

Abstract
Mitochondrial fragmentation from defective fusion or unopposed fission contributes to many neurodegenerative diseases. Small molecule mitofusin activators reverse mitochondrial fragmentation in vitro, promising a novel therapeutic approach. The first-in-class mitofusin activator, 2, has a short plasma t½ and limited neurological system bioavailability, conferring “burst activation”. Here, pharmacophore-based rational redesign generated analogs of 2 incorporating cycloalkyl linker groups. A cyclopropyl-containing linker, 5, improved plasma and brain t½, increased nervous system bioavailability, and prolonged neuron pharmacodynamic effects. Functional and single crystal X-ray diffraction studies of stereoisomeric analogs of 5 containing sulfur as a “heavy atom”, 14A and 14B, showed that 5 biological activity resides in the trans-R/R configuration, 5B. Structural analysis revealed stereoselective interactions of 5 associated with its mimicry of MFN2 Val372, Met376 and His380 side chains. Modification of murine ALS phenotypes in vitro and in vivo support advancement of 5B for neurological conditions that may benefit from sustained mitofusin activation.
Graphical Abstract
INTRODUCTION
Mitochondrial fragmentation caused by an imbalance between mitochondria fusion and fission is a common cause of, and contributor to, neurodegenerative diseases (1, 2). Mitochondrial fusion is initiated by outer mitochondrial membrane-embedded mitofusin (MFN) proteins whose extra-organelle domains extend across cytosolic space to interact with counterparts on neighboring mitochondria, physically linking organelles via MFN-MFN dimers/oligomers (3). Mitofusins subsequently induce outer mitochondrial membrane fusion through a process requiring catalytic GTPase activity (4–6). Multiple mitofusin 2 (MFN2) mutations, most frequently located within the MFN2 GTPase domain, are implicated in a rare untreatable childhood sensory-motor neuropathy, Charcot-Marie-Tooth (CMT) disease type 2A (7, 8). Defects in mitofusin function or expression are also described in other neurodegenerative conditions, including experimental amyotrophic lateral sclerosis (9) and clinical Huntington’s disease (10). For these reasons mitofusins are attractive drug targets (11).
Developing a pharmaceutically acceptable activator of mitofusins to enhance mitochondrial fusion was challenging. The anti-rheumatic drug leflunomide was identified as a transcriptional enhancer of mitofusin mRNA expression, but effective concentrations are high (10-50 μM) (12). A putative small molecule mitofusin activator, 4-chloro-2-(1-(2-(2,4,6-trichlorophenyl) hydrazineylidene) ethyl)phenol, was detected by high-throughput screening for fusogenic compounds, but with an EC50 of 4-5 μM it also has limited utility except as a research tool (13). Franco, et al designed an 18 amino acid cell-permeant mitofusin activating peptide modified from hMFN2 amino acids 367-384 that could be introduced into cells using amino terminal TAT47-57 conjugation (14). This peptide activates mitofusins by competing for and disrupting intramolecular peptide-peptide bonds enforcing the closed (inactive) mitofusin protein conformation and has greater potency for mitofusin stimulation than the above compounds (EC50 ~0.3 μM). However, as peptides their therapeutic utility in clinical disease seems limited. Thus, Rocha et al (15) developed small triazolurea-containing molecules, like 1, that potently induce mitochondrial fusion (EC50 ~5 nM) by mimicking function-critical amino acids of the Franco activator peptide. Unfortunately, mitofusin activators with this chemical structure proved incompatible with in vivo use (16).
Phenylhexanamide mitofusin activators possessing drug-like properties were recently described (16). The lead compound of this chemical series (2) demonstrated in vivo engagement of neuronal mitochondria after intravenous administration; its daily intramuscular administration reversed neuromuscular degeneration in a mouse model of CMT2A (17). However, this compound has a short half-life in mouse plasma (t1/2 1.1h) and brain (t1/2 1.06h), properties most suitable for “burst activation” of mitofusins (17). We posited that mitofusin activators having more prolonged presence in the central nervous system could have greater therapeutic utility in other neurodegenerative conditions.
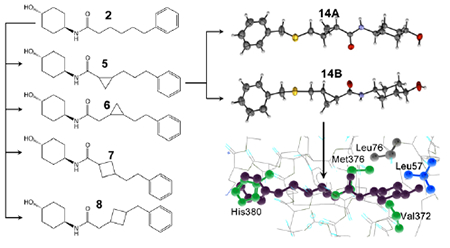
Here, we extended mitofusin activator design by employing the previously described pharmacophore-based strategy (16). We considered that structurally more rigid and metabolically stable compounds resulting from introduction of cycloalkyl modifications into the hexanamide linker (18–21) might have greater engagement of neuronal mitochondria. In particular, introduction of cyclopropyl rings into different compounds has increased potency, selectivity, oral bioavailability, metabolic and chemical stability, or neurological system exposure. Relevant to our pharmacophore-based design strategy, cyclopropyl rings have been employed to restrict the conformation of the serotonin and norepinephrine re-uptake inhibitor Z-Milnacipran and the monoamine oxidase inhibitor Tranylcypramine (21). Here, a pre-clinical lead compound having a linker cyclopropyl group, 5, exhibited potent stereospecific mitofusin activation and longer plasma and brain t1/2 than 2. Single crystal X-ray crystallography of 5 sulfide analogs [14A and 14B] revealed that the (trans-R,R) enantiomer uniquely conferred mitofusin agonist activity. The crystal structure moiety distances and hydrogen bonding characteristics mimicked those of function-critical amino acid side chains within the precursor mitofusin agonist peptide, consistent with the pharmacophore model. In vitro and in vivo proof of concept studies with 5B in murine amyotrophic lateral sclerosis (ALS) support its clinical development for neurodegenerative diseases linked to mitochondrial dysfunction.
RESULTS AND DISCUSSION
Rational re-design of mitofusin activators.
Prototype triazolurea (1) and phenylhexanamide (2) mitofusin agonists share the general structure of a substituted cyclohexyl group (R1) connected to an aromatic moiety (R2) by urea- or carboxamide-containing alkyl linkers (Figure 1A). The linker promotes optimal spacing between R1 and R2, which according to the pharmacophore model (15), respectively mimic human MFN2 His380 and Met376/Val372. The linker can be a source of conformational plasticity, but the published pharmacophore model and reported structure-activity relationships (15, 16) suggest that mitofusin activators can be modified to retain biological activity if R1-R2 separation distance is conserved. Indeed, deletion of one (3) or two (4) carbons from the 2 linker largely abrogated mitofusin activation measured as the ability to promote mitochondrial fusion (increased aspect ratio: length/width) (Figure 1B). Accordingly, we posited that introduction of small cycloalkyl linker groups that do not alter R1-R2 distance could both stabilize the molecule and enhance passive membrane permeability reported to correlate with blood brain barrier penetration (16). Structures and characteristics of parent 2 and nine cycloalkyl linker analogs are shown in Figure 2.
Figure 1. Structures of triazolurea (1) and phenylhexanamide (2) mitofusin activators.
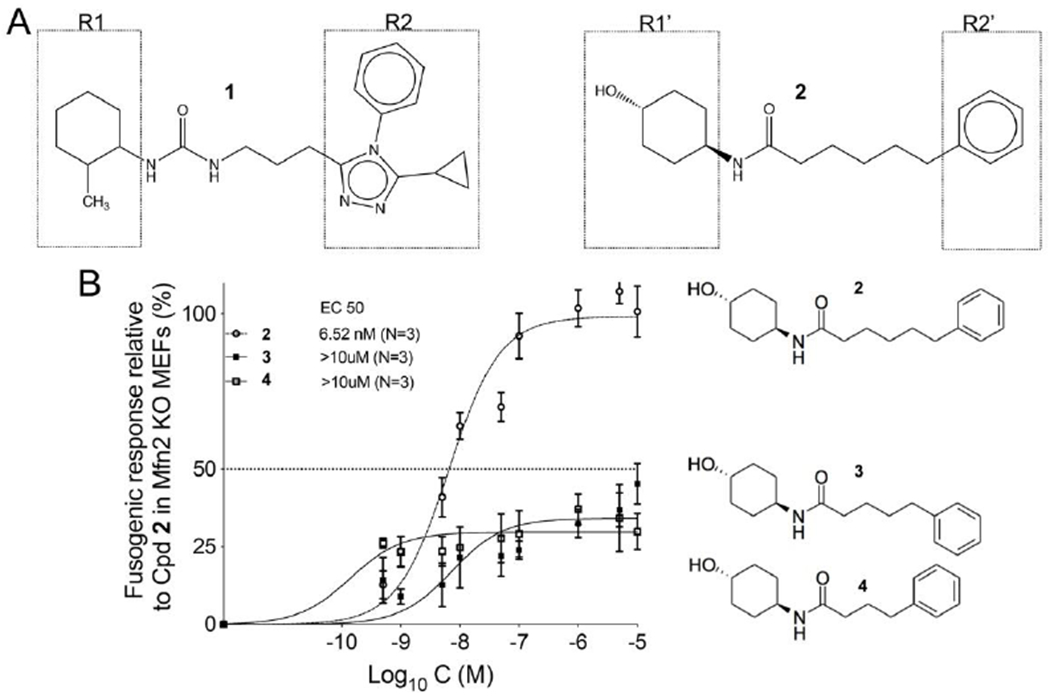
A. Both classes of mitofusin activator share a substituted cyclohexyl (R1, R1’) linked to an aromatic moiety (R2, R2’) by a six or seven member alkyl linker. B. Dose-response relationships for mitochondrial elongation (measured as the increase in mitochondrial aspect ratio) of 2 and its analogs having one (3) or two (4) fewer carbons in the alkyl linker.
Figure 2. Designs for variants of 2 having cycloalkyl linkers.
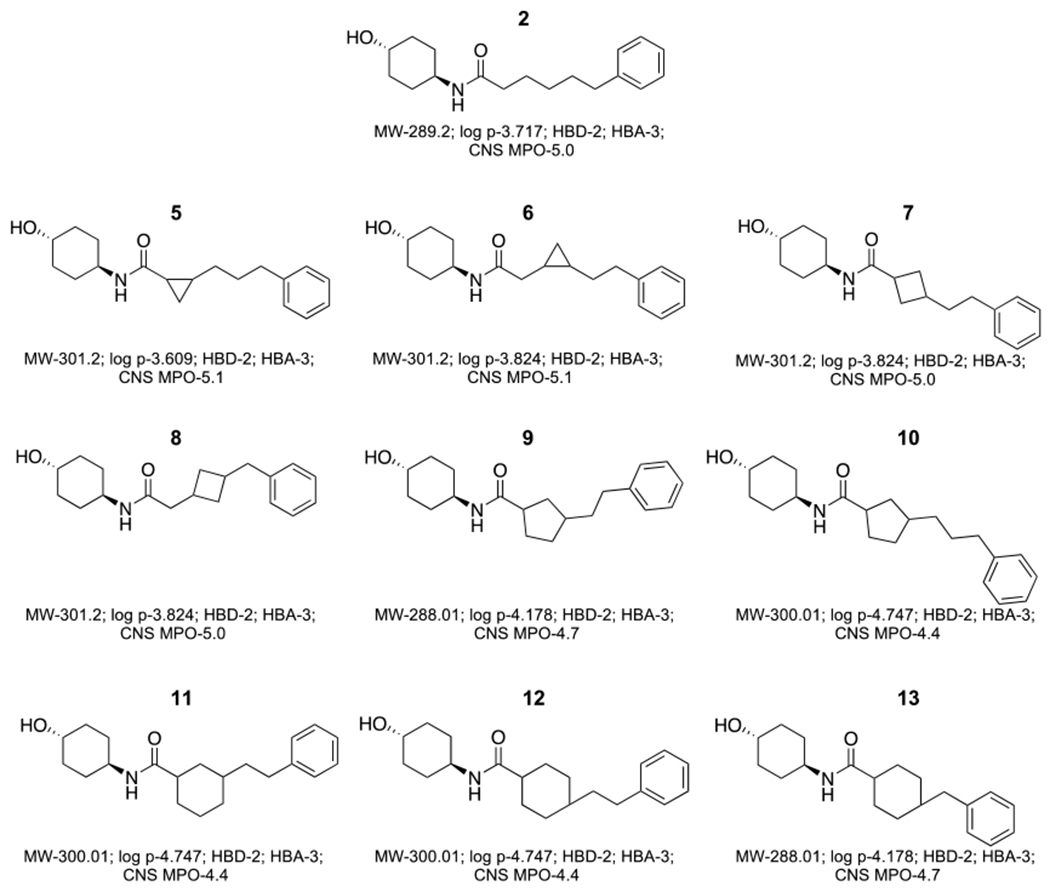
Calculated Lipinski Rule values and central nervous system multiparameter optimization desirability (MPO) factors are shown below. MW = molecular weight; log p = octanol-water partition coefficient; HBD = number of hydrogen bond donors; HBA = number of hydrogen bond acceptors.
In vitro characterization of mitofusin activators with cycloalkyl linkers.
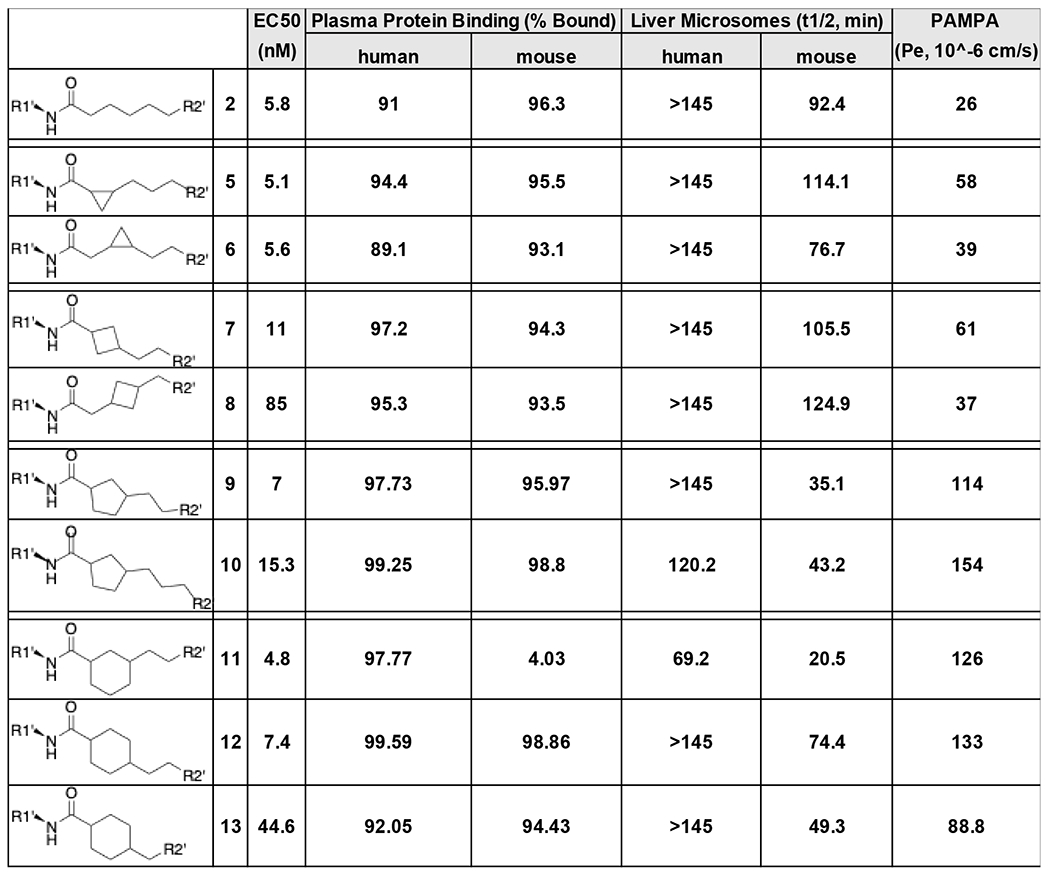
To gauge how pharmacophore modeling conformed to reality, mitofusin activation by 5-13 was compared using the mitochondrial elongation fusogenicity assay in MFN2-deficient cells (14–16). Because the trans-cyclohexanol stereoisomer of parent 2 was biologically active whereas the corresponding cis-analog was not (16), 5-13 were each synthesized as trans stereoisomers at this position. The cyclopropyl analogs, 5 and 6, were potent mitofusin activators having EC50 values for mitochondrial elongation similar to parent 2, i.e. ~5-6 nM (Table 1). There was no clear benefit to increasing the size of linker cycloalkyl groups, which increased PAMPA Pe, but also increased instability in the mouse liver microsomal assay (Table 1, Figure S1).
Table 1.
Functional and pharmacokinetic properties of cycloalkyl linker 2 variants a.
|
In vivo pharmacokinetic properties of 5.
Among the newly synthesized and characterized cycloalkyl linker analogs of 2, in vitro characteristics of 5 appeared pharmaceutically most favorable. Therefore, in vivo PK studies of 5 were undertaken in mice, the species in which all previous PK studies of mitofusin activators have been performed (16). Plasma 5 levels were measured at increasing times after administration of single 10 mg/kg intravenous (IV) doses. Compared to published values for 2 (16), 5 had ~50% longer plasma elimination t1/2 and proportionally greater tissue distribution (Vdss) values, with correspondingly reduced peak plasma and AUC levels (Table 2).
Table 2.
In vivo pharmacokinetic properties of 5 a.
| 5 | 2 | ||
|---|---|---|---|
| Dose (mg/kg) | 10 | 10 | |
| C0 (ng/mL) | 32111 | 50,600 | |
| t1/2 (hours) | 1.64 | 1.1 | |
| AUC0-last (ng*h/mL) | 9178 | 11495 | |
| Vdss (L/kg) | 0.6 | 0.347 |
IV administration in mice, 10 mg/kg; values for 2 are from (16).
