

ABSTRACT
The autophagy-lysosome system is an important cellular degradation pathway that recycles dysfunctional organelles and cytotoxic protein aggregates. A decline in this system is pathogenic in many human diseases including neurodegenerative disorders, fatty liver disease, and atherosclerosis. Thus there is intense interest in discovering therapeutics aimed at stimulating the autophagy-lysosome system. Trehalose is a natural disaccharide composed of two glucose molecules linked by a ɑ-1,1-glycosidic bond with the unique ability to induce cellular macroautophagy/autophagy and with reported efficacy on mitigating several diseases where autophagy is dysfunctional. Interestingly, the mechanism by which trehalose induces autophagy is unknown. One suggested mechanism is its ability to activate TFEB (transcription factor EB), the master transcriptional regulator of autophagy-lysosomal biogenesis. Here we describe a potential mechanism involving direct trehalose action on the lysosome. We find trehalose is endocytically taken up by cells and accumulates within the endolysosomal system. This leads to a low-grade lysosomal stress with mild elevation of lysosomal pH, which acts as a potent stimulus for TFEB activation and nuclear translocation. This process appears to involve inactivation of MTORC1, a known negative regulator of TFEB which is sensitive to perturbations in lysosomal pH. Taken together, our data show the trehalose can act as a weak inhibitor of the lysosome which serves as a trigger for TFEB activation. Our work not only sheds light on trehalose action but suggests that mild alternation of lysosomal pH can be a novel method of inducing the autophagy-lysosome system.
Abbreviations: ASO: antisense oligonucleotide; AU: arbitrary units; BMDM: bone marrow-derived macrophages; CLFs: crude lysosomal fractions; CTSD: cathepsin D; LAMP: lysosomal associated membrane protein; LIPA/LAL: lipase A, lysosomal acid type; MAP1LC3: microtubule-associated protein 1 light chain 3; MFI: mean fluorescence intensity; MTORC1: mechanistic target of rapamycin kinase complex 1; pMAC: peritoneal macrophages; SLC2A8/GLUT8: solute carrier family 2, (facilitated glucose transporter), member 8; TFEB: transcription factor EB; TMR: tetramethylrhodamine; TREH: trehalase
KEYWORDS: Endocytosis, lysosome, MTORC1, TFEB, trehalose
Introduction
Autophagy is a highly conserved evolutionary cellular process by which unwanted intracellular contents, such as protein aggregates and dysfunctional organelles, are degraded by delivery to lysosomes [1,2]. Understanding the intricacies of this degradation system mechanism has become an important area of biomedical research because of an ever-expanding list of human diseases that display accumulation of protein aggregates and dysfunctional organelles as a central feature. For example, neurodegenerative disorders such as Alzheimer, Parkinson, and Huntington disease are defined by accumulation of cytotoxic protein aggregates in the setting of inadequate aggrephagy [3–5]. Generation of deleterious superoxides are a result of accumulating dysfunctional mitochondria and inadequate mitophagy [6]. Fatty liver disease and the development of lipid-rich atherosclerotic plaques have been linked to insufficient lipophagy [7,8]. Therefore, there is intense interest in the development of therapies aimed at inducing cellular autophagy or stimulating the autophagy-lysosome system as a whole.
A well-known autophagy-inducing compound is trehalose, a naturally occurring disaccharide formed by two glucose molecules (in α-1,1 linkage). Found abundantly in a variety of non-mammalian organisms such as yeast, worms, flies, and crustaceans, trehalose is used in nature as a chemical chaperone that prevents protein misfolding, thus protecting the organism against various environmental stressors such as cold, heat, and osmotic shock [9,10]. This property has been exploited by the pharmaceutical industry, where trehalose serves as an excipient for numerous drugs [11,12]. The autophagy-inducing property of trehalose has been the subject of much investigation over the past decade. Trehalose can trigger autophagy in various types of mammalian cells in culture [13,14]. Administration of trehalose in vivo has also been shown to induce autophagy and lessen the neurodegenerative disease burden in murine models of Parkinson, Huntington, and ALS [15–17]. Interestingly, the mechanism by which trehalose induces the autophagic response has remained enigmatic. Even a clear understanding of its fate when added to cells is unknown. A few recent studies have linked trehalose to the activation and nuclear translocation of TFEB (transcription factor EB), which serves as a master regulator of autophagy-lysosomal biogenesis response. Specifically, within neuronal model systems, Rusmini and colleagues demonstrate trehalose induces lysosomal membrane permeabilization, which in turn releases intra-lysosomal calcium that modulates activity of PPP3CB, a calcium-dependent, calmodulin-stimulated protein phosphatase purported to be necessary for TFEB dephosphorylation and nuclear translocation [18]. In cultured fibroblasts and cortical-hippocampal neurons, Palmieri at al propose trehalose promotes nuclear translocation of TFEB by inhibition of AKT, independent of MTORC1 [19]. Our previous work has shown potent trehalose-mediated activation of TFEB in cultured macrophages, as well as reduced atherosclerotic plaque burden in trehalose-treated mice, mirroring the phenotype of macrophage-specific overexpression of TFEB [20].
Following up our extensive work with trehalose in macrophages and atherosclerosis, we now delineate a mechanism by which trehalose triggers TFEB nuclear translocation to stimulate autophagy-lysosomal biogenesis. Using primary macrophages, as well as several commonly used cell lines, we show that trehalose is taken up by cells via endocytic pathways (not dependent upon facilitated transport) and concentrates in lysosomes inducing mild lysosomal stress and altered acidification (mild increase in lysosomal pH). This low-grade lysosomal dysfunction is sufficient to inhibit MTORC1 signaling emanating at the lysosomal surface that relieves MTORC1-mediated inhibition on TFEB, leading to its nuclear translocation and transcriptional activation. These results highlight a novel mechanism by which the autophagy-lysosomal biogenesis response in cells can be leveraged as a therapeutic strategy.
Results
Trehalose induces TFEB nuclear translocation at therapeutically relevant concentrations
In order to evaluate the cellular effects of trehalose, many recent and past studies have used concentrations of trehalose in vitro that are not concordant with those obtainable in vivo. For example, a commonly utilized dose of trehalose in mice to obtain reductions in neurodegenerative markers is 3 g/kg injected intraperitoneally [15,16,21] which results in peak and mean serum trehalose concentrations of ~10 mM and ~2 mM respectively, likely translating to sub-millimolar concentrations in target tissues (Figure S1A) [20]. Yet, numerous publications have utilized concentrations of trehalose in cell culture that are in the 10–100 mM range which are clearly supra-physiologic with regard to the peripheral circulation and confound any mechanistic assessment. We therefore embarked on an evaluation of trehalose and its ability to activate TFEB using concentrations that correspond with therapeutically effective levels in vivo.
We first assessed the effect of trehalose on primary murine macrophages as we have previously shown its ability to robustly activate TFEB and the autophagy-lysosomal biogenesis response [20,22] Trehalose triggered nuclear translocation of TFEB at all concentrations tested (100 uM, 1 mM, and 10 mM) on par with bafilomycin A1, a known potent TFEB activator [20,22,23] (Figure 1A). Nuclear translocation was also seen as early as 6 h post-incubation and persisted up to 24 h (Figure 1B); this was corroborated using immunofluorescence microscopy (Figure 1C). In order to assess the generalizability of this observation, we conducted similar experiments in two commonly used cell lines, HEK 293 T and NIH 3T3. Trehalose triggered nuclear translocation of TFEB in both cell lines even at the lowest tested concentration (100 uM, Figures S1B,C) beginning at approximately 6 h and persisting up to 24 h (Figures S1D,E). Additionally, we found the translocation of TFEB to the nucleus, and the majority of autophagy- and lysosomal-related proteins were quickly up-regulated upon treatment with trehalose, peaking within 24 h followed by a gradual decline over 48 h (Figure 1D and Figure S1F). Overall, these findings demonstrate the capacity of trehalose to activate TFEB in different cell types, suggesting a similar underlying mechanism.
