

ABSTRACT
Thioglycolate-elicited macrophages exhibit abundant conjugation of LC3 with PE (LC3-II). Among other autophagy-related (ATG) proteins, it is proposed that, like in yeast, both ATG5 and ATG7 are essential for LC3 conjugation. Using atg5-deficient (−/-) and atg7−/-macrophages, we provide evidence that loss of ATG5 but not of ATG7 resulted in LC3-II depletion. Accumulation of LC3-II in elicited atg7−/- macrophages in response to bafilomycin A1 validated these data. Furthermore, complete loss of ATG3 in atg7−/- macrophages demonstrated that ATG7 and ATG3 are dispensable for LC3–PE conjugation. In contrast to thioglycolate-elicited macrophages, naïve peritoneal and bone marrow-derived atg7−/- macrophages exhibited no LC3-II, even under inflammatory stimuli in vitro. Hence, the macrophage metabolic status dictates the level of LC3–PE conjugation with a supportive but nonessential role of ATG7, disclosing the eukaryotic exception from the LC3 lipidation model based on yeast data. Abbreviations: ATG: autophagy-related; BM: bone marrow; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; PE: phosphatidylethanolamine.
KEYWORDS: ATG3, ATG5, ATG7, LC3 lipidation, LC3-II, macrophages
Introduction
MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is involved in diverse cellular processes linked to sequestration, transport, and degradation of cellular material such as macroautophagy/autophagy [1], phagocytosis [2], and endocytosis [3]. In all these processes, active LC3 is generated by covalent binding (lipidation) with phosphatidylethanolamine (PE). LC3 is one of the mammalian homologs of yeast Atg8 [4], whose lipidation requires many autophagy-related (Atg) proteins involved in de novo formation of a double-membrane vesicle. Especially Atg5 (substrate), Atg7, Atg3, and Atg10 (enzymes) are critical for ligation with PE [5]. Similar to ubiquitination, Atg12 is first conjugated to Atg5 by Atg7 (E1-like-) and Atg10 (E2-like enzyme). The Atg12–Atg5 complex (E3-like) facilitates conjugation of PE to Atg8 by Atg7 and its corresponding E2-like enzyme Atg3, supporting expansion of phagophore membranes [6]. Out of several mammalian homologs of yeast Atg8, LC3 is the best characterized. It is proposed that LC3 undergoes a modification process similar to Atg8 [4,7]. ATG7 and ATG3 catalyze transformation of cytosolic LC3-I to a membrane-bound form (LC3-II), which corresponds to Atg8–PE in yeast [6].
We investigated the consequences of ATG5 and ATG7 deficiency on LC3-II formation in murine macrophages which utilize LC3-II for canonical and/or non-canonical function(s) [2] and found diverging results between thioglycolate-elicited and non-elicited cells.
Results and discussion
Loss of ATG5 but not ATG7 depletes LC3-II in thioglycolate-elicited macrophages
Loss of ATG5 or ATG7 protein (Figure 1Ai,Aii,Di,Dii) and their respective mRNAs (Figure 1C) in macrophages proves the successful generation of the knockout mice. As expected, deletion of Atg5 resulted in substantial expression of LC3-I and absence of LC3-II (Figure 1Ai, Fig. S1A). However, lack of ATG7 in thioglycolate-elicited macrophages failed to inhibit LC3 lipidation with only minor reduction in LC3-II compared to WT cells (Figure 1Aii, Fig. S1B,C). LC3-II was also found in thioglycolate-elicited atg7−/- macrophages 2 h after isolation, indicating that lipidation was not induced during culturing of the cells (Fig. S1C). After separation of cytosolic and membrane fractions, LC3-II was absent in the membrane fraction of thioglycolate-elicited atg5−/- macrophages (Figure 1Bi), whereas the amount of LC3-II in atg7−/- macrophages was comparable to WT cells. This is an unexpected finding, considering that LC3 lipidation is generally accepted as being dependent on both ATG5 and ATG7 [6].
Figure 1.
