

ABSTRACT
The mechanisms orchestrating recycling of lysosomes through autophagic lysosome reformation (ALR) is incompletely understood. Previous data show that genetic depletion of BLOC1S1/GCN5L1/BORCS1 increases autolysosome (AL) accumulation. We postulated that this phenotype may manifest due to perturbed ALR. We explored this in control and bloc1s1 liver-specific knockout (LKO) mouse hepatocytes, showing that in response to nutrient-deprivation LKO’s fail to initiate ALR due to blunted lysosomal tubulation. As kinesin motor proteins and the intracellular cytoskeleton are requirements for tubular formation from ALs, we explored the interaction of BLOC1S1 with motor proteins and cytoskeletal factors. BLOC1S1 interacts with the ARL8B-KIF5B (GTPase and kinesin motor protein) complex to recruit KIF5B to ALs. Furthermore, BLOC1S1 interacts with the actin nucleation promoting factor WHAMM, which is an essential structural protein in the initiation of lysosomal tubulation (LT). Interestingly, the genetic reintroduction of BLOC1S1 rescues LT in LKO hepatocytes, but not when KIF5B is concurrently depleted. Finally, given the central role of MTORC1 signaling in ALR initiation, it was interesting that MTORC1 activity was increased despite the absence of LT in LKO hepatocytes. Concurrently, inhibition of MTORC1 abolished BLOC1S1 reconstitution-mediated rescue of LT in LKO hepatocytes. Taken together these data demonstrate that the functional interaction of BLOC1S1 with the kinesin binding complex and the actin cytoskeleton are a requirement for LT which, in parallel with MTORC1 signaling, initiate lysosome recycling via ALR.
Abbreviations: 3-MA: 3-methyladenine; AL: autolysosome; ALR: autophagic lysosome reformation; ARL8B: ADP-ribosylation factor-like protein 8B; ARPC2: actin related protein 2/3 complex, subunit 2; ATAT1/αTAT1: alpha tubulin acetyltransferase 1; AVd: autophagic vacuoles, degradative; BLOC1S1/GCN5L1: biogenesis of lysosomal organelles complex-1, subunit 1; CQ: chloroquine; KIF5B: kinesin family member 5B; KLC1: kinesin light chain 1; LAMP1: lysosomal-associated membrane protein 1; LAMP2: lysosomal-associated membrane protein 2; LC3B-I: cytosolic form of LC3B; LC3B-II: lipidated form of LC3B; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; LKO: liver-specific knockout; LIs: lysosome inhibitors; LT: lysosomal tubulation; Ly: lysosome; MTORC1: mechanistic target of rapamycin kinase complex 1; PLEKHM2/SKIP: pleckstrin homology domain containing, family M (with RUN domain) member 2; Snapin: SNAP-associated protein; SQSTM1/p62: sequestosome 1; SVPs: synaptic vesicle precursors; TFEB: transcription Factor EB; TFE3: transcription factor E3; WHAMM: WAS protein homolog associated with actin, golgi membranes and microtubules.
KEYWORDS: Autophagic lysosome reformation, autophagy, gcn5l1, hepatocyte, lysosome, lysosomal tubulation, mtorc1
Introduction
BLOC1S1/GCN5L1, is an enigmatic protein with a myriad of functions, which is reflected in it having multiple alternate nomenclatures, including BLOC1S1, BLOS1 and BORCS1. It was originally identified as a homolog to the nuclear acetyltransferase KAT2A/GCN5, and it contributes to acetylation of lysine residues on mitochondrial and cytosolic proteins [1–4]. At the same time, protein interaction and functional characterization studies support that BLOC1S1 is important in endo-lysosome biology, including playing roles in endosome, vesicular and lysosome positioning [5–7], in vesicular trafficking to lysosomes [8], and in endosomal maturation and function [8,9].
