

ABSTRACT
Recently, we have generated 284 epigenomic maps in melanoma. Using chromatin state profiling we identify an association of NRAS-mutants with bivalent Histone H3 lysine 27 trimethylation (H3K27me3) and broad H3K4me3 domains. Reprogramming of bivalent H3K27me3 occurs on critical invasive-regulators and its resolution using Enhancer of Zeste Homolog 2 (EZH2) inhibition reduces invasive capacity and tumor burden in NRAS-mutant patient samples.
KEYWORDS: Melanoma metastasis, bivalent, broad domains, polycomb repressive complex 2, melanoma therapeutics
Author’s comments
In melanoma, somatic mutations in oncogenes, such as NRAS or BRAF, are well-chronicled drivers of this disease.1 However, the relationship between these genetic alterations and epigenetic elements are not entirely understood. Chromatin profiling is a powerful tool for identifying tissue-specific regulatory elements. Using chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-sequencing) for enhancers (H3K27ac and H3K4me1), promoters (H3K4me3), active transcription (H3K79me2) and polycomb (H3K27me3) or heterochromatin (H3K9me3) repression we generated 284 chromatin maps in melanoma tumors and cell lines. Through integrative analysis of chromatin state profiling coupled with mutation, expression, and methylation data, we identified bivalent Histone H3 lysine 27 trimethylation (H3K27me3) and broad H3K4me3 domains as prospective drivers of an invasive/mesenchymal phenotype, primarily in NRAS-mutants.
Bivalent chromatin, which is defined by the presence of both active (i.e. H3K4me3) and repressive (i.e. H3K27me3) histones, has the potential to rapidly activate or repress lineage-specific genes.2 In adult cells some lineage-specific genes maintain or regain bivalent chromatin,3 potentially leaving them vulnerable to activation. In melanoma, we found bivalent domains display dynamic changes on members of the Epithelial to Mesenchymal Transition Transcription Factor (EMT-TF) network [i.e. Zinc Finger E-Box Binding Homeobox 1 (ZEB1), Twist Family BHLH Transcription Factor 1 (TWIST1) and Cadherin 1 (CDH1)] during the progression from Embryonic Stem Cells (ESCs) > melanocytes > isogenic mutant melanocytes > metastatic melanoma. Albeit melanocytes are not epithelial, during melanocyte development neural crest cells undergo an initial EMT in order to break away from the neural fold and migrate,4 a process involving multiple transitions between alternate cellular states. Interestingly, this process is similar to a model in which melanoma cells undergo a phenotypic switch (from proliferative to invasive) that includes reorganization of EMT-TF network, increased invasion, and poor prognosis in patients.5 Our studies build on these findings and suggest that shifts in bivalent chromatin domains, which contain the ability to rapidly activate or repress genes, may be the mechanism for EMT-TF gene regulation during the progression toward an invasive state.
With an observed shift in bivalent domains, we postulated genes losing bivalent H3K27me3 in melanocytes can retain/acquire distinct H3K4me3 signatures (broad or non-broad) in melanoma. In contrast to “typical” H3K4me3 (200–1000bp long), broad H3K4me3 can span thousands of kilobases (kb) and has been linked to increased gene activation.6 Based on the overall width and density of H3K4me3 we found NRAS- and BRAF-mutants harbor the largest number of broad domains (>30kb in NRAS tumors). Separation of domains > 4kb (4x that of a typical domain) and < 4kb revealed two distinct H3K4me3 signatures, including broad domains spanning outside the transcription start site (TSS) and non-broad domains localized within the TSS. Integration of 1) bivalent domains in melanocytes 2) broad or non-broad H3K4me3 in melanoma 3) active transcription mark H3K79me2 and 4) RNA-seq expression data revealed NRAS-specific broad domains are associated with increased gene activation and enriched on invasive drivers, such as SRY-Box Transcription Factor 9 (SOX9). With little known about H3K4me3 signatures in melanoma, this suggests broad H3K4me3 could be a mechanism for invasive gene activation in cooperation with bivalent domains.
