

ABSTRACT
Mitophagy formed the basis of the original description of autophagy by Christian de Duve when he demonstrated that GCG (glucagon) induced macroautophagic/autophagic turnover of mitochondria in the liver. However, the molecular basis of liver-specific activation of mitophagy by GCG, or its significance for metabolic stress responses in the liver is not understood. Here we show that BNIP3 is required for GCG-induced mitophagy in the liver through interaction with processed LC3B; an interaction that is also necessary to localize LC3B out of the nucleus to cytosolic mitophagosomes in response to nutrient deprivation. Loss of BNIP3-dependent mitophagy caused excess mitochondria to accumulate in the liver, disrupting metabolic zonation within the liver parenchyma, with expansion of zone 1 metabolism at the expense of zone 3 metabolism. These results identify BNIP3 as a regulator of metabolic homeostasis in the liver through its effect on mitophagy and mitochondrial mass distribution.
Abbreviations: ASS1, arginosuccinate synthetase; BNIP3, BCL2/adenovirus E1B interacting protein 3; CV, central vein; GCG – glucagon; GLUL, glutamate- ammonia ligase (glutamine synthetase); HCQ, hydroxychloroquine; LIR, LC3-interacting region; MAP1LC3B/LC3B, microtubule-associated protein 1 light chain 3 beta; mtDNA:nucDNA, ratio of mitochondrial DNA to nuclear DNA; PV, periportal vein; TOMM20, translocase of outer mitochondrial membrane protein 20.
KEYWORDS: BNIP3, glucagon, hepatocyte, LC3B, liver zonation, mitophagy, nutrient deprivation
Introduction
Mitophagy is a selective form of macroautophagy/autophagy in which mitochondria are specifically targeted for degradation in autolysosomes [1,2]. Mitophagy is not limited to the turnover of dysfunctional mitochondria but also reduces overall mitochondrial mass in response to stresses, such as hypoxia and nutrient starvation [1,3]. This may serve to prevent limited nutrients from being consumed inefficiently thereby ensuring cellular survival under conditions of energetic stress. Selective degradation of mitochondria depends on recruitment of phagophore membranes to the outer mitochondria where processed MAP1LC3/LC3-related proteins at nascent phagophores interact with mitochondrial cargo receptor proteins, such as BNIP3, BNIP3L/NIX, FUNDC1, FKBP8, BCL2L13 and targets of PINK1-PRKN/parkin-mediated ubiquitination [1,2].
While PINK1-PRKN are activated by mitochondrial depolarization and play a housekeeping role in eliminating de-polarized mitochondria, there is growing evidence that mitophagy receptors, such as BNIP3, BNIP3L/NIX, FUNDC1, FKBP8 and BCL2L13 are independently activated by different signals [1,2,4–6]. All of these mitophagy receptors act as inducible molecular receptors that interact directly, via conserved LC3-interaction region (LIR) motifs, with processed LC3 at nascent phagophores to selectively target mitochondria for autophagosomal degradation [2,4,6–8]. The identification of multiple mitochondrial cargo receptors that promote mitophagy calls into question whether these molecules are functionally overlapping or whether there is non-redundant regulation of their activity under distinct physiological conditions.
GCG (glucagon) signaling plays a critical role in the response of the liver to fasting [9] but the significance of GCG-induced mitophagy, as originally described by De Duve [10,11] and the mechanism by which it is activated in the hepatic response to nutrient deprivation is not understood. We showed previously that BNIP3 is required for mitochondrial function and mitigation of oxidative stress in the liver [12] but the mechanistic underpinnings of these observations were not investigated. Here we showed that BNIP3 was required for GCG-induced mitophagy in the liver and that expression of BNIP3 was sufficient to induce mitophagy in hepatocytes independent of upstream signaling events. BNIP3-dependent mitophagy relied on the interaction of BNIP3 with LC3B because a point mutant (W18A) that cannot bind LC3B was unable to promote mitophagy. We also identified a role for BNIP3 in promoting the cytoplasmic accumulation of LC3B such that loss of BNIP3 caused nuclear LC3B to predominate. This novel function for BNIP3 was dependent on the ability of BNIP3 to bind LC3B since the W18A mutant form of BNIP3 was unable to promote cytoplasmic accumulation of LC3B. Finally, we showed that BNIP3 was expressed in a zonal manner in the liver, with greatest expression near the less oxygenated central vein of the liver lobule. This in turn contributed to the zonal distribution of mitochondria in the liver, with highest mitochondrial mass near periportal regions of the liver. Loss of BNIP3 caused increased mitochondrial mass throughout the liver, disrupted mitochondrial zonal distribution and was associated with disruption of metabolic zonation in the liver in response to nutrient deprivation. These findings have significance for our understanding of how BNIP3, mitophagy, and mitochondrial mass are regulated to ensure proper functional organization of metabolism in the liver in response to nutrient deprivation.
Results
BNIP3 is required for GCG-induced mitophagy in liver
Expression of Bnip3 mRNA (Figure 1A; FigureS1A) and BNIP3 protein (Figure 1B; FigureS1B) was induced in adult mouse liver following GCG injection and overnight fasting of mice. Bnip3 mRNA expression peaked at 12 h following GCG injection or fasting (Figure 1A; FigureS1A) while BNIP3 protein expression was maximal at 24 h (Figure 1B; FigureS1B). These observations are consistent with previous reports identifying Bnip3 as a downstream transcriptional target of PPARA/PGC-1α in the response of the liver to nutrient deprivation [13].
Figure 1.
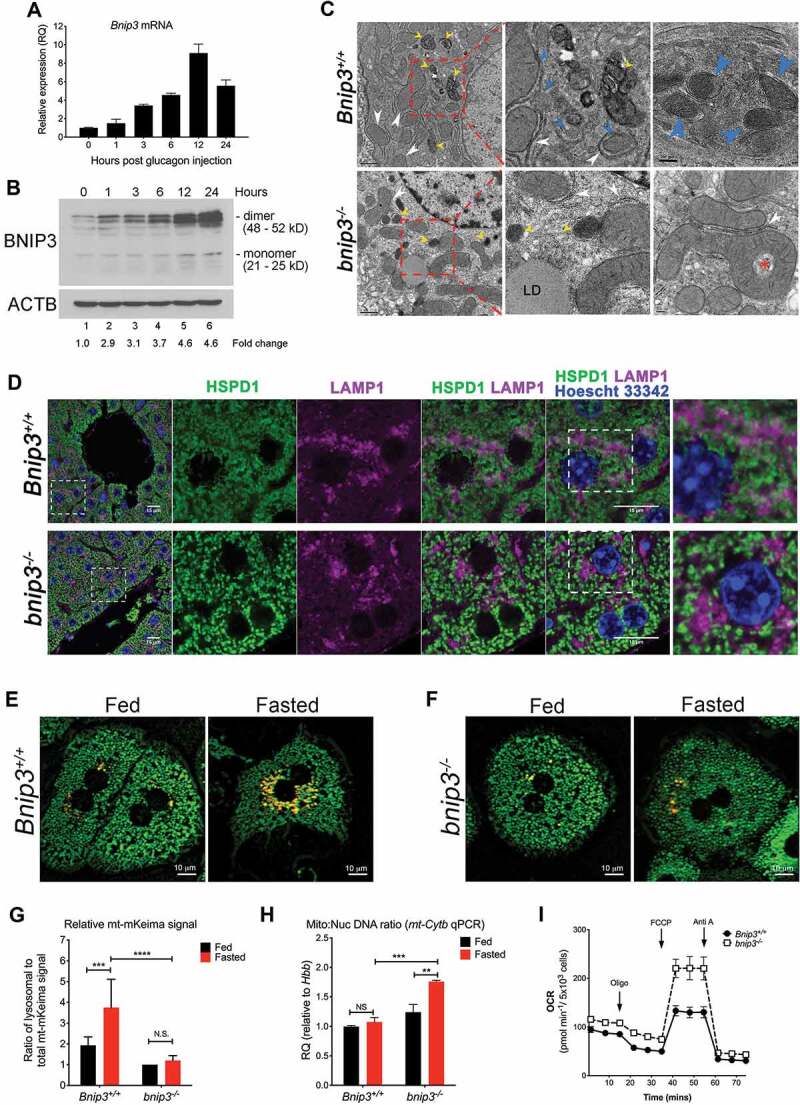
BNIP3 is required for GCG-induced mitophagy in the liver. (A) qPCR was performed using Taqman primers to detect levels of mouse Bnip3 mRNA expression in liver of mice injected with GCG for the indicated timepoints. (B) Western blot analysis for BNIP3 expression in RIPA buffer extracted protein lysates from livers of mice injected with GCG for the indicated timepoints was performed, using rodent-specific anti-BNIP3 antibody. (C) Transmission electron microscopy was performed on glutaraldehyde fixed liver pieces from Bnip3+/+ and bnip3-/- mice that had been injected with GCG and fasted for 16 h. (D) Co-immunofluorescence for HSPD1/HSP60 (green) and LAMP1 (magenta) on fixed liver sections from fasted Bnip3+/+ and bnip3-/- mice; nuclei stained with Hoechst 33,342 are represented in blue. Scale bars: 15 μm. (E, F) mt-mKeima fluorescence in Bnip3+/+ and bnip3-/- hepatocytes under fed and fasted conditions, showing green fluorescence (cytosolic mitochondria at alkaline pH) and yellow fluorescence (mitochondria taken up by lysosomes and at acid pH). Scale bars: 10 μm. (G) Graph quantifying mt-mKeima staining in (E) and (F) was plotted and analyzed using Wilcoxon rank sum analysis (NS – not significant, *** p < 0.001, **** p < 0.0001). (H) qPCR on genomic DNA from livers of fed and fasted Bnip3+/+ and bnip3-/- mice, for mt-Cytb and Hbb/β-globin as a measure of mitochondrial:nuclear genome ratio and analyzed using Wilcoxon rank sum analysis (NS – not significant, ** p < 0.01, *** p < 0.001). (I) Oxygen consumption rates in primary hepatocytes harvested Bnip3+/+ and bnip3-/- mice were measured using the Seahorse XF96 extracellular flux analyzer
De Duve’s original description of GCG-induced mitophagy in rat liver relied on transmission electron microscopy (TEM) to visualize mitochondria inside autolysosomes [10]. When we examined the ultra-structure of mouse liver from Bnip3+/+ mice that had been injected with GCG for 16 h, we observed the abundant presence of lysosomal structures containing cellular content undergoing degradation (Figure 1C, top panels, yellow arrows), including structures that resembled mitochondria with mitochondrial cristae present (Figure 1C, far left top panel, blue arrows). While lysosomes were readily detected in bnip3-/- liver sections (Figure 1C, bottom panels, yellow arrows), they were smaller and visibly void of the same level of content as lysosomes visualized in Bnip3+/+ liver (Figure 1C, top panels, yellow arrows). In addition, extended alignment between mitochondria and endoplasmic reticulum (ER) was clearly visible in Bnip3+/+ fasted liver (Figure 1C, top panels, white arrows) and interestingly, smaller mitochondria appeared frequently to be wrapped in phagophore-like membranes structures, possibly representing early steps in mitophagy (Figure 1C, middle top panel, blue arrows). Again, these structures were not as readily detected in the liver sections from fasted bnip3-/- mice (Figure 1C, bottom panels, white arrows). We did observe unusual donut shaped mitochondria in bnip3-/- fasted liver but not in Bnip3+/+ fasted liver, that appeared to have formed as a result of mitochondria fusing around on itself (Figure 1C, far left bottom panel, red star) although at this time, it is not clear what these unusual mitochondria represent.
