

ABSTRACT
For survival, bacteria need to continuously evolve and adapt to complex environments, including those that may impact the integrity of the DNA, the repository of genetic information to be passed on to future generations. The multiple factors of DNA repair share the substrate on which they operate with other key cellular machineries, principally those of replication and transcription, implying a high degree of coordination of DNA-based activities. In this review, I focus on progress made in the understanding of the protein factors operating at the crossroads of these three fundamental processes, with emphasis on the mutation frequency decline protein (Mfd, aka TRCF). Although Mfd research has a rich history that goes back in time for more than half a century, recent reports hint that much remains to be uncovered. I argue that besides being a transcription-repair coupling factor (TRCF), Mfd is also a global regulator of transcription and a pro-mutagenic factor, and that the way it interfaces with transcription, replication and nucleotide excision repair makes it an attractive candidate for the development of strategies to curb molecular evolution, hence, antibiotic resistance.
What is transcription-coupled repair?
Transcription-coupled DNA repair (TCR) is defined as the preferential repair of DNA damage located on the template strand of actively transcribing genes. This strand is directly read by RNA polymerase (RNAP), and damage located in it, in contrast to damage located on the non-template strand, often leads to transcriptional stalling. TCR has been primarily viewed as a subpathway of nucleotide excision repair (NER), during which subunits of the Uvr(A)BC excinuclease collaborate to recognize helix-distorting lesions and incise the damaged strand both 5' and 3' to the lesion [1]. The excised, damaged oligonucleotide is then removed by UvrD and the gap filled by DNA polymerase I and DNA ligase (Figure 1a). During TCR, in contrast to during global genome NER, damage is initially sensed by the transcription machinery itself rather than dedicated factors, such as bacterial UvrA and UvrB (Figure 1a). Damage-stalled RNAPs have a long half-life , providing the necessary timeframe for the assembly of repair machinery. Moreover, since active transcription elongation complexes survey a large fraction of the genome as part of their function, RNAP is an effective and universal high-specificity damage sensor [2]. The requirement for transcriptional stalling means that TCR operates mainly at sites of bulky or helix-distorting lesions that cannot be easily transcribed through. Several studies have also implicated canonical TCR factors in responses to oxidative damage [3–5], but these responses may well involve additional proteins, and their understanding requires further investigation.
Figure 1.

Overall features of global genome repair, TCR and Mfd proteins. (a) Schematic illustrating the main steps in global genome nucleotide excision repair (inner loop) and Mfd-dependent TCR (outer loop). (b) Schematics of Mfd sequence with labeled domains and sequence motifs. (c–e) Crystal structures of nucleotide-free Mfd from Escherichia coli (Eco, C), Mycobacterium smegmatis (Msm, d) and Mycobacterium tuberculosis (Mtb, e) colored by domain as in (B) and shown as ribbons and transparent molecular surfaces. Rectangles highlight the D2-D7 clamp interaction that masks the UvrA binding determinants. (f–h) Chord plots corresponding to the structures shown in b-d highlighting inter-domain contacts within the Mfd proteins. Structural elements within domains (colored as in Figure 1b) are represented by the arcs of a circle, and arcs are joined by chords whose thickness is determined by the number of interactions between the corresponding elements. Chord plots were obtained using the Protein Contacts Atlas
The preferential repair of active genes was uncovered in Chinese hamster ovary cells [6]. Shortly thereafter, the proposition was made that in active genes, it is the template strand that is specifically prioritized for repair. This led to the first demonstration of TCR [7,8], which in Escherichia coli was observed in the lac operon [8]. It is beyond the scope of this work to detail the early days of bacterial TCR research. The reader is instead referred to several other recent reviews on the topic [9–11]. It suffices to say that attempts at TCR reconstitution demonstrated that the presence of RNAP was inhibitory to repair due to its shielding the lesion from repair enzymes [12]. It was then hypothesized that an additional factor, a transcription-repair coupling factor or TRCF, may be required for the observed preferential repair. The operational definition of transcription-repair coupling used here requires two activities: (1) remodeling and/or removal of the stalled RNAP to expose the damaged site, (2) recruitment of repair systems, primarily NER enzymes to the damaged site to initiate repair. The only microbial factor for which both of these activities have been amply demonstrated is Mfd (Figure 1a), first implicated in the response to UV radiation through genetic manipulation by Evelyn Witkin and coworkers [13], and much later, demonstrated to be a TRCF by in vitro complementation with purified protein [14], subsequent biochemical reconstitution of the entire TCR pathway by the laboratory of Aziz Sancar [15–17] and recently, by next-generation excision repair sequencing (XR-seq) methods [18]. In E. coli, a second pathway relying on RNAP backtracking, UvrD helicase and alarmone ppGpp was also proposed [19,20], but analysis by XR-Seq failed to detect their role in TCR [18]. Additionally, UvrD does not appear to mediate recruitment of repair enzymes, at least not directly.
Mfd – a complex structure–function relationship
Mfd-dependent TCR has proven remarkably complex, occurring via multiple and difficult-to-detect intermediates, whose study was limited by in bulk biochemistry and necessitated structural information and the development of single-molecule tools. Research has been organized around three major questions. 1) How does Mfd recognize RNAP? 2) How does Mfd recruit UvrA? 3) How is damage detection and lesion hand-off different during TCR compared to global genome NER? Instrumental for tackling these questions was the detailed analysis of the complex Mfd structure and associated activities.
Mfd was recognized early on as an ATP-dependent DNA-binding protein with characteristic helicase motifs, but no helicase activity on a variety of substrates tested [15–17]. Work by the laboratory of Jeffrey Roberts showed that, instead, Mfd, like other members of the SF2 ATPase superfamily, is a dsDNA translocase operating at the upstream edge of the transcription bubble [21]. This is an aspect of TCR that is common to both bacterial Mfd and its yeast counterpart, Rad26 [22], perhaps suggesting common relationships in the evolution of TCR. The first structure of a TRCF from any organism was determined for nucleotide-free E. coli Mfd [23], but since then structural models for mycobacterial Mfd homologs have also been reported [24]. As shown in Figure 1, all Mfd homologs are composed of eight domains grouped in five modules: an UvrB-homology module (D1a-D2-D1b), domain D3, the RNAP interacting domain D4 or RID, the bilobal ATP-dependent motor module (D5-D6) and the C-terminal domain D7. The Mfd domains pack together similarly (Figure 1(b–e)) and are linked by highly flexible loops, suggesting potential for conformational rearrangements. Other notable structural elements that were proposed to be mobile and critical for conformational cycling are the relay helix, the hook helices wrapping around the relay helix and the TRG motif located in proximity to ATPase motif VI (Figure 1(b) and 2). The TRG (translocation in RecG) helical hairpin motif was first identified in RecG, the SF2 ATPase most closely evolutionarily related to Mfd [25], and was proposed to serve as a spring-loaded structural element that would change conformation during the ATP hydrolysis cycle to drive translocation [26]. A similar role for this motif (residues 924–965) has also been confirmed experimentally for E. coli Mfd, as TRG mutations within Mfd (Table 1) lead to an impairment of RNAP displacement activity, but modest effects on ATP-dependent DNA binding [27].
