

ABSTRACT
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two clinically distinct classes of neurodegenerative disorders. Yet, they share a range of genetic, cellular, and molecular features. Hexanucleotide repeat expansions (HREs) in the C9orf72 gene and the accumulation of toxic protein aggregates in the nervous systems of the affected individuals are among such common features. Though the mechanisms by which HREs cause toxicity is not clear, the toxic gain of function due to transcribed HRE RNA or dipeptide repeat proteins (DPRs) produced by repeat-associated non-AUG translation together with a reduction in C9orf72 expression are proposed as the contributing factors for disease pathogenesis in ALS and FTD. In addition, several recent studies point toward alterations in protein homeostasis as one of the root causes of the disease pathogenesis. In this review, we discuss the effects of the C9orf72 HRE in the autophagy-lysosome pathway based on various recent findings. We suggest that dysfunction of the autophagy-lysosome pathway synergizes with toxicity from C9orf72 repeat RNA and DPRs to drive disease pathogenesis.
Abbreviation: ALP: autophagy-lysosome pathway; ALS: amyotrophic lateral sclerosis; AMPK: AMP-activated protein kinase; ATG: autophagy-related; ASO: antisense oligonucleotide; C9orf72: C9orf72-SMCR8 complex subunit; DENN: differentially expressed in normal and neoplastic cells; DPR: dipeptide repeat protein; EIF2A/eIF2α: eukaryotic translation initiation factor 2A; ER: endoplasmic reticulum; FTD: frontotemporal dementia; GAP: GTPase-activating protein; GEF: guanine nucleotide exchange factor; HRE: hexanucleotide repeat expansion; iPSC: induced pluripotent stem cell; ISR: integrated stress response; M6PR: mannose-6-phosphate receptor, cation dependent; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MN: motor neuron; MTORC1: mechanistic target of rapamycin kinase complex 1; ND: neurodegenerative disorder; RAN: repeat-associated non-ATG; RB1CC1/FIP200: RB1 inducible coiled-coil 1; SLC66A1/PQLC2: solute carrier family 66 member 1; SMCR8: SMCR8-C9orf72 complex subunit; SQSTM1/p62: sequestosome 1; STX17: syntaxin 17; TARDBP/TDP-43: TAR DNA binding protein; TBK1: TANK binding kinase 1; TFEB: transcription factor EB; ULK1: unc-51 like autophagy activating kinase 1; UPS: ubiquitin-proteasome system; WDR41: WD repeat domain 41.
KEYWORDS: Amyotrophic lateral sclerosis (ALS), autophagy, axonal transport, c9orf72, dipeptide repeat protein (DPR), frontotemporal dementia (FTD), lysosome, smcr8, wdr41
Introduction
The impact of neurodegenerative disorders (NDs), including Alzheimer disease (AD), frontotemporal dementia (FTD), Parkinson disease (PD), Huntington disease (HD), and amyotrophic lateral sclerosis (ALS) on our societies is increasing. The disease mechanisms associated with NDs are complex and are only partially understood. As a result, no cure exists for these conditions. Though clinically distinct, NDs do share several common cellular and molecular features. Among such commonalities is the loss of specific neurons in neuronal networks, along with the accumulation of misfolded proteins and toxic aggregates. Such protein inclusions may originate in distinct regions, but they tend to spread across the central and peripheral nervous systems. Recent findings in several of these NDs point toward alterations in protein homeostasis as a root cause. Impairments in proteostasis results in the accumulation of misfolded proteins and the formation of insoluble protein aggregates. In the current review, we aim to zoom in on a frequent genetic cause of ALS and FTD caused by hexanucleotide repeat expansions (HRE; [G4C2]n) in the C9orf72 gene and review the dysfunctions in the autophagy-lysosome pathway (ALP) that were recently discovered in various C9orf72-ALS and FTD disease models.
Defective protein homeostasis: Leading the way to protein aggregation and neurodegeneration?
Protein misfolding leading to the formation of inter- or intracellular protein accumulations in neuronal and glial cells is a hallmark for NDs [1]. For this reason, NDs are also known as proteinopathies [2,3]. The difference in the aggregating proteins in each ND could account for the selective vulnerability of neuronal subtypes in the different disorders. Aging, the first and primary risk factor in all NDs, is inevitably correlated with a decline in cellular protein quality control systems and a concomitant accumulation of insoluble protein species [4]. Next, neurons, the primary affected cell type in NDs, are incredibly long, polarized, highly differentiated cells and are susceptible to the age-related disturbance in protein homeostasis. In addition, neurons are mature non-dividing cells, and unlike epithelial cells, cannot use cell division to dilute pathological protein aggregates and damaged organelles [4]. Besides the general consensus on toxicity of protein aggregates, it is also hypothesized that soluble forms of the mutated or aggregating protein block the proteasome, sequester part(s) of proteostasis machinery and/or cause direct toxicity to sensitive organelles [5–7]. The concentration of potentially toxic protein species into larger aggregates might, in fact, even protect the cell from these harmful effects and facilitate their clearance by aggrephagy (a selective form of autophagy) [7,8]. However, all this boils down to the everlasting chicken-and-egg question of whether these protein aggregates are a cause or rather the consequence of neurodegeneration.
Amyotrophic lateral sclerosis and Frontotemporal dementia
Amyotrophic lateral sclerosis is an ND characterized by the degeneration of motor neurons located in the motor cortex, the brainstem motor nuclei, and the spinal cord leading to muscle paralysis and atrophy [9–13]. The disease onset is typically late midlife, but survival is often limited to 2–5 years after onset due to the rapid disease progression leading to respiratory failure [14]. Although the disease prevalence is low (10–12 cases per 100 000 individuals), the cumulative lifetime risk of developing ALS is quite high, 1 in 400 [15,16]. Currently, there is no cure for ALS, and the two available FDA-approved drugs (riluzole and edaravone) have only a marginal impact on survival or disease progression [17,18]. About 10% of all ALS cases have a familial form of the disease (fALS), while the remaining 90% report no affected family members are classified as sporadic (sALS). More than 20 genes have been associated with ALS [19]. The most commonly mutated genes are C9orf72 (C9orf72-SMCR8 complex subunit), SOD1 (superoxide dismutase 1), TARDBP/TDP-43 (TAR DNA binding protein), FUS (FUS RNA binding protein), responsible for 30–60%, 20%, ~6% and ~5% of fALS, respectively [10,20].
Frontotemporal dementia (FTD) is an ND characterized by a range of behavioral and language deficits. With an estimated lifetime risk of 1 in 742, FTD is the second most common form of dementia in adults under 60 years of age [21]. The disease typically strikes in late middle age with equal frequency in both sexes and is caused by degeneration of frontal and/or anterior temporal lobes of the brain. The clinical presentation of FTD is diverse. Most patients present with behavioral problems and neuropsychiatric symptoms, while some patients present with language dysfunctions. In about half of FTD patients, the disease runs in their family. In others, the disease is likely caused by the interplay between genetic and environmental factors. At present, three genes (A) MAPT/Tau (microtubule associated protein tau), (B) GRN (granulin precursor), and (C) C9orf72 mutations with GGGGCC repeat expansion in the non-coding region are reported as the most common causes of FTD [22]. At the neuropathological level, protein aggregates containing either MAPT, TARDBP, or FUS are observed in the neurons of FTD patients [21–23]. Although our knowledge of the disease-causing genes and proteins has dramatically improved, many aspects of the disease mechanisms remain unclear, hampering successful therapy developments. Therefore, there is currently no effective treatment for FTD.
ALS and FTD represent two extremes of a disease spectrum, with different intermediate phenotypes, but still, several commonalities between the two diseases exist [24,25]. At the genetic level, C9orf72 mutations are the most common genetic cause of ALS, FTD, and ALS-FTD. Strikingly, several of the other ALS and FTD-causing mutations largely overlap and cluster in endolysosomal pathways. This includes VCP (valosin containing protein), CHMP2B (charged multivesicular body protein 2B), SQSTM1/p62 (sequestosome 1), TBK1 (TANK binding kinase 1), UBQLN2 (ubiquilin 2), OPTN (optineurin), and CCNF (cyclin F) [10,25,26]. Besides, few other risk factors, such as TMEM106B (transmembrane protein 106B) involved in the endo-lysosomal system, have been identified [14]. At the neuropathological level, the presence of TARDBP inclusions in 97% of patients with ALS and about 50% of patients with FTD also links these two NDs [14,23,24]. In addition, the TARDBP inclusions also stain positive for SQSTM1 [27,28], an autophagy receptor protein that gets degraded during the autophagic process. The accumulation of SQSTM1 is an indicator of defective autophagy [29]. The hypothesis that reduced clearance of misfolded TARDBP is underlying the TARDBP proteinopathy in ALS and FTD is emerging [14,24]. Interestingly, the majority of patients with ALS or FTD and TARDBP pathology have no mutations in TARDBP [9]. TARDBP homeostasis is regulated by different proteostasis pathways, including the autophagy pathway. Hence, reduced autophagy is an attractive hypothesis for the accumulation of insoluble TARDBP in ALS-FTD [30,31]. How the cytoplasmatic inclusions composed of TARDBP are formed and how they lead to the degeneration of motor neurons is not known. Whether these inclusions are merely a terminal hallmark of the disease or rather an important aspect of the degenerative process remains to be clarified. As several genes involved in the endolysosomal pathway can cause ALS-FTD, the evidence for a causal role of autophagy defects in ALS-FTD is accumulating [32].
C9orf72 hexanucleotide repeat expansions
In 2011, HREs in the 5ʹ non-coding sequence of the C9orf72 gene was identified as the most common inherited cause of ALS, FTD, and ALS-FTD [33,34]. Healthy individuals carry 2 to 30 hexanucleotide repeats, while ALS-FTD patients typically have over 100s, and repeat lengths of more than 1000 are not exceptional [35]. The exact cutoff for pathogenicity is not known, as repeats in the range of even 30–50 are reported in ALS patients [36].
The pathogenic mechanisms by which repeat expansions in the C9orf72 gene are causing ALS are not fully understood. Three distinct but not mutually exclusive disease mechanisms have been proposed [10,37,38]: 1) Haploinsufficiency of the C9orf72 gene [38,39], 2) Sequestration of RNA-binding proteins (RNPs) by RNA foci containing the C9orf72 HRE RNA [34,38,40] and 3) Repeat-associated non-AUG (RAN) translation of the HRE resulting in dipeptide repeat proteins (DPRs) [40,41]. While the latter two mechanisms propose toxic gain-of-function pathogenic mechanisms in C9orf72 ALS-FTD, the first mechanism opts for a loss-of-function hypothesis, with reduced function of the C9orf72 protein. Although little is known about the physiological functions of C9orf72, it has been implicated in autophagy.
