

ABSTRACT
In humans, TDRD7 (tudor domain containing 7) mutations lead to a syndrome combining congenital cataracts (CCs) and non-obstructive azoospermia (NOA), characterized by abnormal lens development and spermiogenesis. However, the molecular mechanism underlying TDRD7’s functions in eye and testicular development are still largely unknown. Here, we show that the depletion of this gene in mice and humans resulted in the accumulation of autophagosomes and the disruption of macroautophagic/autophagic flux. The disrupted autophagic flux in tdrd7-deficient mouse embryonic fibroblasts (MEFs) was caused by a failure of autophagosome fusion with lysosomes. Furthermore, transcriptome analysis and biochemical assays showed that TDRD7 might directly bind to Tbc1d20 mRNAs and downregulate its expression, which is a key regulator of autophagosome maturation, resulting in the disruption of autophagosome maturation. In addition, we provide evidence to show that TDRD7-mediated autophagosome maturation maintains lens transparency by facilitating the removal of damaged proteins and organelles from lens fiber cells and the biogenesis of acrosome. Altogether, our results showed that TDRD7 plays an essential role in the maturation of autophagosomes and that tdrd7 deletion results in eye defects and testicular abnormalities in mice, implicating disrupted autophagy might be the mechanism that contributes to lens development and spermiogenesis defects in human.
Abbreviations: CB: chromatoid bodies; CC: congenital cataract; CTSD: cathepsin D; DMSO: dimethyl sulfoxide; LAMP1: lysosomal-associated membrane protein 1; LECs: lens epithelial cells; MAP1LC3/LC3/Atg8: microtubule-associated protein 1 light chain 3; MEFs: mouse embryonic fibroblasts; NOA: non-obstructive azoospermia; OFZ: organelle-free zone; RG: RNA granules; SQSTM1/p62: sequestosome 1; TBC1D20: TBC1 domain family member 20; TDRD7: tudor domain containing 7; TEM: transmission electron microscopy; WT: wild type.
KEYWORDS: Acrosome biogenesis, autophagy, lens development, spermiogenesis, tbc1d20, tdrd7
Introduction
Congenital cataracts (CCs), caused by lens opacity resulting from metabolic abnormalities during early development, are a major cause of induced blindness in children, occurring in approximately 1–6 per 10,000 live births worldwide [1]. Inherited cataracts are the major cause of CCs and often constitute one of the multiple phenotypes in syndromic cases [2], such as Down syndrome [OMIM#190,685], myotonic dystrophy [OMIM#602,668], Warburg micro syndrome [OMIM#600,118], and Vici syndrome [OMIM#242,840]. Recently, we found that loss of function mutations in the TDRD7 (tudor domain containing 7) gene caused a novel syndrome in humans manifested by CCs and non-obstructive azoospermia (NOA) [3]. NOA, defined as the absence of spermatozoa in the seminal fluid, is the most severe infertility phenotype resulting from spermatogenesis failure [4,5]. In tdrd7 knockout mice, a loss of TDRD7 protein function leads to failure of lens development due to lens opacity and arrest in spermiogenesis due to the failure of round spermatids to transform into elongating sperm [3,6,7]. CCs and NOA are clinically distinct diseases affecting different organs. Whether TDRD7 has similar or distinct functions in lens development and spermiogenesis needs to be further investigated.
TDRD7 belongs to the TDRD family of proteins and contains three OST-HTH (Oskar-Tdrd7-Helix-Turn-Helix)/LOTUS and three evolutionarily conserved tudor domains [8]. The OST-HTH/LOTUS domains are predicted to regulate multiple aspects of RNA metabolic processes [9,10], while the tudor domains are predicted to bind methylated arginine residues with in other proteins [11]. TDRD7 is abundantly expressed in the eye and testis, but expressed at low endogenous levels in other tissues [7]. In the vertebrate ocular lens, TDRD7 is a component of cytoplasmic RNA granules (RG) that post-transcriptionally regulates the specific genes that are critical for lens development. In male germ cells, TDRD7 is a conserved component of chromatoid bodies (CB), a unique germ cell-specific ribonucleoprotein granule, playing an important role in cellular RNA metabolism [6,7,10]. Loss of function of TDRD7 leads to a syndrome that involves two developmentally and anatomically different organs, namely, the eye and testis, while other organs appear to be unaffected. This suggests that a unique cellular process might be shared between lens development and spermiogenesis.
Recent studies demonstrate that TDRD7, as a new interferon-stimulated gene, exerts its antiviral activity by inhibiting the cellular macroautophagy/autophagy pathway, which is required for paramyxovirus replication [12,13]. Autophagy is an evolutionarily highly conserved lysosomal degradation pathway with important roles in cellular homeostasis, including nutrient provision during fasting or increased metabolic demands, and removal of defective proteins or organelles (e.g., mitochondria) [14,15]. Autophagy plays crucial roles during development and in various diseases, and increasing evidence indicates that autophagy also plays a direct role in lens development and spermiogenesis [16–20]. For example, a defect in EPG5, a key regulator of autophagosome maturation, was found to result in autosomal-recessive CC in individuals with Vici syndrome [20]. Pathogenic mutations in FYCO1, an autophagy receptor directly linking intracellular transport of autophagic vesicles, cause autosomal-recessive CC [16,21]. During spermiogenesis in mice, SIRT1 regulates acrosome biogenesis by modulating autophagic flux [22]. Furthermore, autophagy regulates cytoskeleton organization via degradation of PDLIM1 to facilitate spermatid differentiation [18]. Interestingly, autophagy has a direct role in both lens development and spermiogenesis. For example, TBC1D20 (TBC1 domain family member 20), functional loss of which causes Warburg micro syndrome 4 (WARBM4) with cataracts and male infertility [23], plays an essential role in autophagosome maturation via its RAB1B GAP function, while a TBC1D20 defect is found to result in eye and testicular abnormalities in mice by disrupting the maturation of autophagosomes [24]. Collectively, these data suggest that autophagy is indispensable for eye and testicular development, and that tdrd7 deletion may result in abnormal lens development and spermiogenesis by disrupting the autophagy pathway; however, the underlying molecular mechanisms are still unknown.
In this study, we hypothesized that TDRD7 participates in lens development and spermiogenesis as a mediator of autophagy. To examine this hypothesis, we performed various experiments on tdrd7-deficient mouse cells and patients with TDRD7 mutations. We present the first evidence that TDRD7, by directly bind to Tbc1d20 mRNAs and downregulate its expression, regulates autophagosome maturation, which is required for the maintenance of autophagic flux and degradation of autophagic cargo. Thus, we elucidated why TDRD7-mediated autophagosome maturation is essential for lens fiber cell homeostasis and acrosome biogenesis.
Results
tdrd7-deficient mouse embryonic fibroblasts (MEFs) exhibit accumulation of autophagosomes
To determine whether the autophagy pathway was affected in tdrd7-deficient cells, we first analyzed MEFs generated from embryonic day 13 (E13) of wild type (WT, also termed Tdrd7+/−) and tdrd7-deficient littermates (KO, also termed tdrd7−/−). In tdrd7-deficient MEFs, MAP1LC3/LC3/Atg8 (microtubule-associated protein 1 light chain 3)-labeled autophagosomes were enlarged and accumulated compared with that in WT MEFs (Figure 1A). Transmission electron microscopy (TEM) revealed an accumulation of enlarged electron-light double-membrane structures in tdrd7-deficient MEFs, indicative of autophagosomes (Figures 1B and 1C). Some autophagosomes in tdrd7-deficient MEFs were observed as a part of enlarged multi-lamellar structures (Figure 1B). Furthermore, we examined the expression of endogenous LC3 protein aggregates and found that the protein level of LC3-II was significantly higher in tdrd7-deficient MEFs than that in WT MEFs (Figures 1D and 1E). This finding suggested that there was an accumulation of autophagosomes in tdrd7-deficient MEFs.
Figure 1.
