

ABSTRACT
Long noncoding RNAs (lncRNAs) are an increasing focus of investigation due to their implications in diverse biological processes and disease. Nevertheless, the majority of lncRNAs are low in abundance and poorly conserved, posing challenges to functional studies. The CRISPR/Cas system, an innovative technology that has emerged over the last decade, can be utilized to further understand lncRNA function. The system targets specific DNA and/or RNA sequences via a guide RNA (gRNA) and Cas nuclease complex. We and others have utilized this technology in various applications such as lncRNA knockout, knockdown, overexpression, and imaging. In this review, we summarize how the CRISPR/Cas technology provides new tools to investigate the roles and therapeutic implications of lncRNAs.
KEYWORDS: Lncrna, lincRNA, genome editing, crispr/cas9
INTRODUCTION
As only approximately 2% of the human genome harbors protein-coding genes, there is increased interest in the roles of non-coding regions of the human genome [1]. Using the latest sequencing technologies, various regulatory non-coding RNAs have been identified. These non-coding RNAs (ncRNAs) include small ncRNAs, which are <200 nucleotides (nts) in length, such as miRNAs and piRNAs, and long non-coding RNAs (lncRNAs), which are >200 nts long [2]. Despite being expressed at low levels, being poorly conserved, and showing tissue-specific expression [2,3], lncRNAs have been shown to play important roles in diverse cellular processes such as controling transcription, acting as molecular decoys, guiding localization, and providing protein scaffolding [3]. lncRNAs can be localized to the nucleus and/or the cytoplasm, exerting effects on DNA, RNA, and/or proteins. Due to low abundance, tissue-specific expression, a wide range of cellular roles, and limited research techniques, studying lncRNAs may be complex and challenging.
Often, to interrogate the function of a novel lncRNA, changes are induced to their expression. However, widely used tools such as RNA Interference (RNAi) and antisense oligonucleotides (ASOs) are less effective in lncRNA studies [4–6]. RNAi uses a multi-protein RNA-induced silencing complex (RISC) to induce RNA degradation mediated by small interfering RNA (siRNA) binding to the target RNA [7]. Though useful in studies of cytoplasmic molecules, RNAi is usually less effective in suppressing nuclear lncRNA molecules due to the predominantly cytoplasmic localization of RISC in most cell types. ASOs function by binding to RNA then selectively degrading the ASO-RNA hybrids via the RNase H1 enzyme, which recognizes DNA-RNA hybrids. The use of ASOs relies on exogenous modified oligonucleotides to produce transient effects, making this technique complex. The most significant limitation to these techniques is the resulting degradation of the lncRNAs , limiting their use in mechanism of action studies while not yielding complete loss-of-function.
Beginning in the early 2000s the widespread use of genomic manipulation techniques, such as Zinc Finger Nucleases (ZFNs), changed the scope of molecular biology. Soon after, the use of Transcription Activator-Like Effector Nucleases (TALENs) followed. Almost 20 years later, these techniques remain popular, and now have been utilized in lncRNA studies. For example, Lee et al. (2016) were able to use the TALENs technology, utilizing homology-directed insertion, to insert a Lox-Stop-Lox cassette containing tandem polyadenylation signals into the NORAD locus, silencing expression of the lncRNA [8]. While these systems have accomplished advanced genetic manipulations of lncRNAs, they continue to present issues regarding time, cost, and flexibility, as well as challenges regarding protein engineering, synthesis, and validation [9].
A more powerful method for genetic manipulation of lncRNAs is the Clustered Regularly Interspersed Short Palindromic Repeat (CRISPR) system. The CRISPR system is a genomic editing technique that has emerged as a revolutionary tool in molecular biology due to its ease and flexibility. Because it relies primarily on the use of gRNAs (gRNAs) to direct the catalytic activity of the Cas enzymes [10,11], it is preferable to ZFNs or TALENs that are more laborious. The Cas enzyme creates a double-strand break (DSB) that is often repaired by non-homologous end joining (NHEJ). While the CRISPR system traditionally uses Cas9, it can also be utilized with other enzymes such as dCas9, Cas12, and Cas13, offering great flexibility. Catalytically dead Cas9 (dCas9) disrupts gene function by blocking transcription. Cas12 has two orthologs with known functions; Cas12a targets single-stranded DNA (ssDNA) [12] while Cas12b targets double-stranded DNA [13]. Cas13 selectively targets single-stranded RNA (ssRNA) and has been utilized to edit RNA, image RNA, and knockdown transcripts via RNA degradation [14,15].
Due to its versatility and flexibility, the CRISPR system is especially useful in studies of lncRNAs. The technique presents the ability to target nuclear or cytoplasmic lncRNAs, disrupt the genome to selectively interact with (or avoid) adjacent genetic material, and upregulate or downregulate transcription, all without degrading the lncRNA which allows for advanced mechanism of action studies. The adaptability of the various Cas enzymes presents opportunities to target the gene locus as well as the RNA transcript. By having this flexibility, studies can better address the highly specific and versatile roles of lncRNAs. Although many of these techniques are recent developments, if selected properly, the CRISPR/Cas system can greatly advance our understanding of lncRNA function. In this review, we highlight the various uses of the CRISPR/Cas system in studying lncRNA functions.
1. Targeting lncRNA genes with Cas9
The most basic use of CRISPR/Cas9 is utilizing a gRNA to induce a nonsense or frameshift mutation via a DSB and NHEJ. While an effective method for disrupting proper protein production in protein-coding genes, these mutations are rarely significant enough to inactivate a lncRNA gene. Due to poor evolutionary conservation, it is often unclear which region of a lncRNA is the active domain, preventing efficient selection of gRNA targets. If able to select a target site on the lncRNA gene, it is important to consider off-target effects as well as implications for adjacent genes. lncRNAs may be intergenic or intragenic. Intragenic lncRNAs may overlap with protein-coding genes or may be located within the introns of host genes (intronic lncRNA). lncRNA promoters may be found within adjacent genes or shared with nearby protein-coding genes [16]. The span of lncRNA promoters across the genome poses significant limitations on the use of CRISPR knockout systems because targeting a lncRNA may have significant impacts on nearby genes [16]. As a result of these limitations, new uses of the CRISPR system have emerged. A summary of some of these tools is shown in Table 1.