Overview of the autophagy process
Autophagy is an intracellular lysosome-dependent degradative mechanism that plays a crucial role in maintaining cellular homeostasis [42]. While autophagy was first discovered as a protective pathway activated under starvation conditions, the recent discovery of selective autophagy in nutrient-rich conditions has redefined the definition of autophagy [43]. Three primary types of autophagy have been identified so far in mammalian cells, each of which differs based on the delivery mechanisms of their substrate to the lysosome: macroautophagy (from here on referred to as “autophagy”) (Figure 1), chaperone-mediated autophagy (CMA) and microautophagy [42,44]. The canonical activation route of autophagy is initiated by a protein complex centered around ULK1 (unc-51 like autophagy activating kinase 1) [44]. Two major cellular energy/nutrient-sensing proteins regulate the ULK1 protein complex: the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) kinase and the AMP-activated protein kinase (AMPK). Under starvation or stress conditions, the kinase activity of MTORC1 is inhibited, resulting in the dephosphorylation and activation of the ULK1 complex. ULK1 subsequently phosphorylates the class III phosphatidylinositol 3-kinase (PtdIns3K) complex and recruits intracellular membranous domains (e.g. ER, plasma membrane) giving rise to the omegasome. The omegasome matures further into the phagophore, a process orchestrated by several ATG (autophagy-related) proteins. At the same time, MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) gets conjugated with phosphatidylethanolamine (PE), allowing it to be recruited to the phagophore. The phagophore sequesters the cytoplasmic cargoes and closes upon itself, forming the autophagosome [44]. The sequestration of cytoplasmic components can be either selective or nonselective. Selective autophagy uses autophagy receptors such as SQSTM1 and OPTN to deliver specific proteins/organelles to the phagophores (reviewed in [45,46]). Once autophagosomes are formed, they continue to mature by fusion with early and/or late endosomes, after which they eventually fuse with lysosomes forming the autolysosome [44,47,48]. Finally, cargo inside the autolysosome gets degraded, and resulting metabolites are re-utilized during anabolic cellular activities [44,49]. In the case of microautophagy, the membrane of lysosome invaginates to enwrap cytoplasmic components and transport it to the lumen for degradation [44,50]. CMA uses the HSPA8/HSC70 chaperone to recognize a KFERQ-consensus sequence and deliver it to the LAMP2A complex at the lysosome. This complex oligomerizes and forms a channel in the lysosomal membrane that unfolds and translocates the protein into the lysosome [44,50]. Alongside the ALP, the ubiquitin-proteasome system (UPS) helps to maintain cellular homeostasis. In brief, the UPS uses ubiquitin ligases to add ubiquitin-tags to damaged or unfolded proteins, thereby earmarking them for degradation by the proteasome. Though perceived as independent processes, a balanced connection between the ubiquitin-proteasome system and autophagy exists. The use of ubiquitination as a recognition signal implies a close connection between both pathways. In fact, the UPS mainly clears misfolded proteins and small protein oligomers, while autophagy is crucial for the degradation of larger protein aggregates from the cell [51].
Figure 1.
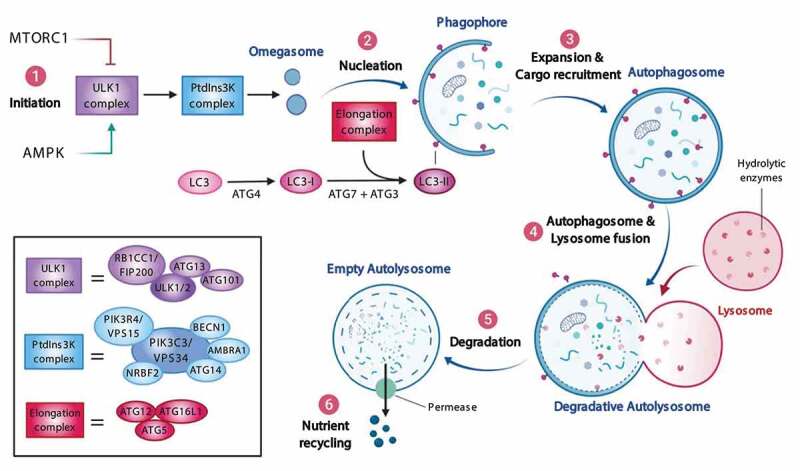
Overview of the autophagy pathway. (1) Bioenergetic stress caused by limited nutrient availability relieves the ULK1 complex from MTORC1 inhibition and activates the ULK1 complex through AMPK. (2) Upon initiation, the ULK1 complex translocates to omegasomes where it activates PtdIns 3-kinase complex to produce PtdIns3P and aided by the ATG12–ATG5-ATG16L1-containing complex matures to form a phagophore. (3) Expansion of the phagophore coincides with the cargo recruitment (such as aggregates, organelles) by autophagy receptors, after which the phagophore closes to form an autophagosome. (4) Retrograde trafficking of the autophagosomes facilitates their fusion with late endosomes/lysosomes converting them to autolysosomes. (5) These autolysosomes contain acid hydrolases that degrade their contents. (6) Finally, permeases present in the autolysosomal membrane mediate the diffusion of nutrients to the cytoplasm where they are re-utilized during anabolic cellular activities. Abbreviations: AMBRA1: autophagy and beclin 1 regulator 1; AMPK: AMP-activated protein kinase; ATG: autophagy-related; BECN1: beclin 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTORC1: mechanistic target of rapamycin kinase complex 1; NRBF2: nuclear receptor binding factor 2; PtdIns3K: phosphatidylinositol 3-kinase catalytic subunit type 3; RB1CC1/FIP200: RB1 inducible coiled-coil 1; ULK1: unc-51 like autophagy activating kinase 1
Mutations in key components of the autophagic pathway cause or enhance the risk for several human diseases, including severe congenital brain disorders (reviewed in [52]), systemic lupus erythematosus, asthma, Crohn’s disease, cancer, and NDs [53,54]. Not surprisingly, dysregulation of different stages (initiation, maturation, and lysosomal degradation) of the autophagic pathway is found in nearly all NDs [3,55]. Finally, accumulations of autophagosomes are found in the cytoplasm of motor neurons (MNs) from both familial and sporadic ALS patients [56]. These studies underscore the potential of a causal link between dysfunction of autophagic pathways and the development of ALS-FTD.
Dysfunction of autophagy at different levels in C9orf72 ALS-FTD
Due to the distinct transcription initiation sites on the C9orf72 gene, three variants (V1, V2, and V3) are transcribed [57]. While V1 and V3 both contain the HRE in their first intron, V2 contains the expansion in its promoter region [57]. One of the proposed mechanisms by which C9orf72 mutations cause ALS is reduced expression of the C9orf72 protein. Multiple studies have endorsed this mechanism and have shown a reduction of C9orf72 transcript levels in both patient iPSC-derived MNs and postmortem brain samples [39,58–65]. Intriguingly, some studies fail to recapitulate these reduced transcript levels [62,66]. The overall observed decrease in C9orf72 transcript levels is mainly due to decreased levels of the V2 transcript variant, probably due to the presence of the HRE in the promoter region of V2 [34,57,61]. Though the mechanism by which the HRE reduces C9orf72 expression is not clear, several factors such as methylation status and reduced binding of transcription factors have been suggested [60,63,68,69]. At the protein level, a relatively small and highly variable (10–75%) decrease of C9orf72 protein levels is noted in postmortem tissue, while C9orf72 levels were mostly unaltered in patient iPSC-derived MNs [33,70]. Therefore, evidence of C9orf72 haploinsufficiency remains incomplete. Further research with the use of recent methods to dissect specific (neuronal) populations from larger (brain) regions and more specific C9orf72 antibodies is needed [71].
Multiple studies have reported two isoforms of the C9orf72 protein: transcripts V2 and V3 encode for a longer isoform of ~50 kDa (C9-L), and translation of transcript V1 result in a smaller one of ~25 kDa (C9-S) [33,34,72]. The C9-S isoform has been localized to the nuclear membrane, and recent findings reveal the protein to have a function in nucleocytoplasmic transport [72]. The C9-L isoform, on the other hand, is localized at endosomes under basal conditions, and upon cellular starvation, the protein translocates to the lysosome [73]. The C9-L isoform is predicted to contain a DENN (differentially expressed in normal and neoplasia) domain [74]. DENN-domain containing proteins are best known for their function as GEFs (guanine nucleotide exchange factors) for RAB GTPases, small regulators of vesicular trafficking in eukaryotes [74–76]. Indeed, numerous recent studies identified a protein complex consisting of C9orf72, SMCR8 (SMCR8-C9orf72 complex subunit; another DENN-domain containing protein), and WDR41 (WD repeat domain 41) that interacts with a variety of RAB proteins, including RAB1A, RAB3, RAB5, RAB7, RAB8, RAB10, RAB11, RAB13, RAB15, RAB29 and RAB39B [29,65,77–85]. Hence, by activating RAB GTPases, C9orf72 is implicated in the regulation of endosomal trafficking events, including autophagy. Also, a proteomic study of the interactome of HA-tagged C9orf72 found several regulators of actin dynamics interacting with C9orf72, explaining the possible defects in axonal outgrowth and growth cones [86]. Two recent studies using cryo-EM revealed the monomeric and dimeric structure of C9orf72-SMCR8-WDR41 complex and showed that the WDR41 subunit bound exclusively to the DENN domain of SMCR8. Interestingly, while both groups attribute a GTPase-activating protein (GAP) function to the complex, the dimeric heterotrimer assembly was characterized as a GAP for RAB8A and RAB11A. In contrast, the monomeric heterotrimer was found to function as a GAP for the ARF family of small GTPases [77,87].
Neurons rely on the proper functioning of the ALP for their survival. Therefore, it is not surprising that dysfunctions in the ALP are implicated in NDs [44,88,89] and that ablation of core autophagy genes leads to progressive neurodegeneration [90,91]. In the following sections, we will focus on the different stages of autophagy and discuss what is known about the detrimental effects of C9orf72 mutations (Figure 2).
Figure 2.
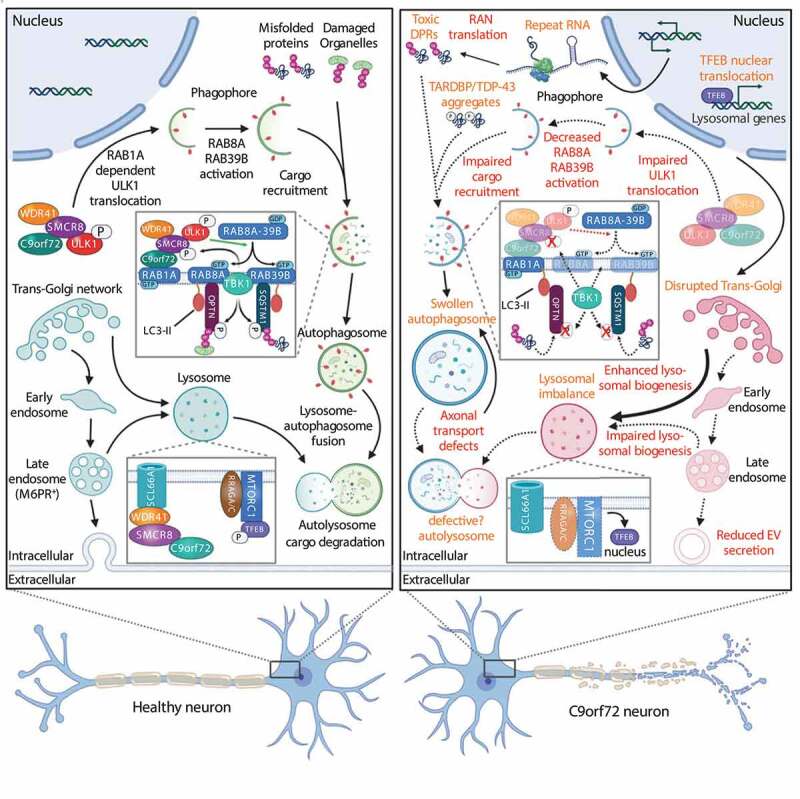
Overview of dysregulations in the autophagy-lysosome pathway in C9orf72 ALS-FTD. Reduced C9orf72 protein levels and toxic DPRs impact the ALP at multiple levels. Autophagy-related processes that are affected in C9orf72 ALS-FTD are indicated in red while cellular hallmarks are depicted in orange. Abbreviations: DPR: dipeptide repeat protein; M6PR: mannose-6-phosphate receptor, cation dependent; MTORC1: mechanistic target of rapamycin kinase complex 1; RRAG: Ras related GTP binding; RAN: repeat-associated non-AUG; SLC66A1/PQLC2: solute carrier family 66 member 1; SMCR8SMCR8-C9orf72 complex subunit; TARDBP/TDP-43: TAR DNA binding protein; TBK1: TANK binding kinase 1; TFEB: transcription factor EB; ULK1: unc-51 like autophagy activating kinase 1; WDR41: WD repeat domain 41
Autophagy initiation
The initiation of autophagy is a highly complex process that involves input from many different proteins and protein complexes (see section 2). While omegasome formation initially takes place on the ER membrane, input from multiple cellular components is required for phagophore growth. The delivery of these proteins/lipids hence requires proper functioning of the vesicular transport machinery [92]. Defects in the initiation of autophagy may thus be due to impairment of upstream autophagy induction mechanisms (e.g., ULK1 or MTORC1-driven), a failure of vesicular transport, or a combination of both. Intriguingly, the connection between autophagy and neurodegeneration is emphasized by knockout studies on Rb1cc1/Fip200, Atg5, or Atg7 genes in mouse models. Knocking out these genes from the central nervous system causes neurodegeneration, axonal dystrophy, and the presence of protein aggregates that stain positive for ubiquitin and SQSTM1 [90,91,93].
