

Abstract
Systemic duress, such as that elicited by sepsis, burns, or trauma, predisposes patients to secondary pneumonia, demanding better understanding of host pathways influencing this deleterious connection. These pre-existing circumstances are capable of triggering the hepatic acute-phase response (APR), which we previously demonstrated is essential for limiting susceptibility to secondary lung infections. To identify potential mechanisms underlying protection afforded by the lung–liver axis, our studies aimed to evaluate liver-dependent lung reprogramming when a systemic inflammatory challenge precedes pneumonia. Wild-type mice and APR-deficient littermate mice with hepatocyte-specific deletion of STAT3 (hepSTAT3−/−), a transcription factor necessary for full APR initiation, were challenged i.p. with LPS to induce endotoxemia. After 18 h, pneumonia was induced by intratracheal Escherichia coli instillation. Endotoxemia elicited significant transcriptional alterations in the lungs of wild-type and hepSTAT3−/− mice, with nearly 2000 differentially expressed genes between genotypes. The gene signatures revealed exaggerated immune activity in the lungs of hepSTAT3−/− mice, which were compromised in their capacity to launch additional cytokine responses to secondary infection. Proteomics revealed substantial liver-dependent modifications in the airspaces of pneumonic mice, implicating a network of dispatched liver-derived mediators influencing lung homeostasis. These results indicate that after systemic inflammation, liver acute-phase changes dramatically remodel the lungs, resulting in a modified landscape for any stimuli encountered thereafter. Based on the established vulnerability of hepSTAT3−/− mice to secondary lung infections, we believe that intact liver function is critical for maintaining the immunological responsiveness of the lungs.
Pneumonia and sepsis are leading global health concerns that are integrally linked (1–3). Sepsis is a highly heterogeneous disease involving many pathways that can lead to life-threatening organ dysfunction. It is the top cause of death in critically ill patients, totaling an estimated 19.7% of all global deaths (4–8). Around half of all sepsis cases stem from respiratory infections, but non-pulmonary-derived sepsis also strongly predisposes patients to hospital-acquired bacterial infections because of host immune dysregulation, with respiratory infections as the most common secondary nosocomial infection (2, 9). Other acute challenges, such as trauma or burn injuries, similarly increase pneumonia susceptibility (3, 10–12), as do more chronic conditions associated with systemic inflammation, such as aging and obesity (13–16). Altogether, these circumstances indicate that disruptions in systemic inflammatory tone render the lungs vulnerable to infection. As the leading cause of infectious disease deaths in the United States, pneumonia is characterized by an influx of fluid and inflammatory cells into the airspaces of the lungs and, like sepsis, has limited treatments, relying mainly on supportive or antibiotic therapies (1, 4, 5). Biological signals related to pneumonia susceptibility during sepsis remain poorly understood but could be useful in advancing medical decisions and identifying patients at risk.
Both sepsis and pneumonia, as well as other homeostatic challenges, cause an acute-phase response (APR), which is a systemic innate reaction resulting in liver activation and characterized by altered circulating levels of acute-phase proteins (APPs). Our laboratory has shown that during pneumonia, the hepatic APR is triggered by induction of the transcription factors STAT3 and NF-κB RelA, which are activated in response to IL-6, TNF-α, and IL-1 (17). Pneumonia susceptibility increases when the lung–liver axis is severed through deletion of STAT3 and RelA in hepatocytes (17–20), and specific hepatic STAT3-dependent liver responses are essential for limiting pneumonia susceptibility in endotoxemic mice (19). To reveal candidate mechanisms whereby hepatic STAT3 activity limits pneumonia susceptibility in this setting, we sought to comprehensively evaluate liver-dependent changes in the lungs in response to endotoxemia followed by pneumonia. Our findings show that both cellular and humoral changes in the lungs are impacted by STAT3-dependent liver activation before and after a localized secondary challenge. This study suggests that in the presence of systemic inflammation, liver activity influences both the lung immune baseline and the airspace environment elicited by subsequent infections, revealing protective liver-directed pathways that likely counter the detrimental consequences of trauma, sepsis, and other forms of non-pulmonary duress.
Materials and Methods
Mice
Experiments were performed in mice with hepatocyte-specific deletion of STAT3 (hepSTAT3−/− mice), which have been described previously (17). In brief, mice with homozygous floxed alleles for STAT3 were crossed with mice expressing albumin-driven Cre-recombinase. Results from hepSTAT3−/− mice were compared with those from littermate controls lacking Cre-recombinase. Both male and female mice were used, all between 6 and 15 wk of age, and results were collected over a minimum of two independent experiments unless otherwise stated. Mice were housed in a pathogen-free environment, provided with food and water, and kept on a 12-h light cycle. All animal protocols were approved by the Boston University Institutional Animal Care and Use Committee.
Experimental infections and stimuli
Mice were given an i.p. injection of saline as a control or 1 mg/kg of body weight of ultrapure LPS (LPS-EB from E. coli O111:B4; catalog code tlrleblps; InvivoGen, San Diego, CA) to induce endotoxemia. After 18 h, intratracheal (i.t.) instillations of bacteria to induce pneumonia were performed as previously described (17). In brief, mice were anesthetized with ketamine (50 mg/kg of body weight) and xylazine (5 mg/kg), administered by i.p. injection. A 24-gauge catheter was inserted into the exposed trachea, and a 50-μL bolus of saline containing 106 CFUs of Escherichia coli (serotype O6:K2:H1, ATCC 19138; ATCC, Manassas, VA) was instilled into the left bronchus. Mice were euthanized for tissue collection using an isoflurane overdose after either 18 h i.p. saline, 18 h i.p. LPS, or 18 h LPS followed by 24 h i.t. E. coli. Where indicated, 6 h i.t. of 107 CFUs of Staphylococcus aureus (ATCC 25923) was substituted for E. coli.
Bronchoalveolar lavage
Bronchoalveolar lavage fluid (BALF) was collected at the indicated time points as previously described (19). In brief, the lung–heart block was removed and tied to a 20-gauge blunted stainless steel catheter via the trachea. Once secured, the lungs were lavaged 10 times with 1 ml of cold PBS. The left lobe and the cell-free supernatant (centrifuged at 300 × g for 5 min at 4°C) from the first lavage were stored at −80°C for further analysis. Pooled cells from all washes were counted using the LUNA-FL Dual Fluorescence Cell Counter (Logos Biosystems). After cytocentrifugation and Camco staining (catalog number 702; Cambridge Diagnostic), differential counts were determined at each time point.
Plasma collection
After collection using a heparinized needle at the indicated time points, blood was centrifuged for 15 min at 1500 × g and 4°C. Plasma was ali-quoted and stored at −80°C before analysis.
Histology
H&E staining was performed on paraffin-embedded lung sections fixed in 4% paraformaldehyde (catalog number 18505; Ted Pella, Redding, CA). In brief, lung tissue was fixed overnight, washed with 1× PBS (10010–023; Life Technologies) followed by 0.9% saline (Baxter Healthcare Corporation), then dehydrated through increasingly concentrated ethanol washes, followed by xylene clearing and paraffin infiltration.
Cytokine and chemokine measurements
Plasma and whole-lung homogenate protein concentrations were measured by using a mouse magnetic Luminex assay (LXSAMSM; R&D Systems) on a Bio-Plex 200 multiplexing analyzer system. The panel included TNF-α, CXCL1, G-CSF, IL-1α, IL-6, urokinase plasminogen activator surface receptor, CCL2, CXCL2, GM-CSF, and IL-10. BALF and plasma concentrations of fibrinogen were measured via ELISA (catalog number ab213478; Abcam).
RNA isolation
Liver and lung tissues were snap frozen in liquid nitrogen and stored at −80°C until RNA isolation. To isolate RNA, we homogenized tissue using seven to nine 2.0-mm RNase-free zirconium oxide beads (catalog number ZROB20-RNA; Next Advance) in the TRIzol reagent (catalog number 15596026; Ambion by Life Technologies) using the manufacturer’s instructions. RNA was cleaned using the RNeasy Mini kit (catalog number 74106; Qiagen). RNA samples were stored at −80°C until needed.
Quantitative real-time PCR
Quantitative RT-PCR (qRT-PCR) was performed with 10 ng of purified RNA using a StepOne Plus real-time PCR system or QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA) and a TaqMan RNA-to-CT 1-step kit (Thermo Fisher Scientific). RNA was probed with primers and fluorescein amidite–labeled probes for Fg-α, Fg-β, Fg-γ, GM-CSF, G-CSF, WNT3A, CXCL1, and LCN2, and VIC-labeled eukaryotic 18S rRNA as an endogenous control (Life Technologies). After normalization to the 18S rRNA content, values are expressed as the fold induction compared with that for the relative control groups.
Lung digestion and flow cytometry
Lungs were enzymatically digested into single-cell suspensions and stained as described previously (21). In brief, lungs were digested at 37°C and 250 rpm for 1 h in a collagenase solution containing type 2 collagenase (1 mg/ml; LS004176; Worthington Biochemical, Lakewood, NJ), DNase I (150 mg/ml; 9003-96-9; Sigma-Aldrich, St. Louis, MO), and CaCl2 (2.5 mM) in PBS, then filtered through a 70-mm cell strainer (Fisher, Grand Island, NY). After using RBC lysis buffer (Sigma-Aldrich, St Louis, MO), cells were counted on the LUNA-FL Dual Fluorescence Cell Counter (Logos Biosystems). Cells were stained for CD45-FITC (Clone: 30-F11, 501129405; Invitrogen), Ly6G-allophycocyanin (501123222; Invitrogen), SiglecF- allophycocyanin/Cy7 (565527; BD Biosciences), CD11b-PE/Cy7 (101215; BioLegend), CD64-PE (50165081; BD Biosciences), and DAPI (D1306; Invitrogen) as a viability stain, with gating adapted from Guillon et al. (21). In brief, samples were gated on singlets and live cells. CD45+ cells were split between Ly6G+ (neutrophils) and Ly6G− cells, which were further gated into alveolar macrophages (CD64+/SigF+), other macrophages (CD64+/SigF−/CD11b+), and monocytes (CD64−/SigF−/CD11b+). Flow cytometry was performed using BD LSR II at the Boston University Flow Cytometry Core Facility. Data were analyzed using FlowJo (version 10) software. Unstained cells, single-stained UltraComp eBeads (01-2222-42; Invitrogen), and fluorescence-minus-one controls were used for each experiment.
RNA sequencing and analysis
Whole-lung RNA was extracted as previously described using the Qiagen RNA Mini Kit. RNA quality, sequencing, and analysis were performed at the Boston University Microarray and Sequencing Resource Core Facility. The libraries were prepared from 200 ng total RNA enriched for mRNA using Illumina’s TruSeq Stranded mRNA Library Preparation kit (Illumina, USA) and sequenced on an Illumina NextSeq 500 instrument (Illumina, USA). Three to four mice per group were used for analysis. Resulting FastQC files were aligned to the mouse genome mm10 using STAR (version 2.5.3a). Ensembl-Gene-level counts were generated for nonmitochondrial genes through featureCounts (Subread package, version 1.6.2) and Ensembl annotation build 92 (uniquely aligned proper pairs, same strand). SAMtools (version 1.8 was used to count reads aligning to proper pairs at least once to either strand of the mitochondrial chromosome or to sense or antisense strands of Ensembl loci of gene biotype “rRNA” or of nonmitochondrial RepeatMasker loci of the class “rRNA” (as defined in RepeatMasker track retrieved from the University of California Santa Cruz Table Browser). FASTQ quality was determined using FastQC (version 0.11.3), alignment quality was determined by RSeQC (version 2.6.4), and principal component analysis was assessed using the prcomp R function with regularized logarithm-transformed expression values that were z-normalized for each sample within a given gene. Differential expression was determined via the Wald test implemented in the DESeq2 R package (version 1.19.49) (22), with correction for multiple-hypothesis testing completed using the Benjamini–Hochberg false discovery rate (FDR). Human homologs of mouse genes were identified using HomoloGene (version 68), and all data analyses were accomplished using the R environment for statistical computing (version 3.5.0). Gene expression differences were considered statistically significant when the FDR q value was <0.05. Ingenuity Pathway Analysis (IPA; Qiagen Bioinformatics) and Mouse Gene Atlas (ENRICHR) (23, 24) were further used for analysis to identify relevant pathways and connections.