Fusogenicity and mitofusin-interactions of 5 are stereoisomer-specific.
Because only the trans-cyclohexanol stereoisomer of 2 has biological activity (16), 5-13 were synthesized as trans-isomers at the cyclohexanol group. However, the linker cyclopropyl group in 5 creates additional chiral centers to either side, with 4 possible stereoisomers (Figure 3, left). Chiral separation of 5 by supercritical fluid chromatography (SFC-H) identified only two isomers, designated -A and -B for the faster and slower eluting forms, respectively (Figure 3, right). Proton NMR of synthetic intermediate 5C revealed a coupling constant of ~19 Hz for the olefin proton at 6.75 ppm, indicating trans-olefin geometry (Figure S2). In agreement with expectations based on known reaction mechanism, the product of the Corey-Chaykovsky reaction leading to 5 (Synthetic Scheme 2) contained only two isomers: trans-R,R and S,S (Figure 3).
Figure 3. Synthesis of 5 results in a ~50:50 mixture of 5 trans- stereoisomers.
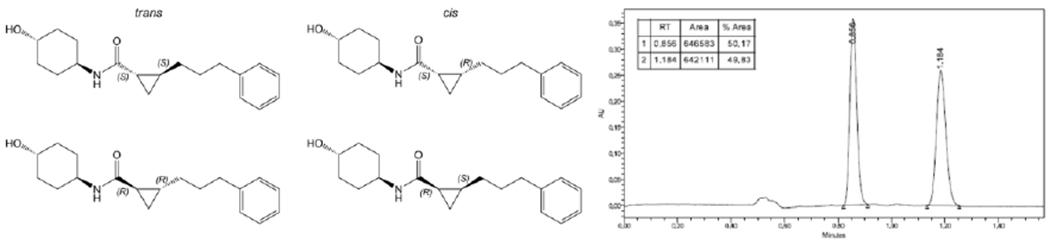
Left, structures of the four possible 5 stereoisomers. Right, chiral separation identifies two forms, shown to be trans- based on 1H-NMR of synthetic intermediate 5C (Figure S2).
Scheme 2.
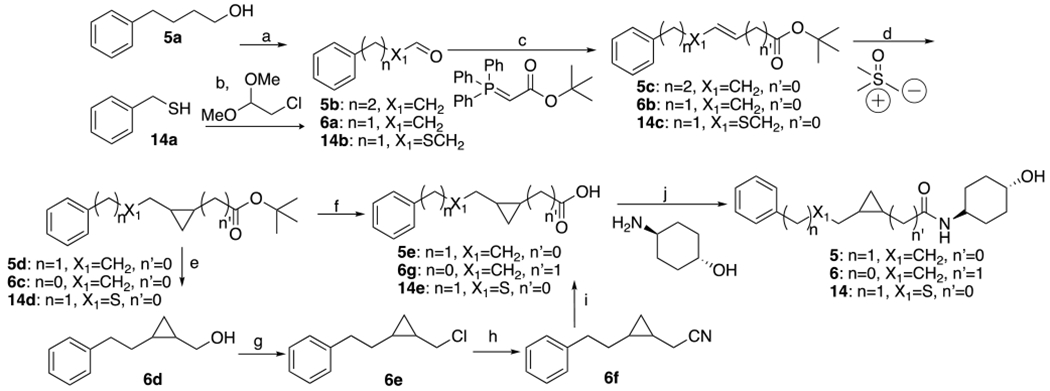
Synthesis of 5, 6 and 14a
aReagents and conditions: (a) oxalyl chloride, DMSO, TEA, DCM, −55-25°C, 20min. (b) i. EtOH, EtONa, KI; ii. 2-chloro-1,1-dimethoxyethane, 80°C, 12h; H2O, H2SO4, 60°C, 12h. (c) THF, 20°C. 5, 14 : 12h, 6: 1h. (d) NaH, DMSO, 20°C, 1.5h. (e) LiAH4, THF, 0-25°C, 3h. (f) TFA, DCM, 25°C, 15h. (g) SOCl2, TEA, CHCl3, 0-70°C, 1h. (h) N(nBu)4CN, THF, 70°C, 12h. (i) KOH, EtOH, H2O, 100°C, 16h. (j) HOBt, EDCI, DIEA, DMV, 25°C. 5, 6: 16h, 14: 2h.
Mitofusin-stimulating activities of 5A and 5B were compared by assaying their effects on mitochondrial elongation and polarization status in Mfn1-null (i.e. expressing Mfn2 alone) or Mfn2-null (i.e. expressing Mfn1 only) murine embryonic fibroblasts (Figure 4). The prototype triazolurea mitofusin activator, 1, was assayed in parallel as a historical comparator. There was little effect produced by 5A (1 μM, 48 hours) on either outcome. By comparison, 5B was equipotent with 1 in cells expressing either Mfn1 or Mfn2. Thus, 5B is a potent mitofusin activator.
Figure 4. Mitochondrial elongation and polarization-enhancing activities of 5 enantiomers.
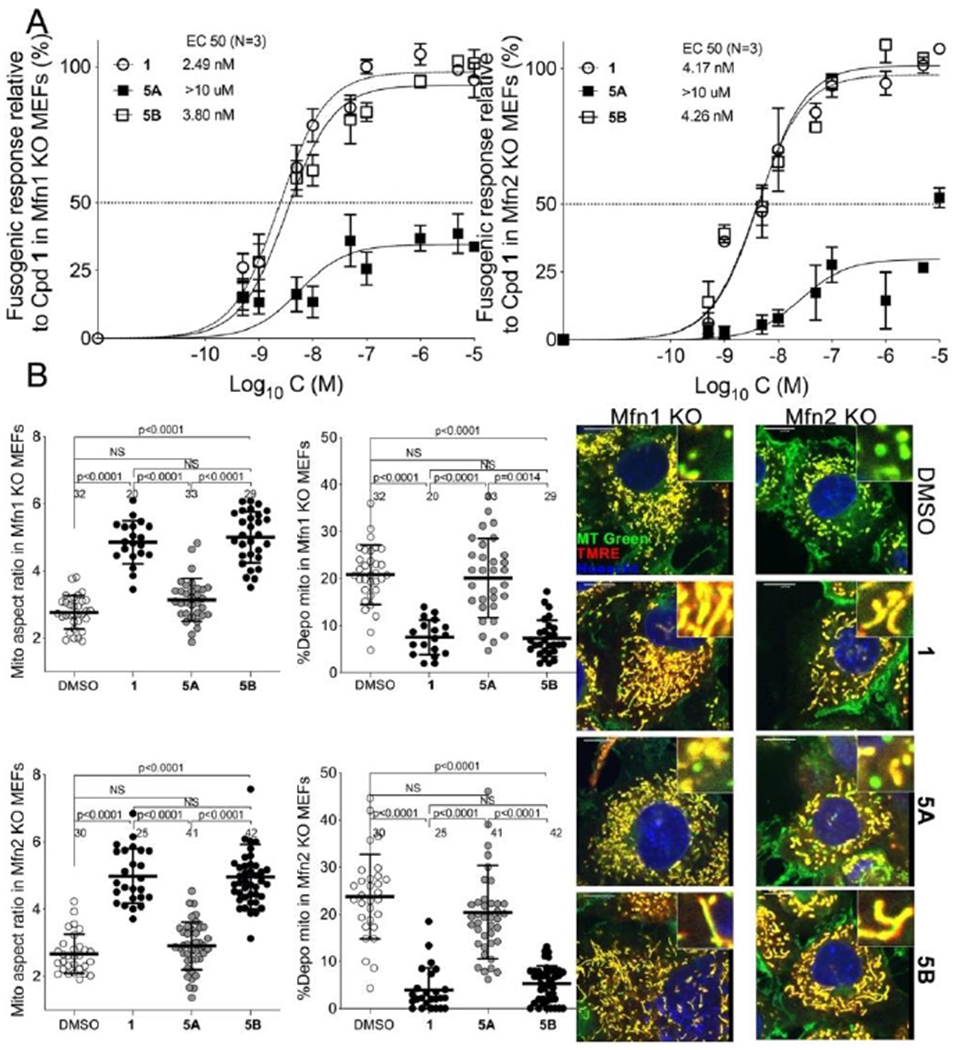
A. Dose-response curves for 5A and 5B to increase mitochondrial aspect ratio (length/width) in cultured fibroblasts lacking Mfn1 (left, 5A: EC50 > 10μM, 5B: EC50 = 3.80nM; 95% confidence limits, 2.70-5.28nM) or Mfn2 (right, 5A: EC50 > 10μM, 5B: EC50 = 4.26nM; 95% confidence limits, 2.99-6.04nM). Parallel studies with prototype mitofusin activator 1 are shown for comparison. B. Group data (left) and representative live cell confocal micrographs (right) from Mfn1 and Mfn2 null cells showing mitochondrial elongation (increase in aspect ratio) and reversal of depolarization with 1 μM test compound added for 48 hours. Enlarged mitochondria are shown in insets. Mitochondria staining yellow are highly polarized, i.e. healthy; green staining mitochondria are depolarized and dysfunctional.
We tested 5A and 5B interactions with MFN2 protein using a previously described FRET assay that measures MFN2 conformational changes provoked by mitofusin activators (15–17). Please note that FRET assays were performed on isolated mitochondria, whereas assessments of mitochondrial elongation were performed in intact cells. A high compound concentration (1 uM) was employed to test whether inactivity of enantiomer 5A to promote mitochondrial elongation in cells corresponded with FRET inactivity for in vitro target engagement. As shown in Figure 5, 5B had the same effect on MFN2 conformation previously described for mitofusin activators 1 15 and 2 16) i.e. it decreased the FRET signal, reflecting separation of the N-terminal mCerulean and C-terminal mVenus fluorophores after unfolding of the protein into its active conformation. By comparison, 5A did not decrease the FRET signal. Thus, 5A did not conformationally activate MFN2.
Figure 5. MFN2 altering activity of 5 is stereoisomer-specific.
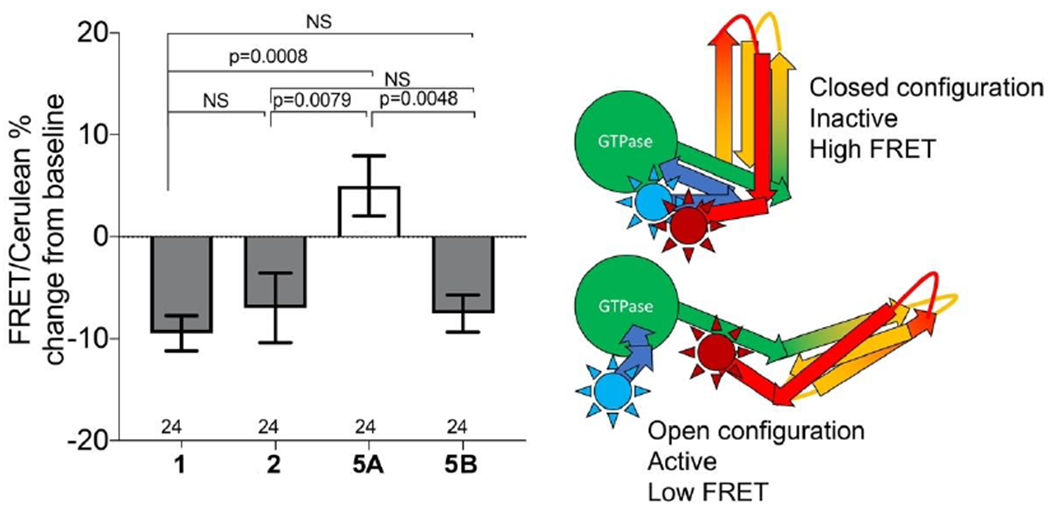
Results of FRET studies comparing MFN2 conformation altering activities of prototype mitofusin activators 1 and 2 with 5 stereoisomers (all compounds added to a final concentration of 1μM as in Figure 4B; assays were performed after 4 hours). Decreased FRET signal reflects MFN2 unfolding as depicted to the right. Blue sun indicates position of amino terminal mCerulean; red sun is C-terminal mVenus. Means ± SEM of 6 separate studies with 4-6 replicates each.
X-ray diffraction reveals enantiomeric structures of 5A and 5B.
Although 5A and 5B were determined to be the trans-cyclopropyl stereoisomers (vide supra), it was not possible to assign R,R vs S,S structures to the stereoisoforms. Thus, we synthesized heavier sulfide analogs of 5A and 5B for X-ray diffraction studies, designated 14A and 14B respectively (Figure 6A). Like their respective parents, 14B potently stimulated mitochondrial elongation in Mfn2 null fibroblasts, whereas 14A had poor biologically activity (Figure 6B).
Figure 6. Mitochondrial fusogenic activities of 5 sulfide analogs.
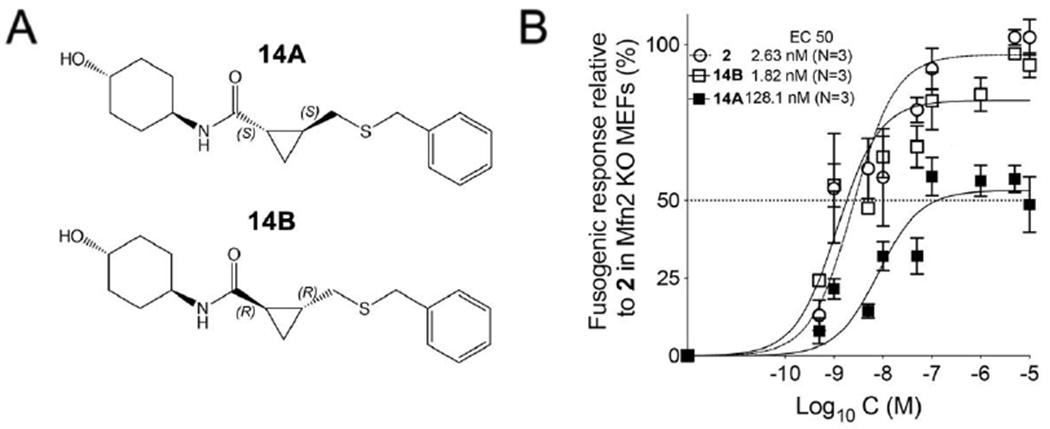
A. Structures of 5 sulfide analogs synthesized for X-ray diffraction studies. B. Dose-response curves showing 14B (slower eluting isoform: EC50 = 1.82 nM; 95% confidence limits, 1.04-2.99 nM) and 14A (faster eluting isoform: EC50 = 128.1 nM; 95% confidence limits, 141.94~ nM) increasing mitochondrial aspect ratio (length/width) in cultured fibroblasts lacking Mfn2. Data are means ± SEM from three independent experiments. 1 dose-response is shown for comparison (EC50 = 2.63 nM; 95% confidence limits, 1.45-4.64 nM).
The X-ray powder diffraction patterns and small rod-like crystal morphologies under polarized light microscopy of 14A and 14B were similar (Figure S3). Crystals used for single-crystal x-ray diffraction (SCXRD) for 14A were obtained by slow evaporation in acetonitrile (Figure S4A), and for 14B by slow evaporation in ethyl acetate (Figure S4B). As anticipated from studies of synthetic intermediates of 5 (see Figure 3), the resulting crystal structures showed that the two compounds are enantiomers, with 14B displaying {R,R} chirality.
Crystalline structures of 14A and 14B displayed no disorder. The asymmetric unit cells contained one molecule of the compound with no solvent molecules present (Figure 7A). Indeed, crystals with identical morphology readily formed out of multiple solvent systems (Table S1). The packing diagram (Figure 7B) and hydrogen bonding network between 14B molecules connecting in two directions (Figure 7C) further support absence of solvent molecules. Thus, 14B molecules form a pseudo-polymeric structure connected by the amide moieties near the center of the molecule. The carbonyl oxygen (O10) forms strong hydrogen bonding interactions with the hydrogens of adjacent molecules’ amide nitrogen (N8).
Figure 7. SCXRD structures of 14A and 14B.
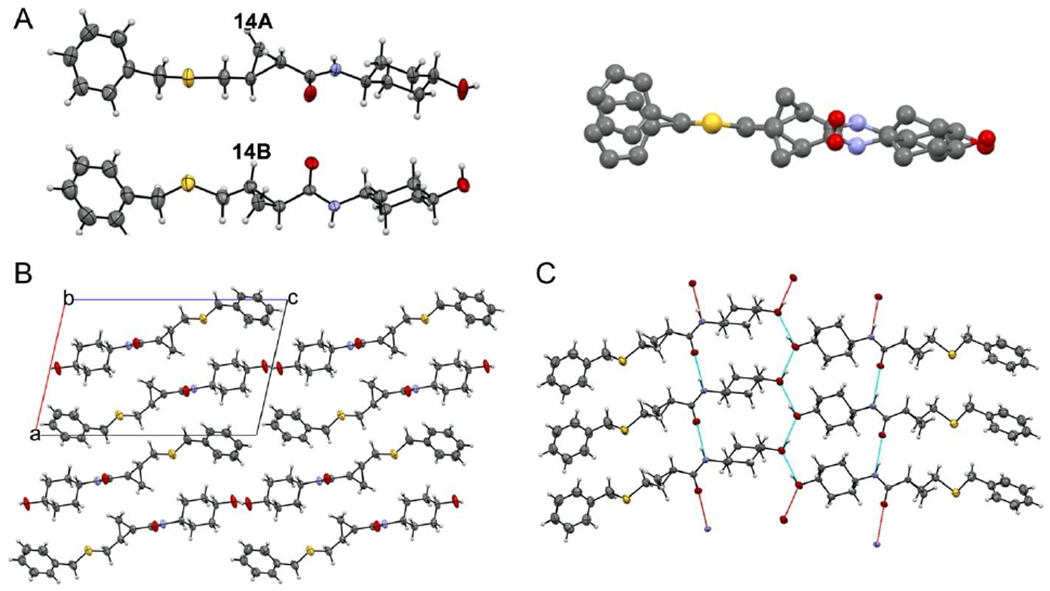
A. (left) ORTEP diagrams of 14A (inactive) and 14B (active). Thermal ellipsoids are shown at 50% confidence intervals. Hydrogen atoms are geometrically idealized. (right) Superposition of 14A and 14B showing mirror structures. Hydrogen atoms are omitted for clarity B. Packing diagram viewed along the b-axis. Unit cell axes are shown and labeled. C. Hydrogen-bonding interactions for 14B. Complete hydrogen bonds are shown in blue; red bonds indicate that only one participating atom is present in the pictured molecules.