Figure 1.
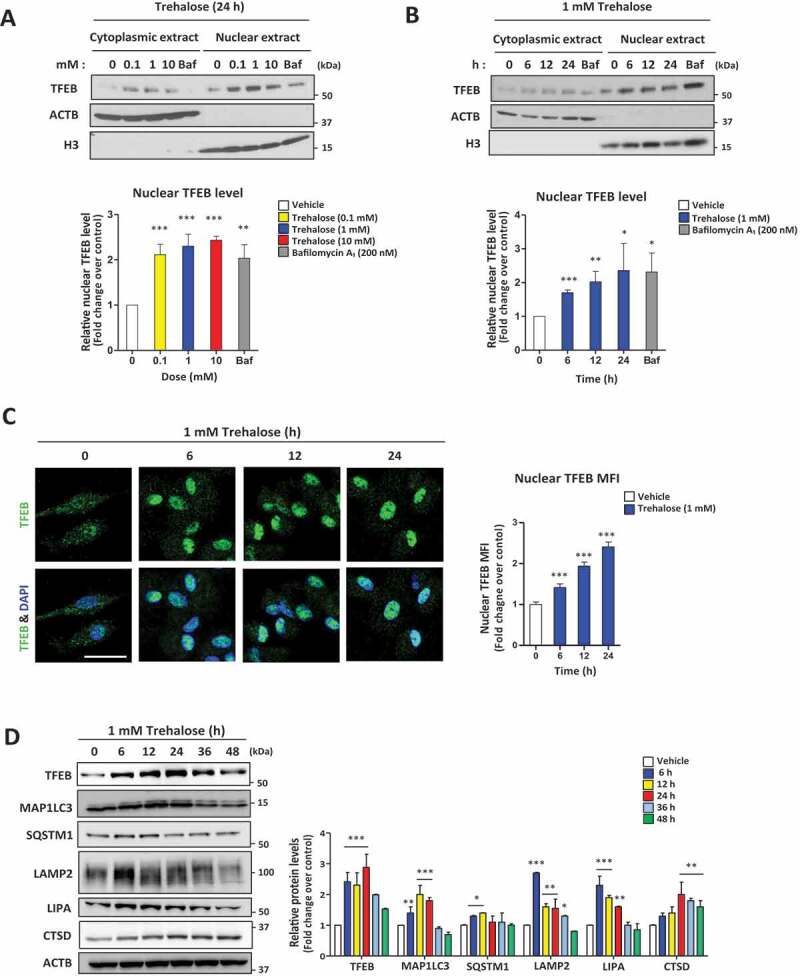
Trehalose induces TFEB nuclear translocation in a dose- and time-dependent fashion
(A-B) Western blot analysis of TFEB in cytoplasmic and nuclear fractions of macrophages after treatment with (A) the indicated concentrations of trehalose for 24 h or (B) 1 mM trehalose for the indicated times. ACTB/β-actin and histone H3 are shown as loading control for cytoplasmic and nuclear fractions respectively. Densitometric quantification from n= 3 independent experiments is shown below each blot. (C) Assessment of TFEB nuclear localization in macrophages by immunofluorescence microscopy after treatment with 1 mM trehalose for the indicated times. Quantification of images is shown to the right graphed as nuclear mean fluorescence intensity (MFI) of TFEB (n ≥ 60 cells per group; scale bar: 10 μm). (D) Western blot analysis of autophagy-lysosomal genes in macrophages after 1 mM trehalose treatment up to 48 h. ACTB/β-actin used as loading control. Densitometric quantification from n = 3 independent experiments is shown. For all graphs are presented as Mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001.
Trehalose is taken up by cells via endocytic pathways
Cell membranes are known to be impermeable to disaccharides including trehalose [24]. Thus, in order for trehalose to stimulate TFEB and the autophagy-lysosome biogenesis response, it either acts at the plasma membrane possibly via receptors or is taken up by cells and has intracellular effects. Since the only known mechanisms of TFEB activation involve intracellular signaling pathways, we first evaluated cellular uptake by incubating trehalose with primary macrophages, HEK 293T, and NIH 3T3 cells.
Utilizing similar escalating doses of trehalose that triggered TFEB nuclear translocation above, we quantified total cellular trehalose levels in macrophages via mass spectrometry. Trehalose was detectable intracellularly even at 100 µM with progressively increasing detection at 1 mM and 10 mM (Figure 2A). Time-course evaluation revealed detectable intracellular trehalose as early as 1 h and again progressively accumulating up to 24 h of incubation (Figure 2B). Similar findings were also observed in the cell-lines HEK 293 T and NIH 3T3 (Figure S2A-D). We confirmed intracellular uptake of trehalose using FACS analysis of macrophages incubated with trehalose conjugated to the FITC fluorochrome (Figure 2C). Again, trehalose uptake was observed in a time-dependent fashion beginning as early as 1 h of incubation (Figure 2C). Trehalose has been implicated in hepatocyte autophagy activation and previous reports have suggested that SLC2A8/GLUT8 (solute carrier family 2, (facilitated glucose transporter), member 8) is specialized trehalose transporter important for maximal trehalose uptake in primary murine hepatocytes and HEK293 cells [25]. To check the contribution of SLC2A8 in macrophages, we used Slc2a8 ASO (antisense oligonucleotide) to reduce SLC2A8 expression. Although expression of SLC2A8 was reduced, TFEB nuclear translocation was not altered in either pMAC (peritoneal macrophages) or BMDM (bone marrow-derived macrophages) treated with trehalose (Figure S2E and S2F). This result shows that SLC2A8 or SLC2A8 facilitated transport is not a requisite for trehalose-mediated TFEB nuclear translocation and autophagosome biogenesis in macrophages. Furthermore, it is known that trehalose can be enzymatically degraded by membrane-bound TREH (trehalase), a glycoside hydrolase enzyme predominately expressed in the kidney and small intestine. Nevertheless, knockdown of TREH using RNA interference (reduction of detectable TREH levels) had no significant effect on intracellular levels of trehalose in macrophages (Figure S2G and S2H).
Figure 2.
