

ABSTRACT
Glioma is the most common primary malignant brain tumor with poor survival and limited therapeutic options. The non-psychoactive phytocannabinoid cannabidiol (CBD) has been shown to be effective against glioma; however, the molecular target and mechanism of action of CBD in glioma are poorly understood. Here we investigated the molecular mechanisms underlying the antitumor effect of CBD in preclinical models of human glioma. Our results showed that CBD induced autophagic rather than apoptotic cell death in glioma cells. We also showed that CBD induced mitochondrial dysfunction and lethal mitophagy arrest, leading to autophagic cell death. Mechanistically, calcium flux induced by CBD through TRPV4 (transient receptor potential cation channel subfamily V member 4) activation played a key role in mitophagy initiation. We further confirmed TRPV4 levels correlated with both tumor grade and poor survival in glioma patients. Transcriptome analysis and other results demonstrated that ER stress and the ATF4-DDIT3-TRIB3-AKT-MTOR axis downstream of TRPV4 were involved in CBD-induced mitophagy in glioma cells. Lastly, CBD and temozolomide combination therapy in patient-derived neurosphere cultures and mouse orthotopic models showed significant synergistic effect in both controlling tumor size and improving survival. Altogether, these findings showed for the first time that the antitumor effect of CBD in glioma is caused by lethal mitophagy and identified TRPV4 as a molecular target and potential biomarker of CBD in glioma. Given the low toxicity and high tolerability of CBD, we therefore propose CBD should be tested clinically for glioma, both alone and in combination with temozolomide.
Abbreviations: 4-PBA: 4-phenylbutyrate; AKT: AKT serine/threonine kinase; ATF4: activating transcription factor 4; Baf-A1: bafilomycin A1; CANX: calnexin; CASP3: caspase 3; CAT: catalase; CBD: cannabidiol; CQ: chloroquine; DDIT3: DNA damage inducible transcript 3; ER: endoplasmic reticulum; GBM: glioblastoma multiforme; GFP: green fluorescent protein; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MTOR: mechanistic target of rapamycin kinase; PARP1: poly(ADP-ribose) polymerase; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; SLC8A1: solute carrier family 8 member A1; SQSTM1: sequestosome 1; TCGA: The cancer genome atlas; TEM: transmission electron microscopy; TMZ: temozolomide; TRIB3: tribbles pseudokinase 3; TRPC: transient receptor potential cation channel subfamily C; TRPV4: transient receptor potential cation channel subfamily V member 4
KEYWORDS: Cannabidiol, cannabinoid, glioblastoma, glioma, mitophagy, trpv4
Introduction
Glioma is a primary brain tumor that is categorized into different malignant grades (by WHO): diffuse astrocytoma (grade II), anaplastic astrocytoma (grade III), and glioblastoma multiforme cell tumor (GBM; grade IV). Despite recent advances in diagnosis and treatment, glioma remains one of the most lethal cancers with a median survival of approximately one year [1]. The standard clinical therapy for glioma is surgery followed by radiation or temozolomide (TMZ)-based chemotherapy. Although various new treatments and techniques have been developed during the past decade, the survival of patients with glioma has not been significantly improved [2]. Therefore, new therapies for this devastating disease are urgently needed.
In recent years, the role of autophagy in malignant progression and response to treatment has become the focus of research in many types of cancer. Autophagy is a well characterized physiological process that is responsible for removing dysfunctional or damaged organelles via lysosomal degradation to sustain cell survival [3]. In tumor cells, however, autophagy has been demonstrated as a double-edge sword [4]; in addition to the above mentioned pro-survival roles, excessive or sustained autophagy may be an inducer of tumor cell death, particularly in apoptosis defective cancer cells. In fact, many anti-cancer drugs have been shown to induce autophagic cell death [5]. Mitophagy, a specific form of autophagy, is the major pathway for the degradation of dysfunctional or damaged mitochondria [6]. Mitophagy plays a crucial role in maintaining cellular homeostasis by sensing metabolic demand, keeping mitochondria quality, and protecting cells against the deleterious effects of damaged mitochondria. There are also increasing evidences for a pro-death function of mitophagy in response to a variety of anti-cancer drugs [7]. Although glioma cells have been shown to be vulnerable to autophagic cell death, the role of mitophagy in the mechanism of anti-glioma drugs is less understood.
Cannabinoids, the major active components of medical cannabis, have been shown to possess a large spectrum of pharmacological activity in a variety of diseases [8]. Several studies have shown that cannabinoids have anti-proliferative and anti-invasive activities in various cancer types [9]. However, most studies were focused on tetrahydrocannabinol (Δ9-THC), the use of which is often limited by unwanted psychoactive effects [10]. Compared to THC, cannabidiol (CBD, Figure 1A), the non-psychoactive isomer of THC, may have better potential as an anti-cancer agent for glioma. Firstly, CBD has no psychotropic activity and low binding affinity to CNR1/CB1 and CNR2/CB2 receptors and is considered as a non-intoxicating drug [11]. Secondly, many antitumor drugs are ineffective against glioma in clinic because they fail to penetrate the blood-brain barrier (BBB). CBD, on the contrary, is a hydrophobic molecule that readily cross the BBB to reach the site of brain tumor [12]. Thirdly, CBD embraces good neuro-physiological activity and has a therapeutic effect on a variety of neurological diseases. It has recently been approved by the FDA for the treatment of epilepsy in children. These advantages of CBD attract us to study the antitumor properties and mechanism of action of CBD in glioma. Although several reports have demonstrated that CBD possesses anti-tumor effects in different cancers, the underlying mechanisms are not fully understood [13]. In addition, molecular targets of CBD in cancer remains elusive.
Figure 1.
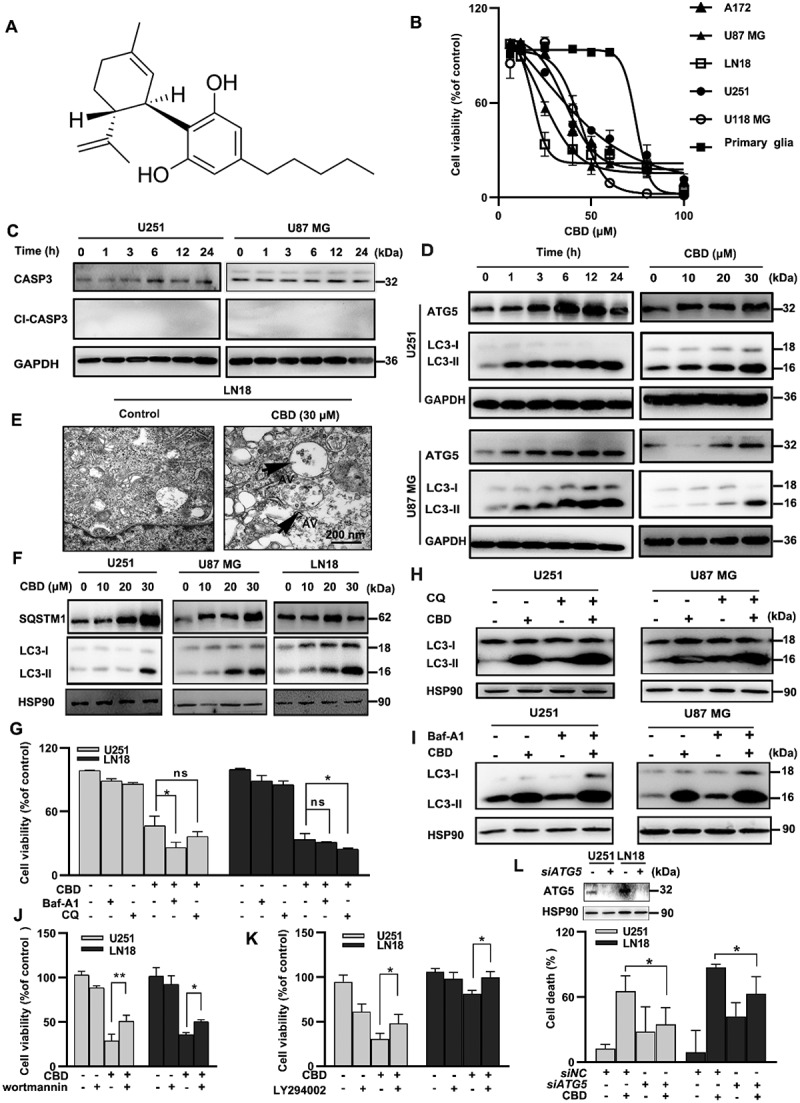
CBD induced lethal autophagy in glioma cells. (A) Molecular structure of CBD. (B) Growth inhibition effect of CBD on A172, U87 MG, LN18, U251, U118 MG and primary glia cells. Cell viability was measured by MTT after 72 h of CBD treatment. (C) Effect of CBD treatment on CASP3 cleavage in glioma cells. Cells were treated with 30 µM CBD for different times and subjected to western blotting. (D) Effect of CBD treatment on autophagy related proteins. Cells were treated with 30 µM CBD for different times (left) or different concentrations for 24 h (right); LC3 and ATG5 levels were analyzed by western blotting. (E) Ultrastructural features of LN18 cells treated with 30 µM CBD for 24 h were analyzed by electron microscopy. Arrows indicate autophagosomes. Scale bar: 2 µm. (F) Effect of CBD treatment on SQSTM1 protein levels in glioma cells. Cells were treated with different concentrations of CBD for 24 h; SQSTM1 and LC3 levels were analyzed by western blotting. (G) Effect of autophagy inhibitors CQ and Baf-A1 on CBD-induced growth inhibition in glioma cells. U251 and LN18 cells were treated with CBD (20 μM for 48 h) alone or in combination with Baf-A1 (100 nM) or CQ (20 μM); cell number was determined by MTT. (H-I) Effect of autophagy inhibitors CQ and Baf-A1 on CBD-induced LC3 expression in glioma cells. Cells were pre-treated with CQ or Baf-A1 for 1 h and then treated with 20 µM CBD for another 24 h; LC3 levels were analyzed by western blotting. (J-K) Effect of autophagy inhibitors wortmannin and LY294002 on CBD-induced cell death in glioma cells. Cells were pretreated with wortmannin (20 μM, J) or LY294002 (5 μM, K) for 1 h and then treated with CBD (20 μM) for 48 h, cell viability was determined by MTT. (L) Effect of ATG5 knockdown on CBD-induced cell death in glioma cells. Cells were transfected with control or ATG5 siRNA for 48 h, followed by treatment with 20 μM CBD for another 48 h, cell viability was determined by MTT. Data are shown as mean ± S.D. and are representative of three independent experiments. *P < 0.05; **P < 0.01
In this study, we dissected the molecular mechanisms underlying the antitumor effects of CBD in glioma. Our results demonstrated that CBD elicited its antitumor activity through the induction of lethal mitophagy in glioma cells. We also identified TRPV4 as a target of CBD in glioma. These investigations shed light on the antitumor potential of CBD and may pave way for the development of novel cannabinoid-based therapeutics for glioma treatment.