Figure 2.

The Mfd functional cycle. (a) Front view of Eco Mfd with highlighted mobile secondary structure elements (the relay helix, hook helices and TRG motif). (b) Bottom view of Eco Mfd with key residues targeted for mutagenesis. The functional relevance of these residues is tabulated in Table 1. (c) color code for Mfd domains. (d) Ordered pathway of intermediates formed upon Mfd loading onto the TEC and upon TEC destabilization. Both loading and remodeling cycle require ATP hydrolysis. Key molecular events, such as the unmasking of the UvrA binding determinants and RNAP clamp repositioning are indicated in cyan and pink, respectively. During the remodeling cycle, Mfd rearrangements are relatively small. Determination of the nucleotide status for L1(adp) and C3(adp) was not possible from the cryo-EM maps alone and this is indicated by the lowercase “adp” nucleotide
Table 1.
Mfd variants described in the literature. Residue numbering corresponds to the E. coli Mfd homolog
| Variant MFD class | Reference |
|---|---|
| Defective in ATP Hydrolysis | |
| K634A | [16,17] |
| E730Q |
[28,50,101] |
| Defective in Binding RNAP | |
| L499R |
[23,50,75] |
| Defective in Binding UvrA | |
| R165A |
[29,75] |
| Defective in Loading DNA | |
| R685A | [44] |
| K690A | [44] |
| H711A | [44] |
| K712A | [44] |
| Y816A | [44] |
| N817A | [44] |
| T868A |
[44] |
| Defective in Translocating on DNA | |
| T710A | [44] |
| H842A | [44] |
| R953A |
[27,100] |
| Hyperactive | |
| R165A R181A F185A | [29] |
| W550A | [54] |
| E1045A D1048A R1049A | [27,28] |
Perhaps most notable in all of the Mfd crystal structures (Figures 1(c–e) and 2(a,b)) as well as in the low-resolution solution structure of the full-length apo protein [28] is the preservation of D2-D7 contacts. This is significant because a core Mfd-UvrA crystal structure revealed that UvrA associates with D2 [28], and that this binding surface within D2 is normally buried at the interface with D7 in the context of the full-length protein (Figure 2c). Therefore, D2 and/or D7 would have to reposition to prevent steric clashes and accommodate UvrA binding. Indeed, disruption of the D2/D7 interaction by site-directed mutagenesis leads to Mfd hyperactivity (i.e., elevated ATPase rates, robust translocation on DNA in the absence of RNAP and the ability to bind UvrA) [29,30] suggesting that the D2-D7 clamp restrains the Mfd motor [23,28,30,31], and likely prevents unscheduled diversion of UvrA from global NER. Consistent with this, deletion of D7 to expose this binding site results in hypersensitivity to UV, likely due to UvrA sequestration and inhibition of NER [16].
It is worthwhile to note that the crystal structure of the core UvrA-Mfd complex may not fully recapitulate the full interaction, particularly since UvrA is thought to be a stable dimer with a dissociation constant in the low nanomolar range [32–34]. Initially, it was thought that UvrA binds UvrB with a 2:1 stoichiometry (i.e., an UvrA2B complex would form). Consistent with this line of thought as well as the UvrA binding surface shared by both UvrB and Mfd [28,35], Selby & Sancar found that binding of Mfd released UvrB, at least partially, from the UvrA2B complex [16]. More sophisticated structural analyses [36], scanning force microscopy [37] and fluorescence work [38,39] later revealed the existence of UvrA2B2 complexes and supported the concept that this heterotetrameric complex can detect and verify damage on both DNA strands. However, in contrast to conclusions derived from in vitro work that readily detected stable UvrA2B2 complexes even in the absence of DNA [36,40], single-molecule imaging in living E. coli cells revealed that free UvrA2 is rarely complexed with UvrB, the latter being recruited later from solution for further damage verification [41].
Unlike Mfd, UvrB contains a well-conserved β-hairpin in its N-terminal domains. This hairpin inserts itself between the two DNA strands and verifies the damage by base flipping [42]. With two copies of UvrB, verification of both strands can thus be achieved as each UvrB copy can locally melt DNA and probe one strand [37]. During TCR, dual verification is not needed, leading to the proposition that TCR may proceed by Mfd displacing one UvrB copy, and by the formation of an asymmetric Mfd-UvrA2-UvrB hand-off intermediate [28,43]. Due to the fluctuating relative levels of UvrA and UvrB, which are environmentally-controlled, it may well be that both assembly pathways (one relying on Mfd displacing one UvrB copy, the other via UvrA2 binding by Mfd followed by UvrB binding) may operate in vivo.
Multi-step remodeling of transcription complexes
For the Mfd translocase activity to be robustly expressed, Mfd needs to be associated with a transcription elongation complex (TEC). Early studies posited that activation of all Mfd activities (e.g., translocation, ATP hydrolysis and UvrA recruitment) is based on a single, well-controlled conformational switch triggered by its association with RNAP [31]. While this proposition was consistent with some data – particularly the weak, non-DNA stimulated ATPase and translocase activity and the hyperactivation of these activities in Mfd variants in which the D2/D7 clamp was disrupted [29,30,44] – it also raised a conundrum because covalently locking D2 to D7 via disulfide crosslinking did not eliminate RNA release from transcription complexes [28]. This pointed to the fact that, at least during early TCR, the D2/D7 interaction is not grossly disrupted. For NER proteins to gain access to the lesion, Mfd has to first remove RNAP or “remodel” it. This could occur through forward translocation of RNAP [21]. Consistent with this, on non-damaged templates, Mfd is able to rescue backtracked RNAP complexes, and transcript release competes ineffectively with transcription elongation [21], suggesting that transcript release happens not from the backtracked RNAP position, but rather after forward translocation of RNAP so that the 3' end of the RNA is poised for elongation. Unexpectedly, following RNA release, Mfd and RNAP maintain their interactions with each other, allowing Mfd to further translocate on the nucleic acid chains and perhaps approach the lesion. It has been proposed that indirect recognition of the lesion – likely through local distortions in the DNA – triggers UvrA recruitment. In fact, Mfd binding to DNA alters its geometry to non B-form, as seen both in the structure of TEC-bound [45] and DNA-bound Mfd [44], consistent with DNA geometry readout being important mechanistically. While Mfd does not participate in the initial DNA damage sensing – the direct sensor is RNAP itself – the requirements for damage sensing and verification by UvrA and UvrB are different during TCR and global NER, with less stringent requirements for damage sensing by the UvrABC excinuclease during TCR [29]. Insightful work using a combination of magnetic tweezer nanomanipulation [46–48], electron cryo-microscopy [44,45,49], in cell fluorescence imaging [50–52] has established many more details of the Mfd remodeling reaction, including the following.
