

ABSTRACT
During spliceosome assembly, interactions that bring the 5′ and 3′ ends of an intron in proximity are critical for the production of mature mRNA. Here, we report synergistic roles for the stem-loops 3 (SL3) and 4 (SL4) of the human U1 small nuclear RNA (snRNA) in maintaining the optimal U1 snRNP function, and formation of cross-intron contact with the U2 snRNP. We find that SL3 and SL4 bind distinct spliceosomal proteins and combining a U1 snRNA activity assay with siRNA-mediated knockdown, we demonstrate that SL3 and SL4 act through the RNA helicase UAP56 and the U2 protein SF3A1, respectively. In vitro analysis using UV crosslinking and splicing assays indicated that SL3 likely promotes the SL4-SF3A1 interaction leading to enhancement of A complex formation and pre-mRNA splicing. Overall, these results highlight the vital role of the distinct contacts of SL3 and SL4 in bridging the pre-mRNA bound U1 and U2 snRNPs during the early steps of human spliceosome assembly.
KEYWORDS: U1 snRNA, Stem-loop, SF3A1, UAP56, Spliceosome, A complex, E complex, U1 snRNP, U2 snRNP, DDX39B, URH49, DDX39A
Introduction
The spliceosome assembles from sequential binding of five small nuclear RNPs (snRNPs: U1, U2, U4, U5, and U6) and many auxiliary proteins [1]. First, binding of the U1 snRNP to the 5′ splice site (5′-ss), splicing factor 1 (SF1) to the branch-point sequence, and the U2 auxiliary factor (U2AF) 65/35 dimer to the polypyrimidine tract and the 3′ splice site (3′-ss), respectively, forms the early (E) complex. The U2 snRNP loosely associates with the E complex by a U2AF65-SF3B1 contact [2,3]. Following this, RNA helicases UAP56 and Prp5 facilitate conversion of the E to A complex, which involves stable binding of U2 to the pre-mRNA by basepairing of the U2 snRNA to the branchpoint sequence [4–6]. The U4/U6.U5 tri-snRNP is then recruited to form the pre-B complex, and the U1 and U4 snRNPs are released by the actions of helicases Prp28 and Brr2, respectively [7–11]. In subsequent steps, complexes containing the U2/U6/U5 snRNPs perform splicing catalysis [12–15]. Recent cryo-EM studies of the budding yeast and human spliceosomal complexes have revealed the nature of molecular contacts and transitions in the later complexes. However, information on early interactions that lead to the formation of a stable A complex is lacking, especially for the human spliceosome.
The human and budding yeast U1 snRNPs differ significantly in their composition. Human U1 small nuclear RNA (snRNA) is 164 nucleotides (nts) long and folds into a structure consisting of four stem-loops. It interacts with the seven-member (B/B′, D1, D2, D3, E, F, and G) Sm-ring and three U1-specific proteins (U1-70k, U1C, and U1A). In the mature U1 particle, the first three stem-loops are separated from the terminal stem-loop 4 (SL4) by the Sm-ring [16,17] (Fig. 1A). The yeast U1 snRNA is much longer (568 nts) than its human paralog and lacks a structure analogous to SL4 downstream of the Sm ring [18–20]. The yeast stem-loop 3 (SL3) region is 15 times the size of SL3 in human U1 and folds into seven stem-loops. Yeast U1 contains seven additional particle-specific proteins, namely Prp39, Prp40 (human Prp40), Prp42, Nam8 (human TIA-1), Snu56, LUC7 (human LUC7L), and Snu71 (human RBM25). In humans, Prp40, TIA-1, LUC7L, and RBM25 act independently as alternative splicing factors and orthologs for the other proteins have not been identified [21–23].
Figure 1.
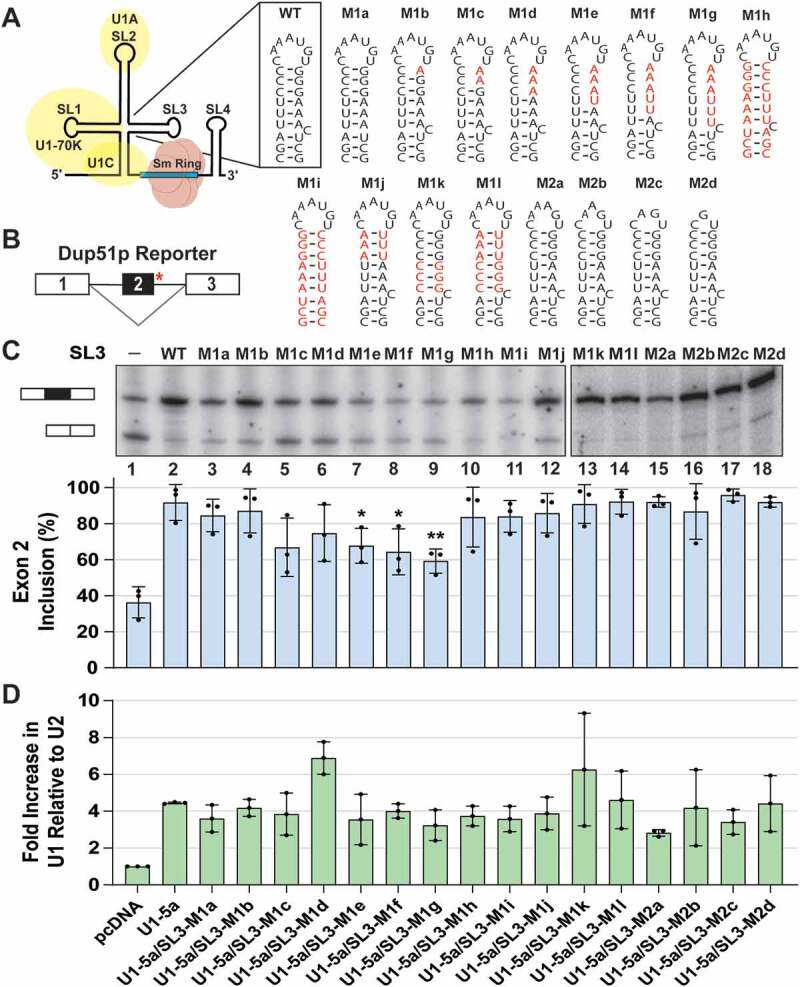
Stem-loop 3 of the U1 snRNA is important for U1 function. (A) Schematic diagram of the U1 snRNP. Secondary structure and sequence of wildtype SL3 and mutations introduced into the U1 snRNA are depicted; nucleotide changes are shown in red. (B) Dup51p pre-mRNA carries a 5′-ss mutation (indicated by the red asterisk) in intron 2 that causes skipping of exon 2 in the mature transcript. (C) Primer extension analysis to monitor splicing of the minigene reporter Dup51p after cotransfections with control (pcDNA) or U1-5a plasmids expressing wildtype or mutant U1 snRNAs. The full-length and exon 2 skipped Dup51p mRNA products are depicted. The percentage of the full-length product (± s.d., n = 3) is represented in the graph below and statistical significance was determined by comparisons to the wildtype control (lane 2) (n = 3; * = p < 0.05, ** = p < 0.01). (D) RT-qPCR analysis of U1 snRNA expression in HeLa cells cotransfected with Dup51p and U1-5a variants carrying wildtype SL3 or mutations. Fold change in U1 snRNA expression was calculated relative to the pcDNA control after normalization to U2; fold change (± s.d.; n = 3) in U1 is graphed. Expression of the Dup51p reporter pre-mRNA upon cotransfection with U1-5a/SL3 mutants M1e, M1f, and M1g (lanes 7–9) appears to be reduced in this experiment. This apparent diminution, however, is not a consistent observation, as the transcript level is not reduced in Fig. 2B, lane 4
Cryo-EM structures of the human E and A complexes are not available, but structures of the yeast complexes provide some insight into early spliceosomal interactions. In the yeast E complex, Prp40 bridges the 5′- and 3′-ss complexes via interactions with U1-70k and SF1 (yeast MSL5) [24]. Other biochemical studies have also reported the occurrence of this contact [25]. The yeast A complex structure identifies two regions of contact between the pre-mRNA bound U1 and U2 snRNPs [26]. The first interface forms from a stable interaction between the U1 protein Prp39 and the core U2 protein U2A′ (yeast Lea1). The second interface involves interactions of SL3 of the U1 snRNA with the U2 proteins SF3B3 (yeast Rse1) and SF3A3 (yeast Prp9). The Prp39-U2A′ contact was found to be preserved in the yeast pre-B complex [27]. Interestingly, proteins SF1, U2AF65 (yeast MUD2), and the RNA helicases UAP56 (yeast SUB2) and Prp5 were not found in the yeast A complex; Prp5 was detected at sub-stoichiometric levels, but not observed in the structure. Thus, the associations of these proteins (and possibly other early factors) with the pre-mRNA are likely transient and precede formation of the stable interfaces observed in the A complex. The dynamic nature of these interactions may present challenges in the structural analysis of the very early spliceosomal complexes. Additionally, differences in compositions of yeast and human U1 snRNPs suggest that the contacts made by human U1 during spliceosome assembly may be different from those observed in yeast.
Previously, we reported a human spliceosome-specific contact between pre-mRNA bound U1 and U2 snRNPs that is crucial for splicing. We found that SL4 of the 5′-ss bound U1 snRNA interacts with the 793 amino acids (aa) long protein SF3A1 of the 3′-ss bound U2 snRNP during the E to A complex transition [28] and identified the C-terminal Ubiquitin-like (UBL) domain of SF3A1 (aa residues 703–793) as the SL4 binding region [29]. The 280 aa long yeast ortholog of SF3A1 (Prp21) lacks a UBL domain and the yeast U1 snRNA lacks a SL4-like structure, thereby explaining the absence of this U1-U2 contact in yeast spliceosomal complexes [26,27,30,31]. Here, we demonstrate that SL3 of the U1 snRNA is also important for U1 function and that optimal U1 activity in humans requires synergistic actions of SL3 and SL4 of the U1 snRNA. We identify the RNA helicase UAP56 as a SL3 interacting spliceosomal protein, and show that spliceosomal interactions of SL3 and SL4 are distinct. UAP56 binds to SL3 but not to SL4 and conversely, SF3A1-UBL binds to SL4 but not to SL3. Additionally, the impact of siRNA-mediated knockdown on U1 activity indicates that the effects of UAP56 and SF3A1 depletion phenocopy SL3 and SL4 mutations, respectively. Finally, analyses using in vitro UV crosslinking and pre-mRNA splicing assays suggest a role for SL3 in promoting the SL4-SF3A1 interaction, thereby enhancing the E to A complex transition and pre-mRNA splicing.
Materials and methods
Plasmid constructs and transfections
The three-exon/two-intron reporter pDUP51p and the U1 snRNA expression plasmid pNS6U1 have been described previously [28]. The constructs expressing U1-5a snRNAs carrying SL3 mutations, SL3/SL4 double mutations, and SL3/SL4 tandem and swap mutations were generated by PCR mutagenesis using oligonucleotides and were verified by DNA sequencing. The sequences of all oligonucleotides used for U1-5a mutagenesis performed in this study are provided in Supplementary Data File Table S6.
HeLa cells, originally purchased from ATCC, were a gift from Kurt Gustin (University of Arizona, College of Medicine-Phoenix). They were cultured in DMEM containing 10% FBS and antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin). Culture supernatants were tested for mycoplasma by PCR using a pool of six mycoplasma-specific primers, and found to be negative [32]. For transfection, 0.5 × 105 cells per well of a six-well plate were transfected with 0.4 μg of Dup51p reporter plasmid and 3.6 μg of control plasmid (pcDNA3.1) or U1 expression plasmid (pNS6U1) using Lipofectamine 2000 according to the manufacturer’s instructions (Thermo Fisher Scientific). Cells were harvested 48 hours post-transfection and total RNA was extracted using TRIzol reagent or prepared using the Direct-zol RNA kit (Zymo Research). For siRNA and reporter double transfection experiments, 0.24 × 105 cells per well of a six-well plate were transfected with 50 nM synthetic siRNA using lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific). After 24 hours, cells were transfected with Dup51p reporter and U1 plasmids as described above. After incubation for another 24 hours, cells were harvested, and total RNA was extracted also as described above. siRNAs targeting SF3A1, UAP56, URH49, and PTBP1 have been described previously [33–36]. All siRNAs, including the non-targeting control (siNT; siGENOME Non-Targeting Pool #1) were purchased from Horizon Discovery; sequences are provided in the Supplementary Data File Table S5. For SL3/SL4 tandem and swap mutants, transfections were performed in a 12-well plate format with a pNS6U1-5a to Dup51p ratio of 7.5:1 (1.5 μg pNS6U1-5a and 0.2 μg Dup51p) with the addition of pNS6U1-WT (0.3 μg) to maintain the total levels of U1 expression plasmid the same as in previous experiments (final ratio of pNS6U1:Dup51p = 9:1). 2.0 × 105 cells per well of a 12-well plate were transfected using Lipofectamine 2000 reagent as before and total RNA was harvested by TRIzol extraction 48 hours post-transfection prior to primer extension analysis of Dup51p reporter transcripts.
