019). Previous reports have noted the association of missense variants with a more severe phenotype, however, no prior study has evaluated the statistical significance of the type of variant with a specific phenotypic feature (Chan et al., 2019; Li et al., 2018).
Although small case series and individual reports of participants from various racial and ethnic backgrounds have been reported, there has not yet been a comprehensive analysis of WSS in diverse populations (Arora et al., 2017; Baer et al., 2018; Grangeia et al., 2019; Li et al., 2018; Ramirez-Montaño & Pachajoa, 2019). Here, we present a diverse cohort of 104 individuals with WSS from five continents to more fully define the molecular spectrum, medical comorbidities, developmental trajectories, and long-term outcomes for individuals with WSS.
2 ∣. MATERIALS AND METHODS
2.1 ∣. Ethics statement
This study was approved by the Institutional Review Board (IRB) at the Children's Hospital of Philadelphia (CHOP) and University of California Los Angeles. Per the protocol, participants were enrolled in the IRB approved study at CHOP or informed consent was obtained locally to avoid the transfer of sensitive information. The participant or participant's parent/guardian provided informed consent for the publication of identifiable photographs. The data are not publicly available due to privacy or ethical restrictions.
2.2 ∣. Inclusion criteria
Inclusion criteria included molecularly confirmed WSS (pathogenic or likely pathogenic variant in KMT2A by ACMG criteria) and evaluation by a medical geneticist, or known familial variant with the phenotype of WSS. Patients with variants of uncertain significance (VUS) were included if WSS was suspected before molecular testing and the phenotype was consistent with WSS. At the time of revisions, all VUSes were reviewed by re-contacting with the clinical laboratory that issued the original report (c.3200G > A, c.7097A > C) or by pathogenicity re-assessment (c.11840A > G, c.3199C > T, c.3508 T > C, c.3539G > T, c.6563C > G). The pathogenicity re-assessment was performed by a molecular laboratory director according to American College of Medical Genetics and Genomics guidelines.
2.3 ∣. Clinical information
The participant's primary geneticist provided de-identified clinical data to the primary investigators for analysis. If a clinician indicated that a particular feature was “possible,” this feature was labeled as not determined. The Fenton growth curve was used for individuals with a history of premature birth, the WHO growth curve was used between birth and 2 years, and the CDC (length, weight) and Nellhaus (head circumference) growth curves were used for participants older than 2 years old. For intelligence and developmental quotient and milestone information, if a range was provided, the mean was used for analysis. In cases where no composite score was given, but some sub-scores (e.g., verbal and nonverbal) were provided, the mean of available scores was used.
2.4 ∣. Growth parameter analysis
Clinicians were asked to provide a birth weight and percentile. To standardize measurements, data were transformed to percentiles. Birth weight measurements with gestational age values were preferred and transformed to percentiles using sex-matched Fenton growth charts. If only a birth weight value without gestational age was available, the data point was omitted. Clinicians were also asked to provide age at last examination along with weight, height, and head circumference measurements. Some provided percentile and Z scores as well. To standardize measurements, each available value was transformed into a corresponding measurement value (kg or cm) using the childsds package in the R Statistical Programing language. If a measurement was provided, this value was preferred, followed by a z-score, and finally percentile. If there were notable inconsistencies between the measurement, percentile, z-score and reported age, the data point was omitted.
2.5 ∣. Statistical analysis
All statistical analysis was performed using the R Statistical Programming Language. p-values were adjusted for multiple comparisons using the method of Bonferroni where appropriate. Due to small sample size, participants were grouped into larger categories of race for statistical significance (Table S1). Frameshift, nonsense, and splice site variants were classified as LoF variants. Missense variants and the single in-frame deletion (indel) were classified as non-LoF variants.
3 ∣. RESULTS
3.1 ∣. Demographics
The study consisted of 104 participants with equal gender participation (Table S2). Individuals ranged from 4 months old to 43 years old (y.o.). The median age was 8.5 y.o. at the time of last evaluation. The majority (77.9%) of participants were less than 18 y.o. The participants were from eight regions (United States, Australia, Mexico, Canada, Hong Kong, Saudi Arabia, West Indies, and Israel). Race and ethnicity were self-reported.
3.2 ∣. Molecular genetics
The majority of the variants were detected by exome sequencing. There were 82 total variants seen in the 104 participants in the study (Figure 1, Table S3), and the majority of them were novel (69 of 82). Novel variants were defined as not previously reported in the literature. These variants were distributed throughout the gene and did not cluster in any specific domain. Recurrent variants were predominantly familial, although there were also novel and previously reported recurrent variants. Most variants were frameshift (37.8%) followed by nonsense (29.3%). Missense made up 20.7% of the variants in the cohort. Splice site variants were seen in 11% of participants. A single participant had an inframe deletion (c.4426_4428del p.[C1476del]). There were eight participants with a VUS in the study (six variants). In these cases, pathogenicity was affected by lack of parental testing (e.g., could not confirm de novo status) (c.11840A > G, c.3199C > T, c.3508 T > C, c.3539G > T) or because the variant was inherited from an affected parent (c.7097A > C; c.3200G > A) (Table S3). A clinical laboratory reported the c.7097A > C variant as a VUS and “likely positive.” Eighty of the 82 variants were not found in gnomAD V2.1.1 (Karczewski et al., 2020.). A Caucasian individual with microcephaly, height and weight less than 10 percentile, mild intellectual disability (ID), hypertrichosis of the back and lower limbs, failure to thrive, constipation, and rib anomalies had the c.10835 + 1G > C variant identified on clinical exome sequencing (parents were unavailable for testing to confirm if the variant was de novo). This variant is present in gnomAD in five European (non-Finnish) individuals at 1.99e-5 allele frequency (Karczewski et al., 2020). A different variant at this site had previously been reported in an individual with WSS (c.10835 + 1G > A[Bogaert et al., 2017]). The missense variant c.6563C > G p.S2188C was present in two Latin individuals in gnomAD at 8.04e-6 allele frequency (Karczewski et al., 2020). This variant was identified in a Mexican father and two sons suspected of having WSS prior to identification of this variant by genome sequencing.