Mfd engages TECs through a multipartite interaction that is based on both protein–protein and protein–DNA contacts. Protein–protein contacts are established between the RID domain (Figures 1 and 3) and the β1 fragment (aka β protrusion) of the β subunit of RNAP [16,21,23,45,52,53], and between D6 and a non-overlapping area of the same β protrusion [45]. Unlike the former interaction, which is nucleotide-independent and serves to tether Mfd to RNAP during both early and late translocation of DNA, the latter is modulated by nucleotide during the loading process, which requires ATP hydrolysis [45,46]. Once Mfd is loaded onto the TEC, more ATP hydrolysis is needed to destabilize the TEC, and structures C1-C5 (Figure 2d) represent Mfd looping through this RNAP remodeling cycle. Mutual conformational changes occur as Mfd wedges itself in between the protrusion and the β’ clamp, forcing the RNAP clamp to open through interaction with both lobes of the Mfd motor module (domains D5 and D6 in Figures 1 and 2). Mfd contacts are also established with upstream dsDNA via both lobes of the motor domain (Figure 3(a–d)) which alternate between weak/strong binding and effectively inchworm along the DNA in 1 bp steps [44,45,49]. Sigma factor binding interferes sterically with both DNA and RNAP binding by Mfd [21,45] explaining why elongation and not initiation complexes are susceptible to Mfd attack [15,21]. Interestingly, Mfd can be rendered hyperactive and indiscriminatory, enabling disruption of both initiation and elongation complexes by a single substitution (W550A, Table 1) mapping to the end of the relay helix [54]. This is coupled to the hook, the TRG motif and ATPase motif VI (Figure 2b), and likely alters DNA-Mfd contacts [45]. It is not surprising that the Mfd pathway involves repositioning of the β’ clamp – a closed clamp is thought to account for the high processivity of TECs, and its opening has been implicated in intrinsic termination where it is not required for transcript release but rather for RNAP dissociation from the DNA [55].
It has been proposed that the Mfd functional cycle can be thought of in terms of an initial, slow loading process followed by a remodeling cycle [45]. This mechanistic scheme is shown in Figure 2d. Loading requires ATP hydrolysis and is exemplified by the L[0], L1[atp], L2[adp] complexes as per the nomenclature of Kang et al. [45]. This initial loading process is followed by more ATP hydrolysis steps associated with complexes C1-C2-C3-C4-C5-C1, etc. Altogether, these later states facilitate translocation of DNA in the downstream direction, effectively “pushing” the RNAP and rescuing transcript elongation when no lesion is encountered on the template strand (i.e., when Mfd is acting on paused or backtracked complexes or on RNAP stalled at a weak roadblock). When a lesion is present on the template strand, the continued action of Mfd pushing against RNAP leads to overwinding of the upstream DNA, leading to gradual reannealing of the transcription bubble [46,56], release of the transcript [12,21] and disruption of specific contacts between RNAP and DNA. Unlike in apo Mfd, the RNAP interacting domain (D4 in Figure 1) is located far away from D7 in many of the Mfd-TEC cryo-EM reconstructions due to the high mobility of the N-terminal region relative to the motor core and D7 [45].
During the loading cycle, UvrA recruitment is sterically hindered due to D2 packing against D7 (in the L[0] and L1[atp] complexes) or DNA (in the L2[adp] complex). Upon the transition L1[adp] -> C1[ATP], the D2 surface becomes exposed due to large-scale motion of the D1-D3 region. Thus, both nucleotide status and binding to the TEC are important for regulating accessibility to the UvrA binding determinants in D2. While the mechanistic scheme of Figure 2d certainly provides a conceptual framework for understanding Mfd action, I note that the ordering of the different conformational states is somewhat speculative as it was based on analysis of Mfd-TEC particles collected from the same heterogeneous specimen spotted onto one electron microscopy grid, and not in a time-resolved manner that would allow for the time-dependent enrichment or appearance of new classes to be correlated with an ordered progression of pathway intermediates. Additionally, due to the use of a synthetic nucleic acid scaffold with a non-complementary bubble allowing for easy reconstitution of transcription complexes, this structural analysis fails to report on bubble remodeling, particularly a long-lived intermediate detected by magnetic trapping by Howan et al., in which 2/3 of the bubble is rewound [46].
Figure 3.

Mfd-TEC Interactions and Conformational Rearrangements. (a–d) Structures of selected Mfd-TEC complexes, with Mfd shown as a ribbon colored by domain as in Figure 2, and RNAP subunits shown in surface representation. (e) color code for the RNAP subunits and Mfd-bound nucleotide. (f–i) Close-up views highlighting the positioning of the motor lobes and D4 of Mfd and the profound structural changes in the relay helix. The TRG motif is colored blue, ATPase motif VI in red and the rest of the Mfd domains have been omitted for clarity. Molecular surfaces represent RNAP subunits. (j–n) Schematic representation of the main inter-domain contacts within the Mfd domains in the Mfd-TEC complexes. State L0, corresponding to the encounter complex, was not experimentally determined and is modeled based on the structure of nucleotide-free Mfd [23]. Adapted from Proteopedia [99]. For simplicity, conformational changes involving the domains are not explicitly shown
The events following RNAP destabilization and involving UvrA and UvrB merit additional consideration. While RNAP contacts with DNA are disrupted by Mfd, contacts with Mfd-D4 are preserved as mentioned above, and so Mfd continues to translocate with RNAP attached to it [47,48,57,58] until arrested by UvrA2 and UvrB binding. This results in formation of a hand-off complex. Notably, this intermediate does not pick up a second copy of UvrB from the cytosol [47]. This would be consistent with Mfd and UvrB having overlapping binding determinants [28]. Repair rate enhancement is primarily due to the more efficient docking of UvrA2 and UvrB to the activated Mfd-DNA-RNAP complex, for which UvrA has dissociation constant which is 20-200 smaller that that for naked DNA, and is essentially diffusion-limited [47]. One may conclude then that TCR is a housekeeping DNA repair mechanism that becomes particularly apparent under non-SOS conditions, when the concentrations of UvrA are quite low [1,59]. For complete release of both Mfd and RNAP, UvrA2B binding is, however, not sufficient. Quantification of the binding lifetimes of fluorescently tagged TCR players in cells revealed that upon expression of a β-hairpin UvrB mutant deficient in DNA loading, the Mfd lifetime is an order of magnitude longer than in cells expressing wild-type UvrB [50]. This points to the ATP-hydrolysis dependent loading of UvrB (and not simple binding) governing dissociation of Mfd from the hand-off complex.