Nucleo-cytoplasmic fractionation
Sub-cellular fractionation of HeLa cells was performed using the protocol by Gagnom et al., with some modifications [37]. Briefly, 300 μl of ice-cold IGEPAL hypotonic lysis buffer (IHLB; 10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 3 mM MgCl2, 0.3% IGEPAL CA-630) was added to each well of a 6-well plate. Cells were lifted by scraping and the lysates were kept on ice for 10 min., vortexed and centrifuged at 5,000 x g at 4°C for 10 min. Supernatants containing the cytoplasmic fractions were treated with SDS/Proteinase K and total cytoplasmic RNA was extracted using phenol:chloroform. For cytoplasmic protein, the fractions were treated with 500 U Nuclease A for 15 min. at room temperature (RT) after addition of SDS-PAGE sample buffer. The nuclear pellets were washed by re-suspension in 50 μl ice-cold IHLB for 10 min. and centrifuged as above. From the nuclear pellet, total RNA was extracted using TRIzol. For nuclear protein, the pellet was re-suspended in 300 μl of PBS and treated with 500 U Nuclease A for 15 min. at RT after addition of SDS-PAGE sample buffer.
Antibodies, Western blotting, and immunoprecipitation
For protein analysis, samples were boiled in SDS-PAGE sample buffer, separated on 10% SDS-PAGE gels and analysed by Western blotting using PVDF membrane. Antibodies against SF3A1, SF3A3, SF3B1, and U1-70k have been described previously [28]. The anti-UAP56 rabbit polyclonal antibody was a gift from Robin Reed (Harvard Medical School). This antibody was raised against GST-UAP56 and cross-reacts with URH49 [38]. Commercial antibodies used in this study included anti-α-Tubulin mouse monoclonal antibody (EMD Millipore; CP06-DM1A), and anti-Prp19 rabbit polyclonal antibody (Bethyl Laboratories; A300-102A). Secondary anti-mouse and anti-rabbit antibodies conjugated to Cy3 and Cy5 fluorophores were purchased from Thermo Fisher Scientific. Proteins were quantified by densitometric scanning of Western blots using ImageQuant.
For U1-70k immunoprecipitation (IP), HeLa cells transiently expressing mutant U1 snRNA from two wells of a 6-well plate were pooled after trypsinization and pelleted by centrifugation at 600 x g for 5 min. at RT. HeLa cell nuclei were then purified as described in the fractionation protocol above. After washing in IHLB, the nuclei were resuspended in 250 μl of Buffer C (20 mM HEPES-KOH pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 1 mM DTT, 0.1 mM PMSF, and 20% glycerol). To extract nuclear components, the Buffer C nuclear suspension was incubated with rotation at 4°C for 30 min. and then centrifuged at 5,000 x g for 10 min. at 4°C. The supernatant was added to 20 μl packed-volume of GammaBind Sepharose beads (Cytiva) that were pre-bound with 5 μg of anti-U1-70k antibody and incubated for 1 hour at RT with rotation. Beads were washed four times in 1x PBS. Bound proteins were extracted by boiling in SDS-PAGE sample buffer and bound RNA was extracted using TRIzol.
Primer extension, RT-qPCR, and Northern blotting
Primer extensions to monitor splicing of the Dup51p reporter and for determining U1-5a snRNA expression were performed using 32P-Dup3r and U17-26R oligonucleotides, respectively, as described previously [28]. Oligonucleotide sequences are provided in the Supplementary Data File Table S5. Spliced products were quantified by densitometric scanning of urea-PAGE images using ImageQuant and percentage of exon 2 inclusion calculated using data obtained from three independent experiments is presented. For RT-qPCRs, reverse transcription was performed using SuperScript III Reverse Transcriptase kit and 1 ng of resulting cDNA was used as a template for qPCR amplification using SYBR Green reagent and StepOnePlus Real-Time PCR Machine; all according to manufacturer specifications (Thermo Fisher Scientific). Primer pairs used for amplification of U1 and U2 snRNA are reported in the Supplementary Data File Table S5. U1 expression across all samples was normalized to U2 snRNA and fold-increase in expression was calculated relative to the pcDNA control using data obtained from three independent experiments.
For Northern blotting, RNA samples were separated on 10% urea-PAGE gels and transferred onto Amersham Hybond nylon membrane (Cytiva) for 1 hour at 15 V, 400 mA using the Trans-blot Turbo semi-dry transfer system (Bio-Rad). Transferred RNA was UV crosslinked to nylon membranes for 10 min. and pre-hybridized in 15 ml of ULTRAhyb hybridization buffer. The membranes were probed with 32P-labelled oligo probes (1.0 x 106 cpm/ml) in hybridization buffer overnight at 42°C. Membranes were washed at the temperature of hybridization once with 2X saline-sodium citrate buffer (2X SSC; 300 mM NaCl, and 30 mM sodium citrate) containing 0.1% SDS for 10 min. and twice with 2X SSC for 10 min. For locked-nucleic acid (LNA)-modified oligonucleotide probes (Exiqon), membranes were incubated at 37°C overnight and washed at RT once with 2X SSC containing 0.1% SDS and twice with 2X SSC alone. Northern blots were visualized using the Typhoon FLA 9500 imager and RNA bands were quantified by densitometric scanning using ImageQuant.
UV crosslinking and pre-mRNA splicing
Nuclear extract from HeLa S3 cells was prepared as described previously [39,40]. The 32P-labelled U1-SL3 and U1-SL4 RNAs were in vitro transcribed from annealed DNA templates, gel purified, and ethanol precipitated. The RNAs were incubated at a final concentration of 20 nM in a splicing reaction containing 2.2 mM MgCl2, 0.4 mM ATP, 20 mM creatine phosphate, 10 U RNaseOUT, and 60% nuclear extract in buffer DG (20 mM HEPES pH 7.9, 80 mM K-glutamate, 0.1 mM EDTA, 1 mM DTT, 20% glycerol). In Fig. 5A and S7, the reactions were preincubated with cold 5′-biotinylated U1-SL3-WT, U1-SL4-WT, U1-SL3-M1g or U1-SL4-M10 RNAs (Integrated DNA Technologies) at the indicated concentrations for 20 min. on ice. After preincubation, 32P-U1-SL4 was added and incubation was continued at 30°C for 30 min. UV crosslinking was performed in a GS Gene Linker (Bio-Rad Laboratories) for a total energy of 1800 mJ. Next, reactions were treated with 100 U of RNase T1 at RT for 5 min. and crosslinked proteins were separated on 10% SDS-PAGE gels and visualized using the Typhoon FLA 9500 imager.
Figure 5.
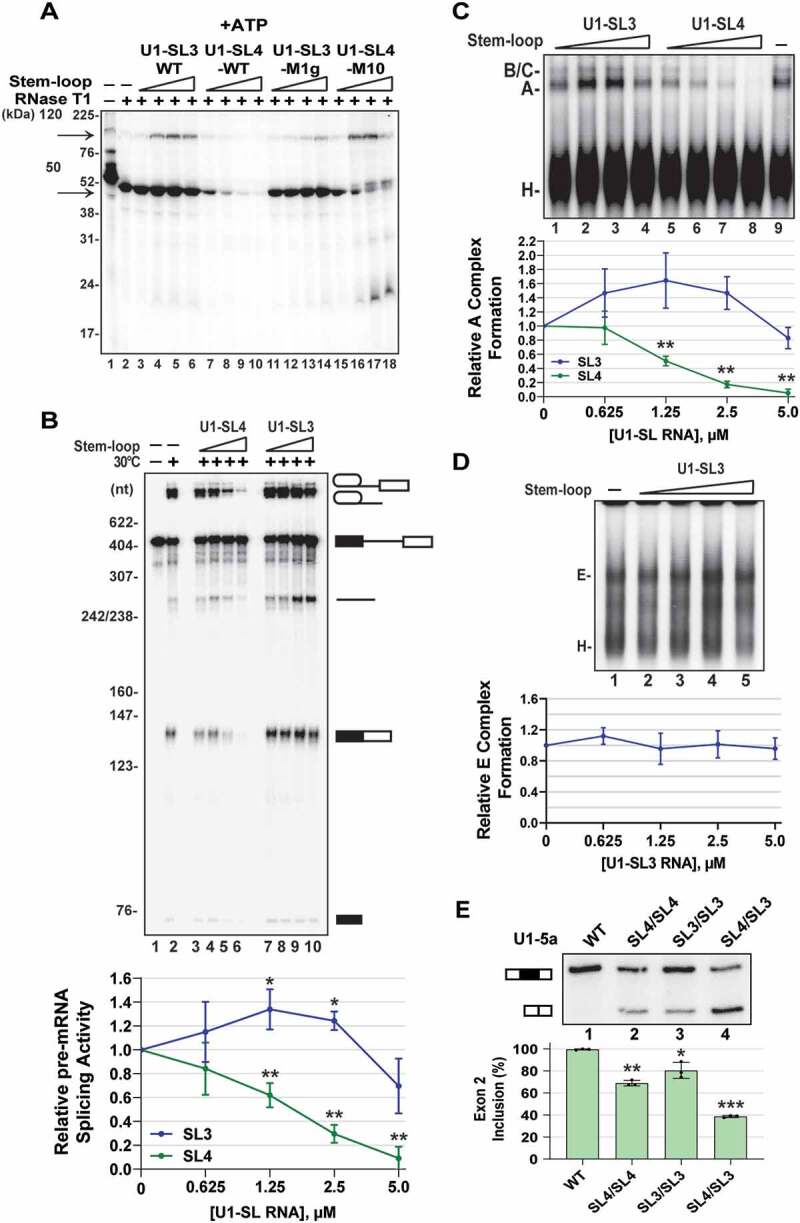
U1-SL3 promotes the U1-SL4-SF3A1 interaction and A complex formation. (A) UV crosslinking of 32P-labelled U1-SL4 RNA in HeLa nuclear extracts in the presence of ATP. To determine the effect of free U1-SL3 and U1-SL4, the reactions were preincubated with 0.625, 1.25, 2.5, and 5.0 μM of the indicated cold stem-loop RNAs prior to addition of 32P-U1-SL4. (B) In vitro splicing of uniformly 32P-labelled AdML pre-mRNA in the absence of stem-loop RNA or in the presence of 0.625, 1.25, 2.5, and 5.0 µM wildtype U1-SL4 or U1-SL3. Splicing intermediates and products are depicted. Fold change in splicing activity is the mRNA/pre-mRNA ratio calculated relative to the no stem-loop control. Statistical analysis compared activity in the presence of U1-SL3 or U1-SL4 to the no stem-loop control (± s.d., n = 4; * = p < 0.05, ** = p < 0.01). (C) Native agarose gel analysis of ATP-dependent spliceosomal complexes assembled on uniformly 32P-labelled AdML pre-mRNA in the absence of stem-loop RNA or in the presence of 0.625, 1.25, 2.5, and 5.0 µM wildtype U1-SL4 or U1-SL3. Fold change in A complex formation is the A complex/H complex ratio calculated relative to the no stem-loop control (± s.d., n = 3, ** = p < 0.01). (D) Native agarose gel analysis of ATP-independent E complex assembled on uniformly 32P-labelled AdML pre-mRNA in the absence or in the presence of 0.625, 1.25, 2.5, and 5.0 µM wildtype U1-SL3. Fold change in E complex formation is the E complex/H complex ratio calculated relative to the no stem-loop control (± s.d., n = 3). (E) Primer extension analysis to monitor splicing of Dup51p after cotransfections with U1-5a plasmids with wildtype U1 or U1 snRNA harbouring tandem SL4 (SL4/SL4), tandem SL3 (SL3/SL3), or swapped SL3 and SL4 (SL4/SL3) structures. The full-length and exon 2 skipped Dup51p mRNA products are depicted. The percentage of the full-length product (± s.d., n = 3) is represented in the graph below and statistical significance was determined by comparisons to the wildtype control (lane 1) (n = 3; * = p < 0.05, ** = p < 0.01, *** = p < 0.001)
For in vitro splicing, uniformly 32P-labelled pre-mRNA substrate was transcribed from the pSPAd plasmid and gel purified and added to splicing reactions containing HeLa nuclear extract and all other components as described above. To examine effects of stem-loop RNAs on AdML splicing, reactions were preincubated with cold wildtype U1-SL3 or U1-SL4 at the indicated concentrations for 20 minutes at RT prior to addition of the pre-mRNA substrate and further incubation at 30°C for 1 hour. Analysis of spliceosomal complexes was performed using native agarose gels as described previously [41]. For analysis of the ATP-independent E complex, native agarose gel electrophoresis was performed at 4°C [42].