Despite these diverse effects, the mechanisms orchestrating the functional contributions of BLOC1S1 are less well characterized. Its role in mitochondrial protein acetylation have been shown to be dependent on acetyl-CoA production [4] or in targeting acetyl-CoA generating enzymes [10], and in the cytosol, BLOC1S1 binds directly to ATAT1 (alpha tubulin acetyltransferase 1) to promote TUBA/α-tubulin acetylation [3]. At the same time, BLOC1S1 appears to function as an adaptor protein and/or an indispensable subunit of multiprotein complexes governing lysosome related organelle (LRO) biology. In this context, BLOC1S1 was initially identified as a component of the multiprotein Biogenesis of Lysosome-related Organelles Complex-1 (BLOC-1) and named BLOC-1 subunit, i.e. BLOS1 or BLOC1S1 [11]. BLOC1S1 was subsequently found to be a component of the BLOC-1 Related Complex (BORC) and here it was named as BORC Subunit 1 (BORCS1). BORC consists of eight subunits, with three shared with BLOC-1 [5,12], and five unique subunits designated as KXD1/BORCS4, and BORCS5 through BORCS8. Genetic knockdown studies support that the disruption of BLOC1S1, BLOC1S2/BORCS2, SNAPIN/BORCS3, KXD1, BORCS5 and BORCS6 regulate lysosome positioning [5] and that the depletion of BORCS5 through BORCS8 disrupt autophagosome lysosome fusion with the concomitant accumulation of autophagy proteins LC3B-II and the autophagy receptor SQSTM1/p62 [12].
An interesting concept that seems to link the role of BORCS1/GCN5L1 in the mitochondria and cytosol, is the nutrient-sensing responsiveness of this protein [10,13–15]. Based on the central autophagosome-lysosomal role in adaptation to nutrient-depletion [16] and the strong association between BLOC1S1 and endo-lysosomal biology we proposed that the direct investigation into the role of BLOC1S1 in lysosomal homeostasis should further enhance our understanding of this protein in cellular biology.
The mechanisms orchestrating fusion of vesicles, endosomes and autophagosomes with lysosomes to facilitate the degradation of extracellular particles and intracellular components is relatively well established. In contrast, the subsequent recycling of the lysosome, where autolysosomes (ALs) are recycled into proto-lysosomes and subsequent mature lysosomes, a program termed autophagic lysosome reformation (ALR), is less well characterized [17–19]. Interestingly, the absence of BLOC1S1 results in the accumulation of autophagosomes [20] and endolysosomes [8], raising the question whether BLOC1S1 is required for the progression to lysosomal reformation. The initial structural requirement for this lysosomal recycling program is termed lysosomal tubulation (LT), where tubulations bud off from ALs prior to the scission off of protolysosomes. The molecular machinery initiating LT is currently being explored [21,22] and requires MTOR and lipid second messenger signaling, cytoskeletal components (microtubules) and the motor protein KIF5B (kinesin family member 5B) to provide the infrastructure and force to facilitate extrusion of tubules from ALs [23]. We reasoned that characterization of the role of BLOC1S1 in autophagosome-lysosome biology and recycling through LT and the ALR may shed further insight into the mechanisms underpinning the cellular roles of BLOC1S1 and enhance our understanding of the lysosomal reformation program.
As the hepatic autophagolysosomal system is exquisitely responsive to nutrient deprivation to synthesize ketones and glucose to sustain higher organisms during starvation, we proposed that the liver would be the ideal organ to explore the ALR. To explore the role of BLOC1S1 in the ALR we focused our studies on primary hepatocytes extracted from bloc1s1 liver-specific knockout (LKO) mice [15] compared to littermate controls. In this study we find that the genetic deletion of BLOC1S1/BORCS1 in the liver partially phenocopies various aspects of the genetic knockdown of other BLOC1 and BORC subunits. The major finding is that the absence of BLOC1S1/BORCS1 completely blocks lysosomal reformation by preventing the initiation of lysosomal tubulation. Here, BLOC1S1 is shown to recruit the motor protein KIF5B, the Arf-like small GTPase ARL8B and the linker protein PLEKHM2/SKIP to the lysosome as a putative structural complex to initiate tubulation. In parallel BLOC1S1 interacts and colocalizes with another critical LT actin cytoskeletal interacting protein WHAMM on lysosomal tubules. Finally, the role of BLOC1S1 appears to function in concert with MTORC1 signaling for the initiation of lysosomal tubulation. This study extends our knowledge of the functioning of a subunit of BLOC-1 and BORC and firmly integrates its role in autolysosomal biology by showing its requirement for lysosomal reformation.