Mfd – a DNA translocase with two modes of translocation?
A defining and unexpected feature of Mfd is that it possesses two modes of translocation of distinct processivities. RNAP-tethered Mfd has an impressive processivity of thousands of base pairs and a speed of ~4bs/sec [48]. On naked DNA, in the absence of RNAP, Mfd can access the active, non-inhibited state through conformational breathing, and can translocate, but it does so at a speed of 6 bp/sec and a modest processivity of only about 200 bp [57].
The structural underpinnings for the RNAP-dependent increase in Mfd processivity are suggested by the recent work of Kang and colleagues [45] and have been attributed to an intriguing order–disorder transition of the relay helix (Figure 3(i–l)). This promotes a topological wrapping of the Mfd molecule around the DNA track ensuring a high processivity length in a manner reminiscent of the use of protein “clamps” encircling the DNA during replication [60]. Relay helix unfolding initiates upon ATP binding in the middle, where it interacts with the hook helices, as seen in the L1[atp] complex (Figure 3i). Upon ATP hydrolysis, the relay helix completely unravels as seen in the L2[ADP] state (Figure 3j), and in the later remodeling intermediates (i.e., C1-C5), it remains unfolded regardless of the nucleotide state. This extensive order–disorder transition was not seen in the structure of Mfd bound to naked DNA, consistent with the above mentioned lower processivity on DNA. However, one caveat to be kept in mind is that structures of Mfd bound to dsDNA in alternative nucleotide states are still missing and so preclude the formulation of a detailed mechanistic model for how Mfd translocates in the absence of RNAP.
Fleshing out the mechanistic details and implications of Mfd binding to naked DNA will require further work, but it is worthwhile to turn to the recent determination of the structure of Mfd bound to dsDNA using electron cryo-microscopy [44]. While this reconstruction was limited in resolution, it nevertheless revealed that Mfd binding locally distorts one end of the bound DNA fragment to a non Watson–Crick pair, with two implications. First, that DNA geometry may be integral to indirect lesion detection and UvrA recruitment during canonical TCR as proposed earlier [28] and, second, that perhaps Mfd-induced DNA distortions might lead to gratuitous DNA repair (e.g., recruitment of NER proteins and excision of undamaged DNA). Gratuitous repair has been previously demonstrated in an in vitro system [61] as well as in vivo upon NER enzyme overexpression and proposed to be a source of spontaneous mutations [62] – it will now be interesting to establish if Mfd has a stimulatory role in this context. Notably, about 20% of cellular Mfd is found on DNA devoid of RNAP [52]. This is despite the low relative numbers of Mfd to RNAP in E. coli (tens versus thousands of molecules [52]; in B. subtilis, the other model organism in which Mfd has been studied some detail, levels are much higher [63]). The modest processivity of Mfd may not be necessarily a disadvantage, keeping in mind that the chromosome is heavily decorated with various roadblocks that would likely reduce processivity length anyway. It has been speculated that this pool of DNA-bound Mfd could be involved in locating RNAP targets via a 1-d search rather than 3-d diffusion [57] and perhaps may exert additional RNAP-independent functions. Details on how Mfd operates on DNA domains free of RNAP, and what other binding factors it collaborates with remain to be better defined.
Mfd – a general transcription factor?
Not long after the discovery of Mfd as a TRCF, it became apparent that this was involved in processes beyond TCR. Early reports implicated Mfd in transcriptional regulation by protein roadblocks, including carbon catabolite repression [64], repression of genes regulated by the global Bacillus subtilis regulator CodY and even exclusion of λ phage by phage HK022. In the case of CodY, Mfd inactivation was shown to lead to relief of CodY repression [65]. In the case of phage HK022, exclusion of superinfecting λ relies on the HK022 protein Nun terminating transcription at certain sites in the DNA in collaboration with Mfd, which is required for releasing Nun-arrested TECs [66]. Due to its additional ability to rescue backtracked RNAP complexes and complexes stalled by nucleotide deprivation and not DNA damage, Mfd was therefore hypothesized to function as a general transcription factor [21]. Indeed, more recent work involving single-molecule fluorescence demonstrated not only that Mfd associates with RNAP in cells in the absence of exogenous damage [52], but also that Mfd can rescue paused complexes, including complexes formed at a physiological ops pause site as shown in the optical trap study of Le et al. [57]. Genome-wide, our knowledge of the effects of Mfd on the cell transcriptome has recently expanded considerably. Using chromatin immunoprecipitation followed by high-throughput sequencing (Chip-seq) on exponentially growing B. subtilis cells, Ragheb et al. showed that Mfd associates with ~500 genes, and that its genomic association pattern correlates well with the occupancy of the RNAP subunit, RpoB [67]. Many sites of Mfd association with DNA and RNAP corresponded to regions encoding for highly structured RNA, including noncoding transcripts, antisense transcripts, riboswitches and pause sites. This is true not only for B. subtilis but also E. coli [67]. Furthermore, Mfd also acts on RNAPs stalled by G quadruplex sequences on the nontemplate strand to promote adaptive mutagenesis [68]. Notably, two of the main loci at which Mfd acts are the type I toxin-antitoxin loci, txpA/ratA and bsrH/as-bsrh. Type I toxin-antitoxins function by inhibiting translation or promoting degradation of the toxin mRNA. They are involved in persister and biofilm formation [69,70] as well as prophage maintenance [71]. Cells overexpressing TxpA or BsrH but lacking Mfd were highly sensitized to the action of the toxin [67], suggesting that Mfd confers resistance to toxin expression, perhaps via transcriptional repression.
In contrast, recent transcriptomic analyses of B. subtilis cells in the stationary phase and upon exposure to diamide-induced disulfide stress found that in untreated cells lacking Mfd, a significant number of genes are both down- and up-regulated [72]. Pathway enrichment analyses highlighted that biological functions affected by Mfd are diverse and include translation, protein folding, pentose metabolism, cellular nitrogen and organonitrogen biosynthesis, phosphorus and toxin metabolism, transport of carbohydrates and organics, ribonucleoside monophosphate and pyrimidine-containing compound biosynthesis as well as flagellum-dependent motility. A similarly complex regulatory pattern was observed in diamide-treated cells, where genes were again both up- and down regulated in the absence of Mfd, affecting processes such as proteolysis, glutamine biosynthesis, isoprenoid biosynthesis, antibiotic and carboxylic acid metabolism, inosine monophosphate biosynthesis, flagellum motility and chemotaxis, transmembrane and sodium transport and cellular nitrogen compound biosynthesis. However, under conditions of stationary phase and unlike in the study of Ragheb et al., no changes in txpA and bsrH expression were seen in untreated cells, suggesting that Mfd-mediated modulation of transcription is dependent on growth and environmental conditions. Mfd was also proposed to be required for protection against double-strand breaks arising due to reactive nitrogen species generated by the immune response upon infection of the host [73,74]. Altogether, this new work extends Mfd’s role from canonical TCR to being a cellular factor involved in coping with multiple stress signals.