Several studies have put forth an adverse effect of C9orf72 downregulation on autophagy initiation [29,39,65,79–82,94-99]. There is, however, no consensus on the role of C9orf72 in the initials steps of the autophagic process as several research groups also reported beneficial effects of C9orf72 knockdown on this process [73,100,101]. These conflicting results may be explained by the multifaceted function of the C9orf72 complex in autophagy. More specifically, C9orf72 does not only interact with several Rab proteins and key regulators of membrane trafficking (reviewed in [102]), it is also found to regulate autophagy at multiple levels as discussed below.
C9orf72 forms a complex with SMCR8 and WDR41 and has GEF activity toward small GTPases. In addition, several independent studies using in vitro binding assays, immunoprecipitation and/or proximity ligation assays (PLA) have identified interactions between C9orf72 complex and the ULK1 complex [65,80,81,97,103,104]. Recently, this interaction was shown to be directed through the DENN domain of the long C9orf72 isoform that interacts with ATG13 (and SMCR8). Moreover, SMCR8 also binds to ATG13 but also directly to the ULK1 kinase [105]. Though it was known that C9orf72 partially localized to lysosomal membrane and nutrient starvation enhances C9orf72 and ULK1 complexes interaction [81], the mechanisms behind it were discovered only recently [105]. SLC66A1/PQLC2 (solute carrier family 66 member 1), a lysosomal amino acid transporter, interacts with the C9orf72 complex member WDR41, and this association is enhanced upon amino acid deprivation [105]. Similarly, under starvation conditions, MTORC1 is inactivated, and its inhibitory effect on ULK1 is lost. Therefore, the coordinated release of the ULK1 complex by MTORC1 and the recruitment of the C9orf72 complex to lysosomes by SLC66A1 (upon starvation) possibly brings the two complexes together and facilitates their interaction.
The method by which the C9orf72 complex regulates ULK1-dependent autophagy initiation remains debated. The knockdown of SMCR8 (but not of its binding partners C9orf72 or WDR41) causes a 3-fold increase in ULK1 protein levels due to increased transcription [103]. On the contrary, a study on c9orf72 knockout neurons revealed defects in the autophagic pathway, together with reduced ULK1 protein levels due to post-transcriptional rather than transcriptional mechanisms [104]. Using a potent ULK1 inhibitor, these researchers recapitulated the autophagy defects and showed that it could be mediated via reduced BECN1/beclin-1 activity [104]. Moreover, a study using MEFs reported a reduction in the inhibitory serine 757 phosphorylation of ULK1 upon knockdown of C9orf72. Interestingly, the same study also reported increased serine 757 phosphorylation levels of ULK1 following SMRC8 knockdown [81]. Differences in the cell types used or in the knockdown efficiency could partially account for the disparities in ULK1 expression and phosphorylation. Besides, knockdown of C9orf72 could also have unpredictable effects, as the loss of a single member from the C9orf72 complex could indirectly influence the expression levels and function of other members [100].
In 2016, the C9orf72 protein complex was implicated in the early stages of autophagy [65]. Using yeast two-hybrid screens, in vitro binding studies, and co-immunoprecipitation assays, an interaction between C9orf72 and RAB1A (involved in autophagy initiation) was discovered [65]. The same group also showed that C9orf72 functions as an adaptor between RAB1A and the ULK1 complex, thereby translocating the latter one to specific sites of autophagosome formation [65]. Unexpectedly, C9orf72 was identified as a RAB1A effector rather than a RAB1A GEF as it preferentially interacted with GTP-RAB1A instead of GDP-RAB1A [65].
RAB5A also plays a crucial role in autophagy initiation by mediating delivery of both the PtdIns3K complex and the ATG7-ATG5-LC3 lipid conjugation complex to the newly formed phagophore [44]. Interestingly, ALS2/Alsin, another GEF involved in ALS-FTD pathogenesis, is a well-characterized activator of RAB5, underscoring the relevance of this interaction in the context of ALS and FTD [106]. In affinity-isolation assays with C9orf72, RAB5 could be detected. The interaction between RAB5 and C9orf72 has been confirmed by a high degree of colocalization (60–80%) of the two proteins in iPSC-derived MNs and human-induced motor neurons (iMNs) [39,79,94,107]. Using the expression of an mRFP-GFP-LC3 fluorescent probe in these cells, a significant reduction in autophagosome numbers was observed, which could be attributed to impaired autophagosome formation [39,94,107]. Interestingly, the expression of constitutively active RAB5A could rescue C9orf72 patient iMN survival and alleviate autophagosome formation deficits [39].
The C9orf72 complex also regulates proteins upstream of ULK1, possibly explaining some of the discrepancies between C9orf72 as a positive or negative regulator of autophagy. As mentioned earlier, the C9orf72 complex regulates nutrient sensing at the lysosome mediated by the WDR41-SLC66A1 interaction [105]. Recently, multiple groups showed a reduction in MTORC1 substrate (RPS6KB1/p70S6K) phosphorylation in C9orf72 knockdown models. This reduction in MTORC1 substrate phosphorylation may contribute to defects in lysosomal nutrient sensing [73,83,100,101]. In addition to regulating ULK1 by direct phosphorylation, inhibition of MTORC1 also transcriptionally regulates autophagy by dephosphorylating transcription factors such as TFEB, TFE3, and MITF [108]. As a consequence, the transcription factors translocate to the nucleus and upregulate the transcription rate of several genes involved in both lysosomal biogenesis and autophagy regulation [108]. Consistent with reduced MTORC1 activity, increased nuclear localization of TFEB, TFE3, and MITF were reported in C9orf72 loss-of-function model systems [83,100,101]. These studies also described an increase in autophagosome and lysosome numbers and a concomitant enhancement in the autophagic flux. Recently, the method by which C9orf72 regulates the nutrient-dependent lysosomal localization was discovered [83]. C9orf72 interacts with inactive RRAG GTPases, but not active RRAG GTPases mediated through its DENN domain. Moreover, the overexpression of active RRAG GTPases rescued the defects in MTORC1 activation and localization and restored the cytoplasmic localization of TFEB. Although this study did not assess the GEF function of C9orf72, the fact that C9orf72 only colocalizes with the inactive GTPases supports that C9orf72 has a GEF function toward these RRAG GTPases [83].
Taken together, loss of C9orf72 can influence the initiation of autophagy in both a positive and negative manner. While loss-of-function of C9orf72 seemingly alters MTORC1 signaling, thereby activating the autophagic pathway, especially in c9orf72 knockout models, C9orf72 haploinsufficiency negatively affects several vesicular trafficking routes (RAB1A, M6PR, RAB5). It inhibits the initiation of autophagy, and this not only in “artificial” models that reduce the C9orf72 protein levels but also in models that use endogenous HRE in C9orf72. Overall, most evidence points toward the inhibition of the initiation of autophagy by subtle but synergistic impairment of several vesicular trafficking events (Figure 2).
Autophagosome maturation and autophagosome-lysosome fusion
The process of autophagosome maturation involves specific or nonspecific recruitment of cellular cargoes that are predestined for degradation. The dynein-dynactin motor protein complex aids this process by mediating the retrograde transport of autophagosomes to the perinuclear region where they fuse with lysosomes to form autolysosomes [109,67,141,162,193,110–112]. Lysosomes derived from the cell soma are delivered to the distal axonal parts where they can fuse with autophagosomes [112]. In axons, the retrograde transport of autophagosomes and the anterograde transport of lysosomes favors early fusion events between the two [113]. Thus, by the time the autophagosomes reach the soma, they become mature and functional. Mutations in motor proteins KIF5A (kinesin family member 5A), DCTN (dynactin subunit), and dynein and in VCP and CHMP2B, two regulators of the autophagosome-lysosome fusion process, cause ALS and/or FTD underscoring the importance of these processes in ALS-FTD pathogenesis. At various stages, several RAB GTPases regulate the fusion processes during the journey of autophagosomes from distal ends to the soma in an axon.
As shown in Figure 2, the C9orf72-SMRC8-WDR41 complex interacts with a variety of Rab proteins. This complex was discovered to exert autophagy-activating GEF activity toward both RAB8A and RAB39B [80]. Immunoprecipitation experiments proved that the interaction (and GEF activity) between the RAB8A-RAB39B-C9orf72 complex is almost exclusively mediated by the SMCR8 subunit [80]. Two recent cryo-EM studies confirmed the interaction between the C9orf72 complex and RAB8A, RAB11A and the ARF family of GTPases [77,87]. However, both studies failed to detect the GEF activity of the C9orf72 complex raising some doubt about the role of the complex in vivo [77,87]. Both RAB8A and RAB39B interact with known ALS-associated proteins such as OPTN, SQSTM1 and TBK1. These proteins are reported to be involved in other NDs as well [80,114]. Of note, the RAB8A GTPase was shown to interact with the OPTN receptor protein, while RAB39B, on the other hand, strongly interacts with SQSTM1 [80,115]. Defects in the recruitment of SQSTM1 to autophagy target proteins were recently confirmed in a cellular model that combined both C9orf72 gain and loss-of-function [99]. The same study also recapitulated autophagy defects upon the lowering of TARDBP levels [99]. Interestingly, overexpression of neither C9orf72, its complex partner SMCR8, nor wild-type or constitutively active RAB8A or RAB39B was able to restore these defects [80]. These results are in agreement with another study that demonstrated that loss of TARDBP disrupted autophagic flux at a later step in the autophagy pathway, namely the fusion of autophagosomes with lysosomes [116].