To further assess the incidence of mitophagy in fasted liver in vivo, we performed co-immunofluorescence for HSPD1/HSP60 (a mitochondrial matrix protein) and LAMP1 (a lysosomal marker) (Figure 1D) on fixed liver sections from Bnip3+/+ and bnip3-/- mice. Fasting induced significant overlap in signal between HSP60 (green) and LAMP1 (magenta) in Bnip3+/+ liver, visualized as pale pink/white merged signal (Figure 1D, top panel; Fig. S2A). While lysosomes were visibly more abundant in bnip3-/- liver than in Bnip3+/+ liver (Fig. S2B), there was no evidence of overlap in HSPD1/HSP60 and LAMP1 staining in the livers from fasted bnip3-/- mice (Figure 1D, bottom panel; Fig. S2B), indicating reduced lysosomal turnover of mitochondria. Taken together with TEM data presented above (Figure 1C), these data suggest that loss of BNIP3 decreases the extent of mitophagy induced in the liver by fasting.
To gain further insight to the role of BNIP3 in GCG-induced mitophagy, we crossed bnip3-/- mice to mt-mKeima transgenic mice [14] to permit quantification of the effects of BNIP3 loss on mitochondrial flux to the lysosome in primary hepatocytes in response to nutrient deprivation. Intact mitochondria expressing pH-sensitive mKeima emit fluorescence at 440 nm (green) due to the alkaline pH of the mitochondrial matrix but this shifts to 586 nm (yellow) when mitochondria are taken up by the lysosome during mitophagy due to the acidic pH of the lysosomal vacuole [15]. We isolated primary hepatocytes from Bnip3+/+ or bnip3-/- mice and cultured them under either fed or fasted conditions before imaging mitophagic flux using mt-mKeima in live hepatocytes. We detected a marked increase in the ratio of yellow to green fluorescence in fasted Bnip3+/+ hepatocytes (Figure 1E, right; Figure 1G) compared to fed Bnip3+/+ hepatocytes (Figure 1E, left; Figure 1G). In contrast, fasted bnip3-/- hepatocytes (Figure 1F, right; Figure 1G) failed to show an increase in yellow fluorescence over fed bnip3-/- hepatocytes (Figure 1F, left; Figure 1G) indicating that BNIP3 loss resulted in a defect in mitochondrial flux to the lysosome in response to fasting. These results show that BNIP3 is required for GCG-induced mitophagy in primary hepatocytes and support the data obtained from whole liver in vivo (Figure 1C, Figure 1D).
To determine how differential rates of mitophagy in Bnip3+/+ and bnip3-/- hepatocytes affected overall mitochondrial mass, we measured the ratio of mitochondrial DNA to nuclear DNA (mtDNA:nucDNA) using two different mitochondrial encoded genes (mt-Cytb and mt-Nd1) relative to the nuclear encoded Hbb/β-globin gene (Figure 1H; Fig. S1C). We determined that despite increased mitophagy in response to fasting, Bnip3+/+ hepatocytes did not show a significant change in mtDNA:nucDNA ratio when either mt-Cytb (Figure 1H) or mt-Nd1 (FigureS1C) copy number was quantified relative to nuclear Hbb. A significant increase in mtDNA:nucDNA ratio was observed in bnip3-/- hepatocytes (Figure 1H; FigureS1C) in both the fed and fasted state compared to Bnip3+/+ fed and fasted hepatocytes (Figure 1H; FigureS1C), consistent with increased mitochondrial mass due to decreased mitophagy in bnip3-/- liver.
Overall mitochondrial mass is determined by the balance between mitophagy that reduces mitochondrial mass and levels of mitochondrial biogenesis that increases mitochondrial mass [16]. Mitochondrial biogenesis is induced in the liver in response to fasting via increased expression of PPARGC1A/PGC-1α [17]. We examined expression of PPARGC1A/PGC-1α protein by western blot in extracts from livers of fed and fasted Bnip3+/+ and bnip3-/- mice and did not detect any significant difference in levels of PPARGC1A/PGC-1α in liver from fasted bnip3-/- mice compared to Bnip3+/+ (FigureS1D). These results indicate that increased mitochondrial mass detected in the livers of fasted bnip3-/- mice compared to fasted Bnip3+/+ mice is due primarily to decreased levels of mitophagy. Consistent with increased mitochondrial mass due to loss of BNIP3-dependent mitophagy, we observed elevated basal and maximal oxygen consumption rates in isolated bnip3-/- hepatocytes compared to Bnip3+/+ hepatocytes (Figure 1I).
Mutation of the BNIP3 LIR abrogates interaction of BNIP3 with LC3 and limits mitophagy
To determine whether BNIP3 expression was sufficient to induce mitophagy in the liver, we over-expressed HA-tagged BNIP3 (HA-BNIP3) in primary hepatocytes from bnip3-/- mice. We also tested the ability of a mutant form of BNIP3, in which amino acid W18 is mutated to alanine (HA-BNIP3W18A), to promote mitophagy in hepatocytes. Previous work identified a conserved LIR motif in BNIP3 from amino acids 18 through 22 [7] (Figure 2A). Here we observed that while HA-BNIP3 was able to bind to GFP-LC3 (Figure 2B, right panel) and rescue mitophagy in bnip3-/- hepatocytes, based on increased mt-mKeima fluorescence shift from green to yellow (Figure 2C; Figure 2D), the W18A mutant was not able to either bind GFP-LC3 effectively (Figure 2B, right) or rescue mitophagy (Figure 2C; Figure 2D). Immunofluorescent staining for HA-tagged BNIP3 confirmed proper mitochondrial localization of exogenously expressed HA-BNIP3 and HA-BNIP3W18A, with HA-BNIP3 localizing to mitochondria that stained positive for TOMM20, a constitutively expressed outer mitochondrial membrane (OMM) protein (Fig. S3A). Exogenous expression of HA-BNIP3 in bnip3-/- hepatocytes was sufficient to induce mitophagy under fed conditions (Figure 2C; Figure 2D) and further increases in mt-mKeima fluorescence shift in response to fasting were not significant (Figure 2D). These observations suggest that BNIP3 is both necessary (in response to fasting) and sufficient to promote mitophagy in the liver in a manner dependent on its ability to bind to LC3.
Figure 2.
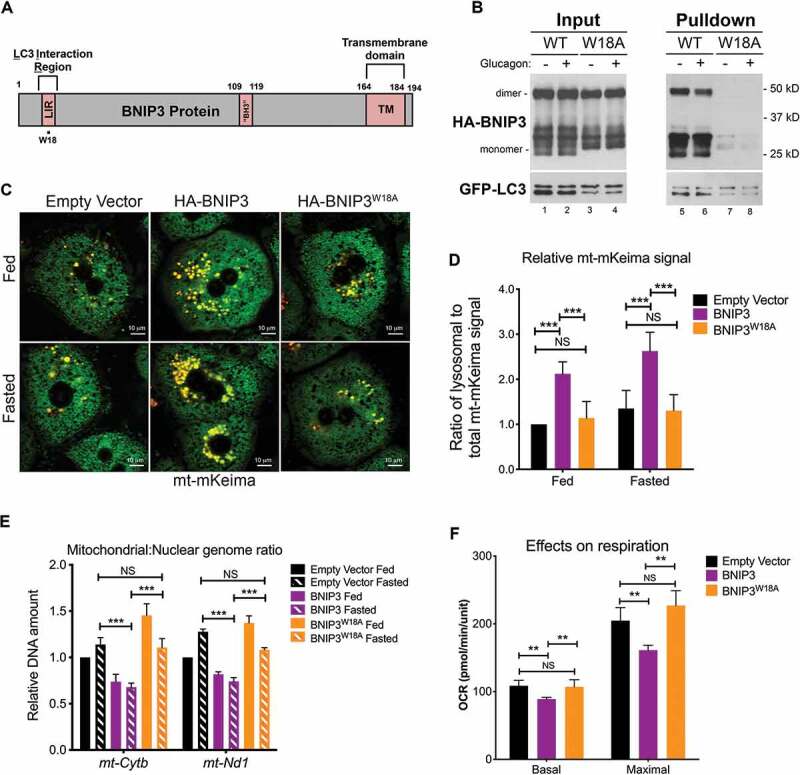
BNIP3 is sufficient to induce mitophagy in primary hepatocytes. (A) Schematic of BNIP3 protein illustrating key functional domains, including the LIR motif (amino acids 18–21), the “BH3” domain (amino acids 109–119) and the transmembrane domain (amino acids 164–184). (B) Pulldown of GFP-LC3 from bnip3-/- hepatocytes (collected from GFP-LC3+; bnip3-/- mice) expressing exogenous HA-BNIP3 or HA-BNIP3W18A (delivered by adenoviral vector), followed by western blot for GFP and HA, with total inputs (left) and pulldown (right). (C) mt-mKeima fluorescence in bnip3-/- hepatocytes reconstituted via adenoviral delivery vector to express EV (control), HA-BNIP3 or HA-BNIP3W18A, under fed or fasted conditions. Scale bars: 10 μm. (D) Graph quantifying mt-mKeima staining in (C) and analyzed using Wilcoxon rank sum analysis (NS – not significant, *** p < 0.001). (E) qPCR on genomic DNA for mt-Cytb or mt-Nd1 and Hbb/β-globin as a measure of mitochondrial:nuclear genome ratio in bnip3-/- hepatocytes reconstituted via adenoviral delivery vector to express exogenous EV (control), HA-BNIP3 or HA-BNIP3W18A, and analyzed using Wilcoxon rank sum analysis (NS – not significant, *** p < 0.001). (F) Basal and maximal oxygen consumption rate (OCR) for bnip3-/- hepatocytes reconstituted via adenoviral delivery vector with EV (control), HA-BNIP3 or HA-BNIP3W18A, and analyzed using Wilcoxon rank sum analysis (NS – not significant, ** p < 0.01)
We also examined whether mutant forms of BNIP3 lacking its putative BH3 domain (amino acids 109–119) or its transmembrane domain (TMD) (amino acids 164 − 184) were able to promote mitophagy (Fig. S3A-C). While HA-BNIP3(∆BH3) localized properly to the mitochondria (Fig. S3A), loss of the TMD caused HA-BNIP3[∆TMD] to localize aberrantly to the nucleus (Fig. S3A). The HA-BNIP3(∆BH3) mutant retained ability to bind GFP-LC3 (Fig. S3B) and to promote mitophagic flux (Figure S3C) indicating that the putative BH3 domain in BNIP3 was not required for BNIP3-dependent mitophagy. By contrast, deletion of the TMD (∆TMD) of BNIP3 impeded both GFP-LC3 binding and mitophagy (Fig. S3B, Fig. S3C) indicating that integration of BNIP3 into the outer mitochondrial membrane (OMM) was required for BNIP3 function in mitophagy.