The hydrogen bonding distance measured by the donor-acceptor distance is 2.887 Å. This contact deviates only slightly from the idealized hydrogen geometry as measured by linearity including the idealized H8A across the O10-N8 angle (171.31°). A second hydrogen bonding interaction dimerizes these pseudo-polymeric structures across the terminal alcohol (O1) with a donor-acceptor distance of 2.745 Å, reflecting an even stronger interaction. This is likely the consequence of every involved alcohol being both donor and acceptor, polarizing each oxygen involved. The resulting zig-zag formation and O-O-O angle of 130.72° is conducive to a trigonal planar type interaction.
Bonds within the 14B cyclopropyl moiety are slightly uneven: the longest interaction is the backbone C11-C13 bond (1.515 Å), while the adjoining bonds are asymmetrical, with a longer bond on the carbon alpha to the electropositive amide carbon (C11-C12, 1.513 Å) and a shorter bond on the carbon beta to the electron-donating sulfur (C13-C12, 1.484 Å). The molecule as a whole, if measured across the two hydrogen atoms idealized upon the two farthest atoms, is 18.415 Å in length. Simulation of the mimicked environment for 14A and 14B within hMFN2 is depicted in Figure 8. Positions of human MFN2 amino acids His380, Met376 and Val372 emulated by small molecule mitofusin activators according to the pharmacophore representation (15, 16) were modelled according to available protein structural information (Figure S5). Compared to 14B, 14A has increased overlap with Leu57. In order for the amide carbonyl to occupy a space similar to Met376, the trans (1r,4r)-4-hydroxycyclohexyl-1-amino group is oriented in such a way that it causes improbable overlap with Leu57. This steric crowding is likely to interfere in the ability of 14A to interact with the proposed adjacent peptide. The direction of rotation with respect to the phenyl moiety caused by the {R,R} cyclopropyl bridging group in 14B better accommodates and avoids this critical steric hindrance.
Figure 8. MFN2 peptide mimicry of 5 sulfide analog enantiomers.
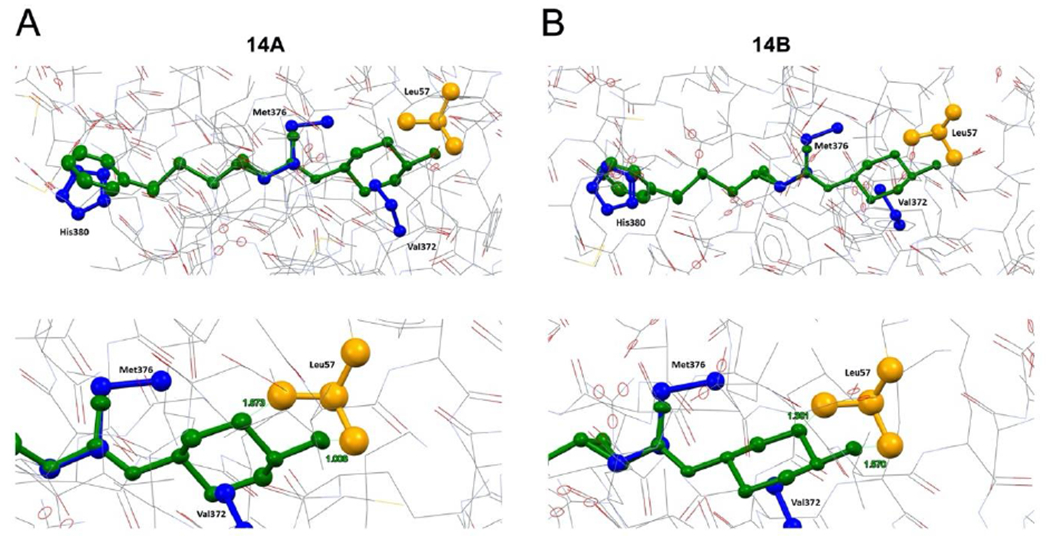
(top) Superimposition of compound. enantiomers (A, 14A, B, 14B; both are green) onto human MFN2 residues Val372, Met376 and His380 (blue) modeled as shown in Figure S5. MFN2 Leu57 is shown in orange. (bottom) Close-up of modeled interactions between compound enantiomers and MFN2 Leu57. Human Mfn2 protein structure was computationally modeled in closed configuration based on structural homology with human Mfn1 (PDB ID: 5GNS) and Arabidopsis thaliana dynamin-related protein (PDB ID: 3T34).
Improved nervous system accumulation of 5B.
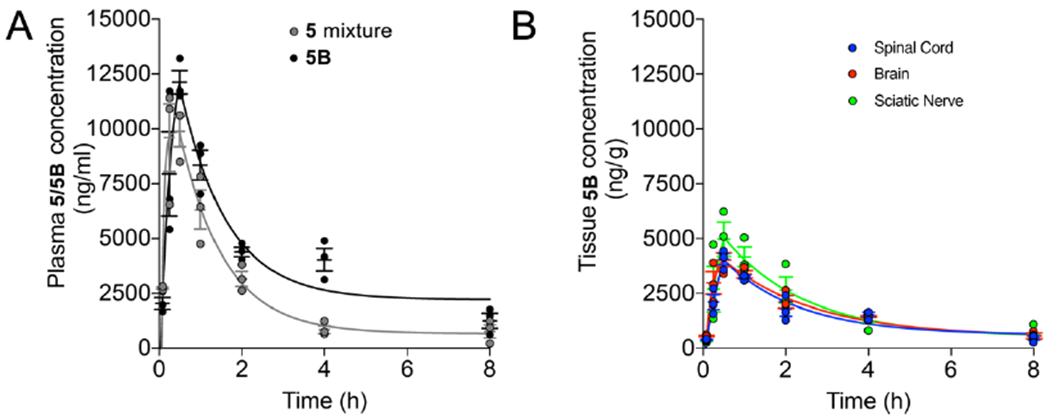
Because a potential clinical application of small molecule peptidomimetic mitofusin activators is neurological disorders (15, 17), 5B levels were measured in plasma and brain tissue at increasing times after a single 50 mg/kg oral dose. Plasma pharmacokinetics after oral 5B administration were similar to those of the 5 isomeric mixture given at an identical dose and route in the same vehicle (10% DMSO, 90% [30% HP-b-CD]): tmax for both was 0.5h, t1/2 was 2.83h and 3.02h respectively, and mean tissue residence times (MRT) were 3.96h and 3.58h respectively (Table 3; compare with 5 values in Table 2) (Figure 9A). The calculated brain/plasma partition coefficient (AUCbrain/AUCplasma; Table 3) was 39.4%, i.e. ~4 fold greater than previously reported for 2 (16).
Table 3.
Pharmacokinetic properties of 5B in plasma and nerve tissue a.
| Compound | Tissue | Dose Route | Actual Mean Dosage (mg/kg) | t1/2 (h) | tmax (h) | Cmax (ng/mL) | AUClast (h*ng/mL) | MRTinf (h) |
|---|---|---|---|---|---|---|---|---|
| 5B | ||||||||
| Plasma | PO | 50 | 2.83 | 0.50 | 12100 | 34000 | 3.96 | |
| Brain | PO | 50 | 3.13 | 0.50 | 4030 | 13400 | 4.30 | |
| Spinal Cord | PO | 50 | 2.71 | 0.50 | 4060 | 12700 | 3.93 | |
| Left Sciatic Nerve | PO | 50 | 3.21 | 0.50 | 4570 | 15300 | 4.62 | |
| Right Sciatic Nerve | PO | 50 | 2.61 | 0.50 | 5370 | 13800 | 3.35 |
Studies were performed in non-fasted mice, resulting in lower plasma 5B levels than reported after the same oral dose in fasted mice (see Table 4).
Figure 9. Plasma and tissue pharmacokinetics of 5B after oral administration to non-fasted mice.
A. Plasma pharmacokinetics of 5 isomeric mixture vs 5B in mice after a single oral dose of 50 mg/kg. B. Comparative neuronal tissue concentrations of 5B after a single oral dose of 50 mg/kg. Vertical axis values are the same in A and B. Data are means ± SEM, each dot represents one mouse. Calculated values are in the text.
The normal central nervous system (brain and spinal cord) is protected by a semipermeable blood brain barrier (BBB) that impedes non-selective solute transport into the cerebrospinal fluid. The blood nerve barrier (BNB) serves an analogous function for peripheral nerves (22), but is uniquely permeable at dorsal root ganglia (DRG) that contain peripheral sensory nerve bodies (23). This raised the possibility that small hydrophobic molecules like 5B can gain access to peripheral nerves through DRGs and be concentrated therein, with therapeutic implications for treating neuropathies. Accordingly, 5B levels were compared in brain and spinal cord (parts of the central nervous system protected by the BBB) and both sciatic nerves (large peripheral nerves innervating the lower limbs that carry sensory neurons originating in DRGs). As reported in Table 3 and depicted in Figure 9B, 5B Cmax, AUC, t1/2, and mean residence time (MRT) were similar in all three neurological tissues. Indeed, the variance between the left and right sciatic nerves and the central and peripheral nervous systems was similar. These results do not indicate a meaningful effect of “leaky” DRG BNBs on peripheral nerve levels of this mitofusin activator.
The above results suggested that 5B might exhibit favorable nervous system pharmacodynamics. Previous studies of mitofusin activators measured disease-relevant pharmacodynamic effects as reversal of mitochondrial dysmotility in mouse disease model sciatic nerve axons (15, 17) (Figure 10A). Peak mitochondrial motility after “burst” mitofusin activation with 2 reportedly occurred 4 hours after administration and was completely extinguished after 24 hours. By contrast, peak mitochondrial motility after oral administration of 5B occurred at 12 hours, and benefits were maintained for greater than 24 hours (Figure 10A–10C). Perhaps as a consequence of these favorable pharmacokinetic and pharmacodynamic properties, 5 partially ameliorated neuromuscular dysfunction in murine amyotrophic lateral sclerosis caused by a mutation of superoxide dismutase 1 (SOD1; 24), whereas 2 showed no significant benefit (Figure 10D).
Figure 10.
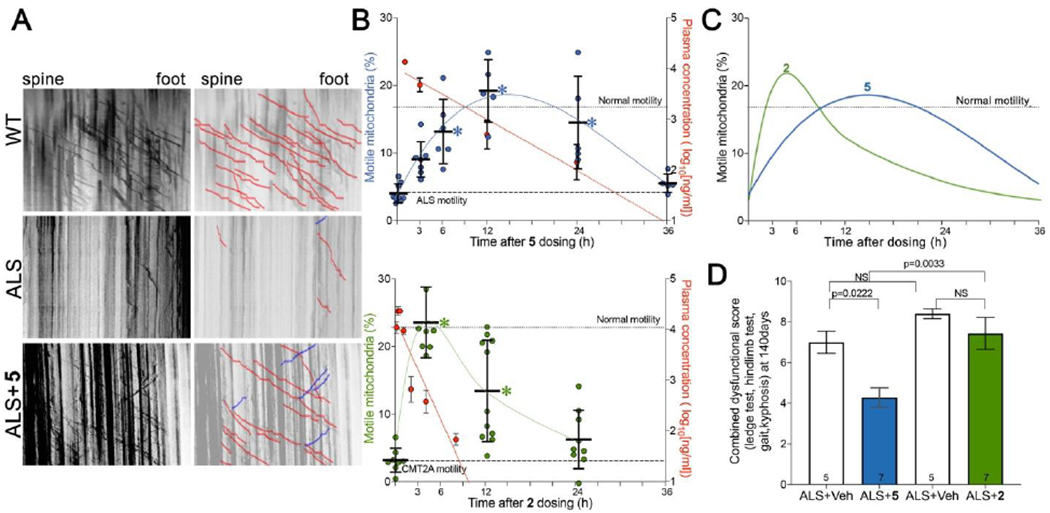
Pharmacodynamic and therapeutic effects of 5 vs 2 in murine ALS. A. Representative kymographs for wild-type (WT) and ALS SOD1G93A mice (ALS) 12 hours after oral administration of 5 or vehicle. B. Time-dependent pharmacokinetics/pharmacodynamics of 5 and 2 after single oral doses (60 mg/kg). (top) Blue values and left vertical axis show mitochondrial motility after 5 in ALS mouse sciatic nerve axons. (bottom) green values and left vertical axis show mitochondrial motility in CMT2A mouse sciatic nerve axons. Each point represents a single neuronal axon, from two or three mice per time point. Red values and right vertical axes of top graph show corresponding plasma 5 levels (n=5 per time point; means ± SD); plasma levels of 2 are from (17). Dotted line designated “Normal motility” is mean value for WT in panel A; dashed line designated “ALS motility” is mean value for untreated ALS in panel A. C. Comparative pharmacodynamics of 5 (blue) and 2 (green). D. Effects of 5 (blue) and 2 (green) on neuromuscular dysfunction score (ledge test, hindlimb test, gait, kyphosis; 25) in a proof-of-concept study of ALS mice. P values by ANOVA.
Stimulation of mitochondrial motility in murine ALS by 5 was delayed in onset, and of longer than expected duration, based on its plasma (and brain, vide infra) levels (Figure 10B). This finding is consistent with the idea that facilitating mitochondrial motility is an indirect effect of mitofusins, mediated via their interactions with Miro proteins that couple mitochondria to the Milton/Trak-dynein/kinesin transport apparatus (26, 27). Mitofusin regulation of mitochondrial fusion, motility and mitophagy is mediated by mitofusin interactions with different effector proteins (28). It seems likely that mitofusin recruitment of Miro proteins to function-critical subcellular domains or an essential multi-molecular complex is the mechanism by which it facilitates mitochondrial coupling to the subcellular transport apparatus. Both Miro recruitment upon mitofusin activation, and its restoration to baseline after mitofusin inactivation, would take time. This can explain the observed temporal discontinuity between mitofusin activator pharmacokinetics and pharmacodynamics.
Together, the above results support superiority of 5 over 2 for extended pharmacodynamic effects and potential therapeutic value in experimental ALS. To better understand the underlying mechanisms we performed a side-by-side comparison of 5B and 2 plasma and brain pharmacokinetics. For these comparative studies the two compounds were administered at the same dose (50 mg/kg) and route (oral gavage), and using the same vehicle (5 mg/ml in 30% SBE-bCD). As shown in Figure 11 and Table 4, greater brain bioavailability (total and free AUCs) and longer plasma and brain t1/2s and MRTs were exhibited by 5B.
Figure 11. Comparative pharmacokinetic properties of 5B and 2 in fasted mice.
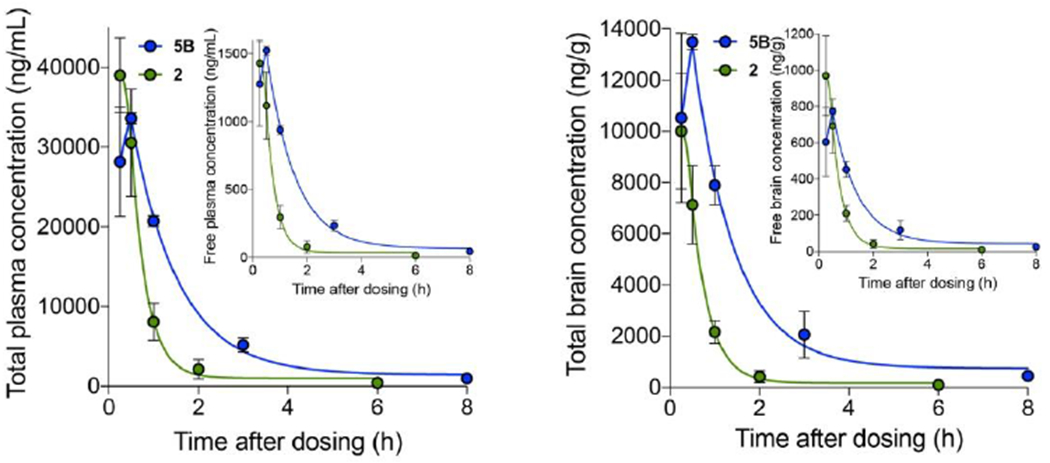
Left panel, total compound levels in plasma for 2 (green) and 5B (blue); inset shows free concentrations. Right panel, total brain compound levels; inset shows free concentrations. Means±SEM of three mice per time point. Calculated values are in Table 4.
Table 4.