Results
CBD induced autophagic cell death in glioma cells
We first evaluated the growth inhibitory effect of CBD (Figure 1A) in human glioma cell lines A172, U251, U87 MG, U118 MG and LN18, with primary glia cells as the normal tissue control. CBD treatment dose-dependently reduced cell viability in all glioma cell lines, with IC50 values ranging from 20 to 30 μM, at which concentration no cytotoxic effect was observed in primary glia cells (Figure 1B). The growth inhibitory effect of CBD was also confirmed by colony formation assays (Fig. S1A) and U87 MG xenograft experiment in vivo (Fig. S1B-D). To examine the nature of CBD-induced cell death, we first measured several apoptosis related biomarkers, the pan-caspase inhibitor Z-VAD-FMK failed to protect glioma cells from CBD-induced cell death (Fig. S1E); 24 h CBD treatment failed to induce CASP3 (caspase-3) cleavage in glioma cells (Figure 1C); apoptosis signal was only measurable after extended treatment (48 or 72 h) or at higher CBD concentrations (Fig. S1F-G). These data suggested that apoptosis is not the primary driving force of cell death. In addition, treatment with necrostatin-1, an inhibitor of necroptosis, did not reverse CBD-induced cell death (Fig. S1H). On the contrary, the expression of autophagy biomarkers (LC3-II, ATG5) in glioma cells were induced by CBD treatment in a time-and dose-dependent manner (Figure 1D). An increasing number of autophagosomes were also found in CBD-treated cells by transmission electron microscopy (TEM) imaging (Figure 1E), confirming the induction of autophagy by CBD in glioma cells.
Autophagy is a dynamic, multi-stage process including autophagosome formation, autophagosome-lysosome fusion, and autophagosome degradation; an increase in the autophagosome marker LC3-II reflects the initiation of the autophagic process but not necessarily its completion; the cellular levels of SQSTM1/p62 protein, which is both an autophagy substrate and an ubiquitin-binding scaffold protein that binds to LC3-II in autophagosomes and is subsequently degraded after the formation of autolysosomes, could better reflect the status of autophagic flux in the cell. The interaction between SQSTM1 and LC3 was readily detected in CBD-treated cells by IP analysis (Fig. S1I); SQSTM1 was also found to colocalize with the lysosome marker LAMP2 (Fig. S1J), indicating the proper initiation of the autophagy flux; however, the total protein level of SQSTM1 was significantly increased after CBD treatment (Figure 1F), indicating the interruption of the final degradation step; moreover, when tandem fluorescent-tagged LC3 (mRFP-EGFP-LC3) was used to monitor the autophagic flux, more puncta dots of yellow or green were observed in the CBD treatment group (Fig. S1K), indicating the impairment of the autolysosome degradation process. Collectively, these results demonstrated that the interruption of autophagic flux occurred in CBD-treated glioma cells, which may contribute to CBD-induced cell death.
To further prove whether CBD-induced autophagy is protective or lethal to glioma cells, we first used two classical autophagy inhibitors CQ and Baf-A1 which prevent the late stage of autophagic flux by inhibiting the fusion of autophagosomes with lysosomes [14]. Autophagy inhibition with either CQ or Baf-A1 significantly enhanced CBD-induced cell death and accumulation of LC3-II protein (Figure 1G-I). On the contrary, growth inhibitory effect of CBD was blocked by the inhibitors of early stage autophagic flux wortmannin or LY294002 (Figure 1J-K). Additionally, blocking the early steps of autophagy via knocking down of ATG5 could also revert growth inhibition induced by CBD (Figure 1L). Altogether, these findings demonstrated that induction of autophagy contributed to glioma cell death after CBD treatment.
CBD induced mitochondrial dysfunction and mitophagy in glioma cells
Next, we investigated the form of autophagy caused by CBD in glioma cell lines. First, we found that CBD treatment led to the reduction in mitochondria quantity (Fig. S2A-B). Second, CBD treatment also led to mitochondria damage as evidenced by decrease in ATP levels, mitochondrial membrane potential (MMP) and increase in mitochondrial reactive oxygen species (ROS) levels (Fig. S2C-F). Notably, primary glia cells are much more resistant to CBD-induced mitochondria damage compared to cancer cells (Fig. S2G-I). Thirdly, CBD-induced mitochondria damage was also confirmed by morphological damage and mitochondrial ultrastructural changes under TEM imaging (Fig. S2J), suggesting the occurrence of mitochondrial fission or even fragmentation after CBD treatment, which was further confirmed by an increase in the expression level of mitochondrial fission protein DNM1L/Drp1 (Fig. S2K) and an decrease of mitochondrial compartmental proteins such as VDAC1, COX4I1/COXIV, and TOMM20 in CBD-treated cells (Fig. S2L). Moreover, when subjected to transcriptome analysis, the 30 most significantly changed genes after CBD treatment were enriched in the first 10 GO terms, the majority of which were associated with mitochondria functions (Fig. S2M). Together, these results indicated that CBD treatment could induced mitochondria loss and impair mitochondrial function in glioma cells.
Previous studies show that mitochondrial dysfunction could induce PINK1 translocation to mitochondrial membrane and induce mitophagy [15]. Hence, we further investigated whether CBD could induce mitophagy in glioma. We first used a reporter for mitophagy, mt-Keima-COX8 (Figure 2A), for detecting mitophagy signal in cells. CBD treatment induced an apparent green-to-red fluorescence shift in glioma cells stably overexpressing mt-Keima, indicating the engulfment of damaged mitochondria into autolysosomes (Figure 2A). We also observed the colocalization between MT-CO2 (mitochondrially encoded cytochrome c oxidase subunit II) and LAMP2 as well as between mitochondrial compartmental protein TOMM20 and LC3 after CBD treatment (Figure 2B, Fig. S3A). These immunofluorescence results indicated the formation of autophagosome containing damaged mitochondria in cells, which was also confirmed by TEM (Figure 2C). To investigate whether mitophagy was the major type of autophagy induced by CBD treatment, the involvement of other types of autophagy (ribophagy, reticulophagy, pexophagy) were then examined. LC3 fluorescence signal were found to predominately co-localize with the mitochondria marker MT-CO2 or TOMM20, but not with RPS3 (ribosome marker), Calnexin (endoplasmic reticulum marker), Catalase (peroxisome marker), or RSC1A1 (Golgi body marker) (Fig. S3B-C). The conversion of LC3-I to LC3-II was observed in a dose-dependent manner along with PINK1 and PRKN enrichment in the mitochondria fraction of CBD-treated glioma cells (Figure 2D). Moreover, the expression of SQSTM1 and LC3-II was also enriched in the mitochondria fraction after CBD treatment (Figure 2E). Collectively, these results suggested mitophagy is the main form of CBD-induced autophagy in glioma.
Figure 2.