Results
Bloc1s1 LKO hepatocytes accumulate autolysosomes and lysosomes and partially retain lysosomal function
We previously showed that bloc1s1/gcn5l1 KO MEFs and LKO hepatocytes had elevated levels of autolysosomes (ALs) and LAMP1 (lysosomal-associated membrane protein 1) [3,20]. Here we firstly quantified the levels of ALs and lysosomal proteins compared to other intracellular organelles in LKO hepatocytes. We confirmed that steady-state levels of LAMP1, and additionally that LAMP2 levels were increased, without changes in canonical endoplasmic reticulum or mitochondrial protein levels in LKO compared to WT hepatocytes (Figure 1A). To validate this, we employed electron microcopy of liver sections from 8-week-old LKO mice. We visualized a greater accumulation of autophagic vacuoles (Avd) with high electron density in the LKO livers and these structures comprised of late ALs as evident by partially degraded cytoplasmic material, and the more electron dense lysosomes (Figure 1B). To further evaluate whether lysosomal proteolytic function was intact in the LKO hepatocytes, we assayed lysosome acidity, the activity of CTSL (cathepsin L) and the enrichment and activity of CTSD (cathepsin D) within lysosomes in vivo using fluorescent indicators, confocal microscopy and by immunoblot analysis. LysoSensor Green, which emits enhanced fluorescence in an acidic pH, was more robust in the LKO hepatocytes (Figure 1C). In parallel, the cellular activity of lysosomal CTSL was measured by its cleavage and subsequent fluorescence of the Magic Red reagent MR-FR2 [24,25]. This emission signal similarly showed increased fluorescence in the LKO hepatocytes (Figure 1D). Furthermore, trafficking of the lysosomal endoprotease CTSD into lysosomes was measured by its interaction with the BODIPY-fluorescent label conjugated to pepstatin A [26]. Here too, we found that the accumulation of bound BODIPY-FL-pepstatin A to CTSD was increased in the LKO hepatocytes (Figure 1E). Although the quantitation of individual hepatocyte LysoSensor, Magic Red and pepstatin A fluorescence suggested increased lysosomal activity per LKO hepatocyte (Figure 1 C-D-left panels), when the intensity of these signals was corrected by lysosome number, the acidity of the LKO hepatocytes were only modestly, albeit significantly increased in LKO hepatocytes, whereas cathepsin activity within each lysosome was similar between genotypes (Figure 1 C-D-right panels). To further characterize the functional capacity of these lysosomes we assayed lysosomal-dependent cleavage of procathepsin D. Denaturing gel electrophoresis was performed on whole cell lysate and probed with an antibody recognizing a cleavage intermediate as well as the mature heavy and light CTSD chains [27]. These data showed that the levels of mature cathepsin were modestly diminished with the retention of excess intermediate forms in LKO hepatocytes. (Fig S1A). These data suggest that in concert with the accumulation of lysosomes, there is excess lysosomal proteins in the LKO hepatocytes. However, the function of these lysosomes may only be modestly disrupted as evident by the changes in pH and CTSD cleavage effects. Overall these data strongly support an increased number of ALs and lysosomes in the absence of BLOC1S1 and suggest that the homeostasis of these vacuolar organelles may be functionally compromised.
Figure 1.