FIGURE 1.

KMT2A variants in this study. (a). The variants were mapped to NM_001197104 using MutationMapper (Cerami et al., 2012). Green variants are missense, black variants are truncating, red variant is indel, and purple variant is other. (b, c, d) tables demonstrating the method to detect variant, the type, and its inheritance. Of the 82 variants, 69 were novel variants of which three occurred at a previously reported location. Thirteen variants were previously reported
The majority of individuals identified in the study had confirmed de novo variants (55.8%). Twelve participants had inherited variants from an affected parent. Ten participants had negative testing from either the mother or the father with the other parent unavailable for testing. About one-quarter of the participants have inheritance undetermined due to parental unavailability (adoption/death/parent not involved), lack of insurance coverage or other unspecified reasons.
3.3 ∣. Facial features
We evaluated the descriptive facial features of the participants in photographs (Figure 2). The majority of participants had vertically narrow palpebral fissures (69.3%), hypertelorism (67.0%), and wide nasal bridge (63.4%) with broad or bulbous tip (63.6%). About half of the participants had a lateral (or other) flare to the eyebrow (47.7%), down-slanting palpebral fissures (49.5%) and blepharoptosis (43.0%). About a quarter of participants had an exaggerated cupid's bow (21.6%) or thin vermillion border (29.0%) to the upper lip and posteriorly rotated ears (28.3%). White individuals were significantly more likely to have bifid uvula (X2 [2, N = 85] = 12.59, p = 0.018, Bonferroni correction) and trended toward being more likely to have a thin upper vermillion border of the lip (X2 [2, N = 92] = 9.78, p = 0.075, Bonferroni correction). Black Indigenous People of Color (BIPOC) participants seemed more likely to have exaggerated cupid's bow of the lip, although this was not significant after correcting for multiple comparisons (X2 [2, N = 93] = 7.34, p = 0.26, Bonferroni correction).
FIGURE 2.




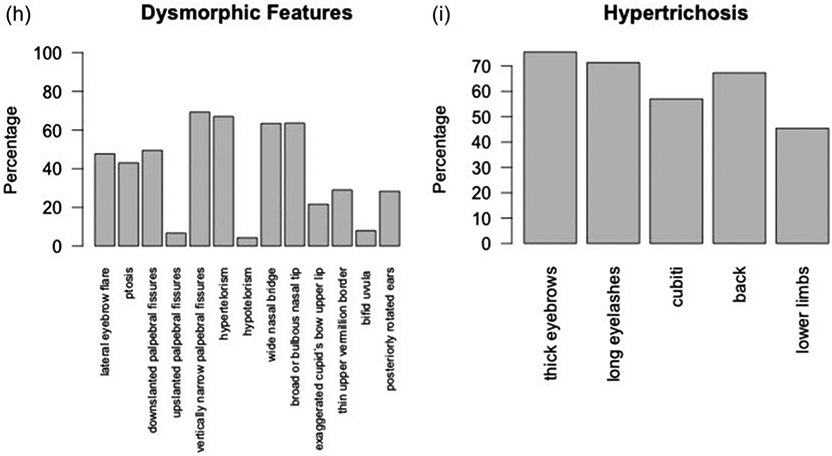
Characteristic features. (a) Front and side photographs of individuals with WSS. (b) Familial groups with WSS. Three Caucasian siblings. Caucasian father and son. Mother and daughter of African descent. Caucasian father and daughter. (c) WSS individuals as they age. Black boxes denote different individuals. Range of hypertrichosis on (d) elbows and knees and (e) backs. (f) Hands. (g) Feet. (h) Facial features include lateral eyebrow flare (42/88), ptosis (40/93), down-slanted palpebral fissures (51/103), up-slanted palpebral fissures (7/103), vertically narrow palpebral fissures (70/101), hypertelorism (63/94), hypotelorism (4/94), wide nasal bridge (64/101), broad or bulbous nasal tip (63/99), exaggerated cupid's bow upper lip (21/97), thin upper vermillion border (47/96), bifid uvula (7/89), and posteriorly rotated ears (28/99). (i) Features of hypertrichosis include thick eyebrows (77/102), long eyelashes (72/101), hypertrichosis cubiti (57/100), hypertrichosis back (68/101), and hypertrichosis lower limbs (44/97)
3.4 ∣. Hypertrichosis
The face was the most common location for hypertrichosis as evidenced by thick eyebrows (75.5%) and long eyelashes (71.3%) (Figure 2). Over half of the participants had hypertrichosis of the back (67.3%) and hypertrichosis cubiti (57%). Hypertrichosis of the lower limbs was seen in 45.4% of participants. Although there were differences in hypertrichosis by race, with overall lower frequency in individuals of African descent and higher frequency in individuals of Asian descent (Table S4); the difference was not statistically significant when compared for white versus BIPOC.
3.5 ∣. Anthropometrics
We examined the anthropometrics for the participants with WSS (Figure 3a,b). The majority of the participants were born at birth weight below the 25th percentile. At the most recent examination, about one-third of participants (37.9%) had a weight less than 5th percentile for age, more than half of the participants (57.4%) had height less than 5th percentile and about one-third of participants (34.6%) had measured head circumference less than the 5th percentile. There were very few participants with anthropometrics greater than 95th percentile (5.3% for weight, 1.0% for height, 3.7% for head circumference). The individual with height greater than 95th percentile had the following KMT2A variant: p.A1030Gfs*9. The individuals with head circumference greater than 95th percentile had the following KMT2A variants: p.R1067W, p.K1751*, p.K1219Vfs*2.
