

ABSTRACT
Macroautophagy/autophagy is known to be important for intracellular quality control in the lens. GJA8 is a major gap junction protein in vertebrate lenses. Mutations in GJA8 cause cataracts in humans. The well-known cataractogenesis mechanism is that mutated GJA8 leads to abnormal assembly of gap junctions, resulting in defects in intercellular communication among lens cells. In this study, we observed that ablation of Gja8b (a homolog of mammalian GJA8) in zebrafish led to severe defects in organelle degradation, an important cause of cataractogenesis in developing lens. The role of autophagy in organelle degradation in lens remains disputable. Intriguingly, we also observed that ablation of Gja8b induced deficient autophagy in the lens. More importantly, in vivo treatment of zebrafish with rapamycin, an autophagy activator that inhibits MAPK/JNK and MTORC1 signaling, stimulated autophagy in the lens and relieved the defects in organelle degradation, resulting in the mitigation of cataracts in gja8b mutant zebrafish. Conversely, inhibition of autophagy by treatment with the chemical reagent 3-MA blocked these recovery effects, suggesting the important roles of autophagy in organelle degradation in the lens in gja8b mutant zebrafish. Further studies in HLE cells revealed that GJA8 interacted with ATG proteins. Overexpression of GJA8 stimulated autophagy in HLE cells. These data suggest an unrecognized cataractogenesis mechanism caused by ablation of Gja8b and a potential treatment for cataracts by stimulating autophagy in the lens.
Abbreviations: 3-MA: 3-methyladenine; ATG: autophagy related; AV: autophagic vacuoles; Dpf: days post fertilization; GJA1: gap junction protein alpha 1; GJA3: gap junction protein alpha 3; GJA8: gap junction protein alpha 8; Hpf: hours post fertilization; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; PtdIns3K: class III phosphatidylinositol 3-kinase; WT: wild type.
KEYWORDS: 3-MA, autophagy, cataract, GJA8, Gja8b, lens, organelle degradation, rapamycin
Introduction
Autophagy plays an essential role in tissue development and cellular homeostasis maintenance [1,2]. Dysregulation of autophagy is implicated in many diseases, including cancer, neurodegeneration and ocular diseases such as cataracts, which is a leading cause of blindness worldwide [2–5]. A cataract is an opacity in the lens. During the vertebrate lens placode formation, lens primary fibers and epithelial cells differentiated from lens anlage at the same time [6,7]. When the lens become mature (after 28 hours post fertilization [hpf]), the lens consists of epithelial and fiber cells. The epithelial cells are anterior and the fiber cells are arcuate with attachments at the anterior epithelia, posterior capsule, and sutures. During lens growth, after the proliferation in the lens proliferation zone, the lens epithelial cells go through epithelial-to-mesenchymal transition, the cells differentiate into lens fiber cells and migrate into inner lens [6,7]. The fiber cells become transparent after programmed degradation of most of the cellular organelles, including the nuclei, endoplasmic reticulum, and mitochondria [6,8]. Degradation of organelles in differentiating lens fiber cells contributes to optical transparency [9]. Defects in cellular proliferation, differentiation and organelle degradation during lens development may cause congenital cataract diseases [10]. In contrast, dysregulation of homeostasis in mature lenses may cause age-related cataract diseases [11].
Currently, how autophagy plays roles in the lens is still a seemingly controversial question. Autophagy is an intracellular process that allows for the degradation of proteins and organelles [1]. Lens-specific ablation of ATG5 (autophagy related 5) induces cataracts in mice older than six months, indicating an important role in cataractogenesis [12]. Numerous autophagosomes are constitutively detected in lens epithelial cells, differentiating primary fiber cells and secondary fiber cells [13,14], implying a potential function of autophagy in programmed organelle degradation during lens development. However, organelles in lens fiber cells were degraded as normal during embryonic periods in lens-specific atg5 or rb1cc1/fip200 knockout mice [12,15]. Given that ATG5 and RB1CC1 are evolutionarily conserved factors that are essential for autophagosome maturation and function [16–18], these studies indicate that autophagy is not indispensable for organelle degradation in lens. Nevertheless, rapamycin, a chemical inhibitor of MAPK/JNK and MTORC1 signaling and an activator of autophagy, can stimulate premature organelle degradation by forced upregulation of autophagy in an ex vivo culture system of chick embryonic lens [19], suggesting a potential role of enhanced autophagy signaling in organelle degradation of lens fiber cells. In addition, mutations in autophagy-related genes in humans, such as FYCO1, EPG5 and CHMP4B, have been reported in patients with congenital cataracts [20–22]. Autophagic defects in lens have also been reported in animal models with cataracts [23,24]. These observations suggest a vital role of autophagy in cataractogenesis. Thus, it is interesting to investigate how autophagy affects lens development and whether cataracts can be treated by stimulating autophagy.
Gap junction proteins (also known as connexins) assemble into gap junction channels, which play an essential role in lens by facilitating intercellular communication among lens cells [25,26]. Three conserved gap junction proteins have been identified in vertebrate lenses, including GJA1/connexin 43 (gap junction protein alpha 1), GJA3/connexin 46 (gap junction protein alpha 3) and GJA8/connexin 50 (gap junction protein alpha 8) [27–29]. GJA3 and GJA8 are the two most abundant gap junction proteins in lens fiber cells, while GJA1 is mainly expressed in lens epithelial cells [30,31]. Another gap junction protein, GJE1/connexin 23 (gap junction protein epsilon 1), has also been identified in lens fiber cells in mice and zebrafish [32,33]. Among these genes, mutations in GJA3 and GJA8 have been identified in humans with inherited recessive cataracts of various different phenotypes. Ablation of either GJA3 or GJA8, but not GJA1, in the lens results in the development of cataracts in mice [34–36]. In addition to assembling gap junction channels, gap junction proteins are also involved in the regulation of other signaling pathways, such as autophagy signaling [37,38]. GJA1 constitutively downregulates autophagy through direct interaction with several ATG proteins [37]. Meanwhile, autophagy is suggested to contribute to the turnover of GJA1 and GJA8 [39].
A few cataractogenesis mechanisms have been invoked to explain how dysfunction of gap junction proteins leads to cataracts [30]. One well-known reason is dysfunction of gap junction proteins reducing intercellular communication of gap junction permeant molecules (including water, ions, and metabolites) between lens cells [40]. Mutant gap junction proteins also contribute to cataracts by altering the trafficking or function of other proteins (non-gap junction proteins) [41]. Nevertheless, given that mutations of gap junction proteins lead to various cataracts, we speculate that other mechanism(s) may be involved in cataractogenesis caused by the dysfunction of gap junction proteins.
In this study, we demonstrated that ablation of Gja8b/connexin 44.1 (a homolog of mammalian GJA8/connexin 50) led to defects in organelle degradation and cataract formation in zebrafish lenses. Intriguingly, we revealed a previously unknown function of GJA8 in modulating autophagy during lens development. GJA8 protein participated in the process of autophagy by interacting with ATG proteins. Ablation of GJA8 downregulated the level of autophagy in lens fiber cells. More importantly, rapamycin treatment stimulated autophagy in zebrafish lenses and consequently relieved the defects in organelle degradation and the cataract phenotype caused by ablation of Gja8b.
Results
Ablation of Gja8b induces cataract/lens defects in zebrafish lenses
Mutations in human and knockout in mice of GJA8 cause cataracts [30,35]. Thus, we first examined whether ablation of Gja8b induces cataracts in zebrafish lenses. Consistent with its lens-specific expression [28], the ablation of Gja8b (Figure 1A-1C) did not significantly affect the general development of the zebrafish (Figures 1E and 1F), whereas we indeed observed significant lens defects in gja8b mutant zebrafish (Figure 1D). The lens defects in gja8b mutants displayed various severities (Figure 1G). Specifically, the lens appeared transparent and similar to the wild type (WT) in 1.2% of gja8b mutants, which is accordingly defined as the WT-like group in this study. In 98.8% of gja8b mutants, an opacity of different sizes was observed in the lens (Figure 1H). The lenses with an opacity smaller than 5% of the whole lens areas were defined as the minor defect group (31.4% of gja8b mutants), and the lenses with an opacity larger than 5% of the whole lens areas were defined as the major defect group (67.4% of gja8b mutants). As controls, only 2.2% of WT and 3.2% of gja8b heterozygous zebrafish displayed light defects in the lens (Figure 1H). These results indicated that similar to humans and mice, Gja8b plays an essential role in improving transparency during zebrafish lens development.
Figure 1.