Consistent with a role in promoting mitophagy in hepatocytes, reconstitution of bnip3-/- hepatocytes with exogenous HA-BNIP3 reduced the mtDNA:nucDNA ratio (Figure 2E, purple bars) compared to control cells expressing empty vector (Figure 2E, black bars), while mitophagy-deficient HA-BNIP3W18A expression did not (Figure 2E, orange bars). Finally, HA-BNIP3 over-expression in bnip3-/- hepatocytes reduced both basal and maximal respiration (Figure 2F), unlike HA-BNIP3W18A which did not significantly affect oxygen consumption in bnip3-/- hepatocytes compared to empty vector expressing control cells (Figure 2F). Together these findings support the conclusion that the BNIP3-LC3B interaction and BNIP3-dependent mitophagy in the liver prevents excess accumulation of mitochondria during the fasting response.
BNIP3 promotes cytosolic localization and turnover of LC3B via mitophagy
To further interrogate the role of BNIP3 in GCG-induced mitophagy in the liver, we examined effects of BNIP3 loss on GFP-LC3 puncta formation and overlap with mitochondria (by staining with MitoTracker Deep Red) following GCG treatment of primary hepatocytes isolated from GFP-LC3 transgenic mice [18]. Fasting with GCG treatment increased numbers of GFP-LC3-positive autophagosomes and increased overlap (yellow) between autophagosomes (green) and mitochondria (red) in the cytosol of Bnip3+/+ hepatocytes (Figure 3A, right panel, white arrows) compared to fed conditions (Figure 3A, left). However, formation of cytosolic GFP-LC3 positive puncta and autophagosome-mitochondrial overlap were strongly diminished in fasted bnip3-/- hepatocytes (Figure 3B, right) compared to fasted Bnip3+/+ hepatocytes (Figure 3A, right panel, white arrows). Consistent with mt-mKeima data (Figure 1C,Figure 1D,Figure 1E), these data indicate that BNIP3 is required for mitophagy in hepatocytes induced by nutrient deprivation.
Figure 3.
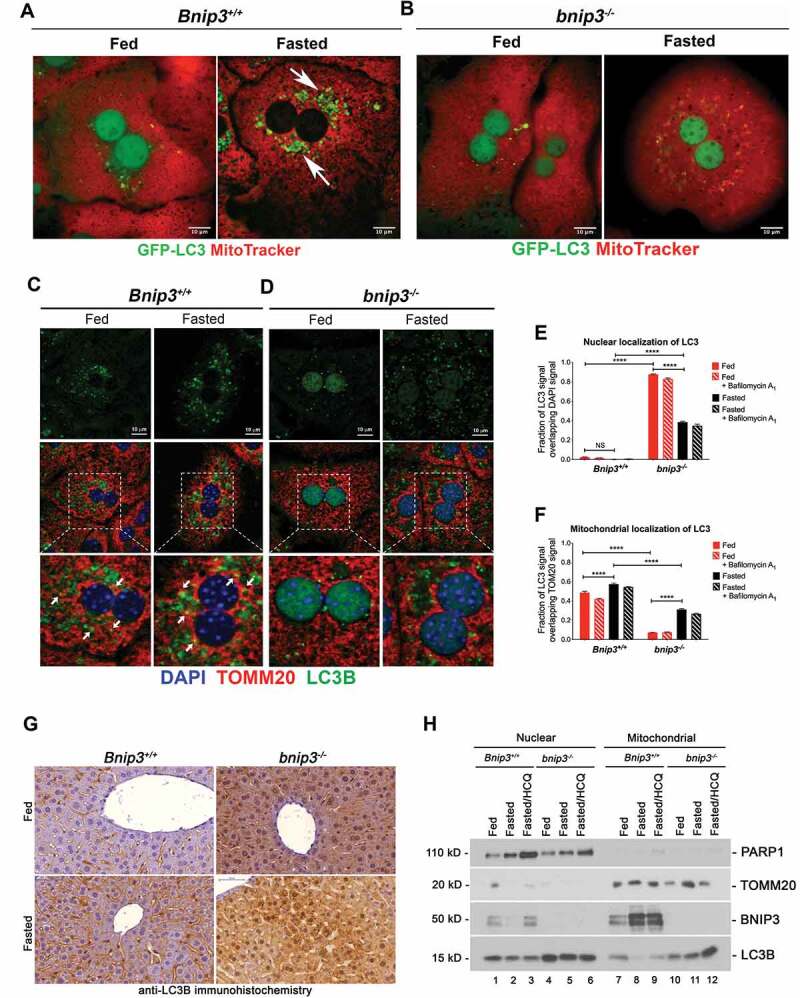
BNIP3 is required for cytosolic localization of LC3 in response to fasting. (A, B) Imaging of live hepatocytes from Bnip3+/+ (A) or bnip3-/- (B) mice collected from compound GFP-LC3+ transgenic mice (Bnip3+/+ or bnip3-/- co-stained with MitoTracker Deep Red to visualize overlap between GFP-LC3+ puncta and mitochondria (red), under fed or fasted conditions. Scale bars: 10 μm. (C, D) Immunofluorescent staining of fixed hepatocytes from Bnip3+/+ (C) or bnip3-/- (D) mice for TOMM20 (mitochondria, red), LC3B (autophagosomes, green) and DAPI (nuclei, blue), under fed and fasted conditions; white arrows in (C and D) indicate areas of overlap between TOMM20 and LC3B staining, seen as yellow puncta. Scale bars: 10 μm. (E) Graph quantifying mitochondrial localization of LC3B in Bnip3+/+ and bnip3-/- hepatocytes under fed and fasted conditions using ImageJ, and analyzed using Wilcoxon rank sum analysis (NS – not significant, **** p < 0.0001). (F) Graph quantifying nuclear localization of LC3B in Bnip3+/+ and bnip3-/- hepatocytes under fed and fasted conditions using ImageJ, and analyzed using Wilcoxon rank sum analysis (NS – not significant, **** p < 0.0001). (G) Immunohistochemistry for LC3B expression around the central vein of livers from fed and fasted Bnip3+/+ and bnip3-/- mice. Hepatocytes are the large blue cells while Kupffer cells are the elongated brown staining cells between hepatocytes, as seen most strikingly in the Bnip3+/+ liver from fed mice (top left). Scale bars (blue, top left): 50 μm. (H) Western blot analysis for PARP1, TOMM20, BNIP3 and LC3B in nuclear (high salt extraction) and mitochondrial enriched protein extracts (sucrose/Triton extraction) from Dounce homogenized whole livers collected from fed and fasted Bnip3+/+ and bnip3-/- mice
We also noticed that GFP-LC3 was predominantly nuclear under fed conditions and only accumulated at cytosolic puncta following nutrient deprivation in Bnip3+/+ hepatocytes (Figure 3A). By contrast, GFP-LC3 remained nuclear in fasted bnip3−/- hepatocytes under fed or fasted conditions (Figure 3B, right). This indicates that, in addition to stimulating mitophagy directly through interaction with LC3, BNIP3 promotes cytoplasmic localization of LC3 in response to starvation.
To determine whether these unexpected findings on localization of LC3 were the consequence of over-expression of the GFP-LC3 transgene, we examined overlap in staining pattern between endogenous TOMM20 and endogenous LC3B under fed and fasted conditions. We observed that endogenous LC3B was not detectable in the nucleus of either fed or fasted Bnip3+/+ hepatocytes (Figure 3C,Figure 3E) but that levels of cytosolic puncta increased significantly in fasted hepatocytes (Figure 3C, arrows). The LC3B-positive puncta in fasted hepatocytes overlapped strongly with perinuclear mitochondria and this pattern was increased by treatment with bafilomycin A1 (Figure 3C, arrows; Figure 3F; Figure S3D), which is consistent with increased formation of mitophagosomes and increased mitophagic flux in response to nutrient deprivation (Figure 1C – Figure 1G).
However, LC3B localized predominantly to the nucleus in fed bnip3-/- hepatocytes in contrast to Bnip3+/+ hepatocytes grown under the same conditions (Figure 3C,Figure 3E). While fasting did increase LC3B localization to the cytoplasm (Figure 3D,Figure 3F), a significant proportion of LC3B remained in the nucleus (Figure 3D,Figure 3E) and significantly fewer LC3B-positive puncta were found in the cytosol of fasted bnip3−/- hepatocytes (Figure 3D,Figure 3E,Figure 3D,Figure 3F) than in fasted Bnip3+/+ hepatocytes (Figure 3C,Figure 3E,Figure 3F). Furthermore, treatment with bafilomycin A1 did not increase the levels of cytosolic LC3B in bnip3-/- hepatocytes under fed or fasted conditions (FigureS3E), suggesting that mitophagosomes were not being actively turned over at the lysosome. Overall, these results indicate that BNIP3 promotes cytosolic localization of LC3B in response to fasting, in addition to acting as a mitophagy receptor.
To assess how BNIP3 affected cytosolic localization of LC3B in vivo, we stained sections of liver from fed and fasted mice for LC3B (Figure 3G; Fig. S4A-B). Levels of LC3B increased in livers from fasted Bnip3+/+ mice compared to livers from fed Bnip3+/+ mice (Figure 3G, bottom left versus top left) where it was detectable in both hepatocytes and in Kupffer cells and localized predominantly to the cytosol (Figure 3G, bottom left; Fig. S4A-B). LC3B levels were markedly higher in liver from bnip3-/- mice compared to Bnip3+/+ mice, under fed or fasted conditions (Figure 3G), possibly due defective mitophagy and reduced LC3B turnover (Figure 1; Figure 2). Significantly, LC3B was localized predominantly in hepatocyte nuclei in bnip3-/- liver in contrast to Bnip3+/+ liver (Figure 3G, right panels). Following fasting, LC3B showed increased cytosolic localization in bnip3-/- liver (Figure 3G, bottom right) but in contrast to Bnip3+/+ liver (Figure 3G, bottom left), a significant proportion of LC3B remained nuclear (Figure 3G, bottom right versus bottom left; Fig. S4A-B). These findings were further supported by immunofluorescent (IF) staining for LC3B on fixed sections of primary liver from fed and fasted Bnip3+/+ and bnip3-/- mice (Fig. S4C) where fasting induced marked loss of nuclear Lc3 in Bnip3+/+ liver (Fig. S4C, bottom left compared to top left) in contrast to bnip3-/- liver, where LC3B remained readily detectable in the nucleus of fasted hepatocytes (Fig. S4C, bottom right). Thus, similar to our observations in primary hepatocytes in vitro, BNIP3 loss appears to cause nuclear LC3B to accumulate in hepatocytes in vivo under both fed and fasted conditions.