The active form of RAB8A can recruit TBK1, a protein kinase that is highly involved in the regulation of antiviral innate immunity and autophagy. Intriguingly, genetic ablation of C9orf72 in rodent models is characterized by immune phenotypes, including increased expression of inflammatory cytokines, splenomegaly, neutrophilia, thrombocytopenia, and severe autoimmunity, providing a possible link between TBK1 and C9orf72 function [117,118]. Although no kinase activity toward C9orf72 was detected for TBK1, the kinase was able to phosphorylate SMCR8 [80], ULK1 [80], as well as OPTN and SQSTM1 [119,120]. The phosphorylation of OPTN and SQSTM1 enhances their interaction with LC3 and hence activates autophagic clearance of target proteins [119,120]. This phosphorylation also proves to be important for the function of SMCR8 in autophagy as phospho-mimetic SMCR8, but not phospho-dead SMCR8 mutant overexpression was able to counteract autophagy defects due to SMCR8 knockdown [80]. In addition, TBK1 depletion also resulted in autophagy dysfunction that could be rescued by expression of phospho-mimetic SMCR8 variant that was specific for TBK1 [80]. The TBK1 loss-of-function autophagy phenotype could also be rescued by expression of constitutively active RAB39B, suggesting a role of TBK1 upstream in the C9orf72-SMCR8-RAB39B-RAB8A-SQSTM1-OPTN pathway [80,115]. This leads to a model where TBK1 and the C9orf72 complex regulate site-specific autophagy initiation at cellular garbage hotspots marked by a high concentration of autophagy receptors. Additional evidence for this interplay comes from the fact that loss-of-function mutations in TBK1 as well as in the autophagy receptors SQSTM1 and OPTN are well-established disease genes in the ALS-FTD field [119,121,122]. Furthermore, the clinical presentation of patients with mutations in TBK1 or C9orf72 share commonalities as both are frequently associated with ALS and FTD, and their neuropathology is highly similar [14,24,119,121].
STX17 (syntaxin 17) is a well-characterized SNARE protein that aids the fusion of autophagosomes and lysosomes. STX17 was recently found to be implicated in the ULK1-dependent initiation of autophagy [123]. Remarkably, STX17 is also a TBK1 substrate and is dependent on this phosphorylation for its translocation to the autophagy initiation complex [123]. It would be interesting to know if the reduction of C9orf72 or SMCR8 levels affects the phosphorylation or, more likely, the translocation of STX17. Currently, we can only speculate on the role of STX17 and other autophagosome-lysosome fusion proteins in the pathogenesis of ALS-FTD. Similarly, autophagy receptors were also found to mediate MYO6 (myosin VI)-dependent retrograde transport of autophagosomes and autophagosomes-lysosome fusion, but whether this is disturbed by C9orf72 mutations is not clear [124].
RAB7A and RAB11A play a vital role in the process of autophagosome maturation [125]. RAB7A is involved in microtubular transport of autophagosomes, late endosomes, and it also regulates their fusion with lysosomes [125,126]. RAB11, on the other hand, plays a role in autophagosome maturation by delivering multivesicular bodies (MVBs) and forming amphisomes [125,127]. At least two independent studies using tagged human C9orf72 have confirmed the endogenous interaction and colocalization between C9orf72 and RAB7-RAB11 in neuronal cell lines, iPSC-derived neurons, or human spinal cord samples [79,80,82]. Knockouts of C9orf72 or RAB7 were able to mimic the defective intracellular trafficking phenotype observed in iPSC-derived motor neurons [82]. Unexpectedly, the C9orf72 complex had a higher affinity for the active RAB7A protein as compared to the inactive GTPase as would be expected for a RAB GEF [82]. Therefore, the C9orf72 complex is thought to function as an effector rather than a GEF for RAB7A. However, this function is hampered in C9orf72 ALS-FTD [82]. On the contrary, while a similar vesicle trafficking phenotype was found in iMNs, the expression of constitutively active RAB5A, but not of constitutively active RAB7A could rescue C9orf72 patient iMN survival [39].
Finally, using immunoprecipitation and immunohistochemistry experiments, an interaction was discovered between C9orf72 and UBQLN2 (ubiquilin-2), an ALS-associated protein that is known to function as a shuttling factor in proteasome-mediated protein degradation [79,128,129]. Upon blockage of the proteasome, a significant increase in the colocalization of C9orf72 and ubiquilin-2 was noted [79]. Though this confirms the interplay between the ALP and UPS in proteostasis, the increased colocalization also implies a dual function for ubiquilin-2 in both UPS and autophagy. Indeed, complete loss or mutations in the ubiquilin-2 proteins affect not only the initiation of autophagy by decreasing MTORC1 signaling but also negatively affect lysosomal pH [130]. In addition, some proteins known to interact with the C9orf72 complex in the ALP (SQSTM1, OPTN, RAB11) have been shown to also influence UBQLN2 [131]. Unfortunately, to date, the exact role of UBQLN2 in C9orf72 ALS-FTD is not known, and the consequences of C9orf72 loss-of-function remain unclear.
Lysosomal function/biogenesis
Indirectly, autophagy could be disturbed by dysfunctional lysosomes or problems in lysosomal biogenesis. Genetic studies support a critical role for lysosomal dysfunction in the pathology of several NDs, including AD (e.g., APP, PSEN1), PD (e.g., PRKN/Parkin, GBA, TMEM175), ALS, and FTD (e.g., TBK1, GRN, VCP) [132]. In fact, the identification of mutations in so many different genes involved in ALP as causes of ALS-FTD or other NDs highlights the importance of this pathway for neuronal function and survival. This raises the question if C9orf72 is involved in lysosomal function as well.
The C9orf72 protein is predominantly localized in the cytosol. However, some studies found that the protein can be found residing on endosomes as well, as shown using colocalization with the RAB5A-endosomal marker protein [39,72,79,94]. Analysis of lysosomal membranes revealed the presence of all C9orf72 complex members at the lysosome [133]. Moreover, the increase in colocalization between C9orf72 and LAMP1 (lysosomal associated membrane protein 1) under starvation condition supports for a function of C9orf72 at the lysosome [73]. The translocation of C9orf72 to the lysosomal membrane is mediated by the interaction between SLC66A1 (a lysosomal cationic amino acid transporter) and WDR41 [85,105]. Loss of C9orf72 results in defective activation of MTORC1 and concomitant upregulation of the ALP [73,83,100,101]. Recently, the mechanisms by which this takes place were elucidated. C9orf72 interacts with RRAG GTPases that, in turn, regulate MTORC1 and TFEB localization [134]. Upon reduction of C9orf72 levels, RRAG GTPase levels are reduced, as does the lysosomal localization of MTORC1 and TFEB [134]. This consequently leads to the increased nuclear localization of TFE3, MITF and TFEB, and is responsible for ALP activation through enhanced transcription of genes involved in both the autophagic process and general lysosomal biogenesis [73,83,100,101,134]. This is supported by the increase in lysosome number (or lysosome size, depending on the study) upon C9orf72 protein reduction seen in a variety of C9orf72 ALS-FTD models, including c9orf72 knockout mice, fibroblasts, macrophages, and primary neurons, patient iPSC-derived motor neurons and C9orf72 knockdown cell models [73,81,83,97,100,101,135,136].
On the contrary, in macrophages and spinal neurons of c9orf72 or smcr8 knockout mice [39,137] or in ALS patient iMNs [39,94,107], a decrease in both endosome and lysosome numbers is reported. This decrease was due to the reduction of C9orf72 protein levels, as ASO-mediated reduction of C9orf72 was able to recapitulate the reduction lysosome numbers, and isogenic control iMNs or forced expression of human C9orf72 protein rescued the endosomal/lysosomal phenotype [39]. Recently, a biochemical study on C9orf72 function in neuronal N2A cells found no changes in the number of lysosomes upon shRNA-mediated C9orf72 knockdown. These data reveal once more that the physiological consequences of C9orf72 loss are highly complex and depend not only on the model system or knockdown/knockout approach used, but could be regulated by other factors as well (e.g., epigenetic regulation, cell state, nutrient availability, protein levels of other C9orf72-complex members).
The interplay between CARM1 (coactivator associated arginine methyltransferase 1) and C9orf72 provides an example for such an epigenetic regulation. CARM1 is a methyltransferase that is involved in epigenetic regulation of lipid metabolism and the ALP. Under normal conditions, Liu et al. 2018 found that CARM1 interacts with the C9orf72 complex and that this interaction drives the delivery of the methyltransferase to the lysosome upon nutrient stress [138]. Subsequently, CARM1 gets degraded in the lysosomes and thus is part of a negative feedback loop that controls ALP activation during starvation [138]. C9orf72 haploinsufficiency causes the aberrant accumulation of CARM1, especially during nutrient stress [138]. Cytoplasmic accumulation of CARM1 results in nuclear translocation of the methyltransferase followed by transcriptional activation of genes involved in the ALP, concurrent with the transcriptional upregulation through MTORC1 effectors TFE3, MITF and TFEB [138].
Finally, C9orf72 deficiency could impede autophagy due to impaired acidification of the lysosomes and autolysosomes. This would not only result in the accumulation of degradative cellular cargo but also in the buildup of regulatory proteins together with an increase in lysosomal biogenesis resulting in the accumulation of dysfunctional lysosomes. However, only one study so far reported blockage of lysosomal acidification in C9orf72 mutant macrophages [137]. In support of a role for impaired lysosomal pH maintenance in the pathophysiology of ALS-FTD, a direct interaction between the C9orf72-interacting protein UBQLN2 and the lysosomal v-ATPase subunits responsible for the acidification was recently established [130]. Furthermore, the enlarged lysosomes found in some model systems are indicative of the accumulation of non-degraded proteins and organelles, possibly due to the combination of enhanced lysosomal biogenesis and dysfunctional lysosomes. Besides, such enlarged autophagosomes are also repeatedly identified in C9orf72 loss-of-function models. Several independent groups attributed this deficiency to alterations in M6PR vesicle trafficking [39,82,139]. These vesicles originate from the trans-Golgi network and transport lysosomal proteins (that have M6P-tags) to endosomes during lysosomal biogenesis [140]. In C9orf72 patient iMNs, fibroblasts or C9orf72 knockout iMNs, M6PR was found in densely packed clusters throughout the cytoplasm, while in control conditions, these vesicles had a perinuclear distribution [39,82]. Ultimately, this may negatively influence endosome maturation, and lysosomal function resulting in a late-stage block of the ALP characterized by swollen autophagosomes and decreased lysosome numbers.
1.1 Impaired autophagy drives the accumulation of TARDBP and DPR proteins, influences stress granule dynamics and contributes to HRE RNA-related toxicity.
Sense and antisense transcripts originating from the HRE in C9orf72 can be RAN-translated in different reading frames to produce poly-GR, poly-GP, poly-GA, poly-PR, and poly-PA DPRs. Among these, the arginine-containing ones appear to be the most toxic, although they are less abundantly expressed in postmortem C9orf72 ALS-FTD patient samples (brain + spinal cord) and patient iPSC-derived motor neurons [39–42,64,107,142–144]. In patients, poly-GA is more abundant than arginine-containing DPRs, and a recent study comparing poly-GA- and poly-PR-expressing mice only revealed a selective neuronal loss in the poly-GA-expressing mice. This suggests that non-arginine containing DPRs are also important for C9orf72 ALS-FTD pathogenesis [144]. Using the proteasome inhibitor MG-132, it has been shown that, with the exception of poly-GP, insoluble DPRs are degraded by the autophagic system and that this system is crucial to counteract the accumulation of DPRs in C9orf72 patients [99,145].
