

ABSTRACT
Human carbonic anhydrase (hCAIX), an extracellular enzyme that catalyzes the reversible hydration of CO2, is often overexpressed in solid tumors. This enzyme is instrumental in maintaining the survival of cancer cells in a hypoxic and acidic tumor microenvironment. Absent in most normal tissues, hCAIX is a promising therapeutic target for detection and treatment of solid tumors. Screening of a library of anti-hCAIX monoclonal antibodies (mAbs) previously identified three therapeutic candidates (mAb c2C7, m4A2 and m9B6) with distinct biophysical and functional characteristics. Selective binding to the catalytic domain was confirmed by yeast surface display and isothermal calorimetry, and deeper insight into the dynamic binding profiles of these mAbs upon binding were highlighted by bottom-up hydrogen-deuterium exchange mass spectrometry (HDX-MS). Here, a conformational and allosterically silent epitope was identified for the antibody-drug conjugate candidate c2C7. Unique binding profiles are described for both inhibitory antibodies, m4A2 and m9B6. M4A2 reduces the ability of the enzyme to hydrate CO2 by steric gating at the entrance of the catalytic cavity. Conversely, m9B6 disrupts the secondary structure that is necessary for substrate binding and hydration. The synergy of these two inhibitory mechanisms is demonstrated in in vitro activity assays and HDX-MS. Finally, the ability of m4A2 to modulate extracellular pH and intracellular metabolism is reported. By highlighting three unique modes by which hCAIX can be targeted, this study demonstrates both the utility of HDX-MS as an important tool in the characterization of anti-cancer biotherapeutics, and the underlying value of CAIX as a therapeutic target.
KEYWORDS: Carbonic anhydrase, hydrogen-deuterium exchange, mass spectrometry, epitope mapping, anti-cancer target, antibody-drug conjugate, allostery, antibody, tumor microenvironment
Introduction
Carbonic anhydrases are integral to the regulation of intracellular pH and fluidics in both physiological and pathological disease states.1 In humans, this family of 15 ubiquitous metallo-enzymes, which exist either as cytosolic, membrane-bound or secreted isoforms, share diverse organ and tissue distribution profiles and catalytic efficiencies.2 All enzymes catalyze the hydration of carbon dioxide (CO2) to protons (H+) and bicarbonate (HCO3−) (Figure 1a) following a two-step mechanism.3 In particular, human carbonic anhydrase IX (hCAIX), one of the four extracellular and membrane-bound zinc-containing carbonic anhydrase isozymes, has long been recognized as a tumor-associated protein. hCAIX expression is, in most cases, triggered by hypoxia (primarily through the H1F-1 transcription factor),4 and the protein can be found in many types of solid tumors where it is correlated with poor prognosis and therapeutic outcome.5 hCAIX is key to the survival of tumor cells in the acidic hypoxic tumor microenvironment, playing a role in cell proliferation, cell adhesion, and other tumor promoting processes.6,7 With the exception of gastric, gall bladder, and intestinal cells, hCAIX is mostly absent in tissues under normoxic conditions.8,9 Taken together, hCAIX can be considered a promising therapeutic target for the detection and treatment of solid tumors.10,11
Figure 1.
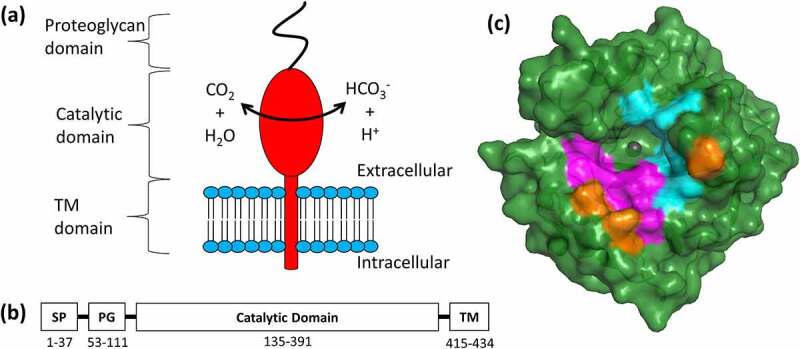
Key structural features of hCAIX. (a) Schematic of hCAIX (red) in the cellular membrane.79 (b) Localization of signal peptide (SP), proteoglycan (PG), catalytic, and transmembrane (TM) domains. (c) Structural representation of ΔPG-CAIXCys38Ser monomer (PDB 3IAI).12 Hydrophilic residues in the catalytic cavity are shown in magenta, while hydrophobic are shown in cyan. Three arginine residues (R58, R60, R130), unique to CAIX within the CA family, that border the active site are highlighted in Orange. View is looking down into the catalytic cavity from above the membrane
Full-length hCAIX exists as a disulfide stabilized membrane-bound dimer with an extracellular domain (ECD) that harbors a proteoglycan (PG) and catalytic (CA) domain (Figure 1a-b),12 both of which are integral to hCAIX’s enzymatic function. The PG domain, a unique structural feature among carbonic anhydrases only found in hCAIX, is an intrinsically disordered motif that has a role in cell adhesion,13 and that has been suggested to act as a “proton antenna” at the entrance of the active site of the catalytic domain.10 The active site itself is a 12 Å wide and 13 Å deep conical cavity bordered by a twisted β-sheet, with a nucleophilic, tetra-coordinated zinc ion located at the bottom.14 This catalytic pocket is characterized by several distinct hydrophobic (L91, V121, V131, L135, L141, V143, L198, P202) and hydrophilic (N62, H64, S65, Q67, T69, Q92) amino acids (Figure 1c), which have been suggested to facilitate efficient substrate turnover.15,16
Several small molecules that inhibit hCAIX’s enzyme activity by targeting the catalytic cleft have been extensively studied.17–19 These include agents that 1) directly bind the catalytically active zinc ion and its coordinated water/hydroxide ion, 2) sterically hinder entrance of the CO2 substrate into the catalytic cleft, or 3) less commonly engage sites distal from the hCAIX active site.20 In contrast to small molecule inhibitors that can lack specificity and may be more likely to cause off-target toxicity, antibody-based therapies generally show improved safety profiles and increased selectivity. Biotherapeutics have emerged as key players in the treatment of cancer.21 There has been growing interest into developing antibody-based therapeutics targeting hCAIX. Two of the most studied anti-hCAIX monoclonal antibodies (mAbs) are the chimeric cG25022, 22which, as an adjuvant treatment, has demonstrated clinical potential against renal cell carcinoma,23,24 and the M75 mouse mAb, which binds the PG domain and is widely used for immunohistochemistry and imaging purposes.25,26 In addition, A3 and CC7, two notable humanized hCAIX’s ECD binding antibodies derived from phage display, have been developed as non-immunogenic imaging tools,27 MSC8, which has demonstrated efficient in vitro enzyme inhibitory activity by modulating the intrinsic enzyme activity of hCA-IX,28 and VII/20 which has been shown to induce significant hCAIX internalization,29 a key characteristic for the development of an antibody-drug conjugate (ADC).30,31
In-depth biophysical characterization of antibody-antigen interactions, both in terms of the binding epitope and downstream effects, is an integral step in the development of biotherapeutics. Methods such as phage display,32 site-directed mutagenesis,33 and synthetic peptide arrays34 have proven invaluable for assigning linear epitopes. In addition, conformational epitopes and allosteric effects can be mapped with high-resolution techniques such as X-ray crystallography, nuclear magnetic resonance, or by using more recently developed techniques such as cryo-electron microscopy.35 However, sample size and protein complexity often limit the routine application of these methods.
Mass spectrometry has recently emerged as a powerful tool for the characterization of biotherapeutics and their interactions with antigens.36,37 In this regard, hydrogen-deuterium exchange mass spectrometry (HDX-MS) has proven its merit. This technology provides detailed insights into protein interactions38–41 and is well-suited for the assignment of both linear and conformational epitopes.42–47 HDX-MS examines conformational changes induced in the antigen upon antibody binding by monitoring the exchange rate of backbone amide-associated hydrogens with solvent deuterons.47–49 The exchange rate is largely determined by the degree of its involvement in secondary protein structure and/or the antigen-antibody binding interface.50 Therefore a comparison of amide hydrogen exchange rates between different protein states (i.e., antibody-bound vs. free), provides a powerful insight into the relative conformational stability of an antigen-antibody complex.51
In this study, we describe the bottom-up HDX-MS characterization of a subset of therapeutic anti-hCAIX antibody candidates, designated c2C7, m4A2 and m9B6, which target the catalytic domain of hCAIX.52 HDX-MS kinetics complemented by an in-depth biophysical profiling of the hCAIX-antibody interactions by surface plasmon resonance (SPR), isothermal calorimetry (ITC) and yeast surface display (YSD), and an in vitro functional assessment of enzymatic inhibition. Our efforts identified three unique modes by which the catalytic activity of hCAIX can be targeted with an antibody-based therapeutic. MAbs that target an epitope distal from the catalytic cleft can be used to develop an ADC (e.g., mAb c2C7), whereas functional inhibition of hCAIX can either be achieved by sterically hindering its substrate (i.e., CO2) from entering the catalytic cleft (e.g., m4A2), or by destabilizing key structural features that decreases the rate of substrate turnover (e.g., m9B6).
Results
Preliminary characterization of antibodies targeting the CAIX catalytic domain
A set of antibodies (c2C7, m4A2 and m9B6) that exclusively bind the hCAIX catalytic (CA) domain were selected from a larger panel of hCAIX binding antibodies for further characterization.52 An initial attempt was made to qualitatively identify the antibody epitopes on hCAIX ECD using YSD by expressing the eight hCAIX fragments as shown in Table 1. Fragments 1–3 consisted of only the proteoglycan (PG) domain, fragments 4 and 5 covered the CA domain with different N-terminal extensions, fragments 6 and 7 covered both the CA similar to fragments 4 and 5 with C-terminal extension just to the transmembrane (TM) domain, and fragment 8 included all domains except for the signal peptide (SP). Antibodies m9B6, m4A2 and c2C7 all demonstrated binding to only those fragments containing the CA domain as highlighted in Table 1, while no binding was observed to fragments containing only the PG domain. Next the structural nature of epitopes, linear versus conformational, was assayed by repeating the YSD assay using heat-denatured hCAIX antigen fragments (Table 1). Interestingly, only m9B6 was able to bind efficiently to the denatured hCAIX fragments, while m4A2 and c2C7 completely lost their binding to these fragments following denaturation. These results provide evidence that m9B6 binds to a contiguous, or linear, epitope, whereas m4A2 and c2C7 recognize conformational epitopes.
Table 1.