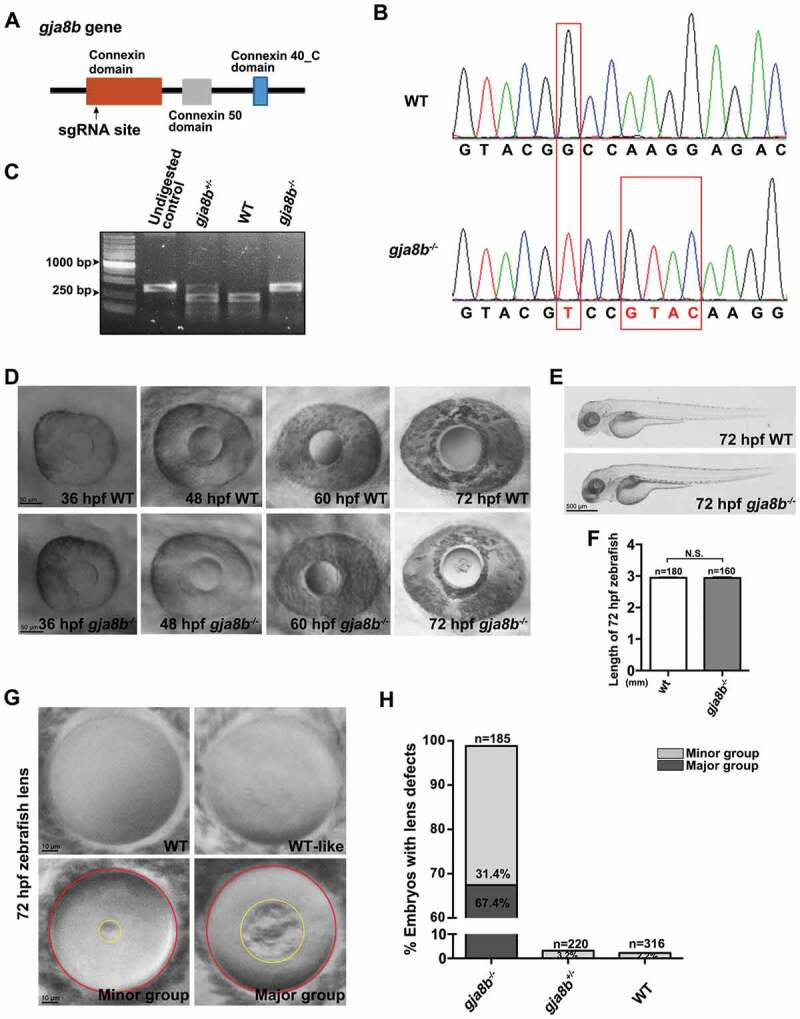
Ablation of Gja8b induces cataracts in zebrafish. (A) Schematic illustration of the position (amino acid 136) of the gja8b gene edited by sgRNA. (B) Sanger sequencing results showed that 1 base pair (bp) was substituted and 4 bp were inserted in gja8b mutant zebrafish, causing the loss of the recognition site for the restriction enzyme StyI. The genome editing led to a truncated Gja8b proteins (amino acid 1–136 followed by 22 mistranslated amino acids). (C) The gja8b mutant zebrafish were genotyped with PCR, which was followed by StyI digestion. The undigested WT PCR products was loaded as a control. The PCR products of WT fish (386 bp) could be digested with StyI to generate two fragments (236 bp and 150 bp). The PCR products of homozygous gja8b mutants could not be digested with StyI. (D) Ablation of Gja8b induced cataracts in zebrafish at 72 hpf. (E and F) Ablation of Gja8b did not affect the overall development of zebrafish. (E) Shows the WT and gja8b mutant larvae at 72 hpf. (F) Statistical analysis of length of larvae at 72 hpf (n ≥ 160 zebrafish for each group). (G and H) The cataract phenotypes displayed various severities in individual gja8b mutant zebrafish at 72 hpf. (H) Shows the statistical analysis of (G). In total, 1.2% of gja8b mutants did not display defects, 31.4% displayed light defects (Minor group), and 67.4% displayed severe defects in lenses (Major group). Yellow circles mark the opacity area in gja8b mutant lens, red circles mark the whole lens. The gja8b mutant embryos with an opaque ratio between the opaque area and the whole lens area is smaller than 5% was designated as minor group. The gja8b mutant embryos with an opaque ratio is larger than 5% was designated as major group. (n ≥ 185 zebrafish for each group). Scale bars: 50 μm (D), 500 μm (E) and 10 μm (G). mean ± SEM, N.S., p > 0.05
Ablation of Gja8b induces severe defects in organelle degradation in lens fiber cells during lens development
Defects in multiple events, such as cell differentiation, organization and organelle degradation, may lead to congenital cataracts during lens development [10,11]. To examine which events are affected by ablation of Gja8b, we performed a series of immunohistochemistry experiments. Gja8b is not expressed in lens epithelial cells in zebrafish [28]. Expectedly, immunohistochemistry for Tjp1a/Zo-1, a tight junction marker, and Prkci/aPKC, an epithelial polarity marker [42], appeared normal in gja8b mutant lens, suggesting that the polarity and organization of lens epithelial cells are normal (Fig. S1). It has been reported that GJA8 is essential for lens cell proliferation in postnatal mice lens [43]. We did not observe a significant difference in total lens cell number or the cell number of lens fiber between WT and mutant zebrafish at 36 hpf and 50 hpf, suggesting that Gja8b did not have a significant effect on the lens cell proliferation at this stage (Fig. S2). One possible reason that caused the difference between the zebrafish and the mice may be the mitotic index in WT mice lenses undergoes a large transient increase on P2 and P3 [43]. We did not observe a similar transient increase in zebrafish between 36 hpf to 50 hpf. Furthermore, we observed that the expression of Zl-1 and Mip/Aqp0, markers of differentiating lens primary fiber cells [44,45], appeared normal in gja8b mutant lens at 36 hpf (Fig. S3).
Intriguingly, we observed that ablation of Gja8b induced severe defects in the organelle degradation in lens fiber cells. Specifically, in zebrafish, programmed organelle degradation begins at ~ 48 hpf in the cells in the center of the lens and spreads outward [8]. By 72 hpf, WT fiber cells, with the exception of the newly differentiating secondary fiber cells at the outermost layers, lacked organelles, including nuclei (labeled by DAPI staining), endoplasmic reticulum (labeled by Kdel staining), and certain cellular proteins (labeled by Zl-1 and Mip staining) (Figure 2A-2C) [8]. The cytoskeleton (labeled by F-actin staining) was also degraded in the center of the lens (Figure 2C). However, all these organelles were significantly retained in lens fiber cells in gja8b mutant zebrafish at 72 hpf (Figure 2A-2C). More importantly, we observed that the nuclei number of lens fiber cells in the WT-like and minor defect group was significantly less than the number in the major defect group (Figure 2D), indicating the tight association between organelle degradation and the severity of the cataract phenotype in gja8b mutant zebrafish. The organelle degradation defects were observed by 4 d post fertilization (dpf) and 5 dpf in gja8b mutant zebrafish (Figures 2E and 2F). Organelle degradation in differentiating lens cells contributes to optical transparency [8,9]. The above data suggested that defect in organelle degradation is a novel cataractogenesis mechanism caused by Gja8b dysfunction.
Figure 2.
