

ABSTRAST
Due to the redundancy of the genetic code most amino acids are encoded by several ‘synonymous‘ codons. These codons are used unevenly, and each organism demonstrates its own unique codon usage bias, where the ‘preferred’ codons are associated with tRNAs that are found in high concentrations. Therefore, for decades, the prevailing view had been that preferred and non-preferred codons are linked to high or slow translation rates, respectively.
However, this simplified view is contrasted by the frequent failures of codon-optimization efforts and by evidence of non-preferred (i.e. ‘slow’) codons having specific roles important for efficient production of functional proteins. One such specific role of slower codons is the regulation of co-translational protein folding, a complex biophysical process that is very challenging to model or to measure.
Here, we combined a genome-wide approach with experiments to investigate the role of slow codons in protein production and co-translational folding. We analysed homologous gene groups from divergent bacteria and identified positions of inter-species conservation of bias towards slow codons. We then generated mutants where the conserved slow codons are substituted with ‘fast’ ones, and experimentally studied the effects of these codon substitutions. Using cellular and biochemical approaches we find that at certain locations, slow-to-fast codon substitutions reduce protein expression, increase protein aggregation, and impair protein function.
This report provides an approach for identifying functionally relevant regions with slower codons and demonstrates that such codons are important for protein expression and function.
KEYWORDS: Protein translation, co-translational folding, codon-usage bias, evolutionary conservation, computational model, experimental validation
INTRODUCTION
Of the 20 canonical amino acids, 18 are coded by multiple codons which are termed ‘synonymous codons’ [1]. Different organisms display a distinct pattern of codon usage preference, or ‘codon usage bias’: for example, the preferred codon for the amino acid arginine in Escherichia coli is CGU while in human it is AGA. The codons that are more abundantly used correspond to tRNAs found at high concentrations, while the less preferred ones largely correspond to tRNAs found at low concentrations [2,3]. The concentration of tRNA that recognizes a certain codon is related to its translation speed, whereupon high or low tRNA concentrations correlate with fast or slow translation rates, respectively [4–7]. Surprisingly, examinations of heterologous gene families from different organisms reveal a wide-spread phenomenon of conservation of rare codons in specific positions [8–10]. Since the codon bias differs between organisms [11], such findings point to the conservation of the codon-rarity trait rather than sequence conservation. This raises the possibility that the use of rare codons, which leads to local translational slowdowns, is of functional importance.
Synonymous mutations may influence the folding of proteins by altering the translation speed: large multi-domain proteins fold co-translationally [12–14], allowing sequential folding of individual segments as they emerge from the ribosome. Sequential folding narrows the search of the folding conformational space by limiting non-native inter-domain interactions. The folded segments can then function as scaffolds to guide the correct folding of the following proteins segments. Correct co-translational folding is suggested to depend on precise timing of events, and synonymous codon changes may impair this process by changing the translation elongation rate at critical checkpoints. For example, in S. cerevisiae and C. elegans modifications of the wobble anticodon uridine (U34) accelerate decoding speed. Loss of these modifications leads to translational stalling at critical points which results in widespread protein aggregation [15].
The effects of synonymous codon replacements have also been studied at the level of the single protein [16–21]. For example, synonymous mutations were reported to change the substrate specificity and solubility of the human ABC transporters MDR1 and CFTR [16,17], reduce the function of firefly luciferase [18], and enhance the expression of the Malaria antigen MSP1 [19]. These reports highlight the importance of codon usage bias and synonymous mutations, but do not provide a broad view of this phenomenon or a means to predict which of the rare codons are important for function.
Using bioinformatics tools, numerous attempts were made to formulate more general rules relating the usage of rare codons to protein folding and function [8,9,22–24]. However, these studies often lead to contradicting results: While some find enrichment of slow codons in domain linkers [22], others see no such tendency [8,9]. Attempts to find a correlation between slow codons and secondary structure motifs also resulted in divergent conclusions [23,24]. Key limitations leading to these controversies appear to be the inherent lack of appropriate controls in most bioinformatics approaches and the paucity of experimental validations of bioinformatics predictions.
Herein, to explore the relation between ‘slow codons’ and protein folding, we developed an approach that combines genome-scale bioinformatics analysis with single-gene experiments.
First, we analysed homologous gene groups from two divergent bacteria, Escherichia coli and Bacillus subtilis, to identify codon positions where the predicted translation rate for a group of homologs is significantly slower than predicted by the corresponding null model. Next, we generated a subset of mutants where conserved ‘slow’ codons were substituted with ‘fast’ ones, and experimentally studied the effects of these codon substitutions.
We report that in 3 out of the 8 tested genes, synonymous codon replacements resulted in deleterious effects in terms of protein expression, aggregation, and function. These results further confirm the functional importance of ‘silent’ mutations, their relation to co-translational folding, and demonstrate the benefits of combining computational and experimental approaches.
MATERIALS AND METHODS
Nucleotide and amino acid sequences for the entire E. coli and B. Subtilis proteome
Nucleotide sequences for all E. coli K12 strain MG1655 genes were retrieved from the Ensembl Genomes database [25]. Nucleotide sequences for all B. Subtilis strain 168 genes were retrieved from the KEGG database [26]. Amino acid sequences for all proteins were inferred from the nucleotide sequences. Genes termed pseudogenes or insertion elements according to UniProt [27] or the EcoGene database [28] annotation were excluded from processing.
Codon scoring indexes used in this work
Two indexes were used to score the codons: 1. tAI – The tRNA adaptation index [29]. This index scores codons based on cognate tRNA availability, taking into account the tRNAs gene copy number and the strength of the codon:anticodon base paring. The Sij weights for E. coli and B. Subtilis were calculated in [30]. Gene copy numbers for the individual tRNAs were used as proxies to concentration levels since a strong correlation was shown for those values in these two organisms [31,32]. 2. TDR – the typical decoding rate index [7]. This index rates codons based on data from ribosome profiling experiments, taking into account the distribution of ribosome dwelling time on each of the 61 codons across all its appearances in the genome. After filtering out noise caused by ribosome pauses which are not associated with the codon itself, the data resembles a normal distribution and its mean constitutes the codon score (see [7], Fig. 3).Fig. 3D).
Figure 3.