Epitope assignment at the domain level by yeast surface display in native and denatured hCAIX fragments
| |
Native |
Denatured |
||||||
|---|---|---|---|---|---|---|---|---|
| Fragment # | Domain | Amino Acid Residues | c2C7 | m4A2 | m9B6 | c2C7 | m4A2 | m9B6 |
| 1 | PG | 52–111 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | +PG+ | 38–136 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | SP+PG+ | 1–136 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | CA | 135–391 | 1.51 | 1.91 | 3.39 | 0 | 0 | 1.61 |
| 5 | +CA | 112–391 | 2.56 | 2.72 | 1.85 | 0 | 0 | 1.5 |
| 6 | CA+ | 135–414 | 2.51 | 1.75 | 2.88 | 0 | 0 | 1.88 |
| 7 | +CA+ | 112–414 | 2.93 | 3.01 | 3.09 | 0 | 0 | 2.05 |
| 8 | +PG+CA+ | 38–414 | 0.38 | 0.43 | 3.34 | 0 | 0 | 2.59 |
In addition, antibody binding was also evaluated by isothermal calorimetry using both the monomeric and dimeric forms of hCAIX. Binding constants ranging from 0.76 to 35 nM (Figure 2a-d) and 0.85 to 96.5 nM (Figure S1), respectively. These calculated binding constants are in close agreement with those previously determined by SPR, for all but m9B6 (KD 0.74 nM), whose KD is weaker by an order of magnitude.52 Nonetheless, both SPR and ITC indicate that the overall binding kinetics of m9B6 are poor compared to the other antibodies.
Figure 2.
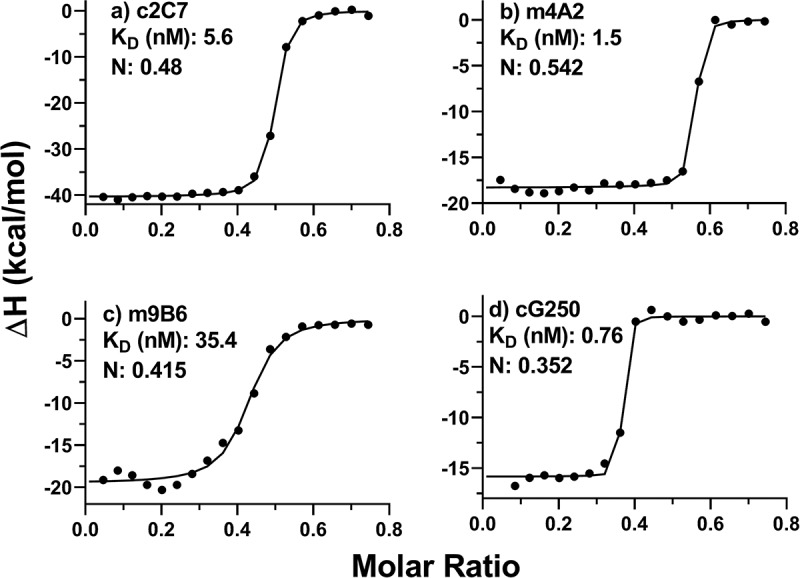
Analysis of solution binding of c2C7, m4A2, m9B6 and cG250 to monomeric hCAIX by ITC. Binding isotherm curves are shown for the interaction of these antibodies to monomeric hCAIX. In all experiments the antibodies were titrated into a cell containing monomeric hCAIX. Concentrations of mAbs in the syringe were 37.5 µM and the concentration of the monomeric hCAIX in the cell was 10 µM
HDX-MS kinetic screening with ΔPG-hCAIXCys38Ser
The binding profile of each of the antibodies was further assessed by HDX-MS. This method is a powerful tool for monitoring changes in the average conformational state of proteins, and is routinely applied in the characterization of antibody-antigen interactions.48,53 To facilitate the HDX-MS interrogation we expressed a monomeric form of hCAIX that is devoid of its PG domain by removing the inter-dimer disulfide bond at Cys38 (Figure 1, ΔPG-hCAIXCys38Ser).12 This construct maintained catalytic activity and structural integrity and allowed for a simplified HDX-MS experimental design to be implemented.12
Overall, peptic digestion of ΔPG-hCAIXCys38Ser resulted in 75 identified peptides with 78% sequence coverage and a redundancy of 4 (Table 2, Figure S2). Gaps in sequence coverage were attributed to N-linked glycosylation (N210) and insufficient reduction of cysteines. Raw deuteration data can be found in Tables S1 and S2, while a complete set of kinetics plots are shown in Figure S3. After 60 min of labeling, normalized deuteration of peptides ranged from minimal deuterium incorporation (~1%), to almost complete labeling ~90%. Technical replicates allowed the calculation of average standard deviation values which range from 0.03–0.07 for all deuteration measurements for each protein state (Table 2). Preliminary data has shown that the mAbs can be separated into two distinct categories: ADC candidates (c2C7 and cG250) and functional enzyme inhibitors (m4A2 and m9B6).52 HDX-MS findings for each category of hCAIX-binding mAbs are described in the following sections. All structural references are based on assignments made by Alterio et al.12 Similar binding profiles to those described below were extracted from preliminary differential HDX-MS using the full-length hCAIX dimer protein, including the PG domain (data not shown) suggesting that neither dimerization nor inclusion of the PG-domain significantly alters the binding mode of c2C7, cG250, m4A2, or m9B6.
Table 2.
Summary of critical parameters for HDX-MS experimentation.80.
| |
Full screen |
Combo |
|||||
|---|---|---|---|---|---|---|---|
| Data Set | Control | 2C7 | 9B6 | 4A2 | G250 | Control | 4A2 + 9B6 |
| # of Peptides found across all samples | 75 | 77 | |||||
| Sequence coverage | 78% | 82% | |||||
| Average peptide length / Redundancy | 13.5/4.0 | 12.1/3.7 | |||||
| Repeatability (average SD, Da) | 0.07 | 0.03 | 0.04 | 0.05 | 0.05 | 0.04 | 0.03 |
| HDX reaction details | Phosphate buffered saline, pD = 6.9, RT | ||||||
| HDX time course (min) | 0.25, 1, 3, 10, 60 | ||||||
| Replicates (biological or technical) | 3 (technical) | ||||||
| Significant differences in HDX | Two-state student T-Test perfomed at each time point (>3 SD, p-value 0.02) | ||||||
c2C7 and cG250 bind an allosterically silent discontinuous epitope
C2C7 and cG250 both bind the CA domain, internalize but do not inhibit CAIXs enzyme activity.52,54 These two mAbs can thus be considered good ADC candidates. We found some striking similarities in the HDX binding profiles of these two antibodies (Figure 3a). First, both cG250 and c2C7 elicited strong stabilizations (decrease in deuteration of the bound state relative to the control) for which the magnitude increased over time in the region spanning amino acid residues L215-L237 (Figure 3b, blue). A smaller stabilization was also observed across residues N238-E251; c2C7 stabilized the complex at shorter timepoints whereas cG250 demonstrated a slightly stronger stabilization at the longest timepoint (Figure 3c, magenta). These stabilized regions encompass the αG-helix (residues S219-T229), as well as a long unstructured region corresponding to residues L229-L251.12 Second, both c2C7 and cG250 stabilized residues L157-S178 (Figure 3d, dark blue), with cG250 inducing a stronger stabilization in I163-S178 (Figure 3e, light blue) at later timepoints. This stabilization covers the C-terminal half of the αE-helix (N150-A164), as well as the βA-sheet feature (E169-V172). Third, stabilization by both c2C7 and cG250 ranging from −4% to −6%, respectively, was also observed in residues H90-E120 (Figure 3f, cyan). This region consists of a buried β-sheet (βD, residues R82-W93) and an unstructured loop that resides immediately adjacent to the αG-helix. cG250, but not c2C7, slightly stabilizes residues R60-Q67 at the longest time point (Figure 3a, purple), covering two β-sheets that sit between the catalytic cleft and αE-helix/βA-sheet. This inference is based on overlapping peptide sequences: significant changes in deuteration were observed in peptides spanning residues L47-Q67 but not those covering residues L47-L59 (Figure S3). No structural relaxation, or increased solvent exposure was observed for either c2C7 or cG250.
Figure 3.
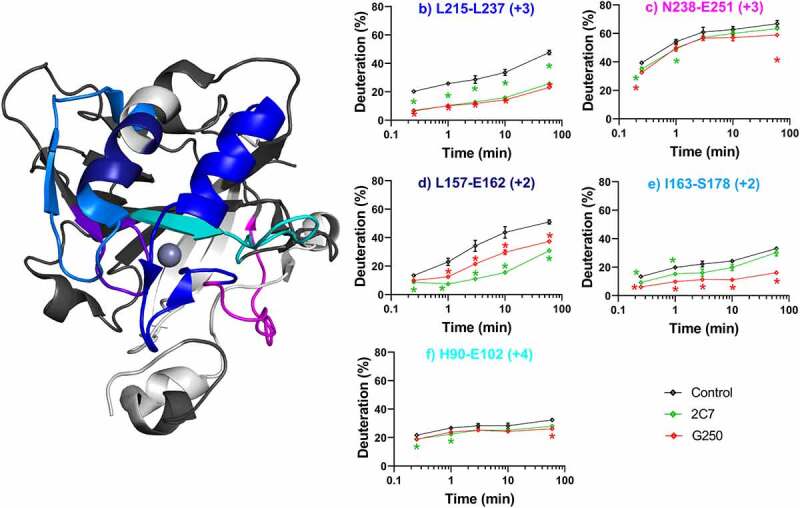
Differential HDX-MS of ADC-candidates c2C7 and cG250 bound to ΔPG- CAIXCys38Ser (PDB 3IAI). Regions demonstrating significant stabilization (3x SD, p = .01) across overlapping peptides at any single time point are color coded for clarity. Zinc is shown as a gray sphere. Regions with no significant change are shown in dark gray, while white corresponds to missing coverage. Kinetics plots corresponding to select peptides within structurally significant regions are shown with arrows indicating associated structural features. Unbound hCAIX control is shown in black, c2C7 in green, and cG250 in red and significant changes in deuteration are marked by asterisks. Data collected in triplicate
It is evident within the timepoints sampled here that the strongest stabilizations are localized to a motif consisting of three structural features (αE/αG/βA) with additional perturbations in the proximal residues. Given the surface-exposure of this motif and its distal location from the catalytic cavity and dimerization interfaces, we hypothesize that the described αE/αG/βA motif represents a discontinuous epitope, where the mAb engagement results in minimal allosteric effects, and no effect on the intrinsic enzymatic activity of hCAIX.
m4A2 and m9B6 inhibit hCAIX through different binding mechanisms
m4A2 and m9B6 both bind the CA domain and inhibit hCAIX activity.52 Three-dimensional projections of significant shifts for both inhibitory antibodies (m4A2 and m9B6) are shown in Figure 4a. First, changes in the region spanning residues F123-L139 bordering the entrance to the catalytic cavity and including the αD-helix (R125-L130), βF-sheet (L136-E145), and an arginine residue (R130) that may be involved in steric control of substrate accessibility12 were observed in response to both antibodies (Figure 4b, magenta). A strong stabilization was observed for all peptides in this region at nearly all labeling timepoints for m4A2. This region has previously been identified in a linear peptide mapping assay as one of two plausible epitopes for m4A2 (1D-5 W or 127D-132 R).55 In contrast, a slight destabilization (4–8%) was observed in this region for m9B6. We also observed a structural destabilization in residues L47-L59 only in response to m4A2 binding (Figure 4c, light blue). This region consists primarily of unstructured loops, but also, importantly, the N-terminus of the βD-sheet, and structurally relevant arginine residues, R58 and R60. Similar to R130, these arginines have been proposed to engage in active-site gating activity.12 m9B6 binding destabilized the regions that were stabilized by cG250 and c2C7. In particular, a strong destabilization at later timepoints was evident in residues L215-L237 (Figure 4d, purple) and in residues Y84-E102 (Figure 4e, cyan).