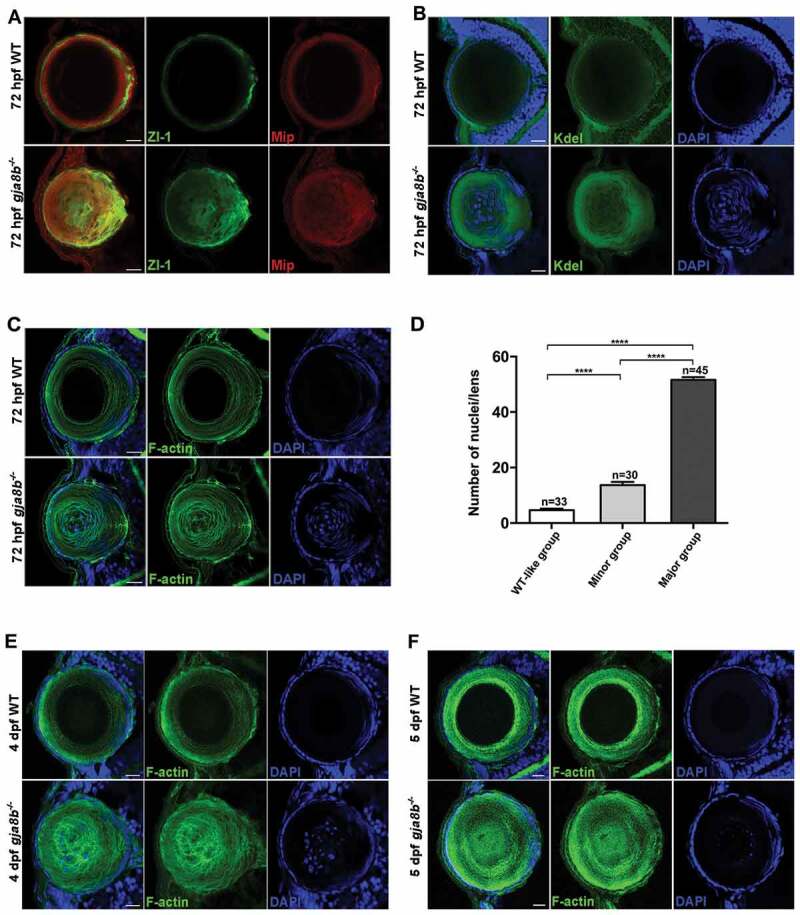
Ablation of Gja8b induces severe defects in organelle degradation of lens fiber cells. (A-C) Immunohistochemistry results showed that lens primary fiber cell markers (A, labeled by Zl-1 and Mip staining), endoplasmic reticulum (B, labeled by Kdel staining), cytoskeleton (C, labeled by F-actin staining), and nuclei (C, labeled by DAPI staining) remained in lens second fiber cells in gja8b mutants at 72 hpf. (D) The quantitative analysis of the average number of lens fiber cell nuclei shown in different gja8b mutants groups (WT-like group, minor group and major group) (mean ± SEM, n ≥ 30 zebrafish for each group, ****p < 0.0001). (E and F) Immunohistochemistry results showed that cytoskeleton (labeled by F-actin staining), and nuclei (labeled by DAPI staining) remained in the center of lens in gja8b mutants at 4 dpf (E) and 5 dpf (F). Scale bars: 50 μm
GJA8 promotes autophagy
Given that autophagy is constitutively active in lens and may play important roles in organelle degradation during lens development [2,13], we then examined whether ablation of Gja8b inhibited autophagy during lens development. Gja8b is only expressed in lens fiber cells in zebrafish [28]. Consistent with this expression pattern, we observed that the number of autophagic vacuoles (AV) was significantly decreased in fiber cells but not in epithelial cells in gja8b mutant zebrafish lenses (Figures 3A and 3B). MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) is a key protein in the phagophore and autophagosome, and it is considered the most specific autophagosomal marker [46,47]. LC3-II accumulates during lysosomal proteolysis inhibition, and the increase in LC3-II results from autophagosome synthesis [46,48]. Consistently, we observed a decrease in Map1lc3b-II levels in gja8b mutant zebrafish (Figures 3C and 3D). The above data suggested that ablation of Gja8b induces the downregulation of autophagy in lens fiber cells.
Figure 3.
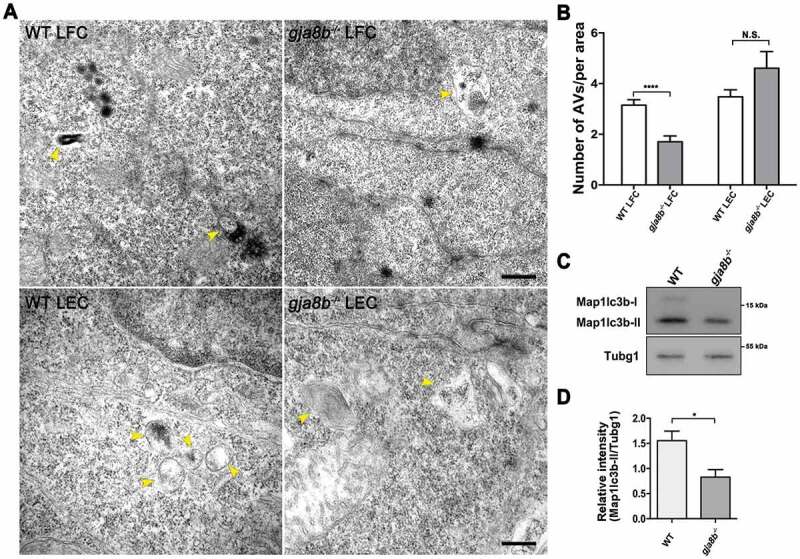
Autophagy is downregulated in lens fiber cells in gja8b mutant zebrafish. (A and B) Electron micrographs of lens fiber cells (LFC) and lens epithelial cells (LEC) in 36 hpf WT and gja8b mutants. Yellow arrowheads: autophagic vacuoles (AV). Scale bar: 100 nm. B is the number of AVs per area (40 μm2) in the lens of WT and gja8b mutants (n ≥ 60 cells from 6 lenses). (C and D) Immunoblot for endogenous Map1lc3b from WT and gja8b mutant zebrafish. (D) The quantitative analysis of the relative intensity of Map1lc3b-II compared to the Tubg1 (tubulin, gamma 1) control (n = 3 independent experiments). Mean ± SEM, N.S., p > 0.05, *p < 0.05, ****p < 0.0001
Next, we further examined the relevance of GJA8 with autophagy in human HLE cells. Interestingly, we observed that the level of GJA8 protein in HLE cells significantly increased when autophagy was activated in starvation medium (Figures 4A and 4B). We further observed that the level of LC3-II increased in HLE cells overexpressing GJA8 in nutrient-rich media, suggesting that overexpression of GJA8 promotes autophagy in HLE cells (Figures 4C and 4D). Furthermore, we observed that GJA8 colocalized with ATG16L1, a marker for phagophore assembly sites and a contributor of plasma membrane proteins participating in autophagy [49], and with LC3 puncta in HLE cells in nutrient-rich medium (Figures 4E and 4F). Activation of autophagy by starvation promoted the number of puncta co-stained with GJA8 and LC3 (Figures 4F and 4H). Consistently, overexpression of GJA8 significantly promoted the number of LC3-positive puncta in HLE cells in nutrient-rich media (Figures 4I and 4J). Taken together, these data suggested that the presence of GJA8 promotes autophagy.
Figure 4.
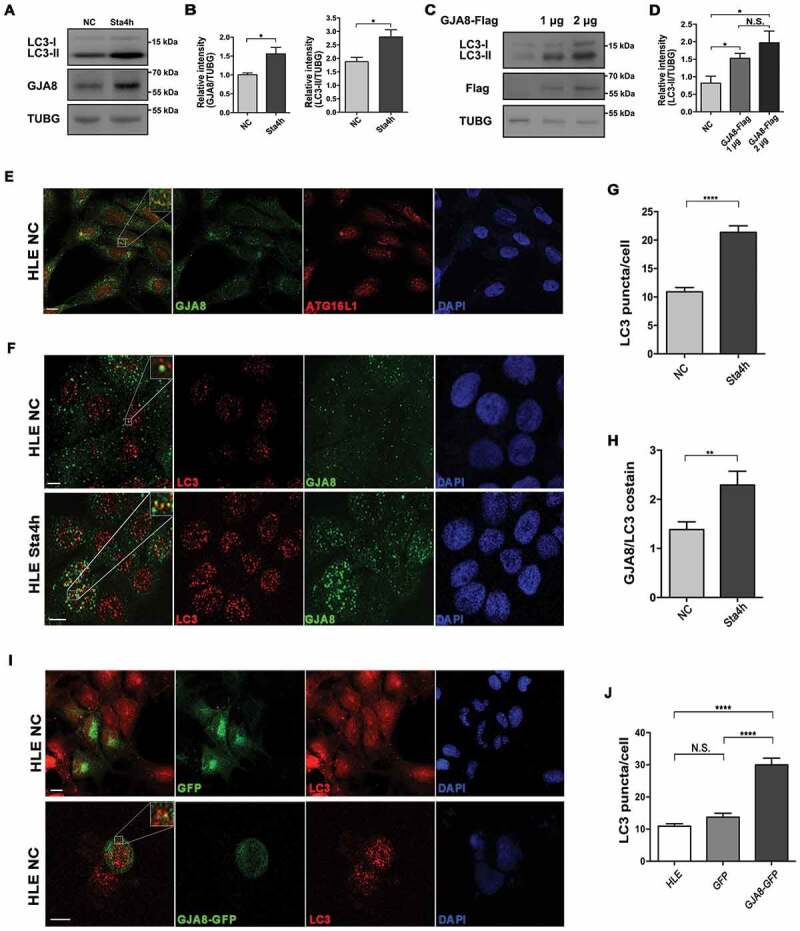
GJA8 promotes autophagy in HLE cells. (A and B) A shows immunoblot for LC3, and GJA8 from HLE cells in normal complete (NC) media and nutrient starvation media for 4 h (Sta4h). B shows the quantitative analysis of the relative intensity of GJA8 and LC3-II normalized to the TUBG control (n = 3 independent experiments). (C and D) Immunoblot for LC3 from HLE cells in NC media transfected with the different doses of GJA8-Flag. (D) Shows the quantitative analysis of the relative intensity of LC3-II compared to the TUBG control (n = 3 independent experiments). (E-H) E shows representative images for endogenous ATG16L1 and GJA8 in the NC group. (F) shows representative images for endogenous LC3 and GJA8 in NC and Sta4h groups. (G) is the quantitative analysis of the average number of endogenous LC3 puncta per cell. H is the quantitative analysis of the average number of colocalized GJA8 and LC3 puncta per cell (n = 3 wells, 3 independent experiments, > 50 cells per experiment). (I and J) I is representative images of LC3 and GJA8-GFP or GFP in HLE cells transfected with GJA8-GFP or GFP-vector. (J) The quantitative analysis of the average number of LC3 puncta per cell (n = 3 wells, 3 independent experiments, > 50 cells per experiment). Scale bars: 10 μm. Mean ± SEM, N.S., p > 0.05, *p < 0.05, **p < 0.01, ****p < 0.0001
Given the colocalization of GJA8 and ATG16L1, we speculated that GJA8 might interact with the ATG12–ATG5-ATG16L1 complex that is involved in the early autophagosome formation [49–51]. Indeed, co-immunoprecipitation results revealed that GJA8 interacts with ATG16L1 and ATG12, but it did not interact with ATG5 (Figure 5A-5C). Domain mapping results revealed that the amino acid 1–229 of GJA8 binds with these ATG proteins (Figure 5D).