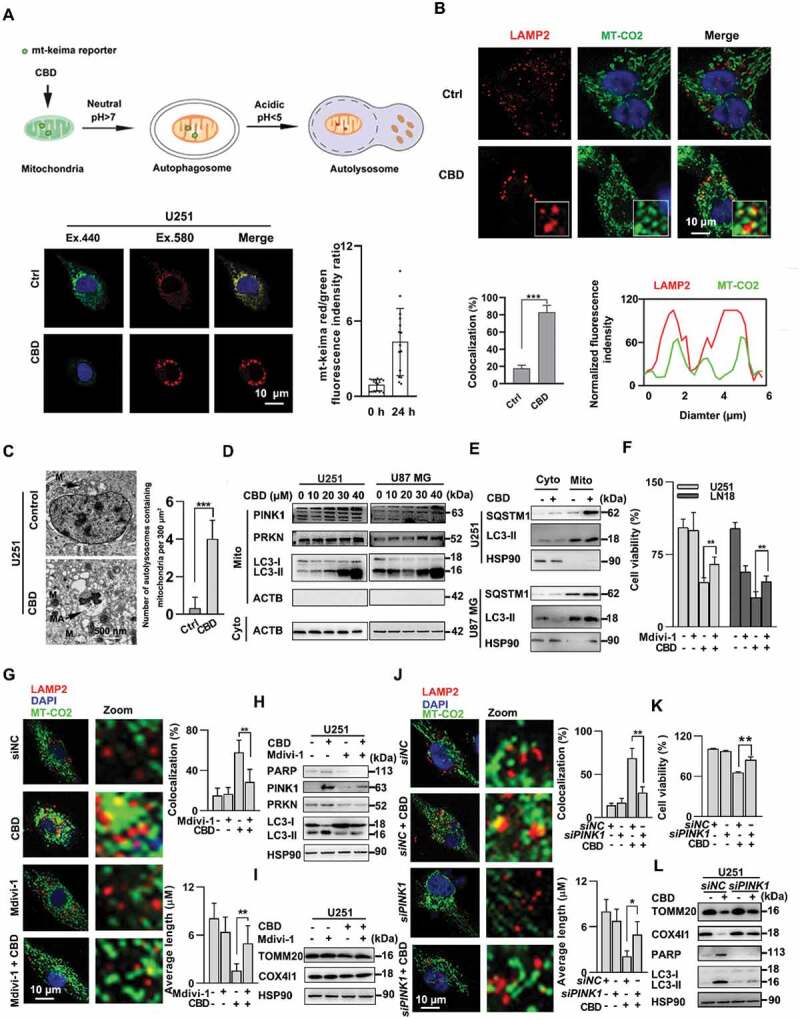
CBD induced mitochondrial dysfuction and mitophagy in glioma cells. (A) Detection of mitophagy induction using the mt-keima reporter assay in CBD treated glioma cells. U251 cells were first transfected with lentivirus carrying the mitochondria-targeting form of Keima (mt-Keima-COX8) for 48 h followed by 30 µM CBD treatment for another 24 h. The amount of mitchondria in the lysosomal compartment was detected using confocal microscopy. The ratio between red and green fluorescence intensities were quantified and shown to the right; n = 10 cells. (B) Detection of mitophagy induction using the LAMP2-MT-CO2 colocalization assay. U251 cells were treated with 30 µM CBD for 24 h, and the intracellular LAMP2 (red) and MT-CO2 (green) were detected by immunofluorescence. Quantification of LAMP2-MT-CO2 colocalization was performed using 50 randomly selected cells. (C) Representative images of transmission electron microscope showing damaged mitochondria was trapped in autophagosomes after 24 h of 30 µM CBD treatment in U251 cells. Scale bar: 500 nm. (D)Immunoblotting of PINK1 and PRKN in the mitochondrial fraction of glioma cells treated with 30 µM CBD for 24 h. (E) Immunoblotting of SQSTM1 and LC3-II in the cytosolic (Cyto) and mitochondrial (Mito) fractions of glioma cells treated with 30 µM CBD for 24 h. (F) Effect of the mitophagy inhibitor Mdivi-1 on CBD-induced cell death in glioma cells. Cells were pretreated with Mdivi-1 (2.5 μM) for 1 h and then treated with CBD (20 μM) for 48 h, cell viability was determined by the MTT assay. (G-I) Effect of Mdivi-1 on CBD-induced mitophagy in glioma cells. U251 cells were pretreated with mdivi-1 (2.5 μM) for 1 h and then treated with CBD (20 μM) for 24 h. Mitophagy induction was evaluated by both LAMP2-MT-CO2 colocalization (G) and immunoblotting of PINK1, PRKN, LC3, TOMM20, and COX4I1 (H, I). (J) Effect of PINK1 knockdown on CBD-induced mitophagy in glioma cells. U251 cells were transfected with control or PINK1 siRNA for 48 h before treatment with CBD (20 μM) for another 24 h, mitophagy induction was evaluated by both LAMP2-MT-CO2 colocalization. (K) Effect of PINK1 knockdown on CBD-induced cell death in glioma cells. U251 cells were transfected with control or PINK1 siRNA for 48 h before treatment with CBD (20 μM) for another 24 h, cell viability was determined by the MTT assay. (L) Effect of PINK1 knockdown on CBD-induced mitophagy related protein in glioma cells. Cells were treated as in J and levels of PARP, LC3, TOMM20, and COX4I1 were analyzed by western blotting. Data are mean ± S.D. and are representative of three independent experiments. *P < 0.05; **P < 0.01, ***P < 0.001
We next asked whether CBD-induced mitophagy is required for its growth inhibition effect in glioma cells. Mdivi-1, a mitochondrial division/mitophagy inhibitor, significantly inhibited the toxicity of CBD in glioma cells (Figure 2F). Mdivi-1 also suppressed the induction of mitophagy as shown by LAMP2-MT-CO2 colocalization as well as western blotting of mitophagy related proteins (Figure 2G-I). Similarly, siRNA against PINK1 could also revert the effect of CBD on mitochondrial damage and cell death (Figure 2J-L, Fig. S3D), suggesting that induction of mitophagy is the major mechanism underlying the antitumor effect of CBD in glioma.
TRPV4 is a potential target of CBD in glioma
We next sought to identify the molecular target of CBD that mediates its mitophagy induction and growth inhibitory effects. At present, more than 65 discrete molecular targets have been proposed for CBD [16], including ion channels, receptors, transporters, and enzymes. However, the target of CBD in glioma is still unclear. We observed that a large number of vacuoles appeared in glioma cells in a time-dependent manner after CBD treatment (Figure 3A; Fig. S4A). Extensive vacuolization have been shown to be characteristic of paraptosis that results from the dilation of the endoplasmic reticulum and mitochondria [17]. However, inhibitors of paraptosis failed to revert the vacuolization induced by CBD, ruling out this possibility (Fig. S4B). Calcium ion has been reported to be positively associated with paraptosis [18]. Surprisingly, when glioma cells were pre-treated with extracellular calcium chelators (BAPTA) and then incubated with CBD for 4 h, vacuolization was significantly reduced (Figure 3B), suggesting that induction of extracellular calcium flux may play a role in CBD-induced cellular vacuolation. Since Ca2+ released from intracellular calcium stores and extracellular Ca2+ influx were two possible causes for an increase in free intracellular Ca2+, we then used the Fluo-4AM probe to monitor calcium flux. CBD induced a much higher level of extracellular Ca2+ influx than intracellular Ca2+ influx (Fig. S4C-E), implying that the increase in intracellular free Ca2+ was mainly caused by the absorption of extracellular Ca2+ rather than a release of endogenous Ca2+ stores. Next, antagonists of various calcium ion channels (TRPV, TRPC, TRPM and CBARP/VGCC), and blockers of GPCRs including CNR1/CB1, CNR2/CB2 and GPR55 were used to pre-treat glioma cells for 1 h before CBD treatment, the GPR55 antagonist (CID16020046), CB1 antagonist (rimonabant), CB2 antagonist (SR144528), VDAC1 inhibitor (DIDS), TRPC blocker (SKF96365), and T-type calcium channel blocker (amiloride) all failed to rescue growth inhibition induced by CBD; however, the TRPV channel blocker RuR could largely reverse the effect of CBD in glioma cells (Figure 3C-D). We then used specific blockers of each ion channel in the TRPV family and found that the TRPV4 antagonist GSK2193874, but not TRPV1 or TRPV2 inhibitors (tranilast), could revert the effect of CBD on glioma cells (Figure 3C-D; Fig. S4F). Subsequently, we found that GSK2193874 could significantly reduce the calcium influx induced by CBD (Figure 3E). In addition, shTRPV4 could also partly reverse the antitumor effect of CBD in U87 MG and U251 cells (Figure 3F-G, Fig. S4G). The above results suggested that TRPV4 is involved in the growth inhibitory effect of CBD in glioma cells. Previous studies have demonstrated that CBD may interact with TRPV2 through a hydrophobic pocket located between S5 and S6 helices [19]. Since TRPV2 and TRPV4 have high similarity in gene sequence, we established a homology model of TRPV4 and simulated the binding between CBD and TRPV4. The docking analyses demonstrated that CBD interacted mainly with residues on S6 helices of TRPV4 by making sigma-π interactions with domains I, III, and IV in the channel’s inner pore (Figure 3H). These results suggested that TRPV4 is a potential target of CBD in glioma.
Figure 3.