Figure 4.
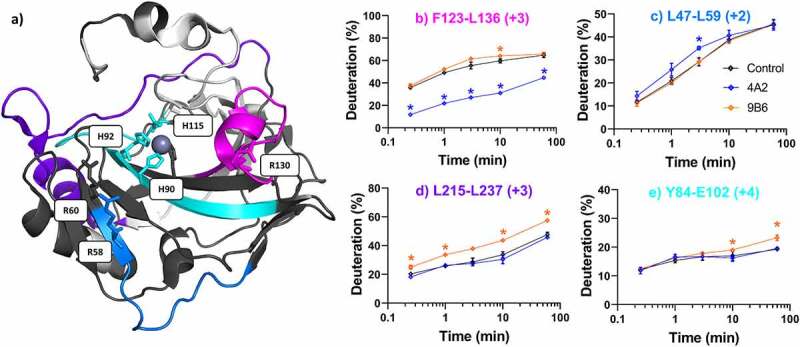
Differential HDX-MS of potential inhibitors m4A2 and m9B6 bound to ΔPG- CAIXCys38Ser (PDB 3IAI). Regions demonstrating significant stabilization (3x SD, p = .01) across overlapping peptides at any single time point are color coded for clarity. Regions with no significant change are shown in dark gray, while white corresponds to missing coverage. Key residues are shown as stick models (R58, R60, R130, H90, H92, H115), and zinc is shown as a gray sphere. Kinetics plots corresponding to select peptides within structurally significant region are shown with arrows indicating associated structural features. Unbound hCAIX control is shown in black, m4A2 in blue, and m9B6 in Orange, and significant changes in deuteration are marked by asterisks. Data collected in triplicate
In summary, binding of the m9B6 antibody resulted in destabilization of multiple ΔPG-CAIXCys38Ser motifs, while the m4A2 antibody stabilized the entrance of the catalytic pocket, likely through direct binding to the αD-helix. To deduce the functional relevance of these conformational “hot-spots”, we performed further biophysical and functional characterization.
m4A2 and m9B6 exhibit synergistic inhibition of hCAIX
Given the contrasting binding profiles and unique inhibitory mechanisms highlighted by HDX-MS for m4A2 and m9B6, we investigated their ability to simultaneously bind and inhibit hCAIX. ADC candidates were excluded from the following assays, due to their demonstrated lack of inhibitory activity.52 Surface plasmon resonance (SPR) experiments, in which both antibodies were either immobilized or flowed, confirmed the non-competing nature of the binding of m9B6 and m4A2 to hCAIX (Figure 5a, b). We next investigated the functional inhibitory effect of m4A2 and m9B6 alone and in combination in an in cell catalytic assay (Figure 5c). This assay assesses hCAIX catalytic activity directly in live 67NR murine breast cancer cells engineered to constitutively express hCAIX by measuring pH changes in the cellular environment resulting from conversion of carbon dioxide (CO2) to bicarbonate (HCO3−) and protons (H+) by hCAIX.56 The presence of either m4A2 or m9B6 inhibited extracellular acidification relative to the IgG2b control. Inhibition was even stronger when the cells were exposed to a combination of m4A2 and m9B6, indicating synergistic action of the two antibodies (Figure 5c). This synergistic behavior was also observed in in vitro enzyme activity assays that used a purified hCAIX dimer and 4-methylumbelliferyl acetate (4-MU-Ac) as a surrogate substrate (Figure S4).
Figure 5.
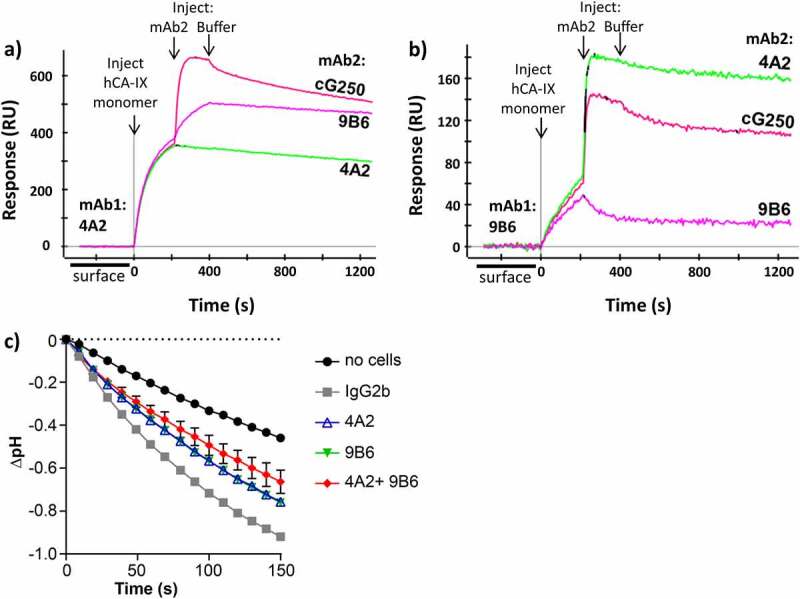
(a-b) Competitive SPR binding demonstrating that mAbs m4A2 and m9B6 bind to independent epitopes. (c) Analysis of CAIX catalytic activity by 67NR murine breast cancer cells expressing hCAIX incubated with anti-CAIX mAb m4A2 and/or m9B6, followed by addition of buffer and CO2-saturated water and monitoring the decrease in pH using the “in-cell” carbonic anhydrase activity assay. Cells incubated in the presence of IgG2b were used as a control. Data show the mean ± SEM of technical replicates (n = 3/group) and are representative of 2 independent experiments
The underlying mechanism of synergistic inhibition was further probed by HDX-MS (Figure 6). As the m4A2/m9B6 antibody combo was not included in the initial screen, an additional peptide map was generated, covering 82% across 77 peptides, and a separate non-binding reference state was collected under similar conditions (Table 2, Table S1–2). Full kinetics plots for the HDX dataset using the mAb combination can be found in Figure S5. Compared to the unbound state, the m9B6/m4A2 combo demonstrated binding profiles that were consistent with the major shifts observed in each of the single antibody configurations. Significant stabilization in residues F123-L136 was observed, despite both 4A2 and 9B6 exerting opposing conformational influences in this region (Figure 4b, magenta). Since we did not correct for intrinsic exchange rates in both HDX-MS assays performed in this report, a quantitative comparison of the stabilization of m4A2 alone or in conjunction with m9B6 is not possible. Further, destabilization observed in residues L215-L237, which can be attributed to m9B6 binding, and residues L47-L59, attributed to m4A2, was preserved. Conformational shifts resulting in destabilizations in response to either m9B6 (residues H90-E102) and m4A2 (residues L68-E75) were not classified as significant in the antibody combination. This is likely attributable to a relative attenuation of the overall mass shifts. Our HDX-MS assay profiles and binding assays provide evidence for simultaneous occupancy and synergistic inhibition of hCAIX by both m4A2 and m9B6, a finding that suggests potential for complementary therapeutic applications.
Figure 6.
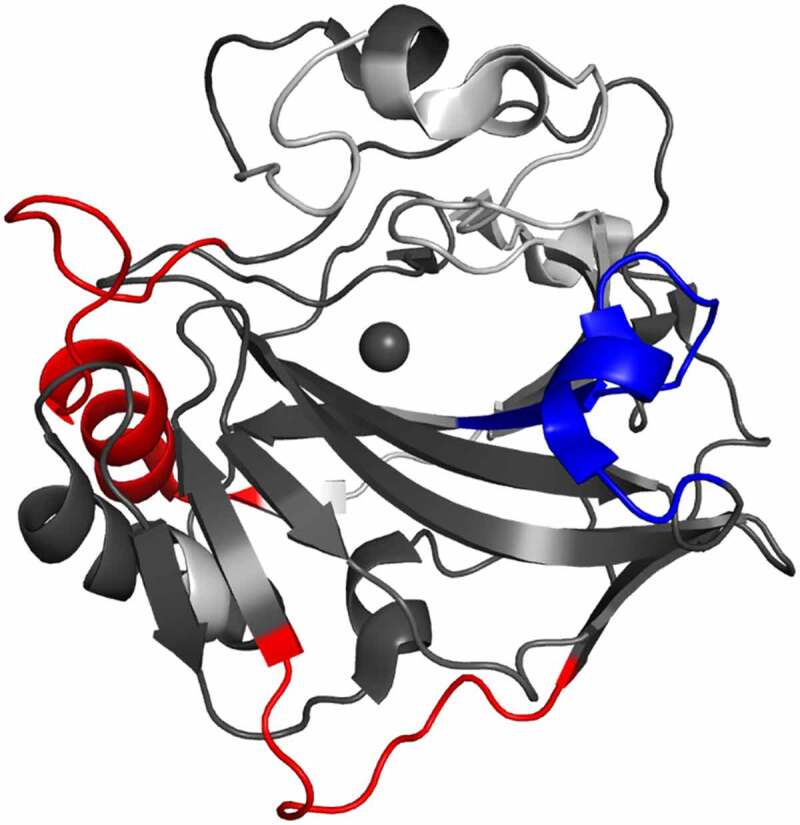
Differential HDX-MS of 4A2 and 9B6 combo bound to ΔPG-CAIXCys38Ser superimposed on PDB 3IAI. Regions demonstrating significant stabilization (3x SD, p = .02) across overlapping peptides at any single time point are shown in blue, while destabilization is shown in red. Regions with no significant change are shown in dark gray, while white corresponds to missing coverage
m4A2 emerges as leading candidate for therapy
A slightly stronger inhibitory effect was observed with m4A2 (Figure 5c and Figure S4) with more favorable binding kinetics (Figure 2 and Figure S1) compared to m9B6, suggesting that the m4A2 binding mechanism may be favorable for the development of this mAb as a CAIX inhibitor. CAIX activity contributes to acidification of the tumor microenvironment, and previous studies have shown that pharmacologic or genetic inhibition of CAIX reduces extracellular acidification and impairs glycolysis by cancer cells.57,58 Therefore, we investigated the effects of m4A2 on extracellular acidification and metabolism. We cultured MDA-MB-231 human breast cancer cells engineered to constitutively express CAIX (CAIX-positive) in the presence of m4A2 and analyzed the extracellular acidification rate (ECAR) and glycolytic metabolism using a Glycolytic Rate Assay. This assay calculates the contribution of mitochondrial-derived CO2 to the overall extracellular acidification rate, allowing for the determination of the proton efflux rate (PER) attributed to glycolysis (glycoPER). We observed reduced basal levels of ECAR in CAIX-positive cells cultured in the presence of m4A2, relative to cells exposed to isotype-matched control IgG (Figure 7a). Consistent with the reduced basal levels of ECAR, analysis of the PER profiles calculated using ECAR and oxygen consumption rate (OCR) measurements demonstrated lower values for both basal total PER and glycoPER by m4A2-treated cells (Figure 7b). Indeed, we found that basal PER and basal glycoPER were significantly reduced in cells cultured with m4A2 (Figure 7c and d), while the mitoOCR to glycoPER ratio was significantly increased (Figure 7e), suggesting that m4A2-mediated inhibition of CAIX activity functions to reduce glycolytic metabolism by CAIX-expressing cancer cells. Similar results were observed using breast cancer cells in which CAIX expression was silenced using CRISPR-mediated gene-editing strategies (Figure 7f-h),56 confirming the effect of 4A2-mediated inhibition of CAIX on extracellular acidification and glycolytic metabolism.