Figure 5.
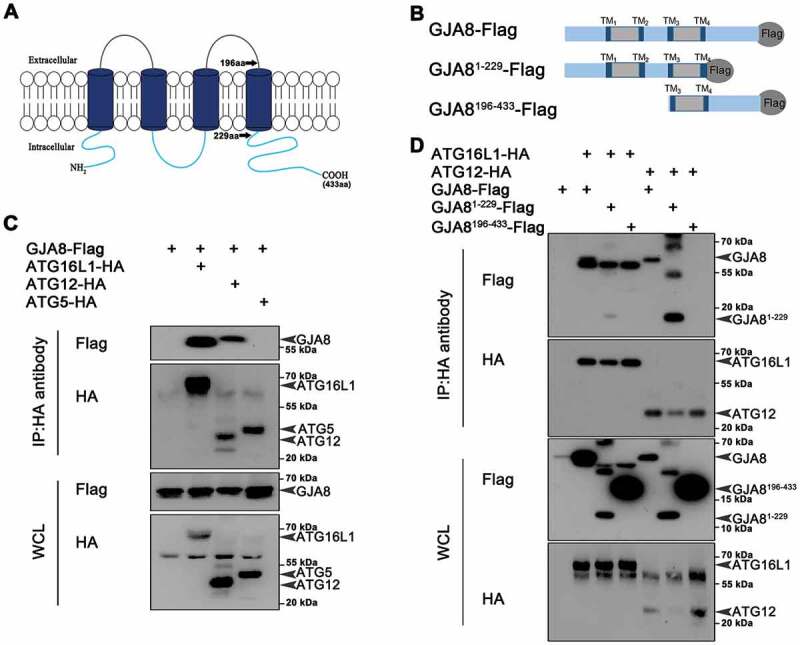
GJA8 interacts with Atg proteins. (A) Diagram of the GJA8 protein that contains 2 extracellular domains, 4 transmembrane domains and 3 intracellular domains. aa: amino acid. (B) Diagram of the different forms of GJA8 (GJA8-Flag, GJA81−229-Flag, GJA8196−433-Flag) tagged with Flag. (C) Immunoblot for immunoprecipitates from GJA8-Flag with Atg16L1-HA, ATG12-HA, or ATG9-HA in HEK293T cells. (D) Immunoblot for immunoprecipitates from ATG16L1 with GJA8-Flag, GJA81−229-Flag, GJA8196−433-Flag in HEK293T cells. WCL: whole cell lysis. WCL is used as a negative control for immunoprecipitates
Overall, the above results support a previously unknown role for GJA8 as an endogenous positive regulator of autophagosome formation. Downregulation of autophagy induced by ablation of Gja8b could be a vital pathogenic mechanism for cataracts in GJA8 mutants.
Rapamycin stimulates autophagy in the lens and relieves cataract/lens defects in gja8b mutant zebrafish.
The pharmacological agent rapamycin is well known to activate autophagy by blocking MAPK/JNK-MTORC1 signaling [52]. Rapamycin treatment in ex vivo chicken lens cultures induces premature loss of organelles by upregulated autophagy via suppressing MAPK/JNK-MTORC1 signaling in lens fiber cells [19]. It is very important to determine whether rapamycin treatment can boost autophagy in animals and rescue cataracts. Thus, we treated gja8b mutant zebrafish with different concentrations of rapamycin. We first examined whether rapamycin promoted autophagy in lens cells. Indeed, we observed that rapamycin treatment significantly promoted both the AV number and the AV area in lens epithelial and fiber cells in gja8b mutant zebrafish (Figure 6A-6C). Immunoblots results consistently revealed that autophagy was promoted in rapamycin-treated zebrafish, which was indicated by the upregulation of Map1lc3b-II levels (Figures 6D and 6E). Furthermore, we observed that the expression of GJA8 increased in rapamycin-treated HLE cells (Figures 6F and 6G). However, the co-localization of GJA8 with ATG16L1 did not increase in rapamycin-treated HLE cells (Figures 6H and 6I).
Figure 6.
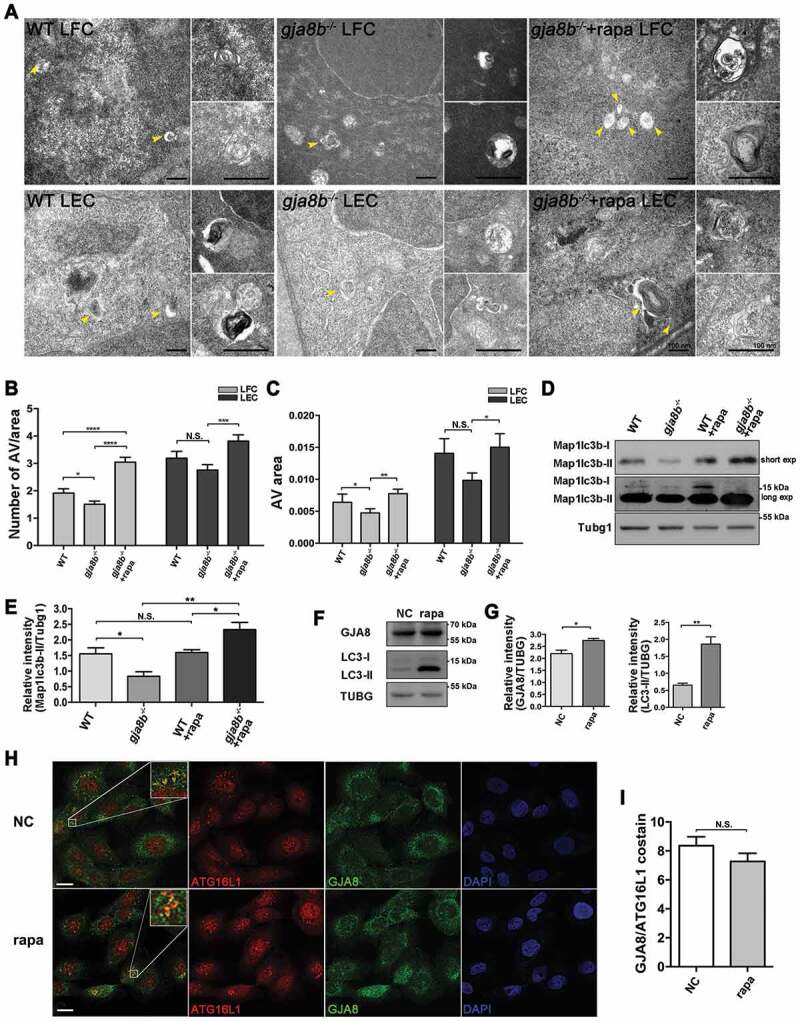
Rapamycin treatment promotes autophagy in gja8b mutant zebrafish lenses. (A-C) Electron micrographs of lens fiber cells (LFC) and lens epithelial cells (LEC) in 72 hpf WT, gja8b mutants, and gja8b mutants treated with 25 μM rapamycin. Yellow arrowheads show AV. (B) The number of AVs per area (40 μm2), and (C) is the average area of AVs in the lens epithelial and fiber cells in WT, gja8b mutants and gja8b mutants treated with 25 μM rapamycin (n ≥ 60 cells from 6 lenses). (D) Shows the concentration of endogenous Map1lc3b-I and Map1lc3b-II in the zebrafish eyes of WT, gja8b mutants, WT treated with 25 μM rapamycin and gja8b mutants treated with 25 μM rapamycin. Short exp and long exp: short exposure and long exposure. (E) shows the quantitative analysis of relative intensity of Map1lc3b-II normalized to the Tubg1 loading control (n = 3 independent experiments). (F) Shows the concentration of endogenous GJA8 and LC3 in NC or rapamycin-treated HLE cells. (G) Shows the quantitative analysis of relative intensity of GJA8 and LC3-II normalized to the TUBG loading control (n = 3 independent experiments). (H) Shows representative images for endogenous ATG16L1 and GJA8 in NC and rapamycin treated HLE cells. (I) Quantitative analysis of the average number of colocalized GJA8 and ATG16L1 puncta per cell. (n = 3 wells, 3 independent experiments, > 50 cells per experiment). Scale bar: 100 nm (A) and 10 μm (H). Mean ± SEM, N.S., p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Intriguingly, we observed that rapamycin treatment effectively relieved lens defects in gja8b mutant zebrafish. The higher concentration of the treatments of this reagent retrieved the better effects (Figure 7A). Consistently, immunohistochemistry results revealed that rapamycin treatment promoted organelle degradation, including denucleation (Figures 7B and 7C), loss of the endoplasmic reticulum and cytoskeleton and decreased expression of markers of primary fiber cells (Figure 7D) in gja8b mutant zebrafish. In summary, the data demonstrated that in vivo rapamycin treatment promotes autophagy in zebrafish lenses and relieves cataracts caused by ablation of Gja8b.