Comparative plasma and brain pharmacokinetic properties of parent 2 and lead compound 5B in fasting mice a.
| Cpd 2 (50 mg/kg PO) | Cpd 5B (50 mg/kg PO) | |||
|---|---|---|---|---|
| total Plasma | free Plasma | total Plasma | free Plasma | |
| Cmax (ng/mL) | 39011 | 1428 | 33619 | 1523 |
| T1/2 (h) | 1.280 | 1.67 | ||
| AUC0-last (ng.h/mL) | 30612 | 1121 | 59567 | 2697 |
| MRT0-last (h) | 1.01 | 1.92 | ||
| total Plasma | free Plasma | total Plasma | free Plasma | |
| Cmax (ng/g) | 10001 | 970 | 13480 | 774 |
| T1/2 (h) | 0.999 | 1.77 | ||
| AUC0-last (ng.h/g) | 7392 | 717 | 23465 | 1347 |
| MRT0-last (h) | 0.959 | 1.98 | ||
Free compound concentrations were calculated from protein binding assays: 2 – mouse plasma 96.7%, mouse brain 90.3%; 5B – mouse plasma 95.5%, mouse brain 94.3%.
Chemistry.
Synthesis of 1 and 2 was previously described (15, 16). Synthesis of novel compounds 3-14 was accomplished as outlined in Schemes 1–5. 3 and 4 were formed by coupling of commercially available trans-4-aminocyclohexan-1-ol with 5-phenylpentanoic acid or 4-phenylpentanoic acid, as shown in Scheme 1.
Scheme 1.
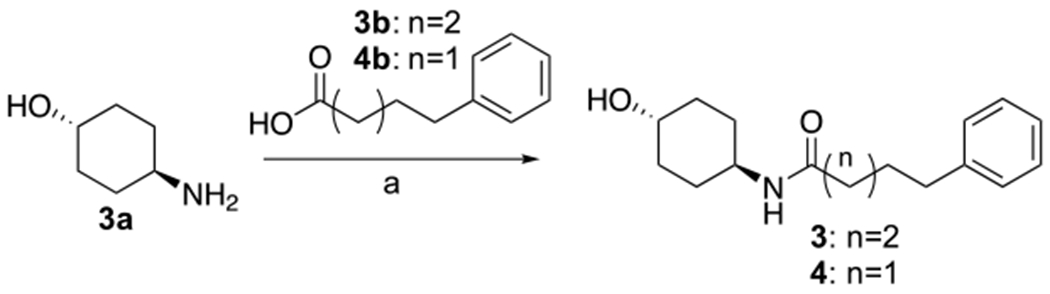
Synthesis of 3, 4a
aReagents and conditions: HATU, Et3N, THF, 25°C, 2h.
Scheme 5.
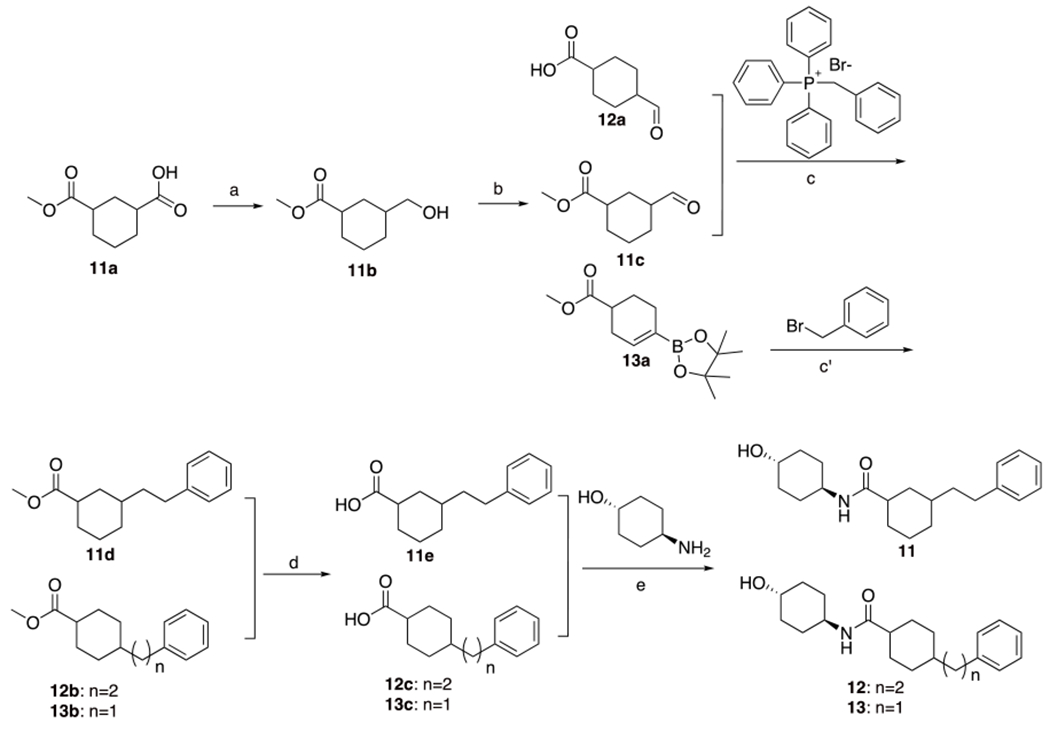
Synthesis of 11, 12 and 13a
aReagents and conditions: (a) BH3-SMe2, THF, −78-25°C, 12.5h. (b) pyridine sulfur trioxide, Et3N, DMSO, 25°C, 12h. (c) i. THF, t-BuOK, N2, −20°C, 2h, ii. Pd/C, H2, MeOH, 25°C, 2h. (c’) i. Pd(dppf)Cl2, dioxane, H2O, K2CO3, N2, 25-100°C, 2h. ii. Pd/C, H2, MeOH, 25°C, 2h. (d) LiOH•H2O, MeOH, THF, H2O, 25-70°C, 2h. (e) HATU, Et3N, THF, 25°C, 12h.
Scheme 2 includes compounds 5, 6 and 14. Compounds 5, and 14 were synthesized by similar procedures, but with the replacement of intermediate 5b with the sulfide 14b. For compounds 5 and 14, the last two steps d and e, to the acid hydrolysis and amide condensation were accomplished by normal conditions from compounds 5d, and 14d. These were obtained from Corey-Chaykovsky cyclopropanation reaction from compounds 5c and 14c. In turn, they were obtained from Wittig coupling with compounds 5b and 14b. Compound 5b was formed by oxidation of 5a. The sequence for compound 6 was similar but had some distinct differences. The aldehyde 6a needed for coupling with the Wittig reagent was commercially available. Following similar cyclopropanation of 6b the ester 6c was reduced to afford alcohol 6d, which was elongated via sequential formation of the chloride 6e, followed by cyanation to yield 6f. Hydrolysis of nitrile 6f yielded the acid 6g needed for the final amide coupling step, affording 6.
7 and 8 were synthesized as shown in Scheme 3. Preparation of 7 commenced with commercially available bromide 7a, which was converted to the Wittig reagent 7b via reaction with Triphenylphosphine. Reaction of 7b via treatment with sodium t-butoxide afforded the intermediate ylide which was coupled with Ethyl 3-oxocyclobutane-1-carboxylate to afford 7c. This intermediate was converted to the final drug candidate 7, by hydrolysis of the ester to an intermediate acid followed b amide formation under standard conditions. Preparation of 8 began by formation of 8b via zinc-copper mediated coupling of trichloracetylchloride with 2-Propen-1-ylbenzene 8a. Removal of the chlorines by treatment with zinc in acetate acid followed by Wittig coupling afforded intermediate 8c which was converted to 8 similarly to 7c to 7.
Scheme 3.
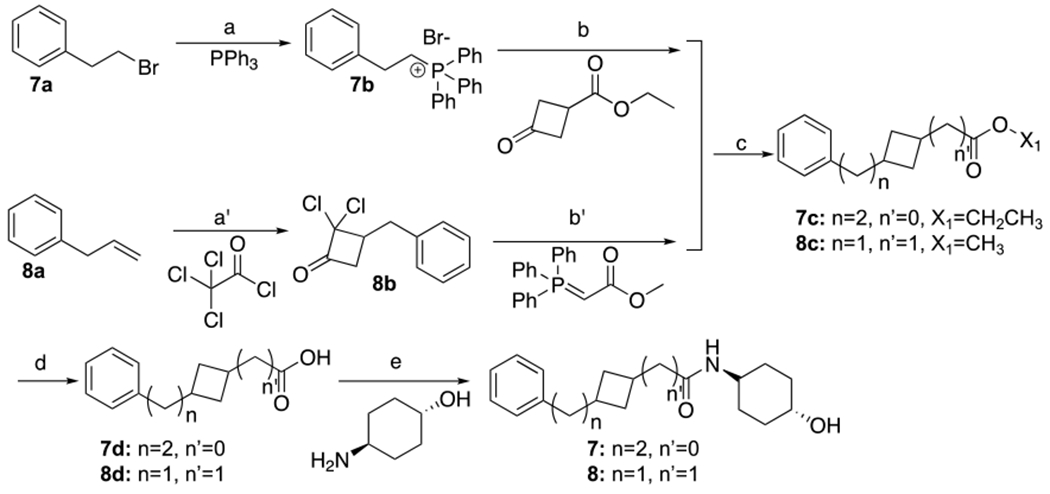
Synthesis of 7 and 8a
aReagents and conditions: (a) 100°C, 16h. (b) sodium 2-methybutan-2-olate, toluene, 20-70°C, 4.5h. (a’) zinc-copper couple, Et2O, 20-35°C, 18h. (b’) acetic acid, zinc, 25-80°C, 3h, toluene, 110°C, 2h. (c) H2, Pd/C. 7: EtOH, 20°C, 16h. 8: MeOH, 25°C 2h. (d) LiOH•H2O, MeOH/THF/H2O. 7: 20°C, 2h. 8: 25°C, 16h. (e) HOBt, EDCI, DIPEA, DMF, 16h. 7: 20°C, 8: 25°C.
9 and 10 were synthesized as shown in Scheme 4. Compound 9 was synthesized starting with commercially available diester 9a via alkaline hydrolysis to obtain acid 9b, which was reduced by reaction with borane to afford alcohol 9c. That alcohol was oxidized with Dess-Martin periodinane to yield aldehyde 9d which was coupled with Wittig reagent 9e to yield an intermediate olefin that was reduced with Pd/C under H2 to obtain ester 9f. This was hydrolyzed to afford acid 9g that upon coupling with the 4-amino-cyclohexanol afforded 9. Compound 10 was synthesized starting with commercially available 10a through the similar procedures with 9d-9 to obtain 10.
Scheme 4.
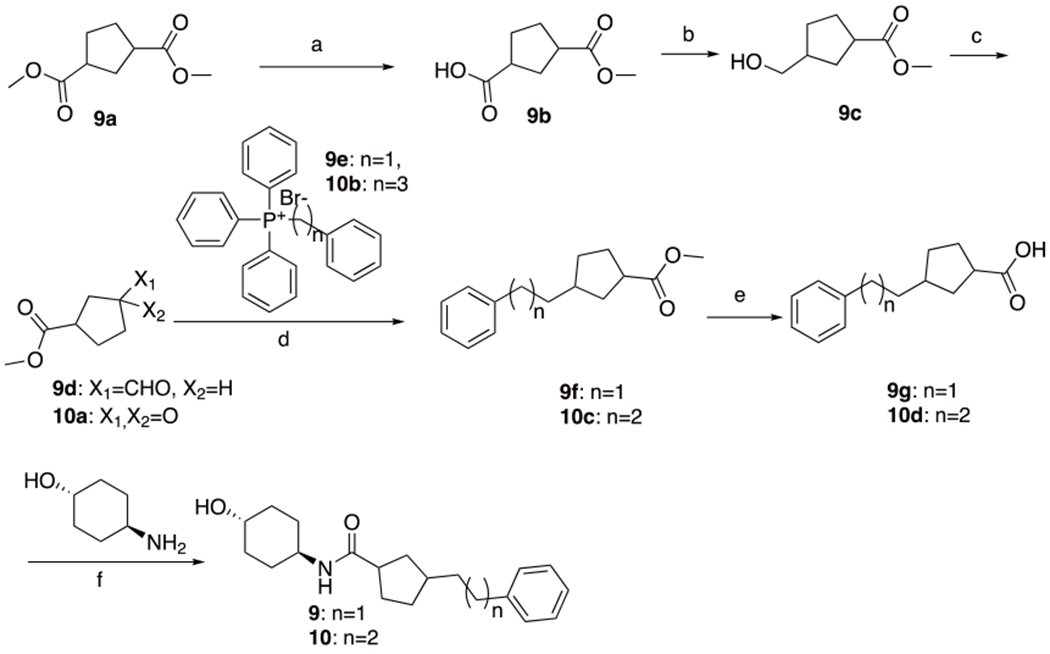
Synthesis of 9 and 10a
aReagents and conditions: (a) NaOH, MeOH, 25°C, 12h. (b) BH3-Me2S, THF, −78-25°C, 12.5h. (c) Dess-Martin, DCM, 25°C, 2h. (d) i. t-BuOK, 9: −20°C, 2h. 10: toluene, 70°C, 17h. ii. Pd/C, H2, MeOH, 25°C, 9: 2h, 10: 4h. (e) LiOH•H2O, MeOH, THF, H2O, 9: 25-70°C, 2h, 10: 70°C, 3h. (f) HATU, Et3N, THF, 25°C, 9: 12h, 10: 16h.
11-13 were synthesized as shown in Scheme 5. Preparation of 11 began with commercially available carboxylic acid 11a which was reacted Borane-dimethylsulfide to obtain 11b. Oxidation of 11b to aldehyde 11c was effected with pyridine-sulfur trioxide. Which after coupling with benzyl(triphenyl)phosphonium bromide afforded olefin 11d. Hydrogenation of this over Pd/C followed by hydrolysis yielded the penultimate acid 11e. Coupling of which with 4-amino-cyclohexanol afforded the target 11. Following a similar sequence 12 was synthesized by starting with commercially available 4-formylcyclohexane-1-carboxylic acid (12a). Compound (13) was made starting with Suzuki coupling of methyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (13a) with benzylbromide followed by hydrogenation to afford (13b). This intermediate was converted to (13) similarly to (11d) and (12b).
Conclusions
In summary, rational re-design of 6-phenylhexanamide mitofusin activators incorporating cycloalkyl-containing linkers generated novel fusogenic compounds having improved pharmaceutical properties. Preclinical lead compound 5, containing a cyclopropyl group in the linker, exhibited potent, stereo-selective mitofusin activation. Single crystal X-ray diffraction studies conducted with sulfide analogs of 5A and 5B showed that MFN2 interactions and biological activity reside exclusively in the trans-(R,R) enantiomer. The structural characteristics of 5B are consistent with mitofusin activation through physico-chemical mimicry of human MFN2 Val372, Met 376 and His380, i.e. the function-critical amino acids in the progenitor mitofusin agonist peptide (14) and provide further support for pharmacophore based rational design of small molecule mitofusin activators. Compared to parent compound 2, 5B exhibited sustained brain levels, markedly prolonged pharmacodynamic effects, and disease modification in a proof-of-concept study of murine amyotrophic lateral sclerosis. Thus, 5B is a strong candidate for preclinical evaluation in diseases wherein sustained activation of mitofusin-mediated mitochondrial fusion and transport might counteract neurological degeneration.
Experimental Section
General procedures and Instrumentation
Compounds are at least 95% pure by HPLC (Column: Kinetex C18 LC, 4.6X50 mm, 5um; Mobile phase A: 0.0375% TFA in water (v/v), B: 0.01875% TFA in Acetonitrile (v/v)) run at 50°C with absorbance at 200nM peaks.
LC-MS/MS (ESI) was performed using multiple systems: 1) SHIMADZU LC-MS-2020 with LabSolution V5.72 analysis software, HPLC used a Chromalith@Flash RP-18E 25*2.0MM column ran at 50°C with PDA (220&254nm) detector, acquired data in scan MS Mode (positive mode) with m/z=100-1000 scan range, drying gas (N2) flow: 15 L/min, DL voltage: 120V and Quarry DC voltage: 20V. 2) Agilent 1200/G6110A instrument with AgilentChemStation Rev. B. 04.03 software. HPLC used a Xbridge C18 2.1*50mm, 5um column ran at 40°C with DAD (220nm)/ELSD detector, acquired data in scan MS Mode (positive mode) with m/z=100-1000 scan range, drying gas (N2) flow: 10 L/min, 350°C, nebulizer pressure: 35psi, capillary voltage: 2500V. NMR spectrometry was carried out on Brucker AVANCE NEO 400MHz with 5mm PABBO BB/19F-1H/D Z-GRD probe.
Synthesis and Characterization of Compounds 1-14
Compounds were synthesized and analyzed at WuXi Apptec Co. Ltd. NMR, analytical HPLC and LC-MS were reported previously. All starting reagents not reported are known compounds purchased by WuXi Apptec Co. Ltd from third party suppliers. The synthesis procedure for compounds 1-2 was reported previously (16).
N-((1r,4r)-4-hydroxycyclohexyl)-5-phenylpentanamide (3).