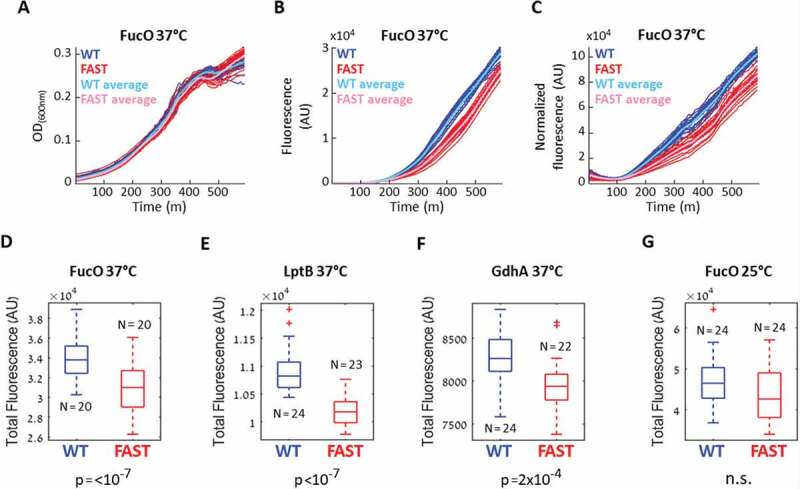
Effects of slow codon replacements on protein expression. (A-D) E. coli cells were transformed with a plasmid carrying either the WT or FAST GFP-fused variants of the Lactaldehyde reductase fucO. The optical density (A) and the GFP fluorescence (B) of the cells were continuously monitored, and the normalized fluorescence (C) was calculated as the Fluorescence/OD ratio. Shown are the values obtained for 20 replicates of the WT (blue) and the FAST (red) fucO variants, with averages shown in cyan and magenta, respectively. (D-G) Box plots of the total normalized fluorescence summarized over the entire growth curves of the WT and FAST variants of fucO (D),lptB (E), gdhA (F) when the experiments were conducted at 37°C, and of fucO when the experiment was conducted at 25°C (G). The indicated number of replicates (N) were compared using the Wilcoxon rank-sum test, n.s. = not significant
The TDR and tAI values from the two organisms were normalized by their standard deviation to bring them to the same scale. Table S1 lists the codon scores used in our calculations.
Orthologue groups of genes from the two organisms
A list of 1,120 Orthologue groups containing genes from both E. coli and B. Subtilis was taken from The Orthologous Matrix database [33].
Alignment, scoring and identification of slow codons
Amino acid sequences in each orthologue group were aligned using the Clustal Omega tool [34]. The DNA sequences were then projected onto the alignments. A Matlab program was used for codon scoring and identification of conserved slow codon stretches. The TDR and tAI indexes were used to score the codons and the process was carried out separately for each of the indexes. Codons along each gene were scored according to their index values. The values were then averaged using a sliding window of 10 codons. Windows containing more than 3 gaps were not given any values and were omitted from further analysis. An average score for the group alignment was then calculated for each position. This average score was termed the ‘real’ group score in that position.
For each group, 1000 altered variations of the sequence alignment were generated, by randomly selecting position pairs along the sequence that code for the same amino acid and swapping the codons used in those positions. This step was repeated until 90% of the codons were swapped. Codons in each randomly altered alignment were then scored, and a group average was calculated. The ‘real’ scores at each codon position were then compared to the 1,000 random ones and a z-score was calculated. A threshold of z-score of −2.8 (corresponding to a p-value of 0.0026) was chosen as a cut-off for ‘real’ scores that are significantly lower than the control random ones. This strict p-value of was chosen to lower the number of false positives. A z-score < −2.3 (corresponding to a p-value of 0.01) was allowed in cases where the same location was identified by both the TDR and tAI indexes. The first 50 codons were excluded from all calculations because they have been suggested to be involved in translation initiation and regulation of early elongation step [35]. Table S2 lists the 57 groups in which conserved slow codons were identified.
Choice of genes for experimental testing and design of the ‘Fast’ variants
The selection of 8 candidates for experimental testing was done by considering the length of the slow codon stretch, the z-score values and the number of genes in the group. Essential proteins (e.g. ribosomal proteins) were excluded to avoid complications of data interpretation due to pleiotropic effects. A list of the 8 groups chosen, the genes chosen to represent them in the experiments, and their attributes are presented in Table 1.
Table 1.
Experimentally tested genes and consequences of codon replacements
| Selected gene | Protein name | Index used | Slow codons positions | Modified codons | Fused GFP fluorescence of FAST variant | Other experimental results |
| fucO | Lactaldehyde reductase | TDR | 63 | 62–80 | Reduced fluorescence | Enhanced aggregation |
| tAI | 242–243 | 243–267 | No change in fluorescence | |||
| gdhA | NADP-specific glutamate dehydrogenase | TDR | 307 | 303–322 | Reduced fluorescence | Enhanced aggregation, reduced protein function |
| msbA | Lipid A export ATP-binding/permease protein MsbA | TDR | 456–462 | 454–473 | Inconsistent results | |
| alr | Alanine racemase, biosynthetic | tAI | 118 | 117–126 | Inconsistent results | |
| TDR | 334–337 | 314–347 | Inconsistent results | |||
| lptB | Lipopolysaccharide export system ATP-binding protein LptB | TDR | 152–154 | 150–166 | Reduced fluorescence | Enhanced aggregation |
| gltA | Citrate synthase | TDR | 227, 231–232 | 225–244 | Inconsistent results | No change in aggregation, no change in protein function |
| dapA | 4-hydroxy-tetrahydrodipicolinate synthase | TDR | 237–241 | 233–252 | No change in fluorescence | |
| mgsA | Methylglyoxal synthase | TDR | 87, 91–93 | 84–105 | Inconsistent results |
Notes : Eight genes representing orthologue groups with conserved slow codons were selected for experimental testing. The table details which index was used to identify the slow codons in each case, the location of the slow codons, the range of codons modified to produce the Fast variants, and whether the codons' modifications had an experimentally detectable deleterious consequence.
For each gene, a ‘Fast’ variant was designed by replacing a stretch of ~20 codons including and flanking the identified conserved slow codons with the highest ranking synonymous codon in each position. The sequence changes introduced to create the Fast variants are indicated in table S3.
Bacterial strains and plasmids
E. coli DH5α strain (Invitrogen) was used for plasmid purification and E. coli NEB5α strain (NEB) was used for cloning. E. coli BW25113 stain was used in the fluorescence and solubility assays and as a positive control in the complementation assays. E. coli stains ΔgltA and PA340 (double deletion ΔgdhA/ΔgltB) were used in the complementation assays of gltA and gdhA, respectively. Stains BW25113, ΔgltA and PA340 were obtained from the CGSC collection at the Yale University. LB media (10% NaCl, 10% Tryptone, and 5% Yeast extract) was used for inoculation of cultures, and Davis minimal media was used for the fluorescence and complementation assays. For solubility assays, Terrific broth (TB) medium was used for FucO, LB medium was used for LptB, and Davis minimal media was used for GdhA and GltA. Unless otherwise noted, 0.4% Glucose was used as a carbon source for growth in minimal media.
λDE3 Lysogenization
The BW25113 and ΔgltA strains were modified to introduce the T7 polymerase into their genome to enable IPTG-induced protein expression. Cloning and verification was done using the λDE3 Lysogenization Kit (Novagen) following the manufacturer instructions.
Plasmid cloning
Cloning of pET21 GFP plasmid
The GFP variant used in this work is enhanced GFP with cycle 3 mutations [36]. The documented mutations are S65T, F64L, F99S, M153T and V163A. Sequencing revealed that the variant also has the mutations H81A, L232H and S209L. The GFP bearing plasmid was a kind gift from the lab of prof. Bibi at the Weizmann institute, Israel. The GFP sequence was amplified from the plasmid adding a 5ʹ sequence containing restriction sites for NehI, KpnI and SacI, and containing a NotI restriction site at the 3ʹ end. The PCR product was cut with NehI and NotI enzymes and ligated to a pET21 vector which was cut with the same enzymes. Transformation and plasmid purification were followed by sequencing.