Mfd – an evolvability factor and target for development of an anti-evolution drug?
While early studies focused on mfd under conditions of UV irradiation followed by inhibition of protein synthesis, conditions under which mfd has a very modest effect on resistance to UV, yet a more pronounced role in reducing mutation rates [13], more recent studies have looked at mfd roles both under active growth and during the stationary phase or upon stress. The picture that has emerged indicates that Mfd functions as a pro-mutagenic factor both during active growth [75–77] and in the stationary phase [78–81], hinting at complex regulation, occurring perhaps via multiple and not necessarily mutually exclusive mechanisms. Analysis has been somewhat complicated by the fact that many studies of Mfd-mediated adaptive (stationary phase) mutagenesis have been carried out in B. subtilis, while Mfd structural biochemistry has been conducted primarily in E. coli.
Mfd-dependent mutagenesis during active growth increases evolvability with direct implications for the development of antimicrobial resistance. This has been documented in multiple bacteria, including Gram-negative and Gram-positive and to antibiotics with distinct mechanisms of action [75,82,83]. Notably, the acquisition of antibiotic resistance appears to be dependent on the interaction of Mfd with the β subunit of RNAP and perhaps UvrA [75] and is evolutionarily conserved, an attribute which makes Mfd an attractive target for the development of an anti-evolution drug. This would be a broad-spectrum agent that could be administered in combination with narrow-spectrum antibiotics to increase the time window of their efficacy and tailor treatment to particular infections. Critically, because Mfd deletion does not lead to a fitness defect in the absence of stress, one would anticipate that development of resistance to the evolution drug itself would be minimal.
While mutation at different chromosomal loci is not the only mechanism for acquisition of antibiotic resistance, critically, for several clinically important pathogens that affect a large fraction of the global population, such as Mycobacterium tuberculosis, plasmid- or transposon-mediated transfer have not been reported. This suggests a lack of horizontally transfer mechanisms in mycobacterial pathogens, and highlights the importance of resistance developed exclusively through mutational events [84,85]. In addition, more than half of the patients infected with Pseudomonas aeruginosa suffering from chronic fibrosis are colonized by hypermutator strains that are associated with reduced lung function and chronic infection [86,87]. In the case of M. tuberculosis, the development of resistance to front-line drugs such as rifampicin is notorious and occurs through the acquisition of mutations within the RpoB subunit of RNAP [88]. Hypermutation also accounts for the evolution of horizontally transferred genes toward resistance to “next generation” antibiotics, arguing that controlling hypermutation is a first and essential step in prolonging the efficacy of front-line antibiotics. The idea that curbing pro-mutagenic pathways, such as the SOS response, could be leveraged for therapeutic purposes is therefore not new. However, the development of anti-evolution drugs for addressing antimicrobial resistance has been fraught with problems, and has achieved limited success so far [89,90]. The wealth of biochemical and structural information on Mfd increases the tractability of developing inhibitors in this system, particularly because distinct activities and multiple Mfd sites with different physicochemical properties can be targeted for structure-based drug design.
The mechanistic details behind Mfd-dependent mutagenesis remain significantly less understood than Mfd-dependent TCR. They were initially attributed to Mfd involvement in resolving conflicts between the transcription and replication machineries, which move along the same DNA track, but at different speeds. These conflicts occur either head-on (when the transcribed strand corresponds to the lagging strand, Figure 4a), but also co-directionally (when the transcribed strand corresponds to the leading strand, Figure 4b). Although the cell has found ways to mitigate head-on collisions – bacterial transcription of highly expressed genes often occurs with the same polarity as replication [91–93], transcription and replication are not well separated spatiotemporally and conflicts of both kinds are bound to occur. Mfd’s role in resolving these conflicts is complex. In vitro, Mfd mediates direct restart of a replisome colliding with RNAP head-on by dismantling the TEC [94], but upon co-directional collisions, replisomes can restart by reinitiating from an mRNA primer without the aid of Mfd [95]. However, these findings are not entirely consistent with in vivo observations. Dutta et al. employed a plasmid-based system relying on Mfd regulation of TECs arrested by the Nun protein to monitor both head-on and co-directional collisions in various genetic backgrounds, including in an Mfd-deficient strain [96]. Co-directional collisions of replisomes with backtracked RNAPs were found to result in double-strand breaks, and, in contrast to the study by Pomerantz et al. [95], were resolved due to the ability of Mfd to release backtracked TECs. The generation of double-strand breaks was attributed to the formation of R-loops, which were proposed to not readily assemble during head-on collisions, but be favored during co-directional collisions due to priming from the nascent transcript and strand jumping [96]. Presumably, the resulting break in the leading strand would likely not be repaired sufficiently fast and would be converted to a double-stranded break at the next round of replication. Work from the Merrikh lab seems to suggest that R-loops form primarily at sites of head-on collisions instead, where they lead to increased mutagenesis, completely interfere with replication, and hinder the expression of stress genes [97]. Why these discrepancies between in vitro and in vivo? The answer may be perhaps connected to the transcriptional status of the gene, characterized by a certain RNAP density, with tightly packed arrays preventing Mfd binding at the upstream edge of the transcription bubble, where Mfd requires approximately 26 bp of naked dsDNA [21,57]. Incidentally, Wimberly et al. implicated Mfd action at sites of R-loop formation in non-replicating cells, where they instigate genomic changes and mutagenesis [98].
Figure 4.