Proteomics and analysis
BALF samples were sent to the Yale MS & Proteomics Resource (New Haven, CT), where they were processed and analyzed via a Label Free Quantification workflow as described by Charkoftaki et al. (25). The sample preparation was slightly modified from the 2019 protocol of Charkoftaki et al. (25) given the samples were BALF. In brief, samples were filtered through a 3-kDa Amicon Ultra filter, and the retentate was SpeedVac dried and used for downstream proteomics preparation. Dried protein pellets were reduced with DTT, alkylated with iodoacetamide, enzymatically digested with LysC and trypsin, and desalted using C18 RP microspin column. High-resolution liquid chromatography mass spectrometry (MS)/MS data were collected on an Orbitrap Fusion mass spectrometer coupled to a NanoACQUITY UPLC and analyzed using Progenesis QI (Waters, Milford, MA) and Mascot search engine. Quantitative data were normalized based on equal total amount of peptides/proteins injected on column, and positive protein identification and quantitation were based on hits with two or more unique peptides per protein. Experimental groups were compared and considered statistically significant when the FDR q value was <0.05, unless otherwise stated. IPA (Qiagen Bioinformatics) was used for further analyses.
Statistics
Statistical analyses were performed using GraphPad Prism software. Graphs are represented as mean, with error bars indicating SEM. Two-group comparisons were made using a Student t test. Multiple-group comparisons used nonparametric 1-way ANOVA (Kruskal–Wallis test) followed by a Tukey multiple-comparison test or 2-way analysis followed by a Sidak test. Data were tested for normality (Shapiro–Wilk test) and homoscedasticity (F-test) and were log transformed if either condition was not met. Data were considered significant if p < 0.05.
Accession numbers
All gene expression data have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus database under Series ID GSE167277 (https://www.ncbi.nlm.nih.gov/geo/). All MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (26, 27) partner repository with the dataset identifier PXD024663 (http://proteomecentral.proteomexchange.org/cgi/GetDataset).
Results
Liver activity modifies the lung transcriptome during endotoxemia
Previously, we found that during endotoxemia, STAT3-dependent hepatocyte activity is essential for limiting pneumonia susceptibility in response to a subsequent intrapulmonary challenge with E. coli (19). As described previously, hepSTAT3−/− mice have decreased survival and increased bacterial burdens in both the lungs and the blood (19). To more comprehensively evaluate the impact of liver activity on lung biological pathways, we performed RNA sequencing (RNAseq) on whole lungs collected from wild-type (WT) or hepSTAT3−/− mice treated for 18 h with i.p. LPS to induce endotoxemia with or without a second 24-h challenge with i.t. E. coli (Fig. 1A). A fifth group of WT mice treated with 18 h i.p. saline alone was included as an unchallenged control group. Unsurprisingly, 18 h of LPS alone had a strong and profound influence on lung gene expression, regardless of genotype (Fig. 1B). However, after LPS alone (without pneumonia), a significant genotype-dependent effect was also observed, as evidenced by the detection of 1898 differentially expressed genes (DEGs; FDR q value < 0.05) in the lungs of hepSTAT3−/− mice when compared with WT controls of the same treatment group (Fig. 1B–D). Interestingly, significant genotype-dependent effects were not present after pneumonia following endotoxemia, suggesting that liver-dependent lung transcriptional responses may be particularly important for shaping lung immunity before bacterial exposure, likely contributing to compromised responses previously observed thereafter (19).
FIGURE 1.
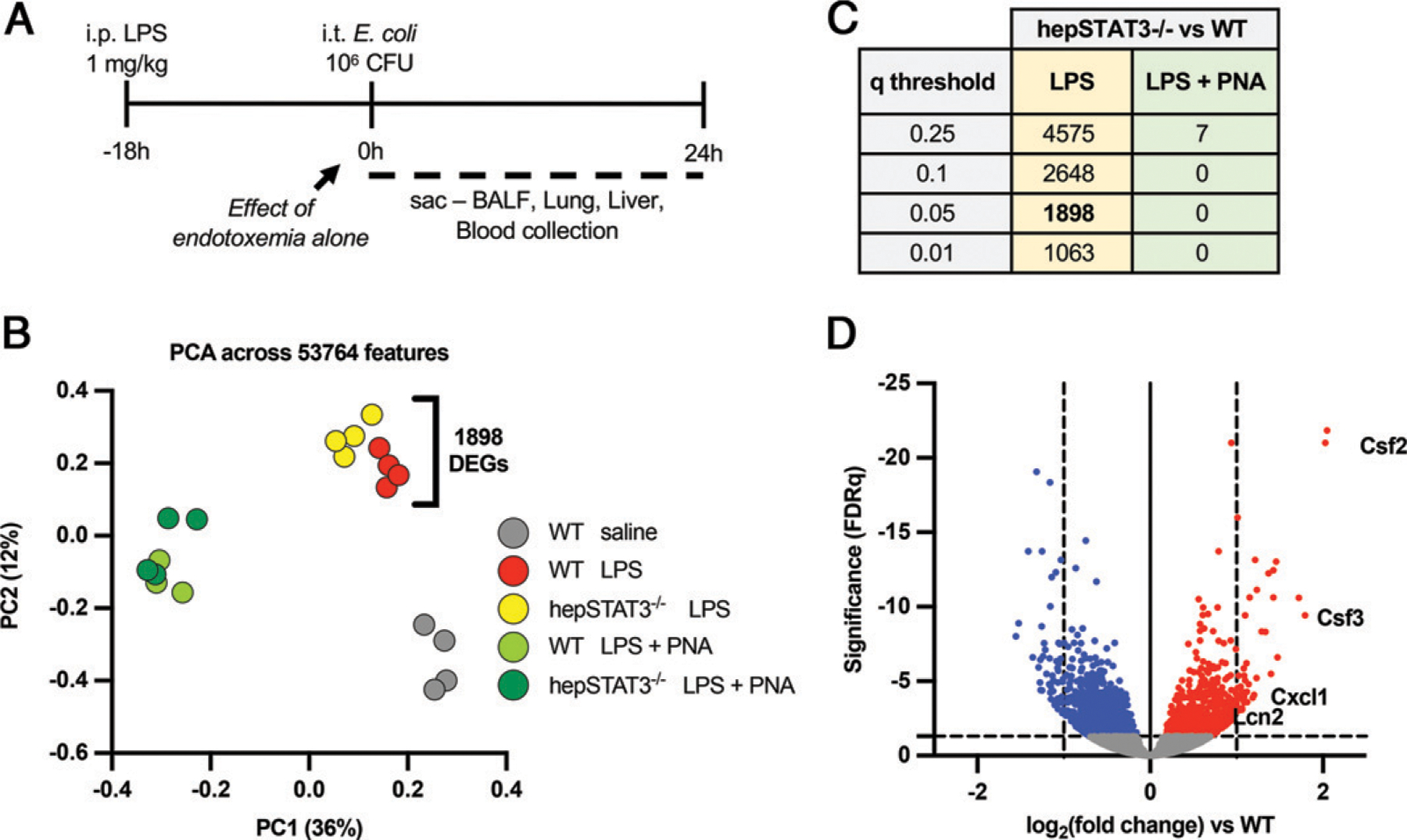
Liver activity modifies the lung transcriptome during endotoxemia in a genotype-dependent manner. (A) Two-hit model of endotoxemia and pneumonia. WT and hepSTAT3−/− mice received i.p. LPS (1 mg/kg) for 18 h and were either collected then (at 0 h) or given a direct pulmonary infection via an i.t. instillation of E. coli (106 CFUs) for an additional 24 h. (B) RNAseq was performed on WT or hepSTAT3−/− mice challenged with LPS for 18 h and either 0 or 24 h of pneumonia (PNA). A control group of WT mice with 18 h i.p. saline was included. The principal component analysis (PCA) plot indicates a distinct genotypic clustering between WT and hepSTAT3−/− mice after endotoxemia alone, with 1898 significant gene changes between the lungs of each group. (C) The table indicates the number of DEGs between hepSTAT3−/− and WT lungs at 18 h i.p. LPS and at 18 h LPS/24 h pneumonia with the corresponding q value. (D) The volcano plot depicts genes in hepSTAT3−/− lungs that are significantly (q < 0.05) increased (red) or decreased (blue) versus WT controls after 18 h i.p. LPS. n = 3–4/group from four independent experiments.
Lungs exhibit a hyperimmune phenotype in the absence of STAT3-dependent liver activity
To determine biological pathways reflected by lung gene expression differences in endotoxemic hepSTAT3−/− lungs, we conducted bioinformatics analyses of changes from the LPS-only groups before pneumonia using IPA. The top 15 upstream regulators identified by IPA consistently suggest elevated responses to known inflammatory triggers in hepSTAT3−/− lungs compared with those from WT mice, based on candidate regulators such as TNF, IFN-γ, IL-1β, and others (Table I). In addition, the top five activated diseases and functions mapped in IPA indicate increased activation and movement of myeloid cells in hepSTAT3−/− lungs, whereas the most decreased categories suggest a dysregulation of cellular dynamics that may be inherent to additional aspects of immune activation (Table II) (28–30). These categories were rated by the predicted activation z-score, a quantitative inference on likely activation of biological function through comparison of the observed dataset and known established directional changes from applicable literature. Validation of RNAseq results using qRT-PCR confirms elevations of GM-CSF (Csf2), G-CSF (Csf3), CXCL1, and LCN2 in lung homogenates from endotoxemic hepSTAT3−/− mice compared with WT after LPS alone, as well as a loss of genotype-dependent differences by 24 h of pneumonia (Figs 2A–D). To more specifically determine whether and how altered immune baselines (after LPS treatment alone) directly influence responses to secondary lung infections, we conducted an alternative analysis on lung qRT-PCR data to specifically calculate the effect of pneumonia within either mouse genotype (Fig. 3). Interestingly, these results revealed almost a complete loss of gene responsiveness in hepSTAT3−/− lungs after the introduction of bacteria (Fig. 3). This was consistent across all immune genes analyzed. WNT3A is included as a representative decreased gene (from RNAseq results), which we validated with qRT-PCR (Figs. 2E, 3E) and which was chosen given its potential protective role in pneumonia (31, 32).
Table I.