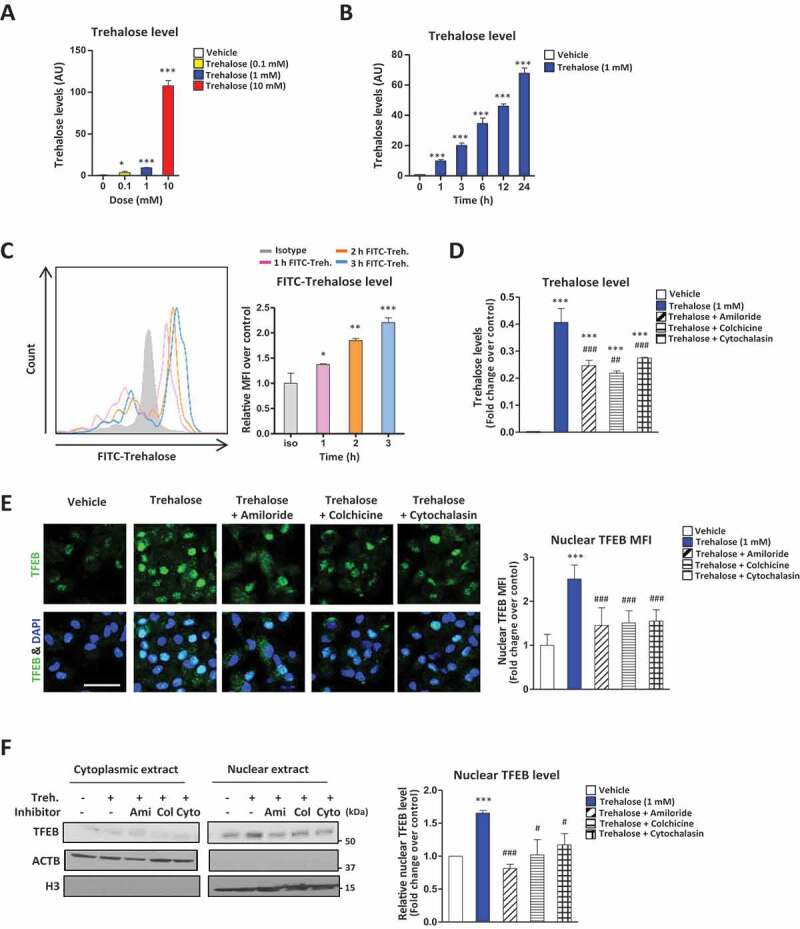
Trehalose undergoes endocytic uptake which is necessary for its stimulatory effect on TFEB. (A-B) Intracellular levels of trehalose measured by mass spectrometry in macrophages (A) treated for 24 h with the indicated concentrations of trehalose or (B) treated with trehalose (1 mM) for the indicated times. AU = arbitrary units. (C) Macrophages treated with FITC-conjugated trehalose for indicated times and fluorescence quantified by FACS analysis. Quantification of intracellular FITC is graphed to the right as MFI. (D) Intracellular levels of trehalose measured by mass spectrometry in macrophages treated for 24 h with trehalose (1 mM) ± endocytosis inhibitors amiloride, colchicine, or cytochalasin B. (E) Assessment of TFEB nuclear localization in macrophages by immunofluorescence microscopy after treatment with 1 mM trehalose for 24 h ± endocytosis inhibitors amiloride, colchicine, or cytochalasin B. Quantification of images is shown to the right graphed as nuclear MFI of TFEB with nuclear marker DAPI (n ≥ 20 cells per group; scale bar: 10 μm). (F) Western blot analysis of TFEB in cytoplasmic and nuclear fractions of macrophages after treatment with 1 mM trehalose for 24 h ± endocytosis inhibitors amiloride, colchicine, or cytochalasin B. Densitometric quantification from n = 3 independent experiments is shown to the right. For all graphs are presented as Mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001, compare with vehicle; #P < 0.05, ##P < 0.01 and ###P < 0.001, compared with trehalose
We next assessed whether endocytic pathways mediate trehalose uptake. Previous work has suggested that fluid-phase endocytosis might be a common mechanism for the uptake of cell-impermeable dissacharides [26–28]. We thus used amiloride, a classic inhibitor of fluid-phase endocytosis [29,30], and two more general endocytosis inhibitors, cytochalasin B and colchicine [30–32] to evaluate their effects on cellular uptake of trehalose. All three inhibitors significantly blocked trehalose uptake in macrophages (Figure 2D), which was sufficient to significantly curb TFEB nuclear translocation as assessed by microscopy and Western blots (Figure 2E,F). Also, these three inhibitors blocked trehalose dependent autophagy initiation (Figure S2I). These results support the notion that trehalose is a cell impermeable disaccharide that is predominantly taken up by nonselective fluid phase endocytosis and macropinocytosis.
Trehalose accumulates in lysosomes and alters lysosomal pH
Lysosomes are generally considered as the final compartment of endocytic pathways where captured cargo is subject to degradation via acidic hydrolases [33,34]. To determine whether the intracellular uptake of trehalose coincides with its accumulation in lysosomes, we isolated CLFs (crude lysosomal fractions) from trehalose-treated cells via centrifugation (Figure S3A). Trehalose levels significantly increased in the CLFs compared than whole cell (Figure S3B), its increase progressively starting at 1 h up to 24 h mirroring the intracellular elevation noted above (Figure 3A). Similar findings were also observed in 293 T and 3T3 cells (Figure S3C, D). Co-staining of cells with FITC-conjugated trehalose and the lysosomotropic dye LysoTracker) confirmed the progressive uptake and colocalization of trehalose with lysosomes (Figure 3B). Finally, the endocytosis inhibitors (amiloride, colchicine and cytochalasin) blocked the lysosomal accumulation of trehalose supporting endocytic-lysosomal trafficking of trehalose uptake (Figure 3C). Overall, these data support the notion that trehalose is taken up via endocytic processes and traffics to lysosomes.
Figure 3.
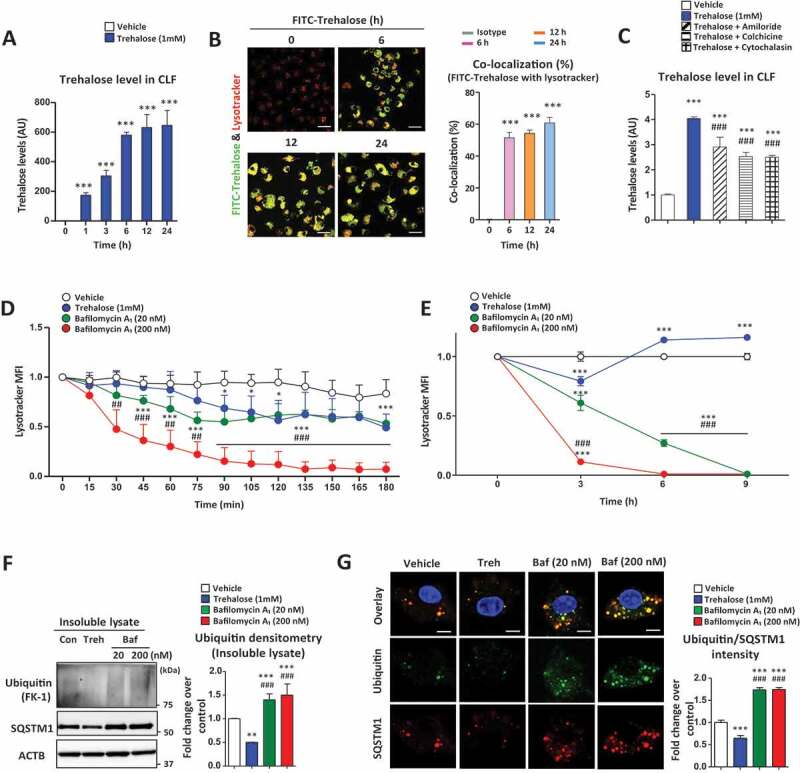
Trehalose accumulates in lysosomes and affects lysosomal acidification. (A) Trehalose levels in the crude lysosomal fraction (CLF) of macrophages were measured by mass spectrometry after treatment with trehalose (1 mM) for the indicated times. AU = arbitrary units. (B) Macrophages treated with FITC-conjugated trehalose for indicated times, stained with the lysosomal marker LysoTracker and imaged by fluorescence microscopy for FITC (green) and LysoTracker Red (Red). Quantification of images is shown to the right graphed as colocalization percent of FITC-conjugated trehalose with LysoTracker Red (n ≥ 20 cells per group; scale bar: 20 μm). (C) Trehalose levels in the lysosomal fraction of macrophages were measured by mass spectrometry after treatment for 24 h with trehalose (1 mM) ± endocytosis inhibitors amiloride, colchicine, or cytochalasin B. (D) Live imaging of macrophages incubated with trehalose (1 mM) or bafilomycin A1 (20 nM or 200 nM) for 180 min after staining with LysoTracker Red to monitor lysosomal acidity. Quantification of LysoTracker Red mean intensity every 15 min is shown (n ≥ 30 cells per group). (E) Confocal imaging of macrophages incubated with trehalose (1 mM) or bafilomycin A1 (20 nM or 200 nM) for indicated times after staining with LysoTracker Red to monitor lysosomal acidity. Quantification of LysoTracker Red mean intensity every 3 h is shown (n ≥ 30 cells per group). (F) Detergent-insoluble lysates were prepared after incubations with trehalose (1 mM) or bafilomycin A1 (20 nM or 200 nM) for 12 h and subjected to Western blot analysis for polyubiquitinated proteins (FK-1 monoclonal antibody) and SQSTM1. ACTB/β-actin used as loading control. Densitometric quantification from (n = 3) independent experiments is shown. (G) Assessment of ubiquitination in macrophages by immunofluorescence microscopy after treatment with trehalose (1 mM) or bafilomycin A1 (20 nM or 200 nM) for 12 h. Quantification of images is shown to the right graphed as ubiquitin intensity fold-change over control (n ≥ 20 cells per group; scale bar: 10 μm). For all graphs are presented as Mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001, compare with vehicle; #P < 0.05, ##P < 0.01 and ###P < 0.001, compared with trehalose