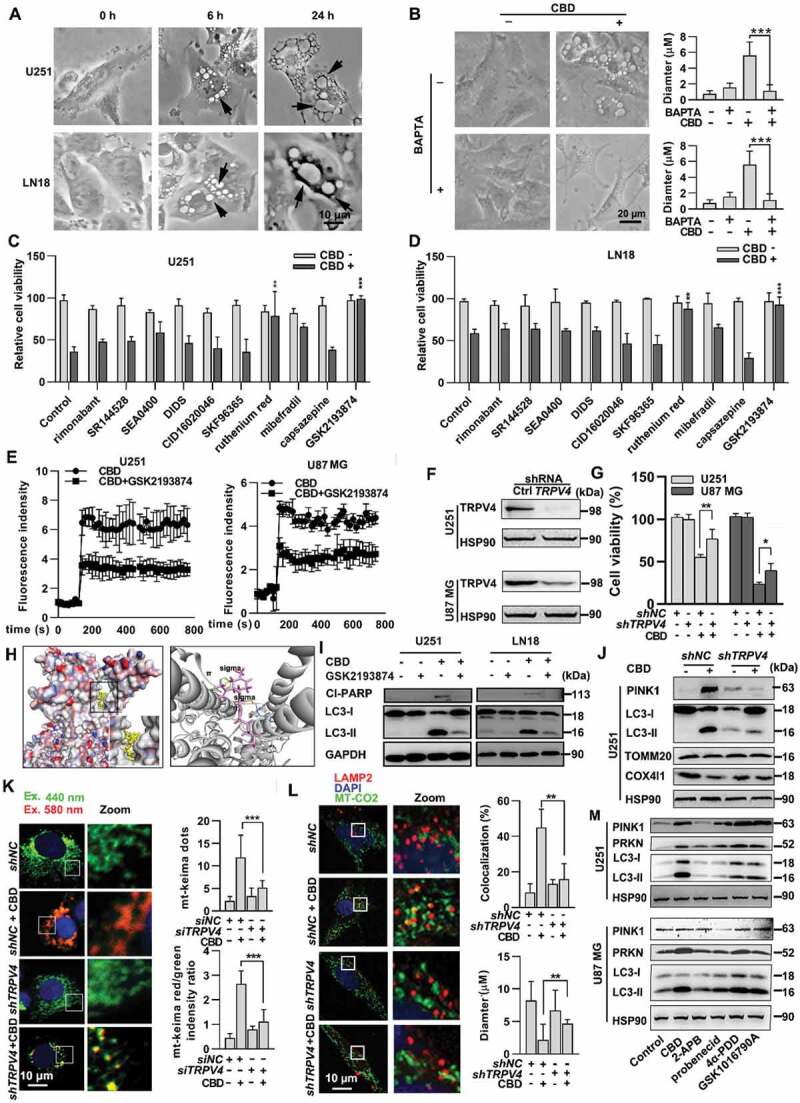
TRPV4 is a potential target of CBD in glioma. (A) Morphological changes in U251 and LN18 cells after treatment with 30 μM of CBD at indicate times. Arrows indicate vacuolation in cells. (B) Effect of the calcium chelator BAPTA on CBD-induced vacuolation in glioma cells. U251 cells were treated with 30 μM CBD for 6 h with or without 10 μM BAPTA (pretreatment for 1 h). Quantification on the number and diameter of vacuoles was performed using 50 cells. (C-D) Effect of various ion channel inhibitors on CBD-induced glioma cell death. U251 or LN18 cells were pretreated with rimonabant (1 μM), SR144528 (1 μM), SEA0400 (10 μM), DIDS (10 μM), CID16020046 (1 μM), SKF96365 (1 μM), RuR (1 μM), mibefradil (2.5 uM), capsazepine (1 μM), or GSK2193874 (5 μM) for 1 h and then treated with CBD (30 μM) for 48 h. Cell viability was determined by the MTT assay. (E) Effect of the TRPV4 inhibitor GSK2193874 on CBD-induced calcium flux in glioma cells. U251 or U87 MG cells were treated with 30 μM CBD with or without GSK2193874; Ca2+ flux was detected by using the Ca2+ indicator Fluro-4AM. (F) Knockdown efficiency of TRPV4 in U251 and U87MG cells was evaluated by western blotting. (G) Effect of TRPV4 knockdown on CBD-induced cell death in glioma cells. Cells with stable TRPV4 knockdown were treated with CBD for 48 h, cell viability was determined by the MTT assay. (H) Molecular docking analysis of the binding between CBD and TRPV4. Left, a homology structure model of TRPV4 in complex with CBD; right, an enlarged snapshot of the proposed binding site in TRPV4, where CBD binds in a hydrophobic pocket located between S5 and S6 helices of adjacent subunits. (I) Effect of TRPV4 inhibitor on CBD-induced mitophagy in glioma cells. U251 or U87 MG cells were treated with 20 μM CBD with or without GSK2193874 pretreatment (5 μM for 1 h), LC3 and Cl-PARP levels were analyzed by western blotting. (J-L) Effect of TRPV4 knockdown on CBD-induced mitophagy in glioma cells. U251 cells stably transfected with shTRPV4 were treated with 30 μM CBD for 24 h. Mitophagy related proteins including PINK1, LC3, TOMM20 and COX4I1 were analyzed by western blotting (J). Mitophagy induction was also evaluated by the mt-Keima reporter assay (K) and LAMP2-MT-CO2 colocalization (L). (M) Effect of TRPV channel agonists on the induction of mitophagy in glioma cells. U251 and U87 MG cells were treated with either 30 μM CBD or different TRPV agonists including 2-APB (20 μM), probenecid (20 μM), 4α-PDD (1.5 μM), or GSK1016790A (2 μM) for 24 h, levels of PINK1, PRKN and LC3 were then analyzed by western blotting. Data are shown as mean ± SD, and are representative of three independent experiments. *P < 0.05; **P < 0.01, ***P < 0.001
To further assess whether CBD-induced mitophagy is TRPV4-dependent, we examined the conversion of LC3-I to LC3-II and the levels of mitophagy-related proteins with TRPV4 inhibition or knockdown. GSK2193874 remarkably reverted LC3-II conversion and suppressed the expression of mitophagy-related proteins (Figure 3I). Similarly, shTRPV4 significantly alleviated CBD induced mitochondrial damage and mitophagy as shown by immunofluorescence and western blotting (Figure 3J-L). Furthermore, we compared the potency of specific agonists of each TRPV family member on mitophagy induction in glioma cells. The TRPV4 agonists (4α-PDD, GSK1016790A), but not 2-APB (TRPV1, TRPV2, TRPV3 agonist) or probenecid (TRPV2 agonist), could phenocopy CBD in terms of mitophagy induction (Figure 3M). Collectively, these results indicated that activation of TRPV4 is a key event for the induction of mitophagy in CBD treated glioma cells.
TRPV4 is a potential biomarker and therapeutic target in glioma
To gain insight into whether TRPV4 can be used as a biomarker for clinical diagnosis and treatment of glioma, bioinformatics analysis was performed to identify the relationship between TRPV4 expression and glioma grades using TGCA datasets (through UALCAN). The expression of TRPV4 was positively correlated with glioma tumor grade and negatively correlated with overall survival (Figure 4A-C). WGCNA analysis was performed in the TCGA dataset to identify the modules related to the expression levels of common markers of glioma. One module (“ME red”), which was significantly associated with TRPV4 expression level, was identified. “ME red” was positively associated with the expression levels of markers (RRTM2, TOP2A, ASPM, PBK, and EZH2) [20,21], and negatively associated with markers (CHD5, AQP4, and PCDH9) (Figure 4D), which suggested that TRPV4 could be a potential prognostic maker and therapeutic target for glioma. The above results were also confirmed in another dataset GSE50161 (Figure 4E). To validate the differential expression of TRPV4 in glioma patients, we analyzed TRPV4 protein levels in a commercial tissue array containing 126 glioma patient samples by immunohistochemistry (IHC) staining, much higher levels of TRPV4 protein expression was detected in patients with higher tumor grades (Figure 4F). Moreover, in an independent set of home-collected glioma patient samples, TRPV4 overexpression was also confirmed by IHC staining in the tumor tissue of glioma patients compared to normal control (Figure 4G-H). Collectively, these data demonstrated that TRPV4 is a potential biomarker for prognosis and a therapeutic target in glioma patients.
Figure 4.
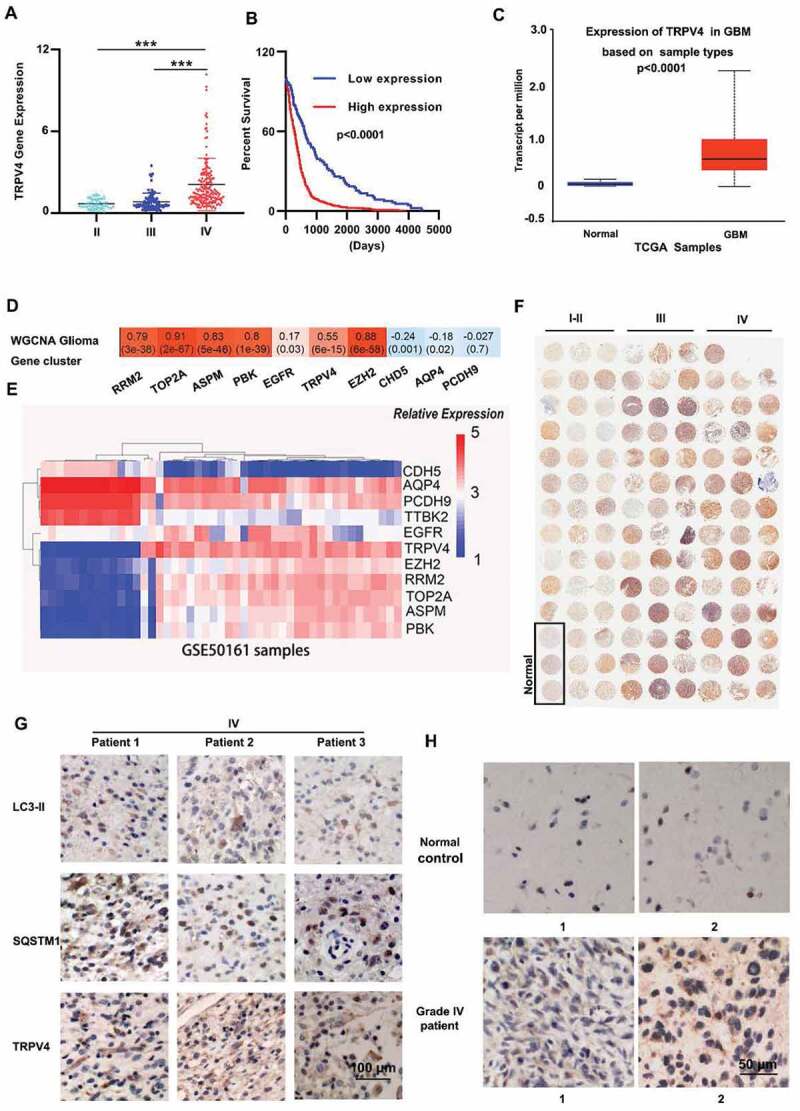
TRPV4 is a potential biomarker for clinical diagnosis and treatment of glioma. (A-B) Correlation analysis of TRPV4 expression and tumor grade (A) or overall survival (B) in The Cancer Genome Atlas (TCGA) dataset. (C) mRNA expression levels of TRPV4 in GBM samples and paired normal control from the UALCAN database. (D) Correlation analysis between module eigengenes and common markers of glioma. WGCNA results demonstrated that TRPV4 gene was in the Red Module (ME Red). Red represents a positive correlation between ME and the specified glioma associated gene, whereas blue represents a negative correlation. Correlation coefficients were calculated using Pearson correlation with P-values in parentheses. (E) Correlation analysis between TRPV4 and glioma related molecular markers in the GSE50161 dataset. (F) Immunohistochemical determination of TRPV4 expression levels in a commercial tissue array of 126 glioma patients. (G) Representative immunohistochemistry images of the expression of TRPV4, SQSTM1, LC3-II in high-grade glioma patient samples. (H) Differential expression of TRPV4 protein in human GBM samples and normal control samples
CBD induced lethal mitophagy via the ATF4-DDIT3-TRIB3-AKT-MTOR axis
Next, to further dissect the molecular mechanism underlying CBD-induced mitophagy in glioma cells, we performed transcriptome analysis in U251 cells. A total of 27,549 genes were analyzed and 653 significant differentially expressed genes (DEGs) were identified, among which 227 genes were downregulated while 426 genes were upregulated compared to control. Specifically, the expression of CHAC1, ATF4, ATF3, DDIT3, TRIB3 and SQSTM1 were significantly upregulated in CBD treated cells (Figure 5A-B). We also identified upregulation of genes involved in EIF2AK3/PERK-mediated unfolded protein response and endoplasmic reticulum (ER) stress response by GO enrichment analysis and cluster analysis (Fig. S5A-B). In agreement with the transcriptome analysis, the levels of ER stress markers including DDIT3/CHOP, EIF2A, ATF4, and EIF2AK3/PERK were also significantly upregulated in a dose- and time-dependent manner in glioma cell lines following CBD treatment (Figure 5C-D).