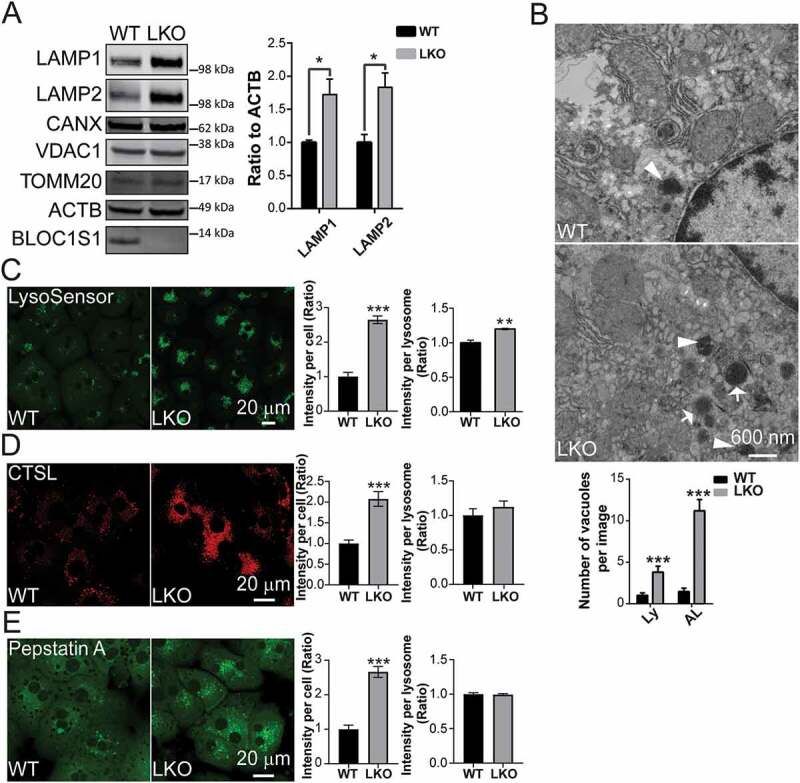
Bloc1s1 LKO hepatocytes exhibit aberrant accumulation of autolysosomes/lysosomes. (A) Immunoblot analysis shows increase LAMP1 and LAMP2 levels in bloc1s1 LKO hepatocytes (LKO) lysates without changes in the levels of mitochondrial and ER proteins compared to WT hepatocytes. ACTB was used as loading control. The accompanying histogram displays the relative quantified ratio of LAMP1 and LAMP2 after normalization to ACTB from three replicates. The endogenous BLOC1S1 expression is depicted with a solid arrowhead. (B) Representative TEM images of WT and LKO liver tissues. The number of autophagic vacuoles (Avd)-like structures with higher electron density were consistently increased in LKO liver sections. Lysosome (Ly) and autolysosomes (AL) are indicated by arrows and arrowheads. The quantification of Ly and AL is shown on the bottom panel. n > 14 cells from each group were analyzed. (C) Fluorescence pH indicator LysoSensor Green shows enhanced acidity in WT and LKO hepatocytes. The relative fluorescence per cell and normalized to cellular lysosomal area from 3 separate experiments are shown in the accompanying histograms. (D) The fluorescence of Magic Red reagent represents the higher CTSL activity in LKO hepatocytes. The relative fluorescence per cell and normalized to cellular lysosomal area from 3 separate experiments are shown in the accompanying histograms. (E) BODIPY FL-pepstatin A (Green) staining representing the excess CTSD lysosomal uptake in LKO hepatocytes with the accompanying histograms showing quantitation. n > 20 cells from each group analyzed in C-E. Bars represent mean SEM, *p < 0.05, **p < 0.01, ***p < 0.001, using the Student’s t test. Scale: 20 μm
Bloc1s1 LKO hepatocytes accumulate autolysosomes and retain autophagic flux
Increased lysosomes could result from increased lysosomal biogenesis, from sustained biogenesis in parallel with delayed fusion of autophagosomes with lysosomes, as evident in disruption of other BORC subunits [12], or from accelerated recycling of lysosomes through the ALR process [17]. Alternatively, a combination of defects in these programs could result in phenotype observed in the LKO hepatocytes. To begin to understand this, we firstly assayed the steady-state protein levels of master regulators of lysosomal biogenesis, TFEB and TFE3, and found that the TFEB steady-state level was attenuated rather than induced in LKO hepatocytes (Fig S2A). However, the evaluation of nuclear translocation of TFEB show that the levels were the same in WT and LKO hepatocytes (Fig S2B). This data would rather suggest that the lysosomal biogenesis activity is similar between the genotypes and cannot, in isolation, explain why there are more lysosomes in LKO hepatocytes.
To further explore the phenotype of excess ALs and lysosomes we began to explore the flux through the autophagy pathway. Interestingly, we had previously found increased levels of the autophagy mediators LC3B-II and SQSTM1 in bloc1s1 KO mouse embryonic fibroblasts (MEFS) [20], which suggested that autophagy may be perturbed in the absence of BLOC1S1. To explore this in the liver and to characterize this at a dynamic level we delineated autophagic flux, by inducing autophagy in response to nutrient deprivation (5 h of incubation in serum, glucose and glutamine free media) [17], and by inhibition of lysosomal proteolysis using pepstatin A and