Figure 7.
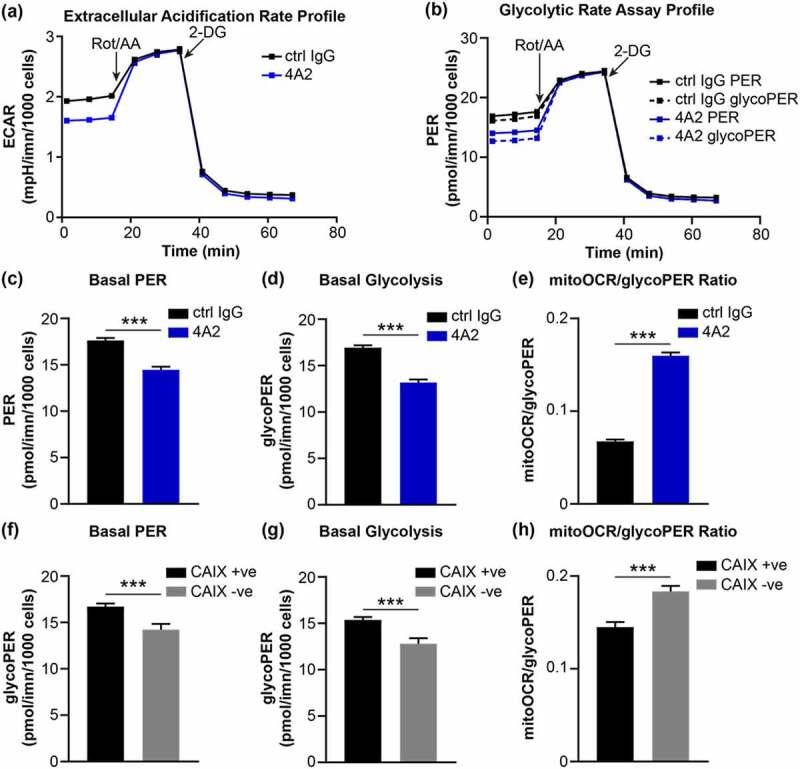
(a) Extracellular acidification profile of MDA-MB-231 human breast cancer cells constitutively expressing CAIX that were cultured with 10 μg/ml of 4A2 and subjected to a Glycolytic Rate Assay. Cells cultured with 10 μg/ml of 4A2 were used as a control. Rot/AA, rotenone with antimycin A; 2-DG, 2-deoxy-D-glucose. (b) Profile of proton efflux rate (PER) and glycolytic PER (glycoPER) of cells cultured as described in panel a. (c-e) Quantification of (c) basal proton efflux rate (PER), (d) basal glycolysis and (e) ratio of mitochondrial oxygen consumption rate to glycolytic proton efflux rate of cells cultured as described in panel a. (f-h) Quantification of (f) basal proton efflux rate (PER), (g) basal glycolysis and (h) ratio of mitochondrial oxygen consumption rate to glycolytic proton efflux rate of MDA-MB-231 human breast cancer cells constitutively expressing CAIX (CAIX +ve) or depleted of CAIX expression (CAIX – ve) and assessed using the Glycolytic Rate Assay. Data show the mean ± SEM of technical replicates (n = 5–6/group) ***P < .001
In addition to changes in metabolism, previous studies have shown that depletion of CAIX expression leads to increased death of cancer cells in hypoxia.56,59 To extend our observations on the biological consequences of inhibiting CAIX function with m4A2 using a model of endogenous CAIX expression, we cultured U87 MG glioblastoma cells in hypoxia, an environment that drives strong upregulation of CAIX,60 in the presence of m4A2 and assessed cell death using a terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (Figure 8a). Quantification of the TUNEL positive cells showed an approximate 1.8-fold increase in the apoptotic cell population in response to m4A2 compared to cells exposed to an isotype control IgG (Figure 8b). Together, the glycolytic rate assay results in combination with the TUNEL assay results indicate that inhibition of CAIX activity leads to decreased glycolysis and an increase in cell death.
Figure 8.
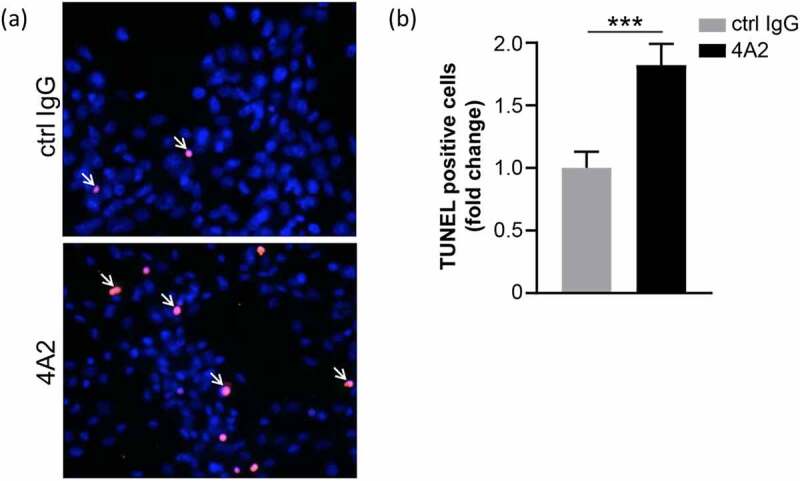
U87 MG glioblastoma cells treated with 100 μg/ml m4A2 or control mouse IgG antibody for 72 hours in hypoxia. After fixation a TUNEL assay was performed. (a) Representative images demonstrating the presence of TUNEL-positive U87MG cells (red, arrows). Nuclei are in blue. (b) Bar graph showing quantification of TUNEL-positive cells. Data show the mean fold change ± s.e.m. of technical replicates and are representative of 2 independent experiments N = 10 fields of view at 20x. ***P < .001
Discussion
Biophysical characterization of antibody-antigen interactions is integral to the design and development of antibody-based therapeutics. Once a library of antibodies with desired binding kinetics and functional activity is selected, the fine-details of their binding modes at the antigen level must be elucidated. Such information is invaluable in classifying mAbs by their binding epitopes and for selecting antibodies with desired binding characteristics. Multiple modes of hCAIX engagement have been observed for anti-hCAIX biotherapeutics, and different modes of engagement lead to different therapeutic outcomes.10,17 This diversity of therapeutic outcomes makes hCAIX an attractive antibody-based therapeutics anti-cancer target.
We have described the selection of a panel of novel antibodies targeting both the catalytic and proteoglycan domains of hCAIX.52 The study presented here focuses on the detailed biophysical characterization of three antibodies selected from this panel that specifically target hCAIX’s catalytic domain. YSD provided data that the epitopes were in the hCAIX and were likely (with the exception of the m9B6 epitope) conformational, but did not provide sufficient granularity to pinpoint the epitope or how the antibodies engage hCAIX differently. A previous report suggested that the binding epitopes of antibodies targeting the catalytic domain are predominantly conformational, while those targeting the PG domain are linear.61 Linear peptide scanning has proven effective for mapping epitopes in the PG-domain,62 but size limitations and the conformational nature of the epitopes on CAIX’s catalytic domain render this technology ineffective in this context.
HDX-MS can efficiently screen antibody-antigen interactions under native conditions with peptide-level resolution.48 We used HDX-MS to interrogate the binding profiles of our three catalytic domain-specific antibodies, in addition to the cG250 mAb. The carbonic anhydrase family has been previously interrogated by HDX-MS, where the catalytic role of zinc in CAII was investigated.63 However, to our knowledge, this is the first report of applying the HDX-MS technology to resolve the binding epitopes of antibodies targeting hCAIX. It is evident that the subtle details in protein dynamics revealed by HDX-MS would have been impossible to obtain with a crystal structure-based epitope determination.
Both c2C7 and cG250 have demonstrated activity as antibody conjugates. For example, c2C7 demonstrated significant reduction in tumor volume in vivo when conjugated to the microtubule inhibitor mertansine.52 cG250-conjugates have shown promise in clinical trials for renal cell carcinoma, either as an imaging tool (124I-Girentuximab)64 or as a radioimmunotherapy (177Lu-Girentuximab).31 A shared binding mode was observed for c2C7 and cG250. The discontinuous nature of this epitope, formed by the αE/αG/βA motif as determined by HDX-MS (Figure 3), is in agreement with the yeast-display findings (Table 1). This epitope is far from the catalytic cavity, dimerization interface, PG- and transmembrane-domain,12and binding to it likely leaves access to the catalytic cavity unimpeded under physiological conditions. This accounts for the lack of inhibitory action, and reinforces the value of these mAbs as ADC candidates. Mapping of the precise residues engaged by c2C7 and cG250 was not possible with our current HDX-MS dataset. Further experiments, such as molecular docking and alanine scanning mutagenesis may be warranted to resolve the epitope.