Figure 5.
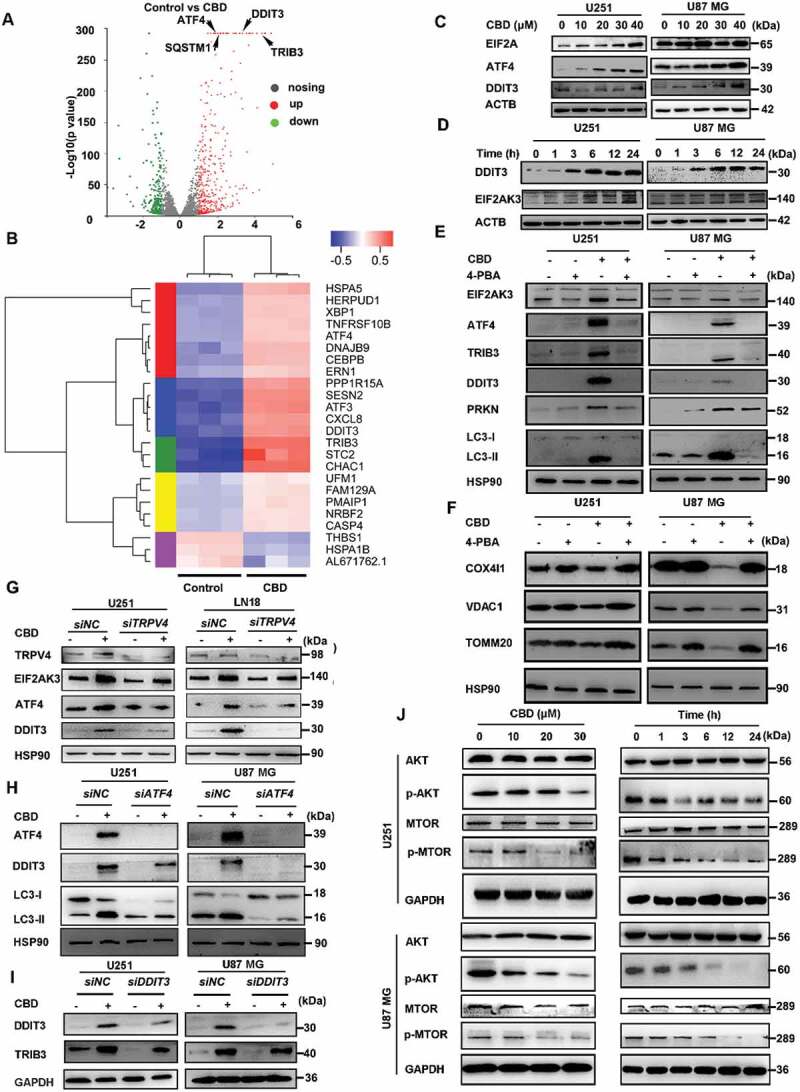
CBD induces ER stress and mitophagy via the ATF4-DDIT3-TRIB3-AKT1-MTOR axis. (A) Transcriptome analysis of CBD-treated U251 cells. Volcano plot showing the up- and down-regulated genes, in red and green colors, respectively. Cells were treated with 30 μM CBD for 12 h. (B) Clustering analysis of stress-related genes in CBD-treated U251 cells. (C) Immunoblotting of ER stress markers EIF2A, DDIT3 and ATF4 in glioma cells after 24 h treatment with indicated concentrations of CBD. (D) Immunoblotting of DDIT3 and EIF2AK3 in glioma cells treated with 30 μM CBD for indicated times. (E-F) Effect of the ER stress inhibitor 4-PBA on CBD-induced mitophagy. Cells were treated with 20 μM CBD for 24 h with or without pretreatment with 4-PBA (1 mM for 30 min); expression levels of ER stress and mitophagy related proteins were analyzed by western blotting. (G) Effect of TRPV4 knockdown on CBD-induced ER stress. Cells with stable TRPV4 knockdown were treated with 30 μM CBD for 24 h, levels of TRPV4, EIF2AK3, ATF4 and DDIT3 were analyzed by western blotting. (H) Effect of ATF4 knockdown on CBD-induced ER stress and mitophagy. Cells were transfected with control or ATF4 siRNA for 48 h before treatment with CBD for another 24 h, express levels of ATF4, DDIT3, and LC3 were analyzed by western blotting. (I) Effect of DDIT3 knockdown on CBD-induced TRIB3 upregulation. Cells were transfected with control or DDIT3 siRNA for 48 h before treatment with CBD for another 24 h, express levels of DDIT3 and TRIB3 were analyzed by western blotting. (J) Effect of CBD treatment on the AKT-MTOR pathway. Cells were treated with CBD at indicated concentrations and times, expression levels of MTOR, p-MTOR, AKT and p-AKT were analyzed by western blotting. Immunoblots shown are representative of three independent experiments
To confirm that CBD-induced ER stress is associated with mitophagy, we pre-treated glioma cells with 4-PBA, an inhibitor of ER stress, before treatment with CBD; the results showed that blocking ER stress could significantly suppress CBD-induced mitochondrial dysfunction and mitophagy (Figure 5E-F, Fig. S5C), suggesting that ER stress was involved in the induction of mitophagy by CBD in glioma cells. Since TRPV channels have been linked to ER stress before, we then investigated the relation between the activation of TRPV4 and ER stress induction; knocking down TRPV4 significantly reduced the induction of ER stress proteins by CBD (Figure 5G). Because transcriptome analysis indicated that ER stress proteins such as ATF4, DDIT3, and TRIB3 were remarkably up-regulated in response to CBD treatment, we hypothesized the induction of ER stress is mediated by the TRPV4-ATF4-DDIT3-TRIB3 axis.
To prove this, we first showed that siRNA against ATF4 could significantly decrease LC3-II and DDIT3 levels in CBD treated glioma cells (Figure 5H). Secondly, CBD treatment induced dose-dependent up-regulation of TRIB3 (Fig. S5D), which was inhibited by siRNA against DDIT3 (Figure 5I). Lastly, we investigated the effect of CBD treatment on AKT-MTOR, a downstream effector pathway negatively regulated by TRIB3. In agreement with KEGG pathway analysis, AKT-MTOR was inhibited by CBD treatment (Figure 5J) and this was rescued by TRIB3 siRNA knockdown (Fig. S5E). Taken together, these data demonstrated that CBD induced mitophagy through activation of ER stress via the TRPV4-ATF4-DDIT3-TRIB3-AKT-MTOR axis.
CBD synergize with TMZ for glioma treatment through induction of autophagy
As the first-line oral chemotherapeutic agent for glioma, the efficacy of TMZ is often limited by the development of drug resistance, thus effective combination strategies are needed to enhance the clinical benefits of TMZ. We first compared the antitumor effects of CBD, TMZ, and CBD-TMZ combination in vitro, a synergic effect on growth inhibition was readily observed in both patient-derived GBM culture and four glioma cell lines U251, U87 MG, LN18, and GL261 (Figure 6A-B, Fig. S6A-C). Higher levels of autophagy protein markers including LC3-II, SQSTM1, PINK1, and PRKN were observed in the combination group compared to either agent alone (Figure 6C; Fig. S6D). Moreover, higher levels of mitophagy induction by CBD-TMZ combination were also confirmed by mt-keima and LAMP2-MT-CO2 colocalization experiments, indicating the antitumor effects of TMZ in glioma cells was reinforced by CBD-induced autophagy (Fig. S6E-F). The synergistic effects of CBD and TMZ were further confirmed in vivo in an orthotopic model of glioma. U87 MG-GFP-Luc cells were implanted by stereotactic intracranial injection into the striatum of nude mice (Figure 6D); mice were then treated with CBD (15 mg/kg) or TMZ (20 mg/kg) or both for 21 consecutive days. Significantly slower growth rate and longer survival was observed in the combination group compared to either agent alone (Figure 6E-G). Mice brain tumor tissues were then probed for PRKN expression as an indicator for mitophagy, PRKN immunofluorescence signal (red, tumor cells in green) was readily induced in the CBD-treatment group and significantly upregulated in the combination group (Figure 6H), indicating maximal induction of mitophagy in the combination group. In addition, the combination group also showed more robust LC3-II and cleaved-caspase 3 IHC staining along with reduced proliferative index as indicated by MKI67/Ki67 staining (Figure 6I). These results demonstrated that CBD and TMZ combination treatment could significantly suppress glioma cells growth and prolong survival compared with each single agent alone, suggesting CBD could effectively enhance the antitumor effect of TMZ in glioma.
Figure 6.