A unique property of trehalose that is not shared by other disaccharides is its relative resistance to acid hydrolysis [35,36]. This property would favor its lysosomal retention in un-degraded form, which is supported by the progressive buildup of trehalose in the lysosomal fraction (Figure 3A). Since TFEB is highly sensitive to lysosomal stress and particularly perturbations in lysosomal pH [37], we were interested in determining the effects of trehalose on lysosomal acidification. Using LysoTracker Red as a real-time gauge of lysosomal pH, we conducted live-cell imaging of macrophages treated with trehalose or bafilomycin A1, a well-known lysosomotropic drug that inhibits lysosomal acidification. The commonly used bafilomycin A1 concentration of 200 nM exerted complete abrogation of LysoTracker fluorescence over a 6-h time course (Figure 3D). Trehalose, at a moderate concentration of 1 mM, was capable of altering LysoTracker fluorescence ~50% of vehicle control, which was on par with low-dose bafilomycin A1 (20 nM, 1:10 concentration; Figure 3D). However, distinction between 1 mM trehalose- and low-dose (20 nM) bafilomycin A1-mediated lysosomal dysfunction becomes apparent in a longer time-course (Figure 3E). As illustrated, the effects of bafilomycin A1, even at lower dose of 20 nM, results in a progressive and permanent disruption of autophagy-lysosomal degradation system after 9 h (Figure 3E). To further elaborate differences between trehalose and bafilomycin A1, we examined SQSTM1/p62 ubiquitin-binding protein (autophagy chaperone) and SQSTM1-conjugated polyubiquitinated proteins (FK-1 monoclonal antibody) within cytoplasmic inclusions. As shown (Figure 3F,G), both concentrations of bafilomycin A1 (20 nM and 200 nM) yielded an accumulation in polyubiquitinated proteins (FK-1 monoclonal antibody) and SQSTM1 indicative of permanent disruption of lysosomal function and autophagy. In contrast, 1 mM trehalose reduction in SQSTM1 and polyubiqutinated proteins within detergent-insoluble fractions (refers to cytoplasmic inclusions) which suggest that collective effect of lysosomal accumulation and disruption of acidification by trehalose is a low-grade, transient phenomenon, which ultimately serves as a trigger for TFEB nuclear translocation and autophagy.
Others have reported that lysosomal accumulation of trehalose exerts lysosomal dysfunction through osmotic stress and lysosomal membrane permeabilization with calcium release [18]. We utilized quantitative assessment of fluorescent dextran to examine lysosomal membrane integrity. Endocytosis of fluorescent dextran leads to quantitative signal correlating to lysosomal accumulation, with a subsequent loss of fluorescence indicative of damage to the lysosomal membrane permeabilization, resulting in release-leakage of lysosomal contents. In pMAC loaded with 70-kDa TMR (tetramethylrhodamine)-conjugated dextran, we did not observe any appreciable lysosomal leakage with treatment of 1 mM trehalose (Figure S3E). These data suggest that lysosomal membrane integrity is not affected by trehalose treatment at this concentration.
Trehalose inhibits lysosomal MTORC1 signaling to trigger TFEB nuclear translocation
MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) is the most prominent regulator of TFEB activation. In fed states where there are ample lysosomal nutrients such as amino acids, MTORC1 is activated on the lysosomal surface and phosphorylates numerous downstream targets including direct phosphorylation/inhibition of TFEB [38,39]. Thus, reduction of MTORC1 signaling is a well-recognized mechanism of TFEB activation as supported by the potent stimulatory effect of classic MTOR inhibitors (e.g., rapamycin and torin) on TFEB nuclear translocation. A less recognized mechanism of MTORC1 inactivation involves disruptions in lysosomal pH as evidenced by the potent inhibitory effect of chloroquine and bafilomycin A1 on cellular MTORC1 signaling [40,41]. Thus, we evaluated the effect of trehalose on MTORC1 signaling by incubating cells with or without amino acids and trehalose. Pre-incubation of macrophages with trehalose inhibited the leucine-induced MTORC1 activation (Figure 4A). This effect was on par with bafilomycin A1 treatment and not observed when trehalose was co-incubated with leucine indicating that the accumulation and progressive disruption of lysosomal pH by trehalose was necessary MTORC1 inhibition (Figure S4A). We further verified this trehalose-MTORC1 link by demonstrating abrogation of MTOR-LAMP2 (lysosomal associated membrane protein2) colocalization by immunofluorescence microscopy when macrophages are pre-incubated with trehalose (Figure 4B). Similar results are recapitulated with LAMP1 (Figure S4B). Finally, we confirmed trehalose-induced abrogation of MTORC1 signaling by using MTOR inhibitors. TFEB nuclear translocation was increased in the presence of torin or rapamycin similar to that observed with trehalose. Importantly, co-treatment of cells with trehalose and either torin or rapamycin led to no synergistic effects on TFEB nuclear translocation (Figure 4C, Figure S4C), supporting the dependence of trehalose action on MTORC1 signaling. Furthermore, we showed that treatment with trehalose (1 mM) is capable of inducing TFEB dephosphorylation and this effect can be neutralized through PPP3/calcineurin inhibition with cyclosporine A, demonstrating MTOR-dependence and phosphatase specificity relating to trehalose-mediated activity (Figure S4D and S4E).
Figure 4.