RNA affinity purification and MS analysis
For RNA affinity purification (RAP), biotinylated wildtype and mutant U1-SL3 RNAs were custom-synthesized (Integrated DNA Technologies). HeLa nuclear extracts were preincubated in the absence or presence of NaCl at 4°C for 20–30 min. in splicing conditions described above. The reaction mix was then added to 20 μl of Neutravidin beads that were pre-bound with 2 nmoles of biotinylated wildtype or mutant SL3 RNA and incubation was continued for 30 min. at RT. Beads were washed four times with 200 μL of buffer DG. Total RNA was extracted from the bound complexes using phenol:chloroform (5:1; pH 4.8), precipitated with ethanol, separated on 8% urea-PAGE gels, and visualized by ethidium bromide staining. For protein analysis, the bound complexes were eluted by treatment with RNase A/T1 cocktail (Life Technology) in 10 mM Tris-HCl (pH 7.2), 1 mM MgCl2, and 40 mM NaCl. In Fig. 3B, nuclear extracts were preincubated with 0, 250, 500, and 1000 mM NaCl prior to addition of biotinylated RNAs. The eluted proteins were analysed either by Mass Spectrometry (MS) or separated on 10% SDS-PAGE gels and analysed by Western blotting.
Figure 3.
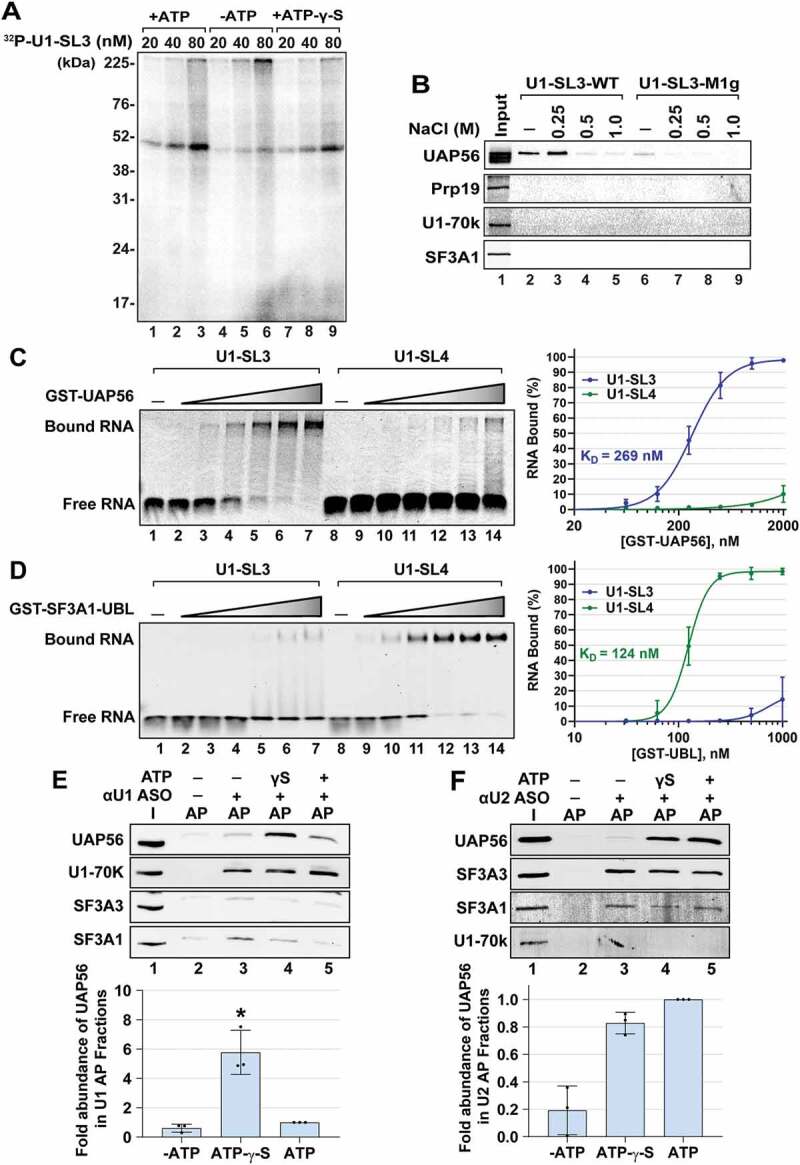
SL3 and SL4 bind to distinct spliceosomal proteins. (A) UV crosslinking analysis for U1-SL3 interacting protein(s). HeLa nuclear extracts were incubated with 20, 40, and 80 nM 32P-U1-SL3 RNA in the presence or absence of ATP and ATP-γ-S. (B) Western analysis of proteins in wildtype and mutant U1-SL3 complexes. HeLa nuclear extracts were preincubated with 0, 0.25, 0.5, and 1.0 M NaCl prior to RNA affinity purification using biotinylated U1-SL3-WT and U1-SL3-M1g RNAs. (C) EMSAs monitoring binding of Cy5-labelled U1-SL3 (lanes 1–7) or U1-SL4 (lanes 8–14) RNAs (10 nM) in the absence and presence of GST-UAP56 (0.0625, 0.125 0.25, 0.5, 1.0, and 2.0 μM). (D) EMSAs monitoring binding of Cy5-labelled U1-SL3 (lanes 1–7) or U1-SL4 (lanes 8–14) RNAs (10 nM) to GST-SF3A1-UBL (0.03125, 0.0625, 0.125 0.25, 0.5, 1.0 μM). Displayed dose-response curves were generated by plotting the average percent of bound U1-SL3 and U1-SL4 RNA (± s.d., n = 3) versus GST-UAP56 or GST-SF3A1-UBL protein concentration and the apparent affinity constant values (KD) are reported. (E) Western analysis of proteins present in input (I) and U1 affinity purified (AP) complexes in the absence and presence of ATP-γ-S, and ATP. The intensity of the UAP56 band was normalized to that of U1-70k in the U1 and U2 complexes, and then fold change was calculated relative to the plus ATP condition (± s.d., n = 3, * = p < 0.05). (F) Western analysis of proteins present in input (I) and U2 AP complexes in the absence and presence of ATP-γ-S, and ATP. Because the signal for SF3A3 was better than SF3A1, the intensity of UAP56 band was normalized to that of SF3A3 protein in the U2 complexes, respectively, and then change was calculated relative to the plus ATP condition (± s.d., n = 3, * = p < 0.05)
For MS, protein samples were reduced, alkylated, and digested using Lys-C and trypsin proteases as previously described [43]. Peptide mixtures were fractionated online using reversed-phase chromatography and then analysed by tandem MS on a Q-Exactive mass spectrometer (Thermo Fisher Scientific) [44]. Data analysis was performed using the IP2 platform (Integrated Proteomics Applications) using the ProLuCID and DTASelect algorithms and filtered at 5% false discovery rate for peptide spectrum matches as calculated using a decoy database approach [45–47]. NSAF values were calculated from the total number of spectrum-matching peptides from the protein (spectrum counts) that were then normalized for protein length [48].
For affinity purification of the U1 and U2 snRNPs, 100 μl splicing reactions containing HeLa S3 nuclear extract and 10 μM U1 or U2 hybridizing, 3′ biotinylated, 2′-O-methyl anti-sense oligonucleotides, U11-13 and U21-21 (Integrated DNA Technologies; Supplementary Data File Table S5) were prepared as described above. These reactions, either lacking ATP or containing 0.5 mM ATP or ATP-γ-S, were incubated at 30°C for 30 min. and then added to 20 μl pre-blocked NeutrAvidin beads and kept at 4°C for one hour with end-over-end rotation [49]. Beads were washed four times with 200 μl of buffer DG and protein was eluted by boiling beads in 1X SDS-PAGE sample buffer. For analysis of RNA in ASO AP complexes, RNA was eluted and purified by standard TRIzol extraction.
GST-UAP56 purification and electrophoretic mobility shift assays
GST-UAP56 construct in the plasmid pGEX-5x was a gift from Robin Reed (Harvard Medical School) and the GST-UBL fusion construct was created by cloning cDNA of SF3A1-UBL domain (SF3A1 aa 704–793) into BamHI and XhoI restriction sites of pGEX-5x. GST, GST-UAP56, and GST-SF3A1-UBL proteins were expressed in Escherichia coli (BL21-DE3) by overnight induction with 0.2 mM IPTG at 18°C. Induced proteins were purified from bacterial lysates using glutathione agarose beads (Thermo Fisher Scientific) according to manufacturer’s protocol, dialysed against two litres of buffer DG, and stored at −20°C. EMSAs were performed as described previously [29]. Briefly, binding reactions contained 10–50 nM 5′-Cy5-labelled U1-SL3 or U1-SL4 RNAs (Integrated DNA Technologies; see Supplementary Data File Table S5 for sequences), 2.2 mM MgCl2, 60% buffer DG, and varying concentrations of purified GST, GST-SF3A1-UBL, and GST-UAP56 protein. ATP-γ-S and other NTPs were added at a final concentration of 0.5 mM. After incubation for 30 min. at RT, binding reactions were loaded onto horizontal 6% native-PAGE gels run at 100 V for 45 minutes at 4°C [50,51]. Native gels were visualized using the Typhoon FLA 9500 Imager.
Statistical analysis
All statistical comparisons were performed using the two-tailed Student t-test in Microsoft Excel. For Figs. 1 and Figs. 2, data obtained from three independent experiments was used for analysis and a difference in exon 2 inclusion of ≥10% with p < 0.05 was considered statistically significant. The analyses for synergy in U1 activity (A; fraction of exon 2 inclusion in the Dup51p reporter mRNA) were performed by STATA version 14 using the linear mixed model and data obtained from three independent experiments. For U1 snRNA mutations, these analyses compared the predicted activity for a particular combination of single SL3 or SL4 mutations (Apred = ASL3*ASL4) to the observed activity (Aobs) of U1 snRNAs carrying double mutations (Table 1). The effects of double mutations were considered synergistic if Aobs<Apred with a difference of ≥ 0.1 and p ≤ 0.01. For combinations of protein knockdown and U1 snRNA mutations, the predicted U1 activity for a combination of siRNA treatment and a particular stem-loop mutation (Apred = AsiRNA*ASL3/SL4) was compared with the observed U1 activity (Aobs) when stem-loop mutations were expressed after siRNA treatment (Table 2). All measurements approximated a normal distribution following a log transformation.
Figure 2.