A conformational epitope on the catalytic domain for cG250 has previously been identified.54,65 A specific epitope identified by linear peptide scanning consisting of A77-L87 and L119-V126 has recently been proposed for cG250.66,67 These sequences are near or overlap with the dimerization interface, and may not be optimal for the binding of a relatively large antibody. Due to the sensitivity of HDX-MS to conformational epitopes and limitations of linear peptide scanning, we propose our findings as the most probable binding mode of cG250. Finally, negatively charged substitutions in hCAIX in residues L157-G167, relative to all α-carbonic anhydrase isoforms, as well as low sequence conservation in the structural features within the discontinuous epitope, may impart specific binding to the hCAIX isoform.2 Indeed, c2C7 was found to be specific for hCAIX ectodomain.52
Antibody-mediated functional inhibition of hCAIX has previously been reported; for example, the MSC8 mAb was suggested to directly inhibit hCAIX activity, rather than induce its internalization.28 Here we report on two non-competing hCAIX enzyme inhibiting mAbs m4A2 and m9B6, whose two binding profiles suggest divergent mechanisms of action. Importantly, both of these antibodies have demonstrated specificity for hCAIX across species and among secreted and membrane-bound isoforms.55
Interestingly, m4A2 epitope shares the lowest homology with other members of the CA family, and has previously been identified as a potential target for rational drug design.12 It is intriguing that conformational perturbations are observed in or near all three of the positively charged arginine residues at the catalytic cavity entrance. Along with the highly negatively charged PG-domain, these residues have been postulated to play an active role in gating the catalytic cavity.12 We therefore hypothesize that m4A2-mediated enzymatic inhibition of hCAIX is driven by steric gating, thereby obstructing the entry of substrate into its catalytic cleft by direct binding to those residues that play a critical role in the function of the catalytic cavity.12,67
m9B6 caused an ensemble of destabilizations of hCAIX and, although this mAb is almost equally potent as m4A2 in inhibiting hCAIX (Figure 5c), it stands out from all the mAbs tested as having the weakest binding kinetics (Figure 2, S1). This likely explains why, contrary to m4A2, only relatively minor destabilizations (~ −5%) were observed in a previously proposed linear epitope (S122 – 128E).55 Despite the lack of a clear canonical binding event, i.e., stabilization of the binding interface, we can make some inferences about the nature of the m9B6 binding mode. First, the YSD experiments show that m9B6 binds a linear epitope (Table 1) that does not overlap with that of m4A2 (Figure 5). Second, m9B6/hCAIX interaction may exhibit a so-called “type-2” binding behavior, described by Sowole et al., where binding energy is reinvested to drive structural destabilization.68 From this point of view one could envision that m9B6 only engages hCAIX after an unfavorable unfolding event,69 where the conformational distribution of the m9B6/hCAIX complex is subsequently biased toward a state with reduced solvent turnover efficiency. This is further supported by the relatively weak binding behavior, and structural loosening of two histidines (H90 and H92; Figure 4d, cyan) within the previously described catalytic triad.12 Importantly, this destabilized state remains accessible to m4A2 binding. The absence of evidence of a direct antibody interaction does not preclude a stable 9B6/hCAIX complex.70 We cannot rule out that m9B6 binds a sequence for which HDX coverage is missing. Further, HDX-MS is limited to changes in the average conformational state of a protein, and therefore this technology is blind to binding events driven purely by side-chain or electrostatic interactions.71
The observation of synergistic behavior and non-competing binding between m4A2 and m9B6 (Figure 5) suggests that both of the outlined inhibitory mechanisms are engaged by non-overlapping epitopes. These findings demonstrate that these antibodies can be used alone or as combinatorial therapy (Figure 5c), or can form the basis for the rational design of biparatopic antibodies. Further studies are required to fully understand what fundamental mechanisms are employed by our antibodies, particularly m9B6, in the inhibition of hCAIX activity. This knowledge, in addition to their possible pH-dependent behavior, is critical for the design of antibody-based biologics that target the acidic tumor microenvironment.
In conclusion, the HDX-MS evaluation of a set of functionally different hCAIX targeting antibodies highlights two groups of mAbs with three modes of binding, and demonstrates power of HDX-MS to deliver conformational insights in combination with complementary biophysical and functional assays. The work presented here thus lays the groundwork for future mAb engineering efforts targeting both hCAIX and other membrane-bound enzymes.
Materials and methods
Recombinant antibody production and purification
Anti-human CAIX mouse mAbs were sequenced and full-size heavy and light human IgG chains were ordered from a commercial vendor (Genscript); VH and VL CDR1-3 regions of 2C7 was grafted on a human IgG1 framework (designated chimeric, ‘c’) whereas those of 4A2 and 9B6 were grafted on a murine IgG2a framework (designated as mouse, ‘m’).52 MAbs were produced by co-transfection of CHO-3E7 cells, which were then grown to a density of 2 ×106 cell/mL in 150 mL of F17 medium in 1 L shaker flasks. Cells were transfected with 2 μg total DNA (containing 500 ng each of the heavy and light chain constructs) using PEI MAX™ (Polysciences, Inc.), maintained at 37°C for 24 h, then fed with Tryptone N1 at 1% w/v, after which they were transferred to 32°C for 6 days. Cell culture supernatant was harvested and analyzed by SDS-PAGE for expression.
MAbs were purified from cell-culture supernatants by protein-A affinity chromatography in batch mode (cG250 and c2C7) or by using packed columns (m9B6 and m4A2). Supernatants were incubated and agitated overnight at 4°C with MabSelect SuRe resin (Cytiva Life Sciences) or loaded onto MabSelect SuRe packed columns, previously equilibrated with HyCloneTM Dulbecco’s phosphate-buffered saline (DPBS; Cytiva Life Sciences) or 10 mM sodium phosphate, 140 mM NaCl buffer pH 7.0. Resin was washed with DPBS or 10 mM sodium phosphate, 140 mM NaCl buffer pH 7.0 and mAbs were eluted with 100 mM citrate buffer pH 3.0 (cG250, c2C7, m4A2) or 100 mM Glycine buffer pH 2.6 (m9B6). Fractions containing mAbs were pooled and neutralized with 1 M HEPES. Protein-A purified material was buffer-exchanged against DPBS by Tangential Flow Filtration using 30 kDa MWCO cassettes (Millipore Sigma: m9B6; Pall Corporation: m4A2), or using 30 kDa MWCO Vivaspin® centrifugal concentrators (Sartorius – cG250 and c2C7). cG250 and c2C7 mAbs were further purified by preparative size exclusion chromatography (SEC) on Superdex-200 columns (Cytiva Life Sciences), using DPBS as mobile phase. Purified mAbs were concentrated to 5–10 mg/mL by ultrafiltration using Vivaspin® centrifugal concentrators (MWCO 30 kDa; Sartorius), and sterilized by filtration through 0.2 μm filters. During the process, the protein concentration was monitored on a NanoDrop™ 2000 spectrophotometer (ThermoFisher Scientific) using absorbance at 280 nm and the calculated specific extinction coefficient of each mAbs. MAbs were kept in aliquots at −80°C for long-term storage, and reevaluated by UPLC-SEC upon each thaw prior to use.
hCAIX ectodomain expression and purification
The hCAIX extracellular domain cDNA (HGNC:1383; a.a. 1–414), was cloned in the PTT5 expression vector, produced as a C-terminal penta-His-tagged protein in CHO-3E7 cells,72,73 purified by immobilized metal affinity chromatography. SDS/PAGE (reducing and non-reducing conditions) indicated the presence of monomeric and dimeric subpopulations which were further purified by preparative SEC to separate dimers from monomers. SEC purification was performed on Superdex-200 Increase column (Cytiva Life Sciences), using DPBS as mobile phase. Preparative SEC fractions were analyzed by SDS-PAGE to select which fractions to pool. Fractions containing monomer or dimer only were pooled together and concentrated by ultrafiltration using 10 kDa MWCO Vivaspin® centrifugal concentrator (Sartorius). Concentrated material was sterilized by filtration through 0.2 μm filters. During the process, the protein concentration was monitored on a NanoDrop™ 2000 spectrophotometer (ThermoFisher Scientific) using absorbance at 280 nm and the calculated specific extinction coefficient of the hCAIX monomer or dimer. Both preparations were kept in aliquots at −80°C for long-term storage, and reevaluated by UPLC-SEC upon each thaw.
Isothermal calorimetry
ITC experiments were performed on a MicroCal Auto-ITC200 (Malvern). In total 19 injections were performed using the following instrument settings: cell temperature 25°C, reference power 10 µCal/second, initial delay 60 seconds, stirring speed 750 rpm, feedback mode/gain high, and injection volume 2 µL for 4 seconds spaced at 120 second intervals with a filter period of 5 seconds. The syringe volume was 120 µL and the cell volume was 400 µL. Data were analyzed using MicroCla PEAQ-ITC Analysis Software v1.1.0.1262 (Malvern).
Domain-level epitope mapping by yeast surface display
To map the epitopes of the anti-hCAIX mAbs, fragments of hCAIX extracellular domain (ECD) were PCR amplified from the hCAIX DNA sequence corresponding to various combinations of hCAIX protein domains and sections between domains. The DNA fragments were then cloned into the YSD vector by in vivo recombination (IVR) in yeast. The hCAIX ECD and its fragments were expressed and covalently displayed on the surface of yeast cells using YSD.74 The YSD vector (pPNL6) was from The Pacific Northwest National Laboratory, USA. The hCAIX fragments were expressed as fusion proteins (Aga2-HA-(CAIX)-MYC) on the yeast cell surface. The binding of the mAbs to yeast cells was performed using a whole yeast cell ELISA. Briefly, the various clones of the induced yeast cells were loaded into a 96-well filter plate (1x107 cells per well), blocked, incubated with the anti-hCAIX antibodies, the binding of these mAbs was revealed by a horseradish peroxidase (HRP)-conjugated secondary antibody followed by incubating with the HRP substrate tetramethyl benzidine (TMB) according to the manufacturer’s conditions and read at OD450. The relative amount of displayed fusion protein was measured by probing with an anti-MYC antibody (ab19233, Abcam), followed by an HRP-conjugated secondary antibody (ab97135, Abcam) to be used to normalize the binding signal for anti-hCAIX mAbs. For determination the nature of epitopes, linear versus conformational, yeast cells with displayed hCAIX fragments were heated at 80°C for 30 min, then chilled on ice for 20 min prior to labeling with antibodies.75 The binding levels for anti-hCAIX mAbs were normalized to the amount of Myc tag (ratio anti-hCAIX/anti-Myc).
HDX-MS
ΔPG-hCAIXCys38Ser was equilibrated with the mAbs at a 1:1 molar ratio in phosphate-buffered saline (PBS, pH 7.4). hCAIX-mAb complexes (4 μL) were diluted with 4 μL of labeling buffer (90% D2O, 0.1x PBS, pD 7.0). The labeling reaction (0.25, 1, 3, 10, 60 min) was quenched with a 5x dilution of reducing buffer (100 mM TCEP, 100 mM Gly-HCl, pH 2.2). Samples (19 pmol) were manually injected into an ice-cooled loop, digested online with a Poroszyme Immobilized pepsin cartridge (Thermo) and trapped on a C18 Pepmap100 (1 x 5 mm, 5 μm, 100 Å) at 100 μL/min for 3 min in mobile phase A (0.23% formic acid). Peptic peptides were eluted from a Halo-Peptide ES analytical column (50 x 0.3 mm, 2.7 μm, 160 Å, Sciex) with a 10–35% acetonitrile gradient (0.23% formic acid) over 7.5 min at 10 μL/min. Data acquisition was performed with a Waters Q-Tof Ultima API MS. Peptides were identified by data-dependent acquisition of an undeuterated sample which subsequently searched against a Mascot database. Data was collected in triplicate and analyzed with MS Studio.76 Significant changes in deuteration were defined by ± 3 SD (p = .02).
Competitive SPR binding assay
All SPR experiments were performed using the ProteOn XPR36 system (Bio-Rad Laboratories Inc.). PBS with 0.05% Tween 20 (Teknova) and with 3.4 mM ethylenediaminetetraacetic acid (EDTA) disodium salt (Teknova), pH 7.4 was used as running buffer and all experiments were performed at 25°C. Reagents used for the antibody immobilizations were obtained from Bio-Rad Laboratories Inc.