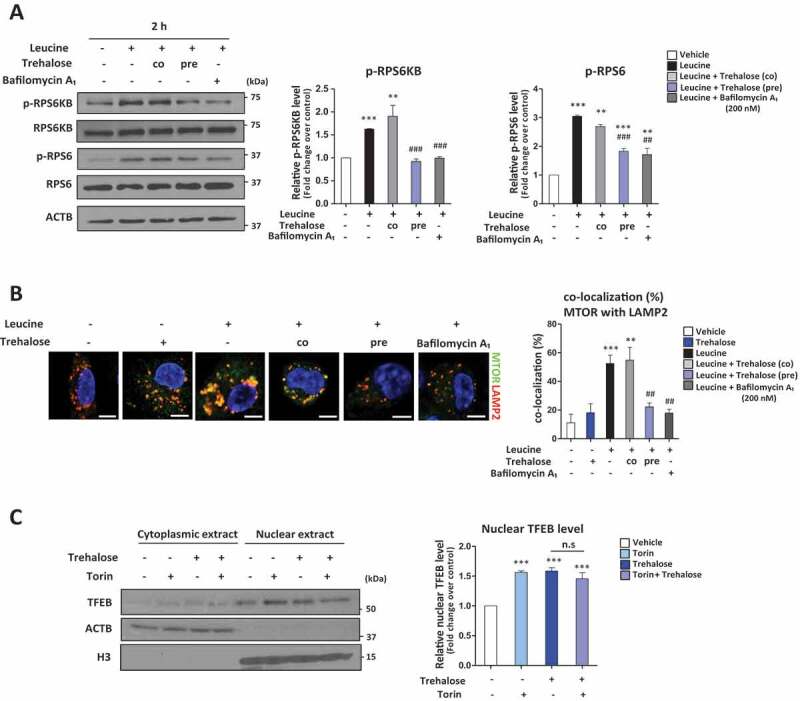
Trehalose affects MTOR activation leading to induction of TFEB nuclear translocation. (A, B) Macrophages were treated with or without Leucine and either trehalose (1 mM) provided 2 h before Leucine (labeled “pre”), trehalose (1 mM) provided at same time as Leucine (labeled “co”), or bafilomycin A1 (200 nM) provided at same time a Leucine followed by: (A) Western blot analysis of MTORC1 downstream targets (phospho-Thr389 and total RPS6KB/S6K; phospho-Ser235/236 and total RPS6 ribosomal protein). Densitometric quantification from three separate experiments is shown to the right. (B) Immunofluorescence staining of MTOR colocalization with lysosome marker (LAMP2). Quantification of images is shown to the right graphed as colocalization percent of MTOR with LAMP2 (n ≥ 10 cells per group; scale bar: 5 μm). (C) Western blot analysis of TFEB in cytoplasmic and nuclear fractions of macrophages after treatment with 1 mM trehalose for 24 h ± MTOR inhibitor Torin. Densitometric quantification from n = 3 independent experiments is shown to the right. For all graphs are presented as Mean ± SEM. **P < 0.01 and ***P < 0.001, compare with vehicle; ## P < 0.01 and ### P < 0.001, compare with leucine only
Discussion
The therapeutic potential of trehalose as an inducing agent of autophagy via lysosomal stress and TFEB activation has been proposed across a variety of model systems aimed at neurodegenerative and cardio-metabolic diseases [18,19,42–44]. These include a few recent studies have highlighted potential trehalose-specific effects on lysosomal stress [18], as well identification of MTORC1-independent regulation of TFEB-mediated autophagy by trehalose [19]. Despite these recent additions, a definitive mechanism for trehalose- and TFEB-mediated autophagy in macrophages has yet to be resolved. In the present work, we have detailed a pathway involving the lysosome and MTORC1 signaling to enact TFEB activation. We find that trehalose utilizes specific endocytic pathways for cellular uptake and trafficking to the lysosome. Accumulation of trehalose imparts mild lysosomal stress and disrupts acidification, leading to inactivation of MTORC1-mediated suppression of TFEB. Removal of this inhibitory phosphorylation allows for liberation of TFEB, which translocates to the nucleus to mediate the autophagy-lysosome transcriptional response. The proposed mechanism of trehalose action in this setting is summarized in Figure 5.
Figure 5.
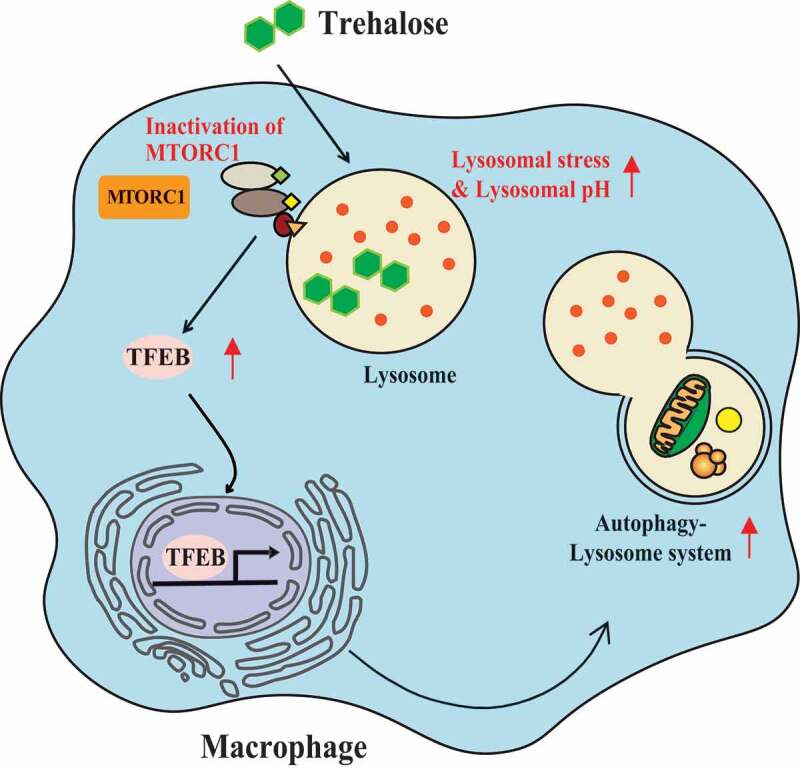
Overview of the proposed mechanism of TFEB activation by trehalose. Trehalose is first taken up by endocytic means and accumulates in lysosomes (note: SLC2A8-independent uptake is only demonstrated in macrophages). The rise in intra-lysosomal trehalose concentration results in modest increases in lysosomal pH, which is sufficient to perturb MTORC1 signaling. Reductions in MTOR activity relieves it suppressive effect on TFEB, resulting in its nuclear translocation and induction of the autophagy-lysosomal biogenesis response. In this context, trehalose functions as a TFEB activator via mild perturbation of lysosomal acidity and function
We have identified several interesting points relating to trehalose-stimulated TFEB-mediated activation of autophagy. First, our observation that endocytosis is the primary means by which trehalose is taken to the intracellular domain and accumulated within lysosomes to exert its activity. This suggests its predominant mode of action is not dependent upon cell surface proteins for binding or signal transduction. Previous reports have suggested that SLC2A8 serves as a specialized trehalose transporter, which is thought to be important for maximal trehalose uptake in primary hepatocytes and HEK293 cells [14,25]. However, the effect of SLC2A8 on trehalose uptake in macrophages has not been established. Moreover, based on our studies employing SLC2A8 ASO to reduce SLC2A8 expression, we observed no alteration in TFEB nuclear translocation in either primary macrophages or derived cell lines in response to treatment with trehalose. Similarly, we did not find evidence to support receptor-mediated endocytosis in trehalose uptake, which was inhibited in response to general endocytic inhibitors. These findings suggest that neither SLC2A8-facilitated transport nor receptor-mediated endocytosis is required for uptake or subsequent trehalose-mediated TFEB nuclear translocation and autophagy-lysosome biogenesis in our various cell models. Bulk endocytic mechanisms, including fluid-phase endocytosis, are likely to be operative across a diverse array of cells, such as primary macrophages, immortalized NIH 3T3 cells (fibroblasts), and transformed HEK 293 T cells (epithelial).