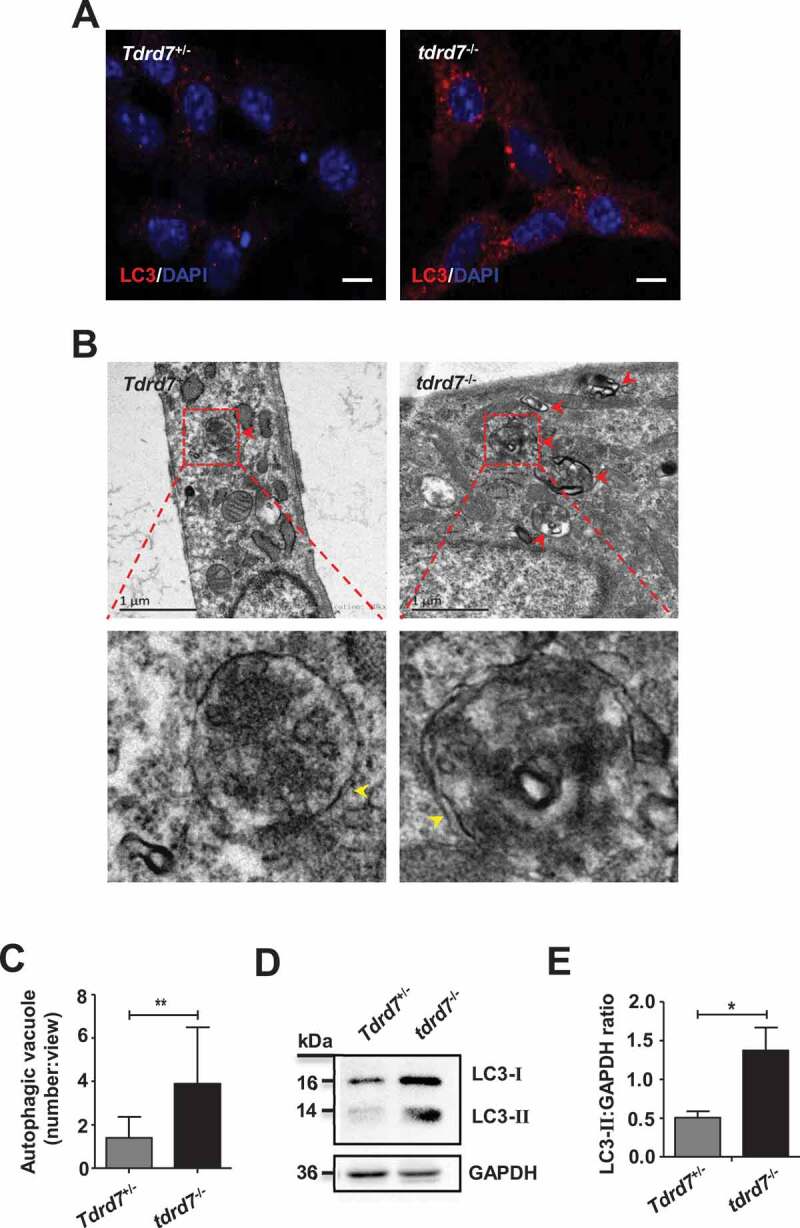
Evaluation of autophagosomes in Tdrd7+/− and tdrd7−/− MEFs. (A) Immunostaining of Tdrd7+/− and tdrd7−/− MEFs for the autophagic marker LC3 (red signal). (B) The accumulation of enlarged electron-light double-membraned structures (red arrows) in tdrd7-deficient MEFs. Some autophagosomes in tdrd7-deficient MEFs were observed as a part of enlarged multi-lamellar structures. Red dotted box: the magnified picture of autophagosomes of which the “double membranes” could be seen faintly (yellow arrows). Scale bar: 1 μm. (C) Quantification of the average number of autophagosomal structures in Tdrd7+/− and tdrd7−/− MEFs using TEM analysis. *P < 0.05. (D) Immunoblotting analysis of LC3 in both Tdrd7+/− and tdrd7−/− MEFs (left). (E) Quantification analysis revealed a significant accumulation of LC3-II (*P < 0.05), GAPDH served as a loading control. Data are representative of three independent assays
Accumulation of autophagosomes in tdrd7-deficient MEFs is caused by disrupted autophagic flux
Autophagosome accumulation could be the result of an increase in the production of autophagosomes, or a disruption in autophagic flux due to the failure of autophagosomes fusion with lysosomes, or both [25]. To confirm whether the TDRD7 defect caused the increase in the production of autophagosomes or disrupted autophagic flux in tdrd7-deficient MEFs, we evaluated the expression level of SQSTM1/p62 (sequestosome 1), whose accumulation is an established indicator of disrupted autophagic flux [26–28], in WT and tdrd7-deficient MEFs. Immunostaining for SQSTM1 demonstrated that the SQSTM1-positive area was significantly greater in tdrd7-deficient MEFs compared with that in WT MEFs (Figure 2A). Western blotting results confirmed that the levels of SQSTM1 were higher in tdrd7-deficient MEFs than in WT MEFs (Figures 2B and 2C). To further investigate autophagic flux, we measured the levels of SQSTM1 in WT and tdrd7-deficient MEFs following a 3-h treatment with dimethyl sulfoxide (DMSO) (vehicle control), hydroxycloroquine (inhibitor of autophagy that inhibits lysosomal function), rapamycin (an inducer of autophagy) [27], or a combination of hydroxycloroquine and rapamycin in complete medium (to both inhibit and induce autophagy) (Figure 2D). Quantification analysis revealed significantly lower levels of SQSMT1 in WT MEFs following rapamycin treatment, which had no effect on tdrd7-deficient MEFs. Additionally, treatment of WT MEFs with hydroxycloroquine and a combination of rapamycin and hydroxycloroquine resulted in significantly higher levels of SQSTM1 than those in vehicle-treated WT MEFs (Figures 2D and 2E). In contrast, the levels of SQSTM1 in vehicle-treated tdrd7-deficient MEFs did not significantly differ from those of SQSTM1 in tdrd7-deficient MEFs following treatment with hydroxychloroquine and a combination of rapamycin and hydroxycloroquine (Figures 2D and 2E). Therefore, in tdrd7-deficient MEFs, SQSTM1 levels were insensitive to treatments with a chemical inhibitor or activator of autophagy.
Figure 2.
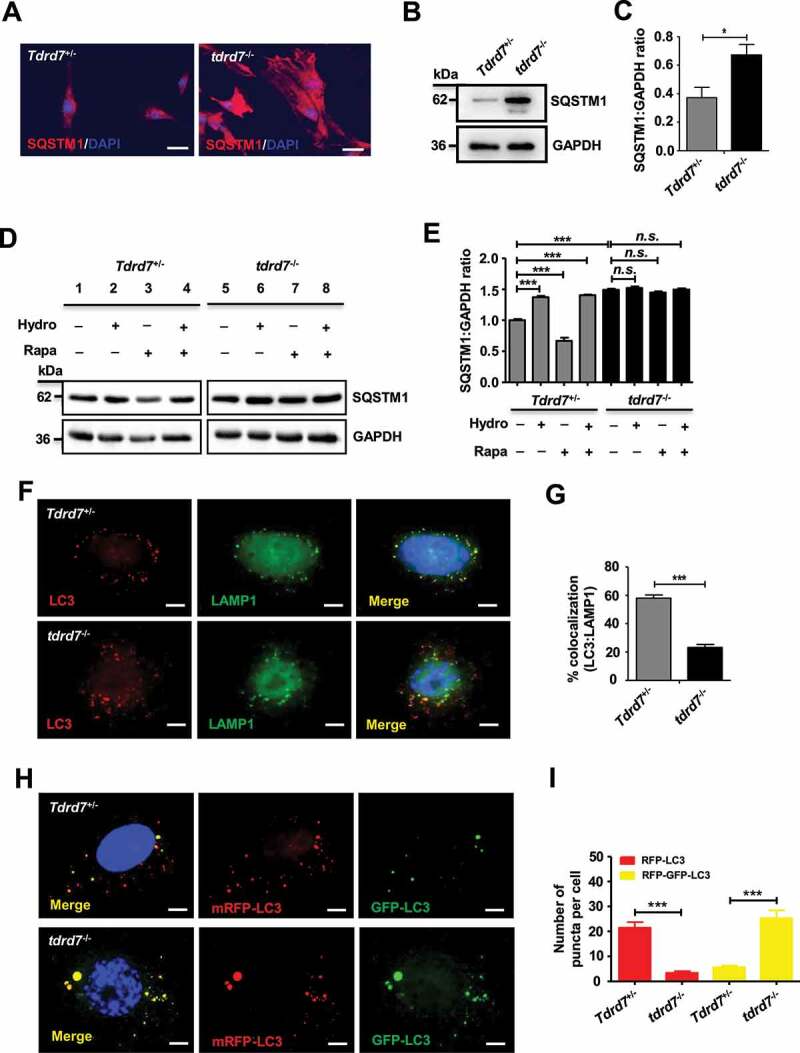
Autophagosome maturation is blocked in tdrd7−/− MEFs. (A) Immunostaining of Tdrd7+/− and tdrd7−/− MEFs for SQSTM1 (red signal), an established indicator of disrupted autophagic flux. (B) Immunoblotting analysis of SQSTM1 in Tdrd7+/− and tdrd7−/− MEFs (left). (C) Quantification analysis revealed a significant accumulation of SQSTM1 (*P < 0.05); GAPDH served as a loading control. Data are representative of three independent assays. (D-E) Immunoblotting of Tdrd7+/− and tdrd7−/− MEF cell lysates for SQSTM1 following a 3-h treatment with DMSO as a vehicle control (lanes 1 and 5), 50 μM hydroxychloroquine (lanes 2 and 6) and 50 nM rapamycin (lanes 3 and 7) in complete medium, and combined treatment with 50 μM hydroxycloroquine and 50 nM rapamycin in complete medium (lanes 4 and 8); GAPDH served as a loading control. The gray level was measured using ImageJ (the signal in Tdrd7+/− MEFs following a 3-h DMSO treatment was set as 1 after normalization to GAPDH blotting). n.s.: not significant, ***P < 0.001. (F) Immunostaining for LC3 (red signal) and LAMP1 (green signal) in Tdrd7+/− and tdrd7−/− MEFs. Scale bar: 5 μm. (G) The colocalization of LC3 and LAMP1 is quantified, n = 20 cells of three independent experiments. **P < 0.01. (H-I) Autophagy flux in Tdrd7+/− and tdrd7−/− MEFs was assessed through an imageing-based co-localization analysis of mRFP-GFP-LC3. RFP positive and GFP negative indicates autolysosomes (red puncta). RFP positive and GFP positive indicates autophgosomes (yellow puncta). The nuclei were stained with 2-(4-amidinophenyl)-1 H-indole-6-carboxamidine (DAPI) (blue signals). Significance was established using the Student’ s t-test. ***P < 0.001. Scale bar: 5 μm
To identify which autophagic step is disrupted as a result of loss of TDRD7 function, we performed immunostaining for LC3 and LAMP1 (lysosome-associated membrane protein 1) in WT and tdrd7-deficient MEFs. The results revealed an obvious decrease in the co-localization of LC3 and LAMP1 in tdrd7-deficient MEFs (Figures 2F and 2G), suggesting that autophagosomes in tdrd7-deficient MEFs were failed to fuse with lysosomes. We further used mRFP-GFP tandem fluorescent-tagged LC3 to investigate whether autophagosome-lysosome fusion is affected in tdrd7-deficient MEFs [29]. As shown in Figure 2H, compared with WT MEFs, tdrd7-deficient MEFs showed a decrease in the number of autolysosomes (RFP positive, GFP negative; red puncta) and an increase in the number of autophagosomes (RFP positive, GFP positive; yellow puncta). Quantification analysis revealed a significantly decreased number of red puncta per cell as well as a significantly increased number of yellow puncta per cell for tdrd7-deficient MEFs compared with that in WT MEFs (Figure 2I). Collectively these findings indicate that functional loss of TDRD7 in tdrd7-deficient MEFs results in the failure of autophagosomes to acidify.