FIGURE 3.

Clinical features of participants, including growth parameter analysis. (a). Birth weight. the reported birth weight percentile is presented with the fill color of the histogram bar indicating the gender of the participant. (b) Growth parameters over time by gender. Each point indicates a participant's growth parameter at the most recent examination. Participants older than 20 years of age at the most recent evaluation were excluded. the black lines indicate in each panel indicate the 1st, 5th, 25th, 50th, 75th, 95th, and 99th percentiles. (c) Clinical features. Each box denotes a single participant. The colored boxes denote affected, the dark gray boxes denote unaffected, the light gray boxes indicate data not available. For quantification, see Table S5
3.6 ∣. Medical issues
We surveyed the medical issues and congenital anomalies in participants (Figure 3c, Table S5).
Neuropsychiatric differences were common in the cohort. Almost all participants had DD or ID (97%). Other common features included hypotonia (72.4%) poor sleep not due to an anatomical issue (46.2%), hyperactivity (44.3%), aggressive behavior (33.0%), and autism spectrum disorder (21.3%). Seizures were seen in 20.0% of individuals surveyed for this feature. A structural brain abnormality was seen in 57.5% of those who underwent brain imaging (n = 52); the most common abnormalities were abnormal corpus callosum (13.5%) or abnormal myelination (13.5%) (Table S5, legend). Sacral anomaly, most commonly a sacral dimple, was seen in 50.5% of participants. There was one participant with spina bifida occulta. Three individuals had a tethered cord that required surgery.
There were a wide variety of ophthalmologic abnormalities in the cohort. The most common ophthalmologic abnormality was strabismus (37.5%), including three individuals who required surgery, followed by astigmatism (20.5%). Surgical correction of blepharoptosis was performed in three individuals.
Over half (57.7%) of participants had a dental issue, the most common of which was advanced dental age (21.8%) characterized by premature loss of deciduous teeth and emergence of secondary teeth at an atypical earlier age.
Almost two-thirds of the participants (63.0%) had a pulmonary or ENT issue. Obstructive sleep apnea was most common, occurring in 24.7% of participants and necessitating tonsillectomy and adenoidectomy in 17.3% of participants.
Cardiac abnormalities were identified in 29 of 81 participants (35.8%) who underwent cardiac evaluation. The majority were structural cardiac anomalies seen in 28.4% of participants, although two participants each had arrhythmia (one who required pacemaker) and pulmonary hypertension.
About two-thirds of the participants had a history of failure to thrive (FTT) (67.7%), with feeding difficulties seen in 66.3% and tube feeds required in 25.5% of participants. Constipation was also seen in 63.8% of participants and was more commonly associated with FTT.
Abnormalities in the genitourinary system were seen in almost half of the participants (46.8%). Renal anomaly was seen in 28.6%, uterine or testicular anomaly seen in 16.9% and an external genital anomaly was seen in 11.7%. Three individuals had macrogenitalia and two individuals had absent uteri.
Endocrine laboratory evaluations were performed in 32 participants. About one-third (34.4%) of participants had premature adrenarche; about one-fifth (18.8%) had growth hormone (GH) deficiency defined by low serum growth hormone, IGF-1 or GH stimulation test. Five participants had normal GH. Three participants received GH therapy, and GH therapy was recommended to three others who did not receive it. Of 11 participants evaluated by pituitary MRI, an abnormality was identified in seven (63.6%) (Table S5, legend).
Bone age X-rays were performed in 29 participants for short stature evaluation. Bone age was normal 55.2%, advanced in 24.1% and delayed in 13.8%. The remainder were disharmonic (e.g., advanced at one point, delayed at another). There was not enough data to draw conclusions about correlation of bone age with growth hormone deficiency.
The most common musculoskeletal anomaly was a vertebral anomaly seen in about half of participants (46.9%), most commonly a fusion anomaly in the cervical spine. Rib anomalies, most commonly fewer number of rib pairs, hypoplastic appearance of ribs, or cervical ribs, were seen in about a third of participants (33.9%). Approximately a quarter of participants had broad first digits (22.4%) and/or tapering fingers (27.7%). One-fifth (21.3%) of participants had scoliosis, and one required surgery. Four participants had pectus excavatum. There were five participants with hip dysplasia (none were breech at birth). Four required surgery and one required a Pavlik harness for treatment. One individual had metopic craniosynostosis.
Thirteen participants completed immunology laboratory evaluations. Seven participants (53.8%) had abnormal immunoglobulin levels. Four participants (30.8%) had insufficient response to pneumococcal vaccinations. There were 18 participants (25.7%) with recurrent infections.
There were two participants with recurrent fevers of unknown origin. No cancers were reported in this cohort.
3.7 ∣. Development
Developmental milestones were delayed for most of the reported cases Figure 4. The median age at sitting was 10 months (range 6–36 months). The median age at standing was 17 months (range 8–60 months). There were six participants (1.25–21 y.o.) who were unable to stand. Two of these individuals had vertebral anomalies. The median age at walking was 20 months (11–60 months). There were nine participants (1.25–21 y.o.) who still were not walking. The median age at first words was 18 months (range 8–60 months). There were 10 participants (2–20 y.o.) who were nonverbal. The 20 y.o. nonverbal, nonambulatory individual had bilateral hip subluxation, extreme varus deformity of feet, bilateral valgus deformity of femurs, and cylindric, and foreshortened thoracic and lumbar vertebral bodies and also epilepsy that may have contributed. Diagnosis for this individual was made by exome sequencing, suggesting a secondary diagnosis was unlikely.
FIGURE 4.