Top 15 predicted upstream regulators of DEGs
| Upstream Regulators | p Value of Overlap |
|---|---|
| TNF | 9.76E–47 |
| IL-1B | 6.69E–31 |
| IFNG | 3.22E–25 |
| TGFB1 | 8.05E–22 |
| NFKBIA | 1.79E–20 |
| IL-4 | 2.78E–18 |
| Ige | 4.94E–18 |
| NF-κB (complex) | 3.37E–16 |
| STAT3 | 4.65E–16 |
| DUSP1 | 8.49E–16 |
| Vegf | 1.41E–15 |
| CSF2 | 1.49E–15 |
| IKBKG | 2.31E–15 |
| HIF1A | 3.97E–15 |
| IKBKB | 6.25E–15 |
The 1898 DEGs (FDR < 0.05) between endotoxemic WT and hepSTAT3−/− lungs were analyzed using IPA, and the predicted top 15 upstream regulators of hepSTAT3−/− lungs (versus WT) after endotoxemia are listed.
Table II.
Top five increased and decreased disease and function categories
| Diseases or Functions Annotation | p Value | Activation z-Score |
|---|---|---|
| Cell movement of myeloid cells | 1.75E–23 | 4.190 |
| Cell movement of phagocytes | 7.42E–25 | 3.985 |
| Chemotaxis of phagocytes | 2.36E–11 | 3.453 |
| Chemotaxis of myeloid cells | 2.87E–10 | 3.452 |
| Cell movement of granulocytes | 2.72E–17 | 3.397 |
| Cell death of T lymphocytes | 3.06E–9 | −2.614 |
| Organization of cytoskeleton | 2.2E–13 | −2.621 |
| Organization of cytoplasm | 1.67E–12 | −2.621 |
| Microtubule dynamics | 2.09E–12 | −2.926 |
| Formation of cellular protrusions | 7.12E–10 | −3.302 |
IPA mapped the significantly different gene changes between endotoxemic WT and hepSTAT3−/− lungs to top diseases and functions categories. The top five increased and decreased categories (rated by predicted activation z-score) are illustrated.
FIGURE 2.
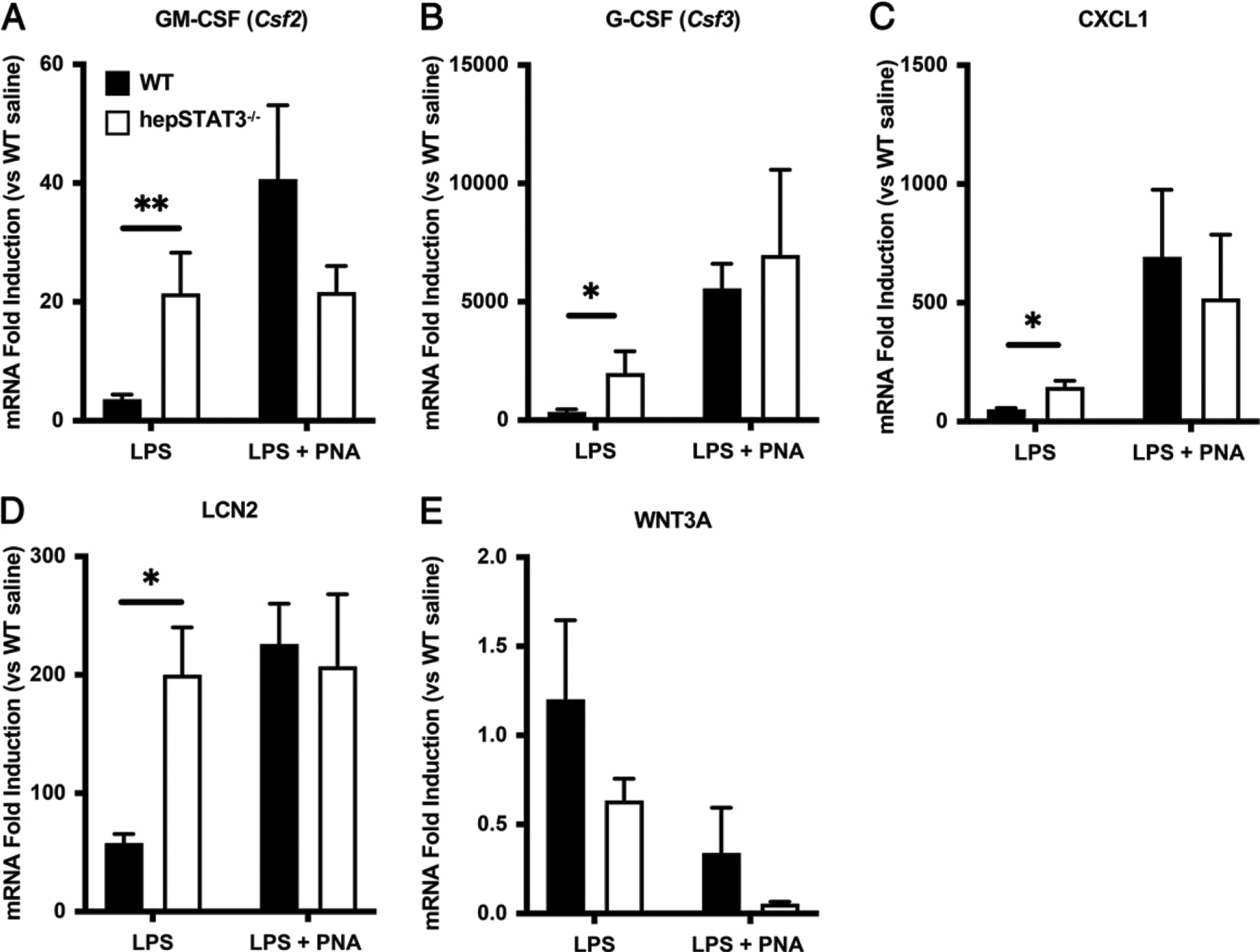
After endotoxemia, immune transcripts are exaggerated in the lungs of hepSTAT3−/− mice before the induction of pneumonia (PNA). Select lung transcripts were quantified using qRT-PCR to validate RNAseq results. Lung mRNA induction was determined for (A) GM-CSF, (B) G-CSF, (C) CXCL1, (D) LCN2, and (E) WNT3A in specimens collected from mice treated for 18 h with i.p. LPS with or without a subsequent i.t. instillation with E. coli (106 CFUs). Fold induction was calculated versus gene expression values detected in WT mice treated with i.p. saline (n = 4) alone. Data are represented as means with SEM, and significance was determined using a 2-way ANOVA followed by a Sidak test for multiple comparisons (*p < 0.05, **p < 0.01). n = 5–8/group from five independent experiments.
FIGURE 3.
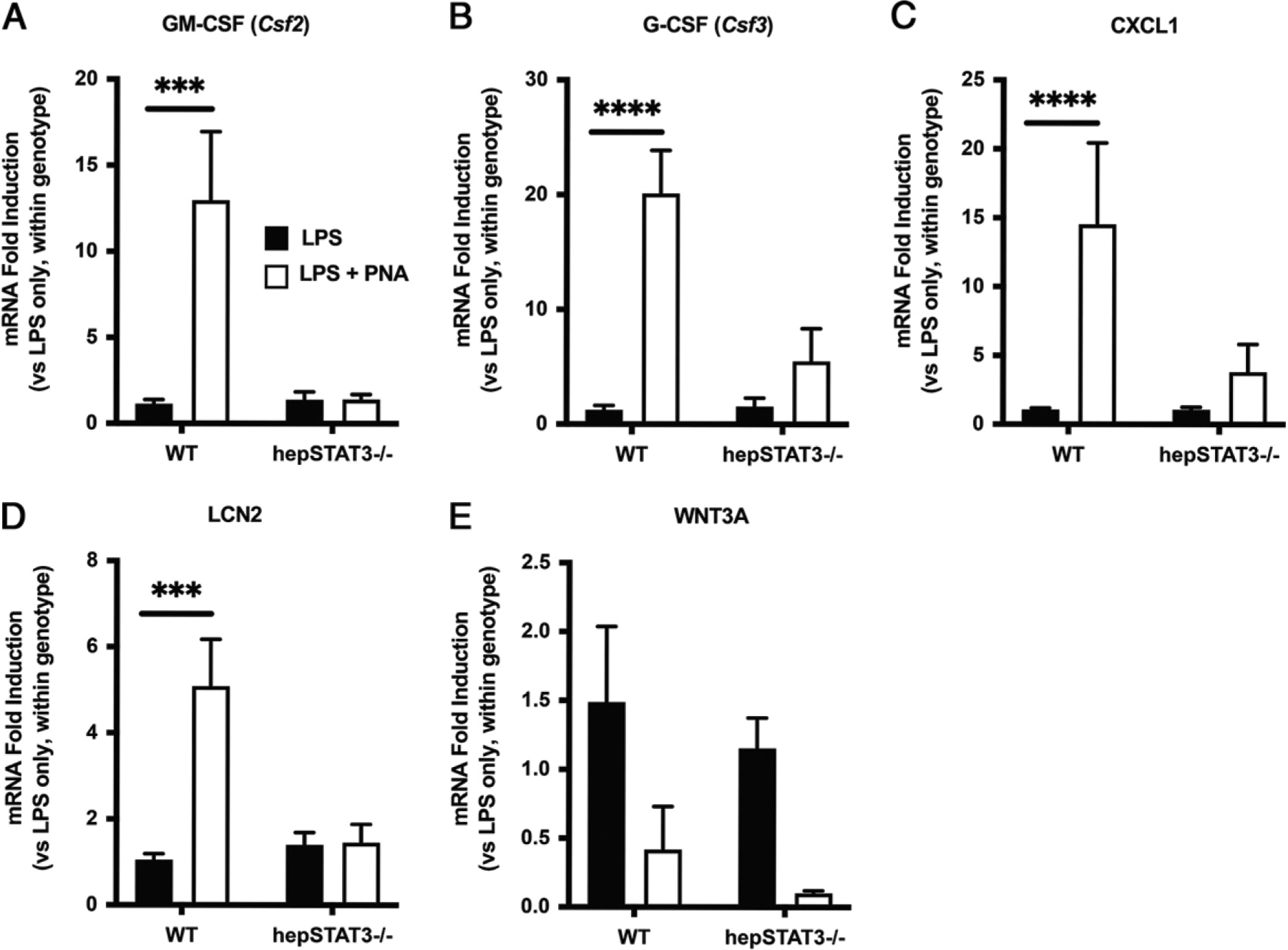
After endotoxemia, lungs from hepSTAT3−/− mice are resistant to pneumonia-induced cytokine changes. Lung mRNA induction was determined for (A) GM-CSF, (B) G-CSF, (C) CXCL1, (D) LCN2, and (E) WNT3A in specimens collected from mice treated for 18 h with i.p. LPS with or without a subsequent i.t. instillation with E. coli (106 CFUs). Fold induction was calculated versus gene expression values detected in uninfected mice of the same genotype. Data are represented as means with SEM, and significance was determined using a 2-way ANOVA followed by a Sidak test for multiple comparisons (***p < 0.001, ****p < 0.0001 versus LPS alone within genotype). n = 5–8/group from five independent experiments.
To establish whether diminished mRNA responsiveness could extend to pathogens other than E. coli, we conducted a separate experiment with S. aureus to determine its capacity to elicit gene expression in WT and hepSTAT3−/− mice beyond that observed with i.p. LPS alone (Supplemental Fig. 1). WT and hepSTAT3−/− mice received i.p. LPS followed by a 6-h challenge with i.t. S. aureus, a time point typically sufficient to detect lung cytokines. After endotoxemia, the impact of pneumonia on CXCL1 and GM-CSF mRNA was reduced in hepSTAT3−/− mice compared with the induction achieved in WT mice. Unlike the values detected for CXCL1 (~10-fold induction; Supplemental Fig. 1B), S. aureus was insufficient to elicit further GM-CSF mRNA in WT mice beyond that stimulated by the preceding LPS challenge, but this response was even further diminished in hepSTAT3−/− mice (Supplemental Fig. 1A). In both cases, therefore, the impact of pneumonia on cytokine induction was reduced in mutant mice, consistent with the notion that hepatocyte STAT3 activity preserves the capacity of lungs to respond to secondary infection.