Consistent with the in situ data shown above, LC3B protein levels were 2–3 fold higher in nuclear extracts from bnip3-/- liver (Figure 3H, lanes 4–6) than in nuclear extracts from Bnip3+/+ livers (Figure 3H, lanes 1–3). LC3B levels were also higher in mitochondrial extracts from bnip3-/- livers (Figure 3H, lanes 7–9) compared to Bnip3+/+ livers (Figure 3H, lanes 10–12). Mitochondrial LC3B decreased markedly upon fasting of Bnip3+/+ mice (Figure 3H, lane 8) and treatment with 60 mg/kg HCQ (to block lysosomal turnover) attenuated the fasting-induced decrease in mitochondrial LC3B levels (Figure 3H, lane 9), indicating that mitochondrial LC3B was being effectively turned over at the lysosome in fasted Bnip3+/+ liver. By contrast, fasting did not decrease mitochondrial LC3B levels in liver from bnip3-/- mice (Figure 3H, lanes 10–12) suggesting lower rates of turnover of mitochondrial Lc3 in bnip3-/- liver compared to Bnip3+/+. Treatment of bnip3-/- mice with HCQ did increase levels of LC3B in fasted liver (Figure 3H, lane 12) indicating that LC3B can get turned over in fasted liver independent of BNIP3, albeit at a reduced rate. Together these results indicate that BNIP3 promotes cytosolic localization of LC3B in the liver and LC3B turnover through mitophagy.
Expressing exogenous HA-BNIP3 in bnip3-/- hepatocytes from GFP-LC3 transgenic mice (Figure 4A, middle panel) was sufficient to promote cytosolic punctate expression of GFP-LC3 and loss of nuclear GFP-LC3, compared to control hepatocytes expressing empty vector (Figure 4A, left panel). By contrast, HA-BNIP3W18A that cannot bind LC3 failed to induce cytoplasmic localization of GFP-LC3 (Figure 4A, right panel) indicating that interaction with BNIP3 was required to induce cytosolic accumulation of GFP-LC3 in fasted hepatocytes. Similar to results obtained with induction of mitophagy measured by mt-mKeima flux (Figure 2C-Figure 2D), over-expressing HA-BNIP3 was sufficient to induce cytosolic localization of GFP-LC3 and puncta formation (Figure 4A).
Figure 4.
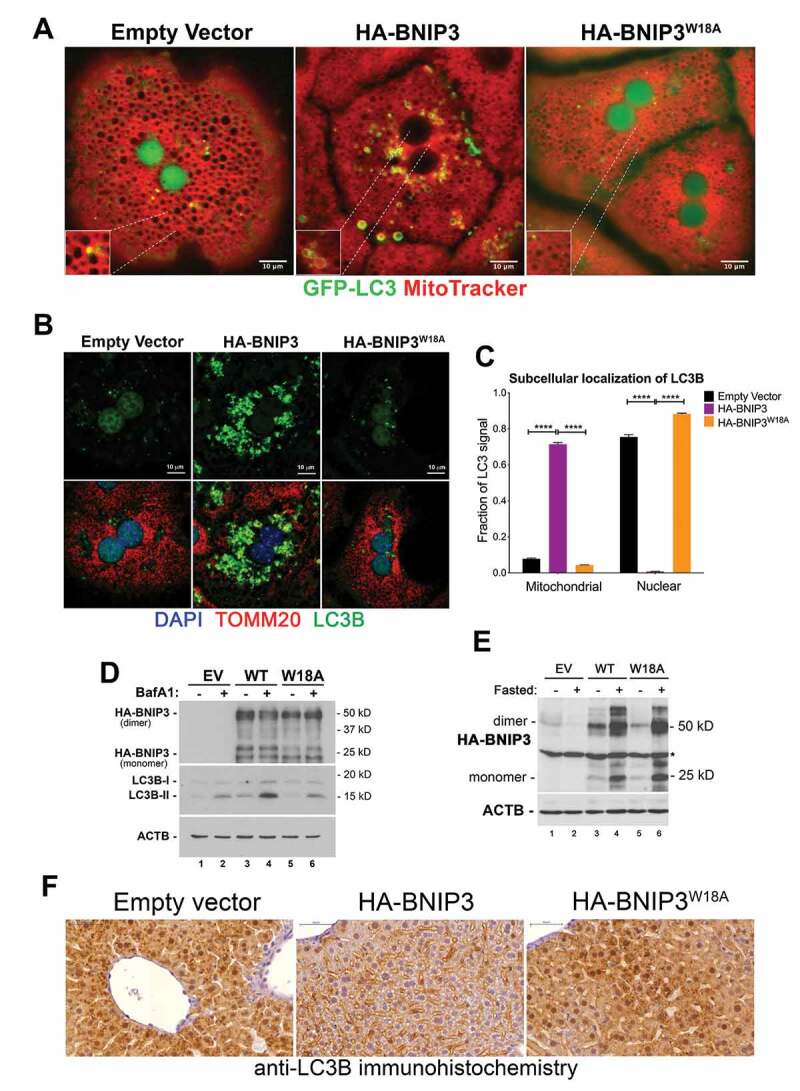
BNIP3 induces cytosolic localization of LC3. (A) Imaging of live hepatocytes from GFP-LC3+;bnip3-/- mice co-stained with MitoTracker Deep Red examining overlap between GFP-LC3+ puncta and mitochondria (red) when hepatocytes are reconstituted using adenoviral delivery vector to express exogenous EV (control), HA-BNIP3 or HA-BNIP3W18A. Scale bars: 10 μm. (B) Immunofluorescent staining for TOMM20 (mitochondria, red), LC3B (autophagosomes, green) and DAPI (nuclei, blue) on fixed bnip3-/- hepatocytes reconstituted using adenoviral delivery vector to express exogenous EV, HA-BNIP3 or HA-BNIP3W18A. Scale bars: 10 μm. (C) Graph quantifying nuclear and mitochondrial localization of LC3B in bnip3-/- hepatocytes reconstituted using adenoviral delivery vector to express exogenous EV (control), HA-BNIP3 or HA-BNIP3W18A using ImageJ, and analyzed using Wilcoxon rank sum analysis (NS – not significant, **** p < 0.0001). (D) Western blot for endogenous LC3B and exogenous BNIP3 (HA) in lysates from bnip3-/- hepatocytes reconstituted using adenoviral delivery vector to express exogenous EV (control), HA-BNIP3 or HA-BNIP3W18A. (E) Western blot for exogenous HA-BNIP3 expressed in vivo in whole liver extracts from bnip3-/- mice 10 days after intra-venous injection with adenoviral delivery vectors expressing exogenous EV (control), HA-BNIP3 or HA-BNIP3W18A. (F) Immunohistochemistry for endogenous LC3B expression around the central vein of livers from fasted bnip3-/- mice reconstituted in vivo using adenoviral delivery vectors to express exogenous EV (control), HA-BNIP3 or HA-BNIP3W18A (see E above). Scale bars (blue, top left): 50 μm
Similar observations were made when examining the effect of BNIP3 expression on localization of endogenous LC3B, where HA-BNIP3 but not HA-BNIP3W18A (Figure 4B) caused LC3B localization to shift from predominantly nuclear to strongly cytosolic (Figure 4B, Figure 4C). Punctate LC3B staining in the cytosol overlapped with staining for TOMM20, indicating that HA-BNIP3 was promoting localization of LC3 to mitochondria (Figure 4B, Figure 4C). Significantly, we do not detect BNIP3 in the nucleus of hepatocytes under fed or fasted conditions (Fig. S4D). Furthermore, when we examined endogenous LC3B levels and processing by western blot, we observed that HA-BNIP3 increased levels of processed LC3B-II (Figure 4D, lane 3 compared to lane 1), which increased further upon treatment with bafilomycin A1 (Figure 4D, lanes 4), consistent with HA-BNIP3 promoting LC3B turnover at the lysosome. HA-BNIP3W18A showed diminished effect on levels of LC3B-II, both before and after bafilomycin A1 treatment (Figure 4D, lanes 5–6). Increased LC3B-II levels induced by HA-BNIP3 over-expression are not explained by increased LC3B transcription since levels of LC3B mRNA were increased to similar levels by fasting in bnip3-/- hepatocytes expressing empty vector, HA-BNIP3 and BNIP3W18A (Fig. S4E). Over-expression of HA-BNIP3, but not empty vector or HA-BNIP3W18A (Figure E), also significantly reduced nuclear accumulation of LC3B in fasted bnip3-/- liver in vivo (Figure 4F, middle panel compared to left and right panels). Interestingly, overnight fasting of mice increased levels of exogenous HA-BNIP3 protein in the liver and also induced a slower migrating form of BNIP3 (Figure 4E, lanes 4 and 6 compared to lanes 3 and 5), suggesting post-translational control of BNIP3 protein stability in vivo in the response to fasting. These results indicate that by promoting localization of LC3B to the cytosol, BNIP3 is promoting LC3B processing and turnover at the mitochondria in primary cultured hepatocytes in vitro and in vivo in the response of the liver to fasting.
BNIP3 controls zonal mitochondrial mass in the liver in response to fasting
We performed immunohistochemistry on liver sections to determine expression levels of the OMM protein TOMM20, as a measure of mitochondrial mass in vivo. While TOMM20 levels increased modestly in Bnip3+/+ liver in response to fasting (Figure 5A), this was not found to be statistically significant (Figure 5C). By contrast, TOMM20 staining was significantly increased in the livers of both fed and fasted bnip3-/- mice (Figure 5B, Figure 5C; Fig. S5A) compared to Bnip3+/+ mice (Figure 5B, Figure 5C; Fig. S5A) with a particularly marked increase in TOMM20 in bnip3-/- liver in response to fasting (Figure 5B, right; Figure 5C), in agreement with measurement of mtDNA:nucDNA ratios (Figure 1H; Fig. S1C). Similar results were obtained when livers sections were stained for the inner mitochondrial membrane protein, Tim23 (FigureS5B). In summary, TOMM20 levels were increased in bnip3−/- liver compared to Bnip3+/+ under both fed and fasted conditions (Figure 5A-Figure 5C), indicating that, due to defective mitophagy, bnip3-/- liver accumulated mitochondrial mass over time, with a further increase in response to fasting.
Figure 5.