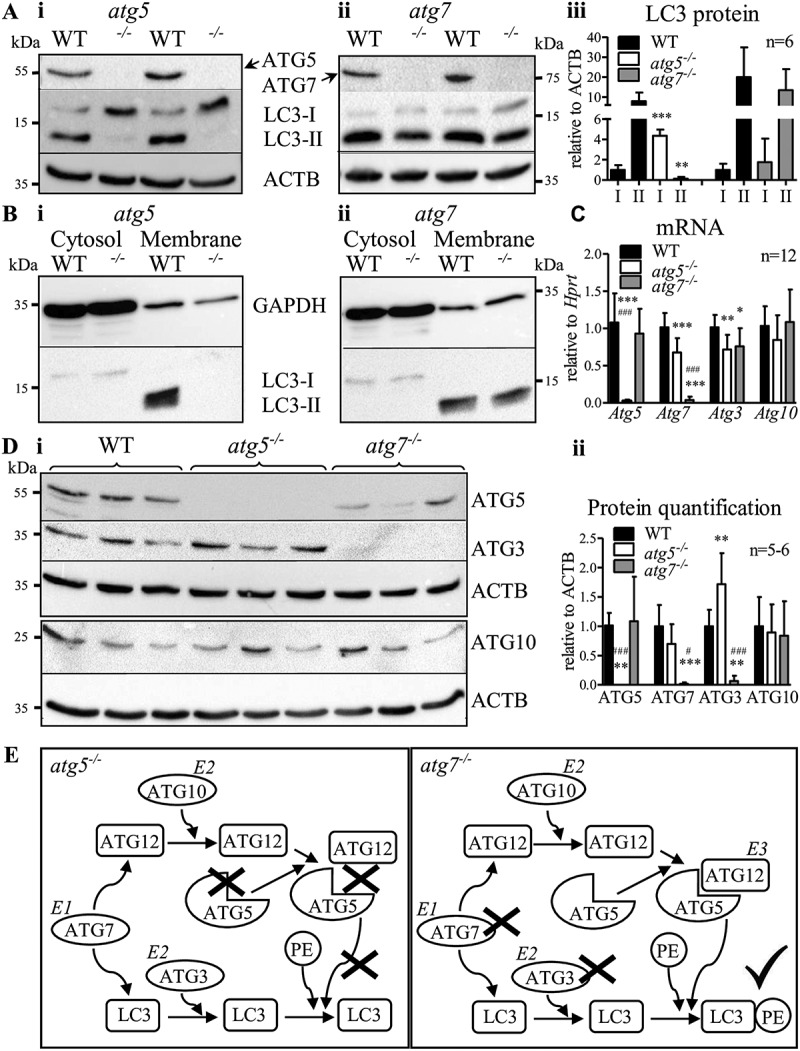
Abolished ATG3 protein expression but comparable LC3–PE lipidation in thioglycolate-elicited atg7−/- macrophages. Representative immunoblotting of LC3, ATG5, ATG7, and ß-actin in whole-cell lysates isolated from thioglycolate-elicited WT, (Ai) atg5−/-, and (Aii) atg7−/- peritoneal macrophages. Each lane represents a macrophage sample from an individual mouse. (Aiii) Quantification of LC3-I and LC3-II in atg5−/-, atg7−/-, and corresponding control macrophages normalized to ACTB/β-actin (n = 6). Statistics were calculated using Student’s t-test (two-tailed) with Welch correction for unequal variances; ** p < 0.01, *** p < 0.001. LC3 protein abundance in cellular fractions of WT, (Bi) atg5−/-, and (Bii) atg7−/- thioglycolate-elicited peritoneal macrophages with GAPDH as loading control. Each lane represents a sample pool from three mice. (C) mRNA expression of Atg5, Atg7, Atg3, and Atg10 in WT, atg5−/-, and atg7−/- macrophages relative to Hprt (n = 12). (Ci) Representative immunoblotting of (ATG12–)ATG5, ATG3 (upper blot), and ATG10 (lower blot), using the same samples obtained from three individual mice. (Dii) Protein quantification relative to ACTB/β-actin (n = 5–6). Data represent mean values + SD. Statistics in (C and Dii) were calculated using one-way ANOVA with Bonferroni correction; * relative to WT; # atg5−/- relative to atg7−/-; *, # p < 0.05, ** p < 0.01, ***/### p < 0.001. (E) Schematic presentation of the summarized results
Atg10 mRNA (Figure 1C) and ATG10 protein expression (Figure 1D) were comparable between all genotypes. However, ATG3 protein was depleted in atg7−/- macrophages (Figure 1D) but only reduced by 25% at mRNA level (Figure 1C), indicating posttranscriptional regulation of ATG3. Despite reduced Atg3 mRNA (Figure 1C), we observed significantly more ATG3 protein in atg5−/- compared to control macrophages (Figure 1Dii). Whether ATG5 protein hinders the translation of Atg3 or whether ATG3, as an E2-like enzyme, increases its activity to compensate for the lack of ATG5 requires further investigation. However, ATG3 depletion in atg7−/- macrophages indicated that the E2-like enzyme ATG3 is redundant without the E1-like enzyme ATG7, similar to Atg7- and Atg3-independent autophagy in Drosophila [8].