To exclude the possibility that the failure of autophagosomes to acidify in tdrd7-deficient MEFs could be due to defective lysosomal functions, we performed immunostaining against RAB5 (as an early endosomal marker) and LAMP1 (as a lysosomal marker) in WT and tdrd7-deficient MEFs. We found that the percentage area for RAB5 and LAMP1 immunostaining per cell did not significantly differ between WT and tdrd7-deficient MEFs (Fig. S1A and S1B). Next, to exclude the possibility that effects on endolysosomal functions were responsible for failed autophagosomes acidification in tdrd7-deficient MEFs, we performed LysoTracker Red staining, which is a reagent that staining lysosomes, in WT and tdrd7-deficient MEFs. As shown in Fig. S1C, the morphology of acidic vesicles in WT and tdrd7-deficient MEFs did not differ following staining with LysoTracker Red, indicating that acidification of organelles in tdrd7-deficient MEFs was not affected. To determine if TDRD7 was required for the functioning of lysosomes, we performed immunostaining and western blots assays for CTSD (cathepsin D; a lysosomal peptidase) in WT and tdrd7-deficient MEFs. The morphology of structures positive for CTSD and levels of mature CTSD revealed no obvious differences between WT and tdrd7-deficient MEFs (Fig. S2A and S2B). To determine whether the functional loss of TDRD7 in tdrd7-deficient MEFs also disrupted the endosomal-lysosomal pathway, we performed western blot assays for analyzing the expression of EGFR (a cell surface tyrosine kinase that upon EGF-mediated ligand binding is rapidly endocytosed and targeted toward lysosomes for degradation [30]) in the cell lysates of WT and tdrd7-deficient MEFs after treatment with 40 ng/mL EGF and 20 µg/mL cycloheximide for 0, 30, 60 and 120 min. As shown in Fig. S2C, the expression of EGFR did not significantly differ between tdrd7-deficient MEFs and WT MEFs, suggesting that the endosomal-lysosomal pathway was not compromised. Collectively, these findings provide evidence that a functional loss of TDRD7 in tdrd7-deficient MEFs results in disrupted autophagic flux due to failed autophagosome-lysosome fusion, and consequently in an accumulation of autophagosomes without disrupting endolysosomal function.
tdrd7 deficiency results in the abnormal expression of metabolism-related genes
TDRD7 is a RNA binding protein and functional as regulators of post-transcriptional gene expression [6]. To further investigate the potential molecular mechanism of TDRD7 mediated autophagosome maturation, we performed transcriptome analysis in both the eyes and testes of WT and tdrd7-deficient mice. A total of 437 differentially expressed genes (DEGs) were detected in the eyes of tdrd7-deficient mice, including 335 upregulated and 102 downregulated genes (Figure 3A, left, Table S1). In tdrd7-deficient mice testes, a total of 378 DEGs were detected, including 75 upregulated and 303 downregulated genes (Figure 3A, right, Table S2). Gene Ontology (GO) analysis of the DEGs was performed. As shown in (Figure 3B), DEGs were specifically enriched for terms, such as eye development, spermatid development, and transcription regulation. Interestingly, DEGs, such as Tbc1d20, Itgav/Cd51, Hipk1, and Spata18, were specifically enriched for metabolism in both the eyes and testes of WT and tdrd7-deficient mice (Figure 3B, Table S3 and S4). To verify the transcriptome data, some genes involved in the different biological processes were randomly selected for quantitative reverse transcription-PCR (qRT-PCR) verification. The expression levels of all the genes correlated well with those observed in the transcriptome data (Figure 3C). Subsequently, we constructed a venn diagram to screen for candidate mRNAs (Fig. S3). We found that five differentially expressed genes, including Tspan6, Timp1, Cd52, Aif1 and Tbc1d20, were downregulated in both tdrd7-deficient eyes and testes. Interestingly, the Tbc1d20, a key regulator of autophagosome maturation, was found to be responsible for both eye development and spermatogenesis [23,24], indicating that TDRD7 might mediate autophagy-dependent disruption of lens development and spermiogenesis by regulating TBC1D20.
Figure 3.
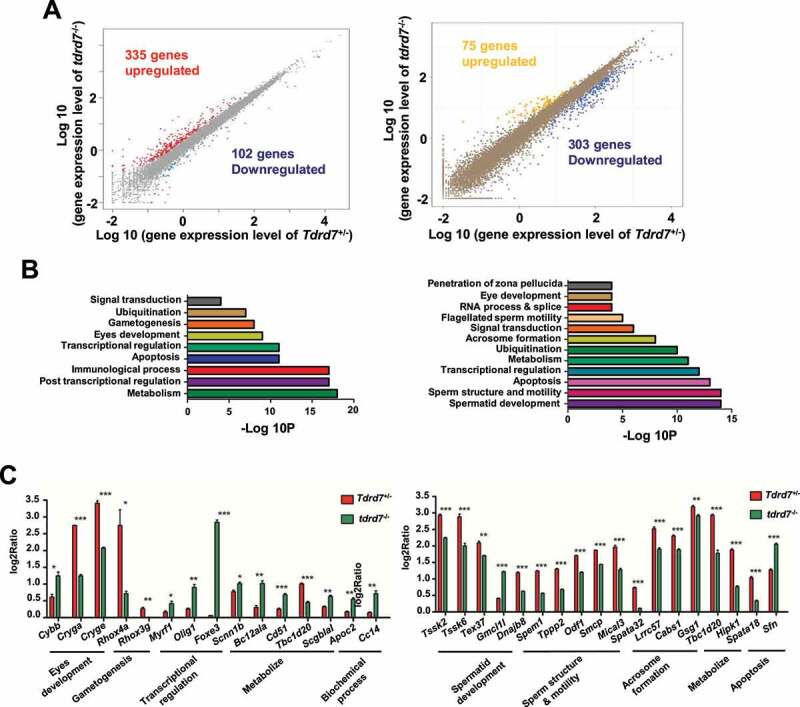
Tdrd7 deficiency results in the abnormal expression of metabolism-related genes. (A) Significantly differentially expressed genes in the eyes (left) and testes (right). The red and blue colors indicate the upregulated and downregulated genes, respectively. (B-C) Gene Ontology term analysis of the significantly differentially expressed genes in the eyes (left) and testes (right). (D-E) Some genes involved in biological processes of the eyes (left) and testes (right) were randomly selected for qRT-PCR verification. Significance was established using the Student’ t test. *P < 0.05; **P < 0.01, ***P < 0.001
TDRD7 mediates autophagosome maturation by directly bind to Tbc1d20 mRNAs
Since TBC1D20 is a key regulator of autophagosome maturation and a TBC1D20 defect results in eye and testicular abnormalities in mice by disrupting the maturation of autophagosomes [24], we hypothesized that tdrd7 deletion disrupted the maturation of autophagosomes by downregulating TBC1D20. To test this hypothesis, we first examined the expression of TBC1D20 in both the eyes and testes of WT and tdrd7-deficient mice. Compared with that in WT mice, the expression level of TBC1D20 was significantly reduced in both the eyes and testes of tdrd7-deficient mice (Figures 4A and 4B). We further examined the expression of Tbc1d20 mRNA and protein in tdrd7-deficient MEFs and found it to be significantly reduced when compared with that in WT MEFs (Fig. S4A and Figure 4C). To test whether TDRD7 is the upstream regulatory molecule of TBC1D20, we established two Tbc1d20 stable knockdown MEF cell lines (Tbc1d20-si1 and Tbc1d20-si2) and a control cell line (Tbc1d20-siNC) using two Tbc1d20 small interfering RNAs (siRNAs) and a control siRNA. qRT-PCR and western blotting revealed that although silencing of TBC1D20 did not change the expression of TDRD7 mRNA and protein (Fig. S4B and Figure 4D), it dramatically increased the accumulation of LC3 and SQSTM1 protein aggregates (Figure 4D). We subsequently conducted a rescue experiment including WT and tdrd7-deficient MEFs with or without overexpression of TBC1D20 (Fig. S4C and Figure 4E). The expression levels of LC3 and SQSTM1 in tdrd7-deficient MEFs with overexpressed TBC1D20 were clearly decreased compared with the tdrd7-deficient MEFs without overexpressed TBC1D20 (Figure 4E). These results indicated that TDRD7 defects might impair autophagosome maturation by downregulating the expression of TBC1D20.
Figure 4.
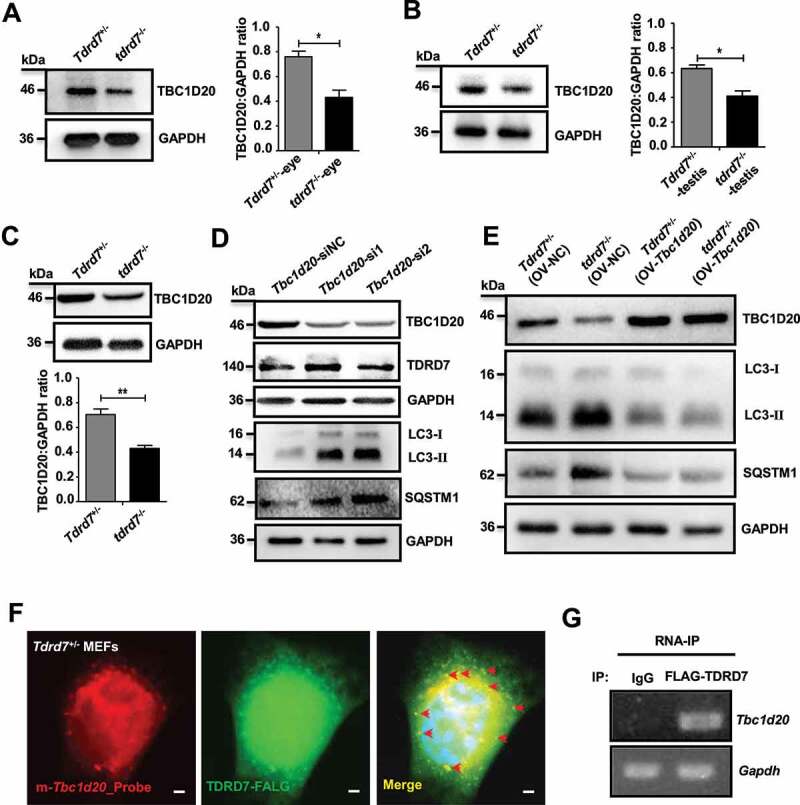
Tdrd7 deletion downregulated TBC1D20 disrupting autophagosome maturation. (A-B) Immunoblotting analysis of TBC1D20 in the eyes and testes of Tdrd7+/− and tdrd7−/− mice (left). Quantification analysis revealed a significantly decreased expression of TBC1D20 (*P < 0.05) (right). (C) Immunoblotting analysis of TBC1D20 in Tdrd7+/− and tdrd7−/− MEFs. Quantification analysis revealed a significantly decreased expression of TBC1D20 (**P < 0.01). GAPDH served as a loading control in (A) to (C). (D) Tdrd7+/− MEFs cells stably expressing Tbc1d20-siRNA1 and Tbc1d20-siRNA2 were generated, and the expression levels of TBC1D20, TDRD7, LC3-II, SQSTM1, and GAPDH (control) in Tbc1d20-silenced Tdrd7+/− MEFs cells were determined by immunoblotting. (E) Tdrd7+/− and tdrd7−/− MEFs transfected with or without Tbc1d20 and control vectors were generated, and the expression levels of TBC1D20, LC3-II, SQSTM1, and GAPDH (control) were determined by immunoblotting. (F) FISH-immunofluorescence assay showed that FLAG-labeled TDRD7 (green dot signals) co-localized with Tbc1d20 mRNAs (red dot signals) in Tdrd7+/− MEFs. The nuclei were stained with 2-(4-amidinophenyl)-1 H-indole-6-carboxamidine (DAPI) (blue signals). Scale bar: 5 µm. (G) RNA immunoprecipitation assay using FLAG-labeled TDRD7 antibody, followed by RNA isolation and RT-PCR, revealed TBC1D20 transcripts enriched in FLAG-labeled TDRD7 complexes. All the data are representative of three independent assays
To determine whether the reduced expression of TBC1D20 resulted from a direct or indirect action of TDRD7, we performed FISH-immunofluorescence and RNA immunoprecipitation (RIP) experiments. Because the commercial TDRD7 antibody is not suitable for immunostaining and immunoprecipitation experiment, we employed WT MEF cells for these experiments by co-transfected with FLAG-labeled TDRD7 and HA-labeled TBC1D20 vectors. We observed that TDRD7 co-localized with Tbc1d20 mRNAs, indicating that Tbc1d20 mRNAs are likely to be direct targets of TDRD7 (Figures 4F and 4G). To further detect whether TDRD7 could interact with TBC1D20 protein, we performed a co-IP assay in WT-MEF cells by co-transfecting the cells with FLAG-labeled TDRD7 and HA-labeled TBC1D20 vectors. No interaction between TDRD7 and TBC1D20 was detected (Fig. S4D). Thus, based on their strong dependence on TDRD7 expression and the evidence for direct binding from the RIP experiments, we conclude that TBC1D20 mRNAs are likely to be the direct targets of TDRD7.