Developmental milestones and IQ. (a) Motor milestones. (b) Speech milestones. (c) IQ distribution
Twenty-nine participants had formal neurodevelopmental evaluations including the full-scale intelligence quotient (FSIQ) or developmental quotient (DQ) in 25 of the 29 (Figure 4). Of the four not included, one participant was “thought to be likely in the normal range,” another was noted to have “mild ID and learning disabilities,” another was noted to be “extremely low” for attention and executive functions, and the fourth had inconclusive testing because lack of cooperation.
3.8 ∣. Adults with WSS
There were 23 participants aged 18 y.o. or older in the study to provide information about school outcome and careers. Of 18 participants with information about schooling, 17 completed high school. One participant who did not complete high school was in mainstream classes at 5 y.o. and then transitioned to special education until 12 y.o. Many of these adult participants had required educational assistance. Eight had modified curriculum or were in special education, one was home schooled, one is nonverbal and nonambulatory (though did crawl at 4 y.o.) and participated in home hospital school, and another participant passed the tests of General Educational Development (a series of four subject tests taken to demonstrate their high school academic knowledge in the United States). Three participants attended some tertiary education (university/college), including one who is still in college and doing well without academic assistance.
We ascertained information about the daytime activities, jobs, or other outcomes for these 23 adult participants. One individual is currently in college and does not have a job yet. Another individual started college, but did not finish and now owns a construction business. Other jobs included stocking shelves at a retail store, stocking shelves at physical therapy, and mailroom delivery. Ten adults did not have jobs. One adult was too impaired for a job, another attends a daytime rehabilitation program, one is a stay at home mother and is able to care for her medically complex son with WSS, one is married and lives independently, but was unable to care for her children. In this case, however, concerns about the participant's ability to care for children may have been complicated by failure to thrive in the childhood, who were later also diagnosed with WSS.
3.9 ∣. Families with WSS
We had eight families with WSS participate in this study including six parent child pairs, one father and two affected sons, and another mother and six affected children. In general, the developmental disabilities seemed to be similar or slightly more delayed in children with an inherited variant. Children trended toward a higher number of congenital anomalies compared with their parents (mean 1.8 vs. mean 0.6) but this was not statistically significant (p = 0.50, two tailed t test with unequal variance).
3.10 ∣. Genotype phenotype correlation
Pearson's chi-squared test was used to identify genotype–phenotype correlation between participants with predicted KMT2A LoF variants (n = 64) and those with other variants (n = 18). Participants with LoF variants were more likely to have hypotonia (X2 [2, N = 87] = 12.57, p = 0.045, Bonferroni correction). In contrast, participants with non-LOF variants were more likely to have seizures (X2 [2, N = 75] = 19.88, p = 0.0012, Bonferroni correction). No significant correlation was identified for birthweight or growth parameters less than 10th percentile; failure to thrive; tube feeding; constipation; vertebral anomaly; rib anomaly; scoliosis; hypertrichosis cubiti; hypertrichosis of the back; hypertrichosis of the lower limbs; any eye abnormality (excluding blepharoptosis); cardiovascular anomaly; endocrine anomaly; abnormal brain or pituitary imaging; immune deficiency, genitourinary anomaly; dental anomaly; pulmonary/otolaryngological anomaly; autism, aggressive behavior; poor sleep; or hyperactivity.
3.11 ∣. Differential diagnosis
The most common conditions in the differential diagnosis of the individuals before molecular analysis in this cohort were WSS, copy number variant (CNV), Cornelia deLange (CdLS), and Noonan syndrome (Figure S1). Within the cohort, there were two individuals with dual diagnoses; one with CNV (hg18 arr12q24.33(130,050,620-130,316,774) x3,19p13.3(1,846,340-2403,185)x1), and one participant with neurofibromatosis 1 (NF1) due to maternally inherited NF1 c.2533 T > C; p. Cys845Arg.
4 ∣. DISCUSSION
We present the largest study of individuals with WSS to date, almost doubling the number of individuals previously reported. Our participants have 69 novel, previously unreported KMT2A variants. Our study included diverse participants from Canada, Israel, West Indies, Saudi Arabia, Mexico, Hong Kong, Australia, and the United States. Approximately 40% of participants were BIPOC. We identified new genotype–phenotype correlations, described correlations between facial features of WSS and race, and delineated new characteristics of the disease that suggest new management recommendations.
Interestingly, two variants found in individuals with classic features of WSS were also present in gnomAD, including one missense variant. There may be individuals with WSS in gnomAD who have no or mild cognitive issues. For example, the missense variant was inherited from a mildly affected parent who did not report any ID until specifically asked about school. Another possibility is that these variants in gnomAD are the consequence of clonal hematopoiesis of indeterminate potential as a result of aging (Jaiswal & Ebert, 2019). Epigenetic signatures may be a way to help interpret missense variants or other variants with unclear significance (Aref-Eshghi et al., 2020).
Evaluation of genetic syndromes in diverse populations is essential for syndrome recognition and diagnosis (Koretzky et al., 2016; Kruszka et al., 2019). A limitation of our study is individuals self-reported ancestry (Kaseniit et al., 2020). Genetically defined ancestry would have been more accurate, but was outside the scope of this investigation (Wang et al., 2015). Nonetheless, participants' photographs from different ethnic backgrounds will be very helpful to the clinician assessing patients in clinic. Importantly, our study is the first to identify features more likely to be seen in white individuals (thin upper vermillion border of the lip and bifid uvula) and BIPOC individuals (exaggerated cupid's bow of the lip). Further work is needed to assess for trends in facial phenotype of indigenous groups, however our study provides a large photographic comparison of the facial features.