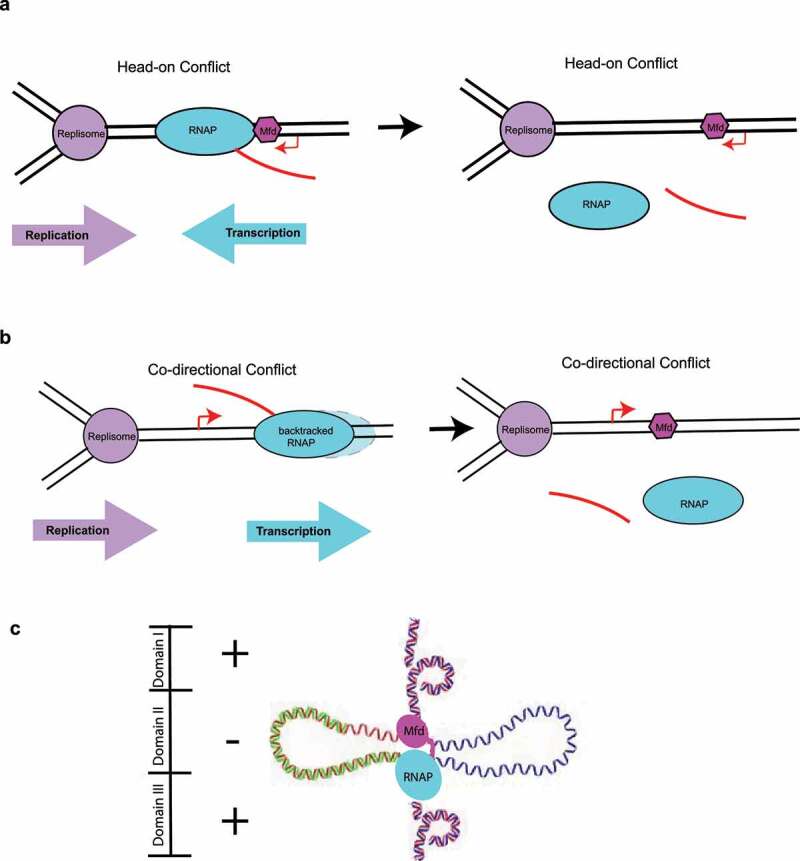
Transcription-Replication conflicts and Mfd-dependent R-loop formation. (a–b) Collisions between the replication and transcription machineries can occur either head-on (when the transcribed strand corresponds to the lagging strand) or co-directionally (when the transcribed strand corresponds to the leading strand). Both types of conflicts have been associated with the generation of double-strand breaks and R-loop formation and may involve the action of the Mfd protein. (c) Mechanism of Mfd-dependent formation of R-loops through topological partitioning of DNA. Figure 4 based on the work of Portman et al. [100]
So, do R-loops prevent Mfd action, or does Mfd action lead to R-loop formation? Recent evidence obtained using single-molecule approaches points to Mfd leading to the formation of R-loops through topological partitioning of DNA even in the absence of a stalled replisome [100]. As described above, Mfd can simultaneously interact with upstream DNA and elongating RNAP, and these associations help partition the DNA into distinct supercoiled domains. Due to the tethering that exists between DNA, the elongating RNAP and Mfd, three topological domains arise, a region of highly negative supercoiling in between Mfd and RNAP, and two flanking positively supercoiled domains, ahead of RNAP and behind Mfd (Figure 4c) [57]. As negative supercoiling accumulates, the DNA becomes single-stranded and a good substrate for R-loop formation, which is thermodynamically favored [73]. Somewhat surprisingly, DNA translocation by Mfd is not essential for this process [100], suggesting a general mechanism for R-loop formation applicable to other DNA- and RNAP tethering factors that do not necessarily have a motor function.
So, after more than half a century of mfd research, one may ask the rather rhetorical question, what more is to know? In the past decades, we have learned a tremendous amount about how it works, and yet, another twist and turn in the Mfd story seems to be coming our way. The mechanism of dsDNA translocation by Mfd in vicinity to the TEC has been generally accepted as underpinning TCR and results in the “pushing” of RNAP in the downstream direction, as described above. Yet a very recent study relying on single-molecule FRET and electron cryo-EM proposes a distinct model in which Mfd loads not on dsDNA upstream but rather at the upstream fork of the transcription bubble, making specific contacts with ssDNA, resulting in scrunching and effectively pulling the DNA from underneath the RNAP [101]. In the years to come, it will be interesting to further explore these concepts, determine structures of Mfd-RNAP – UvrA2-UvrB intermediates, find out if factors other than Mfd also lead to genome diversification and accelerated evolution via a related mechanism involving R-loops, and perhaps, develop Mfd inhibitors to address the worldwide crisis of antimicrobial resistance.
Acknowledgments
The author thanks Ms Lisa Schulmeyer for assistance with figure preparation.
Funding Statement
This work was supported by a grant from the National Institutes of Health (R01GM121975) and a Salomon Research Award from Brown University
Disclosure statement
The author reports no conflict of interest.
References
- [1].Selby, CP, Sancar, A.. Structure and function of the (A)BC excinuclease of Escherichia coli. Mutat Res. 1990;236:203–211. [DOI] [PubMed] [Google Scholar]
- [2].Lindsey-Boltz, LA, Sancar, A.. RNA polymerase: the most specific damage recognition protein in cellular responses to DNA damage? Proc Natl Acad Sci U S A. 2007;104:13213–13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Martin, HA, Porter, KE, Vallin, C, et al. Mfd protects against oxidative stress in Bacillus subtilis independently of its canonical function in DNA repair. BMC Microbiol. 2019;19:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Smith, AJ, Savery, NJ. Effects of the bacterial transcription-repair coupling factor during transcription of DNA containing non-bulky lesions. DNA Repair (Amst). 2008;7:1670–1679. [DOI] [PubMed] [Google Scholar]
- [5].Bregeon. D, Doddridge. ZA, You. HJ, et al. Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell. 2003;12:959–970. [DOI] [PubMed] [Google Scholar]
- [6].Bohr, VA, Smith, CA, Okumoto, DS, et al. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40:359–369. [DOI] [PubMed] [Google Scholar]
- [7].Mellon, I, Spivak, G, Hanawalt, PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51:241–249. [DOI] [PubMed] [Google Scholar]
- [8].Mellon, I, Hanawalt, PC. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature. 1989;342:95–98. [DOI] [PubMed] [Google Scholar]
- [9].Deaconescu, AM, Suhanovsky, MM. From Mfd to TRCF and back again-A perspective on bacterial transcription-coupled nucleotide excision repair. Photochem Photobiol. 2017;93:268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Selby, CP. Mfd Protein and Transcription-Repair coupling in Escherichia coli. Photochem Photobiol. 2017;93:280–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Strick, TR, Portman, JR. Transcription-Coupled repair: from cells to single molecules and back again. J Mol Biol. 2019;431:4093–4102. [DOI] [PubMed] [Google Scholar]
- [12].Selby, CP, Sancar, A. Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J Biol Chem. 1990;265:21330–21336. [PubMed] [Google Scholar]
- [13].Witkin, EM. Radiation-induced mutations and their repair. Science. 1966;152:1345–1353. [DOI] [PubMed] [Google Scholar]
- [14].Selby, CP, Witkin, EM, Sancar, A. Escherichia coli mfd mutant deficient in “mutation frequency decline” lacks strand-specific repair: in vitro complementation with purified coupling factor. Proc Natl Acad Sci U S A. 1991;88:11574–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Selby, CP, Sancar, A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. [DOI] [PubMed] [Google Scholar]
- [16].Selby, CP, Sancar, A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J Biol Chem. 1995;270:4882–4889. [DOI] [PubMed] [Google Scholar]
- [17].Selby, CP, Sancar, A. Structure and function of transcription-repair coupling factor. II. Catalytic properties. J Biol Chem. 1995;270:4890–4895. [DOI] [PubMed] [Google Scholar]
- [18].Adebali, O, Sancar, A, Selby, CP. Mfd translocase is necessary and sufficient for transcription-coupled repair in Escherichia coli. J Biol Chem. 2017;292:18386–18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Epshtein, V, et al. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505:372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kamarthapu, V, et al. ppGpp couples transcription to DNA repair in E. coli. Science. 2016;352:993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Park, J-S, Marr, MT, Roberts, JW. E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. [DOI] [PubMed] [Google Scholar]
- [22].Xu, J, et al. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature. 2017;551:653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Deaconescu, AM, et al. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. [DOI] [PubMed] [Google Scholar]