tdrd7-deficient MEFs exhibit accumulation of ubiquitinated autophagic cargo
Autophagy plays a critical role in facilitating the delivery of ubiquitinated autophagic cargo to the lysosome for degradation, and cells with abrogated autophagy exhibit accumulation of ubiquitinated proteins, peroxisomes, and damaged organelles, resulting in cytoplasmic clarity disorder [31–34]. In tdrd7-deficient mice testes, abnormally accumulated cytoplasm of round sperm cells was observed by immunostaining for α-tubulin compared with that in WT mice testes (Fig. S5). To explore this further, ubiquitin (a marker for autophagic cargo) was immunostained in WT and tdrd7-deficient MEFs. Our results showed an accumulation of ubiquitin in tdrd7-deficient MEFs compared with WT MEFs (Figure 5A). Western blotting of WT and tdrd7-deficient cell lysates confirmed the accumulation of ubiquitinated proteins in tdrd7-deficient MEFs (Figure 5B). Next, we wanted to determine whether other autophagic cargo, such as mitochondria, also accumulate in tdrd7-deficient MEFs. Immunostaining and western blotting for the mitochondrial marker, TOMM20, revealed significant accumulation of mitochondria in tdrd7-deficient MEFs (Figures 5C and 5D), indicating an inhibition of autophagic cargo degradation.
Figure 5.
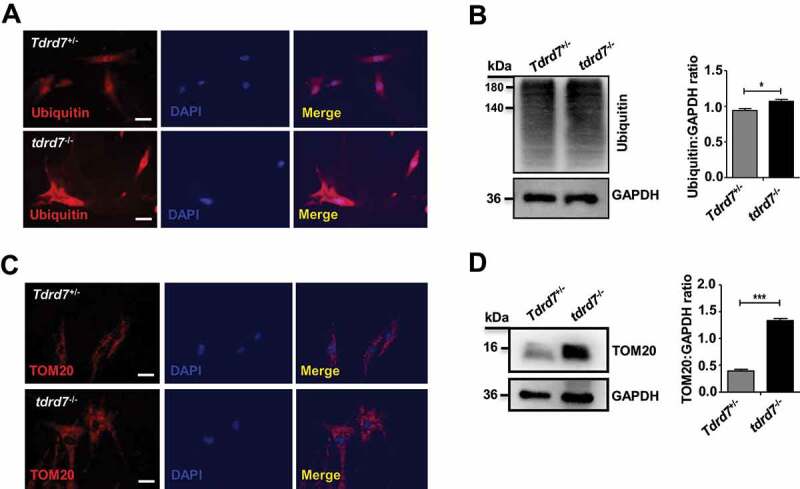
Autophagic cargo in Tdrd7+/− and tdrd7−/− MEFs. (A) Immunostaining of Tdrd7+/− and tdrd7−/− MEFs for ubiquitin (red signal) with DAPI-counterstained nuclei (blue signal). Scale bar: 50 μm. (B) Immunoblotting analysis of Tdrd7+/− and tdrd7−/− MEF lysates for ubiquitin revealed significant accumulation of ubiquitinated proteins in tdrd7−/− lysates (*P < 0.05). (C) Immunostaining of Tdrd7+/− and tdrd7−/− MEFs for TOMM20 (red signal, as a mitochondrial marker) with DAPI-counterstained nuclei (blue signal). Scale bar: 50 μm. (D) Immunoblotting analysis of Tdrd7+/− and tdrd7−/− MEF lysates for TOMM20 revealed significantly greater levels of TOMM20 relative to GAPDH in tdrd7−/− MEF lysates (***P < 0.001)
Lens fiber cells in tdrd7-deficient mice exhibit disrupted autophagic flux and cataracts
We have previously reported that the lenses of both TDRD7-deficient humans and mice with CC manifested as opacity [3,6], To explore this further, we morphologically evaluated WT and tdrd7-deficient mice lenses. The results showed that postnatal day 30 (P30) WT lenses exhibited organized epithelium at the anterior as well as differentiated fiber cells at the cortex, and terminally differentiated anuclear lens fiber cells at the lens core, termed the “organelle-free zone” (OFZ) (Figure 6A). However, the P30 tdrd7-deficient lens fiber cells were highly degenerated and disorganized and showed obviously larger cell gaps within lens epithelial cells (LECs) and retained nuclei in the OFZ (Figure 6A). To determine whether a disrupted TDRD7-mediated autophagic flux can cause CCs in tdrd7-deficient lenses, TEM was used to detect the autophagic vacuole in P30 WT and tdrd7-deficient lenses, and the result showed a significantly accumulated autophagic vacuole in the cytoplasm of tdrd7-deficient lenses (Figures 6B and 6C), which consistent with the result in tdrd7-deficient MEFs (Figures 1B and 1C). Furthermore, immunostaining of P30 WT lenses was positive for LC3 (Figure 6D), SQSTM1 (Figure 6E), and ubiquitin (Figure 6F) in LECs and cortical fibers in a perinuclear pattern, which was absent in the OFZ. In contrast, tdrd7-deficient lenses revealed positive immunostaining for accumulation of LC3 (Figure 6D), SQSTM1 (Figure 6E), and ubiquitin (Figure 6F) in LECs and aberrant accumulation of LC3, SQSTM1, and ubiquitin in fiber cells within the OFZ. Western blotting of LC3 (Figure 6G), SQSTM1(Figure 6H), and ubiquitin (Figure 6I) in P30 WT and tdrd7-deficient lenses confirmed the significantly higher levels of LC3-II, SQSTM1, and ubiquitin in tdrd7-deficient lenses than in WT lenses. This finding suggested that TDRD7 functional loss in tdrd7-deficient lens fiber cells results in disrupted autophagic flux and accumulation of ubiquitinated autophagic cargo.
Figure 6.
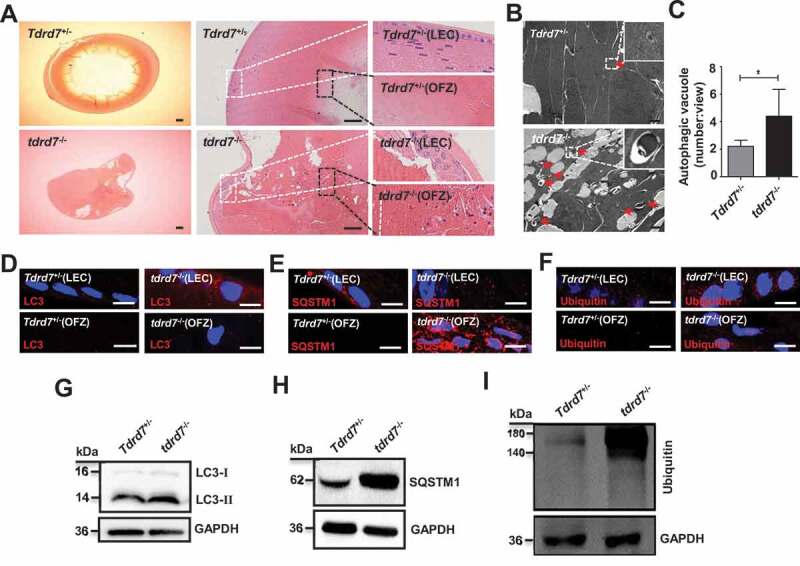
Evaluation of ocular tissues in Tdrd7+/− and tdrd7−/− mice. (A) H&E staining of lenses showed atrophic lens structure in tdrd7−/− mice. There are a number of bubbles in lens epithelial cells (LEC) and lens fiber cells, and degenerative nuclei in the organelle-free zone (OFZ). White box: LECs, black box: OFZ. Scale bar: 200 μm. (B) Detection of Tdrd7+/− and tdrd7−/- mice lenses using TEM. The red arrows indicate the autophagic vacuoles. (C) Quantification of the average number of autophagic vacuoles in Tdrd7+/− and tdrd7−/− mice lenses using TEM analysis. *P < 0.05. (D-F) Immunostaining of Tdrd7+/− mice lenses for LC3 (D), SQSTM1 (E) and ubiquitin (F) (red signal) revealed the presence of LC3-II, SQSTM1, and ubiquitin in LECs without any staining present in the OFZ. By contrast, immunostaining of tdrd7−/− mice lenses revealed accumulation of LC3-II, SQSTM1, and ubiquitin in LECs and the OFZ of tdrd7−/− mice lenses. The nuclei were stained with DAPI (blue signal). Scale bar: 10 µm. (G-I) Immunoblotting analysis of Tdrd7+/− and tdrd7−/− mice lenses for LC3 (G), SQSTM1 (H), and ubiquitin (I) confirmed the accumulation of LC3-II, SQSTM1, and ubiquitin in tdrd7−/− mice lenses
tdrd7-deficient mice testes exhibit defective acrosome biogenesis and disrupted autophagic flux
Previously, we established that spermatogenesis in tdrd7-deficient humans and mice was arrested at the spermiogenesis stage with a failure in the formation of acrosomes [3,7]. The acrosome is a specialized organelle that is formed by the fusion of Golgi-derived vesicles [35], considered to originate from an autolysosome rather than a mere lysosome [36]. To elucidate the nature of defective acrosome biogenesis in the spermiogenesis of tdrd7-deficient mice, we performed immunostaining using PNA (FITC-conjugated peanut agglutinin), which binds to the outer acrosomal membrane. In the Golgi phase during earlier spermiogenesis, the acrosome displays a single vesicle close to the nucleus in control cells (Fig. S6A); however, several acrosomal vesicle-like structures were observed in the Golgi-phase spermatids of tdrd7-deficient mice (Fig. S6B), indicating that disruption of TDRD7 could lead to acrosome biogenesis as early as in the Golgi phase during spermiogenesis. In the cap and acrosome phase during later spermiogenesis, the acrosome grows to form a cap-like structure and then expands to form a moon-shaped structure covering the nucleus of the round spermatids (Fig. S6A). However, in tdrd7-deficient mice spermatids, the cap-like acrosome failed to form, resulting in the formation of a discontinuous acrosomal-like structure adjacent to the nucleus (Fig. S6B). These observations reveal that tdrd7 plays a crucial role in acrosome biogenesis.