We conclude that the E2-like enzyme ATG10 does not require ATG7 for ATG12 activation and conjugation to ATG5 (Figure 1E). Subsequent conjugation of PE to LC3 is possible in the absence of ATG7 and ATG3. Thus, the E3-like ATG12–ATG5 complex is sufficient for conjugation of PE to LC3 in murine thioglycolate-elicited macrophages.
Lipidated LC3 accumulates in thioglycolate-elicited peritoneal atg7−/- macrophages in response to bafilomycin A1
To investigate LC3 lipidation dynamics in thioglycolate-elicited peritoneal WT and atg7−/- macrophages in more detail, we starved (PBS) the cells to induce autophagy and used bafilomycin A1 to inhibit autolysosomal LC3 degradation. We observed time-dependent accumulation of LC3-II in elicited WT and atg7−/- macrophages (Figure 2A), reduction by starvation in both genotypes (Figure 2B), and no induction of LC3-II in atg5−/- macrophages (Figure 2B). To overcome individual differences in LC3 expression between independent experiments, we used LC3-II:LC3-I ratios instead of normalization to a housekeeping protein. Elicited WT and atg7−/- macrophages responded to inhibition and stimulation with comparable increase (1.6- versus 1.5-fold) and decrease (44% versus 66%) of LC3-II:LC3-I ratios (Figure 2B). Thus, we conclude that despite reduced LC3 lipidation capacity in elicited atg7−/- macrophages LC3 lipidation dynamics are functional in response to autophagy inhibition and stimulation, respectively.
Figure 2.
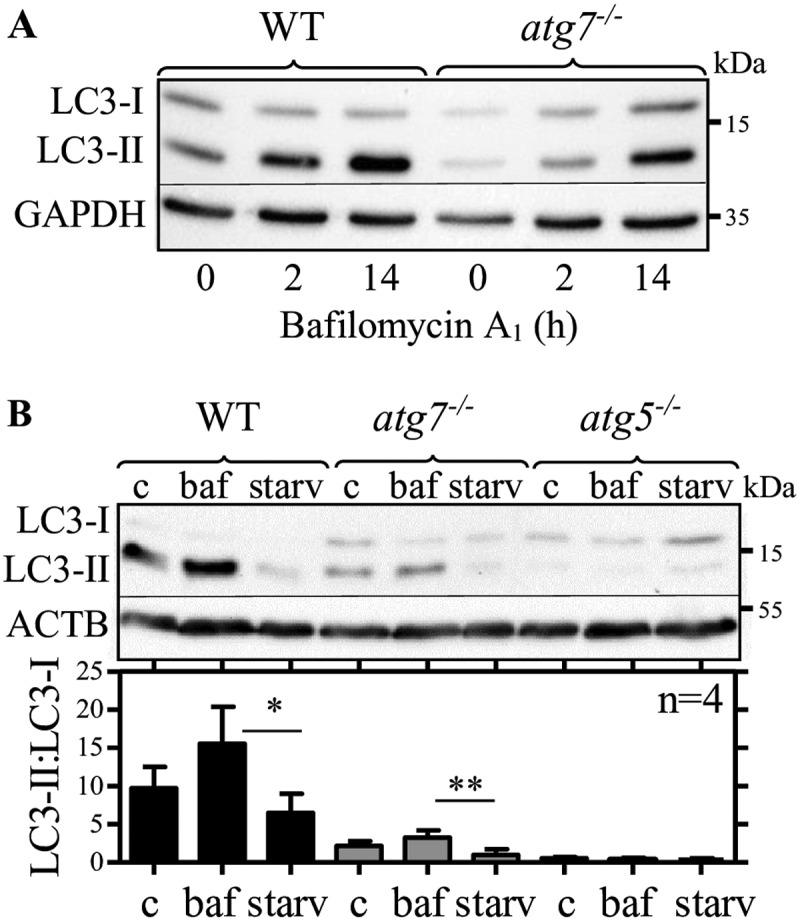