The CAIX antibody clones 4A2 and 9B6 were immobilized on a ProteOn GLC sensor chip using standard amine coupling chemistry (All reagents were obtained from Bio-Rad Laboratories Inc). Using a flow rate of 100 µL/min for 140 s, all ligand channels were activated simultaneously by injecting a fresh mixture of 20 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and 5 mM N-hydroxysulfosuccinimide (Sulfo-NHS) followed by 25 µg/mL of the antibodies diluted in 10 mM sodium acetate buffer (pH 4.5; 25 µL/min for 240 s). The sensor chip surface was then deactivated with 1 M ethanolamine HCl (30 µL/min for 300 s). Approximately 5000 RU each of the 4A2 and 9B6 antibodies were immobilized. One ligand channel was used for a mock immobilization for subsequent reference subtraction. Next, using the Co-Inject Ligand function of the ProteOn ManagerTM software v3.1.0.6, the CAIX monomer (100 nM) and a buffer blank was simultaneously injected (50 µL/min, 210 s) over the two immobilized CAIX antibodies. Immediately after, the 4A2, 9B6 and cG250 (100 nM) and a buffer blank were injected followed by an 800 s dissociation phase (50 µL/min, 180 s). Sensorgrams were aligned, and the resulting sensorgrams were analyzed using ProteOn Manager Software v3.1.0.6.
Cell culture
The MDA-MB-231 human breast cancer cell line and U87 MG human glioblastoma cell line were originally obtained from the American Type Culture Collection (ATCC) and cultured as described previously.56,77 The 67NR murine breast cancer cell line was originally provided by Dr. Fred Miller (Karamanos Cancer Institute, Detroit, MI) and was cultured as described previously.78, The generation of CAIX-negative MDA-MB-231 cells using CRISPR-mediated gene editing and the generation of CAIX-positive MDA-MB-231 cells through the re-introduction of a wild-type CAIX construct have been described previously.56 To generate 67NR cells constitutively expressing human CAIX (67NR-hCAIX), a human wildtype CAIX plasmid (gift from Dr. Jacques Pouyssegur, University of Nice, Nice, France) was transfected into 67NR cells using Lipofectmine 2000 as per the manufacturer’s instructions. Transfected cells were selected using Zeocin and then subcloned for selection of cells with high levels of CAIX expression. For culture in normoxia, cells were incubated in a humidified incubator at 37°C with 5% CO2. For studies in hypoxia, cells were grown at 37°C in an atmosphere of 1% O2, 5% CO2, 94% N2 in a humidified incubator inside a sealed workstation as described previously.58 Cell lines were routinely tested for mycoplasma contamination using the LookOut Mycoplasma PCR detection kit (Sigma; Cat. No. MP0035). The 67NR and MDA-MB-231 cell lines have been authenticated using short tandem repeat DNA profiling (DNA fingerprinting) by a commercial testing facility (Genetica). In addition, the cell lines were routinely tested for viability, morphology and hypoxia-induced endogenous CAIX expression or over-expression of CAIX.
“In cell” CAIX activity assay
In cell CAIX catalytic activity assays were carried out as described previously with the following modifications.56 Assays were performed using 67NR murine breast cancer cells exogenously expressing human CAIX (67NR-hCAIX). For each replicate sample (n = 3/Ab), 5.0 × 105 cells were harvested with 0.5 mM EDTA in PBS and re-suspended in 200 µl CO2-free isotonic buffer (20 mM Hepes, 130 mM NaCl, 5 mM KCl, pH 8.0) in a 2 ml flat-bottom glass vial equipped with an 8 mm magnetic stir bar. 10 µl of a 50 µg/ml stock solution of the anti-CAIX Abs or isotype control Abs were added to the cell suspension and the sample was incubated with gentle stirring at room temperature for 45 min. The remainder of the assay was carried out on ice. 600 µl ice cold buffer was added to the cell-Ab mixture and a narrow pH electrode (Accumet, Cat # 13–620-850) was immersed in the sample and equilibrated for 3–5 min. 200 µl of ice-cold CO2-saturated water was added to initiate the assay and pH readings were immediately recorded at 2 to 5 s intervals. The final Ab concentration in the assay was 0.50 µg/ml. Data were analyzed using Graphpad Prism v8 (Graphpad Software). To determine the rate of spontaneous CO2 hydration, measurements were performed on cell-free samples. The increase in hydration rate above the spontaneous rate is a measure of CA activity.
Glycolytic rate assay
Metabolic flux was assessed using Seahorse XF technology as described previously58 with the following modifications. MDA-MB-231 human breast cancer cells engineered to constitutively express hCAIX (CAIX positive) were seeded at 2.0 × 104 cells/well in a 96-well XF microplate (Agilent Technologies) and allowed to attach overnight. The following day, the cells were washed with complete assay medium (XF DMEM medium, pH 7.4, 10 mM glucose, 1 mM pyruvate, 2 mM glutamine; Agilent Technologies) and incubated in 200 µl/well complete assay medium containing 10 µg/ml m4A2 anti-hCAIX or IgG2b control antibodies for 60 min at 37°C without CO2. Media was then removed, 160 µl/well fresh media containing 10 µg/ml m4A2 anti-hCAIX or IgG2b control antibodies was added, cell layer integrity was verified by microscopic observation and cells were subjected to a Seahorse XF Glycolytic Rate Assay (Agilent Technologies) according to instructions provided by the manufacturer. Assays were carried out using the Seahorse XFe96 Extracellular Flux Analyzer (Agilent Technologies). Data were acquired using Wave v2.6 (Agilent Technologies). After completion of the assay, cell nuclei were stained with HCS NuclearMask Red Stain (ThermoFisher Scientific,), cell layers were imaged using the Incucyte Zoom live cell imaging system (Essen Biosciences, Ann Arbor, MI) and cell nuclei were enumerated as a measure of cell number for normalization of metabolic data. Normalized data were exported to the Glycolytic Assay Report Generator for conversion of experimentally derived ECAR and OCR data into Proton Efflux Rate (PER) and calculation of specific glycolytic rate assay parameters. These parameters include basal PER (last PER measurement prior to injection of Rot/AA), basal glycolysis (last glycoPER measurement prior to injection of Rot/AA) and mitoOCR/glycoPER [(Last OCR measurement before first injection) – (Minimum OCR after Rot/AA injection)]/(Basal Glycolysis). Data for calculated parameters were exported to GraphPad Prism 8 (Graphpad Software) for statistical analysis. Statistical significance between two groups was assessed by unpaired Student’s t-test. Data are presented as mean ± s.e.m. and P values less than 0.05 were considered statistically significant.
TUNEL assay
U87 MG glioblastoma cells cultured for 72 hours in hypoxia to upregulate CAIX expression. Cells were then seeded onto coverslips and incubated with 100 μg/ml m4A2 antibody in media containing 0.1% serum in hypoxia for an additional 72 hours. Mouse IgG2b (100 μg/ml) was used as an isotype control. Cells were then fixed and a TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay (Roche Applied Sciences) was performed according to the instructions provided by the manufacturer to assess cell death. For quantification, 3–4 random fields from triplicate samples were imaged at 20x using a Zeiss Colibri inverted fluorescence microscope. TUNEL-positive cells and total cells were enumerated and the percentage of TUNEL positive cells was calculated using ImageJ (v1.48, National Institutes of Health, USA). Statistical significance between two groups was assessed by unpaired Student’s t-test. Data are presented as mean ± s.e.m. and P values less than 0.05 were considered statistically significant.
Supplementary Material
Funding Statement
This work was supported by funding to SD from Canadian Institutes of Health Research (CIHR grant # FDN-143318).
Abbreviations
ADC antibody-drug conjugate
CAcatalytic domain
ECDextracellular domain
ECARextracellular acidification rate
hCAIXhuman carbonic anhydrase
HDX-MShydrogen-deuterium exchange mass spectrometry
ITCisothermal calorimetry
mAbmonoclonal antibodies
OCRoxygen consumption rate
PERproton efflux rate
PGproteoglycan domain
SPsignal peptide
SPRsurface plasmon resonance
TMtransmembrane domain
YSDyeast surface display
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G.. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–15. doi: 10.1021/cr200176r. PMID: 22607219. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal M, Boone CD, Kondeti B, McKenna R. Structural annotation of human carbonic anhydrases. J Enzyme Inhib Med Chem. 2013;28(2):267–77. doi: 10.3109/14756366.2012.737323. PMID: 23137351. [DOI] [PubMed] [Google Scholar]
- 3.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–81. doi: 10.1038/nrd2467. PMID: 18167490. [DOI] [PubMed] [Google Scholar]
- 4.Wykoff CC, Beasley NJP, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60(24):7075–83. PMID: 11156414. [PubMed] [Google Scholar]
- 5.Tan EY, Yan M, Campo L, Han C, Takano E, Turley H, Candiloro I, Pezzella F, Gatter KC, Millar EKA, et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br J Cancer. 2009;100(2):405–11. PMID: 19165203. doi: 10.1038/sj.bjc.6604844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang JS, Lin CW, Chuang CY, Su SC, Lin SH, Yang SF. Carbonic anhydrase IX overexpression regulates the migration and progression in oral squamous cell carcinoma. Tumor Biology. 2015;36(12):9517–24. doi: 10.1007/s13277-015-3692-8. PMID: 26130414. [DOI] [PubMed] [Google Scholar]
- 7.Yang JS, Lin CW, Hsieh YH, Chien MH, Chuang CY, Yang SF. Overexpression of carbonic anhydrase IX induces cell motility by activating matrix metalloproteinase-9 in human oral squamous cell carcinoma cells. Oncotarget. 2017;8(47):83088–99. doi: 10.18632/oncotarget.20236. PMID: 29137326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pastorekova S, Parkkila S, Parkkila AK, Opavsky R, Zelnik V, Saarnio J, Pastorek J. Carbonic anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology. 1997;112(2):398–408. doi: 10.1053/gast.1997.v112.pm9024293. PMID: 9024293. [DOI] [PubMed] [Google Scholar]