To determine whether the defect of the acrosome in tdrd7-deficient mice is related to autophagy, we performed TEM analysis to evaluate the autophagosomes in WT and tdrd7-deficient seminiferous tubules and observed significantly accumulated autophagic vacuoles in the cytoplasm of tdrd7-deficient seminiferous tubules (Figures 7A and 7B), consistent with the result in tdrd7-deficient MEFs (Figures 1B and 1C). Given that tdrd7-deficient MEFs presented disrupted autophagic flux due to failed fusion of autophagosomes with lysosomes, we proceeded to evaluate the autophagic flux and cargo in tdrd7-deficient seminiferous tubules by immunostaining of LC3, LAMP1, SQSTM1, and ubiquitin. A crescent-shaped staining of LC3, LAMP1, SQSTM1, and ubiquitin, indicative of acrosomes, was detected in the WT seminiferous tubules (Figure 7C, 7D, 7E and 7F). In contrast, immunostaining of tdrd7-deficient seminiferous tubules revealed diffuse and disorganized LC3-positive and LAMP1-positive staining (Figures 7C and 7D) as well as accumulation of SQSTM1 and ubiquitin (Figures 7E and 7F). Furthermore, we examined the expression of LC3, SQSTM1, and ubiquitin and confirmed that the protein levels of LC3, SQSTM1, and ubiquitin protein aggregates in tdrd7-deficient seminiferous tubules (Figure 7G, 7H and 7I). Collectively, these findings suggest that TDRD7-mediated autophagosome maturation is required for the maintenance of autophagic flux and the formation of acrosomes.
Figure 7.
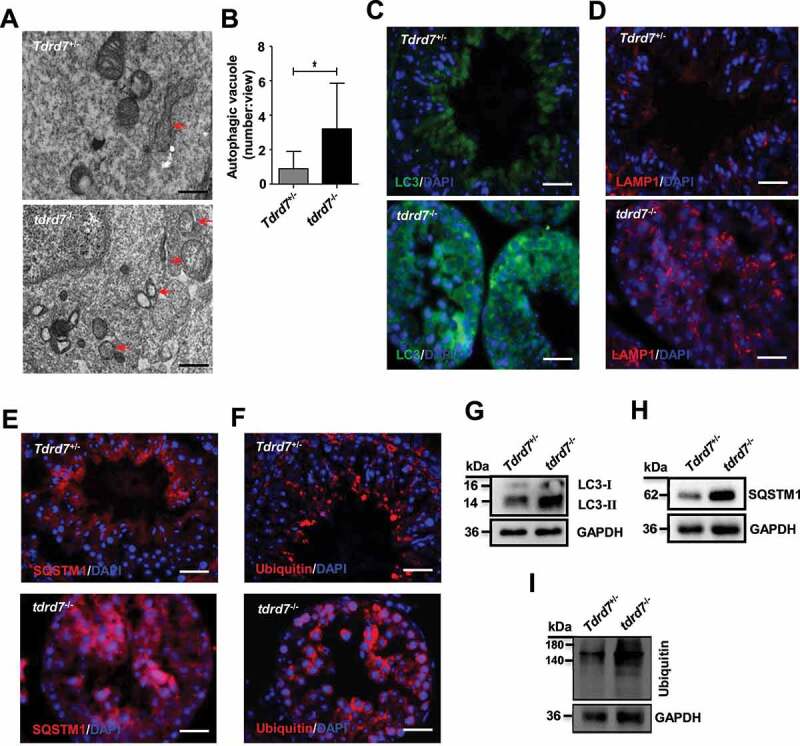
Evaluation of Tdrd7+/− and tdrd7−/− seminiferous tubules. (A) Detection of Tdrd7+/− and tdrd7−/− mice testes using TEM. The red arrows indicate the autophagic vacuoles. (B) Quantification of the average number of autophagic vacuoles in Tdrd7+/− and tdrd7−/− mice testes using TEM analysis. *P < 0.05. (C-F) Immunostaining of Tdrd7+/− and tdrd7−/− seminiferous tubules for LC3 (C), LAMP1 (D), SQSTM1 (E), and ubiquitin (F) (red signal). The nuclei were stained with DAPI (blue signal). Scale bar: 50 μm. (G-I) Immunoblotting analysis of Tdrd7+/− and tdrd7−/− testicular tissue for LC3 (G), SQSTM1 (H), and ubiquitin (I) confirmed the accumulation of LC3-II, SQSTM1, and ubiquitin protein in tdrd7−/− seminiferous tubules
TDRD7 deletion leads to an increase in autophagosomes in patients with TDRD7 mutations
Our previous study found that TDRD7 mutations can lead to a novel syndrome combining CC and NOA in humans [3]. In the present study, we recruited another patient (31 years old) with CC and NOA from a consanguineous family (Figure 8A, Table S5). We speculated that the genetic cause of this patient’s CC and NOA was probably due to a TDRD7 mutation. After a series of genetic analyses, including karyotype analysis, Y chromosome microdeletion detection, whole exome sequencing (WES), bioinformatic and co-separation analysis, we identified another novel rare homozygous frameshift variant (NM_014290, c.3000dupC, p.Asn1001Glnfs*34) in TDRD7, which was likely to be the disease-causing gene (Table S6). These results strengthen the conclusion that TDRD7 mutations lead to a novel syndrome combining CC and NOA in humans.
Figure 8.
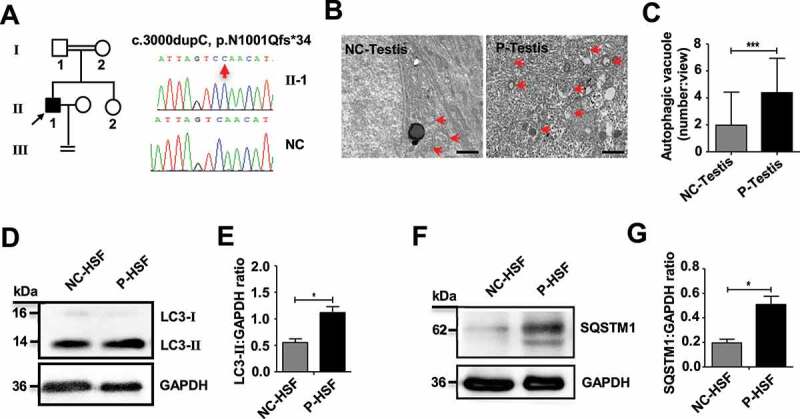
An increase in autophagosomes was found in a patient with a TDRD7 mutation. (A) A homozygous frameshift mutation (c.3000dupC) of TDRD7 in a patient from a consanguineous family. The proband is indicated by the black arrow. The image on the right shows the sequence chromatograms for the proband and normal control (NC). The red arrow indicates the mutation site. (B) TEM detection of the seminiferous tubules in the patient with the TDRD7 mutation (c.324_325 insA) and NC. The red arrows indicate the autophagic vacuoles in the seminiferous tubules of the patient (P-testis) and NC. Scale bar: 2 μm. (C) Quantification of the average number of autophagic vacuoles in the seminiferous tubules of the patient and NC using TEM analysis. ***P < 0.001. (D-G) Western blotting detected the expression of LC3 (D) and SQSTM1 (F) in skin fibroblasts from the patient with a TDRD7 mutation (c.324_325 insA) and NC; GAPDH served as a loading control. Quantification analysis revealed significant accumulation of LC3-II (E) and SQSTM1 (G) (*P < 0.05). The data are representative of three independent assays
To determine whether the phenotype of patients with TDRD7 mutations is related to autophagy, TEM was performed to detect autophagic vacuoles in seminiferous tubules derived from a patient with TDRD7 mutation. Compared with the seminiferous tubules of a normal healthy individual, significantly accumulated autophagic vacuoles were observed in the cytoplasm of the patient’s seminiferous tubules (Figures 8B and 8C). Furthermore, we examined the expression of endogenous LC3 and SQSTM1 protein aggregates in the patient’s skin fibroblasts and observed a significantly increased protein level of LC3-II and SQSTM1 compared with that in skin fibroblasts from a normal healthy individual (Figure 8D, 8E, 8F and 8G), indicating disrupted autophagic flux in skin fibroblasts from the patient with TDRD7 mutation.
Discussion
TDRD7 belongs to a large family of TDRD proteins and contains multiple LOTUS/OST-HTH and tudor domains [6,7,37]. Most of the genes in this family, such as Tdrd1, Tdrd3, Tdrd5, Tdrd6, Tdrd7, and Tdrd12, have a crucial function at the different differentiation stages of spermatogenesis in mice, constituting an essential class of gametogenesis genes [38]. Interestingly, among these genes, only TDRD7 deficiency causes two clinically distinct diseases, namely CCs and NOA, due to defects in lens development and spermiogenesis [3]. Indeed, TDRD7 has been shown to be a component of ribonucleoprotein complexes and RNA granules, and plays an important role in the differentiation of lens and sperm cells [6,7,39]. However, it remains unknown whether TDRD7 has similar or distinct functions in the lens and spermiogenesis. Recent studies demonstrated that TDRD7 exerts antiviral activity by inhibiting the cellular autophagy pathway, which is required for paramyxovirus replication, indicating that TDRD7 is a mediator of autophagy [12,13]. Autophagy is a major intracellular degradation system in which aggregated cytoplasmic proteins and damaged organelles are degraded in the lysosome, which may be particularly important in terminally differentiated cells [25]. However, it is unknown whether TDRD7 is involved in lens development and spermatogenesis through the autophagy pathway. In this study, we found that once TDRD7 is deleted, it downregulates the expression of TBC1D20, disrupts autophagosome maturation (Figure 9), finally leads to the defects of lens fiber cell homeostasis and acrosome biogenesis.