To a mixture of trans 4-aminocyclohexan-1-ol (3a, 500 mg, 4.34 mmol, 1.00 eq) and 5-phenylpentanoic acid (3b, 928 mg, 5.21 mmol, 1.20 eq) in THF (10.0 mL) was added HATU (2.48 g, 6.51 mmol, 1.50 eq), Et3N (1.32 g, 13.0 mmol, 1.81 mL, 3.00 eq) in one portion at 25 °C. The mixture was stirred at 25 °C for 2 hrs. TLC showed the reaction was completed. TLC indicated (Petroleum: Ethyl acetate=3:1) some compound 2a (Rf=0.14) was remained, and one major new spot with larger (Rf=0.29) polarity was detected. The reaction mixture was diluted with H2O 50.0 mL and extracted with Ethyl acetate 60.0 mL (20.0 mL * 3). The combined organic layers were washed with brine 60.0 mL (20.0 mL * 3), dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The crude product was purified by reversed-phase HPLC (0.1% NH3•H2O). 3 (106.02 mg, 0.38 mmol, 8.87% yield) was obtained as a white solid. LC-MS: RT = 0.851 mins, m/z = 276.4 (M+H)+. HPLC: RT = 1.75 mins, 98.7% purity, under 220 nm. SFC: RT = 1.14 mins, ee% = 100%, under 220 nm. 1H NMR: (400 MHz, MeOD) δ 7.25 - 7.22 (m, 2H), 7.16-7.13 (m, 3H), 3.62 – 3.59 (m, 1H), 3.51 - 3.49 (m, 1H), 2.62-2.59 (m, 2H), 2.18 - 2.14 (m, 2H), 1.92 - 1.88 (m, 4H), 1.63 - 1.59 (m, 4H), 1.32-1.27 (m, 4H).
N-((1r,4r)-4-hydroxycyclohexyl)-4-phenylbutanamide (4).
A mixture of trans 4-aminocyclohexan-1-ol (3a, 300 mg, 2.60 mmol, 1.00 eq), 4-phenylbutanoic acid (4b, 513 mg, 3.13 mmol, 1.20 eq) and Et3N (790 mg, 7.81 mmol, 1.09 mL, 3.00 eq), HATU (1.49 g, 3.91 mmol, 1.50 eq) in THF (10.0 mL) was stirred at 25 °C for 2 hrs. TLC indicated (Petroleum: Ethyl acetate=3:1) some Compound 2 (Rf=0.12) was remained, and one major new spot with larger (Rf=0.26) polarity was detected. The reaction mixture was diluted with H2O 50.0 mL and extracted with Ethyl acetate 60.0 mL (20.0 mL * 3). The combined organic layers were washed with brine 60.0 mL (20.0 mL * 3), dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The crude product was purified by reversed-phase HPLC (0.1% NH3•H2O). 4 (108.64 mg, 0.42 mmol, 15.9% yield) was obtained as a white solid. LC-MS: RT = 0.820 mins, m/z = 262.3 (M+H)+. HPLC: RT = 1.604 mins, 97.2% purity, under 220 nm. SFC: RT = 1.05 mins, ee% = 100%, under 220 nm. 1H NMR (400 MHz, MeOD) δ 7.27 - 7.23 (m, 2H), 7.18-7.15 (m, 3H), 3.61 - 3.60 (m, 1H), 3.53 - 3.50 (m, 1H), 2.62-2.58 (m, 2H), 2.18 - 2.14 (m, 2H), 1.92 - 1.87 (m, 6H), 1.36 - 1.24 (m, 4H).
N-(4-hydroxycyclohexyl)-2-(3-phenylpropyl)cyclopropane-1-carboxamide (5).
To a solution of oxalyl chloride (4.65 g, 36.6 mmol, 3.20 mL, 1.10 eq) in DCM (75.0 mL) was cooled to – 55 °C under N2 atmosphere was added drop-wise a added the solution of DMSO (5.72 g, 73.2 mmol, 5.72 mL, 2.20 eq) in DCM (30.0 mL) was, after stirring for 5 min, drop-wise added 4-phenylbutan-1-ol (5a, 5.00 g, 33.2 mmol, 5.08 mL, 1.00 eq) in DCM (15.0 mL) was added drop-wise, after stirring for 15 min, TEA (16.8 g, 166 mmol, 23.1 mL, 5.00 eq) was added, then it was warmed to 25 °C. At that point 100 ml 1 N HCl was added and the suspension was extracted with DCM 200 mL (100 mL * 2). The combined organic layers were washed with water 50 mL, dried over Na2SO4, filtered and concentrated to give 4-phenylbutanal (5b, 5.00 g). To a solution of 5b (5.00 g, 33.7 mmol, 9.80 mL, 1.00 eq) in THF (50.0 mL) was added tert-butyl 2-(triphenyl-λ5-phosphaneylidene)acetate (16.5 g, 43.8 mmol, 1.30 eq), stirred at 20 °C for 12 hrs to give tert-butyl (E)-6-phenylhex-2-enoate (5c, 6.00 g, 24.3 mmol, 72.1% yield). To a solution of NaH (1.17 g, 29.2 mmol, 60.0% purity, 1.20 eq) in DMSO (30.0 mL) was added dimethylmethanesulfinic iodide (6.43 g, 29.2 mmol, 1.20 eq) and the mixture was stirred at 20 °C for 0.5 hr, then added 5c (6.00 g, 24.3 mmol, 1.00 eq) in DMSO (3.00 mL) and stirred at 20 °C for 1 hr to give tert-butyl 2-(3-phenylpropyl)cyclopropane-1-carboxylate (5d, 2.10 g, 8.07 mmol, 33.1% yield). Added TFA (7.70 g, 67.5 mmol, 5.00 mL, 17.5 eq) to a solution of 5d (1.00 g, 3.84 mmol, 1.00 eq) in DCM (5.00 mL), stirred at 25 °C for 15 hrs to give 2-(3-phenylpropyl)cyclopropane-1-carboxylic acid (3e, 800 mg). Added EDCI (1.00 g, 5.22 mmol, 1.50 eq), HOBt (564 mg, 4.18 mmol, 1.20 eq) and DIPEA (1.35 g, 10.4 mmol, 1.82 mL, 3.00 eq), 4-aminocyclohexan-1-ol (580 mg, 3.83 mmol, 1.10 eq, HCl) to a solution of 5e (800 mg, 3.48 mmol, 1.00 eq) in DMF (8.00 mL) and stirred at 25 °C for 16 hrs. The residue was purified by prep-HPLC (column: Waters Xbridge C18 150*50mm* 10um; mobile phase: [water(10mM NH4HCO3)-ACN];B%: 28%−58%, 11.5min) to give 5 (110 mg, 361 umol, 10.3% yield) as a white solid. LC-MS: Rt = 0.904 min, m/z = 302.1 (M+H)+. HPLC: Rt = 2.898 min, purity: 98.6%, under 220 nm. 13C NMR: (400 MHz MeOD) δ 173.97, 142.27, 127.96, 127.89, 125.31, 69.07, 35.14, 33.45, 32.23, 30.87, 30.24, 21.27, 20.55, 13.04. 1H NMR: (400 MHz MeOD) δ 7.27 - 7.24 (m, 2H), 7.18- 7.15 (m, 3H), 3.64 - 3.61 (m, 1H), 3.55 - 3.50 (m, 1H), 2.64 (t, J = 8 Hz, 2H), 1.97 - 1.89 (m, 4H), 1.75- 1.73 (m, 2H), 1.36 - 1.04 (m, 8H), 1.29 - 1.27 (m, 1H), 0.59 - 0.57 (m, 1H).
Separation method of 5A and 5B isoforms from 5 mixture isoforms.
Thar 200 preparative SFC (SFC-7) with Column (ChiralPak IG, 30050mm I.D., 10um), Mobile phase: A for CO2 and B for Methanol(0.1%NH3H20); Gradient: B 35%; Flow rate: 200 mL/min; Back pressure: 100 bar Column temperature: 38°C; Wavelength: 220nm; Cycle time: ~4 min; Sample preparation: Compound was dissolved in ~200 ml methanol; Injection: 10 ml per injection. After separation, the fractions were dried off via rotary evaporator at bath temperature 40°C to get the desired isomers. 5A was the faster eluting isomer by SFC on IG column, and 5B was the slower eluting isomer. Waters UPC2 analytical SFC (SFC-H) with column (ChiralPak IG, 150×4.6mm I.D., 3µm); Mobile phase: A for CO2 and B for Methanol (0.05%DEA); Gradient: B 40%; Flow rate: 2.5 mL/min; Back pressure: 100 bar; Column temperature: 35°C; Wavelength: 220nm. LCMS: 5A: Rt=2.445 min, m/c=302.2 (M+H)+, 5B: Rt=2.445 min, m/c=302.2 (M+H)+. SFC: 5A: Rt=0.852 min, 5B: Rt=1.179 min. 13C NMR: 5A: (400 MHz MeOD) δ 173.97, 142.27, 127.96, 127.89, 125.31, 69.07, 35.14, 33.46, 32.24, 30.86, 30.24, 21.29, 20.55, 13.05. 5B: (400 MHz MeOD) δ 173.97, 142.28, 127.96, 127.90, 125.31, 69.07, 35.14, 33.45, 32.24, 30.86, 30.24, 21.29, 20.55, 13.05.
N-(4-hydroxycyclohexyl)-2-(2-phenethylcyclopropyl)acetamide (6).
Tert-butyl 2-(triphenyl-λ5-phosphaneylidene)acetate (25.2 g, 67.0 mmol, 1.50 eq) to a solution of 3-phenylpropanal (6a, 6.00 g, 44.7 mmol, 5.88 mL, 1.00 eq) in THF (100 mL) and stirred at 20 °C for 1 hr to give tert-butyl (E)-5-phenylpent-2-enoate (6b, 10.0 g, 42.1 mmol, 94.3% yield).
Dimethylmethanesulfinic iodide (10.0 g, 45.5 mmol, 1.20 eq) was added to a solution of NaH (1.82 g, 45.5 mmol, 60.0% purity, 1.20 eq) in DMSO (45.0 mL) and the mixture was stirred at 20 °C for 0.5 hr, then 6b (9.00 g, 37.9 mmol, 1.00 eq) in DMSO (5.00 mL) was added and the reaction mixture stirred at 20 °C for 1 hr to give tert-butyl 2-phenethylcyclopropane-1-carboxylate (6c, 6.00 g, 24.3 mmol, 64.1% yield). LiAlH4 (924 mg, 24.3 mmol, 2.00 eq) at 0 °C was added to a solution of 6c (3.00 g, 12.1 mmol, 1.00 eq) in THF (45.0 mL), stirred at 25 °C for 3 hrs, then the reaction mixture was cooled to 0 °C and 1.00 mL water was drop-wise added, then added 1.00 mL 15.0 % NaOH, next 3.00 ml water and warmed to 25 °C, stirred for 15 mins, added 20.0 g MgSO4 and stirred for 15 mins to give (2-phenethylcyclopropyl)methanol (6d, 2.00 g, 10.1 mmol, 82.9% yield). A solution of (2-phenethylcyclopropyl)methanol (6d, 2.00 g, 10.1 mmol, 1.00 eq) and TEA (2.04 g, 20.2 mmol, 2.81 mL, 2.00 eq) in CHCl3 (20.0 mL) was cooled to 0 °C and drop-wise added SOCl2 (2.40 g, 20.2 mmol, 1.47 mL, 2.00 eq) was added dropwise. The reaction mxture was stirred at 70 °C for 1 hr. The organic phase was separated, the aqueous phase extracted with DCM 40.0 mL (20 mL * 2). The combined organic layers were washed with brine 20.0 mL, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography (SiO2, Petroleum ether / Ethyl acetate = 1 / 0, Plate 2, Petroleum ether / Ethyl acetate = 1 / 0,Rf = 0.50) to give (2-(2-(chloromethyl)cyclopropyl)ethyl)benzene (6e, 1.80 g, 8.69 mmol, 86.0% yield). Stirred a solution of 2-(2-(chloromethyl)cyclopropyl)ethyl)benzene (6e, 1.80 g, 8.69 mmol, 1.00 eq) in THF (9.00 mL) and N(nBu)4CN (2.80 g, 10.4 mmol, 1.20 eq) at 70 °C for 12 hrs to give 2-(2-phenethylcyclopropyl)acetonitrile (6f, 1.40 g, 7.56 mmol, 86.9% yield), added EtOH (7.00 mL) and H2O (7.00 mL), KOH (1.70 g, 30.2 mmol, 4.00 eq), stirred at 100 °C for 16 hrs to give 1-(2-phenethylcyclopropyl)propan-2-one (6g, 1.50 g, 6.95 mmol, 92.0% yield). To a solution of 1-(2-phenethylcyclopropyl)propan-2-one (6g, 1.00 g, 4.64 mmol, 1.00 eq) in DMF (10.0 mL) was added EDCI (1.33 g, 6.95 mmol, 1.50 eq), HOBt (751 mg, 5.56 mmol, 1.20 eq), DIPEA (1.80 g, 13.9 mmol, 2.42 mL, 3.00 eq) and 4-aminocyclohexan-1-ol (773 mg, 5.10 mmol, 1.10 eq, HCl), the mixture was stirred at 25 °C for 16 hrs, LC-MS showed 6g was consumed completely and desired MS (Rt = 0.882 min) was detected, the residue was purified by prep-HPLC (column: Waters Xbridge C18 150*50mm* 10um; mobile phase: [water(10mM NH4HCO3)-ACN];B%: 28%-58%, 11.5min) to give 6 (99.35 mg, 329 umol, 7.11% yield, 100% purity) as a white solid. LC-MS: Rt = 0.895 min, m/z = 302.1 (M+H)+. HPLC: Rt = 2.803 min, purity: 100 %, under 220 nm. 13C NMR: (400 MHz MeOD) δ 173.58, 142.27, 128.05, 127.84, 125.25, 69.03, 40.16, 35.84, 35.34, 33.40, 30.14, 17.77, 15.15, 10.80. 1H NMR: (400 MHz MeOD) δ 7.27 - 7.23 (m, 2H), 7.19- 7.14 (m, 3H), 3.65 - 3.60 (m, 1H), 3.55 - 3.50 (m, 1H), 2.69 (t, J = 7.6 Hz, 2H), 2.09 - 2.02 (m, 2H), 1.97 - 1.89 (m, 4H), 1.75- 1.73 (m, 1H), 1.51 - 1.49 (m, 1H), 1.38 - 1.30 (m, 4H), 0.82 - 0.80 (m, 1H), 0.64 - 0.53 (m, 1H), 0.37 - 0.32 (m, 2H).
N-(4-hydroxycyclohexyl)-3-phenethylcyclobutane-1-carboxamide (7).
A mixture of (2-bromoethyl)benzene (7a, 10.0 g, 54.0 mmol, 7.30 mL, 1.00 eq) and triphenylphosphine (14.2 g, 54.0 mmol, 1.00 eq) was stirred at 100 °C for 16 hrs, then cooled to 20 °C to give compound 2-phenethyl-triphenylphosphonium bromide (7b, 20.0 g, 44.4 mmol, 82.2% yield, 99.4% purity), to this was then added toluene (47.0 mL), sodium 2-methylbutan-2-olate (1.16 g, 10.5 mmol, 1.50 eq), and the mixture stirred at 20 °C for 0.5 hrs under N2, at which point a solution of ethyl 3-oxocyclobutane-1-carboxylate (1.00 g, 7.03 mmol, 1.00 eq) in toluene (10.0 mL) was added dropwise to the mixture and the mixture was stirred at 70 °C for 4 hrs under N2 then concentrated under reduced pressure to give a residue that was purified by Reverse Phase HPLC to give ethyl 3-(2phenylethylidene)cyclobutane-1-carboxylate (0.30 g, 1.14 mmol, 16.2% yield, 87.7% purity), then added EtOH (24.0 mL), Pd/C (0.24 g, 10.0% purity) under N2, the suspension was degassed and purged with H2 for 3 times, the mixture was stirred at 20 °C for 16 hrs under H2 (15 psi) then filtered through kieselguhr and the filter residue was washed with ethyl acetate (60.0 mL), the layer was concentrated under reduced pressure to give ethyl 3-phenethylcyclobutane-1-carboxylate (7c, 1.10 g, 2.46 mmol, 47.1% yield, 51.9% purity). To a solution of ethyl 7c (1.10 g, 2.46 mmol, 1.00 eq) in MeOH (5.00 mL), THF (10.0 mL) and H2O (5.00 mL) was added LiOH•H2O (412 mg, 9.83 mmol, 4.00 eq), stirred at 20 °C for 2 hrs, the mixture was concentrated under reduced pressure to give a residue, the residue was poured into H2O (30.0 mL) and extracted with ethyl acetate (30.0mL * 2), the water phase was adjust pH to 3 ~ 4 with 1 N HCl (10.0 mL) and extracted with ethyl acetate (50.0 mL * 3), the combined layers were dried over Ns2SO4, filtered and concentrated under reduced pressure to give 3-phenethylcyclobutane-1-carboxylic acid (7d, 0.64 g, 1.79 mmol, 72.6% yield, 57.0% purity) as a yellow oil. To a solution of 7d (0.64 g, 1.79 mmol, 1.00 eq) in DMF (5.00 mL) was added HOBt (362 mg, 2.68 mmol, 1.50 eq), EDCI (445 mg, 2.32 mmol, 1.30 eq), DIPEA (692 mg, 5.36 mmol, 93 μL, 3.00 eq) and 4-aminocyclohexan-1-ol (325 mg, 2.14 mmol, 1.20 eq, HCl), stirred at 20 °C for 16 hrs. The mixture was purified by prep-HPLC (column: Waters Xbridge C18 150*50mm* 10um; mobile phase: [water (10mM NH4HCO3) -ACN]; B%: 28%-58%, 11.5min) to give 7 (111.58 mg, 370 umol, 20.7% yield, 100% purity) as a white solid. LCMS: Rt = 0.813 min, m/z = 302.2 (M+H)+; Rt = 0.883 min, m/z = 302.1 (M+H)+. HPLC: purity: 100% under 220nm. 1H NMR: (400 MHz MeOD) δ 7.25 - 7.21 (m, 2H), 7.15 - 7.13 (m, 3H), 3.59 - 3.49 (m, 2H), 2.82 - 2.80 (m, 1H), 2.53 - 2.50 (m, 2H), 2.18 - 2.16 (m, 3H), 1.92 - 1.67 (m, 8H), 1.32 - 1.27 (m, 4H). 13C NMR: (400 MHz MeOD) δ 177.895, 176.889, 143.747, 129.521, 129.422, 126.826, 70.621, 39.844, 39.737, 37.306, 35.031, 34.421, 32.781, 32.435, 31.676, 31.198.