Ribonucleoprotein granules (RNPs) are formed by the association of mRNA and RNA-binding proteins (RBPs) [146,147]. RNPs can develop into stress granules (SGs) by means of low complexity domains or aggregation-prone regions present in these proteins in a complicated process called liquid-liquid phase separation (LLPS) [146,147]. Recently, defects in SG dynamics have been linked to ALS and FTD pathophysiology [146,147]. Interestingly, autophagy proved to be responsible for SG removal and ALP inhibition led to the persistence of SG [148]. Arginine-rich DPRs (poly-GR and poly-PR) itself can undergo LLPS to form SGs but can also interact with RBPs hence influencing the dynamics of SG assembly and disassembly [149]. Excessive stress granule formation, in turn, activates intracellular integrated stress response (ISR) and the associated phosphorylation of EIF2A (eukaryotic translation initiation factor 2a). Besides regulating ISR progression, p-EIF2A was recently found to activate RAN translation of DPRs from the C9orf72 HRE by multiple independent groups [150,151]. Recently, a feed-forward loop between SG assembly and RAN translation mediated through p-EIF2A has been proposed [146,152]. Additionally, the long C9 isoform itself influences SG formation as overexpression of C9-L enhances SG assembly, and C9-L knockdown negatively impacted the assembly of SGs [153]. In the same study, gene expression analysis revealed a downregulation of some SG-related proteins suggesting a combined transcriptional and biochemical regulation of the C9-L isoform on SG formation [153]. Finally, repeat expansion RNA is able to promote LLPS and drive SG assembly in vivo in a repeat-length-dependent manner [153,154].
The correct functioning of the ALP also prevents cytoplasmic accumulation and aggregation of TARDBP, the main hallmark of nearly all ALS cases, and nearly half of FTD cases [6,49,107,155]. The pharmacological induction of the ALP in TARDBP ALS models alleviates the pathogenesis and enhances TARDBP turnover and cell survival [107,156,157]. Interestingly, aggregation and concomitant TARDBP loss-of-function further exacerbates autophagy impairments at multiple levels (reviewed in detail by [49]) [158,159].
However, this does not undermine the loss-of-function effect in C9orf72 ALS-FTD. The impaired degradation of DPRs and TARDBP resulting from disruption of the ALP due to loss of C9orf72 may drive neurodegeneration in the first place. In the last couple of years, several independent research groups reported an increase in the accumulation, aggregation, and toxicity of DPRs upon reduction of C9orf72 protein levels [39,94,99,107,145]. These effects can directly be attributed to alterations in the ALP. Intriguingly, a study that used lactacystin to block UPS-mediated protein degradation found that the C9orf72 protein itself is degraded by the proteasome adding another puzzle piece to the complexity of C9orf72 and protein homeostasis [96].
Finally, few studies do support the link between C9orf72 HRE RNA-related toxicity and ALP dysfunctions. Overexpression of the autophagy receptor protein SQSTM1 alone was enough to attenuate RNA toxicity in a zebrafish model for C9 ALS-FTD [160]. Moreover, a recent study found that inhibition of EXOSC10, the catalytic subunit of the RNA exosome complex that usually degrades HRE-containing RNA, aggravates C9 ALS-FTD pathology [161]. Expression of poly-GR and poly-PR proteins were shown to impair the activity EXOSC10, thus providing a connection between DPR-toxicity and HRE RNA-related toxicity [161].
Taken together, toxic gain-of-function pathology due to DPR proteins, TARDBP, and HRE RNA and loss-of-function mechanisms caused by C9orf72 haploinsufficiency could result in a vicious cycle where one mechanism aggravates the other and vice versa. Ultimately this affects the cellular health status due to the accumulating protein aggregates and the dysregulation of vital cell components (e.g., mitochondria, proteasome, ribosome), processes (e.g., axonal and nucleocytoplasmic transport, translation, apoptosis) (Figure 3) [66,163–166]. This model is consistent with the result of C9orf72 loss-of-function/knockout mice models that show immune phenotypes and early mortality but without clear signs of neurodegeneration. In fact, when these mice were crossed with mice expressing a repeat-containing transgene, autophagy induction upon DPR production was attenuated, and the toxic accumulation of DPRs was accelerated [59]. Moreover, using a transgene expressing poly-PR50 in patient-derived iMNs with reduced C9orf72 protein levels, similar findings were reported [39].
Figure 3.
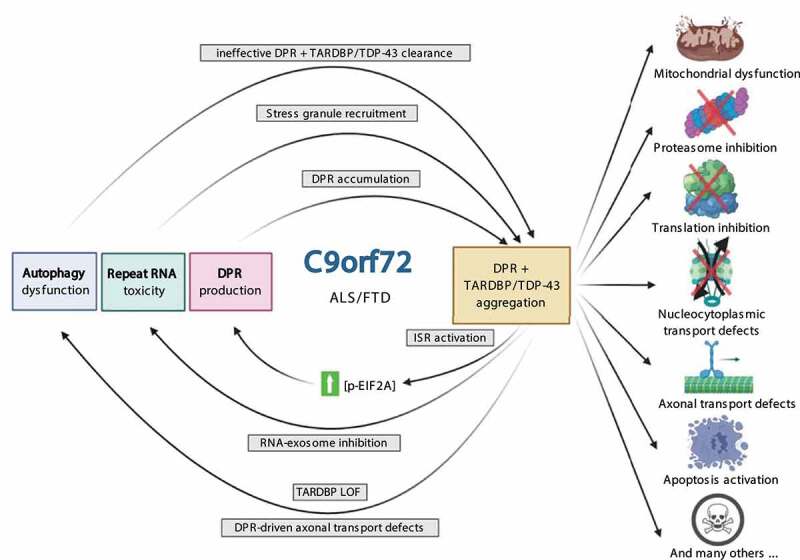
Synergistic pathogenesis of C9orf72 disease mechanisms. Toxic HRE gain-of-function disease mechanisms (repeat RNA toxicity and DPR production) work together with C9orf72-mediated autophagy impairment and result in the aggregation of DPR and TARDBP proteins. These DPR inclusions influence a variety of homeostatic and disease mechanisms, resulting in a vicious feedback cycle. Abbreviations: DPR: dipeptide repeat protein; ISR: integrated stress response; LOF: loss-of-function; p-EIF2A: phosphorylated eukaryotic translation initiation factor 2A
Axonal autophagy: an important player in axonal transport
Active transport of “cargoes” along the axon of a neuron (anterograde or retrograde) is a prerequisite for maintaining neuronal homeostasis and therefore is a tightly regulated process. Kinesin (anterograde) and dynein (retrograde) motors are the driving forces of axonal transport [166]. Defects in the transport along axons result in the dysregulation of multiple cellular processes leading to protein aggregation, ER stress, DNA damage, mitochondrial defects, and the dysregulation of autophagy (reviewed in [167]). Axonal transport is an essential mechanism involved in the ALP as retrograde trafficking of autophagosomes and anterograde trafficking of lysosomes are both crucial for the maturation of autophagosomes [167]. Not surprisingly, impairments in axonal transport are reported in many neurodegenerative diseases underscoring the importance of this mechanism for neuronal maintenance [167,168]. Data obtained in presymptomatic rodent and iPSC-derived models of a variety of ALS subtypes (caused by mutations in TARDBP, FUS, SOD1, DCTN1, and of sporadic ALS) reveals axonal transport defects as one of the early pathological feature [66,167,170–172]. In C9orf72 ALS-FTD, disruption of axonal transport of multiple cargos (mitochondria, RNA granules, lysosomes, and autophagosomes) have been reported in both c9orf72 knockout mice and patient iPSC-derived neurons [66,172]. SMCR8 deficiency alone was also able to cause defects in axonal transport, and the combined loss of SMCR8 and C9orf72 in a C9 ALS-FTD mouse model exacerbated these defects, suggesting the involvement of the larger C9orf72 complex in axonal transport regulation [172].
Interestingly, inhibition of retrograde transport in a poly-GP-expressing cell model of ALS resulted in a reduced aggregation propensity of the DPR in contrast to what would be expected upon ALP disruption [173]. This is achieved by a compensatory cellular mechanism that upregulates UPS-mediated protein degradation [173]. However, this model omits proteasome-inhibiting poly-GA DPRs and does only reflect the early stages of the disease. Over time, disruption of retrograde autophagosomes trafficking will inevitably lead to the aggregation of toxic and misfolded proteins. The upregulation of UPS merely functions as a compensatory mechanism for short-term proteotoxic stress. A recent study reported an inhibitory role of arginine-rich DPRs on microtubule-based axonal transport in C9orf72 patient iPSC-derived motor neurons [66]. Treatment of healthy neurons with synthetic arginine-rich DPRs recapitulated these phenotypes and provided further evidence for a crucial role of toxic-gain-function mechanisms in the pathology of C9orf72 ALS-FTD. These arginine-rich DPRs were able to interact with both the microtubular network and both retrograde and anterograde molecular motors driving axonal transport [66]. Interestingly, in postmortem C9orf72 brain samples, KIF5B (kinesin family member 5B) is found in DPR inclusions, and restoring the levels of functional kinesin-1 proved to be sufficient to reverse the axonal transport defects [66]. Moreover, two other kinesin-1 isoforms (KIF5A, KIF5C), TUBA/α-tubulin and TUBB/β-tubulin subunits as well as members of the dynein and dynactin complexes were co-immunoprecipitated with PR30 aggregates in mouse spinal cord lysate [66]. As mentioned earlier, this fits into the C9orf72 ALS-FTD model where arginine-rich DPR proteins impair axonal transport machinery, which, in turn, aggravates DPR pathology due to ALP impairments and results in a vicious cycle that ultimately culminates in the loss of the most vulnerable neuronal population (Figure 3).
Lastly, nucleocytoplasmic transport defects have been identified as a major source of pathology in various C9orf72 ALS-FTD models [163,174,175]. Although no direct relation between the ALP and nucleocytoplasmic transport dysfunctions has been described so far, possible links between the two pathologies can be drawn. First, protein stress originating from DPR expression and aggregation or proteostasis defects translocates crucial nucleocytoplasmic transport factors to SGs [176,177]. Second, inhibition of nuclear export might be a cellular response to counteract ALP dysregulation, as this can enhance autophagy rates by increased nuclear TFEB levels [178].
Therapeutic strategies for the treatment of C9orf72 ALS-FTD
Currently, there is no cure for ALS or FTD. There is a high unmet medical need for effective treatments and novel drug targets [179]. In this review, we discussed various consequences of C9orf72 mutations on different steps of the ALP. We now will summarize recent progress in drug discovery for C9orf72 patients and the different therapeutic strategies for the treatment of C9orf72 ALS-FTD, focusing on the ALP.