Loss of BNIP3 disrupts zonal pattern of mitochondrial distribution in liver. (A, B) Immunohistochemistry for TOMM20 on 5-μm sections of mouse liver from fed and fasted Bnip3+/+ (A) and bnip3-/- mice (B). Dashed lines are used to highlight the difference in TOMM20 expression in zone 1 and zone 3 surrounding the periportal vein (PV) and central vein (CV) respectively. The PV and CV areas stained and highlighted at low power (100 x) are shown below at higher magnification (400 x). Scale bars (blue, top left): 200 μm for low magnification, 50 μm for high magnification. (C) Quantification of TOMM20 IHC on whole liver sectional areas on each slide from fed and fasted Bnip3+/+ (n = 5) and bnip3-/- mice (n = 6), using Aperio software and analyzed using Wilcoxon rank sum analysis (NS – not significant, *** p < 0.001, **** p < 0.0001). (D) Diagram illustrating metabolic zonation in the liver lobule. (E) Immunohistochemistry for BNIP3 (Sigma Prestige antibody) on 5-μm sections of mouse liver from fed and fasted Bnip3+/+ mice. Scale bars (blue, top left): 200 μm for low magnification, 50 μm for high magnification. (F) Immunohistochemistry for BNIP3 (Sigma Prestige antibody) on healthy human liver. Scale bars (blue, top left): 200 μm for low magnification, 100 μm for high magnification
The liver is made up of functional units called lobules that are organized hexagonally around a central vein, with branches of the periportal vein (PV) and hepatic artery (HA) at the lobule periphery providing hepatic blood supply that flows inwards from the more oxygenated PV/HA to the less oxygenated central vein (CV) at the lobule core (Figure 5D)[19,20]. Liver metabolism is functionally compartmentalized across the liver lobule into zones: zone 1 closest to the more oxygenated PV where gluconeogenesis, urea cycle, fatty acid oxidation amongst other metabolic processes occur, versus zone 3, the less oxygenated zone closest to the CV, where glycolysis and lipogenesis occur (Figure 5D) [20–22]. While aspects of metabolic zonation of the liver are driven by transcriptional events [23], the extent to which mitochondrial mass distribution contributes to metabolic zonation in the liver is not known.
Interestingly, TOMM20 staining appeared zonal in liver from fed Bnip3+/+ mice (Figure 5A, left) with strongest TOMM20 staining in zone 1 around the PV and lower TOMM20 staining around the CV in zone 3 of the liver (Figure 5A, left; regions of low staining around the CV are highlighted with dashed lines). The same zonal pattern of TOMM20 staining was also observed in liver of fasted Bnip3+/+ mice with concentrated TOMM20 expression around the PV and lower TOMM20 expression around the CV (Figure 5A, right). Conversely, we observed that BNIP3 was most strongly expressed in zone 3 of the liver lobule around the CV (Figure 5E) where TOMM20 levels are low (Figure 5A) consistent with lower oxygen levels (BNIP3 is a HIF1A [hypoxia inducible factor 1, alpha subunit]) and increased BNIP3-dependent mitophagy in zone 3. Meanwhile, BNIP3 expression was low in the more oxygenated zone 1 region near the PV (Figure 5E) where TOMM20 levels are highest (Figure 5A). Immunohistochemical staining of primary human liver section showed similarly increased BNIP3 levels around the CV compared to periportal regions (Figure 5F). LC3B also exhibited a zonal expression pattern in Bnip3+/+ liver with strongest expression in zone 1 and lower levels in zone 3, but notably this expression predominated in Kupffer cells, not hepatocytes (Fig. S4A-B). LC3B expression in fasted Bnip3+/+ liver specifically increased in hepatocytes in zone 3 around the CV (Fig. S4A-B), where BNIP3 expression was also highest (Figure 5E) and TOMM20 levels were lowest (Figure 5A), consistent with increased mitophagy in zone 3 around the CV of fasted liver. These results indicate that BNIP3-dependent mitophagy is zonal in the liver and determines mitochondrial mass distribution across the liver lobule, particularly in the response to fasting.
Loss of BNIP3-dependent mitophagy disrupts metabolic homeostasis in the liver
Mitochondria contribute extensively to metabolic processes in the liver and given the importance of separating opposing metabolic processes across the liver lobule [20], we interrogated whether disrupted mitochondrial mass distribution arising from loss of BNIP3-dependent mitophagy affected liver metabolism. We stained Bnip3+/+ and bnip3-/- liver from fed and fasted mice for ASS1 (arginosuccinate synthetase 1), a key enzyme in the urea cycle [24] and also an established zone 1 marker [20,23]. Significantly, while part of the urea cycle is carried out in the cytosol, key steps in the urea cycle are executed in the mitochondrial matrix [24] and thus we postulated that altered mitochondrial mass and mitochondrial distribution in the liver lobule was disrupting urea cycle activity. ASS1 showed highest expression round the PV where the urea cycle is concentrated, and low to undetectable expression immediately around the CV in both Bnip3+/+ and bnip3-/- livers (Figure 6A, Fig. S6A). This zonal pattern of ASS1 expression was maintained in Bnip3+/+ mice under fasting conditions (Figure 6A, Fig. S6A). By contrast, its zonal expression pattern was lost in livers from bnip3-/- mice following fasting, such that strong ASS1 expression was detected in the cell layer closest to the CV where ASS1 is not usually expressed (Figure 6A, Fig. S6A). Thus, similar to what was seen for TOMM20 expression (Figure 5B), loss of BNIP3 resulted in altered zonal ASS1 expression in the liver of fasted mice. Expression of other urea cycle genes (Asl and Arg1) was also upregulated in liver from fasted bnip3-/- mice compared to Bnip3+/+ mice (Figure 6B) and steady state levels of all urea cycle metabolites examined (arginine, urea, ornithine, homocitrulline) exhibited significantly elevated levels in the liver of fasted bnip3-/- mice compared to Bnip3+/+ (Figure 6C). In addition, we detected a marked elevation in levels of the ketone body, 3-hydroxybutyrate (BHBA) that is produced by de-amination of amino acids in the urea cycle (Fig. S6B). Together these results show that loss of BNIP3 increases expression of urea cycle enzymes, including in zones of the liver where urea cycle is not usually active, and increases overall urea cycle activity in the liver of fasted mice.
Figure 6.
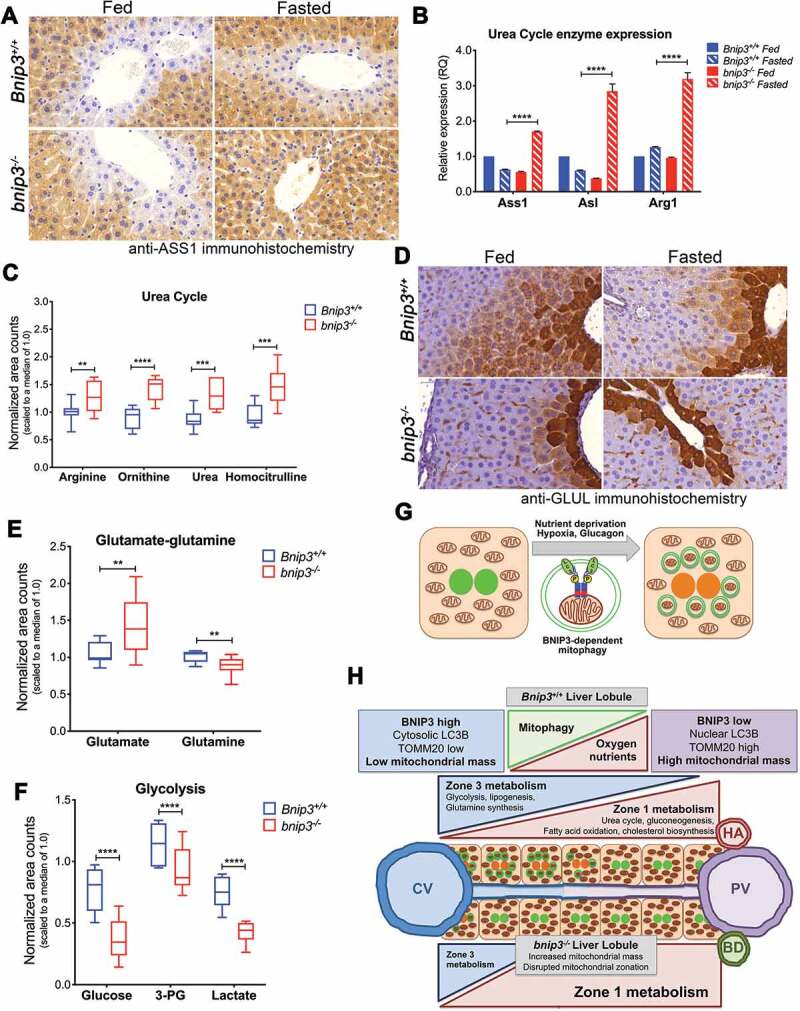
Loss of BNIP3 disrupts metabolic zonation in the liver. (A) Immunohistochemistry for ASS1 (high power magnification) on 5-μm sections of mouse liver from fed and fasted Bnip3+/+ (top) and bnip3-/- mice (bottom). Scale bars (blue, top left): 50 μm. (B) qPCR for levels of urea cycle genes (Ass1, Asl, Arg) expressed in liver from fed and fasted Bnip3+/+ and bnip3-/- mice and analyzed using Wilcoxon rank sum analysis (**** p < 0.0001). (C) Steady state measurement of levels of urea cycle intermediates (arginine, ornithine, urea, homocitrulline) in fasted liver from Bnip3+/+ (n = 8) and bnip3-/- (n = 8) mice performed by LC/MS and analyzed by two-way ANOVA (** p < 0.01, *** p < 0.001, **** p < 0.0001). (D) Immunohistochemistry for GLUL (high power magnification) on 5-μm sections of mouse liver from fed and fasted Bnip3+/+ (top) and bnip3-/- mice (bottom). Scale bars (blue, top left): 50 μm. (E) Steady state measurement of levels of glutamine and glutamate in fasted liver from Bnip3+/+ (n = 8) and bnip3-/- (n = 8) mice performed by LC/MS and analyzed by two-way ANOVA (** p < 0.01). (F) Steady state measurement of levels of glycolytic intermediates (glucose, 3-phosphoglycerate, lactate) in fasted liver from Bnip3+/+ (n = 8) and bnip3-/- (n = 8) mice performed by GC/MS and analyzed by two-way ANOVA (**** p < 0.0001). (G) Diagram summarizing the role of BNIP3 in mitophagy induced by nutrient deprivation, including its ability to promote cytosolic localization of LC3B. (H) Diagram summarizing the role of BNIP3-dependent mitophagy in control of mitochondrial mass distribution and metabolic zonation in Bnip3+/+ liver, and how this is disrupted when BNIP3 is inactivated in bnip3-/- liver
To examine whether zone 3 expression was also disrupted in bnip3-/- liver, we examined expression of GLUL (glutamate-ammonia ligase (glutamine synthetase)), which has the opposite expression pattern as ASS1. GLUL is concentrated around the CV, playing a critical role at the inner mitochondrial membrane (IMM) in converting glutamate to glutamine, to maintain and re-cycle glutamine in the liver [20]. As expected, GLUL protein expression was most highly expressed in zone 3 in the cells surrounding the CV of the liver in Bnip3+/+ fed mice and its expression gradually diminished through zone 2 and was undetectable in zone 1 around the PV (Figure 6D, Fig. S6C). In contrast, GLUL expression was more tightly restricted to zone 3 around the CV in liver from fed bnip3-/- mice and further restricted to the single cell layer around the CV of livers of bnip3-/- mice following fasting (Figure 6D, Fig. S6C). We also detected reduced steady state levels of glutamine and increased levels of glutamate in livers from fasted bnip3-/- mice compared to fasted Bnip3+/+ mice (Figure 6E), consistent with reduced GLUL activity when BNIP3 is lost. Glycolysis is another metabolic pathway that predominates in zone 3 of the liver, and we consistently observed significantly reduced levels of glycolytic intermediates in the liver of fasted bnip3-/- mice compared to fasted Bnip3+/+ mice (Figure 6F). These results show that BNIP3 loss is expanding ASS1 expression and urea cycle activity (zone 1) and restricting GLUL expression, glutamate-glutamine conversion, and glycolysis (zone 3) in the liver of fasted mice, indicating a role for BNIP3 in maintaining metabolic zonation in the liver.