Leupeptin (lysosome inhibitors – LIs) in hepatocytes [25,27]. For the initial confocal microcopy analysis WT and LKO hepatocytes were transduced with mCherry-LC3B. In response to nutrient deprivation we observed a significant accumulation of mCherry-LC3B puncta in LKO hepatocytes (Figure 2A). Concurrent immunostaining for endogenous LAMP1 showed a significant (indicated by Pearson’s coefficient in Fig. S3A), albeit incomplete colocalization of mCherry-LC3B puncta with LAMP1, supporting that the LKO hepatocytes retained the ability to generate ALs (Figure 2A). Quantitative analysis of colocalization suggested increased ALs formation in LKO hepatocytes (Figure 2B). We further validated these data by performing experiments where we substituted mCherry-LC3B with adenoviral vectors harboring GFP-tagged LC3B and obtained similar results (Fig S3C). To confirm this concept, we then transfected primary hepatocytes with a tandem fluorescent pHluorin-mKate2-tagged LC3B expression vector. As the green pH sensitive pHluorin would be quenched by acidification within ALs, this should result in an augmented far red mKate2 signal in the event of intact AL formation [28]. In the overlaid images, the LKO and WT hepatocytes both displayed red organelles consistent with acidified ALs, supporting that the trafficking of LC3B from autophagosomes to ALs was retained in both WT and LKO hepatocytes (Figure 2C). This was further validated by immunofluorescence and immunoblot analysis where the addition of the proteolysis inhibitors (LIs) increased LC3B-II accumulation in both WT and LKO hepatocytes in response to nutrient restriction (Figure 2D and S3B-C). The calculated maximal autophagic flux, as quantified by densitometric changes in signal by immunoblot analysis, in the presence of LIs, was similar in the two genotypes (Figure 2E). To substantiate that the accumulation of autophagosome, AL and lysosome proteins were dependent on the depletion of BLOC1S1, rescue experiments were performed using adenoviral transduction of BLOC1S1 into primary LKO hepatocytes. The restoration of BLOC1S1 in LKO hepatocytes reduced SQSTM1, LC3B-II (Figure 2F) and LAMP1 levels (Figure 2G). Together these data strongly supported that bloc1s1 LKO hepatocytes accumulated autolysosomes, retained autophagic flux and AL generative capacity and appeared to accumulate and maintain lysosome acidification. Collectively these data pose the question as to whether these steps in the complete autophagosomal-lysosomal pathway are constipated by disruption of the recycling through ALR in the LKO liver?
Figure 2.
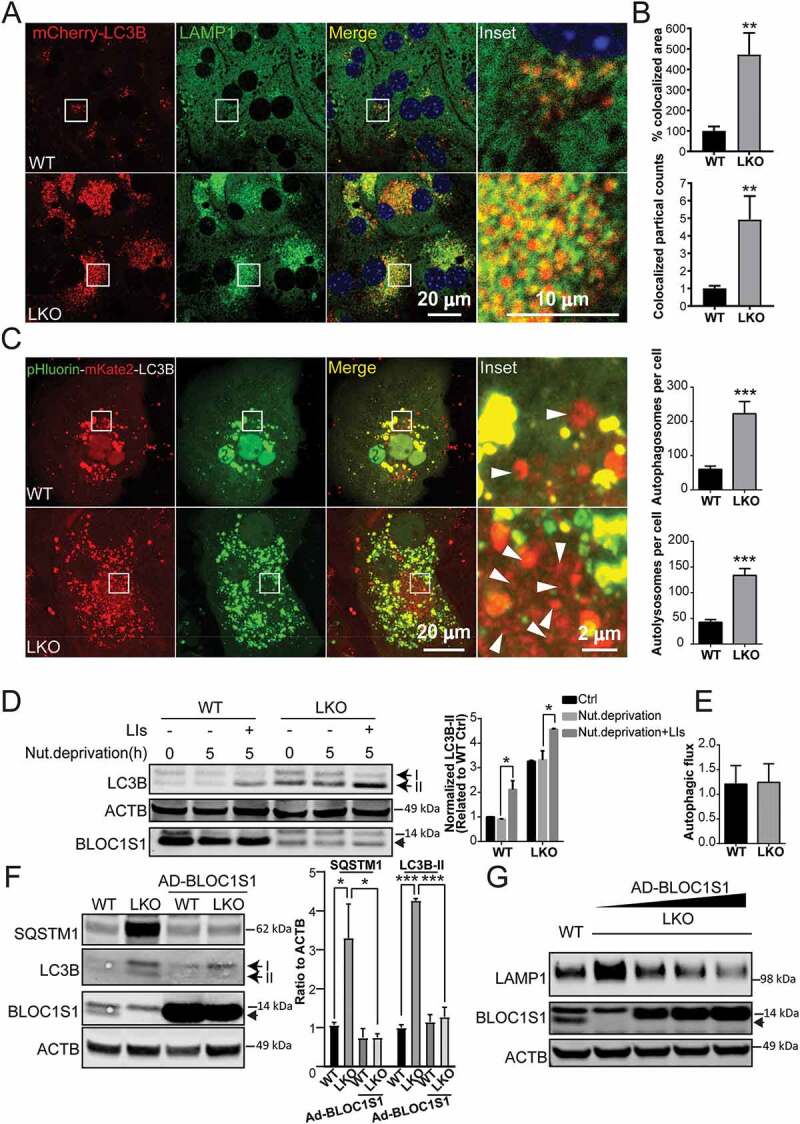
LKO hepatocyte maintain autophagic flux despite impaired clearance of autophagosomes. (A) Confocal microcopy showing autolysosome accumulation in LKO hepatocytes. The autolysosomes were visualized after transfection with transfected with mCherry-LC3B (red) and compared to the endogenous lysosome enriched protein LAMP1 (green). The overlaid puncta (yellow) represent ALs. DNA was visualized with DAPI staining (blue). (B) The colocalized area (yellow) and colocalized puncta (red-green-positive puncta) were determined in whole cell. n > 5 cells from each group were analyzed. (C) Confirmatory confocal microscopy using pHluorin-mKate2, tandem fluorescent protein-tagged LC3B showed accumulation of both autophagosomes (yellow) and acidified (red – mKate2) ALs in LKO