Figure 7.
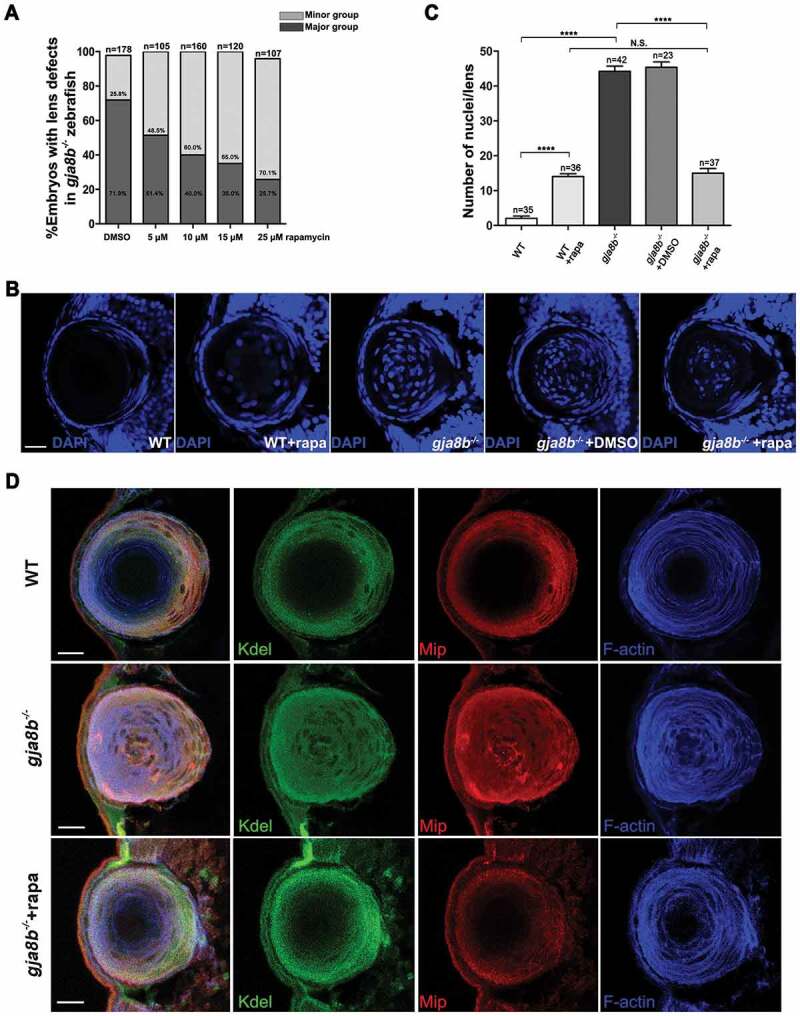
Rapamycin treatment relieves cataract/lens defects in gja8b mutant zebrafish. (A) Statistical analysis of the severity of lens defects in 72 hpf gja8b mutants treated with rapamycin. Horizontal axis labeling shows the final concentration of rapamycin (n > 100 zebrafish for each group). (B) Shows representative images of the nuclei of lens fiber cells in WT, gja8b mutants, gja8b mutants treated with DMSO, and WT and gja8b mutants treated with 25 μM rapamycin at 72 hpf. (C) Shows the quantitative analysis of the average number of lens fiber cell nuclei shown in B (n > 20 zebrafish for each group). (D) Representative images to show the distribution of Kdel, Mip and F-actin in the lens of WT, gja8b mutants, and gja8b mutants treated with 25 μM rapamycin. Scale bars: 50 μm. Mean ± SEM, N.S., p > 0.05, ****p < 0.0001
3-Methyladenine (3-MA) treatment blocks the recovery of cataracts induced by rapamycin in gja8b mutant zebrafish lenses.
To further validate the mechanism through which rapamycin relieves cataracts by promoting autophagy in the lens, we treated zebrafish with 4 mM 3-MA. 3-MA is widely used as an autophagy inhibitor due to its inhibitory effect on class III phosphatidylinositol 3-kinase (PtdIns3K) [53], which is known to be essential for the induction of autophagy. We observed that 3-MA treatment in WT zebrafish induced defects similar to those observed following the ablation of Gja8b, including degradation defects in the endoplasmic reticulum, cytoskeleton and nuclei of lens fiber cells (Figure 8A-8C). More importantly, 3-MA treatment in gja8b mutant zebrafish significantly blocked the recovery effects induced by rapamycin, including the severity of cataract, and organelle degradation (Figure 8D-8G). Taken together, these data suggested that rapamycin treatment relieves cataracts caused by ablation of Gja8b via stimulating autophagy in zebrafish lenses, and 3-MA inhibits this recovery effect via inhibiting autophagy (Figures 8H and 8I).
Figure 8.