2-(3-benzylcyclobutyl)-N-(4-hydroxycyclohexyl)acetamide (8).
To a solution of allylbenzene (8a) (20.0 g, 169 mmol, 22.4 mL, 1.00 eq), ZINC-COPPER COUPLE (65.5 g, 508 mmol, 3.00 eq) in Et2O (200 mL), dropwise 2,2,2-trichloroacetyl chloride (61.6 g, 338 mmol, 37.8 mL, 2.00 eq) and POCl3 (29.2 g, 190 mmol, 17.7 mL, 1.12 eq) in Et2O (100 mL) at 20 °C under N2 for 2 hrs, then the reaction was stirred at 35°C for 16 hrs, the mixture was filtered through a pad of celite and the filtrate was poured into water (300 mL) and extracted with Et2O (200 mL * 3), the organic layer was washed with saturated NaHCO3 (200 mL * 2) and brine (200 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give 3-benzyl-2,2-dichlorocyclobutan-1-one (8b, 46.2 g). To a solution of ZINC (52.8 g, 807 mmol, 4.00 eq) in AcOH (120 mL) was added 8b (46.2 g, 202 mmol, 1.00 eq) in AcOH (120 mL) dropwise at 25 °C for 1 hr, stirred at 25 °C for 1 hr and then heated at 80 °C for 1 hr, the mixture was filtered and poured into water (200 mL), extracted with EtOAc (200 mL * 3), the organic layer was combined and washed with NaHCO3 (saturated, 150 mL * 2) and brine (200 mL), dried over Na2SO4, filtered and concentrated under reduced pressure and was purified by Reverse Phase HPLC (0.1% FA condition) to get 3-benzylcyclobutan-1-one (14.8 g, 78.6 mmol, 38.9% yield, 85.0% purity), to a solution of 3-benzylcyclobutan-1-one (1.00 g, 5.31 mmol, 1.00 eq) in toluene (20 mL) was added methyl 2-(triphenyl-l5-phosphaneylidene)acetate (2.66 g, 7.96 mmol, 1.50 eq), stirred at 110 °C for 2 hrs, the reaction was concentrated under vacuum to get a residue and triturated with petroleum ether (20 mL) at 25 °C for 10 mins, the product was purified by prep-HPLC (column: Phenomenex luna C18 150 * 40mm * 15 μm; mobile phase: [water (0.225% FA) - ACN]; B%: 47% - 77%, 10 mins) to get methyl 2-(3 benzylcyclobutylidene)acetate (943 mg, 4.34 mmol, 81.8% yield, 99.5% purity). To a solution of methyl 2-(3-benzylcyclobutylidene)acetate (943 mg, 4.34 mmol, 1.00 eq) in MeOH (20 mL) was added Pd/C (0.20 g, 10% purity), the reaction was degassed and back filled with H2 (15 psi) and stirred at 25 °C for 2 hrs, the residue was filtered through diatomite and the filtrate was concentrated under vacuum to afford methyl 2-(3-benzylcyclobutyl)acetate (8c, 947 mg, 4.31 mmol, 99.2% yield, 99.3% purity), to a solution of 8c (947 mg, 4.31 mmol, 1.00 eq) in THF (10 mL), H2O (5 mL) and MeOH (5 mL) was added LiOH•H2O (723 mg, 17.2 mmol, 4.00 eq), the reaction mixture was stirred at 25 °C for 16 hrs, then the reaction mixture was concentrated to get a residue, and was poured into H2O (15 mL) and extracted with EtOAc (15 mL * 3), the aqueous layer was adjusted pH = 2 ~ 3 and extracted with EtOAc (15 mL * 3), dried over Na2SO4, filtered and concentrated under reduced pressure to give 2-(3-benzylcyclobutyl)acetic acid (8d, 877 mg, 4.05 mmol, 94.0% yield, 94.4% purity) as a light yellow oil. To a solution of 8d (300 mg, 1.39 mmol, 1.00 eq) in DMF (5 mL) was added HOBt (225 mg, 1.66 mmol, 1.20 eq), EDCI (399 mg, 2.08 mmol, 1.50 eq) and DIEA (538 mg, 4.16 mmol, 724 μL, 3.00 eq), aminocyclohexan-1-ol (241 mg, 1.53 mmol, 1.10 eq, HCl) was added to the mixture. The reaction mixture was stirred at 25 °C for 16 hrs, the reaction mixture was poured into H2O (15 mL) and extracted with EtOAc (10 mL * 3), the organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue and was purified by prep-HPLC (column: Phenomenex luna C18 150 * 40mm * 15 μm; mobile phase: [water (0.225% FA) - ACN]; B%: 23% - 53%, 9 mins) to get 8 (111.58 mg, 366 μmol, 26.3% yield, 98.8% purity) as a white solid. LCMS: Rt = 0.794 min, m/z = 302.1 (M+H)+; Rt = 0.787 min, m/z = 302.2 (M+H)+. HPLC: Rt = 2.682 min, purity: 98.8% under 220 nm.1H NMR: (400 MHz, MeOD) δ 7.24 - 7.20 (m, 2H), 7.14 - 7.12 (m, 3H), 3.60 - 3.51 (m, 2H), 2.74 - 2.63 (m, 3H), 2.30 - 2.28 (m, 2H), 2.20 - 2.18 (m, 2H), 1.94 - 1.84 (m, 6H), 1.45 - 1.42 (m, 1H), 1.32 - 1.24 (m, 4H). 13C NMR: (400 MHz, MeOD) δ 174.65 - 174.39, 142.47 - 142.21, 129.76 - 129.36, 129.90, 70.57, 44.61 - 44.49, 43.73 - 43.46, 35.63, 34.97, 34.36, 33.24, 31.71, 30.65, 30.18.
N-((1r,4r)-4-hydroxycyclohexyl)-3-phenethylcyclopentane-1-carboxamide (9).
A mixture of dimethyl cyclopentane-1,3-dicarboxylate (9a, 2.00 g, 10.7 mmol, 1.00 eq) and NaOH (429 mg, 10.7 mmol, 1.00 eq) in MeOH (10.0 mL) was stirred for 12 hrs at 25 °C, filtered and concentrated under reduced pressure to give a residue, purified by column chromatography to get 3-(methoxycarbonyl)cyclopentane-1-carboxylic acid (9b, 1.20 g, 6.97 mmol, 64.8% yield) as a light yellow oil. To a mixture of 9b (1.10 g, 6.39 mmol, 1.00 eq) in THF (10.0 mL) was added BH3-Me2S (10.0 M, 703 uL, 1.10 eq) at −78 °C, stirred for 0.5 hr, then warmed to 25 °C and stirred for 12 hrs to obtain methyl 3-(hydroxymethyl)cyclopentane-1-carboxylate (9c, 780 g, 4.93 mmol, 77.1% yield) was obtained as a yellow oil. To a mixture of 9c (780 mg, 4.93 mmol, 1.00 eq) in DCM (10.0 mL) was added Dess-Martin (2.72 g, 6.41 mmol, 1.30 eq) at 25 °C and was stirred for 2 hrs, dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue and was purified by column chromatography to obtain methyl 3-formylcyclopentane-1-carboxylate (9d, 283 mg, 1.81 mmol, 36.7% yield) was obtained as a light yellow oil. To a solution of benzyltriphenylphosphonium bromide (9e, 903 mg, 2.08 mmol, 1.15 eq) in THF (10.0 mL) was added t-BuOK (329 mg, 2.94 mmol, 1.62 eq) at −20 °C under N2, stirred for 1 hr at −20 °C and then added 9d (183 mg, 1.81 mmol, 1.00 eq) at −20 °C and stirred another 1 hr, filtered and concentrated under reduced pressure to give a residue and was purified by column chromatography to get methyl (E)-3-styrylcyclopentane-1-carboxylate (124 mg, 538 umol, 29.7% yield) was obtained as a light yellow oil. To a solution of (E)-3-styrylcyclopentane-1-carboxylate (124 mg, 538 umol, 1.00 eq) in MeOH (10.0 mL) was added Pd/C (120 mg, 10% purity) at 25 °C under H2 (15 Psi), stirred for 2 hrs at 25 °C to get methyl 3-phenethylcyclopentane-1-carboxylate (9f, 100 mg, 430 umol, 79.9% yield) was obtained as a light yellow oil. To a mixture of 9f (85.0 mg, 365 umol, 1.00 eq) in MeOH (5.00 mL), THF (5.00 mL) and H2O (2.00 mL) was added LiOH·H2O (30.7 mg, 731 umol, 2.00 eq) at 25 °C, the mixture was heated to 70 °C and stirred for 2 hrs to get 3-phenethylcyclopentane-1-carboxylic acid (9g, 79.0 mg, 361 umol, 98.9% yield) as a light yellow oil. To a mixture of 9g (79.0 mg, 361 umol, 1.00 eq) and (1r,4r)-4-aminocyclohexan-1-ol (65.8 mg, 434 umol, 1.20 eq) in THF (10.0 mL) was added HATU (206 mg, 542 umol, 1.50 eq) and Et3N (727 mg, 7.18 mmol, 1.00 mL) at 25 °C and stirred for 12 hrs. The reaction mixture was concentrated under reduced pressure to give a residue. The reside was purified by prep-HPLC (neutral condition,column: Waters Xbridge 150*25mm*5um;mobile phase: [water( NH4HCO3)-ACN];B%: 43%−73%, 10min). 9 (8.24 mg, 25.2 umol, 6.97% yield, 96.5% purity) was obtained as a white solid. LCMS: RT = 0.933 min, m/z = 316.2 (M+H)+. HPLC: RT = 2.130 mins, purity: 96.5% purity, under 220 nm. 1H NMR: (400 MHz, DMSO-d6) δ 7.52 (d, J = 7.6, 1H), 7.27-7.23 (m, 2H), 7.18-7.15 (m, 3H), 4.52 (d, J = 4.0, 1H), 3.43-3.41 (m, 1H), 2.57-2.53 (m, 2H), 1.87 - 1.57 (m, 12H), 1.18-1.06 (m, 7H).
N-((1r,4r)-4-hydroxycyclohexyl)-3-(3-phenylpropyl)cyclopentane-1-carboxamide (10)
To a solution of triphenyl(3-phenylpropyl)phosphonium bromide (10b, 1.79 g, 3.87 mmol, 1.10 eq) in toluene (10.00 mL) was added t-BuOK (473 mg, 4.22 mmol, 1.20 eq), the mixture was stirred at 70 °C for 1 hr. Then methyl 3-oxocyclopentane-1-carboxylate (10a, 500 mg, 3.52 mmol, 1.00 eq) in toluene (2.00 mL) was added to the mixture and stirred at 70 °C for 16 hrs to give methyl 3-(3-phenylpropylidene)cyclopentane-1-carboxylate (0.100 g, 388 umol, 11.0% yield, 94.8% purity) as a colorless oil, MeOH (10.0 mL), Pd/C (0.10 g, 10% purity) were added at 25 °C, degassed with N2 for 3 times. The resulting mixture was stirred at 25 °C under H2 (15 Psi ) for 4 hrs, the mixture was filtered through celite and the filtrate was concentrated under vacuum to give methyl 3-(3-phenylpropyl)cyclopentane-1-carboxylate (10c, 90.0 mg, 365 umol, 94.1% yield) was obtained as a colorless oil. A solution of 10c (90.0 mg, 365 umol, 1.00 eq) and LiOH·H2O (30.6 mg, 730 umol, 2.00 eq) in MeOH (5.00 mL) and H2O (2.00 mL) was stirred at 70 °C for 3 hrs to obtain 3-(3-phenylpropyl)cyclopentane-1-carboxylic acid (10d, 90.0 mg, crude) was obtained as a yellow oil. A mixture of 10d (90.0 mg, 387 umol, 1.00 eq), (1r,4r)-4-aminocyclohexan-1-ol (49.0 mg, 426 umol, 1.10 eq), HATU (294 mg, 774 umol, 2.00 eq) and Et3N (117 mg, 1.16 mmol, 161 uL, 3.00 eq) in THF (2.00 mL) was stirred at 25 °C for 16 hrs, the mixture was concentrated to give the residue, then purified by column: Phenomenex Gemini-NX C18 75*30mm*3um;mobile phase: [water(0.225%FA)-ACN]; B%: 42%-72%,7min. 11 (23.38 mg, 68.1 umol, 17.6% yield, 96.1% purity) was obtained as an off-white solid. LCMS: RT = 0.859 min, m/z = 330.3 (M+H)+. HPLC: RT = 2.548 mins, purity: 96.1%, under 220 nm. 1H NMR: (400 MHz, DMSO-d6)δ 7.47 (d, J = 8.0, 1H), 7.28-7.24 (m, 2H), 7.18-7.15 (m, 3H), 4.49 (d, J = 4.0, 1H), 3.43-3.37 (m, 1H), 2.58-2.53 (m, 3H), 1.78 - 1.77 (m, 1H), 1.76 - 1.70 (m, 3H), 1.68 - 1.66 (m, 5H), 1.64-1.54 (m, 3H), 1.32-1.29 (m, 3H), 1.18-1.13 (m, 6H).
N-((1r,4r)-4-hydroxycyclohexyl)-3-phenethylcyclohexane-1-carboxamide (11).
To a mixture of 3-(methoxycarbonyl)cyclohexane-1-carboxylic acid (11a, 2.00 g, 10.7 mmol, 1.00 eq) in THF (10.0 mL) was added BH3-Me2S (10.0 M, 1.18 mL, 1.10 eq) at −78 °C and stirred for 0.5 hr at −78 °C, then warmed to 25 °C and stirred for 12 hrs to obtain methyl 3-(hydroxymethyl)cyclohexane-1-carboxylate (11b, 1.79 g, 10.4 mmol, 96.7% yield) was obtained as a light yellow oil. To a mixture of 11b (1.79 g, 10.4 mmol, 1.00 eq) and Et3N (6.31 g, 62.3 mmol, 6.00 eq) in DMSO (20.0 mL) was added pyridine;sulfur trioxide (4.96 g, 31.2 mmol, 3.00 eq) at 25 °C and stirred for 12 hrs, dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography to get methyl 3-formylcyclohexane-1-carboxylate (11c, 710 mg, 4.17 mmol, 40.1% yield) was obtained as a yellow oil. To a solution of benzyltriphenylphosphonium bromide (1.17 g, 2.70 mmol, 1.15 eq) in THF (10.0 mL) was added t-BuOK (427 mg, 3.81 mmol, 1.62 eq) at −20 °C under N2, was stirred for 1 hr at −20 °C and then added 11c (400 mg, 2.35 mmol, 1.00 eq) at −20 °C and stirred another 1 hr. The product was dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography to get intermediate methyl (E)-3-styrylcyclohexane-1-carboxylate (280 mg, 1.15 mmol, 48.7% yield) was obtained as a light yellow oil, then added MeOH (10.0 mL) was added Pd/C (230 mg, 10% purity) at 25 °C under H2 (15 Psi), stirred for 2 hrs to get methyl 3-phenethylcyclohexane-1-carboxylate (11d, 230 mg, 933 umol, 87.7% yield) was obtained as a colourless oil. To a mixture of 11d (165 mg, 669 umol, 1.00 eq) in MeOH (5.00 mL), THF (5.00 mL) and H2O (2.00 mL) was added LiOH·H2O (56.2 mg, 1.34 umol, 2.00 eq) at 25 °C then heated to 70 °C and stirred for 2 hrs to get 3-phenethylcyclohexane-1-carboxylic acid (11e, 150 mg, 645 umol, 96.4% yield) was obtained as a light yellow oil. To a mixture of 11e (150 mg, 645 umol, 1.00 eq) and (1r,4r)-4-aminocyclohexan-1-ol (117 mg, 774 umol, 1.20 eq) in THF (10.0 mL) was added HATU (368 mg, 968 umol, 1.50 eq) and Et3N (1.45 g, 14.3 mmol, 2.00 mL) at 25 °C, stirred at 25 °C for 12 hrs. The reaction mixture was concentrated under reduced pressure to give a residue and was purified by prep-HPLC (neutral condition, column: Waters Xbridge 150*25mm*5um;mobile phase: [water( NH4HCO3)-ACN];B%: 40%−70%,10min). 11 (8.15 mg, 24.3 umol, 3.78% yield, 98.6% purity) was obtained as a white solid. LCMS: RT = 0.930 min, m/z = 330.2 (M+H)+. HPLC: RT = 2.218 mins, purity: 98.6%, under 220 nm. 1H NMR: (400 MHz, DMSO-d6)δ 7.46 (d, J = 8.0, 1H), 7.27-7.24 (m, 2H), 7.19-7.14 (m, 3H), 4.49 (s, 1H), 3.42-3.38 (m, 1H), 2.59-2.55 (m, 2H), 2.04-1.98 (m, 1H), 1.79 - 1.69 (m, 8H), 1.58-1.40 (m, 4H), 1.24-1.11 (m, 6H), 1.09-0.96 (m, 1H), 0.90-0.77 (m, 1H).
N-((1r,4r)-4-hydroxycyclohexyl)-4-phenethylcyclohexane-1-carboxamide (12).