We next determined whether differences in gene expression were also observable on a protein level in lung homogenates and whether these changes were reflected in the systemic circulation. Plasma and lung cytokine concentrations were measured by multiplex bead array after 18 h i.p. LPS alone. Increases in CXCL1, CCL2, CXCL2, and GM-CSF concentrations were observed in both the lungs (Fig. 4A) and plasma (Fig. 4B) of endotoxemic hepSTAT3−/− mice compared with WT, indicating a systemic increase in inflammation in hepSTAT3−/− mice that is potentially tempered by intact liver activity in WT mice. Genotype-dependent increases in factors such as TNF-α, urokinase plasminogen activator surface receptor, and IL-10 (Fig. 4B) suggest additional changes in inflammatory and coagulation mediators, any of which may be a cause or consequence of liver-dependent reprogramming. Protein changes were not observed in the lungs after 18 h i.p. LPS followed by 6 h of pneumonia (data not shown), supporting that early liver-dependent modifications in endotoxemic mice may be more influential before infection as indicated by both RNAseq and qRT-PCR.
FIGURE 4.
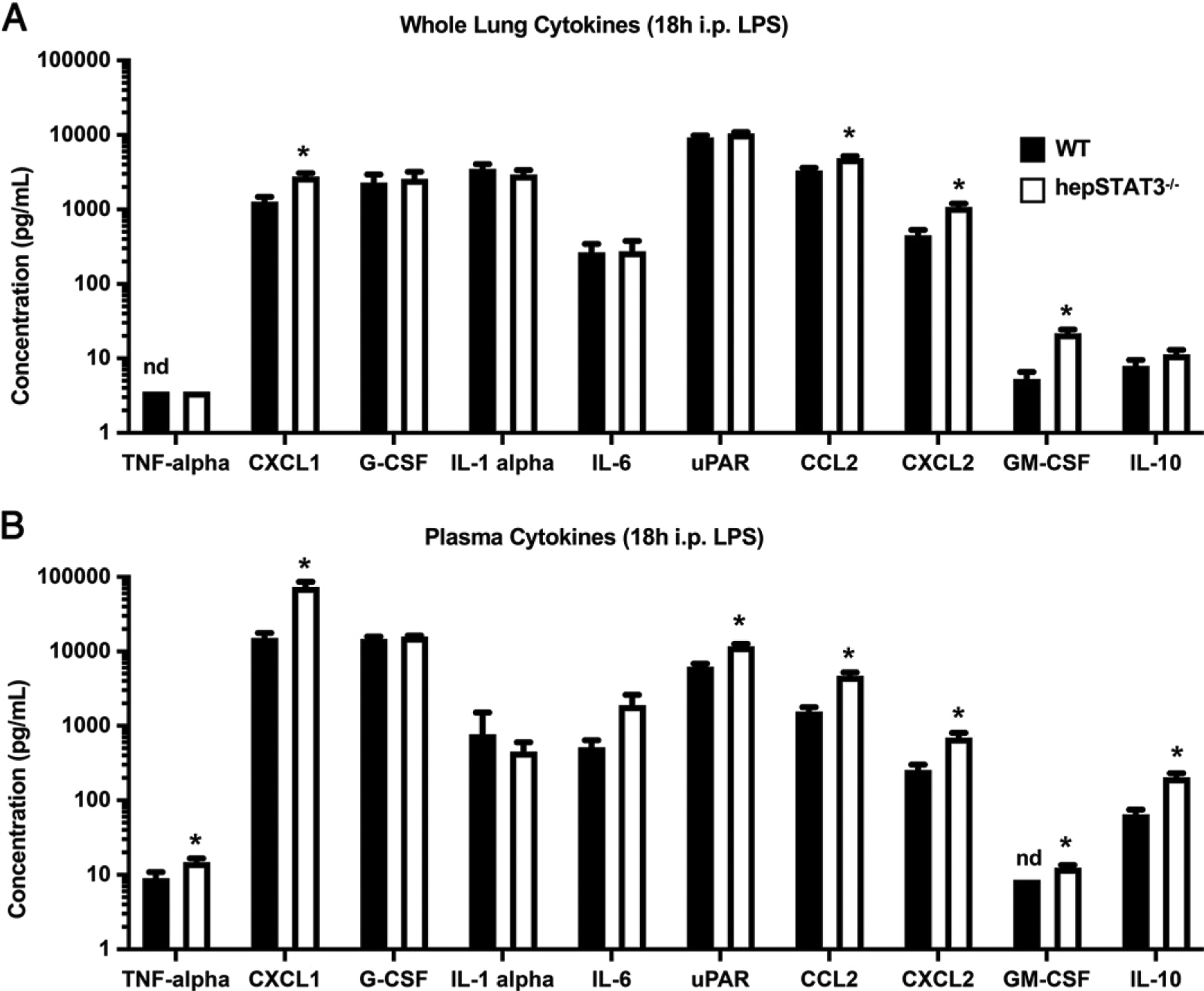
Lung and circulating cytokine concentrations are exaggerated in hepSTAT3−/− mice during endotoxemia. WT and hepSTAT3−/− mice were treated for 18 h with i.p. LPS, and the indicated cytokines were quantified in (A) lungs and (B) plasma by Luminex multiplex bead array. Data are represented as means with SEM, and significance was determined using an unpaired t test; *p < 0.05. n = 7–11/group from two independent experiments. nd, not detectable.
Lungs are histologically unaffected by liver STAT3 deletion
Given the liver-dependent transcriptional and protein changes observed at a whole-lung level after endotoxemia alone, we wanted to determine whether changes in gene expression were related to histopathological differences in hepSTAT3−/− mice challenged with endotoxemia with or without secondary lung infections. Paraffin-embedded sections of WT and hepSTAT3−/− lungs collected after 18 h i.p. saline, after 18 h i.p. LPS, or after 18 h i.p. LPS followed by 24 h of pneumonia (Fig. 5) were stained with H&E. At baseline, there appeared to be no observable differences between lungs, as expected (Fig. 5). Despite the differences to whole-lung transcription and protein concentrations after 18 h of endotoxemia, WT and hepSTAT3−/− lungs were also comparable histologically and remained similar throughout an additional 24 h of pneumonia (Fig. 5), suggesting that established defects in hepSTAT3−/− immunity (19) are not linked to differences in immunopathology during endotoxemia.
FIGURE 5.
Lungs are histologically unaffected by liver STAT3 deletion after endotoxemia with or without pneumonia. WT and hepSTAT3−/− mice were given 18 h i.p. saline (n = 2–5 from two independent experiments), 18 h i.p. LPS (n = 6–7 from two independent experiments), or 18 h i.p. LPS followed by 24 h i.t. E. coli (n = 4–7 from three independent experiments). Nonperfused left lung lobes were collected, paraffin embedded, and stained with H&E. Representative images of all groups from at least two independent experiments illustrate comparable lung architecture under all three conditions between WT and hepSTAT3−/− lungs.
Lung leukocyte numbers are unaffected by liver STAT3 deletion during endotoxemia
Despite the histological similarities observed in WT and hepSTAT3−/− lungs, we remained unsure of whether lung transcriptional differences (Figs. 1–3) reflected altered gene expression signatures in similar populations of lung cells, altered numbers of cells, or a combination therein. Macrophages were of particular interest for several reasons. First, prior results from our group indicated reduced reactive oxygen species (ROS) in macrophages collected from hepSTAT3−/− mice using the current two-hit challenge of endotoxemia followed by pneumonia (19). In addition, our gene expression data revealed increases in hepSTAT3−/− lung cytokines, such as GM-CSF and G-CSF (Figs. 2–4), consistent with potential increases in myeloid activation (Table II). Most importantly, when the significantly upregulated genes were mapped to the Mouse Gene Atlas (https://maayanlab.cloud/Enrichr/), the only significantly represented gene sets were generated from LPS-stimulated macrophages (Table III), further suggesting changes in macrophage number or function in the lungs of hepSTAT3−/− mice. To address this possibility, we used flow cytometry to determine immune cell proportions and numbers after endotoxemia alone, the condition in which both gene expression and protein differences were observed. Lungs from WT and hepSTAT3−/− mice were isolated and stained for flow cytometry using a myeloid panel to enumerate macrophages and neutrophils. Total immune cell numbers (CD45+ cells) were unaffected in hepSTAT3−/− mice, and this outcome was consistent across subpopulations, including alveolar macrophages, lung neutrophils, monocytes, and other lung macrophage populations (comprising interstitial macrophages) (Fig. 6). HepSTAT3−/− lungs showed more variability in cell count than what was observed in WT mice, but no significant increases were detected, suggesting liver-dependent changes in cellular activity rather than changes in cell number as a more prominent driver of transcriptional remodeling.
Table III.
Cell-specific gene sets represented by upregulated lung genes
| Cell Type (Mouse Gene Atlas) | Adjusted p Value |
|---|---|
| Macrophage bone marrow 6 h LPS | 8.557E–12 |
| Macrophage bone marrow 2 h LPS | 4.168E–4 |
| Macrophage peri-LPS thio 7 h | 6.476E–4 |
| T cells FoxP3 + | 1.04E–1 |
| Bone | 1.04E–1 |
The upregulated differentially expressed lung genes between genotypes were mapped to the Mouse Gene Atlas (ENRICHR) to determine overlap with established cell-specific gene sets.
FIGURE 6.