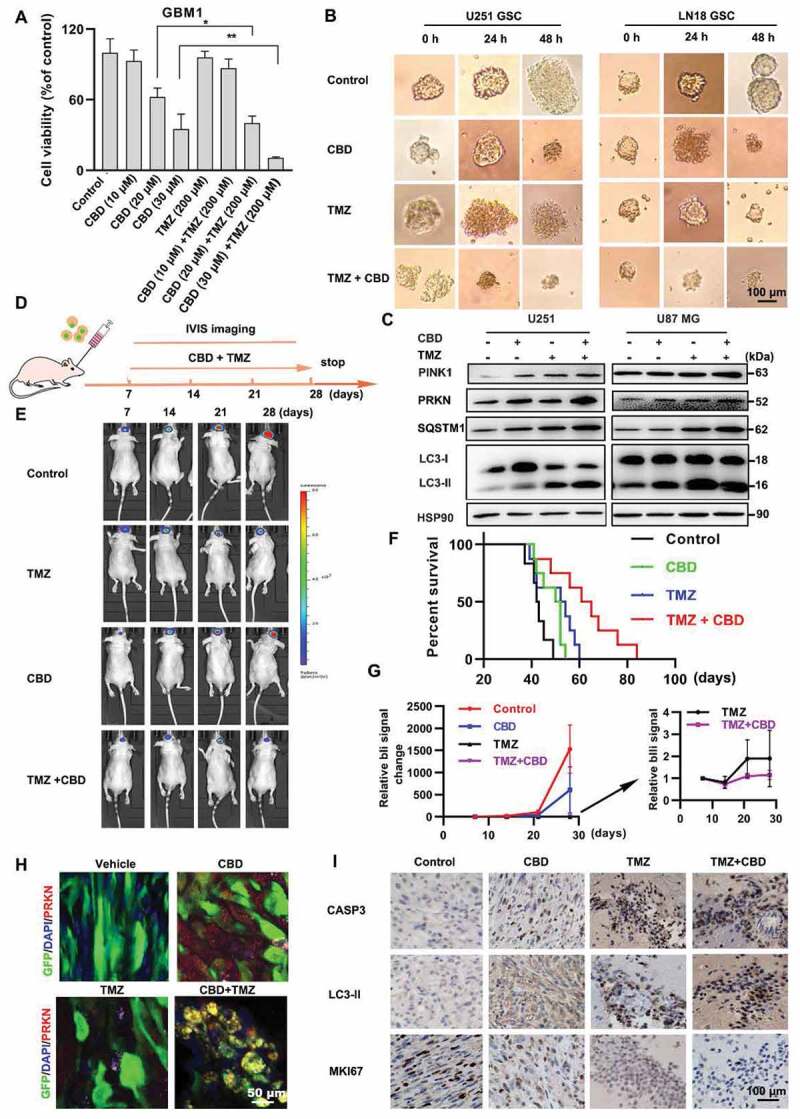
Synergistic effect of CBD and TMZ on glioma in vitro and in vivo. (A) Effect of CBD and TMZ combination on the proliferation of glioma cells in vitro. GBM patient derived primary culture cells were treated with CBD (10, 20 and 30 μM), TMZ (200 μM) or both for 48 h, cell number was determined by the MTT assay. (B) U251 and LN18 glioma sphere culture were treated with CBD (30 μM), TMZ (200 μM) or both for indicated times. (C) Immunoblotting analysis of PINK1, PRKN, LC3, and SQSTM1 in U251 and U87 MG cells treated with CBD (30 μM) in combination with TMZ (500 μM) for 24 h. (D) Scheme of in vivo experiment design. Mice bearing the U87 MG-GFP-luc tumors were treated with control (n = 6), temozolomide (25 mg/kg/once per day, n = 8), CBD (15 mg/kg/once per day, n = 8), or CBD + TMZ (n = 8) for 21 days. IVIS imaging were performed at days 7, 14, 21, 28. (E) Representative bioluminescence images of tumors in mice from control, CBD, TMZ and the combination group 28-day post-transplantation. (F) Kaplan-Meier survival curve of mice in all groups. (G) Quantification of tumor bioluminescence in different treatment arms throughout the course of treatment. (H) Brain tumor tissues were sectioned and subjected to immunofluorescence staining for determination of the expression level of PRKN (red) by confocal microscopy, tumor cells are green because of GFP expression. (I) IHC analysis of MKI67, cleaved-CASP3 and LC3 expression in all groups. Data are shown as mean ± S.D. and are representative of three independent experiments. *P < 0.05, **P < 0.01
Discussion
Although the growth inhibitory effect of CBD on glioma cells has been reported, the molecular mechanism remains elusive. Our results demonstrated that CBD was effective in inhibiting the proliferation of glioma cells both in vitro and in vivo. Mechanistically, the absence of cleaved caspase-3 and PARP suggested the induction of non-apoptotic cell death. The induction of autophagy by CBD was demonstrated by western blotting, confocal imaging and TEM imaging. We then evaluated whether CBD-induced mitophagy is protective or lethal in glioma cells by using specific autophagy inhibitors or siRNA. CBD-induced cell death was reversed by early-stage autophagy inhibitors (wortmannin, LY294002) or ATG5 siRNA but not late-stage autophagy inhibitors (CQ, Baf-A1). These results are in partial agreement with Ivanov et al., who have just published a paper during the preparation of this manuscript, reporting that CBD could induce the death of glioma cells though inhibition of autophagic flux [22]. In this work, we went further to investigated which type of autophagy was induced by CBD and identified mitophagy as the major pathway of CBD-induced glioma cell death. In addition, mitophagy inhibition by either mdivi-1 or siRNAs against PINK1 could partly revert CBD-induced glioma cell death, suggesting that a lethal form of mitophagy contributes to the antitumor function of CBD.
On the molecular level, the target of CBD in glioma was unclear. Previous studies have identified various calcium ion channels such as VDAC, SLC8A1, and GPR55 as the target of CBD in different neurological disorders. To find out the target of CBD in glioma, we screened all reported targets of CBD in literature; inhibitors of TRPV1, TRPV2, GPR55, VDAC1, CNR1/CB1, CNR2/CB2, SLC8A1, TRPC and TRPM all failed to reverse CBD-induced glioma cell death. However, the specific antagonist of TRPV4 significantly alleviated the toxicity of CBD. CBD has been reported to be an agonist for TRPV4 in sebocytes [23] but not in cancer. TRPV4 is a nonselective calcium permeant cation channel; many cellular processes are highly sensitive to the change of calcium ion homeostasis and the abnormal function of these channels have been associated with uncontrolled proliferation and resistance to cell death. Like other TRPV channels, TRPV4 may also be involved in cancer cell proliferation, apoptosis, angiogenesis, migration and invasion and serve as a potential therapeutic target in human cancers [24–26]. Indeed, we found that TRPV4 expression in human GBM tissues correlates with both tumor grade and poor survival, suggesting TRPV4 could be an attractive therapeutic target and biomarker for glioma. However, our knowledge on the connection between TRPV4 activation and ER stress and mitophagy is very limited [27]; our results linked TRPV4 activation to ER stress response in glioma for the first time, which might be the predominant downstream molecular event following CBD treatment in glioma cells. We also identified a new signaling route that links the ER stress response to the activation of mitophagy, our results demonstrated that the activation of TRPV4 by CBD lead to ER stress and subsequently mitophagy via the ATF4-DDIT3-TRIB3-AKT-MTOR axis in glioma cells.
One of the most exciting findings of our study is the synergistic effect of CBD and TMZ against glioma in vivo. The combination of CBD and TMZ significantly prolonged the survival of tumor bearing mice, compared to either agent alone. More importantly, less TMZ related toxic effects and better physical and mental state in mice were observed in the combination group comparing to TMZ alone (data not shown). In line with our result, the co-administration of a 1:1 THC: CBD mixture significantly enhanced the effect of TMZ against glioma stem cells [28]. Because our regimen does not involve the psychoactive THC, and because the mechanism of action of CBD in glioma is clearer comparing to that of THC, it is much easier to translate into clinic. Mechanistically, while CBD induces the initiation of autophagy in glioma cells, TMZ has been shown to induce lethal autophagy arrest in cancer cells via preservation of the autophagic flux [29]. Preclinical studies have shown that the synergistic effect of TMZ and chloroquine is dependent on the inhibition of autophagosome-lysosome fusion [30]. In addition, the antitumor effect of TMZ in glioma cells could also be reinforced by thalidomide-induced autophagy in the combination setting [31]. Moreover, regorafenib was also found to enhance the therapeutic effect of TMZ in glioma by inducing lethal autophagy arrest [32]. Further, autophagy-associated cell death was found to be necessary for the combined antitumor effect of TMZ and radiotherapy [33]. These studies reinforced our observation that inducing mitophagy initiation (with CBD) plus inhibiting autophagosome-lysosome fusion (with TMZ) could be an effective therapeutic strategy in glioma. Since both TMZ and CBD are FDA-approved drugs, these findings could easily translate to the clinic.
In summary, our data presented herein demonstrate for the first time that excessive mitophagy induced by TRPV4-mediated calcium influx may be the primary driving force for glioma cell death in response to CBD treatment. Our study also provides in vitro and in vivo evidence that the combination of TMZ and CBD could produce a strong antitumor effect and significantly prolong the survival of animals. Our results not only provide novel insights into the mechanism of action of CBD, but also lay the foundation for future clinical trials of TMZ and CBD combination therapy for glioma patients.