Regardless of predominant mode of uptake, our data demonstrates eventual accumulation within lysosomes, the terminal stage of most endocytic pathways. Thus, instead of direct, cytoplasmic activation of autophagy, it appears that the lysosome represents the primary target of trehalose to exert its effects on TFEB and autophagy-lysosome biogenesis, presumably through perturbation of lysosomal function and disruption of lysosome-associated MTORC1 signaling. The accumulation of trehalose within lysosomes despite the significant acidity of this compartment signifies one of its most unique chemical characteristics (i.e. the relative resistance of its α-1-1 glycosidic bond acid to hydrolysis compared to other disaccharides) [35]. This relative resistance to degradation likely underlies its progressive accumulation within lysosomes and gradual effects on lysosomal acidity.
Lysosomal stress is known to be a potent trigger for TFEB activation as a result of MTOR inactivation [22,37]. The key feature of any compound that leverages lysosomal stress as a mechanism of TFEB stimulation is that the degree of its impact on lysosomal function cannot supersede the benefits of inducing autophagy-lysosomal biogenesis. In other words, disruptive effects on lysosomes cannot completely or permanently impair lysosomal function without negatively impacting cellular function. In the case of trehalose, we find it partially alters lysosomal acidity on par with low-dose bafilomycin A1 (20 nM), in comparison to 200 nM bafilomycin A1 that is used to fully suppress lysosomal function. This is consistent with the notion that initiation of “low-grade” lysosomal stress is critical in stimulating the autophagy-lysosomal biogenesis response, while not adversely affecting cellular degradative capacity.
Although beyond the scope of our current study, the mechanism(s) by which trehalose disrupts lysosomal function and acidification is likely multifactorial, relating to its unique bio-protective properties. Various studies have shown that trehalose stabilizes membranes and proteins in vivo and in vitro [45]. In aqueous solutions, trehalose acts as a stabilizing co-solvent, which facilitates ordered structure of water around trehalose via hydrogen bonding. Conversely, under conditions of desiccation (absence of solvent), trehalose can mimic water through formation of hydrogen bonds around polar and charged moieties of phospholipids and proteins that promotes structural-conformational stability. The ability to serve as a structure-maker or “kosmotrope” is why trehalose is such an effective preservative that helps prevent protein denaturation and maintain cellular integrity [9].
Trehalose is highly resistant to acid hydrolysis, often requiring high temperatures and/or high hydrogen ion concentrations (pH ~4.0) to initiate decomposition. Such properties would allow for preservation of function within acidic compartments such as the lysosome. In addition, proton (H+) influx along with balanced counter-ion (Cl-, K+) movement are integral to lysosomal acidification and pH homeostasis that is dependent upon a number of key transporter proteins, including V-type ATPase (H+ pump), ClC-7 (Cl−/H+) antiporter, and cation (K+) efflux channels [46]. Studies have shown that trehalose generally improves the thermodynamic stability of proteins, including retention of enzymatic activity at elevated temperatures and denaturing conditions [47]. It is not unreasonable to suspect that trehalose is capable of interacting with luminal side domains of V-type ATPase (perhaps eliciting a bafilomycin-like effect), and/or ClC-7, cation channels, as well as nearby membranes, altering ion transport and acidification of the lysosome. Moreover, trehalose has been shown to augment membrane order and fluidity under disruptive-stress conditions, although higher concentrations of trehalose (300 mM, although dependent on a variety of conditions and variables) can result in a marked reduction of membrane order [45]. Lastly, lysosomal membrane permeabilization was not observed with treatment of 1 mM trehalose on pMAC loaded with 70-kDa TMR-conjugated dextran. Perhaps this reflects a dose-dependent difference between supra-physiologic (100 mM) and therapeutically attainable (1 mM) doses of trehalose, whereby at higher doses intra-luminal accumulation may result in membrane order stabilization, less kosmotropy, and/or more osmotic stress.
Finally, in this study we took great care to examine the mechanisms of trehalose at therapeutically-relevant and -achievable concentrations. In contrast to numerous prior studies where doses of trehalose as high as 100 mM are used to induce autophagy, we used concentrations in the range of 0.1 mM to 10 mM. We based this concentration range on actual peripheral circulating trehalose levels that are measured when mice are administered trehalose at doses which have been shown to be effective at reducing neurodegeneration, atherosclerosis, and other pathologies in mouse models [15,16,21]. At these relevant concentrations, we have shown that trehalose can still accumulate in lysosomes, cause mild perturbations in lysosomal acidity, inhibit MTORC1 signaling, and in turn trigger TFEB nuclear translocation. Our results are on par with results from the known TFEB activator bafilomycin A1, however, key distinctions include a low-grade and transient perturbation of lysosomal function. In comparison to even low-dose bafilomycin A1 (20 nM), we have shown that and MTOR-dependent TFEB activation this provides a more plausible explanation of the TFEB-inducing properties of trehalose and provides a framework by which trehalose action can be mechanistically dissected in vivo.
The ability to modulate the acidity of lysosomes in cells can have therapeutic implications in vivo. In many diseases including atherosclerosis and neurodegeneration, disruption of lysosomal acidity and lysosomal function leads to an inability to degrade both autophagic and extracellular cargo with resultant accumulation of cytotoxic lipids and dysfunctional organelles that disrupt tissue homeostasis and exacerbate disease [16,20,37,48–50]. The salutary effects observed in TFEB overexpression models and with TFEB-activating agents such as trehalose, have raised the prospects of therapies designed to stimulate or rescue such lysosomal dysfunction [20,37,51–53]. Given that the proposed mechanism of trehalose leverages mild perturbations of lysosomal acidity (i.e. low-grade lysosomal stress) to activate TFEB, it remains unclear whether such a route of action can indeed be effective in disease states where lysosomal acidification and dysfunction are already rampant. Although difficult to ascertain the efficacy of trehalose on all cells of a diseased organ system, one can surmise that trehalose action likely exerts optimal efficacy in cells which are involved in earlier phases of the disease where lysosomal dysfunction is not terminal. Studies evaluating the ability of trehalose to ameliorate disease in early, middle, and late phases of pathogenesis will be required to fully evaluate its optimal role.
Trehalose is a safe and naturally occurring disaccharide known for years to have unique autophagy and TFEB-inducing properties. Yet, the mechanism underlying this action has remained enigmatic. Our demonstration of its involvement in lysosome-dependent MTORC1 signaling is not only important for potential uses of trehalose in therapeutics but also in the development of other pharmacologic agents that can harness this a “hormesis effect” (low-dose stimulation, high-dose inhibition) on lysosomes as a therapeutic strategy.
Materials and methods
Cell culture and treatment
For peritoneal macrophage isolation, we used standard techniques. Briefly, mice were injected with 4% thioglycolate media (Sigma, T9032) by intraperitoneal, and after 3 days, peritoneal macrophages were collected from peritoneal lavage. Collected peritoneal macrophages were counted and plated with complementary 10% FBS contained DMEM media. The following treatments were added to cells: Bafilomycin A1 (20 nM and 200 nM; Sigma Aldrich, B1793), amiloride (Sigma Aldrich, A7410), colchicine (Sigma Aldrich, C9754), cytochalasin B (Sigma Aldrich, C6762), trehalose (100 μM, 1 mM and 100 mM; Sigma Aldrich, T0167), torin 1 (Tocris Bioscience, 4247), rapamycin (Enzo Life Sciences, BML-A275-0025). Treated cells were harvested at various times for the subsequent experiments.