Figure 9.
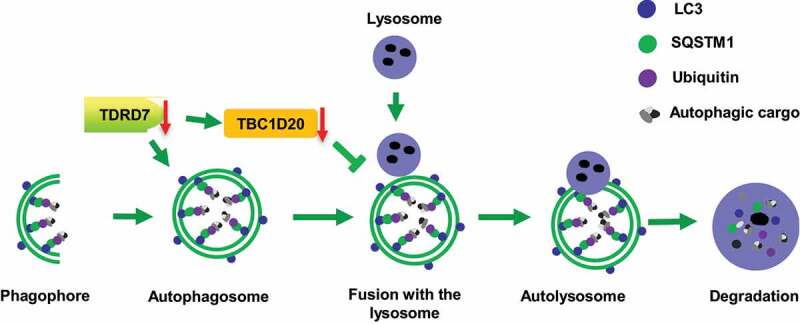
A proposed model for TDRD7 function during autophagy. TDRD7 mediates the maturation of autophagosomes by downregulating the expression of TBC1D20, a process required for the fusion of autophagosomes with lysosomes to form autolysosomes. tdrd7 deletion suppressed the autophagic flux by inhibiting the fusion of autophagosomes with lysosomes and accumulation of ubiquitinated autophagic cargo
The lens of the eye is composed of lens fiber cells that differentiate from lens epithelial cells in which all cytoplasmic organelles are degraded during terminal differentiation to ensure the transparency of the lens [40,41]. Cataracts are defined as a loss of lens transparency with accumulated insoluble opaque protein aggregates [42,43]. To date, multiple potential mechanisms have been reported to result in lens organelle degradation, including 12/15-lipoxygenase [44], the ubiquitin-proteasome system [45], and CDK1-mediated nuclear disassembly [46]. Another potential candidate pathway is autophagy, and previous studies showed that impairment of autophagy is associated with cataracts in humans [47,48]. For example, disrupted autophagy or autophagic flux, as a result of mutations in the key autophagic regulators FYCO1, EPG5 and TBC1D20, causes Mendelian forms of CCs, and CCs in individuals with Vici syndrome and WARBM4, respectively [20,21,24]. Consistently, our results show that TDRD7-mediated autophagic flux maintains lens transparency by facilitating the removal of ubiquitinated autophagic cargo from lens fiber cells. We speculate that lens fiber cells, as terminally differentiated cells, cannot dilute the concentration of damaged proteins and organelles marked for degradation through cell division, and thus require the TDRD7-mediated autophagic flux for continuous removal of autophagic cargo. Therefore, we propose that the disruption of TDRD7-mediated autophagic flux leads to accumulation of damaged proteins, resulting in a loss of lens transparency.
Of note, the role of autophagy machinery in the elimination of organelles during lens OFZ formation has long been debated. For example, selective deletion of autophagy regulators, including Atg5 and Pik3c3, in the lens cause retarded lens growth and cataract formation [49]. These data support the conclusion that autophagy genes are critical for intracellular quality control and lens development, but are not required for programmed degradation of lens organelles. However, recent advances in research provided conflicting evidence regarding the role of autophagy machinery in lens organelle degradation [16,50,51]. For example, inactivation of MAPK/JNK in the ocular lens induces upregulation of autophagy-related proteins by suppressing the MTOR-RPTOR signaling axis, facilitating the premature removal of organelles from the central lens fiber cells to form the OFZ in the lens [51]. Another study detected increased expression of multiple key autophagy genes in lens fiber cells undergoing organelle-elimination [52], and mitochondria contained within autolysosomes were also detected in lens fiber cells during organelle elimination [53], supporting that autophagy and mitophagy may participate in lens fiber cell organelle degradation [54]. However, our new results together with a previous study showed that the degradation of nuclear DNA occurs normally in the lens of tdrd7-deficient mice at embryonic periods and young ages (Fig. S7) [39], indicating that TDRD7 is not primarily required for the degradation of nuclear DNA at young ages. The presence of some condensed nuclei in disorganized lenses of tdrd7-deficient mice at older ages (Figures 6A and 6B) might be secondary to other defects in lens functions [39]. Considering that TDRD7 regulates the expression of several genes important for lens development, further studies are needed to clarify whether the cataracts observed in patients with these conditions are indeed caused by a defect in autophagy or any other function of TDRD7.
The acrosome has been regarded as a modified lysosome or a lysosome-related organelle (LRO) [55], receiving diverse protein cargos from more non-redundant pathways that contribute to acrosome biogenesis [56]. Recent studies have shown that autophagic machinery also participates in acrosome biogenesis by regulating vesicular trafficking. For example, germ cell-specific atg7-knockout mice produce irregular or nearly round-headed spermatozoa due to a malformed acrosome, and autophagy was found to mediate the proacrosomal vesicle transport or fusion in the acrosome [36]. Thus, the acrosome is hypothesized to originate from an autolysosome rather than a mere lysosome [36]. In support of this hypothesis, disrupted autophagic flux caused by SIRT1 ablation in germ cells abrogates acrosome formation through SIRT1-mediated LC3 nucleocytoplasmic transportation [22]. TBC1D20 has also been found to be involved in acrosome biogenesis and regulates autophagic flux [24]. In tdrd7-deficient germ cells, proacrosomal vesicles form and are transported to the perinuclear region, but fail to mature into acrosomes, suggesting that TDRD7 functional loss disrupts the proacrosomal vesicle formation and transport during acrosome biogenesis. Our results show that autophagosome and lysosome formation failed to organize into crescent-shaped acrosome structures in tdrd7-deficient male germ cells. Considering that the function of TDRD7 in MEFs is required for the formation of autolysosomes and that TDRD7 defects reduce the expression of TBC1D20, these findings suggest that disrupted acrosome biogenesis in tdrd7-deficient germ cells occurs because of a failure of autolysosome formation. Therefore, TDRD7-mediated maturation of autophagy may be required for acrosome formation.
RNA granules (RGs) are ribonucleoprotein complexes found in the cytoplasm of eukaryotic cells from yeast to vertebrates that are implicated in the regulation of RNA stability, degradation, and subcellular localization. Germinal RNA granules, also called chromatoid bodies, mediate the post-transcriptional regulation of gene expression in male germ cell differentiation. RGs components are likely to function in the posttranscriptional regulation of gene expression, but the extent of RGs involvement in organogenesis is not well understood. TDRD7 was found in RGs [57] and was considered to control the production of large amounts of lens structural- or spermiogenesis-related genes [7,8,39]. Notably, due to chromatin clearance or compaction during lens fiber cell differentiation and spermatid elongation, the related mRNAs are transcribed earlier and stored in translationally inert messenger ribonucleoproteins (mRNPs) until needed for translation [58–61]. Thus, we hypothesized that TDRD7-containing RGs also use an RG-based mechanism to mediate the post-transcriptional control of gene expression and to acquire their unusual state of differentiation. In this study, we found that both lenses and testes from tdrd7 mutant mice had large numbers of differentially expressed genes, including dozens of metabolism genes, such as Tbc1d20, Itgav/Cd51, Hipk1, and Spata18, supporting the role of Tdrd7 in cellular RNA metabolism [6,7]. Among them, TBC1D20 is a key regulator of autophagosome maturation, resulting in the accumulation of ubiquitinated autophagic cargo marked for degradation [24]. Given that TDRD7 could directly bind to Tbc1d20 mRNAs and downregulating its expression, we proposed that TDRD7 is the upstream regulatory molecule of TBC1D20 that participates in autophagosome maturation.
In summary, we show that TDRD7 plays an essential role in autophagosome maturation and that tdrd7 deletion disrupts autophagic flux by inhibiting the fusion of autophagosomes with lysosomes by directly bind to Tbc1d20 mRNAs and downregulating its expression, which in turn results in the eye and testicular abnormalities in both humans and mice. Based on our findings, we propose that disrupted autophagy is likely to contribute to the pathogenesis of the syndrome combining CC and NOA.
Materials and Methods
MEFs, plasmids, and transfection
Genotyping and maintenance of the Tdrd7 allele have been described previously [3]. In all the experiments, unless otherwise noted, the eyes or testes were dissected from postnatal day 30 (P30) and P60 Tdrd7+/− (WT) and tdrd7−/− mice (KO), respectively. All animal experiments were approved by the Institutional Animal Care and Use Committee and the Animal Ethics Committee of Central South University.
MEFs were isolated from embryonic day 13 (E13) mouse embryos and maintained as previously described [62]. Skin fibroblasts were isolated from a patient with a TDRD7 mutation (c.324_325 insA) as described in our previous study [3]. Fully sequenced Tdrd7 and Tbc1d20 cDNA clones were purchased from the Genechem Company (Shanghai, China) and fused to CMV-MCS-3FLAG-SV40-Neomycin and CMV-MCS-3 HA-SV40-Neomycin (Cloning Vector, pcDNA3.1), respectively. Small interfering RNAs (siRNAs) targeting Tbc1d20 and a negative control (siNC) were purchased from Ribobio Co., Ltd. (Guangzhou, China). Plasmids encoding FLAG/HA-tagged cDNA clones or siRNAs were transfected into cells using Lipofectamine 3000 (Thermo Fisher Scientific, L3000008) according to the manufacturer’s instructions.
Antibodies and reagents
The following primary antibodies were used: LC3 (Cell Signaling Technology, 3868) and LAMP1 (Cell Signaling Technology, 9091), TDRD7 (Abcam, ab241349), TBC1D20 (Santa Cruz Biotechnology, sc-515,697), ubiquitin (AB3237), SQSTM1 (Abways technology, CY5546; Novus, NBP1-48,320), RAB5 (Abways technology, CY5529), CTSD/cathepsin D (Abways technology, CY5339), EGFR (Abways technology, CY5112), TOMM20 (Proteintech, 11,802-1-AP), TUBA1A/α-tubulin (Sigma-Aldrich, T5168), GAPDH (Abcam, ab8245), FITC-labeled PNA (Invitrogen, L32458), FLAG (Abways technology, AB0008), and FLAG (Abways technology, AB0004), rapamycin (Sigma-Aldrich, 553,210), hydroxycloroquine (Sigma-Aldrich, 90,527), DMSO (Sigma-Aldrich, C6295).