Previous work has highlighted a key role for the WNT-CTNNB1/β-catenin pathway in liver zonation, such that loss of the Apc (APC, WNT signaling pathway regulator) tumor suppressor gene in the liver caused genes, such as those encoding glutamine synthetases (GS, of which GLUL is a member) to be aberrantly expressed in the liver [25]. Specifically, expression of perivenous genes (zone 3) such as GLUL, were expanded at the expense of periportal genes (zone 1). Here, we observe the opposite effect with zone 1 genes expanded at the expense of zone 3 genes as a result of BNIP3 loss (Figure 6H). When we examined expression of unphosphorylated CTNNB1/β-catenin in liver from fed and fasted Bnip3+/+ and bnip3-/- mice, we observed normal zonal patterning of CTNNB1/β-catenin in Bnip3+/+ fed and fasted liver (Fig. S6D), consistent with published data [25]. It should be noted however, that expression of CTNNB1/β-catenin underlying its zonal expression pattern appears to be seen primarily in non-hepatocytes and particularly in Kupffer cells in the liver of Bnip3+/+ mice (Fig. S6D). Loss of BNIP3 disrupted CTNNB1/β-catenin staining significantly in livers from fed and fasted mice with a significant increase in cytosolic CTNNB1/β-catenin specifically in hepatocytes in bnip3-/- fasted mice in particular (Fig. S6D), consistent with activation of the WNT-CTNNB1/β-catenin pathway in hepatocytes of liver from fasted bnip3-/- mice, and which will be the focus of future studies.
Taken together, our data supports a model in which increased mitochondrial mass due to loss of BNIP3-dependent mitophagy (Figure 6G) is causing loss of zonation of mitochondrial metabolism (increased zone 1 metabolism and decreased zone 3 metabolism) and altering the balance of key metabolic processes in the liver (Figure 6H). These findings highlight the importance of BNIP3-dependent mitophagy and mitochondrial mass control in metabolic zonation and homeostasis in the liver.
Discussion
The original description of autophagy nearly fifty years ago showed that GCG promotes mitophagy [10,11] but the underlying molecular basis of this observation has remained unexplained. Our work shows for the first time that BNIP3 is a GCG-induced gene that encodes a mitophagy cargo receptor that is both required and sufficient for GCG/fasting induced mitophagy in the liver.
BNIP3, BNIP3L/NIX, FUNDC1, BCL2L13 and FKBP8 all appear to function in mechanistically similar ways to promote mitophagy, through direct interaction with processed LC3 [2,4,6–8]. BNIP3L/NIX in particular is highly related to BNIP3 but while BNIP3L is induced in the liver by fasting (Fig. S1D, lane 1–2), it is not further increased by loss of BNIP3 in the liver (Fig. S1D, lane 3–4). Also, immunohistochemical staining for BNIP3L in liver from fed and fasted Bnip3+/+ and bnip3-/- mice, indicates that BNIP3L/NIX in contrast to BNIP3, is predominantly expressed in Kupffer cells and not in hepatocytes in the liver (Fig. S5C). Thus, it is unlikely that BNIP3L/NIX or other mitophagy cargo receptors can adequately compensate for loss of BNIP3 in this context. Our western blot analysis indicates that some residual mitophagy is retained in fasted liver in the absence of BNIP3 since hydroxychloroquine continued to elicit increased levels of mitochondrial LCB3 in the absence of BNIP3 (Figure 3H, lanes 10–12). However, this may be mitophagy induced by stresses other than fasting/GCG and for example, may be explained by PINK1-PRKN mediated mitophagy in the liver [26]. Thus, similar to the unique role for BNIP3L/NIX-dependent mitophagy in erythroid differentiation that is associated with increased BNIP3L expression during red cell maturation [27–29], elevated BNIP3 expression and BNIP3-dependent mitophagy are integral to the response of the liver to nutrient deprivation.
In addition to its role in mitophagy, we show here for the first time that BNIP3 promotes localization of processed LC3B to mitophagosomes in fasted hepatocytes. Loss of BNIP3 caused LC3B to accumulate in the nucleus under both fed and fasted conditions. Importantly, we showed that HA-BNIP3 but not HA-BNIP3W18A restored cytosolic localization of LC3B. Endogenous LC3B was not evident in the nucleus of Bnip3+/+ hepatocytes from fed mice, in contrast to exogenous GFP-LC3 that did accumulate in the nucleus. This is likely due to Bnip3+/+ hepatocytes expressing sufficient endogenous BNIP3 to promote cytosolic localization of endogenous LC3B, and to high-levels of exogenous GFP-LC3 out-titrating endogenous BNIP3, causing most GFP-LC3 to remain nuclear. This suggests that induction of BNIP3 to high levels by fasting is required to promote cytosolic localization of exogenous GFP-LC3. The fact that BNIP3 is required in fasted hepatocytes to cause LC3B accumulation in the cytosol, suggests that BNIP3 may also be required for other forms of autophagy in the liver that are dependent on LC3B, such as lipophagy.
Previous work reported that acetylation of LC3B promoted nuclear accumulation of LC3B, while deacetylation of LC3B promoted nuclear export of LC3B [30,31]. However, we were unable to demonstrate any change in LC3B acetylation state associated with gain or loss of BNIP3 expression and inhibition of EP300/p300 activity had no effect on LC3B localization in hepatocytes in our hands (data not shown). Previous reports have also suggested that BNIP3 can localize to the nucleus [32], which could suggest a model in which BNIP3 interacts with LC3B in the nucleus to promote its nuclear export. However, using antibodies validated against bnip3-/- tissue, we do not detect BNIP3 in the nucleus of isolated hepatocytes under fed or fasted conditions (Fig. S4D). We have thus postulated that BNIP3 promotes cytosolic LC3 accumulation in response to fasting by indirect means that are yet to be determined.
Our data indicate that the liver induces both mitophagy and mitochondrial biogenesis in response to nutrient deprivation. Increased mitochondrial biogenesis in response to fasting in the liver and other tissues has been previously documented [33] and our work indicates that increased mitophagy in Bnip3+/+ liver is offset by increased mitochondrial mass such that overall mitochondrial mass remains similar before and after fasting. At this time, it is not clear what the adaptive benefit to the liver is of inducing both mitophagy and biogenesis in response to fasting, unless, like other opposing processes, mitophagy and biogenesis are spatially separated in the liver. Alternatively, mitophagy and biogenesis in the same cell may allow the hepatocyte to eliminate mitochondria of a given function and replace them with mitochondria that are functionally distinct and better adapted to the stress at hand. However, our work also does not find any difference in expression of PPRARGC1A/PGC-1α in bnip3−/- liver compared to Bnip3+/+ either before or after fasting (Fig. S1D) indicating that increased mitochondrial mass in bnip3-/- livers is driven primarily by decreased levels of mitophagy.
Our current work was carried out using germline knockouts of BNIP3 generated previously [34], and while BNIP3 is highly expressed in the liver and these mice exhibit minimal phenotype in the absence of imposed stresses, such as fasting [12], it is possible that the phenotype observed in whole liver in vivo under fasting conditions may be affected by consequences of BNIP3 loss in other tissues, such as muscle, adipose tissue and pancreas all of which can affect liver metabolism and stress responses [9]. However, we detected mitophagy defects in cultured primary hepatocytes in the absence of other tissue or cell types present. Moreover, the observed phenotypes can be rescued by exogenous HA-BNIP3 (but not HA-BNIP3W18A) suggesting that the observed mitophagy defects are largely cell autonomous.
While the liver appears macroscopically homogeneous, it is functionally organized into lobules that allow the tissue to separate out opposing metabolic functions, such as urea cycle and glutamine synthesis, fatty acid oxidation and lipogenesis, gluconeogenesis and glycolysis, amongst other functions [20]. This in turn prevents metabolic pathways competing for common substrates and promotes efficient metabolite recycling [20,22]. The oxygen gradient across the liver lobule plays an important role in setting up distinct functional zones and both HIF1A/HIF-1β and EPAS1/HIF2A/HIF-2β are stabilized and more highly expressed in pericentral regions [22,35]. BNIP3 is a validated HIF1A transcriptional target [36] that is rapidly induced as oxygen levels drop. Here we show that BNIP3 is zonally expressed in the liver parenchyma with highest expression in low-oxygen pericentral regions and minimal expression in periportal regions that are better oxygenated. The inverse relationship that we identify between TOMM20 and BNIP3 expression in zone 1 (TOMM20 high, BNIP3 low) and zone 3 (TOMM20 low, BNIP3 high) of the liver lobule in Bnip3+/+ mice, combined with loss of mitochondrial mass zonation in bnip3-/- livers, indicates that BNIP3-dependent mitophagy reduces mitochondrial mass specifically in zone 3 around the CV while low levels of BNIP3 in zone 1 limits mitophagy and allows mitochondrial mass to accumulate (Figure 6H). Increased mitochondrial mass and loss of mitochondrial zonation in fasted bnip3-/- liver disrupts metabolic function in the liver. This is indicated by expanded urea cycle activity and lower levels of glycolysis and glutamate-glutamine recycling, consistent with increased zone 1 metabolism and decreased zone 3 metabolism (Figure 6H). These novel findings showing that mitochondrial mass distribution in the liver modulates metabolism has significance for liver pathologies where BNIP3 expression is altered, such as in hepatocellular carcinoma [37], and may explain how mitochondrial dysfunction contributes to liver disease.
Materials and methods
Mice
All experimental mice (Bnip3+/+, bnip3-/-, GFP-LC3 transgenic, mt-mKeima transgenic mice) were aged 16–20 weeks of age and maintained on an inbred C57bl/6 J background. Mice were housed in specific pathogen-free barrier facilities and provided with water and standard chow ad libitum except under fasting conditions, when animals had no access to chow for 24 h (unless noted otherwise). Unless otherwise stated, all experiments were performed on 3-month-old male mice to control for effects of age and gender on liver phenotypes.