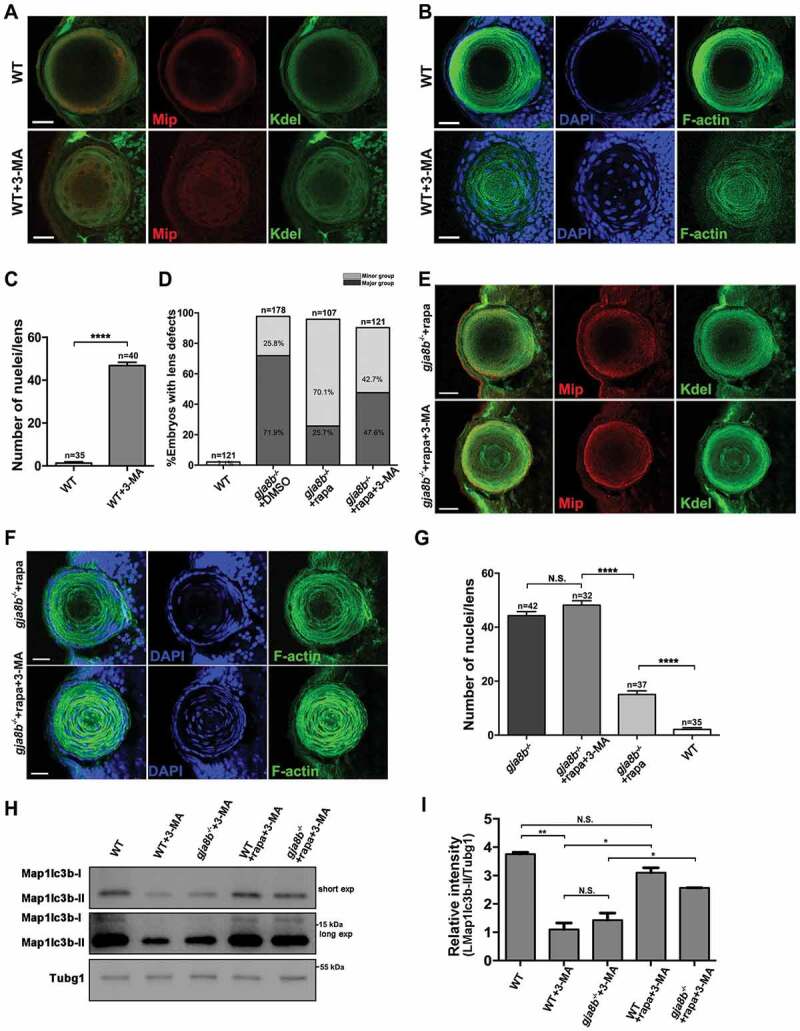
3-MA treatment blocks the rescue effects of rapamycin in gja8b mutant zebrafish lenses. (A) Shows representative images of the distribution of Kdel and Mip, and (B) shows representative images of nuclei and F-actin in the lens of WT and WT zebrafish treated with 4 mM 3-MA at 72 hpf. (C) Average number of lens fiber cell nuclei per lens (n > 30 zebrafish for each group). (D) Statistical analysis of the severity of lens defects in 72 hpf WT, gja8b mutants, gja8b mutants treated with rapamycin, and gja8b mutants treated with 25 μM rapamycin and 4 mM 3-MA (n > 120 zebrafish for each group). (E) Shows representative images of the distribution of Kdel and Mip, and (F) shows representative images of nuclei and F-actin in the lens of gja8b mutants treated with 25 μM rapamycin and gja8b mutants treated with 25 μM rapamycin plus 4 mM 3-MA at 72 hpf. (G) Average number of lens fiber cell nuclei per lens (n > 30 zebrafish for each group). (H) Shows the concentration of endogenous Map1lc3b-I and Map1lc3b-II in WT, WT treated with 4 mM 3-MA, gja8b mutants treated with 4 mM 3-MA, WT treated with 25 μM rapamycin and 4 mM 3-MA, and gja8b mutants treated with 25 μM rapamycin and 4 mM 3-MA. Short exp and long exp: short exposure and long exposure. (I) Shows the quantitative analysis of relative intensity of Map1lc3b-II normalized to the Tubg1 control (n = 3 independent experiments). Scale bars: 50 μm. Mean ± SEM, *p < 0.05, **p < 0.01, ****p < 0.0001
Discussion
Although it is well known that mutations in GJA8 are related to cataract formation [10,30,31], few cataractogenesis mechanisms have been revealed other than abnormal assembly of gap junctions. In this study, we revealed a previously unknown function of GJA8 in the regulation of autophagy that is based on its interaction with ATG proteins. We also proposed that defect in organelle degradation is a novel cataractogenesis mechanism caused by dysfunction of Gja8b. Lens fiber cells in the inner lens, known as the organelle-free zone, lack all organelles and nuclei. Organelle-free fiber cells minimize light scattering and ensure lens transparency [9,54,55]. Our data indicated that GJA8 is required for organelle-free zone formation. Ablation of Gja8b induced severe defects in organelle degradation in zebrafish lens fiber cells. Interestingly, organelle degradation was promoted by stimulating autophagy. Along with the improved organelle degradation, the severity of lens opacity was relieved in gja8b mutant zebrafish.
Unlike many other integral membrane proteins, most gap junction proteins have short half-lives, maybe suiting to the alterations in intercellular communication and cell contacts [56]. Several pathways, including the autophagy pathway, have been reported to be implicated in the rapid turnover of gap junction proteins [39,56]. In HeLa cells stably expressing GJA8, autophagy can regulate the level of GJA8. The cytoplasmic accumulation of a mutant cataract-associated gap junction protein, GJA8P88S that exhibits impaired degradation and forms abnormal accumulations, results from an insufficient degradation capacity of constitutive autophagy [39]. Here, our data demonstrated that GJA8 stimulates autophagy both in zebrafish lenses and in HLE cells. Interestingly, we observed that starve stimulation simultaneously promoted autophagy and increased levels of GJA8 in HLE cells. Taken together, these observations implied that there might be positive feedback between GJA8 and autophagy.
Albeit all gap junction proteins share conservative membrane-spanning structures, it appears that different gap junction proteins may play specific roles [28,57]. In contrast to the specific expression of GJA8 in the lens, GJA1 is the most commonly expressed gap junction protein in multiple tissues [25,57,58]. Interestingly, unlike cataractogenesis due to GJA8 dysfunction in humans and multiple animal species [29], the lenses of mice with a conditional deletion of GJA1 are transparent and develop normally through at least 6 months of age, even though intercellular communication among epithelial cells may be impaired [59]. Mutations in GJA1 have been associated with oculodentodigital dysplasia, a disease that is only rarely accompanied by cataracts [60]. Distinct from the observation that GJA8 stimulated autophagy in the lens in this study, the presence of GJA1 on the plasma membrane negatively modulates autophagosome biogenesis in mouse osteoblasts. Internalization or degradation of GJA1 promotes activation of autophagy in response to nutritional stress [37]. Being involved in autophagy, GJA1 interacts with ATG16L1 via its C terminus, while GJA8 was found to interact with ATG16L1 and ATG12 via its N terminus. More experiments are expected to elucidate the differences between GJA1 and GJA8, including the mechanisms in the regulation of autophagy.
The role of autophagy in organelles degradation in developing lens is still a controversial question. Genetic studies in atg5 knockout mice have demonstrated that autophagy is important for intracellular quality control in the lens but can be dispensable for lens organelle degradation during lens development [12,16]. Similarly, we did not observe defects in organelle degradation and cataract phenotype in the lenses of atg5 mutant zebrafish before the fish died at approximately 10 dpf (data not provided). Nevertheless, in this study, we actually observed a tight association between defects in organelle degradation and downregulation of autophagy in gja8b mutant zebrafish. Importantly, treatment of gja8b mutant zebrafish with rapamycin significantly relieved the defects in organelle degradation and the severity of cataracts. Conversely, 3-MA treatment blocked this recovery effect. A well-known fact that the early initiation of autophagy is negatively regulated by MTOR (mechanistic target of rapamycin kinase) and positively regulated by PtdIns3K pathways [61–63]. Rapamycin, a pharmacological inhibitor of MTOR, functions by reducing MTORC1 phosphorylation and decreasing MTORC2 activity, therefore activating autophagy [64,65], whereas 3-MA treatment inhibits autophagy by suppressing PtdIns3K [66]. Supporting our observation, ex vivo treatment with rapamycin stimulates premature organelle degradation in cultured chick embryonic lenses [19]. Taken together, we speculated that, although autophagy is not necessary for organelle degradation in the lens in the normal physiological state, deficient autophagy might play a critical role in organelle degradation in the pathological state of cataracts.
Despite the clinical success of cataract surgery, patients are still suffering from disturbances in vision quality and have poor performance in functional visual acuity testing [67]. Thus, it is of great significance to screen drugs that can effectively control the development of cataracts. The zebrafish has become an important vertebrate model for in vivo chemical screening in drug development for human diseases [68,69]. Interestingly, in vivo rapamycin treatment significantly relieved the cataract phenotype in gja8b mutant zebrafish. Autophagy plays an essential role in lens development and intracellular quality control to maintain lens transparency. Given the similarities in structure, optical function and metabolism among vertebrate lenses, our study did imply that stimulation of autophagy may be a potential clinical treatment for cataracts caused by GJA8 mutations.
Materials and methods
Zebrafish genotyping, maintenance and breeding
AB WT and gja8b (Ensembl ID: ENSDARG00000015076) mutant zebrafish were used in this study. The gja8b mutant zebrafish was generated in China Zebrafish Resource Center by CRISPR-Cas9 technology. The sgRNA sequence is GGGAAGCGTCCGTACGGCCAagg. The gja8b mutant zebrafish were genotyped by PCR (forward primers, 5ʹ-CCGGGTTGCGAGAACGTTTG-3ʹ, reverse primers, 5ʹ-GAGTGGCAAGATTCGGAAGC-3ʹ) followed by the digestion of StyI restriction enzyme (New England Biolabs, R3500S). In this study, we mated homozygous with WT to obtain heterozygous embryos, and mated homozygous adult zebrafish to obtain homozygous embryos for the phenotype analysis. Zebrafish embryos were raised at 28.5°C in E3 media (5 mM NaCl, 0.33 mM MgSO4, 0.33 mM CaCl2, 0.17 mM KCl, and 0.1% methylene blue). Further, 0.003% PTU (w:v; Sigma-Aldrich, P7629) was added into E3 media to inhibit melanogenesis. Zebrafish were bred in accordance with Zhejiang University Animal Care and Use Committee protocols.