Cloning of GFP fused WT and Fast variants
The WT and Fast variants of gdhA, msbA, fucO, alr1, lptB, gltA, dapA and msgA were synthesized by Twist Bioscience (CA, USA) and inserted into a plasmid that contained a KpnI site at the 5ʹ side of the ORF and a SacI site at the 3ʹ end. The plasmids were then digested with KpnI and SacI and cloned into pET21-GFP vector cut with the same enzymes. Transformation and plasmid purification were followed by sequencing. Sequencing showed that for both variants of the gdhA-gfp gene, a synonymous mutation was introduced into the 100th amino acid of GFP, changing the TTC codon to TTT. This change is not expected to influence the results, as it occurs in both variants.
Cloning His-taged gdhA variants in pBAD plasmid
For the complementation assay of gdhA, the variants were cloned into a pBAD template plasmid with a C’ 6-His tag. The pBAD template and the gdhA-GFP plasmids were cut using NcoI and SacI and a ligation followed. Since the resulting plasmid had the His-tag shifted by (−1) base in reference to the ORF, additional PCR was done to introduce a point mutation adding 1 base to the linker sequence between the SacI site and the His-tag, rendering it in-frame. Cloning was verified by sequencing.
Fluorescence measurement of cells expressing WT and Fast variants
E. coli cells of strain BW25113 were transformed with a plasmid carrying the WT or the Fast variant of a gene. A smear of colonies was grown in LB supplemented with 100 µg/ml ampicillin at 37°C with vigorous shaking for several hours and then diluted to OD600 ~ 0.02 in 3 ml Davis minimal media supplemented with 100 µg/ml ampicillin and grown at 37°C with vigorous shaking for a few hours to allow cells to adjust to the minimal medium. Cells were then diluted to OD600 ~ 0.07 in Davis minimal media and grown in a 96 well plate in a plate reader (Infinite M200 pro, Tecan) for 10 hours. Growth (OD600) and GFP fluorescence (excitation wavelength 465 nm, emission wavelength 515 nm) were measured every 4 minutes. Gene expression was induced by addition of 0.1 mM IPTG after the first hour. The number of replicates varied between assays, ranging from 10 to 36.
Calculating statistical difference between WT and Fast variant fluorescence
For each replicate, normalized fluorescence at each time point was calculated as fluorescence/OD600 at that time point, after first subtracting the background values measured for a sample of pure medium. The area under the normalized fluorescence graph of each replicate was calculated and represented the total normalized fluorescence measured for the replicate. The values obtained for the WT replicates were compared to the values obtained for the Fast replicates using the Wilcoxon rank-sum test. Calculations were done using a homemade Matlab program.
Cell growth for mRNA quantification
Cells were grown and induced in a protocol similar to the one used in the fluorescence assay: E. coli cells of strain BW25113 were transformed with plasmids carrying the WT or Fast variant of a gene. A smear of colonies was grown in LB supplemented with 100 µg/ml ampicillin at 37°C with vigorous shaking for 3 hours and then diluted to OD600 ~ 0.05 in 3 ml Davis minimal media supplemented with 100 µg/ml ampicillin and grown at 37°C with vigorous shaking for a few hours to allow cells to adjust to the minimal medium. Cells were then diluted to OD600 ~ 0.05 in Davis minimal media in 3 separate tubes constituting 3 biological repeats, and grown for 11 hours. Gene expression was induced by addition of 0.1 mM IPTG after the first hour.
mRNA extraction and reverse-transcription PCR
Cells were harvested by centrifugation and washed twice with ice-cold PBS. Total RNA was extractted using RiboPure RNA Purification Kit (Ambion). 0.5–1 μg of purified RNA was reverse transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems). The quality of the extracted RNA was evaluated by agarose gel.
mRNA quantification using real-time PCR
RT-qPCR was performed on 1.6 pg of cDNA using PowerUp SYBR green master mix (Applied Biosystems) and 500 nM forward and reverse primers, designed with the Primer3web software (version 4.0.0) in the QuantStudio™ 3 Real-Time PCR System (Applied Biosystems). The transcription level of each gene of interest was normalized to that of a reference gene, rrsA (16s rRNA). Statistical analysis was performed using the QuantStudio™ Design and Analysis Desktop Software. RT-qPCR primers are described in Table S4.
Solubility assays
E. coli cells of strain BW25113 were transformed with plasmids carrying the WT or Fast variant of a gene. Swipes were grown overnight in 20 ml LB, diluted into TB (FucO), LB (LptB) or Davis minimal media (GdhA, GltA) and grown to mid log phase (OD600 of 2 for rich media, OD600 of 1 for minimal media). Gene expression was induced with 0.5 mM IPTG. After 1–2 hours cells were pelleted and re-suspended in 50 mM TRIS HCl pH7.5, 0.5 M NaCl buffer. Cells were disrupted by sonication (4 cycles of 10 seconds) and cell debris was pelleted by centrifugation at 3000xg. Concentration of supernatant sample was measured and brought to 10 µg/ml and kept as ‘Total’ samples. A volume of 200 µl of each ‘Total’ sample was ultra-centrifuged at 350,000xg. Supernatant samples were kept as ‘Soluble’ samples, and the pellets were re-suspended in 200 µl buffer using a Hamilton syringe and kept as ‘Aggregate’ samples. Samples were then analysed by Western Blot.
Western Blot analysis for solubility assays
Samples with equal amounts of total proteins were separated on polyacrylamide gels (concentration gradient 4%-20%), electro-blotted and probed with the primary antibody anti-GFP pAB 598 (MBL, 1:3000) and the secondary antibody HRP-conjugated anti-rabbit (1:10,000). Western blots were developed by enhanced chemiluminescence.
Evaluation of Western Blot band intensity
Comparison of band intensities in the western blot images was done using Fiji software. For each lane, the area that includes the relevant bands was selected and the total grey intensity value was measured. A value measured for the same area in an empty lane was subtracted from all values as background. The ratio of ‘Aggregate’ and ‘Soluble’ band intensity in each variant was calculated.
Complementation assay for ΔgdhA/ΔgltB strain
E. coli cells of PA340 strain [37] (ΔgdhA/ΔgltB) untransformed or transformed with the pBAD based gdhA variants were grown in LB supplemented with 100 µg/ml streptomycin (+100 µg/ml ampicillin for plasmid baring cells) at 37°C with vigorous shaking up to OD600 ~ 5 and then diluted to OD600 ~ 0.01 in Davis minimal media with 100 µg/ml streptomycin and 4% Glycerol, supplemented with 0.4 mM of the amino acids Threonine, Arginine, Histidine and Leucine, and 20 µg/ml Thiamine. 0.1% Arabinose was used for induction. Growth was monitored continuously in an automated plate reader (Infinite M200 pro, Tecan). BW25113 cells were used as positive control.
Complementation assay for ΔgltA strain
E. coli cells of strain ΔgltA untransformed or transformed with the pET21 based GFP-fused gltA variants were grown in LB supplemented with 30 µg/ml kanamycin (+100 µg/ml ampicillin for plasmid baring cells) at 37°C with vigorous shaking up to OD600 ~ 5 and then diluted to OD600 ~ 0.05 in Davis minimal media supplemented with the same antibiotics. 0.4% glucose or 0.8% acetate were used as a carbon source. Growth was monitored continuously in an automated plate reader (Infinite M200 pro, Tecan). BW25113 cells were used as positive control.