A mixture of benzyltriphenylphosphonium bromide (1.47 g, 3.39 mmol, 1.15 eq) and t-BuOK (535 mg, 4.77 mmol, 1.62 eq) in THF (10.0 mL) at −20 °C under N2 and was stirred for 1 hr, then added 4-formylcyclohexane-1-carboxylic acid (12a, 500 mg, 2.94 mmol, 1.00 eq) at −20 °C and stirred another 1 hr, dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue and was purified by column chromatography to obtain intermediate methyl (E)-4-styrylcyclohexane-1-carboxylate (130 mg, 530 umol, 18.1% yield, 99.7% purity) was obtained as an off-white solid, then added MeOH (10.0 mL), Pd/C (120 mg, 10% purity) at 25 °C under H2 (15 Psi), stirred for 2 hrs to get methyl 4-phenethylcyclohexane-1-carboxylate (12b, 120 mg, 487 umol, 91.5% yield) was obtained as a light yellow oil. To a mixture of 12b (120 mg, 487 umol, 1.00 eq) in MeOH (5.00 mL), THF (5.00 mL) and H2O (2.00 mL) was added LiOH·H2O (40.9 mg, 974 umol, 2.00 eq) at 25 °C, then heated to 70 °C and stirred for 2 hrs to get 4-phenethylcyclohexane-1-carboxylic acid (12c, 110 mg, 473 umol, 97.2% yield) was obtained as a light yellow oil, then added (1r,4r)-4-aminocyclohexan-1-ol (65.4 mg, 568 umol, 1.20 eq) in THF (10.0 mL) was added HATU (270 mg, 710 umol, 1.50 eq) and Et3N (143 mg, 1.42 mmol, 3.00 eq) at 25 °C and stirred for 12 hrs. The reaction mixture was concentrated under reduced pressure to give a residue and was purified by prep-HPLC (neutral condition, column: Waters Xbridge 150*25mm*5um;mobile phase: [water(10mM NH4HCO3)-ACN];B%: 40%-70%,10min). 12 (9.82 mg, 29.3 umol, 6.21% yield, 98.6% purity) was obtained as a white solid. LCMS: RT = 0.859 min, m/z = 330.3 (M+H)+. HPLC: RT = 2.236 mins, purity: 98.6%, under 220 nm. 1H NMR: (400 MHz, DMSO-d6)δ 7.45 (d, J = 8.0, 1H), 7.27-7.24 (m, 2H), 7.18-7.15 (m, 3H), 4.48 (d, J = 4.0, 1H), 3.46-3.36 (m, 2H), 2.59-2.55 (m, 2H), 2.03-1.96 (m, 1H), 1.80 - 1.76 (m, 4H), 1.69-1.64 (m, 4H), 1.45-1.43 (m, 2H), 1.35-1.25 (m, 2H), 1.25-1.13 (m, 5H), 0.91-0.87 (m, 2H).
4-benzyl-N-((1r,4r)-4-hydroxycyclohexyl)cyclohexane-1-carboxamide (13).
A mixture of methyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (13a, 500 mg, 1.88 mmol, 1.15 eq) and (bromomethyl)benzene (385 mg, 2.25 mmol, 1.20 eq) in dioxane (10.0 mL) and H2O (2.00 mL) was added Pd(dppf)Cl2 (274 mg, 375 umol, 0.20 eq) and K2CO3 (778 mg, 5.64 mmol, 3.00 eq) at 25 °C under N2. The mixture was heated to 100 °C and stirred for 2 hrs to get intermediate methyl 4-benzylcyclohex-3-ene-1-carboxylate(300 mg, 1.30 mmol, 69.3% yield) was obtained as a light yellow oil. To a solution of the intermediate (300 mg, 1.30 mmol, 1.00 eq) in MeOH (10.0 mL) was added Pd/C (300 mg, 10% purity) at 25 °C under H2 (15 Psi) and was stirred for 2 hrs at 25 °C to obtain methyl 4-benzylcyclohexane-1-carboxylate (13b, 300 mg, 1.29 mmol, 99.1% yield) was obtained as a light yellow oil. To a mixture of 13b (300 mg, 1.29 mmol, 1.00 eq) in MeOH (5.00 mL), THF (5.00 mL) and H2O (2.00 mL) was added LiOH·H2O (108 mg, 2.58 mmol, 2.00 eq) at 25 °C and then heated to 70 °C and stirred for 2 hrs to get 4-benzylcyclohexane-1-carboxylic acid (13c, 280 mg, 1.28 mmol, 99.3% yield) was obtained as a light yellow oil. To a mixture of 13c (280 mg, 1.28 mmol, 1.00 eq) and (1r,4r)-4-aminocyclohexan-1-ol (147 mg, 1.28 mmol, 1.00 eq) in THF (5.00 mL) was added HATU (731 mg, 1.92 mmol, 1.50 eq) and Et3N (389 mg, 3.85 mmol, 3.00 eq) at 25 °C and stirred for 12 hrs. The reaction mixture was concentrated under reduced pressure to give a residue. The reside was purified by prep-HPLC (neutral condition, column: Waters Xbridge 150*25mm*5um;mobile phase: [water(10mM NH4HCO3)-ACN];B%: 37%-67%,10min). 13 (58.93 mg, 185 umol, 14.4% yield, 99.0% purity) was obtained as a white solid. LCMS: RT = 0.823 min, m/z = 316.3 (M+H)+. HPLC: RT = 2.097 mins, purity: 99.0%, under 220 nm. 1H NMR: (400 MHz, CDCl3-d1)δ 7.21-7.17 (m, 2H), 7.11-7.05 (m, 3H), 5.24 (d, J = 7.6, 1H), 3.72-3.68 (m, 1H), 3.54-3.51 (m, 1H), 2.52 (d, J = 7.6, 2H), 2.15 - 2.13 (m, 1H), 1.93-1.91 (m, 4H), 1.84-1.66 (m, 4H), 1.46-1.36 (m, 7H), 1.12-1.09 (m, 2H).
2-((benzylthio)methyl)-N-((1r,4r)-4-hydroxycyclohexyl)cyclopropane-1-carboxamide (14, 14A and 14B).
Phenylmethanethiol (14a, 25.2 g, 202 mmol, 23.7 mL, 1.00 eq) was added to a solution of EtONa (13.8 g, 202 mmol, 1.00 eq) in EtOH (100 mL), KI (1.68 g, 10.1 mmol, 0.05 eq) and 2-chloro-1,1-dimethoxyethane (30.3 g 243 mmol, 27.8 mL, 1.20 eq), the mixture was stirred at 80 °C for 12 hrs, the reaction mixture was filtered, concentrated under reduced pressure and purified by flash silica gel chromatography to obtain benzyl(2,2-dimethoxyethyl)sulfane (38.0 g, 179 mmol, 88.2% yield), added H2SO4 (17.9 g 179 mmol, 9.74 mL, 98.0% purity, 1.00 eq) and stirred at 60 °C for 12 hrs to obtain 2-(benzylthio)acetaldehyde (14b, 27.0 g). Tert-butyl 2-(triphenyl-λ5-phosphaneylidene)acetate (64.7 g, 172 mmol, 1.30 eq) was added into a solution of 14b (22.0 g, 132 mmol, 1.00 eq) in THF (220 mL), stirred at 20 °C for 12 hrs to obtain tert-butyl (E)-4-(benzylthio)but-2-enoate (14c, 31.3 g). To a solution of NaH (3.87 g, 96.6 mmol, 60.0% purity, 1.20 eq) in DMSO (100 mL) was added dimethylmethanesulfinic iodide (21.2 g 96.6 mmol, 1.20 eq) and the mixture was stirred at 20 °C for 0.5 hr, then added a solution of 14c (21.3 g, 80.5 mmol, 1.00 eq) and stirred at 20 °C for 1 hr to obtain tert-butyl 2-((benzylthio)methyl)cyclopropane-1-carboxylate (14d, 4.10 g 14.7 mmol, 18.2% yield). To a solution of 14d (4.10 g, 14.7 mmol, 1.00 eq) in DCM (20.0 mL) was added TFA (36.1 g, 316 mmol, 23.4 mL, 21.5 eq), the mixture was stirred at 25 °C for 16 hrs to obtain 2-((benzylthio)methyl)cyclopropane-1-carboxylic acid (14e, 2.30 g). To a solution of 14e (2.30 g, 10.3 mmol, 1.00 eq) in DMF (20.0 mL) was added EDCI (2.98 g, 15.5 mmol, 1.50 eq) , HOBt (1.68 g, 12.4 mmol, 1.20 eq) and DIEA (2.68 g, 20.7 mmol, 3.61 mL, 2.00 eq). Then (1r,4r)-4-aminocyclohexan-1-ol (1.73 g, 11.3 mmol, 1.10 eq) was added to the mixture and stirred at 25 °C for 2 hrs. The reaction mixture was poured into water (90.0 mL), stirred at 25 °C for 1 hr, filtered and the filtrate was evaporated under vacuum, the product was triturated with Ethyl acetate (30.0 mL) at 70 °C for 30 mins, then cooled to 25 °C, filtered and the filtrate was concentrated under vacuum, the residue was purified by prep-HPLC (column: Phenomenex luna C18 250*50mm*10 um; mobile phase: [water(0.1%TFA)-ACN]; B%: 20%−50%,20min). To get the individual enantiomers 14A and 14B, the product was purified by prep-SFC (column: DAICEL CHIRALPAK AD-H (250mm*30mm,5um); mobile phase: [0.1%NH3H2O ETOH]; B%: 35%−35%,2.7 min;240 minmin). The product was purified by (column: Phenomenex Gemini-NX C18 75*30mm*3um; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN]; B%: 15%−45%,7min) and (column: Phenomenex Gemini-NX C18 75*30mm*3um;mobile phase: [water(0.225%FA)-ACN]; B%: 30%−60%, 2min). 14A (1.37 g, 4.22 mmol, 40.7% yield, 98.4% purity) was obtained as an off-white solid, 14B (1.18 g, 3.59 mmol, 34.7% yield, 97.3% purity) was obtained as an off-white solid. LCMS: 14A: RT = 0.816 min, m/z = 320.2 (M+H)+; 14B: : RT = 0.645 min, m/z = 319.9 (M+H)+. HPLC: 14A: RT = 1.887 mins, purity: 98.4%, under 220 nm. 14B: RT = 1.880 mins, purity: 97.3%, under 220 nm. 1H NMR: 14A: (400 MHz MeOD) δ 7.33 - 7.21 (m, 5H), 3.76 (s, 2H), 3.60-3.57 (m, 1H), 3.52 - 3.50 (m, 1H), 2.44 - 2.40 (m, 2H), 1.94 - 1.89 (m, 4H), 1.42 - 1.40 (m, 2H), 1.33 - 1.29 (m, 4H), 1.08 (td, J = 4.4, 8.8 Hz, 1H), 0.69 (ddd, J = 4.2, 6.0, 8.4 Hz, 1H); 14B: (400 MHz MeOD) δ 7.32 - 7.28 (m, 5H), 3.76 (s, 2H), 3.61-3.58 (m, 1H), 3.52 - 3.50 (m, 1H), 2.44 - 2.40 (m, 2H), 1.94 - 1.89 (m, 4H), 1.43 - 1.41 (m, 2H), 1.33 - 1.28 (m, 4H), 1.08 (td, J = 4.4, 8.8 Hz, 1H), 0.69 (ddd, J = 4.2, 6.0, 8.4 Hz, 1H).
X-ray diffraction studies of 14A and 14B.
X-ray powder diffraction (XRPD) studies were performed with a Panalytical X’Pert3 Powder XRPD on a Si zero-background holder. The 2θ position was calibrated against a Panalytical Si reference standard disc. The parameters used are shown in Table S2.
TGA/DSCThermogravimetric analysis (TGA) data were collected using a TA Discovery 550 TGA from TA Instrument. Differential scanning calorimetry (DSC) was performed using a TA 2500 DSC from TA Instrument. DSC was calibrated with Indium reference standard and the TGA was calibrated using nickel reference standard. Detailed parameters used are shown in Table S3.
Polarized Light Microscopy crystal images were captured on Nikon DS-Fi2 upright microscope at room temperature.
Single Crystal Data Collection
The X-ray intensity data were measured on a Bruker D8 VENTURE (IμS microfocus X-ray source, Cu Kα, λ =1.54178Å, PHOTON CMOS detector) diffractometer. The strategy was created and optimized with the Bruker Apex3 software, and the frames were integrated with the Bruker SAINT software package. Data were corrected for absorption effects using the Multi-Scan method (SADABS-2016/2). The structure was solved with the ShelXT structure solution program using Intrinsic Phasing and refined with ShelXL (Version 2014/7) refinement package using full-matrix least-squares on F2 contained in the SHELX software suite.24,25
Single Crystal Growth Experiments
Slow Evaporation
HPLC vials with saturated solutions of 14A and 14B were capped with perforated caps. Vials were left at RT to allow slow evaporation of the solvents. A total of 20 experiments were prepared, 4 of which are represented in the table below. The remaining 16 samples were moved to slow cooling due to limited observed solubility. Summary of slow evaporation experiments are shown in Table S4.
Layer Diffusion was performed in HPLC vials with saturated solutions of 14A and 14B. Anti-solvent was layered onto the saturated solutions slowly and the vial capped. Vials were left at room temperature to allow the two solvents to diffuse into one another. A total of 20 experiments were performed with results shown below. Summary of layer diffusion experiments are shown in Table S5.
Slow Cooling
Saturated solutions of 14A and 14B were prepared by slurrying at 35-60 ⁰C in HPLC vials. Suspensions were filtered using a PTFE membrane (pore size 0.20 μm). Filtrates were slowly cooled to 5ºC at a rate of 0.1ºC/min. A total of 15 experiments were performed; results are shown in Table S1.
Dose-response of mitofusin agonist fusogenicity, measured using a mitochondrial elongation assay (aspect ratio) was performed in Mfn1- or Mfn2-deficient MEFs cultured at 37°C, 5% CO2-95% air, cells were seeded on day 1 in 6 well plates at a density of 2x104 cells per well and compounds added at 9 concentrations (0.5nM-10uM dissolved in DMSO) overnight. Mitochondria were then stained with MitoTracker Orange (200 nM; M7510; Invitrogen, Carls-bad, CA, USA), nuclei were stained with Hoechst (10 μg/ml; Invitrogen, Thermo Fisher Scientific Cat: # H3570). Images were acquired at room temperature on a Nikon Ti Confocal microscope using a 60 X 1.3 NA oil- immersion objective in Krebs-Henseleit buffer (138 NaCl, 3.7 nM KCL, 1.2 nM KH2PO4, 15 nM Glucose, 20 nM HEPES pH: 7.2-7.5, and 1 mM CaCl2). Laser excitation was 549 nm with emission at 590 nm for MitoTracker Orange and excitation at 306 nm with emission at 405 nm for Hoechst. Images were analyzed using ImageJ and fusogenicity quantified as mitochondrial aspect ratio (length/width), and were indexed to the maximal response elicited by 1 and response curves interpolated using the sigmoidal model using Prism 8 software. EC50 values are reported as mean with 95% confidence limits for at least 3 independent experiments (16).
Functional evaluation of mitofusin agonist effect on mitochondrial depolarization.
Cultured Mfn2 KO or Mfn1 KO MEFs treated with either DMSO, 1, 5A or 5B (1 μM) for 48 hours, then were stained with Tetramethylrhodamine ethyl ester (TMRE, 200 nM, Invitrogen Thermo Fisher Scientific Cat:# T-669) , MitoTracker Green (200 nM; Invitrogen, Thermo Fisher Scientific Cat:# M7514) and Hoechst (10 ug/ml; Invitro-gen, Thermo Fisher Scientific Cat:# H3570) for 30 min at 37°C in 5% CO2-95% air, washed twice in PBS. Images were acquired at room temperature on a Nikon Ti Confocal microscope using either 60 X 1.3 NA oil- immersion ob-jective, in Krebs-Henseleit buffer (138 NaCl, 3.7 nM KCL, 1.2 nM KH2PO4, 15 nM Glucose, 20 nM HEPES pH: 7.2-7.5, and 1mM CaCl2 ): laser excitation was 488 nm with emission at 510 nm for MitoTracker Green, 549 nm with emission at 590 nm for TMRE, and 306 nm with emission 405 nm for Hoechst. Mitochondrial depolarization was reported as % number of green mitochondria/number of yellow+green mitochondria using Image J. (16).
FRET studies of MFN2 conformation were performed as previously described (15, 16). Briefly, the MFN2 FRET protein engineered with amino terminal mCerulean and carboxy terminal mVenus reporters (14) was expressed with an adenoviral vector in mitofusin null (MFN1/MFN2 knockout) MEFs cultured in UV transparent 96 well plates. 48 hours thereafter 1 μM of each test compound or vehicle (DMSO) was added for 4 hours. Fluorescence signals were acquired on a Tecan Safire II multi-mode plate reader: FRET-excitation 433/8 nm, emission 528/8 nm; Cerulean-excitation 433/8 nm, emission 475/8 nm.
Mitofusin agonist pharmacodynamic effects on sciatic nerve motility were measured using time-lapse imaging (1 frame every 5 s) for 121 frames (10 min, sciatic nerve) at 37°C on a Nikon A1Rsi Confocal Microscope using a 40x oil objective as described. Sciatic nerve axon mitochondria were labeled with TMRE. Kymographs and quantitative data were generated using an Image-J plug-in (17).