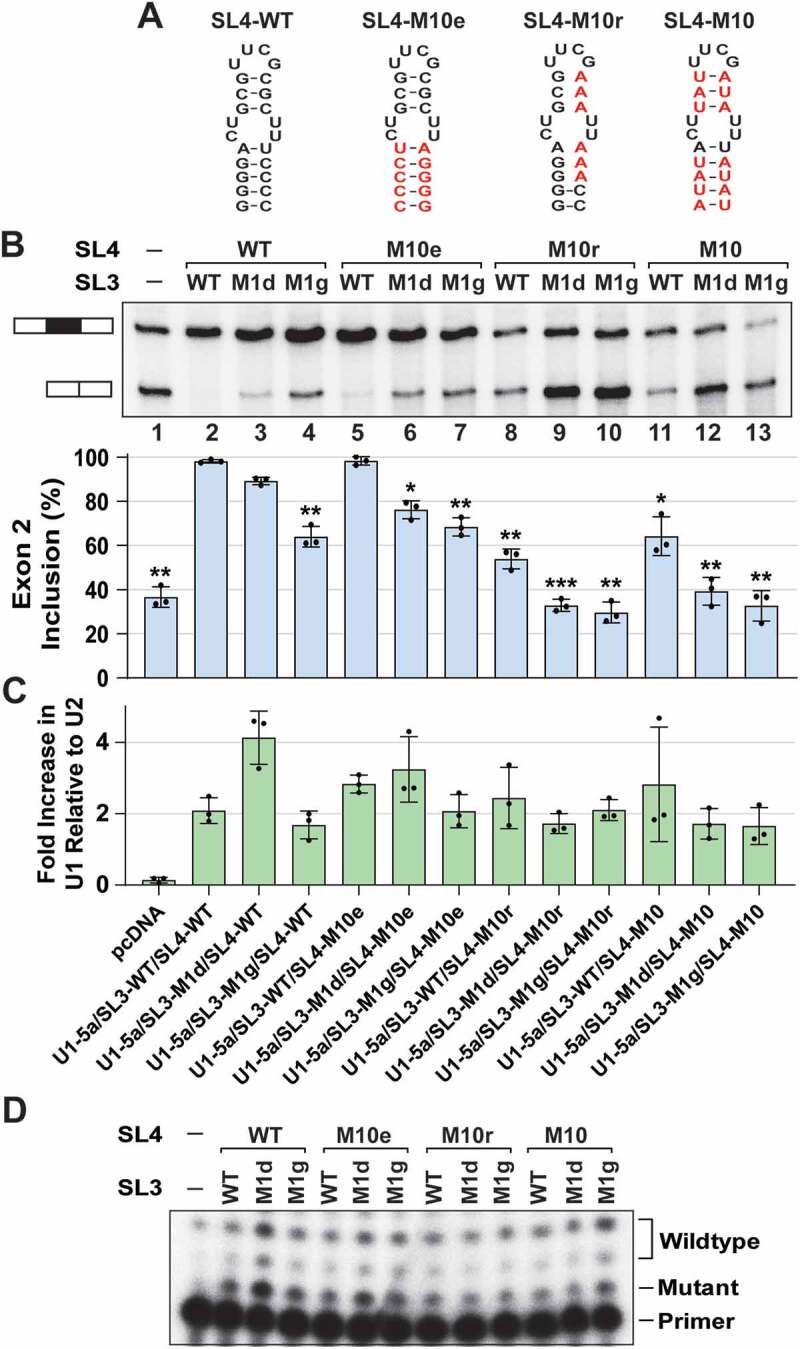
Combined SL3 and SL4 mutations have synergistic effects on U1 function. (A) Schematic of the SL4 secondary structures from wildtype and mutant U1 snRNAs used to create U1-SL3/SL4 double mutants. (B) Primer extension analysis to monitor splicing of the minigene reporter Dup51p after cotransfections with control or U1-5a plasmids for expression of wildtype and mutant U1 snRNA. The full-length and exon 2 skipped Dup51p mRNA products are depicted. The percentage of the full-length product (± s.d., n = 3) is graphed below and statistical significance was determined by comparisons to the wildtype control (lane 2) (n = 3; * = p < 0.05, ** = p < 0.01, *** = p < 0.001). The analysis for synergistic effects is shown in Table 1. (C) RT-qPCR analysis of U1 snRNA expression in HeLa cells cotransfected with Dup51p reporter and U1-5a variant plasmids. Fold change in U1 snRNA expression was calculated relative to the pcDNA control after normalization to U2; fold increase in U1 is graphed (± s.d., n = 3). (D) Primer extension analysis with oligonucleotide 32P-U17-26R (Supplementary Data File Table S5), showing expression of the endogenous U1 and mutant U1-5a snRNAs
Table 1.
Synergy analysis of the impacts of stem-loop 3 and 4 double mutations on the activity of U1 snRNAs
| Combination (SL3 and SL4 mutation) |
Exp # | Apred | Aobs | Coefficient* | 95% Conf. Interval | P-value | Synergy |
|---|---|---|---|---|---|---|---|
| SL3-M1d/SL4-M10e | 1 | 0.867 | 0.793 | 0.113 | 0.074, 0.152 | <0.001 | Yes |
| 2 | 0.869 | 0.716 | |||||
| 3 | 0.888 | 0.776 | |||||
| SL3-M1g/SL4-M10e | 1 | 0.589 | 0.727 | −0.064 | −0.124, −0.003 | 0.04 | No |
| 2 | 0.591 | 0.646 | |||||
| 3 | 0.682 | 0.679 | |||||
| SL3-M1d/SL4-M10r | 1 | 0.439 | 0.322 | 0.196 | 0.146, 0.247 | <0.001 | Yes |
| 2 | 0.507 | 0.244 | |||||
| 3 | 0.514 | 0.304 | |||||
| SL3-M1g/SL4-M10ra | 1 | 0.298 | 0.281 | 0.049 | −0.008, 0.108 | 0.090 | No |
| 2 | 0.344 | 0.349 | |||||
| 3 | 0.395 | 0.258 | |||||
| SL3-M1d/SL4-M10 | 1 | 0.668 | 0.448 | 0.186 | 0.111, 0.262 | <0.001 | Yes |
| 2 | 0.523 | 0.324 | |||||
| 3 | 0.545 | 0.405 | |||||
| SL3-M1g/SL4-M10a | 1 | 0.454 | 0.368 | 0.082 | 0.005, 0.160 | 0.037 | No |
| 2 | 0.356 | 0.247 | |||||
| 3 | 0.419 | 0.365 |
*Average values calculated from three independent experiments were considered synergistic if Aobs < Apred with a difference of ≥ 0.1 and p < 0.01. A positive coefficient value indicates synergy.
aThis combination of stem-loop mutations has a positive coefficient indicating synergy, but does not meet the stringent criteria.
Table 2.
Synergy analysis of the impacts of SF3A1 or UAP56/URH49 knockdown on the activity of U1 snRNAs carrying WT or single stem-loop 3 or 4 mutations
| Combination (SL3 or SL4 mutation and siRNA treatment) |
Exp # | Apred | Aobs | Coefficient* | 95% Conf. Interval | P-value | Synergy |
|---|---|---|---|---|---|---|---|
| WT/siSF3A1 | 1 | 0.529 | 0.557 | −0.027 | −0.078, 0.024 | 0.297 | No |
| 2 | 0.531 | 0.567 | |||||
| 3 | 0.681 | 0.698 | |||||
| SL3-M1d/siSF3A1 | 1 | 0.447 | 0.380 | 0.120 | 0.051, 0.189 | 0.001 | Yes |
| 2 | 0.450 | 0.380 | |||||
| 3 | 0.629 | 0.407 | |||||
| SL4-M10r/siSF3A1a | 1 | 0.308 | 0.323 | 0.036 | −0.054, 0.127 | 0.432 | No |
| 2 | 0.321 | 0.385 | |||||
| 3 | 0.417 | 0.229 | |||||
| WT/siUAP56 | 1 | 0.862 | 0.935 | −0.051 | −0.064, −0.039 | <0.001 | No |
| 2 | 0.880 | 0.926 | |||||
| 3 | 0.900 | 0.936 | |||||
| SL3-M1d/siUAP56 | 1 | 0.781 | 0.823 | −0.053 | −0.066, −0.039 | <0.001 | No |
| 2 | 0.742 | 0.819 | |||||
| 3 | 0.735 | 0.775 | |||||
| SL4-M10/siUAP56 | 1 | 0.540 | 0.392 | 0.137 | 0.087, 0.187 | <0.001 | Yes |
| 2 | 0.431 | 0.314 | |||||
| 3 | 0.481 | 0.335 | |||||
| WT/siURH49 | 1 | 0.802 | 0.956 | −0.063 | −0.118, −0.007 | 0.028 | No |
| 2 | 0.939 | 0.954 | |||||
| 3 | 0.944 | 0.964 | |||||
| SL3-M1d/siURH49a | 1 | 0.858 | 0.832 | 0.041 | 0.005, 0.078 | 0.027 | No |
| 2 | 0.822 | 0.775 | |||||
| 3 | 0.871 | 0.821 | |||||
| SL4-M10/siURH49 | 1 | 0.683 | 0.389 | 0.229 | 0.173, 0.286 | <0.001 | Yes |
| 2 | 0.701 | 0.438 | |||||
| 3 | 0.650 | 0.518 |
*Average values calculated from three independent experiments were considered synergistic if Aobs < Apred with a difference of ≥ 0.1 and p < 0.01. A positive coefficient value indicates synergy.
aThis combination of siRNA treatment and stem-loop mutation has a positive coefficient indicating synergy, but does not meet the stringent criteria.
Results
Stem-loop 3 of the U1 snRNA is important for U1 function
We have developed a genetic complementation assay that uses a 3-exon/2-intron minigene reporter (Dup51p) to examine the role of U1 snRNA in pre-mRNA splicing (Figures. 1B) [28]. When expressed in HeLa cells, 5′-ss mutations in the second intron of Dup51p cause skipping of exon 2 in the mature transcript (Fig. 1C, lane 1). These 5′-ss mutations can be complemented with a compensatory U→A mutation at the 5th position in the U1 snRNA 5′ region that basepairs with the pre-mRNA. In cotransfection assays, the expression of the U1-5a snRNA rescues exon 2 inclusion in the Dup51p transcript (Fig. 1C lane 2). Using this assay, we examined the role of SL3 of the U1 snRNA in pre-mRNA splicing. SL3 consists of a nine basepairs long stem with a single cytidine bulge and a seven nts long terminal loop (Fig. 1A). To evaluate the role of SL3, we created 16 variants of the U1-5a construct carrying SL3 mutations (Fig. 1A). In M1a, the cytidine bulge was deleted. In M1b, M1c, and M1d, G to A changes disrupted basepairing in the stem. Additionally, in M1e, M1f, and M1g, A to U changes were introduced. The strands of the stem were swapped in M1h and in M1i, which also had deletion of the cytidine bulge. The G-C and A-U basepairs were altered partly in M1j and M1k, and completely in M1l. The number of nucleotides in the terminal loop was reduced in M2a, M2b, M2c, and M2d.
The U1-5a variants carrying SL3 mutations were cotransfected with the Dup51p reporter and tested for their ability to rescue exon 2 inclusion (Fig. 1C). The analysis revealed that disruption of three or more basepairs in the upper region of the stem significantly (≥10% reduction in exon 2 inclusion and p < 0.05) affected the rescue of full-length Dup51p splicing (M1e, M1f, and M1g; Fig. 1C lanes 7–9). Disruption of 1–2 basepairs in M1c and M1d had a smaller effect (lanes 5 and 6). Changes that did not exert any effect included deletion of the cytidine bulge (M1a; lane 3), swapping of the two strands of the stem by itself or in combination with deletion of the cytidine bulge (M1h and M1i; lanes 10 and 11) and reducing the size of the terminal loop from six to three nucleotides in M2a, M2b, M2c, and M2d (lanes 15–18). Changing the basepairs in the stem from A-U to G-C and vice versa also did not significantly reduce exon 2 inclusion in M1h, M1i, M1j, M1k, and M1l (lanes 10–14), thereby indicating that the sequence did not matter, as long as the basepairing was maintained. The loss of basepairing in M1e, M1f and M1g displayed the maximum effect and significantly reduced exon 2 inclusion from ~96% to ~60-70% (lanes 7–9). Thus, SL3 of the U1 snRNA plays an important role in U1 function. RT-qPCR quantification showed that expression of variants carrying SL3 mutations was ~4-fold more than the endogenous U1 snRNA (Fig. 1D), thereby indicating abundant expression of U1-5a variants and that loss U1 activity is not due to inefficient expression of the snRNA variants but to a loss of U1 snRNP function induced by the mutations in SL3.
Stem-loops 3 and 4 have synergistic roles in U1 function
In the U1 complementation assays, sequence changes to SL3 caused a decrease in U1 activity to ~60%; however, they did not reduce it to the baseline observed for the pcDNA control (Fig. 1C compare lanes 7–9 with lane 1), thereby indicating that U1 activity was not completely abolished. This observation was similar to our previous analysis of mutations in SL4 of the U1 snRNA, where SL4 mutations were found to compromise, but not completely abrogate U1 activity [28]. We next examined if combining mutations of SL3 and SL4 within the same U1-5a snRNA has larger effects than those observed for a single mutation (Fig. 2). As reported earlier, single SL4 mutants U1-5a/SL4-M10r and U1-5a/SL4-M10 reduced exon 2 inclusion, whereas U1-5a/SL4-M10e did not have a significant effect (Fig. 2A, B lanes 5, 8, and 11) [28]. Relative to U1-5a carrying SL4-wildtype (WT) (lane 2), SL4-M10r and SL4-M10 reduced exon 2 inclusion by ~45% (lane 8; from ~97% to ~52%) and ~33% (lane 11; from ~97% to ~64%), respectively. A double mutant that carries both, SL3-M1d and SL4-M10r, mutations reduced exon 2 inclusion by ~64% (lane 9; from ~97% to ~33%). Thus, the reduction of exon 2 inclusion by the double mutant (~64%) is much larger than the effects seen with the single mutants SL3-M1d (~8%) and SL4-M10r (~45%). Similarly, the double mutant U1-5a/SL3-M1d/SL4-M10 (lane 12) has a much greater influence on exon 2 inclusion (reduced by ~58%) than the effects seen with the single mutants SL3-M1d (~8%) and SL4-M10 (~33%), thereby suggesting that the effects of double mutations may be synergistic.