RESULTS
Identifying conserved slow codons in gene families from the entire genomes of E. coli and B. subtilis
The steps for identifying conserved slow codons are schematically shown in Fig. 1 and are outlined below. A more comprehensive description can be found in the methods section.
Figure 1.
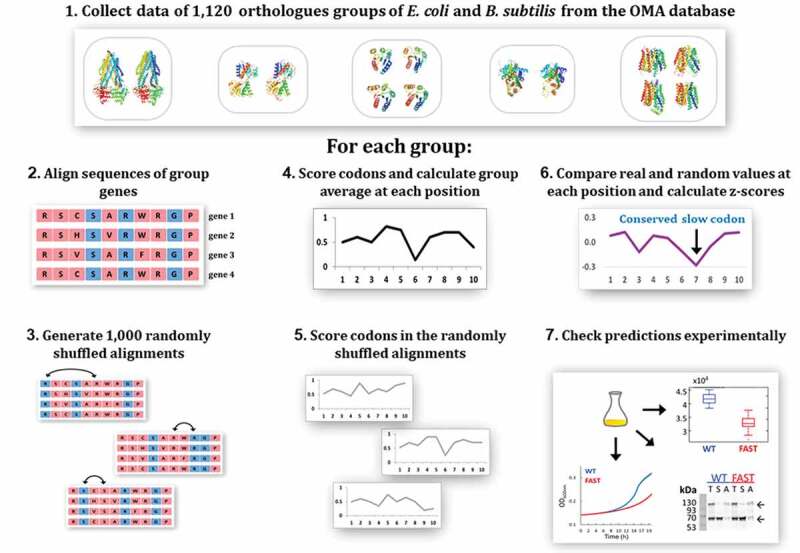
Overview of the methodology. 1. A list of 1,120 orthologue groups containing genes from both E. coli and B. subtilis was retrieved from the OMA database. For each one of these groups, steps 2–6 were performed. 2. The genes in the group were aligned using Clustal Omega. 3. A set of 1,000 randomized variants of the alignment was generated by shuffling synonymous codons along the sequence. 4. Codons in each gene were scored according to their predicted translation rate. The group average at each codon position was calculated. 5. Codon scoring and calculation of group average at each position was also performed for each of the 1,000 randomly-shuffled variants. 6. The ‘real’ codon score at each position was compared to the set of values received in that position for the 1,000 random variants and a z-score was calculated. A threshold of z-score = −2.8 was chosen as the bar under which a real codon score was considered to be significantly lower than control. Codons meeting this criterion are predicted by our model to have a functional role. 7. Out of the 57 homologous groups predicted by our model to contain functionally important slow codons, 8 were chosen for experimental validation. One gene from each of these groups was modified to replace the important slow codons with synonymous fast ones, and the modified gene was compared to the wild type gene in a set of experiments. See main text for details
To identify slow codons that are essential for protein folding we searched for homologous genes that use slow codons in the same positions. Underlying this approach is the hypothesis that homologous proteins are structurally similar, and if folding requires translation slow-down at a specific position we expect to find position-specific conserved usage of slow codons.
We chose to look at homologous proteins from two organisms, E. coli and B. subtilis. These two organisms preferentially use different synonymous codons [11], and therefore conservation of slow translation rate in the same position in homologous genes may suggest that the trait that underwent evolutionary selection was codon ‘speed’ rather than sequence conservation.
To this end, we used the OMA database Orthologous Matrix [33] to extract a list of 1,120 orthologue groups that contain at least one gene from each organism, and then aligned the genes comprising each group using the Clustal Omega tool [34].
A key subsequent step was to score the translation speed of the 61 sense codons. Of the many codon-scoring indexes that were suggested over the years [1,38,39] we chose two that have a direct link to translation rate. The first is the well-established tRNA Adaptation Index (tAI) introduced in 2004 by dos Reis et al. [29], which evaluates the relative translation rates of the 61 codons based on a theoretical model of translation which assumes the tRNA levels are the major determinants of translation speed. This index scores each codon by considering tRNA availability and the strength of codon-anticodon interaction.
The second index that we used was the Typical Decoding Rate (TDR, [7,40]) index. Unlike other available indexes the TDR index does not attempt to score the codons based on theoretical predictions, but rather uses ribosome profiling data [41], which provide a proxy to actual in vivo translation rates. As shown in figure S1, the correlation between the values assigned to each codon by these two indexes is significant but not very high (Spearman’s rank correlation between E. coli tAI and TDR values is ρ = 0.44, p = 3x10−[4]).
We reasoned that the exact location of the slow codons may be slightly shifted in homologous genes, and therefore used a sliding window of 10 codons which is the footprint of the ribosome. As shown in Figure S2, this averaging also smooths the noisy translation rate landscape and allows better visualization of the data.
Next, we calculated the average value in each position of the alignment, to get the group score at each position. A low average score in a certain position represents a tendency of the genes in the group to use slow codons at that position, but does not necessarily mean slow codons were selected for use in that position: possibly, this position underwent selective pressure for an amino acid which is coded only by slow codons. To evaluate if sites of slow codons conservation represent selection for translation speed or are a result of amino acid selection we devised a randomized control: For each group of homologous genes, 1,000 alternative variants of the group alignment were created by randomly switching pairs of synonymous codons along the alignment. For each variant, replacements were carried until a 90% difference from the original sequence was reached. Importantly, this randomization does not change the amino acid sequence, the GC content or the overall codon usage of the genes, but only influences the locations of slow codons along the genes. The score at each codon position in the real alignment was compared to the scores obtained at that position for the 1,000 random control variants. The real score was considered significantly lower than the random ones if the z-score value obtained in the comparison was below a threshold of −2.8 (equivalent to a p value = 0.0026). Positions at which the real codon scores are significantly lower than random controls imply a selective pressure on proteins with that particular structure to use slow codons at this location, presumably assisting correct folding by attenuating the translation speed [42].
In 57 homologous groups we could identify slow codons that met our cut-off threshold selection (Supp. Table S2). We excluded from consideration slow codons that appeared in the first 50 codons as these have been suggested to be involved in translation initiation and regulation of early elongation step [35].
Choosing candidates for experimental testing and designing gene variants
The above-described computational analysis identified 57 groups of homologous that harbour 1–2 sites of conserved slow codons. Of these 57 groups we chose 8 to experimentally test the importance of conserved slow codons for protein expression, folding and structure. The chosen 8 groups were selected based on criteria such as the length of the slow codon stretch, the z-score values, and the number of genes in the group. Essential proteins (e.g. ribosomal proteins) were excluded since we reasoned that alterations in their expression levels would likely lead to pleiotropic effects that would be difficult to interpret. Then, from each group we selected an E. coli representative since this organism was the host for the subsequent experiments (Table 1).