Table 1.
CRISPR/Cas systems used for lncRNA studies
| Editing System | Advantages | Disadvantages | Cas Variant | Approach | Example lncRNAs | Reference |
|---|---|---|---|---|---|---|
| CRISPR/Cas9 | Targeted deletion/mutation of gene to decrease lncRNA expression | Risk impacting the expression of adjacent or related genes | Cas9 | Deletion of whole lncRNA loci | PINCR, Gm26878 | (Chaudhary et al., 2017), (Szafranski et al., 2017) |
| Cas9 | Deletion of transcription factor binding sites | PURPL | (Li et al., 2017) | |||
| Cas9 | Deletion of large fragment of lncRNA locus | Rian | (Han et al., 2014) | |||
| Cas9 | Genome-wide screen via genomic deletion of lncRNA loci | - | (Zhu et al., 2016) | |||
| Cas9 | Genome-wide screen via deletion of lncRNA splice sites | - | (Liu et al., 2018) | |||
| CRISPRa | Overexpress lncRNA genes without permanent alteration of genome | Risk impacting the expression of adjacent genes | dCas9-VP64 | Overexpression | MALAT1, NEAT1 | (Xie et al., 2017), (Yamazaki et al., 2018), (Butler et al., 2019) |
| dCas9-VP64 | Genome-wide screen via CRISPR activation | - | (Bester et al., 2018) | |||
| CRISPRi | Repress lncRNA expression without permanent alteration of genome | Risk impacting the expression of adjacent genes | dCas9-KRAB | Knockdown | DIRC3, roX1, roX2 | (Coe et al., 2019), (Ghosh et al., 2016) |
| dCas9-KRAB, KRAB-dCas9 | Genome-wide screen via knockdown of lncRNAs | - | (Liu et al., 2017), (Cai et al., 2020) | |||
| CRISPR/Cas13 | Directly target lncRNA molecule | Transcription of lncRNA still occurs | Cas13d | Knockdown | HOTTIP, MALAT1 | (Konnerman et al., 2018) |
| Can only target ssRNA | dCas13b | Imaging | NEAT1, SatIII | (Yang et al., 2019) | ||
| Cas13a | Genome-wide knockdown of lncRNAs | - | (Xu et al., 2020) |
1.1. Gene editing with Cas9
One approach to produce an effective lncRNA knockout is to use two paired gRNAs, targeting the beginning and end of the lncRNA gene to excise the full lncRNA locus. It has been found that introducing multiple gRNAs increased the efficiency of this type of knockout [17,18]. Many studies attempt to excise a large fragment of a lncRNA loci rather than the full lncRNA [17,19,20]. For example, Han et al. (2014) deleted up to 23 kb of Rian, a 57.8 kb lncRNA gene, successfully decreasing Rian expression in the offspring of female mice using a Cas9-mediated deletion [17]. Zhu et al. (2016) used this technique for a high-throughput screen, screening over 700 lncRNAs that have oncogenic or tumour suppressive activity [19]. They also found that the paired gRNA system was more efficient when the two gRNAs are introduced in a single lentivirus vector under two separate U6 promoters [19]. Some studies have used this technique to delete full lncRNA loci successfully [5,21,22].
Alternately, Cas9 may be targeted to a transcription factor binding site or promoter of a lncRNA. It has been proposed that the recruitment of transcription factors during lncRNA transcription may impact the transcription of nearby genes [23]. By mutating the promoter of a lncRNA, one can elucidate the effects of lncRNA transcription on adjacent genes, rather than impacts of the lncRNA transcript itself on neighbouring genes [24]. Engreitz et al. (2016) performed a study in which they successfully knocked out promoters of 12 lncRNAs using 2–3 gRNAs to assay for effects on the expression of adjacent genes [25]. They found that in 5 of the 12 lncRNAs, knocking-out the lncRNA promoter impacted genes approximately 5–71 kb from the targeted region [25]. A similar approach has been used to determine the roles of lncRNAs, including UCA1, P14AS, and PURPL, by knocking out transcription factor binding sites in their promoters [26–28]. This type of knockout has been found to be very effective, as demonstrated by Zhen et al. (2017) when they found an 80% reduction in UCA1 expression with a gRNA targeting the UCA1 promoter [26]. When knocking out the promoter-like sequence upstream of P14AS, Ma et al. (2020) found reduced P14AS transcription as well as decreased downstream ANRIL expression [27].
Another location that can be targeted to study lncRNAs are splice sites within the lncRNA gene. Targeting the splice site with ASOs has previously been shown to be effective in knocking down lncRNA expression [29]. Meanwhile, the CRISPR/Cas9 system has been utilized to target pre-mRNA splice sites on the DNA sequence, leading to alternative splicing and gene knockdown through nonsense-mediated decay [30,31]. By combining these techniques, gRNAs can target Cas9 to lncRNA splice sites to produce effective intron retention or exon deletion [32]. Liu et al. (2018) used this technique on a genome-wide scale to screen for essential lncRNAs [32]. It is significant to note that later studies suggest that targeted deletions using this approach may not be as effective as previously suggested [33]. Targeting lncRNA splice sites with CRISPR/Cas9 can be used both in loss of function studies as well as lncRNA screens. Though not commonplace, this method offers a solution to issues associated with deleting a large portion of the DNA while producing an efficient knockout with minimal off-target effects.
Lastly, knock-in models can be utilized to study how lncRNAs function in cells. Due to the large size of lncRNAs, knocking-in a lncRNA is not practical. Studies have knocked-in tags to the lncRNA gene to visualize lncRNA localization, using a system coined CRISPR-mediated Endogenous lncRNA Tracking (CERTIS) [34]. Alternately, Lee et al. (2016), utilizing TALENS, knocked-in a transcriptional stop element to inactivate the NORAD locus [8]. Yin et al. (2015) knocked-in a polyA stop cassette, producing a 55% decrease in Haunt1 lncRNA transcripts using CRISPR/Cas9 [18].