I) Small molecule activators of the autophagic pathway
Activation of autophagic pathway to increase the clearance of misfolded and aggregated proteins would be a promising strategy to protect the cells from the toxic effects of accumulating insoluble proteins. However, drugs targeting upstream regulators of the ALP (i.e., MTORC1, AMPK) may elicit detrimental effects due to the complex intertwinement that exists between various regulators of autophagy and immune pathways [180]. Overexpression of the autophagy receptor SQSTM1 did not lead to enhanced autophagic clearances of aggregates but accelerated and worsened disease onset in SOD1 ALS mouse models [181]. In contrast, many studies have reported beneficial effects of small-molecule autophagy activators (e.g., rapamycin, trehalose, lithium carbonate, fluphenazine, methotrimeprazine, berberine, promethazine, colchicine, spermidine, tamoxifen, and carbamazepine), especially on TARDBP pathology [156,157,182]. An interesting strategy to achieve this is to increase nuclear TFEB levels by inhibiting its nuclear export using selective inhibitors of nuclear export (SINEs) [178]. Alternatively, as C9orf72 has a profound role in RAB5 biology, targeting RAB5 holds promise [39,79,94]. PIKFYVE is a kinase that catalyzes the conversion of PtdIns3P to PtdIns (3,5)P2, thereby inhibiting the fusion between autophagosomes and lysosomes. Inhibition of this kinase by Apilimod or other specific PIKFYVE kinase inhibitors was found to counteract and enhance the fusion of lysosomes and autophagosomes, thereby restoring lysosomal and autophagosomal defects in C9orf72 iMNs [39,94]. An engineered anti-coagulation-deficient form of activated PROC (protein C, inactivator of coagulation factors Va and VIIIa; 3 K3A-APC) could also restore autophagosome formation and increase lysosome numbers in C9orf72 and a subset of sporadic ALS MNs, and this was confirmed in vivo [107]. This lowered the DPR burden in iMNs, corrected TARDBP mislocalization, and enhanced the survival of C9orf72 and sALS iMNs [107]. The therapeutic effects of 3 K3A-APC were found to be mediated through increased expression of autophagy-initiating factors that were dependent on F2R/PAR1 (coagulation factor II thrombin receptor) signaling. Another independent study found that by overexpressing HSPB8 (heat shock protein B8), the autophagy-mediated degradation of DPRs was increased [145]. This approach led to an increase in the turnover rate of both soluble and insoluble protein species and was able to target misfolded and aggregation-prone proteins before they form pathological inclusions [145,183]. Finally, earlier this year, a phase I clinical trial on L-serine dietary supplementation was completed in ALS patients [184]. L-serine could exert neuroprotective effects by upregulating the activity of proteolytic lysosomal enzymes [185]. A phase IIa clinical trial is currently in its recruitment phase [NCT03580616].
Ii) Reverse axonal transport defects
Axonal transport defects are a common feature in NDs and, as discussed earlier, could have a profound role in the pathogenesis of C9orf72 ALS-FTD. In AD models, it was shown that the polyphenol curcumin potently increases autophagy by promoting the expression of retrograde motor proteins and enhancing their affinity toward the microtubular network [186]. Moreover, inhibition of HDAC6 (histone deacetylase 6) was shown to increase TUBA/α-tubulin acetylation and rescue axonal transport defects in patient-derived MNs harboring mutations in the FUS gene [169]. As the autophagy pathway is dependent on axonal transport, strategies that improve axonal transport deficits are indirectly expected to stimulate autophagy as well (reviewed in [167]).
Iii) Gene therapy to activate the autophagy pathway
Genetic modification of autophagy factors could be a useful approach to compensate for proteostasis defects in C9orf72 ALS-FTD. For instance, multiple groups have reported the rescue of observed ALP defects in their respective disease models by overexpressing the C9orf72 protein [39,65]. Another example in ALS-FTD disease context is the overexpression progranulin, which has a neuroprotective effect by enhancing lysosomal function [188–190]. Progranulin is an evolutionary conserved secreted glycoprotein that, among other functions, has a profound role at the lysosome and regulates cell survival, growth, immune response, and proteostasis (reviewed in [190]). Haploinsufficiency due to heterozygous loss-of-function mutations in the GRN gene causes lysosomal deficits, leading to FTD with TARDBP inclusions [191,192]. This suggests that a shortage of progranulin can cause lysosomal dysfunction leading to accumulation of insoluble TARDBP, but also that increasing progranulin levels may stimulate the lysosomal clearance of insoluble proteins. Progranulin has been reported to function as a chaperone of several lysosomal enzymes, including CTSD (cathepsin D) and GBA [188]. Progranulin overexpression could decrease insoluble TARDBP levels and significantly extend the survival of transgenic TARDBPA315T mice [187].
Iv) Targetting upstream disease causes
Genetic strategies to target the upstream cause of C9orf72 ALS-FTD in patients are a promising approach. Such therapies would target not only one disease mechanism, such as autophagy defects but also block various cell death pathways that are activated by pathogenic mutations. Antisense oligonucleotides (ASOs) are single-stranded DNA molecules that selectively target and bind mRNA, thereby earmarking it for RNase H-mediated degradation. It, in turn, prevents the production of toxic protein species (i.e., DPRs) and RNA-toxicity (i.e., RNA foci that sequester RNA binding proteins) [194–196]. After a successful trial in a C9-mouse model in which a C9orf72-ASO managed to alleviate neurodegenerative symptoms, RNA foci, and DPR inclusions, a clinical phase I trial of a C9orf72-ASO therapy is currently taking place [NCT03626012] [196]. As ASOs do not cross the blood-brain barrier, they must be administered directly into the CSF (e.g., by repeated intrathecal infusions). On the other hand, the use of viral vectors, such as those based on the adeno-associated virus (AAV) can circumvent such drawbacks as they safely integrate into the genome and stably express silencing sequences with high efficiency. Recently, two independent groups tested AAV-mediated expression of C9orf72-targeting micro-RNA sequences (Mir) in a C9orf72 BAC transgenic mice model. Expression of the Mir activates the RNA-interference machinery of the cell to silence the C9orf72 HRE transcript and rescue DPR and RNA-foci pathology [197,198].
Another avenue that might provide benefit for C9orf72 ALS-FTD patients is to reduce the production of the toxic DPRs that exert multiple modes of toxicity (reviewed [199]). RAN translation was enhanced upon activation of the cell’s integrated stress response (ISR). The ISR will lead to the phosphorylation of EIF2A, thereby reducing overall levels of mRNA translation. At the same time, ISR-dependent phosphorylation of EIF2A enhances the efficiency of unconventional RAN translation [151,200,201]. This again results in a feedforward cycle where DPRs cause cellular stress that activates the ISR, which upregulates their production [151,200,201]. Drugs that are able to decrease phosphorylation levels of EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3), the kinase responsible for EIF2A phosphorylation, or EIF2A itself are promising drug candidates as they can interrupt this vicious cycle. Recently, an EIF2AK3/PERK inhibitor and an EIF4E phosphorylation inhibitor were identified as potent modifiers DPR production [150]. Lastly, antibody immunization against DPRs recently emerged as potential therapy since poly-GA antibodies were able to effectively reduce poly-GA aggregates in a C9orf72 rodent model [201]. However, arginine-containing DPRs are the most toxic DPR species, and additional experiments are needed to confirm the feasibility and effectiveness of antibody immunization in C9orf72 ALS-FTD therapy.
Concluding remarks
Since the discovery of C9orf72 HRE as a significant cause for ALS and FTD in 2011, an incredible amount of research has been carried out to investigate possible disease mechanisms. Several discrepancies on the role of the C9orf72 protein and the consequences of C9orf72 haploinsufficiency on the ALP still persists. Both increased and decreased autophagy functions have been reported in C9orf72 ALS-FTD models. The multifaceted role of the C9orf72 complex on lysosomal function, autophagy initiation, and vesicular trafficking are only partially understood. Nevertheless, more and more evidence is pointing toward a synergistic 2, or 3-hit model where the C9orf72 mutation disrupts ALP and causes toxicity by a gain of toxic function of the repeat RNA and by the DPRs translated from the repeat RNA. These disease mechanisms appear to be intertwined, leading to a catastrophic feed-forward loop. ALP dysfunctions in C9orf72 ALS-FTD are not a standalone case (i) many ALS-FTD-related genes have a profound role in the ALP and/or vesicular trafficking, (ii) increased age remains as the major risk factor for both ALS and FTD and is accompanied by a well-characterized decrease in cellular proteostasis efficiency and (iii) defects in ALP or other protein quality control systems are also described in sporadic cases of ALS and FTD. Finally, therapeutic strategies to stimulate the ALP might provide functional benefits for C9orf72 ALS-FTD patients and for many other subtypes of ALS-FTD, although potential adverse effects due to the global enhancement of the ALP have to be taken in to account.
Acknowledgments
The authors thank Donya Pakravan for proofreading the manuscript. Figures presented in this manuscript were made using BioRender.com.
Funding Statement
The authors are supported by grants from KU Leuven (C1 - C14-17-107), Opening the Future Fund (KU Leuven), the Fund for Scientific Research Flanders (FWO-Flanders), the ALS Liga Belgium, the KU Leuven funds “Een Hart voor ALS”, “Laeversfonds voor ALS Onderzoek” and the “Valéry Perrier Race against ALS Fund”, the Alzheimer Research Foundation (SAO-FRA 2017/023), the Flemish Government initiated Flanders Impulse Program on Networks for Dementia Research (VIND 135043), Flanders Innovation & Enterpreneurship (IWT grants Project MinE and iPSCAF), the Belgian National Lottery, the Latran Foundation, the European Union’s Horizon 2020 research and innovation programme (755094), the European Union’s ERA-Netfor Research Programmes on Rare Diseases (INTEGRALS). PVD holds a senior clinical investigatorship of FWO-Vlaanderen and is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders. Jimmy Beckers is a PhD fellow at FWO-Vlaanderen(11A2321N).
Disclosure statement
The authors declare no conflict of interest.