To examine if the synergistic effects of combining SL3 and SL4 mutations on U1 activity were statistically significant, we applied the linear mixed model. For this, U1 activity (A) was defined as the fraction of exon 2 inclusion, and the predicted activity for a particular combination of single SL3 and SL4 mutations (Apred = ASL3*ASL4) was compared with the observed activity (Aobs) of the U1 snRNAs carrying double mutations (Table 1). The effects of double mutations were considered synergistic if Aobs < Apred and the difference was ≥0.1 with p ≤ 0.01. In these assays, the magnitude of the effect is limited by the exon 2 inclusion baseline, which is ~35% (lane 1). As a result, the synergistic effects on U1 activity are more apparent for the double mutants carrying the milder SL3-M1d mutation – U1-5a/SL3-M1d/SL4-M10r, U1-5a/SL3-M1d/SL4-M10, and U1-5a/SL3-M1d/SL4-M10e. Notably, the differences between Aobs and Apred for double mutants harbouring the more severe mutation, SL3-M1g, suggested that the effects may be synergistic, but did not meet the stringent statistical criteria (Table 1). Although the U1-5a variant carrying the single SL4-M10e mutation did not affect exon 2 inclusion, the effect of the double mutant U1-5a/SL3-M1d/SL4-M10e was significantly synergistic. Overall, the results demonstrated that combined mutations of SL3 and SL4 exert synergistic effects, indicating that the roles of the two stem-loops are likely interconnected.
RT-qPCR quantification showed that U1-5a variant expression was ~2 to 4-fold higher than the endogenous U1 snRNA (Fig. 2C). Primer extension with U17-26R oligonucleotide confirmed the presence of all mutant U1-5a snRNAs in these samples (Fig. 2D) and thus, the loss of U1 activity is not due to inefficient expression of the snRNA variants but to a loss of U1 snRNP function induced by the mutations in SL3 and/or SL4.
Mutant U1 snRNAs exhibit nuclear localization and normal processing
The current model for biogenesis of the U1 snRNP posits existence of a 213 nt precursor snRNA that transiently traffics from the nucleus to the cytoplasm where 3′-end processing forms the 164 nt snRNA and assembly of the Sm core takes place [52,53]. Subsequent maturation occurs after import of the snRNP intermediate back into the nucleus and involves loading of the U1-specific proteins. To confirm the processing of the U1 snRNA variants to mature length, we carried out Northern blotting using an oligonucleotide complementary to nts 27–46 of the U1 snRNA (U127-46R) to detect both endogenous U1 and U1-5a snRNAs. The results demonstrated that in cells expressing single and double mutants, the U1-5a snRNAs were processed to a length identical to the endogenous U1 snRNA (Fig. S1).
To determine subcellular localization, we performed nuclear-cytoplasmic fractionation of HeLa cells expressing U1-5a variants. Efficiency of the applied protocol was assessed by RNA and protein analysis of the nuclear and cytoplasmic fractions obtained from untransfected cells. Northern blotting confirmed enrichment of the U1 and U2 snRNAs in the nuclear fraction and also demonstrated predominantly cytoplasmic localization of the 5S rRNA (Fig. S2A). Western analysis demonstrated the presence of α-Tubulin only in the cytoplasmic fraction, and predominantly nuclear localization of the U1 protein U1-70k, and the U2 proteins SF3A1 and SF3B1 (Fig. S2B). To specifically detect the SL4-M10r mutation, we designed a locked nucleic acid (LNA) containing oligonucleotide (U1-M10r-LNA) (Supplementary Data File, Table S5). Northern blotting of fractions obtained from transfected cells revealed nuclear localization of the U1 and U2 snRNAs in cells expressing U1-5a variants carrying wildtype stem-loops (Fig. S2C lanes 4–6), single SL4-M10r mutation (lanes 7–9), and double mutations SL3-M1d/SL4-M10r (lanes 10–12) and SL3-M1g/SL4-M10r (lanes 13–15). Probing with U1-M10r-LNA indicated predominantly nuclear localization of the mutant U1 snRNAs (lanes 7–15). In fractions from cells expressing U1 variants, Western analysis confirmed nuclear and cytoplasmic localization of SF3A1 and α-Tubulin, respectively (Fig. S2D).
To assess maturation of the U1-5a variant particles, we used an anti-U1-70k antibody that efficiently immunoprecipitates (IPs) the U1 snRNP (Fig. S3A). Northern blotting of IP complexes from cells expressing U1-5a/SL4-M10r, U1-5a/SL3-M1d/SL4-M10r, and U1-5a/SL3-M1g/SL4-M10r demonstrated the presence of U1 but not U2 snRNA (Fig. S3B compare lanes 3, 6 and 9 with 12, 15, and 18). Probing with the U1-M10r confirmed the presence of U1-5a variants carrying single and double mutations in the IP complexes (lanes 21, 24 and 27).
Overall, the results demonstrated that the U1-5a snRNA variants were expressed efficiently and did not affect integrity of the endogenous U1 snRNA. Although others have reported aberrant cleavage of U1 snRNAs carrying the SL4-M10 mutation [54], we found that variants carrying single and double mutations were the same length as the endogenous U1 snRNA. Therefore, the variant snRNAs were processed to mature length, localized within the nucleus and associated with U1-70K, indicating that they were likely exported to the cytoplasm and then imported back into the nucleus for maturation.
U1-SL3 and U1-SL4 bind distinct spliceosomal proteins
To identify U1-SL3 interacting spliceosomal proteins, we first performed UV crosslinking using uniformly 32P-labelled U1-SL3 RNA and found that in HeLa nuclear extracts, SL3 crosslinks only to a ~50 kDa protein (Fig. 3A). The efficiency of crosslinking was similar in the presence of ATP and ATP-γ-S (Fig. 3A lanes 1–3 and 7–9), but much weaker in the absence of ATP (lanes 4–6). Crosslinking of the mutant U1-SL3-M1g RNA was weaker compared to that of U1-SL3-WT and easily disrupted upon preincubation of the nuclear extract with NaCl (Fig. S4A lane 5–8). In the case of U1-SL3-WT, significant amounts of the ~50 kDa crosslinked product formed even at higher NaCl concentrations (Fig. S4A lanes 2 and 3). These results indicate that wildtype U1-SL3 specifically interacts with a ~50 kDa protein and mutations in the upper region of the stem disrupt this interaction.
We isolated the U1-SL3 complex by RNA affinity purification (RAP) and analysed it for snRNA and protein composition. The snRNA analysis showed that none of the spliceosomal snRNAs were present in either the U1-SL3-WT or the U1-SL3-M1g complexes (Fig. S4B lanes 3 and 4). As reported previously, the U2 snRNA was present in the U1-SL4-WT complex (lane 2) [28]. To analyse protein composition, U1-SL3-WT and U1-SL3-M1g bound proteins were subjected to mass spectrometry (MS). Proteins enriched in either the wildtype or the mutant SL3 complexes and proteins present in both complexes were identified by comparing the normalized spectral abundance factor (NSAF) for each protein [48]. This identified several core spliceosomal proteins in the wildtype and mutant U1-SL3 complexes (Supplementary Data File, Tables S1 and S2). Since U1-SL3 crosslinked to a ~50 kDa protein, we focused on validating proteins in this molecular weight range, including the RNA helicase UAP56 (DDX39B) and Prp19. Lists of peptides for U1-SL3-WT and U1-SL3-M1g complexes are provided in Supplementary Data File Tables S3 and S4, respectively.
Immunoblot analysis confirmed the presence of UAP56 in the U1-SL3-WT complex. In comparison to mutant U1-SL3-M1g, binding of UAP56 to U1-SL3-WT was found to be stronger (Fig. 3B, compare lanes 2 and 6). Similar to UV crosslinking analysis, preincubation with up to 250 mM NaCl did not compete out UAP56 from the U1-SL3-WT complex indicating strong binding (Fig. 3B, lane 3). Prp19 was not found in either the wildtype or the mutant U1-SL3 complexes. U1 and U2 proteins, U1-70k and SF3A1, respectively, were also not detected in either complex.
Previously, we demonstrated that SL4 of the U1 snRNA binds to SF3A1 via the C-terminal UBL domain [29]. To test if SL3 and SL4 can directly interact with UAP56 in vitro, we expressed and isolated glutathione S-transferase-UAP56 fusion protein (GST-UAP56), GST-SF3A1-UBL, and GST alone from Escherichia coli (Fig. S5A) and performed electrophoretic mobility shift assays (EMSAs). Purified GST-UAP56 was incubated with Cy5 labelled U1-SL3 and U1-SL4 RNAs (10 nM) in the presence of ATP. GST-UAP56–bound U1-SL3 with a KD of ~269 ± 43 nM (Fig. 3C, lanes 2–7) but did not bind to U1-SL4 (Fig. 3C, lanes 9–14). An interaction between UAP56 and U1-SL4 was not detected even when a higher concentration of RNA (50 nM) was used in presence of either ATP or ATP-γ-S (Fig. S5B) or at the highest GST-UAP56 concentration (8 μM) used in this study (Fig. S5C, lane 14). The U1-SL3 binding affinity of UAP56 in the presence of ATP-γ-S (KD = ~1,728 ± 243 nM; Fig. S5C) was 6-fold lower than that in the presence of ATP (KD of ~269 ± 43 nM). At the higher U1-SL3 concentration (50 nM), a smear pattern was observed in the presence of ATP, which is likely due to binding and subsequent dissociation of UAP56 from the RNA substrate (Fig. S5D and S5E). Binding reactions in the presence of other NTPs confirmed the ATP specificity of the UAP56-U1-SL3 interaction. Neither UTP, GTP, nor CTP supported formation of the GST-UAP56/U1-SL3 complex (Fig. S5E lanes 5, 6, and 7), thereby indicating that the characteristics of this interaction are consistent with those of an ATP-dependent DEAD box helicase. GST by itself did not bind to either U1-SL3 or U1-SL4 (Fig. S5B, lanes 3 and 6 and Fig. S5D, lanes 6 and 12). Finally, compared to GST-UAP56, GST-SF3A1-UBL demonstrated lack of significant binding to U1-SL3 (Fig. 3D, lanes 2–7), but bound U1-SL4 with a KD of 124 ± 16 nM, as previously reported [29]. Overall, these results demonstrate that distinct spliceosomal proteins interact with SL3 and SL4. UAP56 interacts with U1-SL3 but not with U1-SL4, while SF3A1 interacts with U1-SL4 but not with U1-SL3.
UAP56 association with the U1 snRNP requires ATP
To examine if UAP56 associates with U1 and U2 snRNPs, we performed affinity purification (AP) using 3′-biotinylated, 2′-O-methyl antisense-oligonucleotides (ASO). Nuclear extracts were incubated with the U1-13-ASO or U21-21-ASO in the absence of ATP or in the presence of ATP or ATP-γ-S. RNA and protein analysis of the AP complexes showed the presence of the U1 snRNA and the U1 protein U1-70k in U1 complexes (Fig. S5F and 3E, lanes 3–5) and of the U2 snRNA and the U2 protein SF3A3 in U2 complexes (Fig. S5G and 3 F, lanes 3–5) in all three conditions. Association of UAP56 with U1 and U2 was ATP-dependent. UAP56 was observed in U2 complexes in the presence of both ATP-γ-S and ATP (Fig. 3F, lane 4 and 5), but in the case of U1, UAP56 was present in complexes assembled in the presence of ATP-γ-S but not ATP (Fig. 3E, lane 4 and 5). Thus, UAP56 has the capacity to interact with the U1 and U2 snRNPs. UAP56 has been shown interact with U2 snRNP associated protein U2AF65 and also recruited to the 3′-ss complexes via this interaction [6,55,56]. Our results show that utilizing the energy from ATP hydrolysis, UAP56 can dissociate from U1 but not U2. Notably, ATP requirement is a common feature of U1-SL3 interaction with the ~50 kDa protein in HeLa nuclear extracts and purified GST-UAP56, and for the association of UAP56 with the U1 snRNP.
U1 snRNA acts through UAP56 and SF3A1
To establish that SL3 of the U1 snRNA acts through UAP56, we tested the impact of siRNA mediated UAP56 knockdown on the activity of the U1-5a variants carrying either SL3 or SL4 mutation. Our rationale was that if SL3 was acting through UAP56, UAP56 knockdown would phenocopy SL3 mutations in the U1 complementation assay. Mammalian cells also express a UAP56 paralog called UAP56-related helicase, 49 kDa (URH49; also known as DDX39A). The two proteins are 90% identical, and have redundant functions in pre-mRNA splicing and nuclear export of mature mRNA [57–59]. Treatment of HeLa cells with siUAP56 or siURH49 caused a ~60% decrease in the levels of these proteins, but not of SF3A1 or SF3B1 (Fig. 4A). Simultaneous knockdown of UAP56 and URH49 drastically reduced cell viability and the yield of total RNA from siRNA treated cells. Similar results demonstrating a dramatic loss of cell viability upon simultaneous knockdown of UAP56 and URH49 have been reported by others [34]. Therefore, in determining whether SL3 action involves UAP56 and URH49, we resorted to performing the U1 complementation assay after individual knockdowns.
Figure 4.