Immunofluorescence
For immunofluorescence, WT and tdrd7-deficient MEFs were grown on coverslips until reaching 50%–70% confluency, fixed with 4% paraformaldehyde (PFA) for 15 min, permeabilized using 0.5% Triton X-100 (Sigma-Aldrich, T8787) for 10 min, and blocked in 5% bovine serum albumin (BSA; Sigma-Aldrich, V900933) for 1 h. Next, the cells were incubated with the primary antibodies overnight at 4°C, followed by incubation with Alexa Fluor 488- or 555-conjugated secondary antibodies (1:400; Invitrogen, A21121 and A31572) for 1 h at 37°C. Finally, the cells were stained with 2-(4-amidinophenyl)-1 H-indole-6-carboxamidine (DAPI) for 5 min. Images of the stained cells were obtained using a Leica SP5 II scanning confocal microscope (Leica, Bannockburn, USA). For labeling acidic organelles, WT and tdrd7-deficient MEFs were incubated with 75 nM LysoTracker Red (Beyotime Biotechnology, C1046) in medium for 45 min prior to fixation. Comparison of the levels between WT and tdrd7-deficient cells was performed with at least 20 different cells from each genotype using ImageJ. All experiments were performed in three independent replicates.
Transmission electron microscopy
The lens or seminiferous tubules from a normal individual, a patient with a TDRD7 mutation (c.324_325 insA), WT, and KO mice were treated as described previously [3]. Briefly, biopsy tissues were fixed with glutaraldehyde (Sigma-Aldrich, G5882) and osmium tetroxide, post-fixed with OsO4 and sucrose, and then dehydrated with graded concentrations of ethanol. Subsequently, the samples were embedded in embed-812 (Electron Microscopy Sciences, 14,120), dodecenylsuccinic anhydride, methylnadic anhydride, and dimethylaminomethyl phenol. The 80-nm-thick sections were contrasted with uranyl acetate and lead citrate. An HT7700 Hitachi electron microscope (Hitachi, Tokyo, Japan) and a MegaView III digital camera (Munster, Germany) were used to capture digital images. According to the guidelines [27,63], autophagosomes (also referred to as initial autophagic vacuoles) typically have a double membrane, or a membrane at least partly visible as two parallel membrane bilayers separated by an electron-lucent cleft. At least 20 different fields from each genotype were counted.
RNA sequencing and qRT-PCR
Total RNA was extracted from the eyes and testes of WT and KO mice (litter mate, n = 3) using a TRIzol RNA extraction kit (Invitrogen, 15,596,018), and 1 µg of total RNA was reverse-transcribed using a cDNA reverse-transcription kit (Promega, A5003). cDNA library construction and sequencing were performed by the Beijing Genomics Institute (BGI, Beijing, China) using the BGISEQ-500 platform [64]. High-quality reads were aligned to the Mus musculus reference genome (GRCm38.p5) using Bowtie2 [65]. The expression level of each gene was normalized to the fragments per kilobase of transcript per million mapped reads (FPKM) using RNA-seq by expectation maximization. qRT-PCR was performed using LightCycler 480 SYBR Green I Master (Roche, 4,887,352,001) and a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, USA) according to the manufacturer’s instructions. The relative mRNA expression levels were determined after normalization to the endogenous expression of Gapdh. The primers used are listed in Table S7.
Cell culture and transfection
MEFs were cultivated at 37°C with 5% CO2 in a 6-well plate supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, 30,044,184). Cells were transiently transfected with plasmids encoding FLAG- or HA-tagged cDNA constructs using the Lipofectamine 3000 reagent (Thermo Fisher Scientific, L3000008) and then grown until they reached 70%–80% confluence. The cells were then harvested at 48 h and homogenized using NP40 Lysis Buffer (Beyotime Biotechnology, P0013F) supplemented with a protease inhibitor cocktail (Thermo Fisher Scientific, 78,429). The lysates were centrifuged (12,000 × g, 10 min) at 4°C and used for immunoblot analysis.
Western blotting and EGFR degradation assay
Stimulation and inhibition of autophagy were performed in WT and KO MEFs following a 3 h treatment with: 1) DMSO (control), 2) 50 μM hydroxycloroquine sulfate (Sigma-Aldrich, 90,527), 3) 50 nM rapamycin (Sigma-Aldrich, 553,210), and 4) a combination of 50 μM hydroxychloroquine sulfate and 50 nM rapamycin. The cells were harvested after 48 h treatment completion, and proteins were extracted in three decontamination lysis steps. The protein samples were homogenized and blotted onto a polyvinylidene difluoride membrane, and incubated overnight at 4°C using antibodies against: LC3, SQSTM1, ubiquitin, TDRD7, TBC1D20, TOMM20, FLAG, HA, CTSD, EGFR, and GAPDH. Then, the membranes were incubated with the secondary antibodies, and the blots were evaluated using an ECL western blotting kit (Thermo Fisher Scientific, 32,106). The intensity of the protein fragments was quantified using the Image Lab software (Bio-Rad, Hercules, USA). EGFR degradation assay was performed as described previously [66]. Briefly, WT and tdrd7-deficient MEFs were cultured in serum-free DMEM for 3 h, followed by incubation with serum-free DMEM containing 40 ng/mL EGF (R&D Systems, 236-EG-200) and 20 µg/mL cycloheximide (Sigma-Aldrich, C1988) for 0, 30, 60 and 120 min. After each time point, cells were lysed and protein lysates were subjected to western blotting. The experimental data for WT and tdrd7-deficient MEFs were compared using ImageJ. All experiments were performed in three independent replicates.
RIP assay
RIP assay using FLAG-labeled TDRD7 antibody followed by RNA isolation was performed using EZ-Magna RIP kit (Merck Millipore, 17–701) according to the manufacturer’s instructions. Mouse IgG antibody supplied by the manufacturer was used as the control and the immunoprecipitated RNA was subjected to RT-PCR for analyzing the expression of Tbc1d20 using the following primers: mTBC1D20-F 5ʹ-CATGCAGAGTGCTGAGGTAGG-3ʹ and mTBC1D20-R 5ʹ-ATAGGGCAAGTCCTGAGGGA-3ʹ.
Co-immunoprecipitation assay
For the detection of TDRD7 and TBC1D20 interactions, WT MEF cells co-transfected with HA-tagged Tbc1d20 and FLAG-tagged Tdrd7 vectors were prepared and lysed with NP-40 Lysis Buffer (Beyotime Biotechnology, P0013F) containing a protease inhibitor cocktail (Beyotime Biotechnology, P1005). The cell lysates were incubated with antibodies for 12 h at 4°C and then with Protein G beads (Thermo Fisher Scientific, 20,399) for 4 h at 4°C. After washing the beads and boiling 50 mL of NP-40 Lysis buffer, the lysates were subjected to western blotting.
mRFP-GFP-LC3 transfection
Cells were cultured on slides, transiently transfected with mRFP-GFP-LC3 adenovirus (Hanbio Inc, HB-AP20102304) and treated as indicated. After washing with phosphate-buffered saline (PBS), the cells were fixed with 4% paraformaldehyde, stained with DAPI, and viewed under a Leica SP5 II scanning confocal microscope (Leica, Bannockburn, USA). The number of mRFP and GFP dots was determined by manual counting of fluorescent puncta in at least 20 cells from WT and tdrd7-deficient MEFs.
FISH-immunofluorescence assay
WT MEFs co-transfected with HA-tagged Tbc1d20 and FLAG-tagged Tdrd7 vectors were grown on coverslips, fixed with 4% paraformaldehyde for 15 min, permeabilized using 0.5% Triton X-100 for 10 min, and blocked in pre-hybridization buffer (RiboBio, C10910) for 30 min in 37°C. Next, RiboTM m-TBC1D20 FISH-probe mix (RiboBio, Inc1101879) was added and allowed to hybridize overnight at 37°C, and the cells were washed in different concentrations of saline sodium citrate and 1× PBS in a dark environment. Finally, the cells were blocked in 5% BSA for 30 min, incubated with the primary antibodies overnight at 4°C, followed by incubation with Alexa Fluor 488- or 555-conjugated secondary antibodies (1:400; Invitrogen, A21121 and A31572) for 1 h at 37°C. After staining with DAPI, the FISH and fluorescence signals were observed using an Olympus BX-51 fluorescence microscope (Olympus, Tokyo, Japan). Images were captured using the VideoTesT-FISH 2.0 software (version number 5.0.74.4803, VideoTesT, Petersburg, Russia).
Histology and immunohistochemistry
Tissues from WT and KO mice were fixed in formalin-embedded paraffin, sectioned, processed, and stained with hematoxylin and eosin (H&E) as described previously [3]. For immunohistochemistry, 4-μm sections of tissue were blocked in goat serum for 30 min and incubated with primary antibodies overnight at 4°C. Alexa Fluor 488- or 555-conjugated secondary antibodies (1:400, Invitrogen, A21121 and A31572) were added, and the sections were incubated for 1 h at 37°C, followed by DAPI staining for 5 min. Images of the stained cells or tissue sections were obtained using an Olympus IX51 fluorescence microscope (Olympus, Tokyo, Japan) or a Leica SP5 II scanning confocal microscope (Leica, Bannockburn, USA).
Genetic analysis of a patient with CC and primary male infertility
The proband (31 years old, F1: II-2) of a consanguineous Han Chinese family of first-degree cousins was recruited to identify the genetic causal factor of his infertility at the Reproductive and Genetic Hospital of CITIC Xiangya (Changsha, Hunan, China). The proband was diagnosed with bilateral CC after birth, but the ophthalmic medical records were lost. Three independent routine analyses of semen samples obtained from the proband revealed complete azoospermia with normal volume. The proband’s wife, with normal fertility, failed to conceive after more than five years of marriage, despite unprotected sexual intercourse with ejaculation. Other family members, including parents (F1: I-1 and F1: I-2) and the sister (F1: II-2) did not have any known abnormalities. This study was approved by the institutional ethics committees of the Reproductive and Genetic Hospital of CITIC Xiangya and Central South University. Written informed consent was obtained from all the participants.
Genomic DNA extraction and subsequent WES and data analysis of a DNA sample obtained from the proband was carried out by the BGI at Shenzhen as described previously [3]. The most promising candidate variants were identified using previously described criteria [3]. Briefly, candidate variants were defined as those with a frequency below 5% in three public databases, predicted to be deleterious, homozygous, and relevant to cataracts and/or infertility phenotypes. The PCR primers used were designed to specifically amplify the TDRD7 region containing the variant. The primer sequences were the following: TDRD7-F: 5′-CTTGCCCTTAAAGCAGGTACTGA-3′, TDRD7-R: 5′- ACATACCTAAACTCAAGAGGCCAA-3′. The PCR products were sequenced using a 3730XL sequencer (Applied Biosystems, Foster City, USA) according to the manufacturer’s instructions.