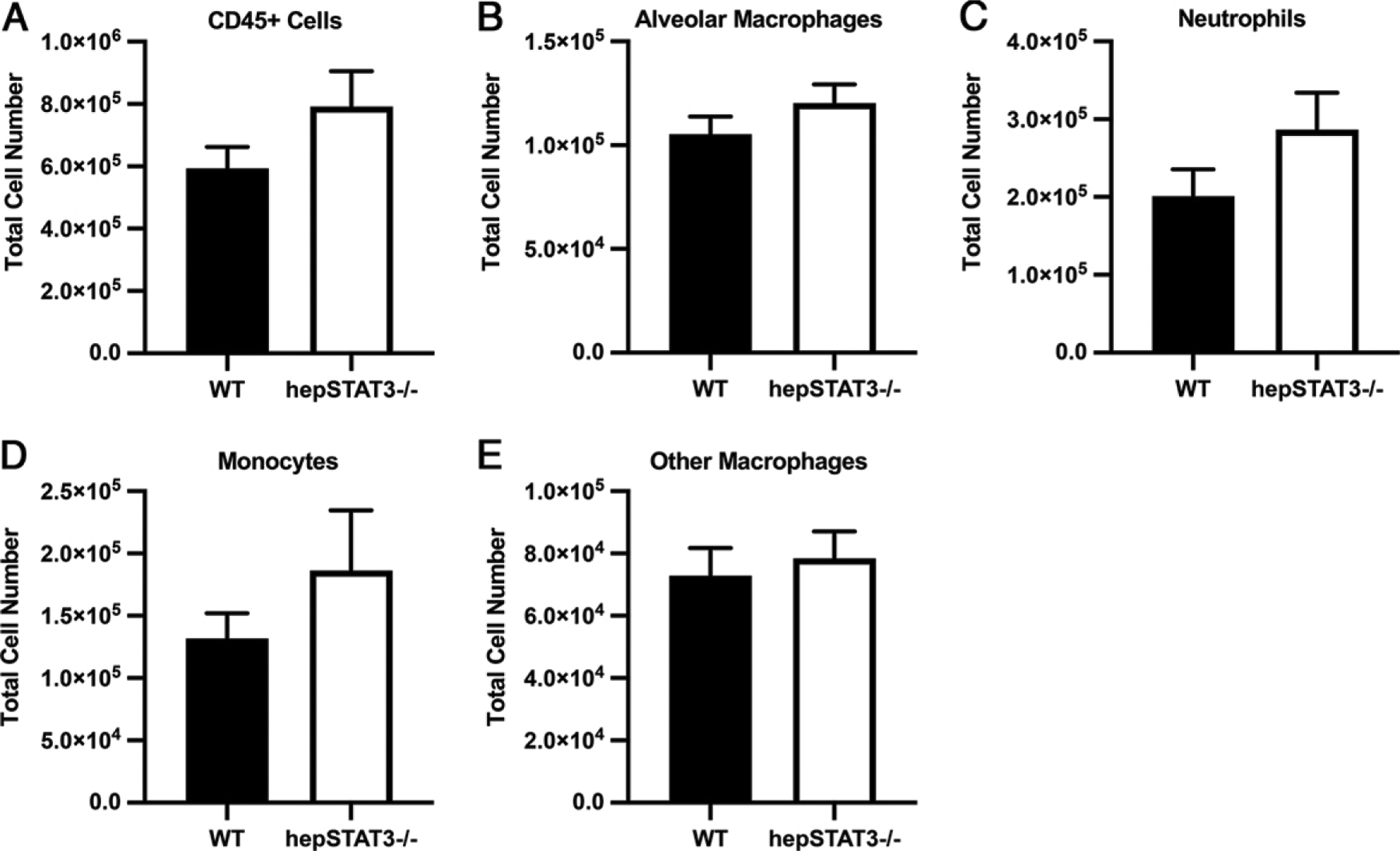
Lung leukocyte numbers are unaffected by liver STAT3 deletion during endotoxemia. Eighteen hours after i.p. LPS, single-cell suspensions were generated from the lungs of WT and hepSTAT3−/− mice and stained for flow cytometric quantification of the indicated populations. Total cell numbers are illustrated for (A) all leukocytes (CD45+), (B) alveolar macrophages (CD45+/Ly6G−/CD64+/SigF+), (C) neutrophils (CD45+/Ly6G+), (D) monocytes (CD45+/Ly6G−/CD64−/CD11b+/SigF−), and (E) other macrophages, including the interstitial population (CD45+/Ly6G−/CD64+/CD11bhi/SigF−). Data are represented as means with SEM. n = 8–15/group from four independent experiments.
Alveolar exudate is shaped by the hepatic APR
We have previously shown that during endotoxemia followed by pneumonia, airspace constituents from hepSTAT3−/− mice (as reflected by cell-free BALF) provide a more favorable environment for bacterial growth (19), indicating important humoral alterations in alveolar exudate that are liver dependent. To determine whether compromised defense may be related to differences in the airspace proteome, we collected BALF from endotoxemic WT and hepSTAT3−/− mice with or without pneumonia and performed label-free quantitative MS. Significant pneumonia-induced proteomic changes were identified in BALF from both genotypes, most of which were shared between WT and hepSTAT3−/− mice. However, 67 pneumonia-induced proteomic changes were exclusive to hepSTAT3−/− mice, whereas 211 proteins were detected in only WT BALF (Fig. 7A). Given that these 211 factors represent those in less susceptible WT mice, they were mapped to canonical pathways using IPA to identify candidate protective factors underlying improved defense in response to secondary lung infection. WT-exclusive protein changes include those indicative of “acute-phase responses,” “production of ROS” (already established as impaired in hepSTAT3−/− mice) (19), “LXR/RXR activation,” and “coagulation system” (Fig. 7B). Corresponding top upregulated and downregulated pneumonia-induced protein changes for each category are shown in Supplemental Table I. We also performed a pairwise comparison between WT and hepSTAT3−/− BALF after pneumonia, and the top 10 analysis-ready genes corresponding to the protein differences between WT and hepSTAT3−/− after endotoxemia and pneumonia corroborate decreases in the APR and coagulation (Table IV). To be considered analysis-ready, genes had to be identifiable and pass any given filters or cutoffs, after which they were ranked by fold change. Within these genes, 9 of the 10 are directly classified as APPs (33–36), with only CPB2 falling short of the classification. Five of these are also involved in the coagulation system (33–35, 37–40), indicating two important liver-driven systems impacting lung airspace content.
FIGURE 7.
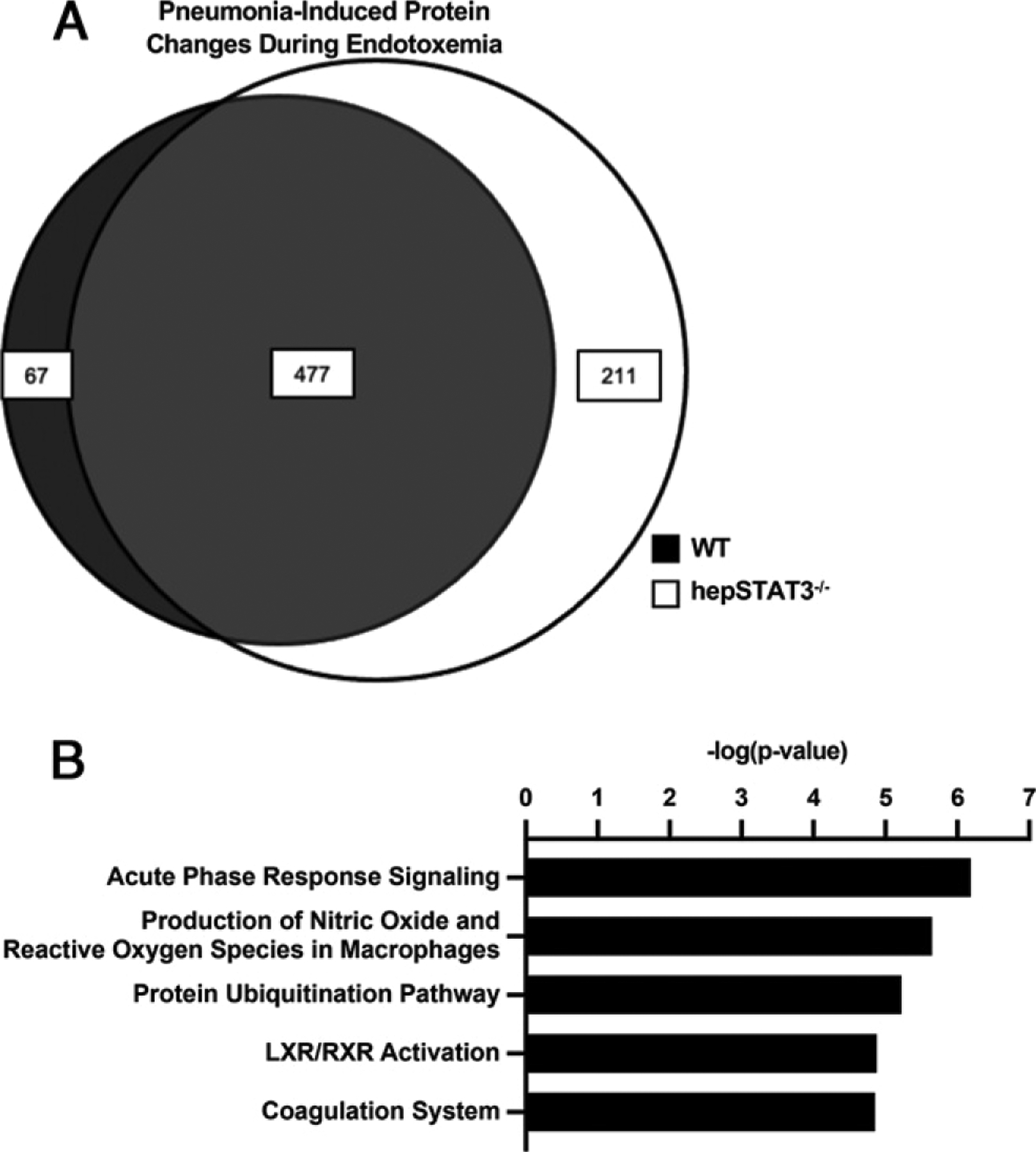
The composition of alveolar exudate is modified by hepatic acute-phase changes. WT and hepSTAT3−/− mice were given 18 h i.p. LPS with or without an additional 24-h challenge with i.t. E. coli. BALF was subjected to MS. (A) Significant pneumonia-induced protein differences (FDR < 0.05) were determined for endotoxemic WT (white circle) and hepSTAT3−/− (gray circle) mice, with shared and genotype-specific differences illustrated as a Venn diagram. (B) Canonical pathways (top five) unique to WT mice were determined using IPA (n = 4/group from two independent experiments).
Table IV.
Top differentially expressed analysis-ready molecules in BALF
| Gene Names | ANOVA (p) | q Value | Fold Change |
|---|---|---|---|
| A2ma,b | 7.2E–5 | 0.11 | −15.69 |
| Apcsa | 1.9E–4 | 0.11 | −11.23 |
| Saa1a | 2.3E–4 | 0.11 | −10.45 |
| Fgga,b | 8.3E–4 | 0.16 | −9.72 |
| Fgaa,b | 7.2E–4 | 0.16 | −8.84 |
| Fgba,b | 9.2E–4 | 0.16 | −8.61 |
| Orm1/2a | 1.9E–3 | 0.22 | −4.32 |
| Cpb2b | 2.0E–3 | 0.22 | −3.77 |
| Itih4a | 3.5E–4 | 0.11 | −3.61 |
| Serpina3na | 8.0E–4 | 0.16 | −3.06 |
Established APPs.
Known coagulation mediators.
Top 10 analysis-ready molecules between WT and hepSTAT3−/− BALF after 18 h i.p. LPS and 24 h i.t. E. coli according to IPA, ranked by fold change.
Interestingly, all three fibrinogen protein chains were identified as WT-exclusive changes in our BALF proteomics analysis, suggesting fibrin and/or its precursors as particularly relevant liver-dependent factors. To study this further, we measured fibrinogen across the liver, blood, and lungs of WT and hepSTAT3−/− mice after endotoxemia and pneumonia. In the liver, mRNA encoding all three protein chains was similarly affected by STAT3 deficiency, with no observable differences in saline controls but a substantial loss of gene induction in hepSTAT3−/− mice after 18 h of LPS that persisted during pneumonia (Fig. 8A). Plasma (Fig. 8B) and BALF (Fig. 8C) protein concentrations (indicative of pan-fibrinogen) also confirmed reduced circulating levels that were ultimately evident in pneumonic lungs, likely resulting from diminished fibrinogen in plasma-derived exudate, which was not present until after the onset of pneumonia. Although the exact roles and contributions of fibrinogen and other liver-dependent APPs remain unclear in the context of our current challenge model, we speculate that such changes are directly linked to the established protective effects when lungs encounter infection after a systemic inflammatory event (19).
FIGURE 8.