1.2. Challenges of Gene editing with Cas9
The major limitation to excising large fragments of lncRNAs using two gRNAs is the significant risk of impacting adjacent genes, such as those that may overlap with the lncRNA gene, those that are regulated by transcription of the lncRNA, or those with DNA elements in the lncRNA locus that may regulate other genes [35]. While targeting Cas9 to transcription factor binding sites, promoters, or splice sites minimizes the risk of deleting otherwise important genetic material, these techniques present issues of their own. When manipulating a lncRNA promoter there is still a possibility of impacting other genes, such as those regulated by the same promoter as the lncRNA or genes that may overlap with the targeted promoter. Regarding knock-ins, these types of studies require extensive research to determine potential off-target effects. The selection of the location where the knock-in will occur is crucial. Specific to lncRNAs, though, the bidirectional relationship that a lncRNA may have with adjacent genes must be considered when determining where to perform a knock-in. Overall, manipulations to the DNA encoding a lncRNA present issues in regard to disrupting surrounding genetic material due to the large size of lncRNAs as well as their overlap and interactions with other genomic elements.
1.3. Silencing or overexpression
Rather than permanently altering the DNA in a knockout or knock-in model, as described in the previous section, the CRISPR interference (CRISPRi) tool offers a method to repress gene expression without permanently mutating the genome. CRISPRi utilizes dCas9 and a gRNA to block RNA Polymerase II transcription, reversibly repressing the functional activity of the gene of interest [36,37]. This technology has been used in lncRNA screens [38,39] as well as silencing studies [40–42]. To further enhance gene repression, transcription repressors such as KRAB may be fused to the dCas9-gRNA system [43]. Despite the widespread use of KRAB in CRISPRi studies, it has been found that alternate transcription repressors, such as the SIN3-interacting domains derived from the MXD1 protein, may be more effective than KRAB in CRISPRi studies of lncRNAs [44].
After the invention of CRISPRi, CRISPR activation (CRISPRa) was developed utilizing dCas9 fused to RNA-polymerase recruiting factors [45]. Transcription activators such as VP64, p65, and Rta are commonly used to induce gene expression [16]. CRISPRa can be used in screens as well as overexpression studies [46]. This technique has been used to elucidate the functions of lncRNAs, such as MALAT1 and NEAT1 [47–49].
1.4. Challenges of the CRISPRi/CRISPRa system
CRISPRi and CRISPRa have similar advantages and disadvantages. First, by not disrupting the DNA sequence or causing any permanent alterations, these methods are less likely to impact neighbouring genes permanently. Nonetheless, the recruitment of transcription activators and repressors may cause changes in expression levels of adjacent genes. For example, when overexpressing lncRNA RP11-326A19.4 in HEK293T cells, Soubeyrand et al. (2019) saw increased expression of cytokine IL6, suggesting that CRISPRa has unpredictable off-target effects [50]. By utilizing transcription repressors or activators, the effects of lncRNA expression on nearby genes can be observed. This is significant as transcription of a lncRNA itself may regulate some adjacent genes. The CRISPRa system induces overexpression of the lncRNA isoforms in the ratio that they are naturally spliced, rather than a knock-in model that may only induce expression of specific isoforms. CRISPRi and CRISPRa may interact with nearby promoters, causing confounding results, a limitation that should be considered if the promoter of the lncRNA of interest is within another gene.
2. Targeting lncRNA molecules with Cas13
Recently, there has been an increase in the use of CRISPR/Cas13 to target RNA. The CRISPR/Cas13 system employs Cas13, an RNase which assembles with CRISPR RNA (crRNA), to form a programmable RNA targeting system. The Cas13 proteins contain two Higher Eukaryotes and Prokaryotes Nucleotide-binding (HEPN) domains with endogenous RNase activity [51]. Presently, there are four known orthologs of the Cas13 protein (Cas13a, Cas13b, Cas13c, and Cas13d), each exhibiting different levels of activity in human cell lines [52]. This system has been utilized mainly in lncRNA knockdown [15,53] and imaging systems [54], although it has the potential to be used in a range of studies focusing on lncRNA function.
2.1. lncRNA knockdown with Cas13
Prior methods of lncRNA knockdown, such as RNAi or ASOs, have the potential to induce significant off-target effects [55–58]. Even CRISPRi can produce non-negligible off-target effects, inducing unintentional transcriptional changes in single-cell clones [59]. The CRISPR/Cas13 approach has been shown to have similar knockdown efficiency, as compared to RNAi, with fewer non-specific effects [60]. Because Cas13 can localize throughout the cell, it has the potential to knockdown both nuclear and cytoplasmic lncRNAs. Using CasRx, a Cas13d ortholog, Konermann et al. (2018) showed that the system allowed for specific, efficient knockdown of lncRNAs in human cell lines [15]. Xu et al. (2020) have also utilized the CRISPR/Cas13 system to perform a high-throughput screen with successful knockdown of very long intergenic non-coding (vlinc) RNAs [53]. This finding suggests that this system can be utilized for phenotypic assays of lncRNAs throughout the cell.
2.2. Challenges of the CRISPR/Cas13 System
Although the CRISPR/Cas13 system provides a robust method to target specific RNAs, it has some limitations. The system utilizes various orthologs of Cas13, each with limitations in their knockdown efficiency. This places restrictions on target locations and sgRNA design for certain model organisms depending on the ortholog. In vitro and bacterial studies using Cas13a have found that the system preferentially targets regions with Protospacer Flanking Sequences (PFSs) [61]. The Cas13b system has also been shown to display a preference for PFSs in bacterial cells [62,63], although the findings regarding PFS requirements have been contradictory in mammalian cells [64]. The secondary structure of the target lncRNA also limits the ability of the CRISPR/Cas13 system. Because Cas13 can only target ssRNA, the system has the potential to function well for lncRNAs without high-order structures [61]. Nevertheless, many lncRNAs do contain regions of high-order or double-stranded structures, complicating sgRNA design [64]. The challenge can be overcome by studying the structure(s) of the lncRNA(s) of interest. Bandaru et al. (2020) developed a workflow that uses Structure-Seq data to strategically determine appropriate sgRNA targets within single-stranded region(s) of a lncRNA, and Wessels et al. (2020) developed a program that predicts gRNA targets for Cas13 [64,65]. Even though there are challenges, the CRISPR/Cas13 system is a powerful and specific tool for knocking down or visualizing lncRNAs.