Cell culture, transfections, co-immunoprecipitations and immunoblots
HLE cells (SRA 01–04) [70] were obtained from the RIKEN Cell Bank (RCB1591), and HEK293T cells were obtained from ATCC (CRL-3216). Cells were cultured in DMEM (Corning, 10–090-CV) with 10% fetal bovine serum (FBS; Gibco, 26,140–079) at 37°C in 5% CO2 (v:v). Nutrient starvation was performed by thoroughly washing the cells with prewarmed 1X phosphate-buffered saline (PBS; Gibco, 10,010–023) and incubating them in starvation medium (1% BSA [w:v; Sigma-Aldrich, A9418], 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose [Sigma-Aldrich, G5500], 20 mM HEPES [Gibco, 15,630–080], pH 7.4) for 4 h at 37°C with 5% CO2 [71]. For the GJA8-Flag overexpression immunoblots, HLE cells were transfected with indicated plasmid using Lipofectamine 2000 (Invitrogen, 11,668,019) according to the manufacturer’s instructions. For the co-immunoprecipitation assays, HEK293T cells were transfected with indicated plasmids using Lipofectamine 2000. Cells transfected for 24 h with specific plasmids were lysed in modified MYC lysis buffer (MLB) (20 mM Tris-Cl, 200 mM NaCl, 10 mM NaF, 1 mM Na3V2O4 [Sigma-Aldrich, 450,243], 1% NP-40 [Thermo Fisher Scientific, 85,124], 20 mM beta-glycerophosphate [Sigma-Aldrich, G9422], and protease inhibitor [Complete Protease Inhibitor Cocktail, Roche, 04693116001], pH 7.5). Cell lysates were then subjected to immunoprecipitation using specific antibodies for the transfected proteins. After 2 ~ 3 washes with MLB, proteins adsorbed on the beads (Repligen, CA-PRI-0100) were resolved in 1X SDS loading buffer and were analyzed by SDS-PAGE and immunoblots with the indicated antibodies. Cell lysates were also analyzed by SDS-PAGE and immunoblots to control for protein abundance. Zebrafish were anesthetized with 1 mM Tricane (Sigma-Aldrich, E10521), then eyeballs were removed, lysed in TBST (1% Triton X-100 [Thermo Fisher Scientific, 28,313]), resolved in 1X SDS loading buffer and analyzed by SDS-PAGE and immunoblots with the indicated antibodies. A serial dilution of the amount of all proteins was firstly used as a scale for creating standard curves for comparative protein quantification. The blots were stained with ECL reagent (Thermo Fisher Scientific, 32,106) and detected with the ChemiDoc Touch Imaging System from Bio-Rad (1,708,371). The densitometry of each band was performed using ImageJ software (https://imagej.nih.gov/ij/). The linear ranges for proteins, including TUBG/γ-tubulin, were determined by creating a standard curve based on the diluted volumes of each protein. The relative level of all proteins was normalized with the level of the TUBG proteins. Three independent experiments were performed for all immunoblots. The following antibodies were used for immunoblots (1:1000): anti-Flag (Millipore, SAB4301135), anti-HA (Cell Signaling Technology, 3724), anti-LC3 (Cell Signaling Technology, 2775), anti-GJA8 (Thermo Fisher Scientific, 334,300) and anti-TUBG (Sigma-Aldrich, T3559).
Expression plasmids
Expression plasmids for FLAG- or hemagglutinin (HA)-tagged pRK5F were kind gifts from Prof. Pinglong Xu (Zhejiang University, Hangzhou, China). The cDNAs of human ATG16L1, ATG12, and ATG5 were provided by Prof. Han jiahuai (Xiamen University, Xiamen, China). Human GJA8 and human GJA8 domains, including GJA8 amino acids 1–229, 196–433, were subcloned into pRK5F-flag. Human ATG16L1, ATG12, and ATG5 were amplified by PCR and cloned into the plasmid pRK5F-HA. All coding sequences were verified by DNA sequencing and detailed information of plasmids is provided in Table S1.
Immunofluorescence and antibodies
The following antibodies were used: anti-GJA8 (1:200; Santa Cruz Biotechnology, sc-20,876), anti-Zl-1/ZL-1 (1:200; Abcam, ab185979), anti-Kdel/KDEL (1:200; Santa Cruz Biotechnology, sc-58,774), anti-ATG16L1 (1:200; MBL, PM040), anti-Mip/Aqp0 (1:200; Millipore, AB3071), anti-LC3 (1:300; Cell Signaling Technology, 2775), anti-Tjp1a/ZO1 (1:200; Thermo Fisher Scientific, 33–9100), anti-Prkci/aPKC (1:200; Santa Cruz Biotechnology, sc-216). Alexa Fluor 488 Phalloidin (1:300; Thermo Fisher Scientific, A12379) was used to visualize F-actin. DAPI (Thermo Fisher Scientific, D3571) was used to stain nuclei. Zebrafish were fixed at the desired developmental stage with 4% paraformaldehyde (Sigma-Aldrich, P6148) for 2 h at room temperature. Immunohistochemistry was performed following the procedure described previously [72]. HLE cell immunofluorescence was performed as previously described [31]. Confocal microscopy was performed using a Nikon A1 confocal microscope. Adobe Photoshop 7.0 was used for subsequent image processing and colocalization calculation.
Severity of cataract/lens defects
Live zebrafish were photographed with a Nikon SMZ18 microscope. The area of the cloudy region in the lens was analyzed with ImageJ. Accordingly, in gja8b mutant zebrafish, the severity of cataract/lens defects caused by the ablation of Gja8b could be classified into three groups (WT-like, minor defect group, and major defect group). We did not select the zebrafish based on the severity of cataract/lens defects for the subsequent experiments, expect the experiments shown in Figure 2D.
Transmission electron microscopy
The embryos at the desired developmental stage were fixed in 2% transmission electron microscopy (TEM) grade glutaraldehyde (SPI Chem, 02608-BA) plus 2% paraformaldehyde (Sigma-Aldrich, P6148) in 0.1 M PBS at 4°C; then, they were rinsed in PBS, postfixed with 1% OsO4 (SPI Chem, 02595-BA) and 0.1% K3Fe(CN)6 (Sigma-Aldrich, 702,587), dehydrated through a graded series of ethanol and absolute acetone, then embedded in Spurr resin (SPI Chem, 02680-AB). The 65-nm ultrathin tissue sections were stained with 2% uranyl acetate (SPI Chem, 02624-AB) and alkaline lead citrate (Sinopharm Chemical Reagent limited corporation, 200,964,701), then examined with a Hitachi Model H-7650 TEM.
Chemical treatments
At 24 hpf, zebrafish were transferred to 10-mm2 plates with E3 media with 0.003% PTU. Chemicals or vehicle controls (DMSO [Sigma-Aldrich, 472,301]) were then added to make a total volume of 10 ml. In the culture media, rapamycin (MedChemExpress, HY-10,219) was dissolved to 25 µM, 3-MA (MedChemExpress, HY-19,312) was dissolved to 4 mM for treatment. All culture media were replaced every 12 h. Then embryos were analyzed by DIC (Nikon SMZ18) or immunofluorescence at 72 hpf.
Nuclei counting and statistical analysis
Number of lens fiber cell nuclei in zebrafish lens shown in Fig. 2D, 7C, 8C, 8G and S2C was counted by cell counter in ImageJ software. The counting area is the center area of lens, including the primary and secondary lens fiber cells, except the outermost layer of the newly differentiating secondary fibers and the epithelium. The total number of lens in different groups was listed on the top of the according bars. Number of whole nuclei in lens (including epithelial and fiber cells) shown in Fig. S2D was counted by the same procedure. Data are expressed as the mean ± SEM. Differences were analyzed by two-tailed Student’s t tests using Prism 6 (GraphPad), and p values < 0.05 were considered significant. No statistical method was used to predetermine sample size. All analyses were performed following a minimum of 3 (n = 3) independent experiments.
Supplementary Material
Acknowledgments
We thank China Zebrafish Resource Center for providing the gja8b mutant zebrafish (ZKO933a). We thank Dr. P.L. Xu (Zhejiang University) and Dr. J.H. Han (Xiamen University) for providing pRK5 plasmid and human ATG5, ATG12, ATG16L1 cDNAs.
Funding Statement
This work was supported by the National Natural Science Foundation of China (Grant no. 81670834, 81970781 to X.S., 81770938 to J.Z., 81800807 to X. T., and 81800869 to X. Y.), Zhejiang Provincial Natural Science Foundation project (LZ15H120001 to J.Z.).