References
- [1].Meyer K, Kaspar BK.. Glia–neuron interactions in neurological diseases: testing non-cell autonomy in a dish. Brain Res. 2017;1656:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hetz C, Mollereau B.. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. [DOI] [PubMed] [Google Scholar]
- [3].Ruegsegger C, Saxena S. Proteostasis impairment in ALS. Brain Res. 2016;1648:571–579. [DOI] [PubMed] [Google Scholar]
- [4].David DC. Aging and the aggregating proteome. Front Genet. 2012;3:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147:498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hergesheimer RC, Chami AA, de Assis DR, et al. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain. 2019;142:1176–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Götzl JK, Lang CM, Haass C, et al. Impaired protein degradation in FTLD and related disorders. Ageing Res Rev. 2016;32:122–139. [DOI] [PubMed] [Google Scholar]
- [8].Park Y, Park J, Ki Y. Crosstalk between translation and the aggresome – autophagy pathway. Autophagy. 2018;14:1079–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Van Damme P, Robberecht W, Van Den Bosch L.. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech. 2017;10:537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Renton AE, Chiò A, Traynor BJ. State of play in ALS genetics. Nat Neurosci. 2014;17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Michael A VE, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:351–360. [Google Scholar]
- [12].Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Prim. 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- [13].Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;10:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10:661–670. [DOI] [PubMed] [Google Scholar]
- [15].Johnston CA, Stanton BR, Turner MR, et al. Amyotrophic lateral sclerosis in an urban setting: A population based study of inner city London [2]. J Neurol. 2006;253:1642–1643. [DOI] [PubMed] [Google Scholar]
- [16].Al-Chalabi A, Calvo A, Chio A, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014;13:1108–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bensimon G, Lacomblez L, Meininger V, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330:153–158. [DOI] [PubMed] [Google Scholar]
- [18].Group TW. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–512. [DOI] [PubMed] [Google Scholar]
- [19].Mejzini R, Flynn LL, Pitout IL, et al. Mechanisms, and therapeutics : where are we now ? Front Neurosci. 2019;13:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Laferrièrea F, Polymenidou M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med Wkly. 2015;145:1–13. [DOI] [PubMed] [Google Scholar]
- [21].ITS C-G, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- [24].Burrell JR, Halliday GM, Kril JJ, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388:919–931. [DOI] [PubMed] [Google Scholar]
- [25].Abramzon YA, Fratta P, Traynor BJ, et al. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Panza F, Lozupone M, Seripa D, et al. Development of disease-modifying drugs for frontotemporal dementia spectrum disorders. Nat Rev Neurol. 2020;16:213–228. [DOI] [PubMed] [Google Scholar]
- [27].Donde A, Sun M, Ling JP, et al. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019;5:813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deng Z, Lim J, Wang Q, et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2019;8627:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Webster CP, Smith EF, Grierson AJ, et al. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases. 2018;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cascella R, Fani G, Bigi A, et al. Partial failure of proteostasis systems counteracting tdp-43 aggregates in neurodegenerative diseases. Int J Mol Sci. 2019;20:3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Webster CP, Smith EF, Shaw PJ, et al. Protein Homeostasis in Amyotrophic Lateral Sclerosis: therapeutic Opportunities? Front Mol Neurosci. 2017;10:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Malik BR, Maddison DC, Smith GA, et al. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain. 2019;12:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dols-Icardo O, Garcia-Redondo A, Rojas-Garcia R, et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet. 2014;23:749–754. [DOI] [PubMed] [Google Scholar]
- [36].Dedeene L, Van Schoor E, Race V, et al. An ALS case with 38 (G4C2)-repeats in the C9orf72 gene shows TDP-43 and sparse dipeptide repeat protein pathology. Acta Neuropathol. 2019;137:855–858. [DOI] [PubMed] [Google Scholar]
- [37].Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14:544–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;6:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shi Y, Lin S, Staats KA, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mori K, Weng S-M, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. [DOI] [PubMed] [Google Scholar]
- [42].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bento CF, Renna M, Ghislat G, et al. Mammalian Autophagy: how Does It Work? Annu Rev Biochem. 2016;85:685–713. [DOI] [PubMed] [Google Scholar]
- [44].Bingol B. Autophagy and lysosomal pathways in nervous system disorders. Mol Cell Neurosci. 2018;91:167–208. [DOI] [PubMed] [Google Scholar]
- [45].Deng Z, Purtell K, Lachance V, et al. Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 2017;27:491–504. [DOI] [PubMed] [Google Scholar]
- [46].Vicencio B, Labrador M, Nassif W. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells. 2020;9:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36:1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Katsumata K, Nishiyama J, Inoue T, et al. Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy. 2010;6:378–385. [DOI] [PubMed] [Google Scholar]
- [49].Budini M, Buratti E, Morselli E, et al. Its Impact on Neurodegenerative Diseases: new Roles for TDP-43 and C9orf72. Front Mol Neurosci. 2017;10:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. 2018;293:5414–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Danieli A, Martens S. p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J Cell Sci. 2018;131:jcs214304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Teinert J, Behne R, Wimmer M, et al. Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J Inherit Metab Dis. 2020;43:51–62. [DOI] [PubMed] [Google Scholar]
- [53].Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cherra SJ, Chu CT, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2011;70:349–359. [DOI] [PubMed] [Google Scholar]
- [57].van Blitterswijk M, Gendron TF, Baker MC, et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015;130:863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mis MSC, Brajkovic S, Tafuri F, et al. Development of therapeutics for c9orf72 als/ftd-related disorders. Mol Neurobiol. 2017;54:4466–4476. [DOI] [PubMed] [Google Scholar]
- [59].Zhu Q, Jiang J, Gendron TF, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;5:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gijselinck I, Van Mossevelde S, Van Der Zee J, et al. The c9orf72 repeat size correlates with onset age of disease, dna methylation and transcriptional downregulation of the promoter. Mol Psychiatry. 2016;21:1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Waite AJ, Bäumer D, East S, et al. Reduced c9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the c9orf72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(1779):e5–1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Donnelly CJ, Zhang P-W, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Belzil VV, Bauer PO, Prudencio M, et al. Reduced c9orf72 gene expression in c9ftd/als is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Webster CP, Smith EF, Bauer CS, et al. The c9orf72 protein interacts with rab1a and the ulk1 complex to regulate initiation of autophagy. Embo J. 2016;35:1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Fumagalli L, Young FL, Boeynaems S, et al. C9orf72 -derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. bioRvix. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu EY, Russ J, Wu K, et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014;128:525–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Xi Z, Zinman L, Moreno D, et al. Hypermethylation of the cpg island near the g4c2 repeat in als with a c9orf72 expansion. Am J Hum Genet. 2013;92:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Selvaraj BT, Livesey MR, Zhao C, et al. C9orf72 repeat expansion causes vulnerability of motor neurons to ca2+-permeable ampa receptor-mediated excitotoxicity. Nat Commun. 2018;9:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Laflamme C, McKeever P, Kumar R, et al. Implementation of an antibody validation procedure: application to the major ALS/FTD disease gene C9ORF72. Elife. 2019;8:e48363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xiao S, MacNair L, McGoldrick P, et al. Isoform-specific antibodies reveal distinct subcellular localizations of c9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78:568–583. [DOI] [PubMed] [Google Scholar]
- [72].Amick J, Roczniak-Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016;27:3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Levine TP, Daniels RD, Gatta AT, et al. The product of c9orf72, a gene strongly implicated in neurodegeneration, is structurally related to denn rab-gefs. Bioinformatics. 2013;29:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Iyer S, Subramanian V, Acharya KR. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ. 2018;6:e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rab SH. GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. [DOI] [PubMed] [Google Scholar]
- [76].Tang D, Sheng J, Xu L, et al. Cryo-em structure of c9orf72–smcr8–wdr41 reveals the role as a gap for rab8a and rab11a. Proc Natl Acad Sci. 2020;20:9876–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Frick P, Sellier C, Mackenzie IRA, et al. Novel antibodies reveal presynaptic localization of c9orf72 protein and reduced protein levels in c9orf72 mutation carriers. Acta Neuropathol Commun. 2018;6:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sellier C, Campanari M, Julie Corbier C, et al. Loss of c9orf72 impairs autophagy and synergizes with polyq ataxin‐2 to induce motor neuron dysfunction and cell death. Embo J. 2016;35:1276–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2016;2:e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Aoki Y, Manzano R, Lee Y, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140:887–897. [DOI] [PubMed] [Google Scholar]
- [82].Wang M, Wang H, Tao Z, et al. 72 associates with inactive rag gtpases and regulates mtorc1-mediated autophagosomal and lysosomal biogenesis. Aging Cell. 2020;19:e13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xiao S, McKeever PM, Lau A, et al. Synaptic localization of c9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol Commun. 2019;7:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Amick J, Tharkeshwar AK, Amaya C, et al. Wdr41 supports lysosomal response to changes in amino acid availability. Mol Biol Cell. 2018;29(mbc):E17–12–0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sivadasan R, Hornburg D, Drepper C, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19:1610–1618. [DOI] [PubMed] [Google Scholar]
- [86].Su M-Y, Zoncu R, Hurley J. Structure of the c9orf72 arf gap complex that is haploinsufficient in als and ftd. Nature. 2020;585:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Meng T, Lin S, Zhuang H, et al. Recent progress in the role of autophagy in neurological diseases. Cell Stress. 2019;3:141–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. [DOI] [PubMed] [Google Scholar]
- [89].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- [90].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- [91].Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Liang -C-C, Wang C, Peng X, et al. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285:3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Staats KA, Seah C, Sahimi A, et al. Small molecule inhibition of PIKFYVE kinase rescues gain- and loss-of-function C9ORF72 ALS/FTD disease processes in vivo. bioRxiv. 2019;685800. [Google Scholar]
- [94].Shi Y, B V Z, Ichida JK. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight. 2019;4(15):e127736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Leskelä S, Huber N, Rostalski H, et al. C9orf72 proteins regulate autophagy and undergo autophagosomal or proteasomal degradation in a cell type-dependent manner. Cells. 2019;8:1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sullivan PM, Zhou X, Robins AM, et al. The als/ftld associated protein c9orf72 associates with smcr8 and wdr41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Cali CP, Patino M, Tai YK, et al. C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 2019;138(5):795–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Boivin M, Pfister V, Gaucherot A, et al. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. Embo J. 2020;39(4):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ugolino J, Ji YJ, Conchina K, et al. Loss of c9orf72 enhances autophagic activity via deregulated mtor and tfeb signaling. PLoS Genet. 2016;12:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ji YJ, Ugolino J, Brady NR, et al. Systemic deregulation of autophagy upon loss of als- and ftd-linked c9orf72. Autophagy. 2017;13:1254–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tang BL. C9orf72’s interaction with rab gtpases—modulation of membrane traffic and autophagy. Front Cell Neurosci. 2016;10:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jung J, Nayak A, Schaeffer V, et al. Multiplex image-based autophagy rnai screening identifies smcr8 as ulk1 kinase activity and gene expression regulator. Elife. 2017;6:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ho WY, Tai YK, Chang JC, et al. The als-ftd-linked gene product, c9orf72, regulates neuronal morphogenesis via autophagy. Autophagy. 2019;15:827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Amick J, Tharkeshwar AK, Talaia G, et al. PQLC2 signals lysosomal cationic amino acid abundance to the C9orf72 complex. J Cell Biol. 2019;219:670034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hadano S, Otomo A, Kunita R, et al. Loss of als2/alsin exacerbates motor dysfunction in a sod1h46r-expressing mouse als model by disturbing endolysosomal trafficking. PLoS One. 2010;5(3):e9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Shi Y, Hung S-T, Rocha G, et al. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight. 2019;4:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. Embo J. 2018;37:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Maday S, Wallace KE, Holzbaur ELF. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Maday S. Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res. 2016;1649:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Stavoe AKH, Holzbaur ELF. Axonal autophagy: mini-review for autophagy in the cns. Neurosci Lett. 2019;697:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Farfel-Becker T, Roney JC, Cheng XT, et al. Neuronal soma-derived degradative lysosomes are continuously delivered to distal axons to maintain local degradation capacity. Cell Rep. 2019;28(51–64):e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Cheng X, Zhou B, Lin M, et al. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].West RJH, Lu Y, Marie B, et al. Rab8, POSH, and TAK1 regulate synaptic growth in a Drosophila model of frontotemporal dementia. J Cell Biol. 2015;208(7):931–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Corbier C, C9ORF SC. 72 is a gdp/gtp exchange factor for rab8 and rab39 and regulates autophagy. Small GTPases. 2017;8:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Xia Q, Wang H, Hao Z, et al. Tdp-43 loss of function increases tfeb activity and blocks autophagosome-lysosome fusion. Embo J. 2016;35:121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Burberry A, Suzuki N, Wang J, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8(347):347ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Mccauley ME, Rourke JGO, Yáñez A, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020;585(7823):96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Majcher V, Goode A, James V, et al. Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci. 2015;66:43–52. [DOI] [PubMed] [Google Scholar]