To test if the identified slow codons have a role in folding we replaced them with synonymous codons predicted to have the highest translation rate. As an example, Fig. 2A shows the codon replacements performed for the gene gdhA, and the resulting TDR-score changes. In gdhA, the modified section spans codons 303–320, and includes 12 synonymous changes. Table S3 specifies the codon replacements performed to generate all of the Fast variants. For two genes, fucO and alr, different locations were suggested by the TDR and tAI indexes (Table 1), and therefore two different Fast variants were generated (Table S3). The sequences coding the wildtype and the modified versions of the 8 genes were inserted into a pET expression vector which harbours an in-frame C-terminal fusion of the green fluorescent protein (GFP, Fig. 2B). The use of a C-terminal GFP fusion as a reporter for protein expression and folding is based on the assumption that the expression level of the in-frame fluorescent protein is related to the expression level of the upstream protein. Misfolding or aggregation of the upstream protein has been shown to affect the expression of the downstream-fused GFP and hence fluorescence [43]. Although GFP has been shown at instances to affect the function, localization, expression or solubility of its fused partners [44–46], it is arguably still the most broadly-used reporter fusion [47]. To curb GFP-related solubility limitations we used the cycle 3 mutant, with its reduced aggregation tendency and improved solubility [36].
Figure 2.
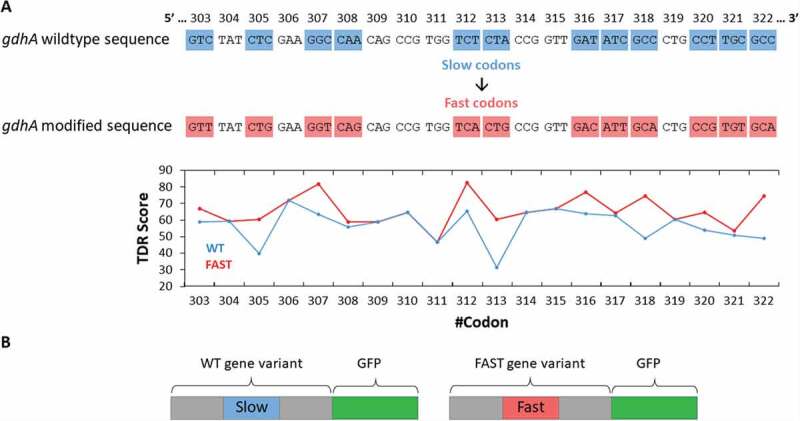
Design of the WT and Fast variants of the selected genes. (A, top) Shown is the nucleotide sequence of a region of gdhA, the location of which was found to be conserved within the group of homologues. In gdhA this region contains 12 slow codons, highlighted in blue (original WT codons) or red (Fast variant). (A, bottom) A plot of the TDR index values of the WT (blue) and Fast (red) variants. (B) An illustration of the two GFP-fused variants created for each gene
Henceforth, the wild type variants (whether gene or protein) are indicated as ‘WT’, and the variants where the conserved slow codons (17–20 codons in length) were replaced with optimized ones are indicated as ‘Fast’ (Table S3).
Fluorescence assays demonstrate impaired folding of the ‘Fast’ protein variants
E. coli WT strain BW25113 cells where transformed with plasmids carrying either the WT or Fast variants. The growth and fluorescence of multiple biological replicates (N > 10) was then continuously measured for 10 hours. The normalized fluorescence at each time point was calculated as the ratio between the fluorescence and optical density, and the total normalized fluorescence was calculated as the area under the curve of the normalized fluorescence graph (See methods). The resulting values obtained for the WT and Fast variants were then compared using the Wilcoxon rank-sum test to evaluate if the two distributions significantly differ. Fig. 3 shows an example of such experiment conducted with the Lactaldehyde reductase FucO [48], where the Fast variant was designed according to the TDR index prediction (Table 1, Table S3). As shown, while the growth of the WT and Fast variants was indistinguishable (Fig. 3A), the GFP fluorescence of the WT variant was higher (Fig. 3B) as was its normalized fluorescence (Fig. 3C), and this difference was found to be extremely significant (p < 10−[7],
The reduced fluorescence observed for the fast variants of fucO could also be the result of reduced mRNA levels. To examine this possibility, we quantified the mRNA levels of fucO using RT-qPCR. Total RNA was extracted from cells grown under the same conditions used in the fluorescence assay, and the mRNA levels of the WT and Fast variants were compared. As shown Supplementary (Fig. 3), the mRNA levels of FucO were unaltered by the codon replacements demonstrating that the lower fluorescence of the Fast variant stems from reduced expression or stability at the protein level.
In addition to fucO, for two other tested genes, the Lipopolysaccharide export system ATP-binding protein LptB and the NADP-specific glutamate dehydrogenase GdhA, we observed reduced normalized fluorescence of the Fast variants compared to the WT variants (p value < 0.001, Fig. 3 E, F). As observed for FucO, the mRNA levels of the Fast variants of LptB and GdhA were similar to those of the WT variants, indicating that their transcription was not compromised by the codon replacements Supplementary (Fig. 3).
The experiments shown in Fig. 3A-Fwere conducted at 37°C, a temperature that supports rapid protein expression [49]. We reasoned that when protein translation is overall fast its slowdown at folding bottlenecks is likely to be consequential. In contrast, when protein translation is overall slow, the importance of local translation speed attenuations is likely to play a more minor role. As such, we expected the adverse effects of the ‘fast’ codon substitutions to be milder when translation speed is already slowed down.
It is well-established that at lower temperatures bacterial growth is slower, and so is protein synthesis [50]. Accordingly, lowering the growth temperature is often used to circumvent folding problems [51].
In line with these predictions, and in contrast to what was observed at 37°C (Fig. 3D), when the experiment was conducted at 25°C we observed no difference in fluorescence between the WT and Fast variants of fucO (Fig. 3G), supporting a role for strategically positioned conserved slow codons in attenuating translation speed under conditions of fast growth/fast translation.
Contradictory to our expectations, for the other 5 tested genes (Table 1) we did not observe a consistent difference in fluorescence between the two variants. The possible reasons for this contradiction are later discussed.
Aggregation tendency of the ‘Fast’ variants is higher
The folding (and therefore fluorescence) of C-terminally fused GFP has been shown to be associated with the expression and/or folding of the upstream protein [43]. Therefore, the reduced fluorescence of the Fast variants of FucO, LptB and GdhA could stem either from reduced protein expression or from a reduction in the fraction of the correctly folded protein. In an attempt to distinguish between these two possibilities, we extracted complete protein lysates from cells expressing the WT or the Fast variants of FucO, LptB and GdhA, and separated the aggregate and soluble fractions by ultracentrifugation at 350,000xg. The fractions were then separated by SDS-PAGE and visualized by immunoblotting with an anti-GFP antibody. Densitometry analysis of the ‘Total’ lanes in Fig. 4A-C suggested a modest ~15% (fucO, gdhA) or a substantial ~250% (lptB) increase in the amount of total GFP-tagged protein that was expressed by the Fast variants. These results suggest that the lower fluorescence observed for the Fast variants of FucO, LptB and GdhA (Figure A-F) in not a result of impaired translational efficiency. However, the Fast variants seemed to be more susceptible to aggregation or degradation. For example, as shown for the Lactaldehyde reductase FucO (Fig. 4A), the aggregate/soluble ratio for the WT is ~1:1, while for the Fast variant it is 2:1. Additional evidence for the suggestion that the Fast variant of FucO is more aggregate-prone may be deduced from a unique property of the fused GFP: in SDS-PAGE gels, GFP does not fully denature, and its folded form runs ~13kDa faster [52]. Accordingly, two prominent bands are visible in all lanes (arrows in left panel of Fig. 4A), representing denatured FucO-denatured GFP (MW~66 kDa) and denatured FucO-folded GFP (MW~53 kDa). In the Fast variant a higher proportion of the higher MW band (unfolded GFP) is observed (compare lanes 3 and 6), supporting the idea that the Fast variant is more aggregation prone.