Materials and methods
Reagents
The following antibodies were purchased from Cell Signaling Technology: CASP3 (9662), cleaved-CASP3/(9664), GAPDH (2118), HSP90 (4874), ACTB (4970); Abcam: AKT (9272), p-AKT (4060), MTOR (2972), p-MTOR (2974), MT-CO2 (ab110258), LAMP2 (ab25631), RPS3 (ab128995), CANX (ab22595), CAT/(ab76024), RSCA1 (ab200348); Proteintech Group: SQSTM1 (18,420-1-AP), MAP1LC3B (14,600-1-AP), ATG5 (10,181-2-AP), EIF2A (11,233-1-AP), DDIT3 (60,304-1-Ig), TRIB3 (13,300-1-AP), EIF2AK3 (24,390-1-AP), VDAC1 (55,259-1-AP), TOMM20 (11,802-1-AP), COX4I1/(11,242-1-AP) TRPV4 (20,987-1-AP), ATF4 (10,835-1-AP). Secondary antibodies used include anti-mouse IgG (Rockland), goat anti-mouse IgG-HRP, and goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology). Cannabidiol was purchased from ZZStandards Reference Materials (ZT-71,836). The following chemicals were purchased from Selleck: hydroxychloroquine (S4430), bafilomycin A1 (S1413), temozolomide (S1237), 3-methyladenine (S2767), Z-VAD-FMK (S7023), necrostatin-1 (S8037); or from MCE: CID16020046 (HY-16,697), rimonabant (HY-14,136), SR144528 (HY-13,439), DIDS (HY-D0086), SKF96365 (HY-100,001), capsazepine (HY-15,640), SEA0400 (HY-15,515), tranilast (HY-B0195), ruthenium red (HY-122,898), GSK2193874 (HY-100,720), 4-PBA (HY-A0281), SB203580 (RWJ64809), PD98059 (HY-12,028), CFSE (HY-D0938), mdivi-1 (HY-15,886), wortmannin (HY-10,197), LY294002 (HY-10,108), TMRM (HY-D0984); or from Sigma: 4α-PDD (P8014), 2-APB (100,065), Probenecid (PHR2608), CHX (C7698); or from Thermo Fisher Scientific: eFluorTM670 (65–0840-90), MitoSOXTM (M36008).
Cell culture
Human glioma cell lines U87 MG, LN18, and U118 MG were purchased from ATCC; A172 and U251 were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Cells were cultured in Dulbecco modified Eagle medium (DMEM; GE Healthcare Life Sciences) supplemented with 10% fetal bovine serum (BIOWST S.A.S, RB35941) in 5% CO2 in a humidified incubator at 37°C. All cell lines were regularly checked for mycoplasma contamination.
Glioma sphere culture
Glioma cell suspensions were seeded in six-well ultra-low attachment plates at a density of 1000 cells per 1 ml in stem cell medium (Invitrogen, 21,103,049) with B27 supplement (Invitrogen,17,504–044), 10 ng/ml EGF (Invitrogen, RP-8661), 10 ng/ml FGF (PeproTech, AF-100-18 C). After 7 days, the images of tumor spheres were captured by microscope and the number was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Tumor sphere with a diameter >50 μm was counted in images of three fields per well.
MTT assay
Cells (3x103 cells/well) were seeded into 96-well plates and incubated at 37°C overnight. CBD (ZZSTANDARD, ZT-71,836) was dissolved in DMSO (Sigma Aldrich, D2650) and diluted to working concentrations in culture medium. After treatment for 72 h; the supernatant was removed and 50 μl of MTT in medium (1 mg/mL; Maklin, C10769164) was added and incubated for 4 h at 37°C. MTT solution were then removed, 100 μl of DMSO was added to each well, and absorbance at 570 nm was measured using a microplate reader (Spectra Max M4, Molecular Devices, USA).
Colony formation assay
Cells were seeded into 6-well plates at a density of 200 cells/well and treated with different concentrations of CBD, medium was changed every three days; 2 weeks later, cells were fixed with 1% glutaraldehyde for 10 min and stained with 0.02% crystal violet for 15 min. After extensive washing with deionized water, colonies were counted using ImageJ software (NIH, Bethesda, MA, USA). For colony formation in soft agar, cell lines were cultured in 6-well plates within a 0.35% agar layer, and 1× 103 cells were seeded to the middle layer of the soft agar (Invitrogen, 16,520,050). The plates were incubated for 14 days (U87 MG, LN18) or 21 days (U251), after which the colonies were inspected and photographed. Assays were conducted in triplicate for a single experiment, and then as three independent experiments.
Apoptosis assay
Cells were harvested and resuspended in binding buffer, and incubated with ANXA5-FITC (Beyotime, C1062S) and PI. Apoptotic cells were detected by flow cytometry (BD Biosciences, San Jose, CA, USA), and results analyzed using Flowjo (Tree Star, Ashland, OR, USA).
Isolation of mitochondria
Isolation of mitochondria was performed using Cell Mitochondria Isolation Kit (Abcam, ab110168) according to the manufacturer’s protocols. Briefly, cells were washed twice with PBS and then harvested, centrifugated, and cell pellets were transferred to a glass homogenizer (Solarbio, YA0853) and homogenized for 10–30 times, followed by centrifugation at 600 g for 10 min at 4°C. Supernatants were carefully removed, pellets (mitochondria) were collected and used for further assays.
Western blotting
Cells were washed with PBS (Hyclone, SH30256.01) and collected into RIPA buffer (Beyotime, P0013 C) containing protease (Thermo Scientific, 36,978) and phosphatase inhibitors (Beyotime, ST640). Protein was collected at 10,000 g and 4°C for 30 min, and the protein concentration was determined using a PierceTM BCA Protein Assay Kit (Thermo Scientific, 23,225). Total cell lysates (20 μg protein) were separated by 8–10% SDS-PAGE. Proteins were transferred onto PVDF membrane and western blotting was carried out following standard protocols with signal detection by chemiluminescence (ECL; Thermo Scientific, TH270252).
siRNA transfection
Gene-specific siRNAs and negative control siRNAs were synthesized by Tuoran (Shanghai, China) and were transfected into U251, U87 MG and LN18 cells for 48 h using Lipofectamine 3000 (Thermo Fisher Scientific, 11,668–027) according to the manufacturer’s protocol. The following siRNA sequences were used to target the RNAs indicated: ATG5, 5ʹ-CCTTTGGCCTAA GAGAA A-3ʹ; ATF4, 5ʹ-TCCCTCAGTGCATAAAGGA-3ʹ; DDIT3, 5ʹ-GCCTGGTATGAGGACCTGC-3ʹ; TRIB3, 5ʹ-CGAGCUCGAAGUGGGCCCC-3ʹ; PINK1, 5′-CGCUGUUCCUCGUUAUGAATT-3′; nontargeting control siRNA, 5ʹ- UUC UCC GAACGUGUCACGA-3ʹ. For shRNA transfections, short hairpin shTRPV4 (5ʹ-CCA GATATTCTGGAATGG AAA-3ʹ) were ligated in the lentiviral vector of HBLV-shRNA-zsgreen-puro and HBLV-shRNA-puro with a puromycin resistant element (Hanbio, HH20190822 MXJ-LV. U251 (CCTCC, TCHu-58) and U87 MG glioma cells (ATCC, HTB-14™) stably infected with lentiviruses expressing shTRPV4 for 24 h, according to the manufacturer’s protocol.
Transfection of RFP-GFP-LC3
Tandem RFP-GFP-LC3 adeno-associated virus was purchased from Hanbio (HB-AP2100001). A suspension of 1 × 104 cells was transfected with Adv-RFP-GFP-LC3 according to the manufacturer’s instructions. Immediately after electroporation, the cells were resuspended in complete medium and incubated for 24 h at 37°C in a humidified atmosphere containing 5% CO2. Cells expressing RFP-GFP-tagged LC3 were used to evaluate autophagy induction.
Transmission electron microscopy (TEM)
Cells were fixed with 4% glutaraldehyde and post fixed with 1% OsO4 in 0.1 M cacodylate buffer for 2 h. The samples were then stained with 1% Millipore-filtered uranyl acetate, dehydrated in increasing concentrations of ethanol, and infiltrated and embedded in epoxy resin (ZXBR, Spon 812). Electron photomicrographs were taken of ultra-structures of GBM cells with a transmission electron microscope (JEM-1200EXII, Tokyo, Japan).
Immunofluorescence
Cells at a density of 3 × 104 were grown in 0.2% gelatin-coated coverslips in 35-mm plates. After treatment, cells were washed with ice-cold PBS, fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) and permeabilized with 0.5% Triton X-100 (MP Blomedicals, 194,854). Cells were then blocked with 3% FCS and followed by incubation with anti-LC3 antibody (Proteintech; diluted 1:100) overnight at 4°C. Next day cells were washed with TBST (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20.) and incubated with Donkey anti-mouse IgG Alexa Fluor 488-labeled secondary antibody (Life Technologies) for 1 h. Cells were then washed with TBST and further incubated with Mito-tracker Deep Red (1:1000; Life Technologies, M22426). Finally, the coverslip was mounted on a slide using Prolong® Gold Antifade Reagent (Beyotime, P0131-5) and images captured using confocal microscopy (Zeiss LSM 880 with Airyscan, Germany).
Immunohistochemistry
Paraffin-embedded samples were sectioned (4 µm) and mounted on microscopic slides. Heat-induced epitope retrieval was performed in 10 mM citric acid buffer at pH 7.2 in a microwave. Sections were incubated with primary antibodies at 4°C overnight (LC3, 1:200; MKI67/Ki67, 1:200; TRPV4, 1:100; PINK1, 1:100), rinsed with PBS, and incubated with horseradish peroxidase-linked goat anti-rabbit secondary antibody (ZSGB-BIO, PV-9000). Visualization was achieved using diaminobenzidine as the substrate, and slides were counterstained with Mayer hematoxylin (Beyotime, C0107).
Mt-keima-based mitophagy assay
For live cell imaging of mt-Keima, cells were transduced with lentivirus encoding mt-Keima (mt-Keima-COX8) at 1 × 108 TU/ml (PPL, ADY190401) for 48 h and treated with CBD. Fluorescence images were captured using confocal microscopy (Zeiss LSM880 with Airyscan, Germany) and quantified using the open source image processing software Fiji (ImageJ).