- 9.Saarnio J, Parkkila S, Parkkila AK, Waheed A, Casey MC, Zhou XY, Pastoreková S, Pastorek J, Karttunen T, Haukipuro K, et al. Immunohistochemistry of carbonic anhydrase isozyme IX (MN/CA IX) in human gut reveals polarized expression in the epithelial cells with the highest proliferative capacity. J Histochem Cytochem: Official J Histochem Soc. 1998;46(4):497–504. PMID: 9524195. doi: 10.1177/002215549804600409. [DOI] [PubMed] [Google Scholar]
- 10.Supuran CT, Alterio V, Di Fiore A, D’ Ambrosio K, Carta F, Monti SM, De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 2018;38(6):1799–836. doi: 10.1002/med.21497. PMID: 29635752. [DOI] [PubMed] [Google Scholar]
- 11.Mboge MY, McKenna R, Frost SC. Advances in anti-cancer drug development targeting carbonic anhydrase IX and XII. Topics in Anti-Cancer Res. 2016;5:3–42. doi: 10.2174/9781681083339116050004. PMID: 30272043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alterio V, Hilvo M, Di Fiore A, Supuran CT, Pan P, Parkkila S, Scaloni A, Pastorek J, Pastorekova S, Pedone C, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci U.S.A. Proc. Natl. Acad. Sci. U.S.A. 2009;106(38):16233–38. doi: 10.1073/pnas.0908301106. PMID: 19805286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Csaderova L, Debreova M, Radvak P, Stano M, Vrestiakova M, Kopacek J, Pastorekova S, Svastova E. The effect of carbonic anhydrase IX on focal contacts during cell spreading and migration. Front Physiol. 2013;4:271. doi: 10.3389/fphys.2013.00271. PMID: 24101905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther. 1997;74:1–20. doi: 10.1016/S0163-7258(96)00198-2. PMID: 9336012. [DOI] [PubMed] [Google Scholar]
- 15.Maupin CM, McKenna R, Silverman DN, Voth GA. Elucidation of the proton transport mechanism in human carbonic anhydrase II. J Am Chem Soc. 2009;131(22):7598–608. doi: 10.1021/ja8091938. PMID: 19438233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domsic JF, Avvaru BS, Chae UK, Gruner SM, Agbandje-mckenna M, Silverman DN, McKenna R. Entrapment of carbon dioxide in the active site of carbonic anhydrase II. J Biol Chem. 2008;283(45):30766–71. doi: 10.1074/jbc.M805353200. PMID: 18768466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kazokaitė J, Aspatwar A, Parkkila S, Matulis D. An update on anticancer drug development and delivery targeting carbonic anhydrase IX. PeerJ. 2017;2017(11). doi: 10.7717/peerj.4068. PMID: 29181278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov. 2017;12(1):61–88. doi: 10.1080/17460441.2017.1253677. PMID: 2778354. [DOI] [PubMed] [Google Scholar]
- 19.De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov. 2013;8(7):793–810. doi: 10.1517/17460441.2013.795145. PMID: 23627619. [DOI] [PubMed] [Google Scholar]
- 20.Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem. 2016;31(3):345–60. doi: 10.3109/14756366.2015.1122001. PMID: 26619898. [DOI] [PubMed] [Google Scholar]
- 21.Castelli MS, McGonigle P, Hornby PJ. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol Res Perspect. 2019;7(6):e00535. doi: 10.1002/prp2.535. PubMed PMID: 31859459; PMID: 31859459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oosterwdk E, Ruiter DJ, Hoedemaeker PJ, Pauwels EKJ, Jonas U, Zwartendijk I, Warnaar SO. Monoclonal antibody G 250 recognizes a determinant present in renal‐cell carcinoma and absent from normal kidney. Int J Cancer. 1986;38(4):489–94. doi: 10.1002/ijc.2910380406. PMID: 2428759. [DOI] [PubMed] [Google Scholar]
- 23.Oosterwijk-Wakka JC, Boerman OC, Mulders PFAM, Oosterwijk E. Application of monoclonal antibody G250 recognizing carbonic anhydrase IX in renal cell carcinoma. Int J Mol Sci. 2013;14(6):11402–23. doi: 10.3390/ijms140611402. PMID: 23759990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belldegrun AS, Chamie K, Kloepfer P, Fall B, Bevan P, Störkel S, Wilhelm O, Pantuck AJ. ARISER: a randomized double blind phase III study to evaluate adjuvant cG250 treatment versus placebo in patients with high-risk ccRCC—Results and implications for adjuvant clinical trials. J Clin Oncol. 2013;31(15_suppl):4507. doi: 10.1200/jco.2013.31.15_suppl.4507. PMID. [DOI] [Google Scholar]
- 25.Závada J, Závadová Z, Pastorek J, Biesovä Z, Ježek J, Velek J. Human tumour-associated cell adhesion protein MN/CA IX: identification of M75 epitope and of the region mediating cell adhesion. Br J Cancer. 2000;82(11):1808–13. doi: 10.1054/bjoc.2000.1111. PMID: 10839295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chrastina A, Závada J, Parkkila S, Kaluz Š, Kaluzová M, Rajčáni J, Pastorek J, Pastoreková S. Biodistribution and pharmacokinetics of 125I-labeled monoclonal antibody M75 specific for carbonic anhydrase IX, an intrinsic marker of hypoxia, in nude mice xenografted with human colorectal carcinoma. Int J Cancer. 2003;105(6):873–81. doi: 10.1002/ijc.11142. PMID: 12767076. [DOI] [PubMed] [Google Scholar]
- 27.Ahlskog JKJ, Schliemann C, Mårlind J, Qureshi U, Ammar A, Pedley RB, Neri D. Human monoclonal antibodies targeting carbonic anhydrase IX for the molecular imaging of hypoxic regions in solid tumours. Br J Cancer. 2009;101(4):645–57. doi: 10.1038/sj.bjc.6605200. PMID: 19623173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murri-Plesko MT, Hulikova A, Oosterwijk E, Scott AM, Zortea A, Harris AL, Ritter G, Old L, Bauer S, Swietach P, et al. Antibody inhibiting enzymatic activity of tumour-associated carbonic anhydrase isoform IX. Eur J Pharmacol. 2011;657(1–3):173–83. PMID: 21315712. doi: 10.1016/j.ejphar.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 29.Zatovicova M, Jelenska L, Hulikova A, Csaderova L, Ditte Z, Ditte P, Goliasova T, Pastorek J, Pastorekova S. Carbonic anhydrase IX as an anticancer therapy target: preclinical evaluation of internalizing monoclonal antibody directed to catalytic domain. Curr Pharm Des. 2010;16(29):3255–63. doi: 10.2174/138161210793429832. PMID: 20819068. [DOI] [PubMed] [Google Scholar]
- 30.Petrul HM, Schatz CA, Kopitz CC, Adnane L, McCabe TJ, Trail P, Ha S, Chang YS, Voznesensky A, Ranges G, et al. Therapeutic mechanism and efficacy of the antibody-drug conjugate BAY 79-4620 targeting human carbonic anhydrase 9. Mol Cancer Ther. 2012;11(2):340–49. PMID: 22147747. doi: 10.1158/1535-7163.MCT-11-0523. [DOI] [PubMed] [Google Scholar]
- 31.Pal SK, Agarwal N. Kidney cancer: findinga niche for girentuximab in metastatic renal cell carcinoma. Nat Rev Urol. 2016;13(8):442. doi: 10.1038/nrurol.2016.115. PMID: 27349371. [DOI] [PubMed] [Google Scholar]
- 32.Rojas G, Tundidor Y, Infante YC. High throughput functional epitope mapping: revisiting phage display platform to scan target antigen surface. mAbs. 2014;6(6):1368–76. doi: 10.4161/mabs.36144. PMID: 25484050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamin DC, Perdue SS. Site-directed mutagenesis in epitope mapping. Meth: A Companion to Meth Enzymol. 1996;9(3):508–15. doi: 10.1006/meth.1996.0058. PMID: 8812706. [DOI] [PubMed] [Google Scholar]
- 34.Buus S, Rockberg J, Forsström B, Nilsson P, Uhlen M, Schafer-Nielsen C. High-resolution mapping of linear antibody epitopes using ultrahigh-density peptide microarrays. Molecular and Cellular Proteomics. 2012;11(12):1790–800. doi: 10.1074/mcp.M112.020800. PMID: 22984286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bianchi M, Turner HL, Nogal B, Cottrell CA, Oyen D, Pauthner M, Bastidas R, Nedellec R, McCoy LE, Wilson IA, et al. Electron-microscopy-based epitope mapping defines specificities of polyclonal antibodies elicited during HIV-1 BG505 envelope trimer immunization. Immunity. 2018;49(2):288–300.e8. PMID: 30097292. doi: 10.1016/j.immuni.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaltashov IA, Bobst CE, Pawlowski J, Wang G. Mass spectrometry-based methods in characterization of the higher order structure of protein therapeutics. J Pharm Biomed Anal. 2020;184:113169. doi: 10.1016/j.jpba.2020.113169. PMID: 32092629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Opuni KFM, Al-Majdoub M, Yefremova Y, El-Kased RF, Koy C, Glocker MO. Mass spectrometric epitope mapping. Mass Spectrom Rev. 2018;37(2):229–41. doi: 10.1002/mas.21516. PMID: 27403762. [DOI] [PubMed] [Google Scholar]
- 38.Sheff JG, Farshidfar F, Bathe OF, Kopciuk K, Gentile F, Tuszynski J, Barakat K, Schriemer DC. Novel allosteric pathway of Eg5 regulation identified through multivariate statistical analysis of hydrogen-exchange mass spectrometry (HX-MS) ligand screening data. Mol Cell Proteom. 2017;16(3):428–37. doi: 10.1074/mcp.M116.064246. PMID: 28062800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang H, Yang H-S, Ki AY, Ko S-B, Kim KW, Shim CY, Kim K, Choi H-J, Chung KY. Conformational dynamics and functional implications of phosphorylated β-arrestins. Structure. 2020;28:314–323.e3. doi: 10.1016/j.str.2019.12.008. PMID: 31948726. [DOI] [PubMed] [Google Scholar]
- 40.Guo C, Steinberg LK, Cheng M, Song JH, Henderson JP, Gross ML. Site-specific siderocalin binding to ferric and ferric-free enterobactin as revealed by mass spectrometry. ACS Chem Biol. 2020;15(5):1154–60. doi: 10.1021/acschembio.9b00741. PMID: 31869199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang RY, Chen G. Higher order structure characterization of protein therapeutics by hydrogen/deuterium exchange mass spectrometry. Anal Bioanal Chem. 2014;406(26):6541–58. doi: 10.1007/s00216-014-7924-3. PubMed PMID: 24948090; PMID: 24948090. [DOI] [PubMed] [Google Scholar]
- 42.Huang RY, Krystek SR Jr., Felix N, Graziano RF, Srinivasan M, Pashine A, Chen G. Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction. MAbs. 2018;10(1):95–103. doi: 10.1080/19420862.2017.1393595. PubMed PMID: 29135326; PMID: 29135326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang RY, Kuhne M, Deshpande S, Rangan V, Srinivasan M, Wang Y, Chen G. Mapping binding epitopes of monoclonal antibodies targeting major histocompatibility complex class I chain-related A (MICA) with hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry. Anal Bioanal Chem. 2020;412(7):1693–700. doi: 10.1007/s00216-020-02409-x. PubMed PMID: 31993727; PMID: 31993727. [DOI] [PubMed] [Google Scholar]