Figure 4.
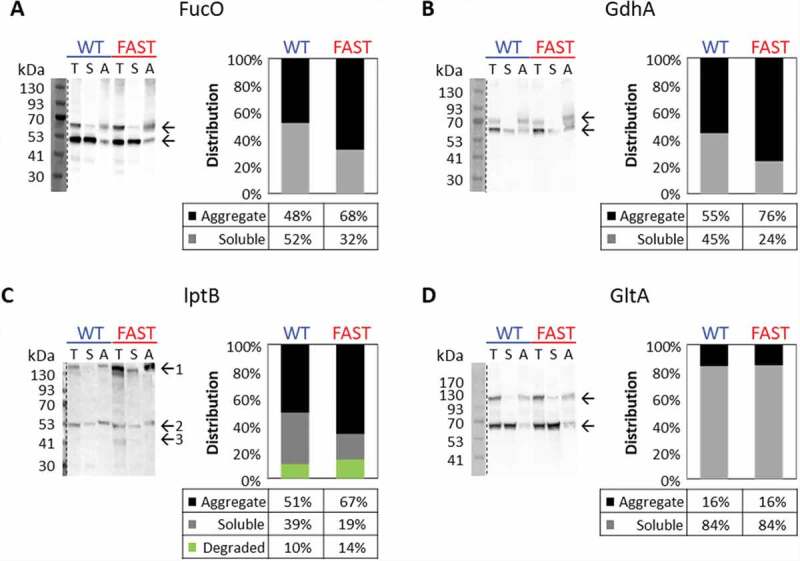
Effects of slow codon replacements on protein aggregation. Left panels: Western blot analysis using an Anti-GFP antibody of the soluble and aggregate fractions of lysates from cells expressing either the WT or FAST variants of FucO-GFP (A), GdhA-GFP (B), LptB-GFP (C), or GltA-GFP (D). In A,B,D, and top and bottom arrows indicate unfolded and folded fused GFP, respectively. Only in C, top, middle, or bottom arrows indicate aggregated, soluble, and degraded fractions, respectively. The intensity of the bands shown in the left panels was calculated using the Fiji software, and the right panels show the distribution (in percentage) of the aggregate (black), soluble (grey) and degraded (green, C only) fractions. Shown are representative results obtained at least three times under multiple conditions (variations in media and length of induction phase)
Fig. 4B shows the western blot analysis for GdhA, another protein suggested by the GFP fluorescence growth experiments (Fig. 3F) to be sensitive to replacements of its slow codons. As shown (Fig. 4B, right), the ratio of aggregate/soluble protein was higher for the Fast variant. In addition, two bands are visible, with the higher molecular one (corresponding to unfolded GFP) appearing only in the aggregated fraction, and more prominently so in the Fast variant. Also for LptB (another protein for which we observed decreased fluorescence of the Fast variant), the ratio of aggregate/soluble protein was higher for the Fast variant (Fig. 4C).
In Fig. 4C, in addition to the band corresponding to the expected size of LptB-GFP (middle arrow), a band with a molecular weight higher than 130kDa is also apparent (top arrow). This band represents a high-oligomer aggregate, and most of it appears in the insoluble fractions. This band is much more prominent in the Fast variant (compare lanes 1 and 4) and is mostly insoluble (Fig. 4C, compare lanes 4 and 6). This observation is in line with our hypothesis that replacement of strategically-positioned slow codons with faster-translating ones may lead to over-expression of misfolded proteins. Finally, a lower molecular-weight protein band (~41kDA, bottom arrow) is observed only in the Fast variant (Fig. 4C, lane 4). Since we have used anti-GFP antibodies in conjunction with a C-terminal GFP fusion construct this band likely represents protein degradation. Fig. 4C shows the densitometry calculation for the bands in the ‘Total’ fraction lanes (lane 1 for the WT variant and lane 4 for the Fast variant), showing the relative distribution of aggregated, soluble and degraded protein. As can be appreciated, the proportion of ‘healthy’ protein is ~2-fold lower for the Fast variant.
To further establish the correlation between the fluorescence (Fig. 3) and solubility (Fig. 4) assays we also tested one protein (GltA), for which we did not detect a changed in fluorescence between the Fast and the WT variants (Table 1). In line with this result, the tendency to aggregate was the same in both variants, suggesting that GltA is resilient to the codons replacement we performed. An explanation for this resilience may be found in the high intrinsic solubility of GltA (Soluble fraction 84%, compared to 39–52% of LptB, GdhA and FucO). This may imply that GltA is an especially good folder, and therefore perhaps more resilient to the codons replacements.
The results of the aggregation/solubility assays described above corroborate the results obtained in the fluorescence assays for these 4 genes, and also perhaps provide a clue regarding the underlying mechanism. While the reduced GFP fluorescence of the Fast variants could have been the result of reduced expression rather than misfolding, the immunoblotting images and its densitometry analysis reveal similar total expression levels for the WT and Fast variants for FucO and GdhA, and even enhanced expression of the Fast variant in LptB, suggesting the reduced fluorescence was a result of protein misfolding/aggregation, rather than simply reduced expression.
Impaired in vivo function of a Fast variant
Next, we wished to test whether ‘slow-to-fast’ synonymous codon replacements also have an effect on in vivo protein function. For this we tested the ability of GdhA (a glutamate dehydrogenase) to complement the growth of the E. coli glutamine auxotroph strain ΔgdhA/ΔgltB [37].
As shown (Fig. 5A), in glutamine-replete media, WT E. coli and the glutamine auxotroph strain grew very similarly, and plasmid-driven expression of either the WT or Fast variants of GdhA had virtually no effect on growth. In contrast, in glutamine-depleted media (Fig. 5B) the deletion strain completely failed to grow while the WT strain grew normally. Importantly, under these conditions of glutamine depletion, the WT variant of GdhA restored growth much more efficiently than the Fast variant (Fig. 5B, p = 3.65x105[5], Wilcoxon rank-sum test). This advantage of the WT variant over the Fast one is consistent with the results obtained for GdhA using the GFP-fluorescence assays (Fig. 3F) and the solubility experiments (Fig. 4B).
Figure 5.