3. Functional characterization of lncRNAs using CRISPR
By properly selecting and executing the various CRISPR techniques, one can perform highly nuanced manipulations of lncRNAs beyond traditional knockouts, knockdowns, and knock-ins.
3.1. lncRNA imaging
Previously, the MS2-Capsid Protein (MS2-MCP) system has been used to visualize RNA [66]. However, this system uses aptamers that have the potential to disrupt RNA structure, expression, and function [54]. Wang et al. (2019) developed a system, entitled CRISPR live-cell fluorescent in situ hybridization (CRISPR LiveFISH) in which oligonucleotides bound to a fluorophore were used in combination with both dCas9 and dCas13 to visualize DNA and RNA in live cells [67]. While the system has not been tested in studies of lncRNA, it offers great promise for future studies. Meanwhile, Yang et al. (2019) visualized lncRNAs using a live-cell imaging method that uses a fluorescently labelled catalytically inactive Cas13 (dCas13) protein to visualize both nuclear and cytoplasmic RNAs [54]. By assaying for functional dCas13 orthologs, they determined that dPspCas13b and dPguCas13b were efficient dCas13 proteins that can label lncRNAs in mammalian cells [54]. This system was used to effectively determine the localization of the nuclear lncRNA, NEAT1. Dual-colour imaging was also performed using two dCas13 proteins to visualize two nuclear lncRNAs, NEAT1 and SatIII, concurrently. This technique allows for dual lncRNA tracking, which has the potential to be expanded to visualize lncRNAs in any cell compartment. The system may also be paired with dCas9 to visualize DNA and RNA simultaneously using a dual-colour system.
3.2. Determining if a lncRNA is cis- or trans-acting
In investigating the function of a lncRNA, it is important to identify its location within the cell. To determine the cellular localization of a lncRNA, subcellular isolation or dCas13 imaging can be performed as previously described [54]. Once the localization of a lncRNA of interest has been identified, one can determine whether the lncRNA is cis-acting or trans-acting. A cis-acting lncRNA regulates the expression and/or chromatin states of surrounding genes, while a trans-acting lncRNA regulates gene expression at a distance [68,69]. To determine whether a lncRNA acts in trans, a rescue experiment can be performed in CRISPR/Cas9 knockout cells. If there is a reversal of the knockout phenotype following the rescue experiment, then the lncRNA likely acts in trans [68]. lncRNA localization in the cytoplasm and/or nucleoplasm further supports that the lncRNA is trans-acting [69]. Nevertheless, it is important to note that if a lncRNA predominantly localizes to the cytoplasm, its coding potential should be interrogated (reviewed in [70]).
Investigating cis-acting lncRNAs may pose challenges due to the ambiguity between the function of the lncRNA and the locus from which it is transcribed [68]. However, the CRISPR/Cas system can provide insight into cis-acting lncRNA function. If the transcription of a lncRNA occurs at a singular promoter, CRISPR/Cas9 knockout of a small region within the first exon of the lncRNA can be performed, and, sequentially, the expression of nearby genes can be determined [68]. A major confound of this type of study occurs due to the direct manipulation of genomic DNA by the CRISPR/Cas9 system. The resultant phenotype may be due to alterations to the mature RNA or the lack of lncRNA transcription completely. A great example of this phenomenon is shown by the investigation of lincRNA-p21. After studying the expression of p21 in lincRNA-p21−/- mouse embryonic fibroblasts (MEFs), lincRNA-p21 was classified as cis-acting because it was found to activate the expression of p21, a neighbouring gene [71]. Alternatively, a study by Groff et al. (2016) suggests that it is not the expression of lincRNA-p21 that mediates its role in the p53 signally pathway, but the DNA enhancer elements that are located within the lincRNA-p21 locus [35]. To better elucidate the function of lincRNA-p21, CRISPRi or CRISPRa techniques could be utilized to further study the effects of cis activation or the enhancer regions of the locus [72–74]. However, these methods may introduce unintentional changes in the chromatin environment which may be falsely attributed to the alterations in lncRNA transcription. Additionally, the lncRNA can be knocked down using CRISPR/Cas13, although the consequences of lncRNA transcription may be indistinguishable from the phenotypes observed as a result of the knockdown.
An alternate method to study lncRNA cis-effects is through tethering the transcript to neighbouring genes. Luo et al. (2016) utilized dCas9 and a gRNA fused to the lncRNA of interest to guide and tether the complex to a nearby locus, determining if the lncRNA regulated the expression of the targeted gene [75]. Another method for investigating lncRNA function is CRISPR-Display (CRISP-Disp) [76,77]. Shechner et al. (2015) engineered this technique to target large lncRNA cargos to endogenous loci [76]. This system uses gRNAs with integrated functional lncRNA domains and a dCas9 molecule fused to VP64 [77]. Shechner et al. (2015) found that the complexes were able to induce significant activation of endogenous loci. This system has the potential to be used for additional applications such as endogenous and fusion protein recruitment as well as affinity tagging [76]. Both dCas9 tethering and CRISPR-Disp can be used to elucidate the functions of cis-acting lncRNAs.