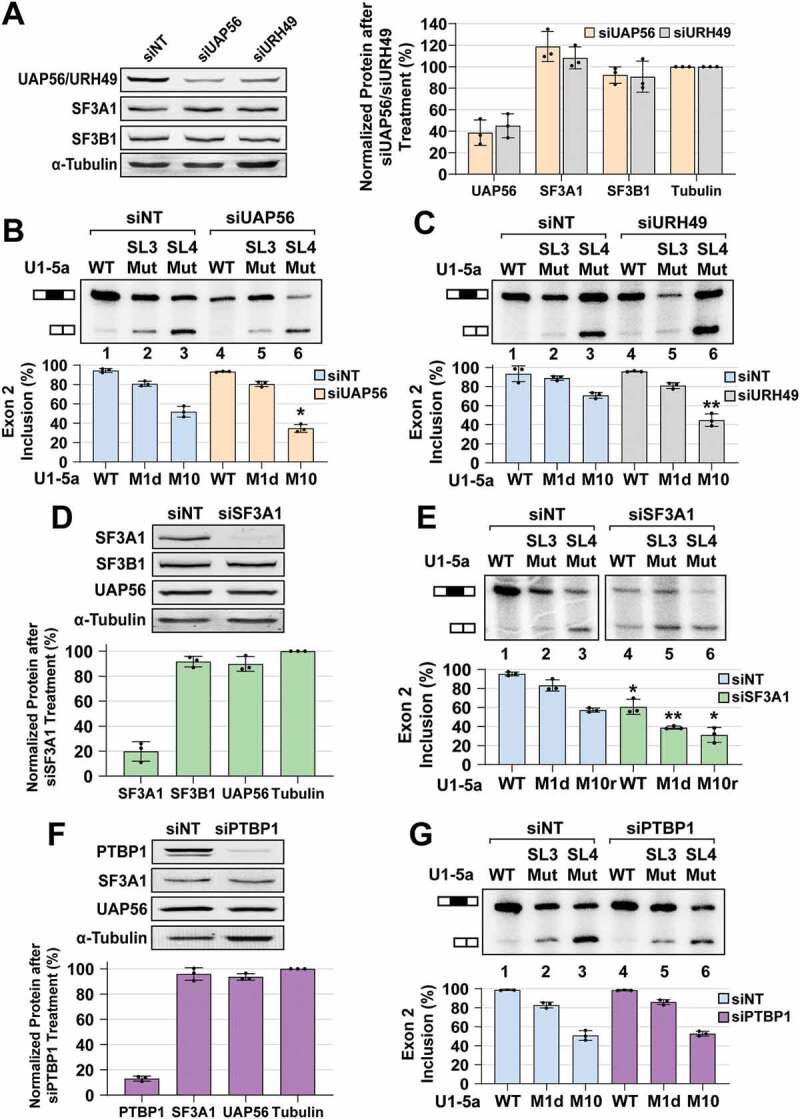
UAP56 and SF3A1 knockdowns phenocopy SL3 and SL4 mutations, respectively. (A) Western analysis of HeLa cell lysates after treatment with non-targeting control (siNT), UAP56 targeting (siUAP56), and URH49 targeting (siURH49) siRNAs. (B) Primer extension analysis of Dup51p reporter transcripts after complementation with U1-5a variants and treatment with control siNT or siUAP56. (C) Primer extension analysis of Dup51p reporter transcripts after complementation with U1-5a variants and treatment with control siNT or siURH49. (D) Western analysis of HeLa cell lysates after treatment with control siNT or SF3A1 targeting (siSF3A1) siRNAs. (E) Primer extension analysis of Dup51p reporter transcripts after complementation with U1-5a variants and treatment with control siNT or siSF3A1. (F) Western analysis of HeLa cell lysates after treatment with control siNT or PTBP1 targeting (siPTBP1) siRNA. (G) Primer extension analysis of Dup51p reporter transcripts after complementation with U1-5a variants and treatment with control siNT or siPTBP1. Average protein expression (± s.d., n = 3) normalized to α-Tubulin is shown. Primer extension products for the full-length and exon 2 skipped Dup51p mRNA are depicted. The average percentage of the full-length product (± s.d., n = 3) is graphed below (n = 3; * = p < 0.05, ** = p < 0.01). Statistical comparisons were performed for each U1-5a snRNA tested under the siNT versus siRNA treatment conditions. Analysis for synergistic effects is shown in Table 2
Treatment with non-targeting siRNA (siNT) did not have any effect on the rescue of exon 2 inclusion by the U1-5a snRNA carrying wildtype SL3 and SL4 (Fig. 4B, lane 1). As before, in comparison to U1-5a/WT, mutants U1-5a/SL3-M1d and U1-5a/SL4-M10 reduced exon 2 inclusion (Fig. 4B, C, lanes 2 and 3). In cotransfections with U1-5a/SL3-M1d, knockdown of UAP56 or URH49 did not significantly exacerbate the effects of the M1d mutation on exon 2 inclusion (Fig. 4B, C compare lane 5 to lane 2). However, in the case of co-transfections with U1-5a/SL4-M10, knockdown of UAP56 or URH49 elicited a much greater decrease in exon 2 inclusion (Fig. 4B, C compare lane 6 to lane 3). To examine if the effects of the combination of UAP56 or URH49 knockdown and SL3 or SL4 mutations were synergistic, we compared the predicted U1 activity for a combination of siUAP56/URH49 treatment and a particular stem-loop mutation (Apred = AsiUAP56/URH49*ASL3/SL4) with the observed U1 activity (Aobs) when stem-loop mutations were expressed after siUAP56/URH49 treatment. This comparison revealed that the effects were synergistic (Aobs < Apred; a difference in exon 2 inclusion of ≥0.1 with p ≤ 0.01) for a combination of UAP56 or URH49 knockdown with U1-5a/SL4-M10, but not with U1-5a/SL3-M1d (Table 2). Effects for the combination of UAP56 or URH49 knockdown with U1-5a/SL4-M10r mutant were also synergistic, but their magnitude (~7%) did not meet our stringent criteria (Fig. S6). Overall, the effects of UAP56 or URH49 knockdown on U1 activity were synergistic only when combined with the U1-SL4 mutation. Therefore, UAP56 or URH49 knockdown and U1-SL3 mutation phenocopy one another, indicating that SL3 of the U1 snRNA likely acts through UAP56 or URH49.
SL4 of the U1 snRNA was previously reported to interact with the U2 protein SF3A1 and also with the polypyrimidine tract binding protein 1 (PTBP1) [28,60]. So, we also examined the impact of knockdown of these two proteins on the activity of U1 variants. The siSF3A1 treatment of HeLa cells caused a ~75% decrease in levels of SF3A1, without affecting UAP56 or SF3B1 (Fig. 4D). The SF3A1 knockdown caused a decrease in exon 2 inclusion in cotransfections with U1-5a carrying wildtype and mutant stem-loops (Fig. 4E, compare lanes 1–3 to 4–6), thereby indicating that loss of SF3A1 caused a general decrease in splicing, which was not observed upon UAP56 or URH49 knockdown. This, in our opinion, is due to the comparatively lower knockdown efficiencies of UAP56 and URH49, and the residual protein levels may be sufficient to support the function of U1-5a snRNAs carrying wildtype stem-loops. Importantly, upon SF3A1 knockdown, effect on exon 2 inclusion in cotransfections with U1-5a/SL3-M1d or U1-5a/SL4-M10r was larger than that for U1-5a/WT (Fig. 4E, compare lanes 5 and 6 to 4). Comparison of the predicted U1 activity (Apred = AsiSF3A1*ASL3/SL4) with the observed activity (Aobs) revealed that the effects on U1 activity were synergistic (Aobs < Apred; a difference in exon 2 inclusion ≥ 0.1 with p ≤ 0.01) for a combination of SF3A1 knockdown with U1-5a/SL3-M1d, but not with U1-5a/SL4-M10r (Table 2). Thus, the effects of SF3A1 knockdown on U1 activity phenocopy loss-of-function SL4 mutations in the U1 snRNA. The siPTBP1 treatment of HeLa cells caused a ~80% reduction in PTBP1 levels; however, it did not exacerbate the effects of either SL3 or SL4 mutations, and synergistic effects were not observed for a combination of PTBP1 knockdown and SL3 or SL4 mutations (Fig. 4F, G; Table 2). These results emphasize context dependence and indicate that the capacity to interact with the U1 snRNA may not be sufficient, and that pre-mRNA binding by PTBP1 is a prerequisite for regulation of cassette exon splicing. Previous work has demonstrated that the Dup51 pre-mRNA lacks PTBP1 binding sites and its splicing is not regulated by this protein [61]. Overall, the results show that SF3A1 knockdown phenocopies SL4 mutations and UAP56 knockdown phenocopies SL3 mutations. The lack of synergy, when SL3 mutations were combined with UAP56/URH49 knockdown or when SL4 mutations were combined with SF3A1 knockdown, indicated that each member of the stem-loop/protein pairs contributes to the same phenotype, i.e., an epistatic relationship exists between SL3 and UAP56, and between SL4 and SF3A1. This epistatic relationship enables the synergistic effects of SL3 and SL4 double mutations on U1 activity to be replicated by combining SL3 mutations with SF3A1 knockdown or SL4 mutations with UAP56 knockdown and indicates that the stem-loop/protein pairs likely act together during constitutive pre-mRNA splicing.
U1-SL3 promotes the SL4-SF3A1 interaction and A complex assembly
We next examined if free U1-SL3 could directly influence the interaction between U1-SL4 and its interacting U2 protein SF3A1 [28]. For this, uniformly 32P-labelled U1-SL4-WT RNA was incubated in HeLa nuclear extract under splicing conditions, UV crosslinked, and then analysed by SDS-PAGE. In HeLa nuclear extracts, U1-SL4 crosslinks to two proteins of ~120 kDa and ~50 kDa (Fig. 5A lanes 1 and 2). In a previous work, by a combination of crosslinking and immunoprecipitation, the ~120 kDa protein was identified as SF3A1 [28]. To test the effect of U1-SL3 on the U1-SL4-SF3A1 interaction, the reactions were preincubated with free cold U1-SL3-WT, U1-SL4-WT, U1-SL3-M1g or U1-SL4-M10 RNAs (see Figs. 1 and Fig. 2 for sequences). Preincubation with U1-SL4-WT competed out crosslinking of the ~120 kDa SF3A1 band and the ~50 kDa protein, indicating the specificity of crosslinking (Fig. 5A, lanes 7–10). In contrast, preincubation with U1-SL3-WT enhanced U1-SL4-SF3A1 crosslinking but did not affect the ~50 kDa band (Fig. 5A, compare lane 2 to lanes 3–6). U1-SL3-M1g, which was found to reduce U1 activity in the complementation assay, did not have the same effect (lanes 11–14). The U1-SL4-M10 mutant carries changes to the upper and lower stems but retains the CU-rich bulge and loop regions (Fig. 2A). We have previously shown that these changes lead to loss of SF3A1 binding [28]. Preincubation with U1-SL4-M10 competed out the ~50 kDa product but did not affect binding to SF3A1 (Fig. 5A, lanes 15–18). U1-SL4-M10 slightly enhanced U1-SL4-SF3A1 crosslinking. This could be due to depletion of the ~50 kDa protein(s) by the excess SL4 mutant, leading to increased availability of the 32P-U1-SL4 for interaction with SF3A1. Preincubation with U1-SL4-M10 also led to crosslinking of 32P-U1-SL4 with two additional proteins of ~52 kDa and ~20 kDa that were not observed with U1-SL4-WT or U1-SL3-M1g. These crosslinked products are likely non-specific, and like the enhanced crosslinking of SF3A1 observed in the presence of U1-SL4-M10, and the loss of binding to the ~50 kDa protein(s) allows their interaction with 32P-U1-SL4. Previously, analysis by RNA affinity purification and immunoblotting indicated that PTBP1 may be the ~50 kDa protein that binds to U1-SL4-WT [28]. However, it is likely that other ~50 kDa RNA binding proteins are also present in the U1-SL4-WT complex.
Since the U1 snRNA-SF3A1 interaction during spliceosome assembly was found to be ATP-dependent [28], we examined if the enhancement of U1-SL4-SF3A1 interaction by U1-SL3 was ATP-dependent. UV crosslinking in the absence of ATP or in the presence of ATP-γ-S revealed that under both conditions, wildtype U1-SL4 was able to compete out crosslinking (Fig. S7A and S7B, lanes 7–10), but wildtype U1-SL3 did not enhance U1-SL4-SF3A1 crosslinking (lanes 3–6), thereby indicating that ATP-hydrolysis is required for the U1-SL3 mediated effect. These results suggest that the wildtype U1-SL3, but not the M1g mutant, may be promoting the interaction between U1-SL4 and SF3A1 in an ATP-dependent manner.