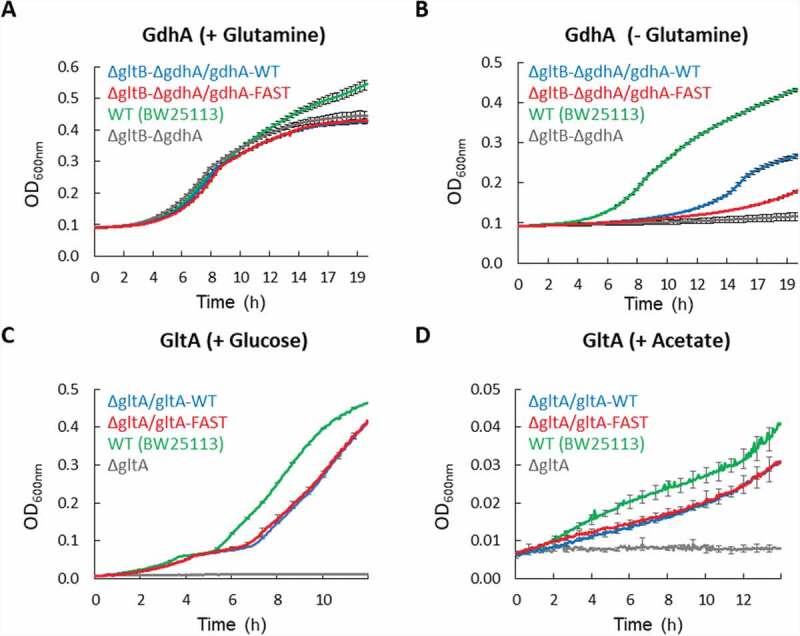
Effects of slow codon replacements on protein function. (A, B) WT E. coli (green) or a strain deficient in utilization of glutamine (ΔgdhA/ΔgltB, all other curves) were grown in minimal media in the presence (A) or absence (B) of 0.4% glutamine. Cells of the deletion strain were transformed with plasmids encoding the Fast or WT variants of gdhA, as indicated. Shown are mean values of 12 biological replicates, and error bars represent standard deviations. (C, D) WT E. coli cells (green) or a strain deficient in carbohydrate metabolism (ΔgltA, all other curves) were grown in minimal media supplemented with 0.4% Glucose (C) or 0.8% Acetate (D) as a sole carbon source. Cells of the deletion strain were transformed with plasmids encoding the Fast or WT variants of gltA, as indicated. Shown are mean values of 3 biological replicates, and error bars represent standard deviations
We next conducted similar growth-complementation assays with GltA, a citrate synthase and a key enzyme in carbohydrate metabolism. As demonstrated above, we observed GltA to be an especially soluble protein that is insensitive to ‘slow-to-fast’ synonymous codon replacements. In line with these observations, under multiple experimental conditions, the expression of either the WT or Fast variants of GltA equally compensated for the lack of the endogenous gltA gene (Fig. 5 C, D), in line with the suggested robust folding/solubility of this protein.
DISCUSSION
Diverging from the central dogma, synonymous mutations have been reported to have dramatic effects on protein expression and function [16,17]. These unexpected observations inspired extensive research of codon usage. To date, most studies applied either a genome scale bioinformatics approach or a single case experimental one.
Here we aimed to resolve this gap by combining computations and experiments. We demonstrate the ability to detect regions of slow codons, mutations in which affect the functionality of the translated proteins. The results reported here suggest that the effect of the slow codons on protein functionality varies and may be condition specific, highlighting the complexity of the co-translational folding process. We hope that our study will promote further research on this topic in the future.
The aim of this study was to establish an approach for detecting slow codons that are functionally important. In the future, the approach used here can be improved both computationally and experimentally. For example, we included orthologue gene groups only from two organisms, E. coli and B subtilis, and our model found conserved slow codons in ~ 5% of the groups (57 out of the 1,120 groups). In a more comprehensive computational study, Chaney et al. [8] examined homologue groups from 76 organisms across the 3 domains of life, and found conserved slow codons in 15% of the groups. Using a larger set of organisms should improve the predictions of our model.
In addition, the models we used are highly simplified, and neglect important aspects that are not codon-centric. For example, the tAI index does not consider factors that affect protein translation and folding such as mRNA secondary structure [53,54], interactions of positively charged amino acids with the ribosome’s exit tunnel [55,56], mRNA-rRNA hybridization [57,58], and chaperones [59,60]. Similarly, the TDR index is inherently biased by the exclusion of polysomes, which likely play an important part in translation attenuation. Moreover, the models assume constant tRNA levels. However, tRNA isodecoders levels can vary in a cellular specific manner, during the cellular life cycle, and depend on environmental conditions [61]. Accurate modelling of such aspects is likely to give more significant results than the ones we reported. Unfortunately, at present, the relevant parameters of such models in the analysed organisms are unavailable and therefore could not be included in the model. Developing more accurate computational models of translation rates in many organisms is expected to improve the performances of our approach.
From an experimental perspective, the experiments that we performed did not directly measure protein folding and function and could not detect small changes in protein conformation. Thus, a mutation with a small but biologically significant effect on protein folding can be selected for during evolution but its effect on co-translational folding may be too small to be detected by our methods. In an elegant study, Buhr et al. [62] used NMR, protease-digestion and Förster resonance energy transfer (FRET) to directly evaluate the effects of synonymous mutations on the folding of gamma-B crystallin. Incorporating such direct measurements of protein structure and folding with the approach we present in this work is likely to provide a deeper understanding of the effects of synonymous mutations on protein translation, structure and function.
An additional experimental point to consider is throughput: the effect of the slower codon on the protein’s functionality may be relevant only under certain conditions. Thus, improving our screening pipeline by incorporating high throughput screening methods will increase the likelihood of identifying more cases where slow codons have a functional role.
Nevertheless, despite the above-mentioned limitations of our model and experimental design, experimental validation revealed that in 3 out of the 8 tested cases we correctly predicted the functional importance of slow codons. This relative success rate demonstrates the usefulness of this model and supports the hypothesis that even short stretches of slow codons can be very important for correct protein expression and function. Possibly, the other 5 tested cases are also functionally relevant yet were undetected due to the computational and experimental limitations mentioned above.
Finally, of the 1,120 orthologue groups we analysed we identified only 57 harbouring conserved slow codons. This low number could be explained by our choice of the threshold (α < 0.0026). Choosing a more permissive threshold (α < 0.01) leads to identification of conserved slow codons in about 350 groups. However, we preferred to err on the side of identifying fewer cases, rather than identifying false positives. It is possible that many of these cases are also functional and can be detected in the future with improved computational models and experimental screening abilities.
Supplementary Material
Acknowledgments
This work was supported by grants from NATO Science for Peace and Security Program (SPS Project G4622, OL, NBT), the Israeli Academy of Sciences Project 1006/18 (OL).
Funding Statement
This work was supported by the Ministry of Science and Technology, Israel; North Atlantic Treaty Organization; Israel Science Foundation.