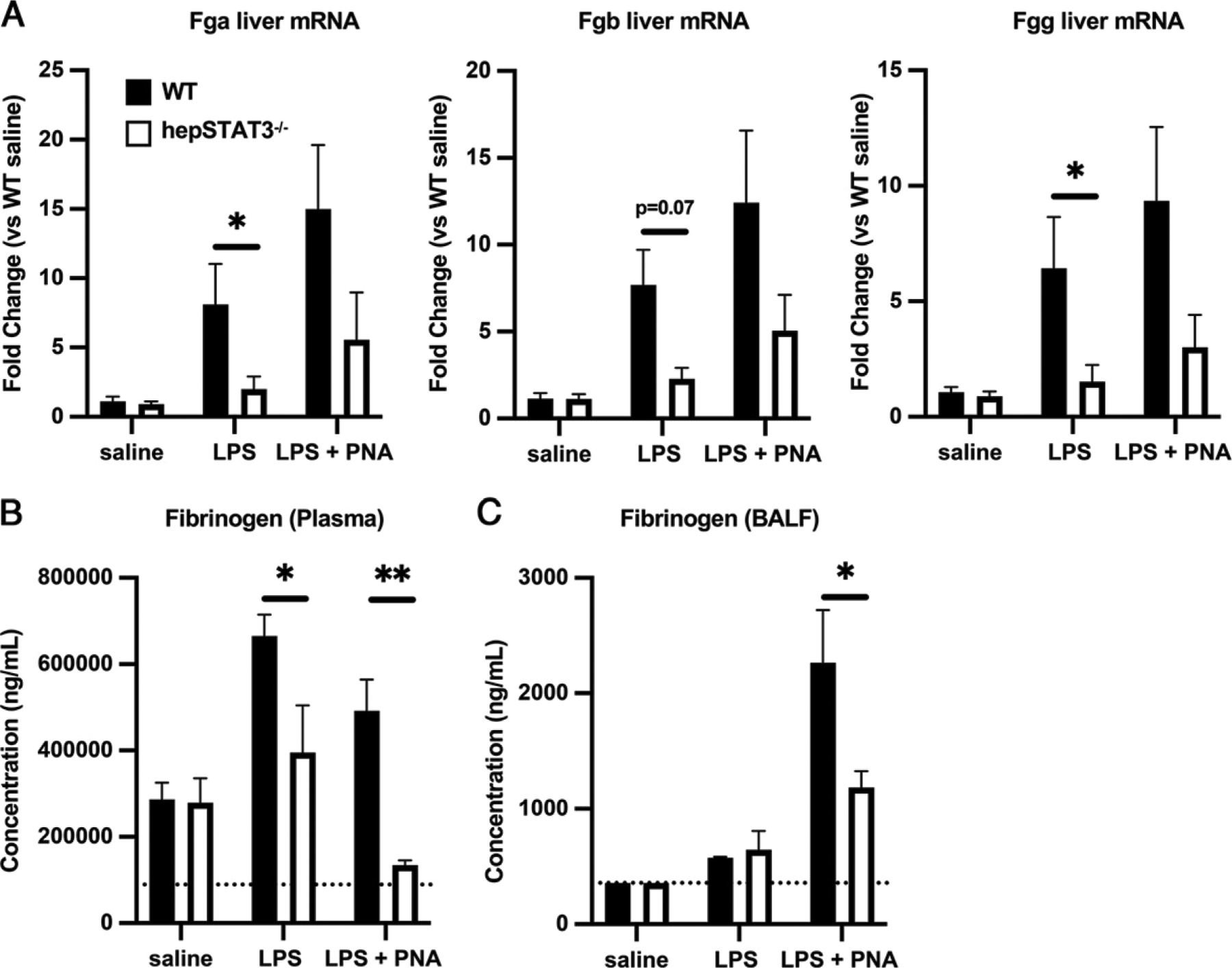
Synthesis and delivery of fibrinogen to pneumonic airspaces is compromised in hepSTAT3−/− mice in response to endotoxemia followed by pneumonia (PNA). WT and hepSTAT3−/− mice were given 18 h i.p. saline, 18 h i.p. LPS, or 18 h i.p. LPS followed by a 24-h challenge with i.t. E. coli (106 CFUs). (A) qRT-PCR was used to determine liver mRNA induction for all three fibrinogen peptide chains. ELISAs were performed to determine fibrinogen concentrations in (B) plasma and (C) BALF from the indicated experimental groups. Data are represented as means with SEM, and significance was determined using a 2-way ANOVA followed by a Sidak test for multiple comparisons (*p < 0.05, **p < 0.01). n = 3–6/group from eight independent experiments.
Discussion
The results of this study, to our knowledge, are the first to reveal liver-dependent remodeling of the lungs, here initiated in response to a systemic challenge with LPS. Together with our prior work (19), we believe that the lung’s response to liver-initiated signals is essential for maintaining its ability to respond to secondary bacterial challenges, likely influencing the degree to which systemic duress, such as that elicited by trauma, burn, and sepsis (1–3, 10–12), impacts pneumonia susceptibility. Our laboratory previously established the presence, activation mechanics, and functional significance of liver acute-phase changes across multiple settings, including lung infections with several pathogens and the two-hit challenge involving endotoxemia followed by pneumonia as implemented here (17–20). Although these prior studies consistently revealed hepatocyte STAT3 activation as a requirement for full liver activity, the direct influence of this response on biological pathways in the lungs has been relatively underdeveloped. This study advances our understanding of the lung–liver axis by comprehensively detailing liver-dependent transcriptional and protein changes to the lung, depicting kinetic differences in response between genotypes, implicating myeloid and macrophage activation as liver dependent, and revealing the strong influence of liver activity on coagulation mediators and other humoral changes within the pneumonic airspaces, many of which likely collaborate to promote defense and tissue homeostasis.
In our transcriptional profiling studies, we were surprised to detect a far greater influence of the liver after endotoxemia alone compared with that observed after the secondary intrapulmonary challenge, with nearly 2000 significant gene changes between genotypes present before the introduction of bacteria to the lungs. This result demonstrates a profound impact of the liver on the pulmonary environment in response to a systemic inflammatory challenge, which, based on our prior study (19), is essential for limiting vulnerability to secondary bacterial infections. Given the substantially compromised immune response that we previously reported in hepSTAT3−/− mice under these circumstances (19), we were initially surprised to discover an exaggeration of proinflammatory gene programs in endotoxemic mice lacking an intact liver response. Indeed, the predicted upstream regulators based on this gene set include quintessential proinflammatory triggers, many of which promote innate pulmonary defense in response to bacterial infection (41–44), seemingly contradicting the impaired responses observed in this context (19). Activated functional pathways predicted for hepSTAT3−/− lungs after endotoxemia were also consistent with an exaggerated immune state, including evidence of elevated myeloid cell function and mobility, as well as diminished cytoskeletal organization (perhaps attributable to migration/engulfment), again implicating liver function as a regulatory measure for lung immune responsiveness. This notion was further supported by increased protein concentrations of numerous lung cytokines, many of which promote myeloid cell activation and migration (43, 45–48).
Although the heightened immune status of hepSTAT3−/− lungs was not sufficient to cause detectable changes in cell numbers or histopathology, we believe that it modulates the activation capacity of the lungs to further stimulation, such that immune “bandwidth” is compromised by the inappropriately elevated baseline. This indicates that liver remodeling of the lung during endotoxemia (and perhaps other settings of systemic inflammation) tailors the immunological tone of the lungs, rendering an environment that is appropriately responsive to secondary bacterial challenges. In support of this, we observed substantial pneumonia-induced effects in WT lungs that were virtually absent in the lungs of hepSTAT3−/− mice (Fig. 5), in which inflammatory cytokine responses peaked after endotoxemia alone (Fig. 4). Ultimately, these findings raise the question of whether a heightened immune baseline is detrimental to the lung’s capacity to respond further when needed. This concept has, in fact, been observed across multiple conditions that increase pneumonia susceptibility, including age, obesity, and chronic diseases (1, 13–16, 49). For instance, older patients, a group associated with higher pneumonia risk, showed increased inflammatory cytokine levels and immune cells in BAL compared with younger patients (50), indicating a lingering level of inflammation at baseline. In addition, age-associated inflammation has been shown to increase pulmonary bacterial ligand expression while also delaying immune responses (13, 51); particularly, the aged lung microenvironment has been implicated in dysfunctional alveolar macrophage responses (52). Moreover, premature and unbalanced inflammation may elicit biological changes similar to those observed after endotoxin tolerance, which also precedes blunted responses to secondary infection (53). Here we have established the liver as an important checkpoint through which the lungs and likely other tissues are calibrated in response to systemic inflammatory triggers.
As discussed earlier, changes in lung gene expression profiles did not correspond to changes in cellularity, indicating that the observed alterations in lung immune activity are more likely attributable to differences in cell function. Although the identity of lung cell types influenced by the liver remains an active area of investigation for our laboratory, we speculate that macrophage populations may be particularly reliant on hepatic input for several reasons. First, this is supported by our prior results in which alveolar macrophages from hepSTAT3−/− lungs produced significantly less ROS after endotoxemia followed by pneumonia (19). In this study, bioinformatics analysis of upregulated genes in hepSTAT3−/− lungs (after endotoxemia) revealed significant alignment with other gene sets generated from stimulated macrophages, whereas such effects were not identified for any other cell type. Meanwhile, at the mRNA and/or protein levels, we detected exaggerated expression of cytokines, such as GM-CSF, CCL2, and CXCL1/2, which serve known roles either upstream or downstream of macrophage activity in the lungs (54–56). Our own studies, however, are not the first to consider responses of lung macrophages to remote stimuli. For instance, macrophage defects in models of sepsis-induced immunosuppression have been previously described (57–61), and recent evidence suggests that alveolar macrophage immunoparalysis may be because of epigenetic modifications after a systemic inflammatory challenge (62). Similar circumstances have been described for interstitial macrophages, which are reprogrammed by endothelial production of Rspondin3 during endotoxemia to dampen inflammatory responses (63). Sepsis, stroke, and myocardial infarction were also recently shown to have substantial effects on tissue macrophages, including, but not limited to, those in the lungs (64). These observations combined with our own substantiate lung macrophages as a target of nonpulmonary duress. However, the precise degree to which this impacts pneumonia susceptibility in a liver-dependent manner remains to be determined.
Although gene expression differences were detected in hepSTAT3−/− lungs before pneumonia (after endotoxemia), seemingly dictating responses to bacteria thereafter, we also detected significant modifications to the alveolar proteome during pneumonia, with results demonstrating liver activation as a requirement for the accumulation of coagulation mediators and other APPs. Inflammation and coagulation are integral for tissue homeostasis but require delicate coordination, as evidenced recently by the specific pathology seen in patients with coronavirus disease 2019 (65). Poorly regulated coagulation can be pathological, especially given its significance in sequestering pathogens within tissues such as the lungs to prevent spread of infection (66–72). Yet, the manner in which coagulation mediators and other liver-derived APPs collaborate in the airspaces to control pneumonia outcome is poorly understood. Our data highlight fibrinogen as a potential APP of interest, given substantial reductions in all three fibrinogen peptides across the liver, circulation, and lungs of hepSTAT3−/− mice during endotoxemia and pneumonia. Fibrinogen and its downstream products are known to exhibit direct antimicrobial functions, constrain bacterial dissemination, and facilitate immune function independent of other coagulation factors (71–74). We also identified A2M and CPB2 as prominent liver-dependent constituents of pneumonic lung. These factors cannot only control coagulation through fibrin stabilization, but they also have immunomodulatory effects through their respective interactions with complement proteins and urokinase plasminogen activator, whose receptor is implicated as a liver-dependent change during endotoxemia (Fig. 4B) (37–39, 75–81). Together, these findings implicate fibrinogen as a key liaison for liver-dependent protection in the lungs, aided by supporting factors, such as A2M, CPB2, and likely others, that constitute important avenues for future investigation.