Due to syndromic short stature, hypertrichosis with long eyelashes and prominent eyebrows, WSS has phenotypic overlap with CdLS, Kabuki syndrome, Rubinstein-Taybi syndrome, and Coffin-Siris syndrome (Aoi et al., 2019; Bramswig et al., 2015; Homma et al., 2019; Negri et al., 2019). Homozygous LoF variants in TASP1, which cleaves and activates KMT2A, should be considered in the differential diagnosis of WSS due to the overlapping phenotype of DD, hypotonia, microcephaly, feeding difficulties with failure-to-thrive, recurrent respiratory infections, cardiovascular malformations, happy demeanor, and distinctive facial features (Suleiman et al., 2019). Two patients in this cohort had previously been provisionally diagnosed with Dubowitz syndrome, a well-established recognizable malformation syndrome, that has increasingly been recognized to be clinically diverse and etiologically heterogeneous (Innes et al., 2018). Features frequently associated with Dubowitz syndrome including mild to moderate growth and developmental issues, distinctive facial features (including short or narrow palpebral fissures), behavioral issues including hyperactivity and nonspecific immunological findings area also over-represented in this WSS patient cohort, and therefore pathogenic variants in KMT2A can be added to the list of potential diagnoses when a physician is considering a diagnosis of Dubowitz syndrome.
As reported previously, among individuals with WSS, in this cohort prenatal and postnatal growth failure were common issues. Growth hormone deficiency as well as feeding difficulties may be responsible for the short stature seen in over half the cohort, though it is possible that a distinct mechanism is involved in the growth retardation/short stature in WSS, similar to Kabuki syndrome 1 (Fahrner et al., 2019). We confirmed macrocephaly as a rare feature in three participants, as was previously reported in one individual (Li et al., 2018). The individual with a coexisting diagnosis of NF1 did not have macrocephaly.
Neurodevelopmental disabilities were prominent in our cohort and at a higher incidence compared with a previous meta-analysis (Chan et al., 2019). Our population showed DD or ID (97%), hypotonia (72.4%), abnormal brain imaging (57.5%), poor sleep (46.2%), and hyperactivity (44.3%), and aggressive behavior (33.0%). In comparison, a previous meta-analysis of 127 individuals in the literature found ID or DD in 59.1% and 48.8%, respectively, hypotonia in 30.7% of individuals, and central nervous system malformations in 16.5%, poor sleep in just over 10%, hyperactivity and ADHD in less than 10%, and aggressive behavior in less than 15% of individuals. This discrepancy may be due to the lack of detailed information available in case reports and case series compared with the primary records used in our study. Many of the children are described as sweet and happy by their caregivers; however, this was not formally surveyed. Our findings suggest that these neurodevelopmental features are prominent in individuals of WSS with all ages. Future work including formal neurodevelopmental testing and brain imaging will evaluate these components in an unbiased manner. Notably, we also confirm the prior association with periventricular nodular heterotopia, and the variants are only a few base pairs apart (Baer et al., 2018). Importantly, we were able to identify genotype–phenotype associations for hypotonia and seizures. Finally, our study provides detailed information about neurodevelopmental, educational, and occupational outcomes in adults with WSS to add to the three prior cases in the literature (Feldman et al., 2019; Li et al., 2018).
Other medical issues recognized in this large cohort include osteopenia, high incidence of tube feeding and GI motility issues, cardiac arrhythmia including one participant requiring a pacemaker, macrogenitalia, absent uteri, hip dysplasia requiring surgery, tethered cord requiring surgery, and early loss of deciduous teeth and emergence of secondary dentition. A comparison of the features in our cohort to previous studies is available in Table S5. Notably the frequency of cardiac arrhythmia and hip dysplasia in our WSS cohort is higher than the general population (0.02% vs. 1.9%; 0.6% vs. 4.8%) (Reidy, Collins, MacLean, & Campbell, 2019; Turn & Wren, 2013). Thus, we suggest modifications to the previous clinical care recommendations (Table 1) (Baer et al., 2018).
TABLE 1.
Clinical care recommendations to consider
| Neurology | EEG if signs of seizure, non-LoF variants, or other clinical concern; consider brain MRI |
| Ophthalmology | To screen for strabismus (esotropia or exotropia), astigmatism, cataract; consideration of oculoplastic surgery for blepharoptosis |
| Otolaryngology/sleep study | To evaluate for sleep apnea |
| Immunology | To assess immunoglobulins and response to vaccines, and further evaluation if recurrent infections |
| Cardiology | Echocardiogram to evaluate for congenital heart disease, EKG to screen for arrhythmia |
| Endocrinology | Growth hormone assessment, thyroid assessment, and evaluation for metabolic bone disease |
| Gastrointestinal | Monitor growth and feeding difficulties, constipation evaluation; Abdominal ultrasound |
| Developmental Pediatrics | To monitor development and facilitate additional services |
| Physical therapy, occupational therapy, speech therapy, early intervention | To maximize developmental outcome |
| Orthopedics | To evaluate for vertebral anomalies by dedicated spine imaging with particular attention to C-spine; consideration of evaluation for hip dysplasia |
| Neurosurgery | MRI for tethered cord if concerns |
| Pediatric dentistry | To evaluate early loss of deciduous teeth and emergence of secondary dentition |
| Genitourinary | Abdominal ultrasound including survey of pelvis to screen for genitourinary anomalies |
In our cohort of 104 participants (including 23 adults with the oldest in their 40s), there have not been any reported cancers. Murine knockout studies demonstrate a role for KMT2A in hematopoiesis and axial skeletal patterning (Hess et al., 1997; Yu et al., 1995). Over 90 fusions involving KMT2A have been implicated in about 10% of all leukemias in both the pediatric and adult population (Meyer et al., 2018; Winters & Bernt, 2017). While seen in all age groups, leukemias with KMT2A translocations have a predilection to occur in infants and young children and as secondary leukemia after chemotherapy for a first cancer with a Topoisomerases II poison. The translocations cause disturbances in epigenetic regulation and disordered gene transcription. The translocations in the infant cases are in utero events with short latency to overt leukemia(Ford et al., 1993; Megonigal et al., 1998). The resulting fusion proteins are potent oncoproteins and few, if any, additional somatic mutations are required for leukemogenesis (Andersson et al., 2015; Bolouri et al., 2018). Cooperation between the fusion oncoprotein and its wild-type counterpart has been suggested (Thiel et al., 2010). WSS is likely due to haploinsufficiency. The finding of immunological disturbances in patients with WSS is consistent with the role of KMT2A in hematopoietic development (Rao & Dou, 2015). While the mutations in WSS are heterogeneously distributed throughout the gene, the leukemia-associated translocations cluster in a breakpoint cluster region between exons 7 and 13 (NCBI numbering system). While a complete blood count could be considered at diagnosis for screening or if symptoms arise, there is no current evidence to support or negate recommendations surrounding cancer screening.