Disclosure statement
The authors declare no conflict of interest.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the Ecoli translational system. J Mol Biol. 1981;151:389–409. [DOI] [PubMed] [Google Scholar]
- [2].Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2(1):13–34. [DOI] [PubMed] [Google Scholar]
- [3].Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10(22):7055–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sørensen MA, Kurland CG. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 1989;207(2):365–377. [DOI] [PubMed] [Google Scholar]
- [5].Andersson SGE, Kurland CG. Codon preferences in free-living microorganisms. Microbiol Rev. 1990;54(2):198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sørensen MA, Pedersen S. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J Mol Biol. 1991;222(2):265–280. [DOI] [PubMed] [Google Scholar]
- [7].Dana A, Tuller T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014;42(14):9171–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chaney JL, Steele A, Carmichael R, et al. Widespread position-specific conservation of synonymous rare codons within coding sequences. PLoS Comput Biol. 2017;13:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chartier M, Gaudreault F, Najmanovich R. Large-scale analysis of conserved rare codon clusters suggests an involvement in co-translational molecular recognition events. Bioinformatics. 2012;28(11):1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jacobs WM, Shakhnovich EI. Evidence of evolutionary selection for cotranslational folding. Proc Natl Acad Sci U S A. 2017;114(43):11434–11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kanaya S, Yamada Y, Kudo Y, et al. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238(1):143–155. [DOI] [PubMed] [Google Scholar]
- [12].Netzer WJ, Hartl FU. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature. 1997;388(6640):343–349. [DOI] [PubMed] [Google Scholar]
- [13].Chaney JL, Clark PL. Roles for Synonymous Codon Usage in Protein Biogenesis. Annu Rev Biophys. 2015;44(1):143–166. [DOI] [PubMed] [Google Scholar]
- [14].Thommen M, Holtkamp W, Rodnina MV. Co-translational protein folding: progress and methods. Curr Opin Struct Biol. 2017;42:83–89. [DOI] [PubMed] [Google Scholar]
- [15].Nedialkova DD, Leidel SA. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell. 2015;161(7):1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kim SJ, Yoon JS, Shishido H, et al. Translational tuning optimizes nascent protein folding in cells. Science. 2015;348(6233):444–448. (80-). . [DOI] [PubMed] [Google Scholar]
- [17].Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:(5811):525–528. (80-). [DOI] [PubMed] [Google Scholar]
- [18].Yu CH, Dang Y, Zhou Z, et al. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol Cell. 2015;59(5):744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Angov E, Hillier CJ, Kincaid RL, et al. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS One. 2008;3(5):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cortazzo P, Cerveñansky C, Martín M, et al. Silent mutations affect in vivo protein folding in Escherichia coli. Biochem Biophys Res Commun. 2002;293(1):537–541. [DOI] [PubMed] [Google Scholar]
- [21].Zhong C, Wei P, Zhang YHP. Enhancing functional expression of codon-optimized heterologous enzymes in Escherichia coli BL21(DE3) by selective introduction of synonymous rare codons. Biotechnol Bioeng. 2017;114(5):1054–1064. [DOI] [PubMed] [Google Scholar]
- [22].Zhang G, Ignatova Z. Generic algorithm to predict the speed of translational elongation: implications for protein biogenesis. PLoS One. 2009;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saunders R, Cm D. Synonymous codon usage influences the local protein structure observed. Nucleic Acids Res. 2010;38:6719–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pechmann S, Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol. 2013;20(2):237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kersey PJ, Lawson D, Birney E, et al. Ensembl Genomes: extending Ensembl across the taxonomic space. Nucleic Acids Res. 2009;38(suppl_1):563–569. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aoki KF, Kanehisa M. Using the KEGG Database Resource. Curr Protoc Bioinforma. 2005;11(1):1–54. [DOI] [PubMed] [Google Scholar]
- [27].Bateman A, Martin MJ, O’Donovan C, et al. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rudd KE. EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res. 2000;28(1):60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32(17):5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Renana S, Tamir T. Modelling the efficiency of codon-tRNA interactions based on codon usage bias. DNA Res. 2014;21(5):511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol. 1996;260(5):649–663. [DOI] [PubMed] [Google Scholar]
- [32].Percudani R, Pavesi A, Transfer OS. RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol. 1997;268(2):322–330. [DOI] [PubMed] [Google Scholar]
- [33].Altenhoff AM, Glover NM, Train CM, et al. The OMA orthology database in 2018: retrieving evolutionary relationships among all domains of life through richer web and programmatic interfaces. Nucleic Acids Res. 2018;46(D1):D477–85. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tuller T, Carmi A, Vestsigian K, et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141(2):344–354. [DOI] [PubMed] [Google Scholar]
- [36].Fukuda H, Arai M, Kuwajima K. Folding of Green Fluorescent Protein and the Cycle3 Mutant. Biochemistry. 2000;39(39):12025–12032. [DOI] [PubMed] [Google Scholar]
- [37].Lozoya E, Sanchez-Pescador R, Covarrubias A, et al. Tight linkage of genes that encode the two glutamate synthase subunits of Escherichia coli K-12. J Bacteriol. 1980;144(2):616–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sharp PM, Li WH. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].The WF. apos;effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. [DOI] [PubMed] [Google Scholar]
- [40].Dana A, Tuller T. Mean of the typical decoding rates: a New translation efficiency index based on the analysis of ribosome profiling data. G3 Genes, Genomes, Genet. 2015;5::73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ingolia NT, Ghaemmaghami S, Newman JRS, et al. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:(5924):218–223. (80-). . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Komar AA. A pause for thought along the co-translational folding pathway. Trends Biochem Sci. 2009;34(1):16–24. [DOI] [PubMed] [Google Scholar]
- [43].Waldo GS, Standish BM, Berendzen J, et al. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17(7):691–695. [DOI] [PubMed] [Google Scholar]
- [44].Huang L, Pike D, Sleat DE, et al. Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS One. 2014;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Norris SR, Núñez MF, Verhey KJ. Influence of fluorescent tag on the motility properties of kinesin-1 in single-molecule assays. Biophys J. 2015;108(5):1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Swulius MT, Jensen GJ. The helical mreb cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-terminal yellow fluorescent protein tag. J Bacteriol. 2012;194(23):6382–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Day RN, Davidson MW. The Fluorescent Protein Revolution. Boca Raton: CRC Press; 2014. [Google Scholar]
- [48].Boronat A, Aguilar J. Metabolism of L-fucose and L-rhamnose in Escherichia coli: differences in induction of propanediol oxidoreductase. J Bacteriol. 1981;147(1):181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yamamori T, Ito K, Nakamura Y, et al. Transient regulation of protein synthesis in Escherichia coli upon shift-up of growth temperature. J Bacteriol. 1978;134:1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Farewell A, Neidhardt FC. Effect of temperature on in vivo protein synthetic capacity in Escherichia coli. J Bacteriol. 1998;180(17):4704–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schein CH, MHN . Formation of soluble recombinant proteins in Escherichia coli is favored by lower growth temperature. Nat Biotechnol. 1988;6:291–294. [Google Scholar]
- [52].Schlegel S, Löfblom J, Lee C, et al. Optimizing membrane protein overexpression in the Escherichia coli strain Lemo21(DE3). J Mol Biol. 2012;423(4):648–659. [DOI] [PubMed] [Google Scholar]
- [53].Faure G, Ogurtsov AY, Shabalina SA, et al. Role of mRNA structure in the control of protein folding. Nucleic Acids Res. 2016;44(22):10898–10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tuller T, Veksler-Lublinsky I, Gazit N, et al. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011;12(11):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sabi R, Tuller T. A comparative genomics study on the effect of individual amino acids on ribosome stalling. BMC Genomics. 2015;16(S10):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lu J, Deutsch C. Electrostatics in the Ribosomal Tunnel Modulate Chain Elongation Rates. J Mol Biol. 2008;384:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Elitzur SB, Cohen-Kupiec R, Yacobi D, et al. Prokaryotic rRNA-mRNA interactions are involved in all translation steps and shape bacterial transcripts. bioRxiv. 2020;2020(07):24.220731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484(7395):538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70(1):603–647. [DOI] [PubMed] [Google Scholar]
- [60].Ellis RJ, Hartl FU. Principles of protein folding in the cellular environment. Curr Opin Struct Biol. 1999;9:102–110. [DOI] [PubMed] [Google Scholar]
- [61].Torres AG, Reina O, Attolini CSO, et al. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc Natl Acad Sci U S A. 2019;116(17):8451–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Buhr F, Jha S, Thommen M, et al. Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol Cell. 2016;61(3):341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.