- 44.Huang RY, Wang F, Wheeler M, Wang Y, Langish R, Chau B, Dong J, Morishige W, Bezman N, Strop P, et al. Integrated approach for characterizing bispecific antibody/antigens complexes and mapping binding epitopes with SEC/MALS, native mass spectrometry, and protein footprinting. Anal Chem. 2020;92(15):10709–16. PubMed PMID: 32639723; PMID: 32639723. doi: 10.1021/acs.analchem.0c01876. [DOI] [PubMed] [Google Scholar]
- 45.Zhang MM, Huang RY, Beno BR, Deyanova EG, Li J, Chen G, Gross ML. Epitope and paratope mapping of PD-1/Nivolumab by mass spectrometry-based hydrogen-deuterium exchange, cross-linking, and molecular docking. Anal Chem. 2020;92(13):9086–94. doi: 10.1021/acs.analchem.0c01291. PubMed PMID: 32441507; PMID: 32441507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grauslund LR, Calvaresi V, Pansegrau W, Norais N, Rand KD. Epitope and paratope mapping by HDX-MS combined with SPR elucidates the difference in bactericidal activity of two anti-NadA monoclonal antibodies. J Am Soc Mass Spectrom. 2021;32(7):1575–82. doi: 10.1021/jasms.0c00431. PubMed PMID: 33683906; PMID: 33683906. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Q, Yang J, Bautista J, Badithe A, Olson W, Liu Y. Epitope mapping by HDX-MS elucidates the surface coverage of antigens associated with high blocking efficiency of antibodies to birch pollen allergen. Anal Chem. 2018;90(19):11315–23. doi: 10.1021/acs.analchem.8b01864. PMID: 30170487. [DOI] [PubMed] [Google Scholar]
- 48.Zhu S, Liuni P, Ettorre L, Chen T, Szeto J, Carpick B, James DA, Wilson DJ. Hydrogen deuterium exchange epitope mapping reveals distinct neutralizing mechanisms for two monoclonal antibodies against diphtheria toxin. Biochemistry. 2018;acs.biochem.8b01123. doi: 10.1021/acs.biochem.8b01123. PMID: 30560647. [DOI] [PubMed] [Google Scholar]
- 49.Lim XX, Chandramohan A, Lim XYE, Crowe JE, Lok SM, Anand GS. Epitope and paratope mapping reveals temperature-dependent alterations in the dengue-antibody interface. Structure. 2017;25(9):1391–1402.e3. doi: 10.1016/j.str.2017.07.007. PMID: 28823471. [DOI] [PubMed] [Google Scholar]
- 50.Hvidt A, Linderstrøm-Lang K. Exchange of hydrogen atoms in insulin with deuterium atoms in aqueous solutions. Biochim Biophys Acta. 1954;14(C):574–75. doi: 10.1016/0006-3002(54)90241-3. PMID: 13198919. [DOI] [PubMed] [Google Scholar]
- 51.Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 2011;8(1):43–59. doi: 10.1586/epr.10.109. PMID: 21329427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenferink AG, McDonald PC, Gosselin M, Baardsnes J, Robert A, Cepero-Donates Y, Radinovic S, Salois P, Parat M, L’Abbé D, et al. Isolation and characterization of monoclonal antibodies against human carbonic anhydrase-IX. mAbs. 2021; in press. PMID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puchades C, Kűkrer B, Diefenbach O, Sneekes-Vriese E, Juraszek J, Koudstaal W, Apetri A. Epitope mapping of diverse influenza Hemagglutinin drug candidates using HDX-MS. Sci Rep. 2019;9(1):1–10. doi: 10.1038/s41598-019-41179-0. PMID: 30894620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zatovicova M, Jelenska L, Hulikova A, Ditte P, Ditte Z, Csaderova L, Svastova E, Schmalix W, Boettger V, Bevan P, et al. Monoclonal antibody G250 targeting CA IX: binding specificity, internalization and therapeutic effects in a non-renal cancer model. Int J Oncol. 2014;45(6):2455–67. PMID: 25230982. doi: 10.3892/ijo.2014.2658. [DOI] [PubMed] [Google Scholar]
- 55.Lenferink A, O’Connor M, inventors HIGH AFFINITY MONOCLONAL ANTIBODIES (MABS) AGAINST CELL SURFACE EXPRESSED HUMAN CARBONIC ANHYDRASE IX (HCA-IX), AND USES THEREOF patent WO/2019/204939. 2019.
- 56.Swayampakula M, McDonald PC, Vallejo M, Coyaud E, Chafe SC, Westerback A, Venkateswaran G, Shankar J, Gao G, Laurent EMN, et al. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene. 2017;36(45):6244–61. PubMed PMID: 28692057; PMID: 28692057. doi: 10.1038/onc.2017.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chafe SC, McDonald PC, Saberi S, Nemirovsky O, Venkateswaran G, Burugu S, Gao D, Delaidelli A, Kyle AH, Baker JHE, et al. Targeting hypoxia-induced carbonic anhydrase IX enhances Immune-checkpoint blockade locally and systemically. Cancer Immunol Res. 2019;7(7):1064–78. PubMed PMID: 31088846; PMID: 31088846. doi: 10.1158/2326-6066.CIR-18-0657. [DOI] [PubMed] [Google Scholar]
- 58.McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, Nemirovsky O, Gillespie JA, Karasinska JM, Kalloger SE, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology. 2019;157(3):823–37. PubMed PMID: 31078621; PMID: 31078621. doi: 10.1053/j.gastro.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, Auf Dem Keller U, Leung S, Huntsman D, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011;71(9):3364–76. PubMed PMID: 21415165; PMID: 21415165. doi: 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- 60.McIntyre A, Patiar S, Wigfield S, J-l L, Ledaki I, Turley H, Leek R, Snell C, Gatter K, Sly WS, et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clinical Cancer Research. 2012;18(11):3100–11. PMID: 22498007. doi: 10.1158/1078-0432.Ccr-11-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zatovičová M, Tarábková K, Švastová E, Gibadulinová A, Mucha V, Jakubíčková L, Biesová Z, Rafajová M, Gut MO, Parkkila S, et al. Monoclonal antibodies generated in carbonic anhydrase IX-deficient mice recognize different domains of tumour-associated hypoxia-induced carbonic anhydrase IX. J Immunol Meth. 2003;282(1–2):117–34. PMID: 14604546. doi: 10.1016/j.jim.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 62.Lenferink A, O’Connor MD, Marcil A, Durocher Y, inventorCARBONIC ANHYDRASE IX-SPECIFIC ANTIBODIES AND USES THEREOF patent WO2016/199097. 2016. 15/12/2016.
- 63.Avvaru BS, Busby SA, Chalmers MJ, Griffin PR, Venkatakrishnan B, Agbandje-mckenna M, Silverman DN, McKenna R. Apo-human carbonic anhydrase II revisited: implications of the loss of a metal in protein structure, stability, and solvent network. Biochemistry. 2009;48(31):7365–72. doi: 10.1021/bi9007512. PMID: 19583303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Divgi CR, Uzzo RG, Gatsonis C, Bartz R, Treutner S, Yu JQ, Chen D, Carrasquillo JA, Larson S, Bevan P, et al. Positron emission tomography/computed tomography identification of clear cell renal cell carcinoma: results from the REDECT trial. J Clin Oncol. 2013;31(2):187–94. PMID: 23213092. doi: 10.1200/JCO.2011.41.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warnaar SO, Ullrich S. inventorsCO-ADMINISTRATION OF CG250 AND IL-2 OR IFN-ALPHA FOR TREATING CANCER SUCH AS RENAL CELL CARCINOMA. United States patent US 8,828,381 B2. 2014.
- 66.Wöhl T, Böttger V, inventorsBINDING EPITOPES FOR G250 ANTIBODY patent US 2014/0017252 A1. 2014.
- 67.Ciani L, Cecchi A, Temperini C, Supuran CT, Ristori S. Dissecting the inhibition mechanism of cytosolic versus transmembrane carbonic anhydrases by ESR. Journal of Physical Chemistry B. 2009;113(42):13998–4005. doi: 10.1021/jp906593c. PMID: 19778001. [DOI] [PubMed] [Google Scholar]
- 68.Sowole MA, Konermann L. Effects of protein-ligand interactions on hydrogen/deuterium exchange kinetics: canonical and noncanonical scenarios. Anal Chem. 2014;86(13):6715–22. doi: 10.1021/ac501849n. PMID: 24904985. [DOI] [PubMed] [Google Scholar]
- 69.Konermann L, Rodriguez AD, Sowole MA. Type 1 and Type 2 scenarios in hydrogen exchange mass spectrometry studies on protein-ligand complexes. Analyst. 2014;139(23):6078–87. doi: 10.1039/c4an01307g. PubMed PMID: 25319399; PMID: 25319399. [DOI] [PubMed] [Google Scholar]
- 70.Wildes D, Marqusee S. Hydrogen exchange and ligand binding: ligand-dependent and ligand-independent protection in the Src SH3 domain. Protein Sci. 2005;14(1):81–88. doi: 10.1110/ps.04990205. PubMed PMID: 15576569; PMID: 15576569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Percy AJ, Rey M, Burns KM, Schriemer DC. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry-A review. Anal Chim Acta. 2012;721:7–21. doi: 10.1016/j.aca.2012.01.037. PMID: 22405295. [DOI] [PubMed] [Google Scholar]
- 72.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30(2):E9. doi: 10.1093/nar/30.2.e9. PubMed PMID: 11788735; PMID: 11788735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tom R, Bisson L, Durocher Y. Culture of HEK293-EBNA1 cells for production of recombinant proteins. CSH Protoc. 2008;2008:pdb prot4976. doi: 10.1101/pdb.prot4976. PubMed PMID: 21356792; PMID: 21356792. [DOI] [PubMed] [Google Scholar]
- 74.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21(2):163–70. PubMed PMID: 12536217; PMID: 12536217. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 75.Cochran JR, Kim YS, Olsen MJ, Bhandari R, Wittrup KD. Domain-level antibody epitope mapping through yeast surface display of epidermal growth factor receptor fragments. J Immunol Methods. 2004;287(1–2):147–58. doi: 10.1016/j.jim.2004.01.024. PubMed PMID: 15099763; PMID: 15099763. [DOI] [PubMed] [Google Scholar]
- 76.Rey M, Sarpe V, Burns KM, Buse J, Baker CA, Van Dijk M, Wordeman L, Bonvin AM, Schriemer DC. Mass spec studio for integrative structural biology. Structure. 2014;22(10):1538–48. doi: 10.1016/j.str.2014.08.013. PubMed PMID: 25242457; PMID: 25242457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edwards LA, Woo J, Huxham LA, Verreault M, Dragowska WH, Chiu G, Rajput A, Kyle AH, Kalra J, Yapp D, et al. Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of Integrin-linked kinase (ILK). Mol Cancer Ther. 2008;7(1):59–70. PMID: 18202010. doi: 10.1158/1535-7163.Mct-07-0329. [DOI] [PubMed] [Google Scholar]
- 78.Lou Y, Preobrazhenska O, Auf Dem Keller U, Sutcliffe M, Barclay L, McDonald PC, Roskelley C, Overall CM, Dedhar S. Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev Dyn. 2008;237(10):2755–68. doi: 10.1002/dvdy.21658. PMID: 18773493. [DOI] [PubMed] [Google Scholar]
- 79.Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol. 2014;4 JAN(January):1–14. doi: 10.3389/fphys.2013.00400. PMID: 24409151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, Bou-Assaf GM, Engen JR, Englander SW, Faber J, et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Meth. 2019;16(7):595–602. PMID: 31249422. doi: 10.1038/s41592-019-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.