Previously, we demonstrated that the addition of free U1-SL4 to HeLa nuclear extracts inhibits pre-mRNA splicing by blocking the formation of the A complex and not affecting the E complex [28]. We rationalized that if SL3 had a role in promoting the interaction between SL4 of pre-mRNA–bound U1 snRNA and SF3A1, the effect of free U1-SL3 RNA addition would be to enhance in vitro splicing. To test this, HeLa nuclear extract was preincubated with increasing concentrations of free wildtype U1-SL3 and U1-SL4 RNAs prior to the addition of 32P-labelled adenovirus major late (AdML) pre-mRNA. Analysis of the spliced product showed that in contrast to U1-SL4-WT, U1-SL3-WT enhanced splicing by ~1.4 fold (Fig. 5B compare lanes 7–10 with 3–6; U1-SL4-WT IC50 = ~2.0 μM). Preincubation with U1-SL3-M1g mutants RNA neither enhanced nor inhibited splicing (Fig. S7C). Analysis of ATP-dependent spliceosomal complexes demonstrated that in contrast with U1-SL4-WT, U1-SL3-WT caused an increase in A complex formation (Fig. 5C, compare lanes 5–8 to 1–4). The reduction in splicing activity and A complex formation at 5 μM U1-SL3 was notable and was also observed with U1-SL3-M1g (Fig. 5B lane 10, and Fig. 5C lane 4). Although the cause of this reduction is not clear and could be non-specific, the trend of enhanced splicing activity and A complex formation in the presence of lower U1-SL3 concentrations is consistent. Analysis of the ATP-independent complexes showed that like U1-SL4 [28], U1-SL3 did not affect E complex formation (Fig. 5D), thereby suggesting that U1-SL3 likely promotes the E to A transition. Overall, these results underscore the ATP-dependence of U1-SL3 functions. Similar to its ability to bind UAP56, the ability of free wildtype U1-SL3 to promote the interaction between the U1-SL4 RNA and the U2 protein SF3A1 is also ATP-dependent. These observations, along with the fact that the association of UAP56 with the U1 snRNP also requires ATP, strongly suggest that the ability of U1-SL3 to promote the E to A complex transition and enhance pre-mRNA splicing may be due to its association with UAP56.
Spatial orientation of SL3 and SL4 is important for U1 function
In the U1 snRNP, SL3 and SL4 are located diagonally opposite to each other and do not interact with any of the U1-specific proteins [16,17]. To determine if the relative orientation of these stem-loops within the U1 snRNP was important for activity, we created U1-5a snRNA constructs carrying tandem SL3 (SL3/SL3), tandem SL4 (SL4/SL4), and also swapped the positions of the stem-loops (SL4/SL3), and tested their activity in the Dup51p reporter assay (Fig. 5E). Primer extension with U17-26R oligonucleotide confirmed the expression of U1-5a snRNAs carrying tandem and swapped stem-loops in HeLa cells (Fig. S7D). U1-5a constructs carrying tandem SL3 or SL4 decreased exon 2 inclusion to levels observed for single SL3 or SL4 mutations (Fig. 5E, lanes 2 and 3). The swap-construct, on the other hand, caused a much larger decrease in exon 2 inclusion (lane 4), suggesting that exchanging the positions of SL3 and SL4 leads to complete loss of U1 activity as observed for SL3/SL4 double mutants. Thus, the inability of the swap-construct to support U1 activity suggests that the three-dimensional orientation of the stem-loops relative to other U1 components may be important for their recognition by spliceosomal proteins and critical for optimal U1 activity.
Discussion
Our studies demonstrate that in addition to recognizing the 5′-ss, the human U1 snRNA has the key functional role of bringing intron ends in proximity during pre-mRNA splicing. This U1 function is enabled by the stem-loops 3 and 4 of the U1 snRNA that are not bound by any of the U1-specific proteins, and are available to interact with other factors during the early steps of spliceosome assembly. Previously, we reported that an interaction between SL4 of the U1 snRNA and SF3A1 of the U2 snRNP occurred during the transition of the E to A complex [28]. In this study, mutation analysis by the U1 complementation assay revealed that SL3 mutations affect pre-mRNA splicing and that combining mutations of SL3 and SL4 synergistically compromises U1 snRNP activity, indicating that the roles of SL3 and SL4 are partially redundant. Binding analysis demonstrated selective interactions of SL3 with UAP56 and SL4 with SF3A1. Additionally, U1 complementation assays in combination with siRNA-mediated knockdown confirmed that SL3 and SL4 of the U1 snRNA act through UAP56 and SF3A1, respectively. Finally, the addition of free U1-SL3 to nuclear extracts was found to promote the U1-SL4-SF3A1 interaction in an ATP-dependent manner, and enhance pre-mRNA splicing in vitro by promoting the E to A complex transition. Based on these observations, we propose that interactions of SL3 and SL4 of the pre-mRNA bound U1 with UAP56 and SF3A1 of the pre-mRNA bound U2, respectively, bridge the 5′- and 3′-ss complexes during the early steps of spliceosome assembly (Fig. 6A). The SL3-UAP56 complex stabilizes the interaction between SL4 and SF3A1 in an ATP-dependent manner, thereby enhancing pre-mRNA splicing. In the absence of the U1 snRNA-UAP56 contact, due to SL3 mutations or UAP56 knockdown, the SL4-SF3A1 interaction occurs, but is weaker or less efficient, overall leading to reduced splicing (Fig. 6B). Similarly, in the absence of the U1 snRNA-SF3A1 contact, due to SL4 mutations or SF3A1 knockdown, splicing is reduced (Fig. 6C). In this scenario, the 5′- and 3′-ss complexes are likely bridged by interactions of UAP56 with SL3 of the U1 snRNA and with U2AF65. UAP56 was shown to be essential for A complex formation, and interact with U2AF65 in an ATP-dependent manner in yeast and humans [6,55,56]. The primary role of the SL4-SF3A1 contact in cross-intron bridging is underscored by the observation that the addition of free U1-SL4 prevents the E to A complex transition and is sufficient for inhibition of pre-mRNA splicing in vitro (Fig. 6D) [28]. The addition of free U1-SL3, on the other hand, has the profoundly different effect of enhancing splicing. This suggests that the SL3-UAP56 complex may be stabilizing the cross-intron contact between SL4 of the U1 snRNA and the U2 protein SF3A1 during A complex formation (Fig. 6E). This idea is supported by the observations that the addition of U1-SL3 in trans promotes the U1-SL4-SF3A1 contact in an ATP-dependent manner and that free U1-SL3 enhances pre-mRNA splicing by promoting the E to A complex transition. It is very likely that these U1 snRNA contacts occur in addition to the other reported contacts between U1 and 3′-ss complex components including interactions between U1-70k and U2AF65, Prp40 and SF1, and an indirect contact between U1A and SF3B1 [62–66].
Figure 6.
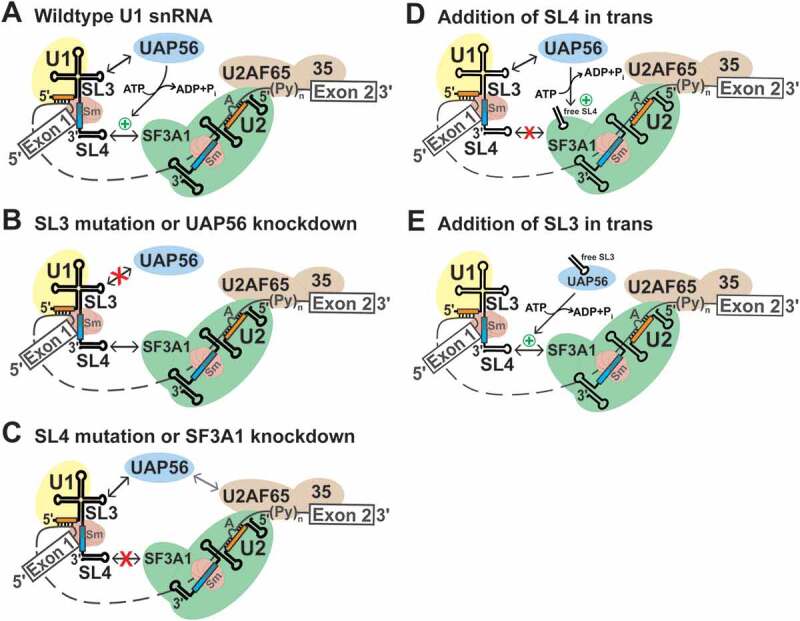
Model for the role of the U1 snRNA during early spliceosome formation. (A) During the early steps of spliceosome assembly, SL3 and SL4 of the U1 snRNA interact with the RNA helicase UAP56 and the U2 snRNP specific protein SF3A1, respectively (double headed black arrows). The SL4-SF3A1 contact bridges the 5′- and 3′-ss complexes. The SL3-UAP56 complex, directly or indirectly, promotes the SL4-SF3A1 interaction (green plus symbol) in an ATP-dependent manner, leading to enhancement of pre-mRNA splicing. (B) Disruption of the SL3-UAP56 contact by either SL3 mutations or UAP56 knockdown prevents stabilization of the SL4-SF3A1 interaction, resulting in reduced splicing. (C) Disruption of the SL4-SF3A1 interaction by either SL4 mutations or SF3A1 knockdown reduces but does not completely abrogate splicing as the SL3-UAP56 interaction can occur. In the absence of the SL4-SF3A1 contact, interaction of UAP56 with U2AF65 likely bridges the 5′- and 3′-ss complexes (grey double headed arrow) [6,55,56]. (D) The addition of excess U1-SL4 in trans competes out the interaction of SF3A1 with endogenous U1 snRNA, reducing A complex formation and inhibiting splicing in vitro [28]. (E) By contrast, addition of excess U1-SL3 in trans enhances pre-mRNA splicing by binding to endogenous UAP56. The U1-SL3-UAP56 complex promotes the SL4-SF3A1 interaction in an ATP-dependent manner, enhancing A complex formation and splicing
Selective binding of UAP56 to U1-SL3, but not to U1-SL4 is intriguing. UAP56 belongs to the helicase super-family 2 and has been shown to exhibit single and double stranded (ss and ds) RNA-dependent ATPase activity, and to unwind dsRNA, without sequence specificity, in an ATP-dependent manner [67,68]. There are a few examples of DEAD-box helicases that exhibit selective binding to RNA substrates. The E. coli DEAD-box protein A (DbpA) and its Bacillus subtilis ortholog (YxiN) are RNA-dependent ATPases with an exceptional specificity for a short hairpin (H92) in the bacterial 23S rRNA [69,70]. Recently, a human DEAD-box helicase DDX55 was shown to interact with domain IV of 28S rRNA in nuclear pre-ribosomal complexes with some specificity and was also reported to exhibited higher affinity for dsRNA than ssRNA [71]. Interestingly, ATPase activity of the yeast RNA helicase Prp5, the other helicase required for A complex formation, was demonstrated to be stimulated by U2 snRNA [72,73]. To understand how UAP56 discriminates between free SL3 and SL4 and their orientation in the intact U1 snRNP, and the mechanism underlying the action of the SL3-UAP56 complex in the context of early spliceosome assembly, a systematic analysis of the binding of wildtype and mutant SL3 RNAs by UAP56 and their impact on its ATPase and helicase activities would be required. It will also be important to determine if SL3-bound UAP56 has the ability to interact with SF3A1 (or another SF3A subunit).
The U1 snRNP has been reported as a target of hnRNP proteins in normal regulation of alternative splicing, and in pathogenesis of diseases associated with RNA-binding proteins. SL4 of the U1 snRNA is targeted by the PTBP1 protein during repression and enhancement of cassette exon inclusion. In the Src pre-mRNA, PTBP1 bound to the intronic sequence upstream and downstream of exon N1 interacts with SL4 of the pre-mRNA associated U1 snRNP [41,60,74]. This precludes U1 snRNA interactions with U2 snRNP components present in the downstream 3′-ss complex, thereby blocking the formation of an active spliceosome. Studies on the enhancement of exon inclusion by binding of PTBP1 to sites that are present only in the intron downstream of the regulated exon also imply a role for the pre-mRNA bound U1 snRNA [75]. Repression of exon 4 of CD45 pre-mRNA by the combined actions of hnRNPs L and A1 induces extended basepairing of 5′-ss bound U1 snRNA [76], thereby stabilizing the U1/pre-mRNA association, which prevents the displacement of U1 by U6 and subsequent spliceosome assembly. It is likely that the non-canonical basepairing of U1 with exon 4 of the CD45 pre-mRNA, and the interaction between pre-mRNA bound U1 snRNA and PTBP1 in the Src pre-mRNA sterically prevent the optimal orientation of the U1 snRNA for an interaction with UAP56 and/or SF3A1. Recently, SL3 of the U1 snRNA was identified as a contact of the fused in sarcoma (FUS) protein [77]. Aberrant cytoplasmic interactions between FUS and the U1 snRNA were shown to disrupt U1 biogenesis and was suggested as an underlying mechanism in FUS-induced amyotophic lateral sclerosis. Our work shows that the unique contacts made by SL3 and SL4 of the human U1 snRNA during the early steps of spliceosome assembly are crucial for the formation of a functional spliceosome, and their inhibition by competing splicing regulators is a potential mechanism in modulation of alternative splicing.