In summary, we present the largest study of WSS participants including 69 novel variants, developmental milestones and detailed neurodevelopmental outcomes in adults, genotype–phenotype associations, and novel medical findings leading to modified clinical care recommendations.
Supplementary Material
ACKNOWLEDGMENTS
We thank the participants and their families. We also thank the members of the Center for Applied Genomics, CHOP Division of Human Genetics, Roberts Collaborative, and UCLA Clinical Genomics Center. We thank Jessica Douglas MS CGC of Boston Children's Feingold Center and Harvard Undiagnosed Disease Network and Hessa Saad Alsaif of King Faisal Specialist Hospital and Research Centre. Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number TL1TR001880 (S. E. S), and by the Institute for Translational Medicine and Therapeutics of the Perelman School of Medicine at the University of Pennsylvania (S. E. S). J. A. F. acknowledges support from Hartwell Foundation, and the NIH (K08HD086250). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The work was partially supported by the University of California, Los Angeles California Center for Rare Diseases. Clinical whole genome sequencing (cWGS) was provided to four patients through the iHope Program. iHope is a philanthropic initiative at Illumina, Inc. in which cWGS is provided to individuals who are suspected of having a genetic etiology to their disease but lack access to this testing. This article reflects the views of the author and should not be construed to represent FDA's views or policies. This work was partially presented at the 40th Annual David W Smith Workshop on Malformations and Morphogenesis, August 23–27, 2019, at the Snowbird Resort in Snowbird, Utah.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Aggarwal A, Rodriguez-Buritica DF, & Northrup H (2017). Wiedemann-Steiner syndrome: Novel pathogenic variant and review of literature. European Journal of Medical Genetics, 60(6), 285–288. 10.1016/j.ejmg.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. (2015). The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nature Publishing Group, 47(4), 330–337. 10.1038/ng.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi H, Mizuguchi T, Ceroni JR, Kim VEH, Furquim I, Honjo RS, Iwaki T, Suzuki T, Sekiguchi F, Uchiyama Y, Azuma Y, Hamanaka K, Koshimizu E, Miyatake S, Mitsuhashi S, Takata A, Miyake N, Takeda S, Itakura A, … Matsumoto N (2019). Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. Journal of Human Genetics, 89, 584–978. 10.1038/s10038-019-0643-z [DOI] [PubMed] [Google Scholar]
- Aref-Eshghi E, Kerkhof J, Pedro VP, Barat-Houari M, Ruiz-Pallares N, Andrau JC, Lacombe D, van-Gils J, Fergelot P, Dubourg C, Cormier-Daire V, Rondeau S, Lecoquierre F, Saugier-Veber P, Nicolas G, Lesca G, Chatron N, Sanlaville D, Vitobello A, … Sadikovic B (2020). Evaluation of DNA methylation Episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. American Journal of Human Genetics, 106(3), 356–370. 10.1016/j.ajhg.2020.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora H, Falto-Aizpurua L, Cortés-Fernandez A, Choudhary S, & Romanelli P (2017). Connective tissue nevi: A review of the literature. The American Journal of Dermatopathology, 39(5), 325–341. 10.1097/DAD.0000000000000638 [DOI] [PubMed] [Google Scholar]
- Baer S, Afenjar A, Smol T, Piton A, Gérard B, Alembik Y, Bienvenu T, Boursier G, Boute O, Colson C, Cordier MP, Cormier-Daire V, Delobel B, Doco-Fenzy M, Duban-Bedu B, Fradin M, Geneviève D, Goldenberg A, Grelet M, … Morin G (2018). Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: A study of 33 French cases. Clinical Genetics, 94 (1), 141–152. 10.1111/cge.13254 [DOI] [PubMed] [Google Scholar]
- Beighton P (1970). Familial hypertrichosis cubiti: Hairy elbows syndrome. Journal of Medical Genetics, 7(2), 158–160. 10.1136/jmg.7.2.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert DJ, Dullaers M, Kuehn HS, Leroy BP, Niemela JE, de Wilde H, de Schryver S, de Bruyne M, Coppieters F, Lambrecht BN, de Baets F, Rosenzweig SD, de Baere E, & Haerynck F (2017). Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes. Scientific Reports, 7(1), 3702. 10.1038/s41598-017-02434-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, Ma Y, Moore R, Mungall AJ, Marra MA, Zhang J, Ma X, Liu Y, Liu Y, Auvil JMG, Davidsen TM, Gesuwan P, Hermida LC, Salhia B, … Meshinchi S (2018). The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nature Medicine, 24(1), 103–112. 10.1038/nm.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramswig NC, Lüdecke H-J, Alanay Y, Albrecht B, Barthelmie A, Boduroglu K, Braunholz D, Caliebe A, Chrzanowska KH, Czeschik JC, Endele S, Graf E, Guillén-Navarro E, Kiper PÖS, López-González V, Parenti I, Pozojevic J, Utine GE, Wieland T, … Wieczorek D (2015). Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of coffin-Siris and Nicolaides-Baraitser syndromes. Human Genetics, 134(6), 553–568. 