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [2].Morishita H, Mizushima N.. Autophagy in the lens. Exp Eye Res. 2016;144:22–28. [DOI] [PubMed] [Google Scholar]
- [3].Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. [DOI] [PubMed] [Google Scholar]
- [5].Bourne RRA, Stevens GA, White RA, et al. Causes of vision loss worldwide, 1990-2010: a systematic analysis. Lancet Glob Health. 2013;1(6):E339–E49. [DOI] [PubMed] [Google Scholar]
- [6].Chow RL, Lang RA. Early eye development in vertebrates. Annu Rev Cell Dev Biol. 2001;17(1):255–296. [DOI] [PubMed] [Google Scholar]
- [7].Greiling TM, Clark JI. Early lens development in the zebrafish: a three-dimensional time-lapse analysis. Dev Dyn. 2009;238(9):2254–2265. [DOI] [PubMed] [Google Scholar]
- [8].Greiling TMS, Clark JI. New insights into the mechanism of lens development using zebra fish. Int Rev Cel Mol Bio. 2012;296:1–61. [DOI] [PubMed] [Google Scholar]
- [9].Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res. 2009;88(2):133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pichi F, Lembo A, Serafino M, et al. Genetics of congenital cataract. Dev Ophthalmol. 2016;57:1–14. [DOI] [PubMed] [Google Scholar]
- [11].Shiels A, Hejtmancik JF. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2017;156:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Morishita H, Eguchi S, Kimura H, et al. Deletion of Autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem. 2013;288(16):11436–11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Costello MJ, Brennan LA, Basu S, et al. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res. 2013;116:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Matsui M, Yamamoto A, Kuma A, et al. Organelle degradation during the lens and erythroid differentiation is independent of autophagy. Biochem Biophys Res Commun. 2006;339(2):485–489. [DOI] [PubMed] [Google Scholar]
- [16].Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152(4):657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luo S, Rubinsztein DC. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 2007;14(7):728–741. [DOI] [PubMed] [Google Scholar]
- [19].Basu S, Rajakaruna S, Reyes B, et al. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy. 2014;10(7):1193–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen JJ, Ma ZW, Jiao XD, et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet. 2011;88(6):827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45(1):83–U122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sagona AP, Nezis IP, Stenmark H. Association of CHMP4B and autophagy with micronuclei: implications for cataract formation. Biomed Res Int. 2014. DOI: 10.1155/2014/974393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wignes JA, Goldman JW, Weihl CC, et al. p62 expression and autophagy in alpha B-crystallin R120G mutant knock-in mouse model of hereditary cataract. Exp Eye Res. 2013;115:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Makley LN, McMenimen KA, DeVree BT, et al. Pharmacological chaperone for alpha-crystallin partially restores transparency in cataract models. Science. 2015;350(6261):674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saez JC, Berthoud VM, Branes MC, et al. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–1400. [DOI] [PubMed] [Google Scholar]
- [26].Jiang JX. Gap junctions or hemichannel-dependent and independent roles of connexins in cataractogenesis and lens development. Curr Mol Med. 2010;10(9):851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Beyer EC, Berthoud VM. Connexin hemichannels in the lens. Front Physiol. 2014;5. DOI: 10.3389/fphys.2014.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng SH, Christie T, Valdimarsson G. Expression of connexin48.5, connexin44.1, and connexin43 during zebrafish (Danio rerio) lens development. Dev Dynam. 2003;228(4):709–715. [DOI] [PubMed] [Google Scholar]
- [29].Rong P, Wang X, Niesman I, et al. Disruption of Gja8 (alpha 8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development. 2002;129(1):167–174. [DOI] [PubMed] [Google Scholar]
- [30].Beyer EC, Ebihara L, Berthoud VM. Connexin mutants and cataracts. Front Pharmacol. 2013;4. DOI: 10.3389/fphar.2013.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].XN Y, XY P, Zhang X, et al. The impact of GJA8 SNPs on susceptibility to age-related cataract. Hum Genet. 2018;137(11–12):897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sonntag S, Sohl G, Dobrowolski R, et al. Mouse lens connexin23 (Gje1) does not form functional gap junction channels but causes enhanced ATP release from HeLa cells. Eur J Cell Biol. 2009;88(2):65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Iovine MK, Gumpert AM, Falk MM, et al. Cx23, a connexin with only four extracellular-loop cysteines, forms functional gap junction channels and hemichannels. FEBS Lett. 2008;582(2):165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gong XH, Li E, Klier G, et al. Disruption of alpha(3) connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91(6):833–843. [DOI] [PubMed] [Google Scholar]
- [35].White TW, Goodenough DA, Paul DL. Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol. 1998;143(3):815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mathias RT, White TW, Gong XH. Lens gap junctions in growth, differentiation, and homeostasis. Physiol Rev. 2010;90(1):179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bejarano E, Yuste A, Patel B, et al. Connexins modulate autophagosome biogenesis. Nat Cell Biol. 2014;16(5):401–U55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Su V, Lau AF. Connexins: mechanisms regulating protein levels and intercellular communication. FEBS Lett. 2014;588(8):1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lichtenstein A, Minogue PJ, Beyer EC, et al. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci. 2011;124:910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Evans WH, Martin PEM. Gap junctions: structure and function (review). Mol Membr Biol. 2002;19(2):121–136. [DOI] [PubMed] [Google Scholar]
- [41].Wei CJ, Xu X, Lo CW. Connexins and cell signaling in development and disease. Annu Rev Cell Dev Biol. 2004;20(1):811–838. [DOI] [PubMed] [Google Scholar]
- [42].Chen J, Zhang MJ. The Par3/Par6/aPKC complex and epithelial cell polarity. Exp Cell Res. 2013;319(10):1357–1364. [DOI] [PubMed] [Google Scholar]
- [43].Sellitto C, Li LP, White TW. Connexin50 is essential for normal postnatal lens cell proliferation. Invest Ophthalmol Vis Sci. 2004;45(9):3196–3202. [DOI] [PubMed] [Google Scholar]
- [44].Greiling TMS, Aose M, Clark JI, et al. Differentiation of the developing ocular lens. Invest Ophthalmol Vis Sci. 2010;51(3):1540–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Korol A, Pino G, Dwivedi D, et al. Matrix metalloproteinase-9-null mice are resistant to TGF-beta-induced anterior subcapsular cataract formation. Am J Pathol. 2014;184(7):2001–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kabeya Y, Mizushima N, Uero T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19(21):5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kaminskyy V, Zhivotovsky B. Proteases in autophagy. Bba-Proteins Proteom. 2012;1824(1):44–50. [DOI] [PubMed] [Google Scholar]
- [49].Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19(5):2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Bi. 2011;27(1):107–132. [DOI] [PubMed] [Google Scholar]
- [51].Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(9):1679–1688. [DOI] [PubMed] [Google Scholar]
- [52].Jung CH, Ro SH, Cao J, et al. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wu YT, Tan HL, Shui GH, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bassnett S, Costello MJ. The cause and consequence of fiber cell compaction in the vertebrate lens. Exp Eye Res. 2017;156:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cvekl A, Ashery-Padan R. The cellular and molecular mechanisms of vertebrate lens development. Development. 2014;141(23):4432–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Berthoud VM, Minogue PJ, Laing JG, et al. Pathways for degradation of connexins and gap junctions. Cardiovasc Res. 2004;62(2):256–267. [DOI] [PubMed] [Google Scholar]
- [57].Willecke K, Eiberger J, Degen J, et al. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383(5):725–737. [DOI] [PubMed] [Google Scholar]
- [58].Beyer EC, Kistler J, Paul DL, et al. Antisera directed against connexin43 peptides react with a 43-kD protein localized to gap junctions in myocardium and other tissues. J Cell Biol. 1989;108(2):595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].DeRosa AM, Mese G, Li LP, et al. The cataract causing Cx50-S50P mutant inhibits Cx43 and intercellular communication in the lens epithelium. Exp Cell Res. 2009;315(6):1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Paznekas WA, Boyadjiev SA, Shapiro RE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72(2):408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273(7):3963–3966. [DOI] [PubMed] [Google Scholar]
- [63].Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2013;115(4):263–273. [DOI] [PubMed] [Google Scholar]
- [64].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29(26):3733–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Jaber N, Dou ZX, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. P Natl Acad Sci (USA). 2012;109(6):2003–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yamaguchi T, Negishi K, Tsubota K. Functional visual acuity measurement in cataract and intraocular lens implantation. Curr Opin Ophthalmol. 2011;22(1):31–36. [DOI] [PubMed] [Google Scholar]
- [68].MacRae CA, Peterson RT. Zebrafish as tools for drug discovery. Nat Rev Drug Discov. 2015;14(10):721–731. [DOI] [PubMed] [Google Scholar]
- [69].DS W, SE R, LI Z. Chemical screening in zebrafish for novel biological and therapeutic discovery. Methods Cell Biol. 2017;138:651–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ibaraki N, Chen SC, Lin LR, et al. Human lens epithelial cell line. Exp Eye Res. 1998;67(5):577–585. [DOI] [PubMed] [Google Scholar]
- [71].Huang R, Xu YF, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–466. [DOI] [PubMed] [Google Scholar]
- [72].Wei XY, Zou J, Takechi M, et al. Nok plays an essential role in maintaining the integrity of the outer nuclear layer in the zebrafish retina. Exp Eye Res. 2006;83(1):31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.