- [24].Sivasankar Putta, SP, Vinayak Bhat, GC, Fox, MA, et al. Structural Insights into the molecular mechanisms of the Mycobacterium evolvability factor Mfd. Biorxiv. 2019. [Google Scholar]
- [25].Fairman-Williams, ME, Guenther, UP, Jankowsky, E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mahdi, AA, Briggs, GS, Sharples, GJ, et al. A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J. 2003;22:724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chambers, AL, Smith, AJ, Savery, NJ. A DNA translocation motif in the bacterial transcription–repair coupling factor, Mfd. Nucleic Acids Res. 2003;31, 6409–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deaconescu, AM, Sevostyanova, A, Artsimovitch, I, et al. Nucleotide excision repair (NER) machinery recruitment by the transcription-repair coupling factor involves unmasking of a conserved intramolecular interface. Proc Natl Acad Sci U S A. 2012;109:3353–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Manelyte, L, Kim, YI, Smith, AJ, et al. Regulation and rate enhancement during transcription-coupled DNA repair. Mol Cell. 2010;40:714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith, AJ, Szczelkun, MD, Savery, NJ. Controlling the motor activity of a transcription-repair coupling factor: autoinhibition and the role of RNA polymerase. Nucleic Acids Res. 2007;35:1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Murphy, MN, et al. An N-terminal clamp restrains the motor domains of the bacterial transcription-repair coupling factor Mfd. Nucleic Acids Res. 2009;37:6042–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mazur, SJ, Grossman, L. Dimerization of Escherichia coli UvrA and its binding to undamaged and ultraviolet light damaged DNA. Biochemistry. 1991;30:4432–4443. [DOI] [PubMed] [Google Scholar]
- [33].Jaciuk, M, Nowak, E, Skowronek, K, et al. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat Struct Mol Biol. 2011;18:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pakotiprapha, D, et al. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol Cell. 2008;29:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Truglio, JJ, et al. Interactions between UvrA and UvrB: the role of UvrB’s domain 2 in nucleotide excision repair. Embo J. 2004;23:2498–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pakotiprapha, D, Samuels, M, Shen, K, et al. Structure and mechanism of the UvrA-UvrB DNA damage sensor. Nat Struct Mol Biol. 2012. [DOI] [PubMed] [Google Scholar]
- [37].Verhoeven, EE, Wyman, C, Moolenaar, GF, et al. The presence of two UvrB subunits in the UvrAB complex ensures damage detection in both DNA strands. Embo J. 2002;21:4196–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Malta, E, Moolenaar, GF, Goosen, N. Dynamics of the UvrABC nucleotide excision repair proteins analyzed by fluorescence resonance energy transfer. Biochemistry. 2007;46:9080–9088. [DOI] [PubMed] [Google Scholar]
- [39].Kad, NM, Wang, H, Kennedy, GG, et al. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol Cell. 2010;37:702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pakotiprapha, D, Jerusalmi, JD. Small-angle X-ray scattering reveals architecture and A(2)B(2) stoichiometry of the UvrA-UvrB DNA damage sensor. Proteins. 2013;81:132–139. [DOI] [PubMed] [Google Scholar]
- [41].Stracy, M, et al. Single-molecule imaging of UvrA and UvrB recruitment to DNA lesions in living Escherichia coli. Nat Commun. 2016;7:12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Truglio, J, et al. Structural basis for DNA recognition and processing by UvrB. Nat Struct Mol Biol. 2006;13:360–364. [DOI] [PubMed] [Google Scholar]
- [43].Deaconescu, AM, Artsimovitch, I, Grigorieff, N. Interplay of DNA repair with transcription: from structures to mechanisms. Trends Biochem Sci. 2012;37:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brugger, C, et al. Molecular determinants for dsDNA translocation by the transcription-repair coupling and evolvability factor Mfd. Nat Commun. 2020;11:3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kang, JY, et al. Structural basis for transcription complex disruption by the Mfd translocase. Elife. 2021;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Howan, K, et al. Initiation of transcription-coupled repair characterized at single-molecule resolution. Nature. 2012;490:431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fan, J, Leroux-Coyau, M, Savery, NJ, et al. Reconstruction of bacterial transcription-coupled repair at single-molecule resolution. Nature. 2016. [DOI] [PubMed] [Google Scholar]
- [48].Graves, ET, et al. A dynamic DNA-repair complex observed by correlative single-molecule nanomanipulation and fluorescence. Nat Struct Mol Biol. 2015;22:452–7. [DOI] [PubMed] [Google Scholar]
- [49].Shi, J, et al. Structural basis of Mfd-dependent transcription termination. Nucleic Acids Res. 2020;48:11762–11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ho, HN, van Oijen, AM, Ghodke, H. Single-molecule imaging reveals molecular coupling between transcription and DNA repair machinery in live cells. Nat Commun. 2020;11:1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ghodke, H, Ho, HN, van Oijen, AM. Single-molecule live-cell imaging visualizes parallel pathways of prokaryotic nucleotide excision repair. Nat Commun. 2020;11:1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ho, HN, van Oijen, AM, Ghodke, H. The transcription-repair coupling factor Mfd associates with RNA polymerase in the absence of exogenous damage. Nat Commun. 2018;9:1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Butland, G, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. [DOI] [PubMed] [Google Scholar]
- [54].Smith, AJ, Pernstich, C, Savery, NJ. Multipartite control of the DNA translocase, Mfd. Nucleic Acids Res. 2012;40:10408–10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bellecourt, MJ, Ray-Soni, A, Harwig, A, et al. Movement aids dissociation from DNA but is not required for RNA release at intrinsic terminators. J Mol Biol. 2019;431:696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Park, JS, Roberts, JW. Role of DNA bubble rewinding in enzymatic transcription termination. Proc Natl Acad Sci U S A. 2006;103:4870–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Le, TT, et al. Mfd dynamically regulates transcription via a release and catch-up mechanism. Cell. 2018;172:344–357 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Haines, NM, Kim, YI, Smith, AJ, et al. Stalled transcription complexes promote DNA repair at a distance. Proc Natl Acad Sci U S A. 2014;111:4037–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Taniguchi, Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bruck, I, O’Donnell, M. The ring-type polymerase sliding clamp family. Genome Biol. 2001;2:REVIEWS3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Branum, ME, Reardon, JT, Sancar, A. DNA repair excision nuclease attacks undamaged DNA. A potential source of spontaneous mutations. J Biol Chem. 2001;276:25421–25426. [DOI] [PubMed] [Google Scholar]
- [62].Hasegawa, K, Yoshiyama, K, Maki, H. Spontaneous mutagenesis associated with nucleotide excision repair in Escherichia coli. Genes Cells. 2008;13:459–469. [DOI] [PubMed] [Google Scholar]