10.1007/s00439-015-1535-8 [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, & Schultz N (2012). The cBio cancer genomics portal: An open platform for exploring multi-dimensional cancer genomics data. Cancer Discovery, 2(5), 401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AJS, Cytrynbaum C, Hoang N, Ambrozewicz PM, Weksberg R, Drmic I, Ritzema A, Schachar R, Walker S, Uddin M, Zarrei M, Yuen RKC, & Scherer SW (2019). Expanding the neurodevelopmental phenotypes of individuals with de novo KMT2A variants. NPJ Genomic Medicine, 4(1), 9. 10.1038/s41525-019-0083-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner JA, Lin W-Y, Riddle RC, Boukas L, DeLeon VB, Chopra S, Lad SE, Luperchio TR, Hansen KD, & Bjornsson HT (2019). Precocious chondrocyte differentiation disrupts skeletal growth in kabuki syndrome mice. JCI Insight, 4(20), 2013. 10.1172/jci.insight.129380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HR, Dlouhy SR, Lah MD, Payne KK, & Weaver DD (2019). The progression of Wiedemann-Steiner syndrome in adulthood and two novel variants in the KMT2A gene. American Journal of Medical Genetics. Part A, 179(2), 300–305. 10.1002/ajmg.a.60698 [DOI] [PubMed] [Google Scholar]
- Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, & Greaves M (1993). In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature, 363(6427), 358–360. 10.1038/363358a0 [DOI] [PubMed] [Google Scholar]
- Genome Aggregation Database Consortium, Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangiobbe S, Caraffi SG, Ivanovski I, Maini I, Pollazzon M, Rosato S, Trimarchi G, Lauriello A, Marinelli M, Nicoli D, Baldo C, Laurie S, Flores-Daboub J, Provenzano A, Andreucci E, Peluso F, Rizzo R, Stewart H, Lachlan K, … Garavelli L (2020). Expanding the phenotype of Wiedemann-Steiner syndrome: Craniovertebral junction anomalies. American Journal of Medical Genetics. Part A, 177(4), 406–2886. 10.1002/ajmg.a.61859 [DOI] [PubMed] [Google Scholar]
- Grangeia A, Leão M, & Moura CP (2019). Wiedemann-Steiner syndrome in two patients from Portugal. American Journal of Medical Genetics. Part A, 198, 25–28. 10.1002/ajmg.a.61407 [DOI] [PubMed] [Google Scholar]
- Hess JL, Yu BD, Li B, Hanson R, & Korsmeyer SJ (1997). Defects in yolk sac hematopoiesis in Mll-null embryos. Blood, 90(5), 1799–1806. [PubMed] [Google Scholar]
- Homma TK, Freire BL, Honjo Kawahira RS, Dauber A, Funari MFDA, Lerario AM, Nishi MY, Albuquerque EV, Vasques GA, Collett-Solberg PF, Miura Sugayama SM, Bertola DR, Kim CA, Arnhold IJP, Malaquias AC, & Jorge AAL (2019). Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. The Journal of Pediatrics, 215, 192–198. 10.1016/j.jpeds.2019.08.024 [DOI] [PubMed] [Google Scholar]
- Innes AM, McInnes BL, & Dyment DA (2018). Clinical and genetic heterogeneity in Dubowitz syndrome: Implications for diagnosis, management and further research. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 178(4), 387–397. 10.1002/ajmg.c.31661 [DOI] [PubMed] [Google Scholar]
- Jaiswal S, & Ebert BL (2019). Clonal hematopoiesis in human aging and disease. Science (New York, N.Y.), 366(6465), eaan4673. 10.1126/science.aan4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, Irving M, Saggar AK, Smithson S, Trembath RC, Deshpande C, & Simpson MA (2012). De novo mutations in MLL cause Wiedemann-Steiner syndrome. American Journal of Human Genetics, 91(2), 358–364. 10.1016/j.ajhg.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaseniit KE, Haque IS, Goldberg JD, Shulman LP, & Muzzey D (2020). Genetic ancestry analysis on >93,000 individuals undergoing expanded carrier screening reveals limitations of ethnicity-based medical guidelines. Genetics in Medicine, 22(10), 1694–1702. 10.1038/s41436-020-0869-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig R, Meinecke P, Kuechler A, Schafer D, & Muller D (2010). Wiedemann-Steiner syndrome: Three further cases. American Journal of Medical Genetics. Part A, 152A(9), 2372–2375. 10.1002/ajmg.a.33587 [DOI] [PubMed] [Google Scholar]
- Koretzky M, Bonham VL, Berkman BE, Kruszka P, Adeyemo A, Muenke M, & Hull SC (2016). Towards a more representative morphology: Clinical and ethical considerations for including diverse populations in diagnostic genetic atlases. Genetics in Medicine, 18(11), 1069–1074. 10.1038/gim.2016.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruszka P, Tekendo-Ngongang C, & Muenke M (2019). Diversity and dysmorphology. Current Opinion in Pediatrics, 31(6), 702–707. 10.1097/MOP.0000000000000816 [DOI] [PubMed] [Google Scholar]