3.3. Determining potential domains within a lncRNA
Determining the structure of a lncRNA can elucidate its potential functions and aid in gRNA design. Many lncRNAs have complex secondary and tertiary structures that provide interfaces for protein, RNA, or DNA interactions [78]. Unlike many protein-coding genes, many lncRNAs are low in abundance [79] and poorly conserved [80]. This poses challenges to the identification of functional domains within lncRNAs. Furthering complicating matters, RNA-binding proteins may bind to small, conserved regions along a lncRNA transcript, making it difficult to distinguish whether the lncRNA or the protein are mediating the observed phenotypes. CRISPR-Disp can be used to analyse these domains. Using this system, uncharacterized lncRNA domains can be targeted to loci of interest to investigate their potential roles. Tiling CRISPR can also be used to examine functional lncRNA domains. Wang et al. (2019) developed this novel CRISPR/Cas9 approach to characterize the functional domains of Xist, a lncRNA involved in X-chromosome inactivation [81,82]. By introducing over 1,500 gRNAs, tiling the entire Xist transgene, the authors probed for clustered mutations from overlapping gRNAs, indicating a functional domain and successfully identifying essential regions of the gene [81]. Screens that utilize the Tiling CRISPR technology will have the potential to characterize functional domains of lncRNAs.
3.4. In vivo studies
One of the most comprehensive methods of determining the function of a lncRNA is studying its role in animal models. Traditional in vivo studies use the Cre-LoxP system, in which tissue-specific Cre recombinase (Cre) performs targeted deletions of genomic DNA between two LoxP sites [83]. Although this system does provide temporal and tissue-specific control, it can be a costly and time-consuming process [84]. The CRISPR/Cas9 system offers an alternate method of genetic modification in organisms. As previously described, this has been achieved using pairs of gRNAs to precisely delete up to 23kb of a lncRNA locus in a murine model [17]. Silencing lncRNA expression has also been performed by inserting a polyA cassette within an exon via the CRISPR/Cas9 system to promote cleavage of the transcript and terminate transcription [85]. Additionally, CRISPRa has been developed to activate lncRNA loci in vivo using a combination of the piggyBac transposon and Cre-inducible dCas9 systems [86].
Non-specific gRNA targets must be taken into account when creating in vivo systems to study lncRNAs [87]. When whole genome sequencing was performed on mice edited with the CRISPR/Cas9 system, a surprising number of single nucleotide variants were identified [87]. Although the system can induce significant off-target mutations, a recent study developed a method that provides verification of in vivo off-targets (VIVO) [88]. The VIVO workflow consists of two steps: in vitro reporting of cleavage effects by sequencing to identify potential off-target cleavage sites and off-target mutation validation in the CRISPR/Cas9-edited target tissues by deep sequencing [88]. This method can be combined with in silico techniques, which design gRNAs with the lowest number of similar genomic sites, to assess the accuracy of gRNAs [88]. This emerging technology shows promise for our future ability to perform functional studies of lncRNAs in animal models by minimizing potential off-target effects.
3.5. CRISPR library screens
CRISPR library screens introduce a pool of gRNAs into a cell population, knocking out (as occurs in the traditional case of Cas9) various genes in order to identify how genes may be enriched or depleted in response to a selective pressure. CRISPR library screens can be done in vitro and in vivo, using paired gRNAs to knockout lncRNAs or single gRNAs to target splice sites [19,32,89,90]. Screens have also utilized both the CRISPRi and CRISPRa systems [89,91]. These studies offer opportunities to identify novel lncRNA roles and interactions in an unbiased and genome-wide scale, elucidating both cytoplasmic and nuclear interactions. However, genome-wide lncRNA screens can be difficult due to the size of lncRNAs and inability of point mutations to yield the same inactivations as screens for protein-coding genes.
CONCLUSIONS
Despite low expression [79] and poor conservation [80], the CRISPR/Cas system provides innovative ways to investigate the potential roles of lncRNAs through knockout, knockdown, overexpression, and imaging approaches (Fig. 1). In addition to many of the techniques mentioned in this review, various other advancements are posed to greatly increase our knowledge of both RNAs and lncRNAs. The catalytically inactive CasRx (dCasRx) system has been employed in the manipulation of RNA splicing [15]. Because lncRNAs undergo splicing similar to mRNAs [92], this system can be used to perform splice isoform engineering on these transcripts. Additionally, dCas13 has been shown to effectively edit RNA when fused with adenosine deaminase acting on RNA type 2 (ADAR2) enzymes [63]. This system, termed RNA Editing for Programmable A to I Replacement (REPAIR), is not limited by sequence restrictions and has the potential to support lncRNA editing. A similar system entitled RESCUE (RNA Editing for Specific C-to-U Exchange) expands upon the REPAIR system, allowing for RNA editing of A to I and C to U [93]. The CRISPR/Cas13 systems have also been engineered to perform sensitive RNA detection through methods such as Specific High-Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK) [94]. These detection methods can likely be utilized in lncRNA studies due to the programmable nature of the system. Gao et. al (2021) were able to manipulate the CRISPR system to reliably quantify low-copy-number lncRNA expression levels using an endogenous transcription-gated switch with a CRISPR-activator-associated reporter [95]. The CRISPR/Cas9 technology may also be useful in manipulating the epigenetic regulation of lncRNA genes by utilizing dCas9 proteins fused with the catalytic domain of acetyltransferase [96]. This would allow for the controlled transactivation of various lncRNA loci, thus providing another systematic method to interrogate lncRNA functions.
Figure 1.