Although our current studies were mostly performed using E. coli as a secondary challenge after systemic LPS, we anticipate that our findings are similar in response to other secondary infections given: (1) similar results in cytokine mRNA induction in response to S. aureus (Supplemental Fig. 1); and (2) the abundance of liver-dependent changes that occur before the introduction of E. coli as a result of LPS alone (Figs. 1, 2, 4), any number of which could potentially impact secondary challenges of any type. That said, we acknowledge that the experimental conditions used here, including our choices for inducing systemic inflammation (LPS), respiratory infection (E. coli/S. aureus), or any combination therein, may not reflect the conditions of other important biological contexts, which will require further consideration.
Another limitation of our study relates to the challenge of delineating precisely which targets are most responsible for the pulmonary effects observed in hepSTAT3−/− mice. By targeting hepatocyte STAT3, which was originally named “acute phase response factor” on its discovery in 1994 (82), we have blocked the induction of myriad gene programs constituting the liver APR. This approach importantly benefits from not eliciting baseline changes in hepatic gene expression (17). Thus, although hepSTAT3−/− mice lack the ability to respond to inflammatory stimulation, circulating levels of APPs, such as fibrinogen (Fig. 8) and others (17), are intact. This differs from mouse models in which specific liver-derived factors are deleted (83–86), which have their own strengths and limitations. For instance, mice lacking the gene encoding serum amyloid P (Apcs) are susceptible to pneumonia, providing an important example whereby a specific factor contributes to lung immunity (83). Similar results have been reported for other APP-specific knockout mice (86, 87), but this strategy neither accounts for baseline APP concentrations nor does it specify the importance of hepatocytes as the origin of gene expression. In all likelihood, the benefits of STAT3-driven hepatocyte activity observed in our prior and current studies are due to integrated contributions from multiple factors that alter lung immunity in a complex and coordinated fashion. Further investigations using conditional liver-specific mutant mice will be required to more precisely understand which factors affect which lung cell types in which ways.
Overall, we have identified biological pathways in the lung that rely on an intact liver response during systemic inflammation. These changes involve myriad transcriptional programs and an altered airspace proteome. We believe that this response calibrates lung immunity to respond more appropriately and efficiently to secondary challenges, resulting in a scenario whereby hepatic acute-phase changes counter the deleterious consequences of systemic inflammation. Further studies are needed to determine the extent to which lung transcriptional programs, macrophage activity, coagulation modulators, and other pathways responsive to hepatic input coalesce to reduce the vulnerability of lungs to infection.
Supplementary Material
Acknowledgments
We thank the Boston University Microarray and Sequencing Resource Core Facility for RNAseq and analysis and the Boston University Flow Cytometry Core Facility for help with flow cytometry. We would also like to thank Weiwei Wang and Jean Kanyo from Yale MS & Proteomics Resource for sample preparation and liquid chromatography MS/MS data collection, respectively.
This work was supported by the Department of Health and Human Services (HHS), National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) Grant R01-GM120060; HHS, NIH, National Heart, Lung, and Blood Institute (NHLBI) Grants R01-HL111449 and K08-HL130582; HHS, NIH, National Center for Advancing Translational Sciences (NCATS) Grant TL1-TR001410. The Orbitrap Fusion mass spectrometer utilized was supported in part by NIH SIG Grant 1S10OD019967-0 and the Yale School of Medicine.
Abbreviations used in this article:
- APP
acute-phase protein
- APR
acute-phase response
- BALF
bronchoalveolar lavage fluid
- DEG
differentially expressed gene
- FDR
false discovery rate
- HepSTAT3−/−
mice with hepatocyte-specific deletion of STAT3
- IPA
Ingenuity Pathway Analysis
- i.t.
intratracheal
- MS
mass spectrometry
- qRT-PCR
quantitative RT-PCR
- RNAseq
RNA sequencing
- ROS
reactive oxygen species
- WT
wild-type
Footnotes
The gene expression data presented in this article have been submitted to the National Center for Biotechnology Information’s Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) database under accession number GSE167277. The mass spectrometry proteomics data have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset) via the PRIDE partner repository under accession number PXD024663.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Quinton LJ, Walkey AJ, and Mizgerd JP. 2018. Integrative Physiology of Pneumonia. Physiol. Rev 98: 1417–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alberti C, Brun-Buisson C, Burchardi H, Martin C, Goodman S, Artigas A, Sicignano A, Palazzo M, Moreno R, Boulmé R, et al. 2002. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. [Published erratum appears in 2002 Intensive Care Med. 28: 525–526.] Intensive Care Med. 28: 108–121. [DOI] [PubMed] [Google Scholar]
- 3.Chastre J, and Fagon JY. 2002. Ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med 165: 867–903. [DOI] [PubMed] [Google Scholar]
- 4.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esper AM, Moss M, Lewis CA, Nisbet R, Mannino DM, and Martin GS. 2006. The role of infection and comorbidity: Factors that influence disparities in sepsis. Crit. Care Med 34: 2576–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, and Remick DG. 2013. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol. Rev 93: 1247–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paoli CJ, Reynolds MA, Sinha M, Gitlin M, and Crouser E. 2018. Epidemiology and Costs of Sepsis in the United States—An Analysis Based on Timing of Diagnosis and Severity Level. Crit. Care Med 46: 1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, et al. 2020. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395: 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotchkiss RS, Monneret G, and Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lachiewicz AM, Hauck CG, Weber DJ, Cairns BA, and van Duin D. 2017. Bacterial Infections After Burn Injuries: Impact of Multidrug Resistance. Clin. Infect. Dis 65: 2130–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delano MJ, and Ward PA. 2016. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J. Clin. Invest 126: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bronchard R, Albaladejo P, Brezac G, Geffroy A, Seince PF, Morris W, Branger C, and Marty J. 2004. Early onset pneumonia: risk factors and consequences in head trauma patients. Anesthesiology 100: 234–239. [DOI] [PubMed] [Google Scholar]
- 13.Boyd AR, and Orihuela CJ. 2011. Dysregulated inflammation as a risk factor for pneumonia in the elderly. Aging Dis. 2: 487–500. [PMC free article] [PubMed] [Google Scholar]
- 14.Kornum JB, Nørgaard M, Dethlefsen C, Due KM, Thomsen RW, Tjønneland A, Sørensen HT, and Overvad K. 2010. Obesity and risk of subsequent hospitalisation with pneumonia. Eur. Respir. J 36: 1330–1336. [DOI] [PubMed] [Google Scholar]
- 15.Jain S, Self WH, Wunderink RG, Fakhran S, Balk R, Bramley AM, Reed C, Grijalva CG, Anderson EJ, Courtney DM, et al. ; CDC EPIC Study Team. 2015. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Adults. N. Engl. J. Med 373: 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelton SI, Shea KM, Farkouh RA, Strutton DR, Braun S, Jacob C, Klok R, Gruen ES, and Weycker D. 2015. Rates of pneumonia among children and adults with chronic medical conditions in Germany. BMC Infect. Dis 15: 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinton LJ, Blahna MT, Jones MR, Allen E, Ferrari JD, Hilliard KL, Zhang X, Sabharwal V, Alg€ul H, Akira S, et al. 2012. Hepatocyte-specific mutation of both NF-κB RelA and STAT3 abrogates the acute phase response in mice. J. Clin. Invest 122: 1758–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinton LJ, Jones MR, Robson BE, and Mizgerd JP. 2009. Mechanisms of the hepatic acute-phase response during bacterial pneumonia. Infect. Immun 77: 2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilliard KL, Allen E, Traber KE, Kim Y, Wasserman GA, Jones MR, Mizgerd JP, and Quinton LJ. 2015. Activation of Hepatic STAT3 Maintains Pulmonary Defense during Endotoxemia. Infect. Immun 83: 4015–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hilliard KL, Allen E, Traber KE, Yamamoto K, Stauffer NM, Wasserman GA, Jones MR, Mizgerd JP, and Quinton LJ. 2015. The Lung-Liver Axis: A Requirement for Maximal Innate Immunity and Hepatoprotection during Pneumonia. Am. J. Respir. Cell Mol. Biol 53: 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillon A, Arafa EI, Barker KA, Belkina AC, Martin I, Shenoy AT, Wooten AK, Lyon De Ana C, Dai A, Labadorf A, et al. 2020. Pneumonia recovery reprograms the alveolar macrophage pool. JCI Insight 5: e133042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. [Published erratum appears in 1994 Genome Biol. 24: 1949.] Genome Biol. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44(W1): W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charkoftaki G, Thompson DC, Golla JP, Garcia-Milian R, Lam TT, Engel J, and Vasiliou V. 2019. Integrated multi-omics approach reveals a role of ALDH1A1 in lipid metabolism in human colon cancer cells. Chem. Biol. Interact 304: 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47(D1): D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deutsch EW, Bandeira N, Sharma V, Perez-Riverol Y, Carver JJ, Kundu DJ, Garćıa-Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS, et al. 2020. The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 48(D1): D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivanov AI, Parkos CA, and Nusrat A. 2010. Cytoskeletal regulation of epithelial barrier function during inflammation. Am. J. Pathol 177: 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAleer JP, and Vella AT. 2008. Understanding how lipopolysaccharide impacts CD4 T-cell immunity. Crit. Rev. Immunol 28: 281–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakravortty D, and Nanda Kumar KS. 2000. Bacterial lipopolysaccharide induces cytoskeletal rearrangement in small intestinal lamina propria fibroblasts: actin assembly is essential for lipopolysaccharide signaling. Biochim. Biophys. Acta 1500: 125–136. [DOI] [PubMed] [Google Scholar]
- 31.Chen K, Fu Q, Li D, Wu Y, Sun S, and Zhang X. 2016. Wnt3a suppresses Pseudomonas aeruginosa-induced inflammation and promotes bacterial killing in macrophages. Mol. Med. Rep 13: 2439–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Y, Mishra A, Weng T, Chintagari NR, Wang Y, Zhao C, Huang C, and Liu L. 2014. Wnt3a mitigates acute lung injury by reducing P2X7 receptor-mediated alveolar epithelial type I cell death. Cell Death Dis. 5: e1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cray C 2012. Acute phase proteins in animals. Prog. Mol. Biol. Transl. Sci 105: 113–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan JJ, Wang PY, Zhang Y, Qin Z, Sun Y, Hu BH, Su DF, Xu DP, and Liu X. 2019. Role of acute-phase protein ORM in a mice model of ischemic stroke. J. Cell. Physiol 234: 20533–20545. [DOI] [PubMed] [Google Scholar]