Lipidated LC3 accumulates in thioglycolate-elicited peritoneal atg7−/- macrophages in response to bafilomycin A1. Representative LC3 immunoblotting in WT and atg7−/- macrophages. (A) GAPDH and (B) ACTB/β-actin were used as loading controls. Cells were treated for (A) 2 h and (A and B) 14 h with bafilomycin A1 (baf), (B) starved for 1 h in PBS (starv) or left untreated (C). Bars represent mean LC3-II:LC3-I ratios (+ SD) obtained from four individual mice. One-way ANOVA with Bonferroni correction were used to calculate differences between the different treatments; * p < 0.05, ** p < 0.01
LC3-II is absent in non-elicited peritoneal and bone marrow-derived atg7−/- macrophages
To examine whether other inflammatory agents stimulate LC3 lipidation in atg7−/- macrophages in vivo, we compared LC3 protein expression in peritoneal macrophages elicited by thioglycolate, proteose-peptone, and concanavalin A (Figure 3A). LC3-II was hardly detectable in proteose-peptone elicited cells but we observed LC3-II in atg7−/- macrophages elicited by concanavalin A (Figure 3Ai). However, if compared with LC3-II from thioglycolate-elicited atg7−/- cells, LC3-II expression was significantly reduced in concanavalin A-elicited macrophages (Figure 3Aii). Moreover, only thioglycolate-elicited atg7−/- macrophages exhibited significantly higher LC3-II than LC3-I expression, in contrast to WT macrophages showing increased LC3-II by all eliciting agents. We conclude that among investigated inflammatory stimuli peritoneal injection of thioglycolate induces LC3 lipidation in atg7−/- macrophages most effectively. However, low expression of LC3-II in cells isolated after concanavalin A injection may be a result of a reduced macrophage response, considering that thioglycolate is more potent than other eliciting agents to induce peritonitis and yield a greater number of cells in the peritoneum [9]. These results indicated that pronounced inflammation is necessary to boost LC3 lipidation in atg7−/- macrophages.
Figure 3.