- [119].Heo JM, Ordureau A, Paulo JA, et al. The pink1-parkin mitochondrial ubiquitylation pathway drives a program of optn/ndp52 recruitment and tbk1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. [DOI] [PubMed] [Google Scholar]
- [121].Shen WC, Li HY, Chen GC, et al. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy- mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy. 2015;11:685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kumar S, Gu Y, Abudu YP, et al. Phosphorylation of syntaxin 17 by tbk1 controls autophagy initiation. Dev Cell. 2019;49(130–144):e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Tumbarello DA, Waxse BJ, Arden SD, et al. Autophagy receptors link myosin vi to autophagosomes to mediate tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012;14:1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ao X, Zou L, Wu Y. Regulation of autophagy by the rab gtpase network. Cell Death Differ. 2014;21:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hyttinen JMT, Niittykoski M, Salminen A, et al. Maturation of autophagosomes and endosomes: a key role for rab7. Biochim Biophys Acta Mol Cell Res. 2013;1833:503–510. [DOI] [PubMed] [Google Scholar]
- [126].Campa CC, Rab HE. 11 and phosphoinositides : A synergy of signal transducers in the control of vesicular traf fi cking. Adv Biol Regul. 2017;63:132–139. [DOI] [PubMed] [Google Scholar]
- [127].E-N N, Debnath J, Brown EJ. Ubiquilins accelerate autophagosome maturation and promote cell survival during nutrient starvation. Autophagy. 2009;5:573–575. [DOI] [PubMed] [Google Scholar]
- [128].Hjerpe R, Bett JS, Keuss MJ, et al. Ubqln2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell. 2016;166:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Şentürk M, Lin G, Zuo Z, et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol. 2019;21(3):384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Osaka M, Ito D, Yagi T, et al. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:1617–1629. [DOI] [PubMed] [Google Scholar]
- [131].Wallings RL, Humble SW, Ward ME, et al. Dysfunction at the centre of parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci. 2019;42:899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Schröder B, Wrocklage C, Pan C, et al. Integral and associated lysosomal membrane proteins. Traffic. 2007;8:1676–1686. [DOI] [PubMed] [Google Scholar]
- [133].Ji YJ, Ugolino J, Zhang T, et al. 72/alfa-1 controls tfeb/hlh-30-dependent metabolism through dynamic regulation of rag gtpases. PLOS Genet. 2020;16:e1008738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Lan Y, Sullivan PM, Hu F. SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy. 2019;15:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Liu Y, Wang T, Ji YJ, et al. A c9orf72–carm1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes Dev. 2018;32:1380–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Shao Q, Yang M, Liang C, et al. c9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy. 2019;16(9):1635–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Liu Y, C9orf WJ.72-dependent lysosomal functions regulate epigenetic control of autophagy and lipid metabolism. Autophagy. 2019;15:913–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Burk K, Pasterkamp RJ. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019;137(6):859–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. [DOI] [PubMed] [Google Scholar]
- [140].Yamada SB, Gendron TF, Niccoli T, et al. Rps25 is required for efficient ran translation of c9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat Neurosci. 2019;22(9):1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Ash PEA, Bieniek KF, Gendron TF, et al. Unconventional translation of c9orf72 ggggcc expansion generates insoluble polypeptides specific to c9ftd/als. Neuron. 2013;77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Kramer NJ, Haney MS, Morgens DW, et al. Crispr-cas9 screens in human cells and primary neurons identify modifiers of c9orf72 dipeptide-repeat-protein toxicity. Nat Genet. 2018;50:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].LaClair KD, Zhou Q, Michaelsen M, et al. Congenic expression of poly-ga but not poly-pr in mice triggers selective neuron loss and interferon responses found in c9orf72 als. Acta Neuropathol. 2020;140(2):121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Cristofani R, Crippa V, Vezzoli G, et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones. 2018;23:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Baradaran-Heravi Y, Van Broeckhoven C. van der Zee J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol Dis. 2020;134:104639. [DOI] [PubMed] [Google Scholar]
- [146].Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20:649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Buchan JR, Kolaitis RM, Taylor JP, et al. Eukaryotic stress granules are cleared by autophagy and cdc48/vcp function. Cell. 2013;153:1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Boeynaems S, Bogaert E, Kovacs D, et al. Phase separation of c9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell. 2017;65(1044–1055):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Westergard T, McAvoy K, Russell K, et al. Repeat‐associated non‐ aug translation in c9orf72‐ als/ftd is driven by neuronal excitation and stress. EMBO Mol Med. 2019;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Cheng W, Wang S, Mestre AA, et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat Commun. 2018;9(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Hartmann H, Hornburg D, Czuppa M, et al. C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci Alliance. 2018;1:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Maharjan N, Künzli C, Buthey K, et al. 72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol Neurobiol. 2017;54:3062–3077. [DOI] [PubMed] [Google Scholar]
- [153].Fay MM, Anderson PJ, Ivanov P. Als/ftd-associated c9orf72 repeat rna promotes phase transitions in vitro and in cells. Cell Rep. 2017;21:3573–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Scotter EL, Vance C, Nishimura AL, et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 2014;127:1263–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Wang I-F, Guo B-S, Liu Y-C, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109:15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ying Z, Xia Q, Hao Z, et al. TARDBP/TDP-43 regulates autophagy in both MTORC1-dependent and MTORC1-independent manners. Autophagy. 2016;12:707–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Leibiger C, Deisel J, Aufschnaiter A, et al. 43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum Mol Genet. 2018;27:1593–1607. [DOI] [PubMed] [Google Scholar]
- [159].Swinnen B, Bento-Abreu A, Gendron TF, et al. A zebrafish model for c9orf72 als reveals rna toxicity as a pathogenic mechanism. Acta Neuropathol. 2018;135:427–443. [DOI] [PubMed] [Google Scholar]
- [160].Kawabe Y, Mori K, Yamashita T, et al. Rna exosome complex degrades expanded hexanucleotide repeat rna in c9orf72 ftld/als. Embo J. 2020;39(19):e102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Kanekura K, Yagi T, Cammack AJ, et al. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. 2016;25:1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Guo Q, Lehmer C, Martı A, et al. In situ structure of neuronal c9orf72 poly-ga aggregates reveals proteasome recruitment article in situ structure of neuronal c9orf72 poly-ga aggregates reveals proteasome recruitment. Cell. 2018;172(4):696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Lopez-gonzalez R, Lu Y, Gendron TF, et al. Mitochondrial function and increases oxidative stress and dna damage in ipsc-derived motor neurons poly (gr) in c9orf72 -related als/ftd compromises mitochondrial function and increases oxidative stress and dna damage in ipsc-derived motor neurons. Neuron. 2016;92:383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Maday S, Twelvetrees AE, Moughamian AJ, et al. Axonal Transport: cargo-Specific Mechanisms of Motility and Regulation. Neuron. 2014;84:292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Guo W, Stoklund Dittlau K. Van Den Bosch L. Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin Cell Dev Biol. 2019;99:133–150. [DOI] [PubMed] [Google Scholar]
- [167].Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. [DOI] [PubMed] [Google Scholar]
- [168].Guo W, Naujock M, Fumagalli L, et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat Commun. 2017;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Kreiter N, Pal A, Lojewski X, et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol Dis. 2018;115:167–181. [DOI] [PubMed] [Google Scholar]
- [170].Bilsland LG, Sahai E, Kelly G, et al. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci. 2010;107:20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Liang C, Shao Q, Zhang W, et al. Smcr8 deficiency disrupts axonal transport-dependent lysosomal function and promotes axonal swellings and gain of toxicity in C9ALS/FTD mouse models. Hum Mol Genet. 2019;28:3940–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Cristofani R, Crippa V, Rusmini P, et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy. 2017;13:1280–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Kim HJ, Taylor JP. Lost in Transportation: nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron. 2017;96:285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Jovičič A, Mertens J, Boeynaems S, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Zhang YJ, Gendron TF, Grima JC, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Zhang K, Daigle JG, Cunningham KM, et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell. 2018;173(958–971):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Silvestrini MJ, Johnson JR, Kumar AV, et al. Nuclear Export Inhibition Enhances HLH-30/TFEB Activity, Autophagy, and Lifespan. Cell Rep. 2018;23:1915–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].McDermott CJ. Clinical trials in amyotrophic lateral sclerosis. Curr Opin Neurol. 2019;32:758–763. [DOI] [PubMed] [Google Scholar]
- [179].Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Mitsui S, Otomo A, Nozaki M, et al. Systemic overexpression of SQSTM1/p62 accelerates disease onset in a SOD1H46R-expressing ALS mouse model. Mol Brain. 2018;11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Ramesh N, Pandey UB. Autophagy Dysregulation in ALS: when Protein Aggregates Get Out of Hand. Front Mol Neurosci. 2017;10:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Cristofani R, Rusmini P, Galbiati M, et al. The Regulation of the Small Heat Shock Protein B8 in Misfolding Protein Diseases Causing Motoneuronal and Muscle Cell Death. Front Neurosci. 2019;13:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Levine TD, Miller RG, Bradley WG, et al. Phase I clinical trial of safety of L-serine for ALS patients. Amyotroph Lateral Scler Front Degener. 2017;18:107–111. [DOI] [PubMed] [Google Scholar]
- [184].Dunlop RA, Carney JM. Mechanisms of L -Serine-Mediated Neuroprotection Include Selective Activation of Lysosomal Cathepsins B and L. Neurotox Res. 2020. [DOI] [PubMed] [Google Scholar]
- [185].Liang J, Zhou F, Xiong X, et al. Enhancing the retrograde axonal transport by curcumin promotes autophagic flux in N2a/APP695swe cells. Aging (Albany NY). 2019;11:7036–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Beel S, Herdewyn S, Fazal R, et al. Progranulin reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice 11 Medical and Health Sciences 1109 Neurosciences. Mol Neurodegener. 2018;13:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Beel S, Moisse M, Damme M, et al. Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet. 2017;26:2850–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Kao AW, McKay A, Singh PP, et al. Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci. 2017;18:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- [191].Cruts M, Gijselinck I, Van Der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. [DOI] [PubMed] [Google Scholar]
- [192].Cappella M, Ciotti C, Cohen-tannoudji M, et al. Gene therapy for als — a perspective. Int J Mol Sci. 2019;20(18)4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense c9orf72 rna foci as therapy for als and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):4530–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Riboldi G, Zanetta C, Ranieri M, et al. Antisense oligonucleotide therapy for the treatment of c9orf72 als/ftd diseases. Mol Neurobiol. 2014;50:721–732. [DOI] [PubMed] [Google Scholar]
- [195].Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from als/ftd-linked repeat expansions in c9orf72 is alleviated by antisense oligonucleotides targeting ggggcc-containing rnas. Neuron. 2016;90:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Peters OM, Cabrera GT, Tran H, et al. Human c9orf72 hexanucleotide expansion reproduces rna foci and dipeptide repeat proteins but not neurodegeneration in bac transgenic mice. Neuron. 2015;88:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Martier R, Liefhebber JM, García-Osta A, et al. Targeting rna-mediated toxicity in c9orf72 als and/or ftd by rnai-based gene therapy. Mol Ther Nucleic Acids. 2019;16:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Goodman LD, Bonini NM. Progress in neurobiology repeat-associated non-aug (ran) translation mechanisms are running into focus for ggggcc-repeat associated als/ftd. Prog Neurobiol. 2019;183:101697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Green KM, Glineburg MR, Kearse MG, et al. Ran translation at c9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun. 2017;8(1):2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Sonobe Y, Ghadge G, Masaki K, et al. Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol Dis. 2018;116:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Zhou Q, Mareljic N, Michaelsen M, et al. Active poly-ga vaccination prevents microglia activation and motor deficits in a c9orf72 mouse model. EMBO Mol Med. 2019;12(2):e10919. [DOI] [PMC free article] [PubMed] [Google Scholar]