Nuclear and cytoplasmic extraction
The nuclear and cytoplasmic extract was isolated using an NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Thermo Scientific, 78,833) according to the manufacturer’s protocol. Briefly, the treated cells were washed with DPBS (Life Technologies, 14,190,235) and centrifuged at 500 g for 3 min. The pellet was suspended in 100 μl of cytoplasmic extraction reagent I, vortexed for 15 s and incubated on ice for 10 min. Cytoplasmic extraction reagent II (5.5 μl) was added, vortexed for 5 s, incubated on ice for 1 min and centrifuged at 15,500 g for 5 min at 4°C. The supernatant fraction (cytoplasmic extract) was transferred to a pre-chilled tube. The insoluble pellet fraction, which contains crude nuclei, was washed with PBS, and resuspended in 50 μl of nuclear extraction reagent by vortexing during 15 s every 10 min (for a total of 40 min), then centrifuged at 15,500 g for 10 min at 4°C. The resulting supernatant, constituting the nuclear extract, was used for the subsequent experiments. Relative changes in the expression of nuclear TFEB protein was measured by forming the ratio of the densitometric values of TFEB protein versus histone H3.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde, blocked and permeabilized using Roth buffer (0.25% milk powder [BD Biosciences, 232100], 1% BSA [Sigma Aldrich, A7906], 0.3% Triton X-100 [Sigma Aldrich, X100] in TBS [20 mM Trizma hydrocholride {Sigma Aldrich, T3253} and 150 mM NaCl {Sigma Aldrich, S3014}], pH 7.4) for 1 h. The following primary antibodies were used: TFEB (Bethyl, A303-673A), MTOR (Cell Signaling Technology, 2983), LAMP2 (Abcam, ab13524) and DAPI (4,6-diamidino-2-phenylindole; Invitrogen, D21490). Species-specific fluorescent secondary antibodies were obtained from Life Technologies (A11001, A11007, A11008 and A11076). FITC-conjugated trehalose (2 µM; gift from Dr. Brian J. DeBosch, Washington University School of Medicine) was incubated with cells for the indicated time. LysoTracker Red DND-99 (Life Technologies, L7528) was used according to the manufacturer’s protocol. A Zeiss LSM-700 confocal microscope was used for imaging. Signal over the threshold was quantified using ZEN microscope software (Carl Zeiss Microscopy) after determination of regions of interest and thresholds.
Western blotting
Cells were lysed in a standard RIPA lysis buffer. Standard techniques were used for protein quantification, preparation and blotting. The following primary antibodies were used: TFEB (Bethyl, A303-673A), LAMP1 (Abcam, ab24170), LAMP2 (Abcam, ab13524), LIPA/lysosomal acid lipase (Origene, TA309730), phospho-RPS6KB/S6K (Cell Signaling Technology, 9234S), RPS6KB/S6K (Cell Signaling Technology, 2708S), phospho-RPS6/S6 (Cell Signaling Technology, 4856S), RPS6/S6 (Cell Signaling Technology, 2217S), phospho-TFEB (Cell Signaling Technology, 86,843), TREH/trehalase (Santa Cruz Biotechnology, sc-390,034), ACTB/β-actin (Sigma Aldrich, A2066) and histone H3 (Abcam, ab1791). Relative changes in the expression of nuclear TFEB protein was measured by forming the ratio of the densitometric values of TFEB protein verse histone H3. Protein expression was quantified using densitometric analyses (ImageJ software).
Trehalose measurement
For measuring serum trehalose level, mice were fasted for 4 h, and injected with trehalose (3 g/kg bodyweight) by intraperitoneal. Blood were collected at indicated times centrifuged, transferred the supernatant (serum) to clear tube for measurement. Serum and intracellular trehalose levels were measured using a trehalose assay kit (Megazyme, K-TREB) by manufacturer’s protocol.
Gas chromatography-mass spectrometry (GC-MS)
Quantification of trehalose levels in the cells were measured by GC-MS. The isotope [13C] trehalose (Omicron Biochemicals, TRE-002) was used as an internal standard for quantitation of the trehalose during the sample preparation. Samples were extracted into isopropanol:acetonitrile:water (3:3:2), centrifuged at 18,000 g for 15 min at 4°C and dried under N2 gas. N-Methyl-N-(trimethysilyl) trifluoroacetamide with 10% pyridine in CH3CN was then used to derivatize samples for analysis by GC–MS using Agilent 7890A gas chromatograph interfaced to Agilent 5975 C mass spectrometer and HP-5 ms gas chromatography column (30 m per 0.25-mm internal diameter per 0.25-mm film coating). A linear temperature gradient was used. The initial temperature of 80°C was held for 2 min and increased to 300°C at 10°/min. The temperature was held at 300°C for 6 min. The samples were run by electron ionization (EI) and the source temperature, electron energy and emission current were 250°C, 70 eV and 300 uA, respectively. The injector and transfer line temperatures were 250°C. Quantitation was carried out by monitoring the ions at m/z 361 (trehalose) and 367 (trehalose-13C12). Since cellular or lysosomal volumes are not known, actual trehalose concentrations are not known and values reported in all figures are denoted with AU indicative of arbitrary units.
L-amino acid assay
Intracellular L-amino acids were measured in macrophages using a colorimetric L-amino acid assay kit (Abcam, ab65347) according to the manufacturer’s protocol.
FACS analysis
FACS analysis for checking uptake of FITC-conjugated trehalose by peritoneal macrophages. Macrophages were plated, incubated with FITC-conjugated trehalose (2 μM) for indicated times, collected with Cellstripper (Corning, 25056), and transferred to 96 well v-bottom plate (Fisher Scientific, 7000142). Macrophages blocked with FCGR3/CD16 (low affinity immunoglobulin gamma Fc region receptor III) and FCGR2/CD32 (low affinity immunoglobulin gamma Fc region receptor II) FcBlock (BD Bioscience, 553141, 1:100) and stained with Pacific Blue-conjugated PTPRC/CD45 (protein tyrosine phosphatase receptor type C; BioLegend 103126), PerCP-Cy5.5-conjugated ITGAM/CD11b (integrin alpha M; BioLegend 101228) and APC-conjugated ADGRE1/F4/80 (adhesion G protein-coupled receptor E; (BioLegend, 123116) antibodies. All samples were analyzed using the BD Biosciences LSR II flow cytometer and quantified using FlowJo software.
Isolation of lysosome enriched fraction
Cells were harvested using PBS, centrifuged and the pellet was resuspended in a lysosomal buffer (0.1% BSA and 0.5 μM EDTA) containing protease inhibitors (Sigma Aldrich, 4693132001). Cells were broken up by sonication, with an aliquot of lysate reserved for whole cell lysate analysis. The remaining lysate was centrifuged at 2,000 g for 12 min at 4°C to pellet the pre-lysosomal fraction. The supernatant was transferred to a fresh tube and centrifuged at 15,000 g for 60 min at 4°C to pellet the lysosomal fraction lysate, which was re-suspended in RIPA buffer for analysis.