In vitro pharmacokinetic analyses of mitofusin agonists were performed in duplicate using standard methods by WuXi Apptec Co. Ltd. (Shanghai, China). Plasma protein binding was measured by equilibrium dialysis; % bound = (1-[free compound in dialysate]/[total compound in retentate]) X 100. Plasma stability of 2 μM compounds in clarified freeze-thawed plasma was assessed by LC-MS/MS of supernatants after protein precipitation; 120 min data are reported for studies including 0, 10, 30, 60, and 120 min. Liver Microsome stability of 1 μM compounds in liver microsomes (0.5 mg/ml) after 0, 5, 10, 20, 30, 60 min. incubation was assessed by LC/MS/MS of reaction extracts. Passive artificial blood brain barrier membrane permeability assay (PAMPA-BBB) were performed using 150 μL of 10 μM compounds (5% DMSO) added to PVDF membranes pre-coated with 5 μL of 1% brain polar lipid extract (Porcine)/dodecane mixture and incubated for 4h at room temperature with shaking at 300 rpm. Donor and acceptor samples were analyzed by LC-MS/MS.
In vivo pharmacokinetic analyses were performed in triplicate at WuXi Apptec Co. Ltd. (Shanghai, China) or Frontage Laboratories, Inc (Exton PA, USA). Except where specifically noted, compounds were administered orally to mice fasted overnight with food returned 4 hours post-dosing. Compound 2 and 5B (5mg/mL) were dissolved in 30% SBE-β-CD and administered by oral gavage (50 mg/kg) to 7-9 week male CD-1 mice, plasma was collected and brains were prepared by homogenizing with 4 volumes (w:v) of homogenizing solution (MeOH/15 mM PBS (1:2, v:v)) followed by further diluting in blank matrix to obtain dilution factor of 40 and 10. A 40 uL aliquot of the study sample was quenched with 800 μL of IS1 (6 in 1 internal standard in ACN (Labetalol & tolbutamide & Verapamil & dexamethasone & glyburide & Celecoxib 100 ng/mL for each)) respectivelly, and then the mixture was vortex-mixed well (at least 15 s) and centrifuged for 15 min at 12000 × g, 4 °C, an aliquot of 60 μL supernatant was transferred to the 96-well plate and centrifuged for 5 min at 3220 × g, 4 °C, then the supernatant was directly injected for HPLC analysis. Compound 5 enantiomeric mixture was dissolved in 10%DMSO/90%(30%HP-b-CD) and administered intravenously (10 mg/kg) or 5B (5mg/mL) was dissolved in 10%DMSO/90%(30%HP-b-CD) and administered by oral gavage (50 mg/kg) to 7-9 week male CD-1 mice. Plasma was collected, brain, nerve and spinal cord samples were homogenized in water with 4, 8, and 4 dilution factors, respectively. Plasma, spinal cord, and nerve homogenates were further diluted in control mouse plasma to achieve a total dilution factor of 10, 20, and 40, respectively. For mouse study samples, 30 μL of each sample was added to separate wells of the 96-well plate and 30 μL of diluent (ACN:H2O, 50:50) was added to each sample. To all samples, 200 μL of internal standard (200 ng/mL warfarin) solution in ACN was added. The plate was vortexed vigorously for 10 minutes and then centrifuged for 10 minutes at 4000 rpm (Sorvall Legend X1R centrifuge, Thermo Scientific) at 15°C. Supernatants were further diluted 1:1 in H2O prior to HPLC analysis. Time-concentration curves were generated using non-compartmental approaches and Phoenix WinNonlin 6.3 software. Data are presented as mean±SEM from 3 mice for each condition.
Measuring effects of 2 and 5 on neuromuscular dysfunction in murine ALS.
ALS (SOD1 G93A) mice (24) were randomly assigned to oral treatment with vehicle (10% Me2SO/90% (30% 2-hydroxypropyl)-β-cyclodextrin (HP-BCD; Sigma, Cat :#332607) or 5 (60mg/kg twice daily) from age 60 through 140 days. Disease severity was assessed by investigators blind to treatment status using an integrated multi-test scoring system (25) as follows:
Ledge test:
Score 0 = effectively use its hind legs while walking along the ledge of the cage; Score 1= loses its footing some times while walking along the ledge, but appears coordinated; Score 2= it does not effectively use its hind legs; Score 3= refuses to move along the ledge or falls off from the ledge while walking;
Hindlimb clasping:
Score 0 = hindlimbs completely splayed outward while being lifted by grasped its tail; Score 1= one hindlimb is partially collapsed toward the abdomen; Score 2= both hindlimbs are partially collapsed toward the abdomen; Score 3= hindlimbs are entirely touching the abdomen;
Gait:
Score 0 = it appears normal gait when the mice is allowed to walk; Score 1= it is tremor or appears to limp while walking; Score 2= feet point away from the body while waking; Score 3= it has difficulty moving forward;
Kyphosis:
Score 0 = it is able to straighten its spine while walking, no kyphosis is observed; Score 1= it shows mild kyphosis but is able to straighten its spine; Score 2= it is unable to straighten its spine but mild kyphosis; Score 3= severe kyphosis while walking and sitting.
The aggregate dysfunction score as reported is the sum of all four individual test scores.
Lipinski’s rule and Central Nervous System Multiparameter Optimization calculation.
Lipinski’s rule related compounds properties were calculated using DruLiTo software. Compounds chemical properties were obtained using ACD/Laboratories, version 12.1, for ClogD at pH 7.4, pKa. For calculation of TPSA and ClogP were obtained using Chemdraw 19.1. The CNS MPO scores were calculated as described. (29)
PAINS Analysis.
All reported compounds were screened for PAINS through FAF-Drugs4 website tool (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#welcome) and none were identified as pan assay interference compounds.
Animals.
ALS B6.SOD1-G93A mice (24) were purchased from The Jackson Laboratory (Stock No: 004435). Mouse procedures were approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis Protocol ID: 19-0910, by IACUC-SH for the WuXi Corporate Committee for Animal Research Ethics, by Confluence Discovery Technologies Animal Care and Use Committee, and by Frontage Labs Animal Care and Use Committee.
Supplementary Material
Funding:
X.D. was supported in part by the China Scholarship Council and GWD by NIH R41NS113642, R41NS115184, R35135736 and Research Grant 628906 from the Muscular Dystrophy Association. GWD is the Philip and Sima K. Needleman-endowed Professor at Washington University in St. Louis and a Scholar-Innovator awardee of the Harrington Discovery Institute.
Abbreviations
- ACN
acetonitrile
- ALS
Amyotrophic Lateral Sclerosis
- CDI
1,1’-carbonyldiimidazole
- CMT2A
Charcot-Marie-Tooth Disease type 2A
- DCC
N,N’-dicyclohexylcarbodiimide
- DCM
dichloromethane
- DEAD
Diethyl azodicarboxylate
- DIPEA
N,N-Diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDCI
N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride
- EtOAc
ethyl acetate
- EtOH
ethanol
- FRET
förster resonance energy transfer
- HP-b-CD
2-Hydroxyproryl-ß-cyclodextrin
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- PA
isopropyl alcohol
- IMIBK
4-methyl-2-pentanone
- IPAc
isopropyls acetate
- LC-MS
liquid chromatography tandem mass spectrometry
- MEFs
Mouse Embryonic Fibroblasts
- MEK
methyl ethyl ketone
- MeOH
methanol
- 2-MeTHF
2-methyltetrahydrofuran
- MFN
mitofusin
- MTBE
methyl tert-butyl ether
- NMR
nuclear magnetic resonance
- PAMPA-BBB
passive artificial blood brain barrier membrane permeability assay
- PBS
phosphate-buffered saline
- PK
pharmacokinetic
- PPH3
triphenylphosphine
- r.t.
room temperature
- SFC
supercritical fluid chromatography
- TEA
triethylamine
- THF
Tetrahydrofuran
- tPSA
topological polar surface area
Footnotes
Supporting Information
Figure S1 (Dose response curves and EC50 values of 1-6); Figure S2 ( 1H-NMR spectrum of intermediate 5C); Figure S3 (Characteristics of 9 and 10 starting material); Figure S4 (Crystalization of 9 and 10 for single crystal X-ray diffraction studies); Figure S5 (Structural modeling of positions for human MFN2 amino acids mimicked by small molecule mitofusin activators); Figure S6 (HPLC, 1H NMR and 13C NMR chromatography of 5); Figure S7 (HPLC, 1H NMR and 13C NMR chromatography of 6); Figure S8 (HPLC, 1H NMR and 13C NMR chromatography of 7); Figure S9 (HPLC, 1H NMR and 13C NMR chromatography of 8); Figure S10. HPLC and 1H NMR chromatography of 9; Figure S11. HPLC and 1H NMR chromatography of 10; Figure S12. HPLC and 1H NMR chromatography of 11; Figure S13. HPLC and 1H NMR chromatography of 12; Figure S14. HPLC and 1H NMR chromatography of 13; Figure S15 (SFC, 1H NMR and 13C NMR chromatography of 5A and 5B); Figure S16. HPLC and 1H NMR chromatography of 14A and 14B. Table S1. Solvents used in slow cooling experiments. Table S2. Parameters used in X-ray powder diffraction. Table S3. Parameters used in thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Table S4. Summary of slow evaporation experiments. Table S5. Summary of layer diffusion experiments. Molecular formula string for all the final compounds (CSV). 3D PDB files for Figure S5 (PDB).
Accession Codes
CCDC 2057833-2057834 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
PDB ID Code
Human Mfn1 (PDB ID: 5GNS) and Arabidopsis thaliana dynamin-related protein (PDB ID: 3T34) were used to computationally model human Mfn2 closed configuration based on structural homology has been described previously (15). And this hypothetical human Mfn2 structure and hMFN2 found in the protein database (PDB; code 6JFM) were used for molecular modeling with compound 14A and 14B, authors will release the atomic coordinates and experimental data upon article publication.
Competing interests: G.W.D. is an inventor on patent applications PCT/US18/028514 submitted by Washington University and PCT/US19/46356 and PCT/US20/14784 submitted by Mitochondria Emotion, Inc that cover the use of small molecule mitofusin agonists to treat chronic neurodegenerative diseases, and is the founder of Mitochondria in Motion, Inc., a Saint Louis based biotech R&D company focused on enhancing mitochondrial trafficking and fitness in neurodegenerative diseases. The other authors declare no competing interests. Studies were performed under terms of an MTA between Mitochondria in Motion, Inc. and Washington University in St. Louis.
REFERENCES
- 1.Knott AB; Perkins G; Schwarzenbacher R; Bossy-Wetzel E Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci 2008, 9, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan DC Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet 2012, 46, 265–287. [DOI] [PubMed] [Google Scholar]
- 3.Koshiba T; Detmer SA; Kaiser JT; Chen H; McCaffery JM; Chan DC Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [DOI] [PubMed] [Google Scholar]
- 4.Chen H; Detmer SA; Ewald AJ; Griffin EE; Fraser SE; Chan DC Mitofusins mfn1 and mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol 2003, 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eura Y; Ishihara N; Yokota S; Mihara K Two mitofusin proteins, mammalian homologues of fzo, with distinct functions are both required for mitochondrial fusion. J. Biochem 2003, 134, 333–344. [DOI] [PubMed] [Google Scholar]
- 6.Dorn GW, 2nd. Evolving concepts of mitochondrial dynamics. Annu. Rev. Physiol 2019, 81, 1–17. [DOI] [PubMed] [Google Scholar]
- 7.Feely SM; Laura M; Siskind CE; Sottile S; Davis M; Gibbons VS; Reilly MM; Shy ME Mfn2 mutations cause severe phenotypes in most patients with cmt2a. Neurology 2011, 76, 1690–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuppia G; Rizzo F; Riboldi G; Del Bo R; Nizzardo M; Simone C; Comi GP; Bresolin N; Corti S Mfn2-related neuropathies: Clinical features, molecular pathogenesis and therapeutic perspectives. J. Neurol. Sci 2015, 356, 7–18. [DOI] [PubMed] [Google Scholar]
- 9.Wang L; Gao J; Liu J; Siedlak SL; Torres S; Fujioka H; Huntley ML; Jiang Y; Ji H; Yan T; Harland M; Termsarasab P; Zeng S; Jiang Z; Liang J; Perry G; Hoppel C; Zhang C; Li H; Wang X Mitofusin 2 regulates axonal transport of calpastatin to prevent neuromuscular synaptic elimination in skeletal muscles. Cell Metab 2018, 28, 400–414.e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J; Moody JP; Edgerly CK; Bordiuk OL; Cormier K; Smith K; Beal MF; Ferrante RJ Mitochondrial loss, dysfunction and altered dynamics in huntington’s disease. Hum. Mol. Genet 2010, 19, 3919–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zorzano A; Sebastian D; Segales J; Palacin M The molecular machinery of mitochondrial fusion and fission: An opportunity for drug discovery? Curr Opin Drug Discov Devel 2009, 12, 597–606. [PubMed] [Google Scholar]
- 12.Miret-Casals L; Sebastián D; Brea J; Rico-Leo EM; Palacín M; Fernández-Salguero PM; Loza MI; Albericio F; Zorzano A Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism. Cell Chem Biol 2018, 25, 268–278.e264. [DOI] [PubMed] [Google Scholar]
- 13.Wang D; Wang J; Bonamy GM; Meeusen S; Brusch RG; Turk C; Yang P; Schultz PG A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl 2012, 51, 9302–9305. [DOI] [PubMed] [Google Scholar]
- 14.Franco A; Kitsis RN; Fleischer JA; Gavathiotis E; Kornfeld OS; Gong G; Biris N; Benz A; Qvit N; Donnelly SK; Chen Y; Mennerick S; Hodgson L; Mochly-Rosen D; Dorn GW II. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocha AG; Franco A; Krezel AM; Rumsey JM; Alberti JM; Knight WC; Biris N; Zacharioudakis E; Janetka JW; Baloh RH; Kitsis RN; Mochly-Rosen D; Townsend RR; Gavathiotis E; Dorn GW 2nd. Mfn2 agonists reverse mitochondrial defects in preclinical models of charcot-marie-tooth disease type 2a. Science 2018, 360, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang X; Zhang L; Franco A; Li J; Rocha AG; Devanathan S; Dolle RE; Bernstein PR; Dorn GW, 2nd. Discovery of 6-phenylhexanamide derivatives as potent stereoselective mitofusin activators for the treatment of mitochondrial diseases. J. Med. Chem 2020, 63, 7033–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franco A; Dang X; Walton EK; Ho JN; Zablocka B; Ly C; Miller TM; Baloh RH; Shy ME; Yoo AS; Dorn GW 2nd. Burst mitofusin activation reverses neuromuscular dysfunction in murine cmt2a. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan L; Huo P; Hale JJ; Mills SG; Hajdu R; Keohane CA; Rosenbach MJ; Milligan JA; Shei GJ; Chrebet G; Bergstrom J; Card D; Mandala SM Sar studies of 3-arylpropionic acids as potent and selective agonists of sphingosine-1-phosphate receptor-1 (s1p1) with enhanced pharmacokinetic properties. Bioorg. Med. Chem. Lett 2007, 17, 828–831. [DOI] [PubMed] [Google Scholar]
- 19.Thorarensen A; Wakefield BD; Romero DL; Marotti KR; Sweeney MT; Zurenko GE; Rohrer DC; Han F; Bryant GL Jr. Preparation of novel anthranilic acids as antibacterial agents. Extensive evaluation of alternative amide bioisosteres connecting the a- and the b-rings. Bioorg. Med. Chem. Lett 2007, 17, 2823–2827. [DOI] [PubMed] [Google Scholar]
- 20.Shiozaki M; Maeda K; Miura T; Ogoshi Y; Haas J; Fryer AM; Laird ER; Littmann NM; Andrews SW; Josey JA; Mimura T; Shinozaki Y; Yoshiuchi H; Inaba T Novel n-substituted 2-phenyl-1-sulfonylamino-cyclopropane carboxylates as selective adamts-5 (aggrecanase-2) inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 1575–1580. [DOI] [PubMed] [Google Scholar]
- 21.Talele TT The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 22.Poduslo JF; Curran GL; Berg CT Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc. Natl. Acad. Sci. U. S. A 1994, 91, 5705–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinhold AK; Rittner HL Characteristics of the nerve barrier and the blood dorsal root ganglion barrier in health and disease. Exp. Neurol 2020, 327, 113244. [DOI] [PubMed] [Google Scholar]
- 24.Heiman-Patterson TD; Deitch JS; Blankenhorn EP; Erwin KL; Perreault MJ; Alexander BK; Byers N; Toman I; Alexander GM Background and gender effects on survival in the tgn(sod1-g93a)1gur mouse model of als. J. Neurol. Sci 2005, 236, 1–7. [DOI] [PubMed] [Google Scholar]
- 25.Guyenet SJ; Furrer SA; Damian VM; Baughan TD; La Spada AR; Garden GA A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misko A; Jiang S; Wegorzewska I; Milbrandt J; Baloh RH Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the miro/milton complex. J. Neurosci 2010, 30, 4232–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruppa AJ; Buss F Motor proteins at the mitochondria-cytoskeleton interface. J. Cell Sci 2021, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorn GW 2nd. Mitofusins as mitochondrial anchors and tethers. J. Mol. Cell. Cardiol 2020, 142, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lacivita E; Niso M; Mastromarino M; Garcia Silva A; Resch C; Zeug A; Loza MI; Castro M; Ponimaskin E; Leopoldo M Knowledge-based design of long-chain arylpiperazine derivatives targeting multiple serotonin receptors as potential candidates for treatment of autism spectrum disorder. ACS Chem. Neurosci 2021, 12, 1313–1327. [DOI] [PubMed] [Google Scholar]
- 24.ldrick GM; SHELXTL-2014, Crystallographic Computing System; Bruker Analytical X-Ray Instruments: Madison, WI, 2014 [Google Scholar]
- 25.Sheldrick GM; A short history of SHELX; Acta. Cryst. A, 2008, A64, 112–122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.