- [63].Muntel, J, et al. Comprehensive absolute quantification of the cytosolic proteome of Bacillus subtilis by data independent, parallel fragmentation in liquid chromatography/mass spectrometry (LC/MS(E)). Mol Cell Proteomics. 2014;13:1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zalieckas, JM, Wray, LVJ, Ferson, AE, et al. Transcription-repair coupling factor is involved in carbon catabolite repression of the Bacillus subtilis hut and gnt operons. Mol Microbiol. 1998;27:1031–1038. [DOI] [PubMed] [Google Scholar]
- [65].Belitsky, BR, Sonenshein, AL. Roadblock repression of transcription by Bacillus subtilis CodY. J Mol Biol. 2011;411:729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Washburn, RS, Wang, Y, Gottesman, ME. Role of E. coli transcription-repair coupling factor Mfd in Nun-mediated transcription termination. J Mol Biol. 2003;329:655–662. [DOI] [PubMed] [Google Scholar]
- [67].Ragheb, MN, Merrikh, C, Browning, K, et al. Mfd regulates RNA polymerase association with hard-to-transcribe regions in vivo, especially those with structured RNAs. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ermi, T, et al. Non-B DNA-forming Motifs promote Mfd-Dependent stationary-phase mutagenesis in bacillus subtilis. Microorganisms. 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dorr, T, Vulic, M, Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010;8:e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Domka, J, Lee, J, Bansal, T, et al. Temporal gene-expression in Escherichia coli K-12 biofilms. Environ Microbiol. 2007;9:332–346. [DOI] [PubMed] [Google Scholar]
- [71].Durand, S, Jahn, N, Condon, C, et al. I toxin-antitoxin systems in Bacillus subtilis. RNA Biol. 2012;9:1491–1497. [DOI] [PubMed] [Google Scholar]
- [72].Martin, HA, et al. Mfd affects global transcription and the physiology of stressed bacillus subtilis cells. Front Microbiol. 2021;12:625705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Darrigo, C, Guillemet, E, Dervyn, R, et al. The bacterial Mfd protein prevents DNA damage induced by the host nitrogen immune response in a NER-Independent but RecBC-Dependent pathway. PLoS One. 2016;11:e0163321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Guillemet, E, et al. The bacterial DNA repair protein Mfd confers resistance to the host nitrogen immune response. Sci Rep. 2016;6:29349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ragheb, MN, et al. Inhibiting the evolution of antibiotic resistance. Mol Cell. 2019;73:157–165.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Million-Weaver, S, et al. An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis. Proc Natl Acad Sci U S A. 2015;112:E1096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Paul, S, Million-Weaver, S, Chattopadhyay, S, et al. Accelerated gene evolution through replication-transcription conflicts. Nature. 2013;495:512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gomez-Marroquin, M, et al. Stationary-Phase mutagenesis in stressed bacillus subtilis cells operates by Mfd-Dependent mutagenic pathways. Genes (Basel). 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Martin, HA, Pedraza-Reyes, M, Yasbin, RE, et al. Transcriptional de-repression and Mfd are mutagenic in stressed Bacillus subtilis cells. J Mol Microbiol Biotechnol. 2011;21:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Pybus, C, et al. Transcription-associated mutation in Bacillus subtilis cells under stress. J Bacteriol. 2010;192:3321–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ross, C, et al. Novel role of mfd: effects on stationary-phase mutagenesis in Bacillus subtilis. J Bacteriol. 2006;188:7512–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Han, J, Sahin, O, Barton, YW, et al. Key role of Mfd in the development of fluoroquinolone resistance in Campylobacter jejuni. PLoS Pathog. 2008;4:e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lee, GH, et al. The Helicobacter pylori Mfd protein is important for antibiotic resistance and DNA repair. Diagn Microbiol Infect Dis. 2009;65;454–456. [DOI] [PubMed] [Google Scholar]
- [84].Pepperell, CS, et al. The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathog. 2013;9:e1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Eldholm, V, Balloux, F. Antimicrobial resistance in mycobacterium tuberculosis: the odd one out. Trends Microbiol. 2016;24:637–648. [DOI] [PubMed] [Google Scholar]
- [86].Oliver, A, Canton, R, Campo, P, et al. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. [DOI] [PubMed] [Google Scholar]
- [87].Waine, DJ, Honeybourne, D, Smith, EG, et al. Association between hypermutator phenotype, clinical variables, mucoid phenotype, and antimicrobial resistance in Pseudomonas aeruginosa. J Clin Microbiol. 2008;46:3491–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Molodtsov, V, Scharf, NT, Stefan, MA, et al. Structural basis for rifamycin resistance of bacterial RNA polymerase by the three most clinically important RpoB mutations found in Mycobacterium tuberculosis. Mol Microbiol. 2017;103:1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Crane, JK, Cheema, MB, Olyer, MA, et al. Zinc blockade of SOS response inhibits horizontal transfer of antibiotic resistance genes in enteric bacteria. Front Cell Infect Microbiol. 2018;8:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Domenech, A, et al. Proton motive force disruptors block bacterial competence and horizontal gene transfer. Cell Host Microbe. 2020;27:544–555 e3. [DOI] [PubMed] [Google Scholar]
- [91].Brewer, BJ. When polymerases collide: replication and the transcriptional organization of the E. coli chromosome. Cell. 1988;53:679–686. [DOI] [PubMed] [Google Scholar]
- [92].Blattner, FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. [DOI] [PubMed] [Google Scholar]
- [93].Rocha, EP, Danchin, A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34:377–378. [DOI] [PubMed] [Google Scholar]
- [94].Pomerantz, RT, O’Donnell, M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pomerantz, RT, O’Donnell, M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008;456:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Dutta, D, Shatalin, K, Epshtein, V, et al. RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lang, KS, et al. Replication-Transcription conflicts generate R-Loops that orchestrate bacterial stress survival and pathogenesis. Cell. 2017;170:787–799 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wimberly, H, et al. R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat Commun. 2013;4:2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Prilusky, J, et al. Proteopedia: a status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. J Struct Biol. 2011;175:244–252. [DOI] [PubMed] [Google Scholar]
- [100].Portman, JR, Brouwer, GM, Bollins, J, et al. Cotranscriptional R-loop formation by Mfd involves topological partitioning of DNA. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Paudel, BP, et al. Mechanism of transcription modulation by the transcription-repair coupling factor. bioRxiv. 2021;2021(4):02.438179. [DOI] [PMC free article] [PubMed] [Google Scholar]