Calcium measurement
Real-time calcium flux detection was conducted in glioma cells incubated in a physiological Tyrode solution (120 mM NaCl, 4.7 mM KCl, 1 mM MgCl2, 5 mM HEPES, 1.8 mM CaCl2 and 11 mM glucose, pH 7.4). Cells were loaded with the Fluo-4AM dye (Beyotime, S1060) at 37°C for 15 min and observed with a LSM510 Meta microscope (Zeiss). The experiment was independently performed twice, and 32 cells were monitored for each condition and each experiment. To detect calcium flux in response to CBD treatment, cells were loaded with Fluo-4 AM (Beyotime, S1060) at 37°C for 30 min in dark by changing the recording medium (120 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 1.4 mM KH2PO4, 25 mM HEPES, pH 7.4) and then treated with CBD (30 μM). Changes in fluorescence (494 nm excitation, 516 nm emission) were recorded over 15-min period using a microplate reader (Spectra Max M4, Molecular Devices, USA). Data were expressed as fluorescence units in CBD-treated cells minus the readings in control media.
Tissue microarray of patient samples
Glioma tissue microarray slides including 126 samples of formalin-fixed, paraffin-embedded GBMs was used. Samples were purchased from Shanghai Outdo Biotech Co. Ltd. (Shanghai, China). In addition, ten glioma specimens from patients who underwent brain tumor resection between 2018 and 2020 at the Department of Neurosurgery, The Affiliated Drum Tower Hospital, School of Medicine, Nanjing University were collected. The study was approved by the local ethical committee of The Affiliated Drum Tower Hospital, Nanjing University, and was in accordance with Helsinki Declaration. Immunohistochemistry was performed with antibodies against TRPV4 (1:100), SQSTM1 (1:100), LC3 (1:100) and PINK1 (1:100).
RNA-sequencing and transcriptome analysis
U251 cells treated with DMSO or CBD (30 µM) for 12 h were used as control and treatment group, respectively. Triplicate samples of each group were collected and total RNA extracted and sent to Shanghai Majorbio Bio-Pharm Technology Co., Ltd for transcriptome sequencing by Illumina HiSeqTM 2500 sequencer. Data were analyzed using the free online Majorbio Cloud Platform.
The Cancer Genome Atlas (TCGA) database
The mRNA expression and survival data from 961 glioblastoma samples were downloaded from The Cancer Genome Atlas (TCGA) dataset (https://cancergenome.nih.gov/). The UALCAN platform was used for analyzing TRPV4 expression between normal tissue and glioma tissue.
Ethics and animal use statement
This study was conducted in strict accordance to the recommendations in the Guide for the Care and Use of Laboratory Animals in China. Animals were housed at 25°C with 12 h light/dark cycles and free access to water and standard rodent chow in the animal facility of Nanjing University School of Life Sciences. All experiments using animals were conducted under the Institutional Animal Care and Use Committee (IACUC)-approved protocols at Nanjing University in accordance with NIH and institutional guidelines.
Animal experiments
Subcutaneous xenograft assays were conducted using nude mice (male, 6–8 weeks old, 20–25 g, SPF Biotechnology Co. Ltd., Beijing, China). After the cells (1x106) were injected, mice were randomly separated into two groups (n = 4). Mice were treated with 10% DMSO, or CBD (20 mg/kg) by i.p. injection every other day for a total of 21 days. Tumor volume were measured twice a week and calculated using the formula (width)2 × length/2. After 4 weeks, mice were sacrificed and tumors were excised. To establish intracranial xenograft model of glioma, mice were anesthetized and placed in stereotactic frames with their skulls exposed. A 1 mm-diameter hole was drilled 2 mm lateral to the bregma. U87 MG glioma cells stably overexpressing luciferase (1 × 106) in 5 μl of PBS were injected with a Hamilton syringe implanted 4 mm into the right striatum. Tumor growth was monitored by bioluminescence imaging with IVIS system (IVIS Lumina XR, Caliper, USA) every week.
Statistical analysis
Unless otherwise noted, data were expressed as mean ± standard deviation (SD) and represent the results of at least three independent experiments. Statistical significance was determined using the unpaired t-test for two-group experiments. For comparison among 3 or more groups, two-way analysis of variance (ANOVA) and Bonferroni posttests were used. P value lower than 0.05 were considered statistically significant and were indicated as follows: *P < 0.05; ** P < 0.01; *** P < 0.001.
Supplementary Material
Funding Statement
This work was supported by the Fundamental Research Funds for the Central Universities [090314380027]; National Natural Science Foundation of China [21877060]; National Natural Science Foundation of China [31900824]; National Natural Science Foundation of China [81802921]; Postdoctoral Research Foundation of China [2019M651779].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Aldape K, Brindle KM, Chesler L, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019;16(8):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhou W, Wahl DR.. Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers (Basel). 2019;11(9):1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Huang Q, Zhan L, Cao H, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12(6):999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen R, Jiang M, Li B, et al. The role of autophagy in pulmonary hypertension: a double-edge sword. Apoptosis. 2018;23(9–10):459–469. [DOI] [PubMed] [Google Scholar]
- [5].Elshaer M, Chen Y, Wang X, et al. Resveratrol: an overview of its anti-cancer mechanisms. Life Sci. 2018;207:340–349. [DOI] [PubMed] [Google Scholar]
- [6].Wang Y, Tang C, Cai J, et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018;4:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lleonart ME, Grodzicki R, Graifer DM, et al. Mitochondrial dysfunction and potential anticancer therapy. Med Res Rev. 2017;37(6):1275–1298. [DOI] [PubMed] [Google Scholar]
- [8].Zecchini S, Proietti S, Catalani E, et al. Dysfunctional autophagy induced by the pro-apoptotic natural compound climacostol in tumour cells. Cell Death Dis. 2018;10(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pisanti S, Malfitano AM, Ciaglia E, et al. Cannabidiol: state of the art and new challenges for therapeutic applications. 2017;175:133–150.Pharmacol therape [DOI] [PubMed] [Google Scholar]
- [10].Iffland K, Grotenhermen F.. An update on safety and side effects of cannabidiol: a review of clinical data and relevant animal studies. Canna cannabin res. 2017;2(1):139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hind WH, England TJ, O’Sullivan SE. Cannabidiol protects an in vitro model of the blood-brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT1A receptors. Brit j pharmacol. 2016;173(5):815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Patra PH, Barker M, White HS, et al. Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia. 2019;60(2):303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Reddy PM, Maurya N, Velmurugan BK. Medicinal use of synthetic cannabinoids—a mini review. Curr Pharmacol Rep. 2019;5(1):1–13. [Google Scholar]
- [14].Hoshino A, Wang W, Wada S, et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature. 2019;575(7782):375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li H, Ham A, Ma TC, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15(1):113–130. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ibeas C, Chen T, Nunn AV, et al. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics. 2015;12(4):699–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fontana F, Raimondi M, Marzagalli M, et al. The emerging role of paraptosisin tumor cell biology: perspectives for cancer prevention and therapy with natural compounds. Biochim Biophys Acta Rev Cancer. 2020;1837(2):188338. [DOI] [PubMed] [Google Scholar]
- [18].Bury M, Girault A, Mégalizzi V, et al. Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis. 2013;4(3):e561. J Mole Neurosci, 2019; 70: 6–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pumroy RA, Samanta A, Liu Y, et al. Molecular mechanism of TRPV2 channel modulation by cannabidiol. Elife. 2019;8:e48792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Suvà ML, Riggi N, Janiszewska M, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009;69(24):9211–9218. [DOI] [PubMed] [Google Scholar]
- [21].McKallip RJ, Jia W, Schlomer J, et al. Cannabidiol-induced apoptosis in human leukemia cells: a novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol Pharmacol. 2006;70(3):897–908. [DOI] [PubMed] [Google Scholar]
- [22].Ivanov VN, Grabham PW, Wu CC, et al. Inhibition of autophagic flux differently modulates cannabidiol-induced death in 2D and 3D glioblastoma cell cultures. Sci Rep. 2020;10(1):2687. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [23].Oláh A, Tóth BI, Borbíró I, et al. Cannabidiol exerts sebostatic and anti-inflammatory effects on human sebocytes. J Clin Invest. 2014;124(9):3713–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De Petrocellis L, Orlando P, Moriello AS, et al. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf). 2012;204(2):255–266. [DOI] [PubMed] [Google Scholar]
- [25].Lee WH, Choong LY, Mon NN, et al. TRPV4 regulates breast cancer cell extravasation, stiffness and actin cortex. Sci Rep. 2016;6(1):27903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bahari N, Jamaludin S, Jahidin A, et al. The emerging roles of TRPV4 in cancer[J]. Biomedical & Pharmacology Journal. 2017;10(4):1757–1764. [Google Scholar]
- [27].Yang W, Wu PF, Ma JX, et al. TRPV4 activates the Cdc42/N-wasp pathway to promote glioblastoma invasion by altering cellular protrusions. Sci Rep. 2020;10(1):14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].López V, Saiz L, Torres S. et cl. Targeting Glioma Initiating Cells with A combined therapy of cannabinoids and temozolomide. Biochem pharmacol. 2018;157:266–274. [DOI] [PubMed] [Google Scholar]
- [29].Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10):e838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin C-J, Lee -C-C, Shih Y-L. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitroand in vivo by inhibiting autophagy. Free Radic Biol Med. 2012;52(2):377–391. [DOI] [PubMed] [Google Scholar]
- [31].Gao S, Yang XJ, Zhang WG, et al. Mechanism of thalidomide toenhance cytotoxicity of temozolomide in U251-MG glioma cells in vitro. Chin Med J (Engl). 2009;122(11):1260–1266. [PubMed] [Google Scholar]
- [32].Jiang J, Zhang L, Chen H, et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy. 2020;16(1):106–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Palumbo S, Pirtoli L, Tini P, et al. Different involvement of autophagy in human malignant glioma cell line sunder going irradiation and temozolomide combined treatments. J Cell Biochem. 2012;113(7):2308–2318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.