Although genes retaining/acquiring broad H3K4me3 in melanoma were associated with increased transcription, this bivalent switch occurred on a small number of genes (n = 142) compared to the total number of broad domains (n = 1206) in NRAS-mutants. Hence, we postulated lengthening of H3K4me3 may be associated with increased gene activation whereas shortening may be associated with repression. Unlike that of bivalent transitions, we found H3K4me3 displayed preferential shortening (<2kb) and associated gene repression compared to melanocytes in both NRAS- and BRAF-mutants, suggesting this may be a common response to oncogenic activation. Interestingly, promoters harboring the largest broad H3K4me3 domain losses included critical melanocyte regulators involved in the transition to an invasive state [i.e. Premelanosome Protein (PMEL) and Melanocyte Inducing Transcription Factor (MITF)]. For example, genes such as MITF, a melanocyte “master regulator,” has been described as a molecular rheostat in which, depending on its activity levels, is critical for switches between cellular states.7 With marked shortening of H3K4me3 on melanocyte- and proliferative-regulators, this suggests a subset of broad H3K4me3 domains may be a mode of gene regulation to maintain normal melanocyte function (or a proliferative state) which is lost upon oncogenic activation (or during the switch to an invasive state).
Within bivalent chromatin domains an active PRC2 [Enhancer of Zeste Homolog 2 (EZH2), SUZ12 polycomb repressor complex 2 subunit and Embryonic Ectoderm Development (EED)], through trimethylation of H3K27, is essential for maintaining transcriptional repression.8 In melanoma, we observed an association between NRAS-mutants, bivalent chromatin and PRC2, on a gene, protein and genome-wide level. NRAS-mutants displayed increased expression of SUZ12 compared to BRAF-mutants, and bivalent H3K27me3 colocalized with PRC2 in ESCs, which are known to harbor functional bivalent domains. Removal of H3K27me3 through disruption of PRC2 [via EZH2 inhibition (GSK-126)]9 was able to activate the expression of key EMT-TF (i.e. ZEB1) in melanomas retaining a bivalent configuration after oncogenic activation, suggesting a subset of these domains are functional in cancer. By taking advantage of this epigenetic information, we found EZH2 inhibition markedly impacted the invasive, but not proliferative, capacity of NRAS-mutant cells. With impact on invasion, but not proliferation, we next tested whether EZH2 inhibition could be combined with proliferation-blocking Mitogen-activated protein kinase kinase (MEK) inhibitor (trametinib) to block tumor growth. While mono-therapy of EZH2 or MEK inhibition induced a modest effect, the combinatorial application of EZH2+ MEK markedly decreased tumor burden in NRAS-mutants, suggesting this may be a promising therapeutic strategy for this patient population.
Our results suggest reprogramming of bivalent H3K27me3 and broad H3K4me3 domains may function as a mode of gene regulation during the transition to an invasive state in melanoma cells, particularly in NRAS-mutants (Figure 1). Moreover, we show that combination of EZH2+ MEK inhibition markedly decreases tumor burden in NRAS-mutant melanomas (Figure 1). Taken together, this study provides a potential strategy and conceptual advancement for future therapeutics to focus on targeting these two processes in this patient population.
Figure 1.