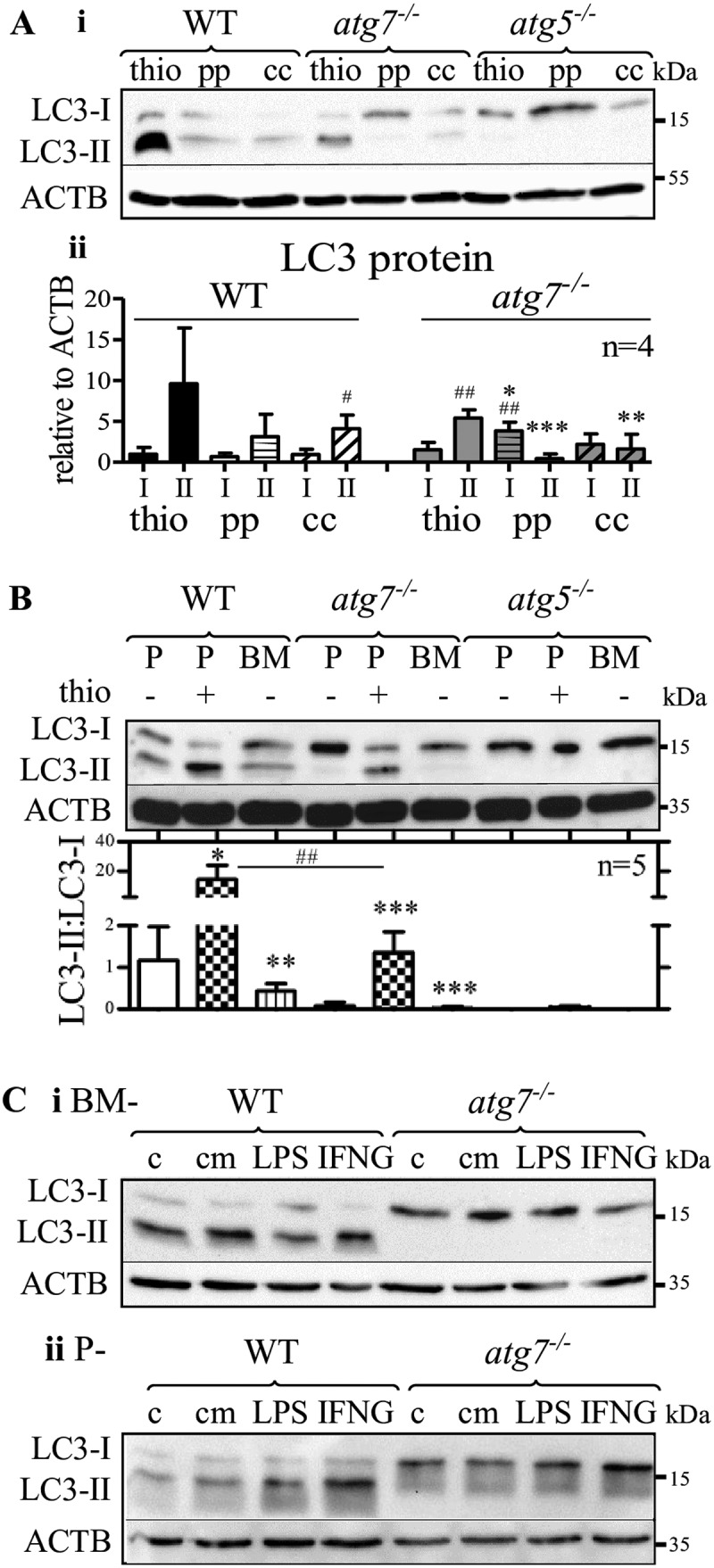
Non-elicited peritoneal and BM-derived atg7−/- macrophages do not conjugate PE to LC3. (A) Comparison of LC3 protein expression in WT, atg7−/-, and atg5−/- peritoneal macrophages elicited by thioglycolate (thio), proteose-peptone (pp), and concanavalin A (cc). (Ai) Representative LC3 immunoblot with ACTB/β-actin as a loading control. (Aii) Mean LC3 expression values (+ SD) obtained from four individual mice. One-way ANOVA with Bonferroni correction were used to calculate differences between the different treatments; * p < 0.05, ** p < 0.01, *** p < 0.001. Student’s t-test (two-tailed) with Welch correction was used to compare LC3-II with LC3-I; # p < 0.05, ## p < 0.01. (B) Representative LC3 immunoblotting in non-elicited peritoneal (P-), thioglycolate (thio)-elicited (P+), and BM-derived (BM-) WT, atg5−/-, and atg7−/- macrophages, respectively. Bars represent mean LC3-II:LC3-I ratios + SD (n = 5). Statistics were calculated using one-way ANOVA with Bonferroni correction. * differences by treatment; # differences by genotype; * p < 0.05, **/## p < 0.01, *** p < 0.001. LC3 immunoblotting in (Ci) BM- and (Cii) P- WT and atg7−/- macrophages treated with lipopolysaccharide (LPS), IFNG (interferon gamma), thioglycolate (thio), conditioned media (cm) collected after incubation of thioglycolate-elicited WT macrophages for 24 h, or left untreated (C). ACTB/β-actin was used as loading control
In line with these results, non-elicited peritoneal and bone marrow (BM)-derived atg7−/- macrophages were unable to conjugate PE to LC3 (Figure 3B). As expected, LC3-II was absent in all types of atg5−/- macrophages. If compared with non-elicited peritoneal and BM-derived macrophages, the LC3-II:LC3-I ratios were drastically higher in thioglycolate-elicited WT and atg7−/- macrophages. Consistent with data in Figure 2B, the LC3-II:LC3-I ratio was markedly reduced in elicited atg7−/- compared with WT macrophages.