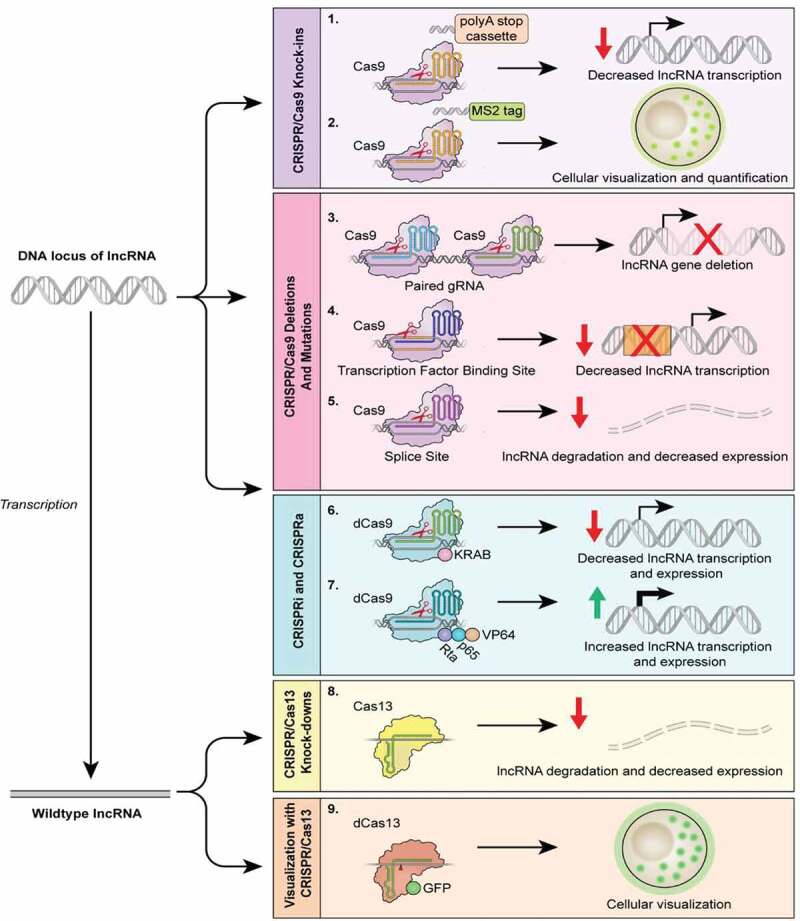
CRISPR uses in studies of lncRNAs. The CRISPR/Cas9 technology can be used to target various regions within a lncRNA gene, manipulating the lncRNA expression to gain functional insights. The CRISPR/Cas9 technology can be used to produce knock-in phenotypes (1,2), inserting a polyA stop cassette downstream of the transcription start site to decrease transcription or to encode a MS2 tag, as used in the CERTIS system, to localize lncRNAs. Additionally, the Cas9-gRNA complex can be directed to the beginning and end of the locus (3), splice sites (5), as well as transcription factor binding sites within the promoter region (4). These techniques will likely result in the partial or complete reduction of lncRNA levels within a cell. CRISPR activation, CRISPRa (7), and CRISPR interference, CRISPRi (6), can be used to overexpress or silence lncRNA expression via catalytically dead Cas9 (dCas9) without permanent genomic editing. CRISPRa employs a dCas9-gRNA complex fused with transcription activators (i.e. VP64, p65, and Rta) to robustly activate the transcription of the lncRNA gene. Conversely, the transcription of a lncRNA gene can be inhibited by CRISPRi, which utilizes a dCas9-gRNA complex fused with KRAB, a transcription repressor. Various orthologs of Cas13 can be utilized in lncRNA knockdown (8) in which a gRNA targets catalytically active Cas13 to single-stranded lncRNAs, leading to degradation of the molecule. Cas13 can also be used to visualize lncRNAs (9) by fusing dCas13 to a fluorescent protein
In addition to these incredible opportunities the CRISPR system offers to further our understanding of lncRNAs, with futher research and development, the CRISPR system could theoretically be clinically utilized to manipulate lncRNAs in a therapeutic context. CRISPR has corrected point mutations in vivo in mice and has been used ex vivo in the treatment of cancers, suggesting that CRISPR could be used to treat disease in vivo in the future [97–99]. Because lncRNAs are lowly-expressed and often tissue- and cell-specific, they could be good options as therapeutic targets. The major concern when considering CRISPR use in patients is off-target effects, though this may be less of an issue when targeting lncRNA compared to protein-coding genes. In addition to this serious concern, studying in vivo targeting of lncRNAs is complex due to the lack of conservation between species. Lastly, the prospect of using CRISPR to target lncRNAs therapeutically does present ethical concerns that need to be addressed prior to clinical use of this technology. From manipulating the traditional Cas9 knockout to utilizing inventive approaches such as CRISPR-Disp, the CRISPR/Cas system provides endless opportunities for innovative studies regarding the function on lncRNAs.
Acknowledgments
This research was supported by the Intramural Research Program (M.S.Z., C.C.R.H., and A.L.) of the National Cancer Institute (NCI), Center for Cancer Research (CCR), NIH. We thank Yannis Grammatikakis from the Lal lab (NCI, NIH) for his comments on the manuscript. We also thank the Medical Arts Branch of the NIH for their assistance. We apologize to those whose work we were unable to cite due to space limitations.