Supplementary Material
Acknowledgments
We thank Xiaoxuan Yang from the Central South University for her experimental assistance including western blotting and EGFR degradation assay. We also thank Xing Wang from Chinese Academy of Sciences for kindly providing scholarly discussions.
Funding Statement
This work was supported by the national key research & developmental program of China (2018YFC1004900), the national natural science foundation of China (81771645 and 81971447), the key grant of prevention and treatment of birth defect from Hunan province (2019SK1012), the research grant of CITIC-Xiangya (YNXM-201915, YNXM-201913, YNXM-201912, YNXM-201916), the China Postdoctoral Science Foundation Funded Project (2019M662786), and the Hunan Provincial Natural Science Foundation of China (2020JJ5993).
Statistical analysis
The statistical significance of the results was calculated using a Student’s t-test and one-way ANOVA using the SPSS software, version 19.0 (SPSS, Chicago, USA). All the experiments were repeated three times, and a P-value of < 0.05 was considered statistically significant.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Sheeladevi S, Lawrenson JG, Fielder AR, et al. Global prevalence of childhood cataract: a systematic review. Eye (Lond). 2016;30(9):1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pichi F, Lembo A, Serafino M, et al. Genetics of Congenital Cataract. Dev Ophthalmol. 2016;57:1–14. [DOI] [PubMed] [Google Scholar]
- [3].Tan YQ, Tu C, Meng L, et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet Med. 2019;21(5):1209–1217. [DOI] [PubMed] [Google Scholar]
- [4].Cervan-Martin M, Castilla JA, Palomino-Morales RJ, et al. Genetic Landscape of Nonobstructive Azoospermia and New Perspectives for the Clinic. J Clin Med. 2020;9(2):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wosnitzer M, Goldstein M, Hardy MP.. Review of Azoospermia. Spermatogenesis 2014;4(1):e28218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lachke SA, Alkuraya FS, Kneeland SC, et al. Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science 2011;331(6024):1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tanaka T, Hosokawa M, Vagin VV, et al. Tudor domain containing 7 (Tdrd7) is essential for dynamic ribonucleoprotein (RNP) remodeling of chromatoid bodies during spermatogenesis. Proc Natl Acad Sci USA. 2011;108(26):10579–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hosokawa M, Shoji M, Kitamura K, et al. Tudor-related proteins TDRD1/MTR-1, TDRD6 and TDRD7/TRAP: domain composition, intracellular localization, and function in male germ cells in mice. Dev Biol. 2007;301(1):38–52. [DOI] [PubMed] [Google Scholar]
- [9].Callebaut I, Mornon JP.. LOTUS, a new domain associated with small RNA pathways in the germline. Bioinformatics 2010;26(9):1140–1144. [DOI] [PubMed] [Google Scholar]
- [10].Cui G, Botuyan MV, Mer G. (1)H, (15)N and (13)C resonance assignments for the three LOTUS RNA binding domains of Tudor domain-containing protein TDRD7. Biomol NMR Assign. 2013;7(1):79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen C, Nott TJ, Jin J, et al. Deciphering arginine methylation: tudor tells the tale. Nat Rev Mol Cell Biol. 2011;12(10):629–642. [DOI] [PubMed] [Google Scholar]
- [12].Subramanian G, Kuzmanovic T, Zhang Y, et al. A new mechanism of interferon’s antiviral action: induction of autophagy, essential for paramyxovirus replication, is inhibited by the interferon stimulated gene, TDRD7. PLoS Pathog. 2018;14(1):e1006877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Subramanian G, Popli S, Chakravarty S, et al. The interferon-inducible protein TDRD7 inhibits AMP-activated protein kinase and thereby restricts autophagy-independent virus replication. J Biol Chem. 2020;295(20):6811–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8(1):105–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen J, Ma Z, Jiao X, et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet. 2011;88(6):827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yefimova MG, Buschiazzo A, Burel A, et al. Autophagy is increased in cryptorchid testis resulting in abnormal spermatozoa. Asian J Androl. 2019;21(6):570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shang Y, Wang H, Jia P, et al. Autophagy regulates spermatid differentiation via degradation of PDLIM1. Autophagy 2016;12(9):1575–1592. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oz-Levi D, Ben-Zeev B, Ruzzo EK, et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet. 2012;91(6):1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45(1):83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Da Ros M, Lehtiniemi T, Olotu O, et al. FYCO1 and autophagy control the integrity of the haploid male germ cell-specific RNP granules. Autophagy 2017;13(2):302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu C, Song Z, Wang L, et al. Sirt1 regulates acrosome biogenesis by modulating autophagic flux during spermiogenesis in mice. Development 2017;144(3):441–451. [DOI] [PubMed] [Google Scholar]
- [23].Liegel RP, Handley MT, Ronchetti A, et al. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am J Hum Genet. 2013;93(6):1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sidjanin DJ, Park AK, Ronchetti A, et al. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 2016;12(10):1759–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008;4(2):151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012;8:445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bjorkoy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. [DOI] [PubMed] [Google Scholar]
- [29].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007;3(5):452–460. [DOI] [PubMed] [Google Scholar]
- [30].Sorkin A, Duex JE. Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr Protoc Cell Biol. 2010; Chapter 15:Unit. ; . 15 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009;458(7242):1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53(2):167–178. [DOI] [PubMed] [Google Scholar]
- [33].Deosaran E, Larsen KB, Hua R, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126(4):939–952. [DOI] [PubMed] [Google Scholar]
- [34].Kim PK, Hailey DW, Mullen RT, et al. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA. 2008;105(52):20567–20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Berruti G, Paiardi C. Acrosome biogenesis: revisiting old questions to yield new insights. Spermatogenesis 2011;1(2):95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang H, Wan H, Li X, et al. Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell Res. 2014;24(7):852–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pek JW, Anand A, Kai T. Tudor domain proteins in development. Development 2012;139(13):2255–2266. [DOI] [PubMed] [Google Scholar]
- [38].Gan B, Chen S, Liu H, et al. Structure and function of eTudor domain containing TDRD proteins. Crit Rev Biochem Mol Biol. 2019;54(2):119–132. [DOI] [PubMed] [Google Scholar]
- [39].Barnum CE, Al Saai S, Patel SD, et al. The Tudor-domain protein TDRD7, mutated in congenital cataract, controls the heat shock protein HSPB1 (HSP27) and lens fiber cell morphology. Hum Mol Genet. 2020;29(12):2076–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res. 2009;88(2):133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wride MA. Lens fibre cell differentiation and organelle loss: many paths lead to clarity. Philos Trans R Soc Lond B Biol Sci. 2011;366(1568):1219–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moreau KL, King JA. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med. 2012;18(5):273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Boscia F, Grattagliano I, Vendemiale G, et al. Protein oxidation and lens opacity in humans. Invest Ophthalmol Vis Sci. 2000;41(9):2461–2465. [PubMed] [Google Scholar]
- [44].Van Leyen K, Duvoisin RM, Engelhardt H, et al. A function for lipoxygenase in programmed organelle degradation. Nature 1998;395(6700):392–395. [DOI] [PubMed] [Google Scholar]
- [45].Imai F, Yoshizawa A, Fujimori-Tonou N, et al. The ubiquitin proteasome system is required for cell proliferation of the lens epithelium and for differentiation of lens fiber cells in zebrafish. Development 2010;137(19):3257–3268. [DOI] [PubMed] [Google Scholar]
- [46].Chaffee BR, Shang F, Chang ML, et al. Nuclear removal during terminal lens fiber cell differentiation requires CDK1 activity: appropriating mitosis-related nuclear disassembly. Development 2014;141(17):3388–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Morishita H, Mizushima N. Autophagy in the lens. Exp Eye Res. 2016;144:22–28. [DOI] [PubMed] [Google Scholar]
- [48].McWilliams TG, Prescott AR, Villarejo-Zori B, et al. A comparative map of macroautophagy and mitophagy in the vertebrate eye. Autophagy 2019;15(7):1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Morishita H, Eguchi S, Kimura H, et al. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem. 2013;288(16):11436–11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brennan LA, McGreal-Estrada R, Logan CM, et al. BNIP3L/NIX is required for elimination of mitochondria, endoplasmic reticulum and Golgi apparatus during eye lens organelle-free zone formation. Exp Eye Res. 2018;174:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Basu S, Rajakaruna S, Reyes B, et al. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy 2014;10(7):1193–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brennan LA, Kantorow WL, Chauss D, et al. Spatial expression patterns of autophagy genes in the eye lens and induction of autophagy in lens cells. Mol Vis. 2012;18:1773–1786. [PMC free article] [PubMed] [Google Scholar]
- [53].Chauss D, Basu S, Rajakaruna S, et al. Differentiation state-specific mitochondrial dynamic regulatory networks are revealed by global transcriptional analysis of the developing chicken lens. G3: Genes|Genomes|Genetics. 2014;4(8):1515–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Costello MJ, Brennan LA, Basu S, et al. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res. 2013;116:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Moreno RD, Alvarado CP. The mammalian acrosome as a secretory lysosome: new and old evidence. Mol Reprod Dev. 2006;73(11):1430–1434. [DOI] [PubMed] [Google Scholar]
- [56].Khawar MB, Gao H, Li W. Mechanism of Acrosome Biogenesis in Mammals. Front Cell Dev Biol. 2019;7:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10(6):430–436. [DOI] [PubMed] [Google Scholar]
- [58].Wang X, Garcia CM, Shui YB, et al. Expression and regulation of alpha-, beta-, and gamma-crystallins in mammalian lens epithelial cells. Invest Ophthalmol Vis Sci. 2004;45(10):3608–3619. [DOI] [PubMed] [Google Scholar]
- [59].Vourekas A, Zheng Q, Alexiou P, et al. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat Struct Mol Biol. 2012;19(8):773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lee TL, Pang AL, Rennert OM, et al. Genomic landscape of developing male germ cells. Birth Defects Res C Embryo Today. 2009;87(1):43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dai P, Wang X, Gou LT, et al. A Translation-Activating Function of MIWI/piRNA during Mouse Spermiogenesis. Cell 2019;179(7):1566–1581 e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hassemer EL, Le Gall SM, Liegel R, et al. The waved with open eyelids (woe) Locus Is a Hypomorphic Mouse Mutation in Adam17. Genetics 2010;185(1):245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mak SST, Gopalakrishnan S, Caroe C, et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 2017;6(8):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Langdon WB. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015;8(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sarkar S, Carroll B, Buganim Y, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5(5):1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.