The induction of an inflammatory response by thioglycolate [10] is accompanied by increased macrophage phagocytosis and lysosomal activity [11]. Besides, granulocyte-macrophage colony-stimulating factor induces LC3 lipidation in human monocytes [12]. We therefore investigated whether pro-inflammatory stimuli [13,14] trigger the lipidation of LC3 in atg7−/- macrophages in vitro. BM-derived (Figure 3Ci) and non-elicited peritoneal (Figure 3Cii) atg7−/- macrophages showed only non-lipidated LC3-I after incubation with lipopolysaccharide and interferon gamma. Moreover, LC3-II bands were absent in atg7−/- macrophages after in vitro treatment with conditioned medium from thioglycolate-elicited WT macrophages (Figure 3C) or with thioglycolate in vitro [10] (Fig. S2).
It is likely that inflammatory responses to thioglycolate trigger the LC3 lipidation in nonresident atg7−/- macrophages differentiated from recruited monocytes. Though additional experiments are required to understand the mechanism of LC3-lipidation in atg7−/- macrophages after i.p. injection of thioglycolate in atg7−/- mice, this process likely occurs only in vivo. It remains unclear whether the peritoneal architecture and the interaction with the surrounding tissues and the extracellular matrix or the presence of other cells in the peritoneal cavity are required. Numerous myeloid and lymphoid cells found in the peritoneal inflammatory exudate [15] might be involved. Neutrophils, which are plentiful in the peritoneum 24 h after thioglycolate injection [10] and 6 h after administration of concanavalin A [16], are the most interesting cells for future investigation.
We conclude that the macrophage activation status dictates the level of LC3–PE conjugation. In contrast to ATG5, the role of ATG7 and ATG3 in this process is supportive but not essential.
Materials and methods
Reagents
The following reagents were used: thioglycolate broth containing (per liter of deionized water) 17 g peptone from casein (1654) and 3 g peptone from soy (1657) from GERBU Biotechnik GmbH, 6 g glucose (X997), 2.5 g NaCl (3957), 0.1 g Na2SO3 (P033) from Carl Roth, 0.5 g sodium thioglycolate (T0632) and 0.7 g agar (A1296) from Sigma-Aldrich; RIPA buffer containing 150 mM NaCl, 1% Triton® X-100 (3051), 0.1% SDS (CN30), 50 mM Tris, pH 8 (9140), 1 mM DTT (6908) from Carl Roth, 0.5% Na-deoxycholate (Merck, 1065040250), and protease inhibitor cocktail (Sigma-Aldrich, P8340); fractionation lysis buffer containing 20 mM HEPES (HN77), 10 mM KCl (HN02), 2 mM MgCl2 (A537) and 1 mM DTT (6908) from Carl Roth, 1 mM Invitrogen™ UltraPure™ EDTA, pH 8.0 (Thermo Fisher Scientific, 15575020), 1 mM EGTA (GERBU Biotechnik GmbH, 1310) and protease inhibitor cocktail; Gibco™Bacto™ proteose peptone (Thermo Fisher Scientific, 211684); bafilomycin A1 (B1793), concanavalin A (C5275), DMEM (41966029), IFNG/interferon-gamma (SRP3058), lipopolysaccharide (L4391), PBS, pH 7.4 (10010), and penicillin-streptomycin (P4333) from Sigma-Aldrich.