Funding Statement
This work was supported by the National Cancer Institute [ZIA BC 011646].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1]. Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature 2001;409(6822):860-921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- [2].Charles Richard JL, Eichhorn PJA.. Platforms for Investigating LncRNA Functions. SLAS Technol. 2018;23(6):493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang KC, Chang HY.. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zong X, Huang L, Tripathi V,et al. Knockdown of nuclear-retained long noncoding RNAs using modified DNA antisense oligonucleotides, in nuclear bodies and noncoding RNAs: methods and protocols. Nakagawa S, Hirose T. Editors 2015.Springer New York: New York, NY. Vol. 1262:321–331. [DOI] [PubMed] [Google Scholar]
- [5].Hao Q, Zong X, Sun Q, et al. The S-phase-induced lncRNA SUNO1 promotes cell proliferation by controlling YAP1/Hippo signaling pathway. Elife; 2020;9:e55102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sun Q, Hao Q, Lin YC, et al. Antagonism between splicing and microprocessor complex dictates the serum-induced processing of lnc-MIRHG for efficient cell cycle reentry. RNA. 2020;26(11):1603–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. [DOI] [PubMed] [Google Scholar]
- [8].Lee S, Kopp F, Chang TC, et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016;164(1–2):69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. [DOI] [PubMed] [Google Scholar]
- [10].Lino CA, Harper JC, Carney JP, et al. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 2018;25(1):1234–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jansen R, Embden JD, Gaastra W, et al. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43(6):1565–1575. [DOI] [PubMed] [Google Scholar]
- [12].Leroy E, Calvo CF, Divine M, et al. Persistence of T8+/HNK-1+ suppressor lymphocytes in the blood of long-term surviving patients after allogeneic bone marrow transplantation. J Immunol. 1986;137(7):2180–2189. [PubMed] [Google Scholar]
- [13].Strecker J, Jones S, Koopal B, et al. Engineering of CRISPR-Cas12b for human genome editing. Nat Commun. 2019;10(1):212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yan F, Wang W, Zhang J. CRISPR-Cas12 and Cas13: the lesser known siblings of CRISPR-Cas9. Cell Biol Toxicol. 2019;35(6):489–492. [DOI] [PubMed] [Google Scholar]
- [15].Konermann S, Lotfy P, Brideau NJ, et al. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018;173(3):665–676. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Goyal A, Myacheva K, Gross M, et al. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017;45(3):e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Han J, Zhang J, Chen L, et al. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014;11(7):829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yin Y, Yan P, Lu J, et al. Opposing roles for the lncrna haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell. 2015;16(5):504–516. [DOI] [PubMed] [Google Scholar]
- [19].Zhu S, Li W, Liu J, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol. 2016;34(12):1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zare K, Shademan M, Ghahramani Seno MM, et al. CRISPR/Cas9 KNOCKOUT strategies to ablate CCAT1 lncRNA gene in cancer cells. Biol Proced Online. 2018;20:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Szafranski P, Karolak JA, Lanza D, et al. CRISPR/Cas9-mediated deletion of lncRNA Gm26878 in the distant Foxf1 enhancer region. Mamm Genome. 2017;28(7–8):275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chaudhary R, Gryder B, Woods WS, et al. Prosurvival long noncoding RNA PINCR regulates a subset of p53 targets in human colorectal cancer cells by binding to Matrin 3. Elife; 2017;6:e23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Long Y, Wang X, Youmans DT, Cech TR. How do lncRNAs regulate transcription? Sci Adv. 2017;3(9):eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ebisuya M, Yamamoto T, Nakajima M, et al. Ripples from neighbouring transcription. Nat Cell Biol. 2008;10(9):1106–1113. [DOI] [PubMed] [Google Scholar]
- [25].Engreitz JM, Haines JE, Perez EM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539(7629):452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhen S, Hua L, Liu YH, et al. Inhibition of long non-coding RNA UCA1 by CRISPR/Cas9 attenuated malignant phenotypes of bladder cancer. Oncotarget. 2017;8(6):9634–9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma W, Qiao J, Zhou J, et al. Characterization of novel LncRNA P14AS as a protector of ANRIL through AUF1 binding in human cells. Mol Cancer. 2020;19(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li XL, Subramanian M, Jones MF, et al. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep. 2017;20(10):2408–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ulitsky I, Shkumatava A, Jan CH, et al. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147(7):1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tang JX, Chen D, Deng SL, et al. CRISPR/Cas9-mediated genome editing induces gene knockdown by altering the pre-mRNA splicing in mice. BMC Biotechnol. 2018;18(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Garcia-Tunon I, Alonso-Perez V, Vuelta E, et al. Splice donor site sgRNAs enhance CRISPR/Cas9-mediated knockout efficiency. PLoS One. 2019;14(5):e0216674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu Y, Cao Z, Wang Y, et al. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat Biotechnol. 2018;36(12):1203–1210. [DOI] [PubMed] [Google Scholar]
- [33].Horlbeck MA, Liu SJ, Chang HY, et al. Fitness effects of CRISPR/Cas9-targeting of long noncoding RNA genes. Nat Biotechnol. 2020;38(5):573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen B, Deng S, Ge T, et al. Live cell imaging and proteomic profiling of endogenous NEAT1 lncRNA by CRISPR/Cas9-mediated knock-in. Protein Cell. 2020;11(9):641–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Groff AF, Sanchez-Gomez DB, Soruco MML, et al. In Vivo Characterization of Linc-p21 Reveals Functional cis-Regulatory DNA Elements. Cell Rep. 2016;16(8):2178–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Larson MH, Gilbert LA, Wang X. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8(11):2180–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu SJ, Horlbeck MA, Cho SW, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355(6320):eaah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cai P, Otten ABC, Cheng B, et al. A genome-wide long noncoding RNA CRISPRi screen identifies PRANCR as a novel regulator of epidermal homeostasis. Genome Res. 2020;30(1):22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cho SW, Xu J, Sun R, et al. Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element. Cell. 2018;173(6):1398–1412. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Coe EA, Tan JY, Shapiro M, et al. The MITF-SOX10 regulated long non-coding RNA DIRC3 is a melanoma tumour suppressor. PLoS Genet. 2019;15(12):e1008501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ghosh S, Tibbit C, Liu JL. Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic Acids Res. 2016;44(9):e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].MacLeod RS, Cawley KM, Gubrij I, et al. Effective CRISPR interference of an endogenous gene via a single transgene in mice. Sci Rep. 2019;9(1):17312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Raffeiner P, Hart JR, Garcia-Caballero D, et al. An MXD1-derived repressor peptide identifies noncoding mediators of MYC-driven cell proliferation. Proc Natl Acad Sci U S A. 2020;117(12):6571–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tanenbaum ME, Gilbert LA, Qi LS, et al. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159(3):635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bester AC, Lee JD, Chavez A, et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell. 2018;173(3):649–664. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xie H, Liao X, Chen Z, et al. LncRNA MALAT1 inhibits apoptosis and promotes invasion by antagonizing miR-125b in bladder cancer cells. J Cancer. 2017;8(18):3803–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Butler AA, Johnston DR, Kaur S, et al. Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci Signal. 2019;12(588):eaaw9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yamazaki T, Fujikawa C, Kubota A, et al. CRISPRa-mediated NEAT1 lncRNA upregulation induces formation of intact paraspeckles. Biochem Biophys Res Commun. 2018;504(1):218–224. [DOI] [PubMed] [Google Scholar]