Primary hepatocyte preparation and culture
Primary hepatocytes were isolated from anesthetized mice using a modified two-step non-recirculating perfusion method. While mice were anesthetized with isoflurane, the portal vein (PV) was cannulated using a 23-gauge needle and pre-warmed Hanks’ Balanced Salt Solution (HBSS: 5 mM glucose, 0.5 mM EGTA, 25 mM HEPES, pH 7.4 [Thermo Fisher, 14,175]) was perfused at a flow rate of 2 mL/min. Successful cannulation was confirmed upon immediate blanching of the liver, followed by cutting of the inferior vena cava (IVC) and increasing of the flow rate to 8 mL/min. Approximately 50 mL of HBSS was perfused through the liver, for approximately 6 min. During this time, the IVC was periodically clamped for 5-second intervals, ensuring proper flow of the solution through the liver. Pre-warmed digestion medium (HEPES buffered low-glucose DMEM) [Gibco, 12320032], supplemented with 100 U/mL penicillin and 0.1 mg/mL streptomycin (Pen/Strep) [Gibco, 15140122], and 100 U/mL type IV collagenase [Worthington, CLS-4] was then perfused at 8 mL/min for approximately 8 min or until the liver appeared fully digested, with periodic clamping of the IVC to aid in digestion and maximize cell yield. Digestion was considered complete upon loss of liver elasticity and little to no change in liver size upon IVC clamping. The digested liver was excised and transferred to a culture dish containing 20 mL of the digestion medium, and gently ripped and shaken using forceps to release the cells. The remainder of the procedure was performed in a tissue culture hood. Cells were gently triturated 3 times using a 25 mL pipet and filtered through a 74-μm fine mesh stainless steel strainer (Dual Manufacturing Co, US12-170HS). Cells were centrifuged at 50 g for 2 min at 4°C followed by three washes with 25 mL of cold isolation medium (HEPES buffered high glucose DMEM [Gibco, 10313021], supplemented with 1 mM sodium lactate [Sigma Aldrich, L1750], 2 mM L-glutamine [Gibco, A2916801], 0.1 μM dexamethasone [Sigma Aldrich, D2915], 1X Pen/Strep, and 10% fetal bovine serum). Following the final wash, the pellet was resuspended in 25 mL of isolation medium and the viability and total yield were assessed by 0.4% trypan blue exclusion and counting using a hemocytometer. Hepatocytes were diluted to 3 × 105 cells/mL and approximately 4.5 × 105 cells were added per well of 6 well tissue culture plates. Tissue culture plates (overlayed with glass coverslips if being used for immunofluorescence) were previously coated with 5 μg/cm2 rat tail collagen (type I) [Becton Dickinson, 356236] in 0.02 N acetic acid, dried, and sterilized in a hood under UV light for at least 4 h, and washed with sterile PBS [Thermo Fisher, 100100] stainless steel strainer (Dual Manufacturing prior to plating hepatocytes. Cells were evenly distributed by shaking of the plates gently in a linear fashion. Following a 1-h incubation in a tissue culture incubator (37°C, 5% CO2), media were aspirated and replaced with fresh, pre-warmed isolation medium, followed by an additional 4 h of incubation. After this recovery period, the medium was replaced with serum-free culture medium (low-glucose DMEM supplemented with 10 mM sodium lactate, 2 mM L-glutamine, 5 mM HEPES, pH 7.4, 10 nM dexamethasone, and 1X Pen/Strep). For starvation experiments used for qPCR, cells were incubated overnight in HEPES-buffered Kreb’s Ringer solution (KRH: 115 mM NaCL, 0.5 mM KCl, 0.1 mM CaCl2, 0.1 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM Na-HEPES, pH 7.4) and 1 μM GCG (glucagon; [Sigma Aldrich, G2044]). For experiments requiring exogenous overexpression, adenoviruses encoding empty vector (EV), HA-BNIP3-WT, or HA-BNIP3-W18A were added a dose of 2.9 × 1010 viral particles in 1 mL of media. For experiments involving immunofluorescence or western blots, viral media was aspirated the next morning, and wells were washed 2X with PBS followed by incubation in experimental media conditions.
Analysis of oxygen consumption rates
Primary hepatocytes were seeded in Seahorse XF96 microplates at a density of 0.75 × 104 cells/well. Primary hepatocytes were pre-coated with collagen as previously described. The next day, the cellular mito stress test was performed according to the manufacturer’s protocol (1 μm oligomycin [Sigma Aldrich, 495455], 0.75 μm FCCP [Sigma Aldrich, C2920], 5 μM antimycin A [Sigma Aldrich, A8674]), using the Seahorse XF96 analyzer in the Biophysics Core at the University of Chicago. Briefly, 2X DMEM base media was used to make 1X DMEM supplemented with 4.5 g/L glucose, 2 mM glutamine, and 1 mM sodium pyruvate [Gibco, 11360070], with a pH adjusted to 7.35. Cells were rinsed with PBS prior to addition of 175uL of 1X DMEM and the plate was incubated in the absence of CO2 for approximately 1 h. Data were normalized by cell number using Hoechst 33342 [Thermo Fisher, H3570] nuclear counterstain and fluorescence quantification using a microplate reader. Normalized OCR data was then analyzed using Agilent Seahorse Wave software.
RNA extraction
Cells in a 6-well plate were washed twice with 2 mL of DPBS [Gibco, 14190250] followed by addition of 1 mL Trizol [Invitrogen, 15596018]. Wells were incubated for 5 min at room temperature (RT) and collected in eppendorf tubes. At this step, samples could be frozen at −80°C or immediately extracted for RNA. For extraction, 200uL of chloroform was added to each sample, followed by vigorous shaking for 15 seconds and incubation for 3 min at RT. Tubes were centrifuged at 12,000xg, 15 min, at 4°C. The aqueous upper phase (~400 μL) was transferred into a fresh tube, followed by addition of 1 volume of 70% EtOH (~400uL) and vigorous shaking. Samples were incubated for 5 min and then applied to RNeasy columns. The remainder of the extraction was performed according to the RNeasy Mini kit protocol [Qiagen, 74034], and included on-column DNase-I digestion. RNA was eluted in 50 μL of RNase-free water, concentrations were measured using the NanoDrop Spectrophotometer, and samples were stored at −80°C.
Quantitative PCR
To make cDNA, 1–2 μg of RNA was reverse transcribed using the High Capacity RNA-to-cDNA kit [Applied Biosystems, 4388950]. The concentration of cDNA was measured by NanoDrop and samples were stored at −20°C. For gene expression analysis, we performed quantitative real-time PCR on 250 ng of cDNA per sample using Taqman gene-specific fluorogenic probes (Applied Biosystems/Thermo Fisher). Primers used for qPCR included in this manuscript are as follows:
Genomic copy number (Mt:Nuc gDNA):
Cyba: Mm00241140_cn
Ndufa1: Mm00526370_cn
Hbb-bh1: Mm00216612_cn
Gene expression:
Bnip3: Mm01275601_g1
Map1lc3b: Mn00782868_sh
Ass1: Mm00711256_m1
Arg1: Mm00475988_m1
Asl: Mm01197741_m1
qPCR was performed on a StepOne Plus thermocycler (Applied Biosystems). Samples were assayed in triplicate and normalized to their respective endogenous control (ACTB). Gene expression levels were quantified using the ΔΔCT method and relative expression of each sample was normalized to a control and/or untreated sample.
Immunohistochemistry
Immunohistochemistry on mouse liver sections was carried out as described previously [38] using heat denaturation in citrate buffer pH 6.0 to expose epitope. Stained slides were digitized using an Allied Vision Technologies Stingray F146 C color slide scanner and quantified using the Spectrum Plus Image analysis software (Aperio). Antibodies were used in IHC as follows: anti-TOMM20 (Santa Cruz Biotechnology, sc-11,415) 1:500; anti-BNIP3 (Sigma Prestige, HPA003015) 1:100; anti-LC3B (Nanotools 0231–100/LC3-5F10) 1:250; anti-ASS1 (Proteintech, 16,210-1-AP) 1:4,000; anti-GLUL (Abcam 49,873) 1:20,000; anti-TIMM23 (Proteintech, 11,123-1- AP) 1:100;βanti-BNIP3L (Thermo, ZN-002) 1:400; anti-CTNNB1/β-catenin (Sigma, clone 05–655) 1:500.
Protein extraction
For harvesting of cells, plates were washed in ice-cold DPBS followed by scraping in 1 mL of DPBS containing protease inhibitors (0.5 mM PMSF [Sigma Aldrich, P7626], 1 μg/mL aprotinin [Sigma Aldrich, A6103], 1 μg/mL leupeptin [Sigma Aldrich, L2884], 1 mM Na3VO4 [Sigma Aldrich, S6508]). Cells were pelleted at 3,000xg for 3 min at 4°C and resuspended in RIPA lysis buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% sodium deoxycholate [Sigma Aldrich, S6750], 0.1% SDS, 1% Triton X-100 [Sigma Aldrich, X100]) containing protease and phosphatase inhibitors (Roche PhosSTOP inhibitor cocktail tablet). Samples were incubated on ice for 15 min with vortexing every 5 min, and centrifuged at full speed for 15 min at 4°C. The supernatant was transferred to pre-chilled microcentrifuge tubes and protein concentration was measured on a NanoDrop spectrophotometer and stored frozen at −80°C.
Mitochondrial and nuclear protein fractionation
Whole liver was isolated, washed in cold PBS to remove blood and gall bladder was removed. The liver was then chopped quickly on ice using razor blades and transferred to a Dounce homogenizer in 2 ml of isolation buffer (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 250 mM sucrose [Sigma Aldrich, S0389], 0.25% Triton X-100 [Sigma Aldrich, X100], 1 mM EDTA, 1 mM EGTA, 1 mM DTT plus protease and phosphatase inhibitors (Roche cOMPLETE Ultra [Sigma Aldrich, 5892970001] and PhosSTOP inhibitor cocktail tablet [Sigma Aldrich, PHOSSRO]). Liver homogenates were generated and nuclei pelleted at 1,000 g for 10 min at 4°C. Supernatant was recovered and pelleted at 12,000 g for 15 min at 4°C. Both nuclear and mitochondrial pellets were washed three times in isolation buffer. After the final wash, pellets were extracted in RIPA buffer (see above). Mitochondrial and nuclear fraction protein concentration was measured using a NanoDrop. Samples were prepared for western blot analysis using the provided 5X sample buffer and boiled for 10 min. To determine fraction enrichment, TOMM20 was used as a mitochondrial marker and PARP1 was used as a nuclear marker.