Supplementary Material
Acknowledgments
The authors would like to thank Robin Reed for the anti-UAP56 antibody and the GST-UAP56 plasmid, and Eckhard Jankowsky for discussion.
Funding Statement
This work was supported by funds from the National Institutes of General Medical Sciences [GM127464 to SS and GM089778 to JAW], The American Cancer Society [The Institutional Research Grant 74-001-34-IRG to SS], and The Valley Research Partnership Program [P1-4009 to SS and WM]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Will CL, Luhrmann R.. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3. DOI: 10.1101/cshperspect.a003707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Das R, Zhou Z, Reed R.. Functional association of U2 snRNP with the ATP-independent spliceosomal complex E. Mol Cell. 2000;5:779–787. [DOI] [PubMed] [Google Scholar]
- [3].Gozani O, Potashkin J, Reed R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol. 1998;18:4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Perriman R, Ares M Jr.. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell. 2010;38:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu YZ, Query CC. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell. 2007;28:838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shen H, Zheng X, Shen J, et al. Distinct activities of the DExD/H-box splicing factor hUAP56 facilitate stepwise assembly of the spliceosome. Genes Dev. 2008;22:1796–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boesler C, Rigo N, Anokhina MM, et al. A spliceosome intermediate with loosely associated tri-snRNP accumulates in the absence of Prp28 ATPase activity. Nat Commun. 2016;7:11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bertram K, Agafonov DE, Dybkov O, et al. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell. 2017;170:701–13 e11. [DOI] [PubMed] [Google Scholar]
- [9].Plaschka C, Lin PC, Nagai K. Structure of a pre-catalytic spliceosome. Nature. 2017;546:617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Haselbach D, Komarov I, Agafonov DE, et al. Structure and conformational dynamics of the human spliceosomal B(act) complex. Cell. 2018;172:454–64 e11. [DOI] [PubMed] [Google Scholar]
- [11].Zhang X, Yan C, Zhan X, et al. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018;28:307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhan X, Yan C, Zhang X, et al. Structure of a human catalytic step I spliceosome. Science. 2018;359:537–545. [DOI] [PubMed] [Google Scholar]
- [13].Fica SM, Oubridge C, Galej WP, et al. Structure of a spliceosome remodelled for exon ligation. Nature. 2017;542:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yan C, Wan R, Bai R, et al. Structure of a yeast step II catalytically activated spliceosome. Science. 2017;355:149–155. [DOI] [PubMed] [Google Scholar]
- [15].Galej WP, Wilkinson ME, Fica SM, et al. Cryo-EM structure of the spliceosome immediately after branching. Nature. 2016;537:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Weber G, Trowitzsch S, Kastner B, et al. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J. 2010;29:4172–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kondo Y, Oubridge C, van Roon AM, et al. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5ʹ splice site recognition. Elife. 2015;4. DOI: 10.7554/eLife.04986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kretzner L, Rymond BC, Rosbash M. S. cerevisiae U1 RNA is large and has limited primary sequence homology to metazoan U1 snRNA. Cell. 1987;50:593–602. [DOI] [PubMed] [Google Scholar]
- [19].Siliciano PG, Kivens WJ, Guthrie C. More than half of yeast U1 snRNA is dispensable for growth. Nucleic Acids Res. 1991;19:6367–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li X, Liu S, Jiang J, et al. CryoEM structure of Saccharomyces cerevisiae U1 snRNP offers insight into alternative splicing. Nat Commun. 2017;8:1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Carlson SM, Soulette CM, Yang Z, et al. RBM25 is a global splicing factor promoting inclusion of alternatively spliced exons and is itself regulated by lysine mono-methylation. J Biol Chem. 2017;292:13381–13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Izquierdo JM, Majos N, Bonnal S, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19:475–484. [DOI] [PubMed] [Google Scholar]
- [23].Becerra S, Montes M, Hernandez-Munain C, et al. Prp40 pre-mRNA processing factor 40 homolog B (PRPF40B) associates with SF1 and U2AF65 and modulates alternative pre-mRNA splicing in vivo. RNA. 2015;21:438–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li X, Liu S, Zhang L, et al. A unified mechanism for intron and exon definition and back-splicing. Nature. 2019;573:375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abovich N, Liao XC, Rosbash M. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev. 1994;8:843–854. [DOI] [PubMed] [Google Scholar]
- [26].Plaschka C, Lin PC, Charenton C, et al. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559:419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bai R, Wan R, Yan C, et al. Structures of the fully assembled Saccharomyces cerevisiae spliceosome before activation. Science. 2018. DOI: 10.1126/science.aau0325 [DOI] [PubMed] [Google Scholar]
- [28].Sharma S, Wongpalee SP, Vashisht A, et al. Stem-loop 4 of U1 snRNA is essential for splicing and interacts with the U2 snRNP-specific SF3A1 protein during spliceosome assembly. Genes Dev. 2014;28:2518–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martelly W, Fellows B, Senior K, et al. Identification of a noncanonical RNA binding domain in the U2 snRNP protein SF3A1. RNA. 2019;25:1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin PC, Xu RM. Structure and assembly of the SF3a splicing factor complex of U2 snRNP. EMBO J. 2012;31:1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kramer A, Mulhauser F, Wersig C, et al. Mammalian splicing factor SF3a120 represents a new member of the SURP family of proteins and is homologous to the essential splicing factor PRP21p of Saccharomyces cerevisiae. RNA. 1995;1:260–272. [PMC free article] [PubMed] [Google Scholar]
- [32].Pisal RV, Hrebikova H, Chvatalova J, et al. Detection of mycoplasma contamination directly from culture supernatant using polymerase chain reaction. Folia Biol (Praha). 2016;62:203–206. [DOI] [PubMed] [Google Scholar]
- [33].Tanackovic G, Kramer A. Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol Biol Cell. 2005;16:1366–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kapadia F, Pryor A, Chang TH, et al. Nuclear localization of poly(A)+ mRNA following siRNA reduction of expression of the mammalian RNA helicases UAP56 and URH49. Gene. 2006;384:37–44. [DOI] [PubMed] [Google Scholar]
- [35].Yamazaki T, Fujiwara N, Yukinaga H, et al. The closely related RNA helicases, UAP56 and URH49, preferentially form distinct mRNA export machineries and coordinately regulate mitotic progression. Mol Biol Cell. 2010;21:2953–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang C, Norton JT, Ghosh S, et al. Polypyrimidine tract-binding protein (PTB) differentially affects malignancy in a cell line-dependent manner. J Biol Chem. 2008;283:20277–20287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gagnon KT, Li L, Janowski BA, et al. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat Protoc. 2014;9:2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dias AP, Dufu K, Lei H, et al. A role for TREX components in the release of spliced mRNA from nuclear speckle domains. Nat Commun. 2010;1:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chan RC, Black DL. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol Cell Biol. 1995;15:6377–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dignam JD. Preparation of extracts from higher eukaryotes. Methods Enzymol. 1990;182:194–203. [DOI] [PubMed] [Google Scholar]
- [41].Sharma S, Falick AM, Black DL. Polypyrimidine tract binding protein blocks the 5ʹ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol Cell. 2005;19:485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Das R, Reed R. Resolution of the mammalian E complex and the ATP-dependent spliceosomal complexes on native agarose mini-gels. RNA. 1999;5:1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kaiser P, Wohlschlegel J. Identification of ubiquitination sites and determination of ubiquitin-chain architectures by mass spectrometry. Methods Enzymol. 2005;399:266–277. [DOI] [PubMed] [Google Scholar]
- [44].Kelstrup CD, Young C, Lavallee R, et al. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res. 2012;11:3487–3497. [DOI] [PubMed] [Google Scholar]
- [45].Xu T, Venable JD, Park SK, et al. ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Mol Cell Proteomics. 2006;5:S174–S. [Google Scholar]
- [46].Tabb DL, McDonald WH, Yates JR 3rd.. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Florens L, Carozza MJ, Swanson SK, et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 2006;40:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Blencowe BJ, Lamond AI. Purification and depletion of RNP particles by antisense affinity chromatography. Methods Mol Biol. 1999;118:275–287. [DOI] [PubMed] [Google Scholar]
- [50].Ryder SP, Recht MI, Williamson JR. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol Biol. 2008;488:99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dowdle ME, Imboden SB, Park S, et al. Horizontal gel electrophoresis for enhanced detection of protein-RNA complexes. J Vis Exp. 2017. DOI: 10.3791/56031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yong J, Kasim M, Bachorik JL, et al. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol Cell. 2010;38:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fischer U, Englbrecht C, Chari A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip Rev RNA. 2011;2:718–731. [DOI] [PubMed] [Google Scholar]
- [54].Rogalska ME, Tajnik M, Licastro D, et al. Therapeutic activity of modified U1 core spliceosomal particles. Nat Commun. 2016;7:11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fleckner J, Zhang M, Valcarcel J, et al. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 1997;11:1864–1872. [DOI] [PubMed] [Google Scholar]
- [56].Kistler AL, Guthrie C. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for sub2, an essential spliceosomal ATPase. Genes Dev. 2001;15:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Luo ML, Zhou Z, Magni K, et al. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature. 2001;413:644–647. [DOI] [PubMed] [Google Scholar]
- [58].Zhang M, Green MR. Identification and characterization of yUAP/Sub2p, a yeast homolog of the essential human pre-mRNA splicing factor hUAP56. Genes Dev. 2001;15:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pryor A, Tung L, Yang Z, et al. Growth-regulated expression and G0-specific turnover of the mRNA that encodes URH49, a mammalian DExH/D box protein that is highly related to the mRNA export protein UAP56. Nucleic Acids Res. 2004;32:1857–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sharma S, Maris C, Allain FH, et al. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol Cell. 2011;41:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Amir-Ahmady B, Boutz PL, Markovtsov V, et al. Exon repression by polypyrimidine tract binding protein. RNA. 2005;11:699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Abovich N, Rosbash M. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell. 1997;89:403–412. [DOI] [PubMed] [Google Scholar]
- [63].Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. [DOI] [PubMed] [Google Scholar]
- [64].Boukis LA, Liu N, Furuyama S, et al. Ser/Arg-rich protein-mediated communication between U1 and U2 small nuclear ribonucleoprotein particles. J Biol Chem. 2004;279:29647–29653. [DOI] [PubMed] [Google Scholar]
- [65].Staknis D, Reed R. SR proteins promote the first specific recognition of Pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Mol Cell Biol. 1994;14:7670–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cho S, Hoang A, Sinha R, et al. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shen J, Zhang L, Zhao R. Biochemical characterization of the ATPase and helicase activity of UAP56, an essential pre-mRNA splicing and mRNA export factor. J Biol Chem. 2007;282:22544–22550. [DOI] [PubMed] [Google Scholar]
- [68].Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Diges CM, Uhlenbeck OC. Escherichia coli DbpA is an RNA helicase that requires hairpin 92 of 23S rRNA. EMBO J. 2001;20:5503–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kossen K, Karginov FV, Uhlenbeck OC. The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J Mol Biol. 2002;324:625–636. [DOI] [PubMed] [Google Scholar]
- [71].Choudhury P, Kretschmer J, Hackert P, et al. The DExD box ATPase DDX55 is recruited to domain IV of the 28S ribosomal RNA by its C-terminal region. RNA Biol. 2020;1–12. DOI: 10.1080/15476286.2020.1829366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wu G, Adachi H, Ge J, et al. Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly. EMBO J. 2016;35:654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].O’Day CL, Dalbadie-mcfarland G, Abelson J. The Saccharomyces cerevisiae Prp5 protein has RNA-dependent ATPase activity with specificity for U2 small nuclear RNA. J Biol Chem. 1996;271:33261–33267. [DOI] [PubMed] [Google Scholar]
- [74].Sharma S, Kohlstaedt LA, Damianov A, et al. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol. 2008;15:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hamid FM, Makeyev EV. A mechanism underlying position-specific regulation of alternative splicing. Nucleic Acids Res. 2017;45:12455–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chiou NT, Shankarling G, Lynch KW. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell. 2013;49:972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jutzi D, Campagne S, Schmidt R, et al. Aberrant interaction of FUS with the U1 snRNA provides a molecular mechanism of FUS induced amyotrophic lateral sclerosis. Nat Commun. 2020;11:6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.