- [50].Soubeyrand S, Lau P, Peters V, et al. Off-target effects of CRISPRa on interleukin-6 expression. PLoS One. 2019;14(10):e0224113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Grynberg M, Erlandsen H, Godzik A. HEPN: a common domain in bacterial drug resistance and human neurodegenerative proteins. Trends Biochem Sci. 2003;28(5):224–226. [DOI] [PubMed] [Google Scholar]
- [52].O’Connell MR. Molecular mechanisms of RNA targeting by cas13-containing type VI CRISPR-cas systems. J Mol Biol. 2019;431(1):66–87. [DOI] [PubMed] [Google Scholar]
- [53].Xu D, Cai Y, Tang L, et al. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci Rep. 2020;10(1):1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang LZ, Wang Y, Li SQ, et al. Dynamic imaging of rna in living cells by CRISPR-Cas13 systems. Mol Cell. 2019;76(6):981–997. e7. [DOI] [PubMed] [Google Scholar]
- [55].Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. [DOI] [PubMed] [Google Scholar]
- [56].Persengiev SP, Zhu X, Green MR. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). Rna. 2004;10(1):12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yoshida T, Naito Y, Yasuhara H, et al. Evaluation of off-target effects of gapmer antisense oligonucleotides using human cells. Genes Cells. 2019;12:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253–1261. [DOI] [PubMed] [Google Scholar]
- [59].Stojic L, Lun ATL, Mangei J, et al. Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res. 2018;12:5950–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Abudayyeh OO, Gootenberg JS, Konermann S, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353(6299):aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Smargon AA, Cox DBT, Pyzocha NK, et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell. 2017;65(4):618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cox DBT, Gootenberg JS, Abudayyeh OO, et al. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bandaru S, Tsuji MH, Shimizu Y, et al. Structure-based design of gRNA for Cas13. Sci Rep. 2020;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wessels HH, Mendez-Mancilla A, Guo X, et al. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat Biotechnol. 2020;38(6):722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Vera M, Tutucci E, Singer RH. Imaging single mRNA molecules in mammalian cells using an optimized MS2-MCP system. Methods Mol Biol. 2019;2038:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang H, Nakamura M, Abbott TR, et al. CRISPR-mediated live imaging of genome editing and transcription. Science. 2019;365(6459):1301–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bassett AR, Akhtar A, Barlow DP, et al. Considerations when investigating lncRNA function in vivo. eLife. 2014;3:e03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hartford CCR, Lal A. When long noncoding becomes protein coding. Mol Cell Biol. 2020;40(6):e00528–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dimitrova N, Zamudio JR, Jong RM, et al. LincRNA-p21 activates p21 in cis to promote polycomb target gene expression and to enforce the G1/S checkpoint. Mol Cell. 2014;54(5):777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gil N, Ulitsky I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet. 2020;21(2):102–117. [DOI] [PubMed] [Google Scholar]
- [73].Fulco CP, Munschauer M, Anyoha R, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354(6313):769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Simeonov DR, Gowen BG, Boontanrart M, et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. 2017;549(7670):111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Luo S, Lu JY, Liu L, et al. Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell. 2016;18(5):637–652. [DOI] [PubMed] [Google Scholar]
- [76].Shechner DM, Hacisuleyman E, Younger ST, et al. Multiplexable, locus-specific targeting of long RNAs with CRISPR-display. Nat Methods. 2015;12:664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Perez-Pinera P, Jones MF, Lal A, et al. Putting non-coding RNA on display with CRISPR. Mol Cell. 2015;59(2):146–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zampetaki A, Albrecht A, Steinhofel K. Long non-coding RNA structure and function: is there a link? Front Physiol. 2018;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cabili MN, Dunagin MC, McClanahan PD, et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015;16(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Pang KC, Frith MC, Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 2006;22(1):1–5. [DOI] [PubMed] [Google Scholar]
- [81].Wang Y, Zhong Y, Zhou Y, et al. Identification of a Xist silencing domain by Tiling CRISPR. Sci Rep. 2019;9(2408). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Brown CJ, Ballabio A, Rupert JL, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349(6304):38–44. [DOI] [PubMed] [Google Scholar]
- [83].Kim H, Kim M, Im S-K, et al. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab Anim Res. 2018;34(4):147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huang J, Chen M, Whitley MJ, et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat Commun. 2017;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ballarino M, Cipriano A, Tita R, et al. Deficiency in the nuclear long noncoding RNA Charme causes myogenic defects and heart remodeling in mice. Embo J. 2018;37(18):e99697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhou H, Liu J, Zhou C, et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat Neurosci. 2018;21:440–446. [DOI] [PubMed] [Google Scholar]
- [87].Schaefer KA, Wu WH, Colgan DF, et al. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat Methods. 2017;14:547–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Akcakaya P, Bobbin ML, Guo JA, et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Liu S, Harmston N, Glaser TL, et al. Wnt-regulated lncRNA discovery enhanced by in vivo identification and CRISPRi functional validation. Genome Med. 2020;12(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Esposito R, Bosch N, Lanzos A, et al. Hacking the cancer genome: profiling therapeutically actionable long non-coding RNAs using CRISPR-Cas9 screening. Cancer Cell. 2019;35(4):545–557. [DOI] [PubMed] [Google Scholar]
- [91].Joung J, Engreitz JM, Konermann S, et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548(7667):343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. [DOI] [PubMed] [Google Scholar]
- [93].Abudayyeh OO, Gootenberg JS, Franklin B, et al. A cytosine deaminase for programmable single-base RNA editing. Science. 2019;365(6451):382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gootenberg JS, Abudayyeh OO, Lee JW, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017; 356(6336):438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gao N, Hu J, He B, et al. Endogenous promoter-driven sgRNA for monitoring the expression of low-abundance transcripts and lncRNAs. Nat Cell Biol. 2021;23(1):99–108. [DOI] [PubMed] [Google Scholar]
- [96].Hilton IB, D'Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33(5):510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32(6):551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Koblan LW, Erdos MR, Wilson C, et al. In vivo base editing rescues hutchinson-gilford progeria syndrome in mice. Nature. 2021;589(7843):608–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]