Immunoprecipitation
Cells were transiently transfected and/or treated prior to protein extraction as described above. Following scraping and centrifugation, cell pellets were resuspended in 150 μl NP-40 IP lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Igepal [Sigma Aldrich, I8896], 0.01% β-mercaptoethanol), and sonicated at 10% power for 5 s using a Fisher Sonic Dismembrator Model 500 while keeping samples cold in an ethanol-ice bath. Samples were incubated on ice for 5 min and centrifuged at full speed for 15 min at 4°C. The supernatant was transferred to pre-chilled Eppendorf tubes and protein concentration measured as above. For immunoprecipitation of exogenously expressed GFP-tagged proteins, 25 μl GFP-Trap or agarose control-Trap magnetic beads [Chromotek, gtma-100] were washed twice in 500 μl IP lysis buffer using a magnetic bead rack, followed by resuspension in 1 ml IP lysis buffer and addition of 1.5 mg protein lysate. Lysates were incubated on beads for 1 hr at 4°C on a rotator followed by three 1 ml washes with IP lysis buffer using a magnetic bead rack. Beads were transferred to a fresh Eppendorf tube for the final wash and resuspended in 50 μl 2x sample loading buffer (1:2:2 10x SDS:5xBPB:ddH2O) and boiled for 10 min. Supernatant containing denatured IP lysate was transferred to a fresh tube and frozen at −80°C.
Western blot
Protein samples were denatured by boiling for 5 min with SDS reducing sample buffer (400 mM Tris pH 6.8, 10% SDS, 500 mM β-mercaptoethanol) and sample loading dye (60% glycerol and bromophenol blue). The amount of protein loaded per sample varied depending on the proteins being probed, but typically 75 μg was loaded onto SDS-PAGE gels, followed by transfer to nitrocellulose (0.2 μm pore) [GE Healthcare, 45–004-001] or PVDF (0.45 μm pore) [GE Healthcare, 45–004-113] membranes. Membranes were blocked in 5% nonfat milk in TBS, 0.05% Tween 20 [Sigma Aldrich, P1379], for 30 min at room temperature with shaking and incubated with primary antibodies overnight at 4°C on a rocker, in 5% BSA [Sigma Aldrich, B6917], in TBST for antibodies from Cell Signaling Technology and in 5% nonfat milk in TBST for all others. The next day, membranes were washed 3 times with TBST and incubated with HRP-conjugated secondary antibody (Agilent Dako, P0448) in 5% nonfat milk in TBST for 2 h at room temperature on a shaker. Membranes were washed 3 times in TBS-T and proteins were visualized by chemiluminescence and exposure on X-ray film. Antibodies for western blots were used as follows: anti-HA (Cell Signaling Technology, 3724) 1:2,000, anti-GFP (Cell Signaling Technology, 2956) 1:500, anti-TOMM20 (Abcam, 56783) 1:500, anti-LC3B (Cell signaling Technology, 83506) 1:500, anti-BNIP3 (Cell Signaling Technology, 3769) 1:500, anti-PARP1 (Santa Cruz Technology, sc-7150) 1:250.
Steady state metabolite analysis
Mouse liver was harvested and snap-frozen in liquid nitrogen from four treatment groups: fed Bnip3+/+ (n = 8), fed bnip3-/- (n = 8), fasted Bnip3+/+ (n = 8), and fasted bnip3-/- (n = 8). Biochemicals present in pulverized frozen tissue samples were analyzed using GC/MS and LC/MS/MS platforms (Metabolon). Biochemicals that differed significantly between experimental groups were identified via ANOVA contrasts and Two-way ANOVA with statistical significance set at p ≤ 0.05 while approaching significance was set at 0.05 < p < 0.1.
Immunofluorescence & confocal microscopy
Hepatocytes were seeded onto collagen-coated glass-coverslips in 6-well tissue culture plates as previously described. Wells were treated with full media or KRH supplemented with 1 μM GCG (KRHg) for 6 h, with the addition of 100 nM bafilomycin A1 [Sigma Aldrich, B1793] or DMSO vehicle [Sigma Aldrich, D2650] for the final 4 h of incubation. At experimental endpoint, media were aspirated and wells washed in DPBS followed by fixation in 4% paraformaldehyde (PFA) [Sigma Aldrich, 100,496] for 20 min at RT. Wells were washed 3 times in DPBS followed by permeabilization in ice-cold 100% methanol for 5 min at −20 C. Coverslips were washed with TBST 3 times and blocked in 10% goat serum [Vector Labs, S-1000] in PBS for 30 min. Coverslips were incubated with primary antibodies in 10% goat serum in PBS overnight at 4°C. The next day, wells were washed in TBST for 3 × 5 min, followed by incubation in appropriate fluorescent secondary antibodies in 10% goat serum/PBS for 1 h at RT, protected from light. Wells were washed in PBS, rinsed in ddH2O, and mounted onto slides with 18 μL ProlongGold containing DAPI [Thermo Fisher, P36931]. Slides were allowed to cure for 24 h in the dark at room temperature, with subsequent storage at 4°C. Antibodies for immunofluorescence were used as follows: anti-TOMM20 (Abcam, ab-56783) 1:100;βanti-TOMM20 (Cell Signaling Technology, 42406) 1:200; anti-HA (Cell Signaling Technology, 3724) 1:1,600, anti-LC3 (Cell Signaling Technology, 83506) 1:400; anti-BNIP3 (Sigma Aldrich, B7931) 1:500; anti-HSP60 (Cell Signaling Technology, 12165) 1:400; anti-LAMP1 (Abcam, ab25245) 1:200. Live cell imaging of GFP-LC3+ hepatocytes was performed after staining with 500 nM MitoTracker Deep Red FM [Invitrogen, M22426]. Live cell imaging of mt-mKeima was performed at an excitation wavelength of 436 nm (for neutral pH) and 565 nm (for acidic pH) with emission spectra collected at 620 nm, in accordance with published protocols [14]. Imaging was performed using the Olympus DSU Spinning Disk Confocal microscope.
Staining of formalin fixed whole liver sections was performed following heat denaturation of epitopes for in retrieval solution (IHC Tek, IW-1100) and overnight incubation with primary antibody. Antibodies used were: anti-TOMM20 (Cell Signaling Technology, 42406) 1:200;βanti-HSPD1/HSP60 (Cell Signaling Technology, 12165) 1:400; anti-LAMP1 (Abcam, ab25245) 1:200; anti-LC3B (Nanotools, clone 5 F10) 1:250. Nuclei were stained with Hoechst 33342 (10 μg/ml). Imaging was performed using the Olympus DSU Spinning Disk Confocal microscope and the Leica TCS SP8 laser scanning Confocal microscope in the Integrated Microscopy Core Facility at the University of Chicago. All images were collected using a 100X oil-immersion objective. Ten representative images per sample were obtained and images were background subtracted and thresholded using ImageJ software.
ImageJ quantification of colocalization
For quantification of colocalization (overlap) between two channels, a macro was generated in ImageJ to automatically subtract background, threshold each channel, and select the cell of interest in each image. The JACoP plugin was then used to calculate the M1 and M2 coefficients, representing the overlap between two channels. Averages and SEM were then calculated for each set of images.
Statistical Analysis
All statistical analyses were carried out using GraphPad Prism of raw data. The data were analyzed using two-way ANOVA with Tukey’s posttest with a 95% confidence interval for data sets involving single parameters or single groups of data. Other datasets involving comparisons among multiple groups used Wilcoxon rank sum analyses with a 95% confidence interval. Data are shown as the mean ± s.e.m. Values of p ≤ 0.05 are considered significant. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
Supplementary Material
Acknowledgments
This work was supported by funding from NIH R01 CA200310 (KFM), R01 CA216242 (KFM) and NIH T32 CA009594 (KFM that supported MZS, LPP, ABH, AGS). This work made use of the Human Tissue Resource Center and the Digital Light Microscope Facility that are supported by the University of Chicago Cancer Center Support Grant (P30 CA014599). Use of mt-mKeima is covered by an MTA signed with Dr. Atsushi Miyawaki of the RIKEN Brain Science Institute and mt-mKeima mice were kindly provided by Dr. Toren Finkel [14].
Funding Statement
This work was supported by the National Cancer Institute [R01 CA200310]; National Cancer Institute [T32 CA009594]; National Cancer Institute [R01 CA216242].
Disclosure statement
All authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Palikaras K, Lionaki E, Tavernarakis N.. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–1022. [DOI] [PubMed] [Google Scholar]
- [2].Macleod KF. Mitophagy and mitochondrial dysfunction in cancer. Ann Rev Cancer Biol. 2020;4:41–60. [Google Scholar]
- [3].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. [DOI] [PubMed] [Google Scholar]
- [5].Bhujabal Z, Birgisdottir ÅB, Sjøttem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with BNip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol. 1967;35:C11–C16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Glick D, Zhang W, Beaton M, et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32(13):2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516(7529):112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun N, Yun J, Liu J, et al. Measuring In Vivo Mitophagy. Mol Cell. 2015;60(4):685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Katayama H, Kogure T, Mizushima N, et al. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. [DOI] [PubMed] [Google Scholar]
- [16].Palikaras K, Lionaki E, Tavenarakis N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Diff. 2015;22:1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rodgers JT, Lerin C, Gerhart-Hines Z, et al. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stanger BZ. Cellular homeostasis and repair in the mammalian liver. Annu Rev Physiol. 2015;77:179–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol. 2019;16(7):395–410. [DOI] [PubMed] [Google Scholar]
- [21].Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. [DOI] [PubMed] [Google Scholar]
- [22].Kietzmann T. Metabolic zonation of the liver: the oxygen gradient revisited. Redox Biol. 2017;11:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Halpern KB, Shenhav R, Matcovitch-Natan O, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017;542(7641):352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Keshet R, Szlosarek P, Carracedo A, et al. Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer. 2018;18(10):634–645. [DOI] [PubMed] [Google Scholar]
- [25].Benhamouche S, Decaens T, Godard C, et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006;10(6):759–770. [DOI] [PubMed] [Google Scholar]
- [26].Wang H, Ni H-M, Chao X, et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol. 2019;22:101148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of red cells. Nature. 2008;454:232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Diwan A, et al. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA. 2007;104:6794–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–466. [DOI] [PubMed] [Google Scholar]
- [31].Song T, Su H, Yin W, et al. Acetylation modulates LC3 stability and cargo recognition. FEBS Lett. 2019;593(4):414–422. [DOI] [PubMed] [Google Scholar]
- [32].Metukuri MR, Beer-Stolz D, Namas RA, et al. Expression and subcellular localization of BNIP3 in hypoxic hepatocytes and liver stress. Am J Physiol Gastrointest Liver Physiol. 2009;296(3):G499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Puigserver P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-alpha. Int J Obes (Lond). 2005;29(Suppl 1):S5–9. [DOI] [PubMed] [Google Scholar]
- [34].Diwan A, Krenz M, Syed FM, et al. Inhibition of ischemic cardiomycoyte apoptosis through targeted ablation of BNip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kietzmann T. Liver Zonation in Health and Disease: hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters. Int J Mol Sci. 2019;20(9):2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kasper LH, Boussouar F, Boyd K, et al. Two transcriptional mechanisms cooperate for the bulk of HIF-1-responsive gene expression. Embo J. 2005;24:3846–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Calvisi DF, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117(9):2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Macleod K, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and -independent cell death pathways in the developing mouse nervous system. Embo J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.