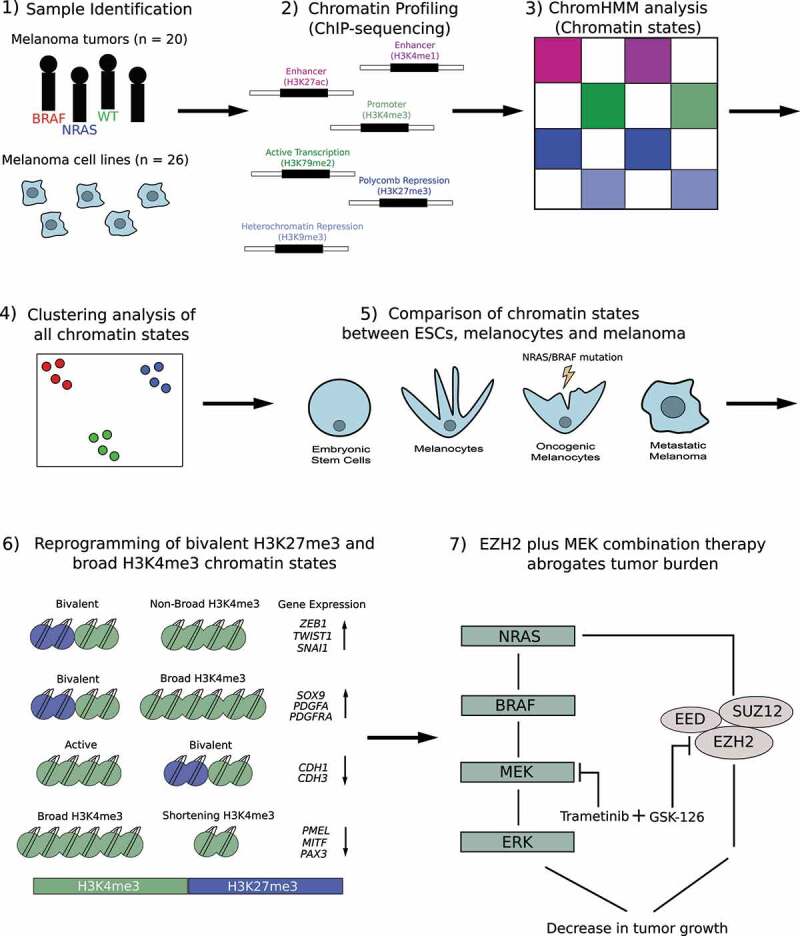
Overview of chromatin state analysis in melanoma tumor samples and cell lines. 1) Samples used in this study included 20 melanoma tumors (BRAF, NRAS, Wild-Type [WT]), 10 melanoma short-term cultures and 16 commercially available melanoma cell lines. 2). Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-sequencing) was performed in melanoma tumors and cell lines using histone modifications for H3K4me1, H3K27ac, H3K4me3, H3K79me2, H3K27me3 and H3K9me3. 3) ChromHMM analysis was used to identify combinatorial chromatin state definitions and histone mark probabilities in 20 metastatic melanoma tumor samples. 4) Multidimensional Scaling (MDS) and differential analysis was performed for each chromatin state annotated by mutational subtype. 5) Chromatin states were compared between embryonic stem cells (ESCs), melanocytes, oncogenic melanocytes harboring an NRAS- or BRAF-mutation and melanoma samples harboring an NRAS- or BRAF-mutation. 6) Model describing potential mechanism in which switches of bivalent H3K27me3 domains on Epithelial to Mesenchymal Transition Transcription Factor (EMT-TF) and invasive genes, and shortening of H3K4me3 domains on melanocyte regulatory and proliferative genes, controls their expression during the transition to an invasive state. 7) Model describing potential therapeutic strategy for targeting the Mitogen-activated protein kinase (MEK) pathway (trametinib) and Enhancer of Zeste Homolog 2 (EZH2) (GSK-126) to block tumor burden in NRAS-mutant melanoma
Funding Statement
Funding for the work described here was supported by grants from the National Institutes of Health (CA160578; CA016672), Center for Cancer Epigenetics at MDACC and MD Anderson Cancer Center Start-up funds, UTMDACC CCSG-funded Characterized Cell Line Core performed STR DNA fingerprinting (CA016672). Support for the molecular characterization of the short-term culture tumor lines was provided by generous philanthropic contributions to the Moon Shots Program at The University of Texas MD Anderson Cancer Center.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Tsao H, Chin L, Garraway LA, Fisher DE.. Melanoma: from mutations to medicine. Genes Dev. 2012;26:1–3. doi: 10.1101/gad.191999.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meer A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 3.Jadhav U, Nalapareddy K, Saxena M, O’Neill NK, Pinello L, Yuan GC, Orkin SH, Shivdasani RA. Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell. 2016;165:1389–1400. doi: 10.1016/j.cell.2016.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang W, Bronner ME. Neural crest lineage analysis: from past to future trajectory. Development. 2020;147. doi: 10.1242/dev.193193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G, et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell. 2013;24:466–480. doi: 10.1016/j.ccr.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goding CR, Arnheiter H. MITF-the first 25 years. Genes Dev. 2019;33:983–1007. doi: 10.1101/gad.324657.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]