- 35.Dalby MJ, Aviello G, Ross AW, Walker AW, Barrett P, and Morgan PJ. 2018. Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci. Rep 8: 15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura N, Hatano E, Iguchi K, Sato M, Kawaguchi H, Ohtsu I, Sakurai T, Aizawa N, Iijima H, Nishiguchi S, et al. 2019. Elevated levels of circulating ITIH4 are associated with hepatocellular carcinoma with nonalcoholic fatty liver disease: from pig model to human study. BMC Cancer 19: 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Xie ZH, Lv YX, Tang QP, Zhang H, Zhang JY, Wu B, and Jiang WH. 2016. A proteomics analysis reveals that A2M might be regulated by STAT3 in persistent allergic rhinitis. Clin. Exp. Allergy 46: 813–824. [DOI] [PubMed] [Google Scholar]
- 38.Leenaerts D, Aernouts J, Van Der Veken P, Sim Y, Lambeir AM, and Hendriks D. 2017. Plasma carboxypeptidase U (CPU, CPB2, TAFIa) generation during in vitro clot lysis and its interplay between coagulation and fibrinolysis. Thromb. Haemost 117: 1498–1508. [DOI] [PubMed] [Google Scholar]
- 39.Rehman AA, Ahsan H, and Khan FH. 2013. α−2-Macroglobulin: a physiological guardian. J. Cell. Physiol 228: 1665–1675. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, He J, Zhao J, Xu M, Lou D, Tso P, Li Z, and Li X. 2017. Effect of ApoA4 on SERPINA3 mediated by nuclear receptors NR4A1 and NR1D1 in hepatocytes. Biochem. Biophys. Res. Commun 487: 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin CF, Tsai CC, Huang WC, Wang CY, Tseng HC, Wang Y, Kai JI, Wang SW, and Cheng YL. 2008. IFN-gamma synergizes with LPS to induce nitric oxide biosynthesis through glycogen synthase kinase-3-inhibited IL-10. J. Cell. Biochem 105: 746–755. [DOI] [PubMed] [Google Scholar]
- 42.Bradley JR 2008. TNF-mediated inflammatory disease. J. Pathol 214: 149–160. [DOI] [PubMed] [Google Scholar]
- 43.Hamilton JA 2020. GM-CSF in inflammation. J. Exp. Med 217: e20190945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasembeli MM, Bharadwaj U, Robinson P, and Tweardy DJ. 2018. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci 19: 2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawlor KE, Campbell IK, Metcalf D, O’Donnell K, van Nieuwenhuijze A, Roberts AW, and Wicks IP. 2004. Critical role for granulocyte colony-stimulating factor in inflammatory arthritis. Proc. Natl. Acad. Sci. USA 101: 11398–11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsantikos E, Lau M, Castelino CM, Maxwell MJ, Passey SL, Hansen MJ, McGregor NE, Sims NA, Steinfort DP, Irving LB, et al. 2018. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J. Clin. Invest 128: 2406–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boro M, and Balaji KN. 2017. CXCL1 and CXCL2 Regulate NLRP3 Inflammasome Activation via G-Protein-Coupled Receptor CXCR2. J. Immunol 199: 1660–1671. [DOI] [PubMed] [Google Scholar]
- 48.De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, and Hogg N. 2013. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121: 4930–4937. [DOI] [PubMed] [Google Scholar]
- 49.Sethi S 2010. Infection as a comorbidity of COPD. Eur. Respir. J 35: 1209–1215. [DOI] [PubMed] [Google Scholar]
- 50.Meyer KC, Ershler W, Rosenthal NS, Lu XG, and Peterson K. 1996. Immune dysregulation in the aging human lung. Am. J. Respir. Crit. Care Med 153: 1072–1079. [DOI] [PubMed] [Google Scholar]
- 51.Hinojosa E, Boyd AR, and Orihuela CJ. 2009. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J. Infect. Dis 200: 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, Lu Z, Sichizya L, Aillon RP, Chen CI, et al. 2021. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J. Clin. Invest 131: e140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cavaillon JM, and Adib-Conquy M. 2006. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care 10: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herbold W, Maus R, Hahn I, Ding N, Srivastava M, Christman JW, Mack M, Reutershan J, Briles DE, Paton JC, et al. 2010. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect. Immun 78: 2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell BC, Carey BC, Uchida K, and Suzuki T. 2009. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr. Opin. Immunol 21: 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maus UA, Waelsch K, Kuziel WA, Delbeck T, Mack M, Blackwell TS, Christman JW, Schlöndorff D, Seeger W, and Lohmeyer J. 2003. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J. Immunol 170: 3273–3278. [DOI] [PubMed] [Google Scholar]
- 57.Reddy RC, Chen GH, Newstead MW, Moore T, Zeng X, Tateda K, and Standiford TJ. 2001. Alveolar macrophage deactivation in murine septic peritonitis: role of interleukin 10. Infect. Immun 69: 1394–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, and Standiford TJ. 1999. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J. Immunol 162: 392–399. [PubMed] [Google Scholar]
- 59.Peters van Ton AM, Kox M, Abdo WF, and Pickkers P. 2018. Precision Immunotherapy for Sepsis. Front. Immunol 9: 1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson SQ, Modi HN, Balk RA, Bone RC, and Casey LC. 1991. Reduced alveolar macrophage production of tumor necrosis factor during sepsis in mice and men. Crit. Care Med 19: 1060–1066. [DOI] [PubMed] [Google Scholar]
- 61.Smith PD, Suffredini AF, Allen JB, Wahl LM, Parrillo JE, and Wahl SM. 1994. Endotoxin administration to humans primes alveolar macrophages for increased production of inflammatory mediators. J. Clin. Immunol 14: 141–148. [DOI] [PubMed] [Google Scholar]
- 62.Roquilly A, Jacqueline C, Davieau M, Mollé A, Sadek A, Fourgeux C, Rooze P, Broquet A, Misme-Aucouturier B, Chaumette T, et al. 2020. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. [Published erratum appears in 2020 Nat. Immunol. 21: 962.] Nat. Immunol 21: 636–648. [DOI] [PubMed] [Google Scholar]
- 63.Zhou B, Magana L, Hong Z, Huang LS, Chakraborty S, Tsukasaki Y, Huang C, Wang L, Di A, Ganesh B, et al. 2020. The angiocrine Rspondin3 instructs interstitial macrophage transition via metabolic-epigenetic reprogramming and resolves inflammatory injury. Nat. Immunol 21: 1430–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoyer FF, Naxerova K, Schloss MJ, Hulsmans M, Nair AV, Dutta P, Calcagno DM, Herisson F, Anzai A, Sun Y, et al. 2019. Tissue-Specific Macrophage Responses to Remote Injury Impact the Outcome of Subsequent Local Immune Challenge. Immunity 51: 899–914.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iba T, Levy JH, Connors JM, Warkentin TE, Thachil J, and Levi M. 2020. The unique characteristics of COVID-19 coagulopathy. Crit. Care 24: 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Esmon CT, Xu J, and Lupu F. 2011. Innate immunity and coagulation. J. Thromb. Haemost 9(Suppl 1): 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delvaeye M, and Conway EM. 2009. Coagulation and innate immune responses: can we view them separately? Blood 114: 2367–2374. [DOI] [PubMed] [Google Scholar]
- 68.Levi M, and van der Poll T. 2017. Coagulation and sepsis. Thromb. Res 149: 38–44. [DOI] [PubMed] [Google Scholar]
- 69.Franchini M, and Lippi G. 2012. Fibrinogen replacement therapy: a critical review of the literature. Blood Transfus. 10: 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeaman MR 2014. Platelets: at the nexus of antimicrobial defence. Nat. Rev. Microbiol 12: 426–437. [DOI] [PubMed] [Google Scholar]
- 71.Ko YP, and Flick MJ. 2016. Fibrinogen Is at the Interface of Host Defense and Pathogen Virulence in Staphylococcus aureus Infection. Semin. Thromb. Hemost 42: 408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Påhlman LI, Mörgelin M, Kasetty G, Olin AI, Schmidtchen A, and Herwald H. 2013. Antimicrobial activity of fibrinogen and fibrinogen-derived peptides—a novel link between coagulation and innate immunity. Thromb. Haemost 109: 930–939. [DOI] [PubMed] [Google Scholar]
- 73.Yang H, Ko HJ, Yang JY, Kim JJ, Seo SU, Park SG, Choi SS, Seong JK, and Kweon MN. 2013. Interleukin-1 promotes coagulation, which is necessary for protective immunity in the lung against Streptococcus pneumoniae infection. J. Infect. Dis 207: 50–60. [DOI] [PubMed] [Google Scholar]
- 74.Claushuis TA, de Stoppelaar SF, Stroo I, Roelofs JJ, Ottenhoff R, van der Poll T, and Van’t Veer C. 2017. Thrombin contributes to protective immunity in pneumonia-derived sepsis via fibrin polymerization and platelet-neutrophil interactions. J. Thromb. Haemost 15: 744–757. [DOI] [PubMed] [Google Scholar]
- 75.Komissarov AA, Stankowska D, Krupa A, Fudala R, Florova G, Florence J, Fol M, Allen TC, Idell S, Matthay MA, and Kurdowska AK. 2012. Novel aspects of urokinase function in the injured lung: role of α2-macroglobulin. Am. J. Physiol. Lung Cell. Mol. Physiol 303: L1037–L1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurdowska AK, Geiser TK, Alden SM, Dziadek BR, Noble JM, Nuckton TJ, and Matthay MA. 2002. Activity of pulmonary edema fluid interleukin-8 bound to alpha(2)-macroglobulin in patients with acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol 282: L1092–L1098. [DOI] [PubMed] [Google Scholar]
- 77.Leung LLK, and Morser J. 2018. Carboxypeptidase B2 and carboxypeptidase N in the crosstalk between coagulation, thrombosis, inflammation, and innate immunity. J. Thromb. Haemost 16: 1474–1486. [DOI] [PubMed] [Google Scholar]
- 78.Morser J, Shao Z, Nishimura T, Zhou Q, Zhao L, Higgins J, and Leung LLK. 2018. Carboxypeptidase B2 and N play different roles in regulation of activated complements C3a and C5a in mice. J. Thromb. Haemost 16: 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shao Z, Nishimura T, Leung LL, and Morser J. 2015. Carboxypeptidase B2 deficiency reveals opposite effects of complement C3a and C5a in a murine poly-microbial sepsis model. J. Thromb. Haemost 13: 1090–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Plug T, and Meijers JC. 2016. Structure-function relationships in thrombinactivatable fibrinolysis inhibitor. J. Thromb. Haemost 14: 633–644. [DOI] [PubMed] [Google Scholar]
- 81.Del Rosso M, Margheri F, Serratì S, Chillá A, Laurenzana A, and Fibbi G. 2011. The urokinase receptor system, a key regulator at the intersection between inflammation, immunity, and coagulation. Curr. Pharm. Des 17: 1924–1943. [DOI] [PubMed] [Google Scholar]
- 82.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, and Kishimoto T. 1994. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77: 63–71. [DOI] [PubMed] [Google Scholar]
- 83.Yuste J, Botto M, Bottoms SE, and Brown JS. 2007. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLoS Pathog. 3: 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Umans L, Serneels L, Overbergh L, Lorent K, Van Leuven F, and Van den Berghe H. 1995. Targeted inactivation of the mouse alpha 2-macroglobulin gene. J. Biol. Chem 270: 19778–19785. [DOI] [PubMed] [Google Scholar]
- 85.Suh TT, Holmbäck K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, and Degen JL. 1995. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 9: 2020–2033. [DOI] [PubMed] [Google Scholar]
- 86.Fan Y, Zhang G, Vong CT, and Ye RD. 2020. Serum amyloid A3 confers protection against acute lung injury in Pseudomonas aeruginosa-infected mice. Am. J. Physiol. Lung Cell. Mol. Physiol 318: L314–L322. [DOI] [PubMed] [Google Scholar]
- 87.Michels KR, Zhang Z, Bettina AM, Cagnina RE, Stefanova D, Burdick MD, Vaulont S, Nemeth E, Ganz T, and Mehrad B. 2017. Hepcidin-mediated iron sequestration protects against bacterial dissemination during pneumonia. JCI Insight 2: e92002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.