Animals and cells
Mice with a targeted deletion of Atg5 or Atg7 in myeloid cells were generated by crossing [17] Lyz2/LysM-Cre with Atg5flox/flox mice (RIKEN BRC) or Atg7flox/flox mice [18] (M. Komatsu). Mice were kept in a clean environment with unlimited access to chow diet (Altromin Spezialfutter, 1324) and water. All experiments were approved by the Austrian Federal Ministry of Education, Science, and Research (BMWFW-66010/0076-WF/II/3b/2014, BMWFW-66010/0153-WF/V/3b/2015 and BMBWF-66.010/0023-V/3b/2018).
Peritoneal macrophages were obtained 3 days after intraperitoneal injection of 2 ml of 3% thioglycolate broth, 2 ml of 3% proteose peptone or 25 µg/ml of concanavalin A. Macrophages were isolated by peritoneal lavage with 10 ml ice-cold PBS/EDTA and cultured in high glucose (25 mM) DMEM supplemented with 10% LPDS (Sigma-Aldrich, S5394), 1% penicillin-streptomycin for 2 h to adhere to the non-treated culture dish. Thereafter, cells were washed twice with PBS and cultured in DMEM for 48 h prior to further use. For the bafilomycin A1 treatment, cells were incubated as described before for 46- or 34 h and then co-incubated with 10 nM bafilomycin A1 for 2 or 14 h, respectively. PBS-starved cells were incubated as described before for 47 h and then incubated in PBS for 1 h. Cells were harvested simultaneously with untreated cells. BM-derived cells were differentiated to macrophages for 7 days in DMEM containing 10% of conditioned medium from L929 cells (ECACC, 85011425). Cells were lysed in RIPA buffer in the presence of 1 mM DTT and protease inhibitor cocktail. For subcellular fractionation, cells were lysed in fractionation lysis buffer by triturating cells through a 27 G needle with visual inspection of successful lysis under the microscope. After separation of the nuclear fraction (2,655 x g), the membrane fraction was obtained by centrifugation at 100,000 x g, while the cytosolic fraction was collected as supernatant. For in vitro treatments of naïve peritoneal and BM-derived macrophages, 10 nM bafilomycin A1, 100 ng/ml lipopolysaccharide, 5 ng/ml interferon-gamma or 0.3% thioglycolate broth were used. Conditioned media were collected from thioglycolate-elicited WT macrophages after culturing for 24 h in DMEM.
Western blotting
Protein samples were separated by 15% SDS-polyacrylamide gel electrophoresis transferred to PVDF membranes and incubated with rabbit polyclonal antibodies against ATG3 (Thermo Fisher Scientific, PA5-17018), ATG10 (Abclonal, A7390), ATG5, ATG7, LC3B (Cell Signaling Technology, 4445), GAPDH/glyceraldehyde 3-phosphate dehydrogenase (Cell Signaling Technology, 2118), and mouse monoclonal ACTB/β-actin (Sigma-Aldrich, A2228). HRP-conjugated rabbit anti-mouse (Dako, P0260) and goat anti-rabbit (Thermo Fisher Scientific, 31460) were visualized with ECL (BioRad, 1705061) using the ChemiDoc™ Imaging System (Bio-Rad Laboratories, Hercules, CA).
Real-time PCR
RNA was isolated using TriFast™ (VWR, 30–2010) and reverse transcribed using the Applied BiosystemsTM High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, 4368813). qPCR was performed on a Bio-Rad CF X96 (Bio-Rad Laboratories, Hercules, CA) with GoTaq® qPCR Mastermix (Promega, A6002), normalized to Hprt (hypoxanthine guanine phosphoribosyl transferase) and calculated using the 2−ΔΔCT method. Primer sequences are available upon request.
Supplementary Material
Acknowledgments
This work was supported by the BioTechMed-Graz-funded flagship project “Lipases and Lipid Signaling”, the Austrian Science Fund FWF (DK-MCD W1226, SFB F73, P30882, P32400, DP-iDP DOC 31), and the PhD program “Molecular Medicine” of the Medical University of Graz. The authors thank A. Absenger and I. Hindler (Medical University of Graz, Austria) for mice care.