- Li N, Wang Y, Yang Y, Wang P, Huang H, Xiong S, Sun L, Cheng M, Song C, Cheng X, Ding Y, Chang G, Chen Y, Xu Y, Yu T, Yao RE, Shen Y, Wang X, & Wang J (2018). Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet Journal of Rare Diseases, 13, 178. 10.1186/s13023-018-0909-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megonigal MD, Rappaport EF, Jones DH, Williams TM, Lovett BD, Kelly KM, Lerou PH, Moulton T, Budarf ML, & Felix CA (1998). T(11;22)(q23;q11.2) in acute myeloid leukemia of infant twins fuses MLL with hCDCrel, a cell division cycle gene in the genomic region of deletion in DiGeorge and velocardiofacial syndromes. Proceedings of the National Academy of Sciences of the United States of America, 95(11), 6413–6418. 10.1073/pnas.95.11.6413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, Sutton R, Venn NC, Emerenciano M, Pombo-de-Oliveira MS, Barbieri Blunck C, Almeida Lopes B, Zuna J, Trka J, Ballerini P, Lapillonne H, de Braekeleer M, Cazzaniga G, Corral Abascal L, … Marschalek R (2018). The MLL recombinome of acute leukemias in 2017. Leukemia, 32(2), 273–284. 10.1038/leu.2017.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri G, Magini P, Milani D, Crippa M, Biamino E, Piccione M, Sotgiu S, Perrìa C, Vitiello G, Frontali M, Boni A, di Fede E, Gandini MC, Colombo EA, Bamshad MJ, Nickerson DA, Smith JD, Loddo I, Finelli P, … Gervasini C (2019). Exploring by whole exome sequencing patients with initial diagnosis of Rubinstein-Taybi syndrome: The interconnections of epigenetic machinery disorders. Human Genetics, 60, 285–269. 10.1007/s00439-019-01985-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavone V, Testa G, Falsaperla R, & Sessa G (2015). Syringomyelia and bone malformations in the setting of hypertrichosis Cubiti (hairy elbow syndrome). Journal of Orthopaedic Case Reports, 5(2), 32–34. 10.13107/jocr.2250-0685.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Montaño D, & Pachajoa H (2019). Wiedemann-Steiner syndrome with a novel pathogenic variant in KMT2A: A case report. Colombia Medica (Cali, Colombia), 50(1), 40–45. 10.25100/cm.v50i1.3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RC, & Dou Y (2015). Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nature Reviews. Cancer, 15(6), 334–346. 10.1038/nrc3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy M, Collins C, MacLean JGB, & Campbell D (2019). Examining the effectiveness of examination at 6–8 weeks for developmental dysplasia: testing the safety net. Archives of Disease in Childhood, 104(10), 953–955. 10.1136/archdischild-2018-316520. [DOI] [PubMed] [Google Scholar]
- Steel D, Salpietro V, Phadke R, Pitt M, Gentile G, Massoud A, Batten L, Bashamboo A, Mcelreavey K, Saggar A, & Kinali M (2015). Whole exome sequencing reveals a MLL de novo mutation associated with mild developmental delay and without “hairy elbows”: Expanding the phenotype of Wiedemann-Steiner syndrome. Journal of Genetics, 94(4), 755–758. [DOI] [PubMed] [Google Scholar]
- Steiner CE, & Marques AP (2000). Growth deficiency, mental retardation and unusual facies. Clinical Dysmorphology, 9(2), 155–156. 10.1097/00019605-200009020-00021 [DOI] [PubMed] [Google Scholar]
- Stellacci E, Onesimo R, Bruselles A, Pizzi S, Battaglia D, Leoni C, Zampino G, & Tartaglia M (2016). Congenital immunodeficiency in an individual with Wiedemann-Steiner syndrome due to a novel missense mutation in KMT2A. American Journal of Medical Genetics. Part A, 170(9), 2389–2393. 10.1002/ajmg.a.37681 [DOI] [PubMed] [Google Scholar]
- Strom SP, Lozano R, Lee H, Dorrani N, Mann J, O'Lague PF, Mans N, Deignan JL, Vilain E, Nelson SF, Grody WW, & Quintero-Rivera F (2014). De novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Medical Genetics, 15(1), 49. 10.1186/1471-2350-15-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman J, Riedhammer KM, Jicinsky T, Mundt M, Werner L, Gusic M, Burgemeister AL, Alsaif HS, Abdulrahim M, Moghrabi NN, Nicolas-Jilwan M, AlSayed M, Bi W, Sampath S, Alkuraya FS, & el-Hattab AW (2019). Homozygous loss-of-function variants of TASP1, a gene encoding an activator of the histone methyltransferases KMT2A and KMT2D, cause a syndrome of developmental delay, happy demeanor, distinctive facial features, and congenital anomalies. Human Mutation, 40(11), 1985–1992. 10.1002/humu.23844 [DOI] [PubMed] [Google Scholar]
- Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, & Hua X (2010). MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell, 17(2), 148–159. 10.1016/j.ccr.2009.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CJ, & Wren C (2013). The epidemiology of arrhythmia in infants: A population-based study. Journal of Paediatrics and Child Health, 49(4), 278–281. 10.1111/jpc.12155. [DOI] [PubMed] [Google Scholar]
- Wang C, Zhan X, Liang L, Abecasis GR, & Lin X (2015). Improved ancestry estimation for both genotyping and sequencing data using projection procrustes analysis and genotype imputation. American Journal of Human Genetics, 96(6), 926–937. 10.1016/j.ajhg.2015.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann HR, Kunze J, Grosse F-R, & Dibbern H (1989). Atlas of clinical syndromes: A visual aid to diagnosis for clinicians and practicing physicians (2nd ed.). London: Wolfe Publishing Ltd. [Google Scholar]
- Winters AC, & Bernt KM (2017). MLL-rearranged leukemias-an update on science and clinical approaches. Frontiers in Pediatrics, 5(9), 4. 10.3389/fped.2017.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hess JL, Horning SE, Brown GA, & Korsmeyer SJ (1995). Altered Hox expression and segmental identity in Mll-mutant mice. Nature, 378(6556), 505–508. 10.1038/378505a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are not publicly available due to privacy or ethical restrictions.