Live cell image
For live imaging, Cells were plated on glass-bottom culture dishes (Mattek Corporation, P35G-1.5–10-C). Cells were imaged using a Nikon A1Rsi Confocal Microscope with Tokai-hit stage-top incubator (37°C and 5% CO2). Reagents were added after the first image, and following images were captured every 15 min. Image analysis and quantification was performed using ImageJ software.
Assessment of lysosomal function by FACS
Cells were pre-treated with 70-kDa tetramethyl-rhodamine (TMR)-conjugated dextran (25 µg/mL; Life Technologies, D1818) for 2 h at 37°C before incubation with 1 mM trehalose. Cells were washed with FACS buffer (2% FBS [Life Technologies, 26140079] and 2% EDTA [Sigma Aldrich, E5134] in DPBS [Life Technologies, 14190235]) and analyzed using the BD Biosciences LSR II flow cytometry and quantified using FlowJo software.
Statistics
Statistical significance of differences was calculated using the two-tailed Student’s unpaired t-test or ANOVA for parametric data. Data presented as the Mean ± SEM.
Supplementary Material
Funding Statement
This work was supported by NIH R01 HL125838, VA MERIT I01 BX003415, American Diabetes Association [#1-18-IBS-029], Washington University Diabetic Cardiovascular Disease Center and Diabetes Research Center Grant [P30 DK020579], Washington University Mass Spectrometry core grants [P41GM103422 and P30DK056341], the Foundation for Barnes-Jewish Hospital, and NIH [1R21 AT010520], Integrative Systems Biology of Cardiovascular Disease Training Grant, Center for Cadiovascular Research at Washington University School of Medicine [5T32HL134635].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol [Internet]. 2009;10:458–467. [DOI] [PubMed] [Google Scholar]
- [2].Mizushima N, Komatsu M.. Review autophagy : renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [3].Sa´nchez I, Mahlke C, Yuan J.. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–379. [DOI] [PubMed] [Google Scholar]
- [4].Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. [DOI] [PubMed] [Google Scholar]
- [5].Polanco JC, Li C, Bodea L, et al. Amyloid‑β and tau complexity — towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2018;14:22–39. [DOI] [PubMed] [Google Scholar]
- [6].Caspi M, Sasai M, Kyu H, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. PNAS. 2009;106:2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Razani B, Feng C, Coleman T, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jain NK, Roy I. Effect of trehalose on protein structure. Protein Sci. 2009;18:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Garg AK, Kim JK, Owens TG, et al. Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proc Natl Acad Sci U S A. 2002;99:15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee J, Lin E-W, Lau UY, et al. Trehalose glycopolymers as excipients for protein stabilization. Biomacromolecules. 2013;14:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Teramoto N, Sachinvala ND, Shibata M. Trehalose and trehalose-based polymers for environmentally benign, biocompatible and bioactive materials. Molecules. 2008;13:1773–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J Biol Chem. 2007;282:5641–5652. [DOI] [PubMed] [Google Scholar]
- [14].DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2017;9:ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Castillo K, Nassif M, Valenzuela V, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9:1308–1320. [DOI] [PubMed] [Google Scholar]
- [16].Tanaka M, Machida Y, Niu S, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. [DOI] [PubMed] [Google Scholar]
- [17].Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–438. [DOI] [PubMed] [Google Scholar]
- [18].Rusmini P, Cortese K, Crippa V, et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2019;15:631–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Palmieri M, Pal R, Nelvagal HR, et al. MTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 2017;8:14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim J, Cheon H, Jeong YT, et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J Clin Invest. 2014;124:3311–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science (80-). 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42–ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang NJ, Hinner MJ. Getting across the cell membrane: an overview for small molecules, peptides, and proteins. Methods Mol Biol. 2016;1266:29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mayer AL, Higgins CB, Heitmeier MR, et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep. 2016;6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Oliver AE, Jamil K, Crowe JH, et al. Loading human mesenchymal stem cells with trehalose by fluid-phase endocytosis. CELL Preserv Technol. 2004;2:35–49. [Google Scholar]
- [27].Ma X, Jamil K, Macrae TH, et al. A small stress protein acts synergistically with trehalose to confer desiccation tolerance on mammalian cells. Cryobiology. 2005;51:15–28. [DOI] [PubMed] [Google Scholar]
- [28].Oldenhof H, Zhang M, Narten K, et al. Freezing-induced uptake of disaccharides for preservation of chromatin in freeze-dried stallion sperm during accelerated aging †. BiologyofReproduction. 2017;97:892–901. [DOI] [PubMed] [Google Scholar]
- [29].Koivusalo M, Welch C, Hayashi H, et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. JCB. 2010;188:547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang J, Jiang G, Song Q, et al. Lipoprotein-biomimetic nanostructure enables efficient targeting delivery of siRNA to Ras-activated glioblastoma cells via macropinocytosis. Nat Commun. 2017;8:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sandvig K, Van DB. Selective modulation of the endocytic uptake of ricin and fluid phase markers without alteration in transferrin endocytosis *. J Biol Chem. 1990;265:6382–6388. [PubMed] [Google Scholar]
- [32].Inoue K, Ishizawa M, Kubota T. Monoclonal anti-dsDNA antibody 2C10 escorts DNA to intracellular DNA sensors in normal mononuclear cells and stimulates secretion of multiple cytokines implicated in lupus pathogenesis. Clin Exp Immunol. 2019;199:150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–632. [DOI] [PubMed] [Google Scholar]
- [34].Huotari J, Helenius A. Endosome maturation. Embo J. 2011;30:3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Richards AB, Krakowka S, Dexter LB, et al. Trehalose : a review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem Toxicol. 2002;40:871–898. [DOI] [PubMed] [Google Scholar]
- [36].Russ N, Zielbauer BI, Vilgis TA. Impact of sucrose and trehalose on different agarose-hydrocolloid systems. Food Hydrocoll. 2014;41:44–52. [Google Scholar]
- [37].Emanuel R, Sergin I, Bhattacharya S, et al. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol. 2014;34:1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Napolitano G, Esposito A, Choi H, et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018;9:3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012;31:1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science (80-). 2011;334:678 LP– 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang Y, Higgins CB, Mayer AL, et al. TFEB-dependent induction of thermogenesis by the hepatocyte SLC2A inhibitor trehalose. Autophagy. 2018;14:1959–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sciarretta S, Yee D, Nagarajan N, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2018;71:1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2016;9:ra21–ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Al-Ayoubi SR, Schinkel PKF, Berghaus M, et al. Combined effects of osmotic and hydrostatic pressure on multilamellar lipid membranes in the presence of PEG and trehalose. Soft Matter. 2018;14:8792–8802. [DOI] [PubMed] [Google Scholar]
- [46].Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. [DOI] [PubMed] [Google Scholar]
- [47].Kaushik JK, Bhat R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J Biol Chem. 2003;278:26458–26465. [DOI] [PubMed] [Google Scholar]
- [48].Zhang X, Sergin I, Evans TD, et al. High-protein diets increase cardiovascular risk by activating macrophage mTOR to suppress mitophagy. Nat Metab [Internet]. 2020;2:110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- [50].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. [DOI] [PubMed] [Google Scholar]
- [51].Leeman DS, Hebestreit K, Ruetz T, et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science (80-). 2018;359:1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pastore N, Huynh T, Herz NJ, et al. TFEB regulates murine liver cell fate during development and regeneration. Nat Commun [Internet]. 2020;11. DOI: 10.1038/s41467-020-16300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Settembre C, De CR, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation- induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.