Funding Statement
This work was supported by the Austrian Science Fund [P30882]; Austrian Science Fund [DK-MCD W1226]; Austrian Science Fund [DP-iDP DOC 31]; Austrian Science Fund [P32400]; Austrian Science Fund [SFB F73].
Disclosure statement
The authors declare no competing interests.
Data availability statement
All data are available from the corresponding author branislav.radovic@medunigraz.at upon reasonable request.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000. Nov 1;19(21):5720–5728. PubMed PMID: 11060023; PubMed Central PMCID: PMC305793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Heckmann BL, Boada-Romero E, Cunha LD, et al. LC3-associated phagocytosis and inflammation. J Mol Biol. 2017. Nov 24;429(23):3561–3576. PubMed PMID: 28847720; PubMed Central PMCID: PMCPMC5743439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Heckmann BL, Teubner BJW, Tummers B, et al. LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2019. Jul 25;178(3):536–551e14. PubMed PMID: 31257024; PubMed Central PMCID: PMCPMC6689199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004. Jun 1;117(Pt13):2805–2812. PubMed PMID: 15169837. [DOI] [PubMed] [Google Scholar]
- [5].Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000. Nov 23;408(6811):488–492. PubMed PMID: 11100732. [DOI] [PubMed] [Google Scholar]
- [6].Geng J, Klionsky DJ.. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008. Sep 9;9:859–864. PubMed PMID: 18704115; PubMed Central PMCID: PMC2529362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tanida I, Tanida-Miyake E, Komatsu M, et al. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002. Apr 19;277(16):13739–13744. PubMed PMID: 11825910. [DOI] [PubMed] [Google Scholar]
- [8].Chang TK, Shravage BV, Hayes SD, et al. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol. 2013. Sep;15(9):1067–1078. PubMed PMID: 23873149; PubMed Central PMCID: PMC3762904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang X, Goncalves R, Mosser DM.. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008. Nov;83. Chapter 14: Unit14 1. 10.1002/0471142735.im1401s83. PubMed PMID: 19016445; PubMed Central PMCID: PMCPMC2834554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baron EJ, Proctor RA. Elicitation of peritoneal polymorphonuclear neutrophils from mice. J Immunol Methods. 1982. Mar 26;49(3):305–313. PubMed PMID: 7040554. [DOI] [PubMed] [Google Scholar]
- [11].Pavlou S, Wang L, Xu H, et al. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J Inflam. 2017;14:4. PubMed PMID: 28203119; PubMed Central PMCID: PMC5301433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang Y, Morgan MJ, Chen K, et al. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012. Mar 22;119(12):2895–2905. PubMed PMID: 22223827; PubMed Central PMCID: PMC3327464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ngkelo A, Meja K, Yeadon M, et al. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Gialpha dependent PI-3kinase signalling. J Inflam. 2012. Jan 12;9(1):1. PubMed PMID: 22239975; PubMed Central PMCID: PMCPMC3293082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang F, Zhang S, Jeon R, et al. Interferon gamma Induces Reversible Metabolic Reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine. 2018. Apr;30:303–316. PubMed PMID: 29463472; PubMed Central PMCID: PMCPMC5953001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hermida MDR, Malta R, de SSM, et al. Selecting the right gate to identify relevant cells for your assay: a study of thioglycollate-elicited peritoneal exudate cells in mice. BMC Res Notes. 2017. Dec 6;10(1):695. PubMed PMID: 29208049; PubMed Central PMCID: PMCPMC5718147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Loyola W, Gaziri DA, Gaziri LC, et al. Concanavalin A enhances phagocytosis and killing of Candida albicans by mice peritoneal neutrophils and macrophages. FEMS Immunol Med Microbiol. 2002. Jul 12;33(3):201–208. PubMed PMID: 12110482. [DOI] [PubMed] [Google Scholar]
- [17].Clausen BE, Burkhardt C, Reith W, et al. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999. Aug 8;8(4):265–277. PubMed PMID: 10621974 [DOI] [PubMed] [Google Scholar]
- [18].Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005. May 9;169(3):425–434. PubMed PMID: 15866887; PubMed Central PMCID: PMC2171928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding author branislav.radovic@medunigraz.at upon reasonable request.