hepatocytes in response to 5 h of nutrient deprivation. The arrowhead indicated the formation of autolysosome (red puncta). (D) Biochemical analysis showing equivalent autophagic flux as shown by the accumulation of LC3B-II in both genotypes in response to the combination of nutrient deprivation and LIs (leupeptin and pepstatin A, 20 µM each). Cell lysates were immunoblotted with antibodies targeting LC3B, BLOC1S1, and ACTB. The intensity of LC3B-II bands was normalized to ACTB and expressed as a ratio relative to WT Ctrl (*p < 0.05, n = 4 experiments). (E) Autophagic flux in WT and LKO hepatocytes was calculated by using the normalized intensity of LC3B-II with LIs minus the intensity of LC3B-II without LIs after 5 h starvation and expressed as a ratio relative to WT Starvation.(F) Representative immunoblot analysis showing restoration of autophagic turnover in LKO hepatocytes following the rescue of BLOC1S1 levels by the measurement of autolysosome substrates SQSTM1 and LC3B-II (n = 3). (G) Immunoblot analysis showing the BLOC1S1-dose responsive dissipation of LAMP1 levels in LKO hepatocytes in response to increasing amounts of Ad-BLOC1S1 transduction (n = 3). Endogenous BLOC1S1 immunoblot expression is depicted with a solid arrowhead
Bloc1s1 LKO hepatocytes have impaired LT and ALR
To explore lysosomal tubulation (LT) as the initial step in ALR, primary hepatocytes were transduced with adenoviral RFP-LAMP1. Five hours after nutrient deprivation, confocal microcopy in WT hepatocytes showed abundance of RFP-LAMP1 labeled tubules extending from autolysosomal bodies (Figure 3A). In contrast, there was a paucity of these tubulations in the LKO hepatocytes (Figure 3A). The average length and number of the tubules in cells showed that the formation of lysosomal tubules was significantly impaired by BLOC1S1 deficiency (Figure 3A). To confirm that this phenomenon arose from lysosomes, the cells were loaded with Dextran-647, which has been shown to localize to lysosomes [29]. Here, GFP-LAMP1 was colocalized with the Dextran Alexa Fluorophore loaded LTs in the WT, which again was not evident in the LKO cells (Fig S4A). This finding was further confirmed using LAMP1 antibody, which similarly overlaid with lysosomes loaded with LysoTracker Red (Fig S4B). To validate that these LTs arose from autolysosomes hepatocytes were transfected with the RFP-LC3B and loaded with Dextran-647. Here too, the WT hepatocytes showed extensive LTs from LC3B and Dextran double-labeled autolysosomes with an absence of these protrusions in LKO hepatocytes in response to nutrient deprivation (Figure 3B). To validate the role of BLOC1S1 in this process, reconstitution studies were performed. Overexpression of BLOC1S1 with the adenoviral transduction restored LT protrusions in LKO hepatocytes (Figure 3C). To further explore the dynamic formation of LT and the progression of ALR, primary hepatocytes were starved for 5 and 10 h and the distribution of endogenous LAMP1 was imaged by confocal microcopy. In WT hepatocytes, 5 h after starvation LT lengths were increased along with the progressive loss of autolysosomes and ALR was completed by 10 h of starvation (Figure 4A). This process was markedly retarded in the LKO hepatocytes with residual autolysosomes still present after 10 h of starvation (Figure 4A). This defective ALR processing was similarly rescued following adenoviral-expressing BLOC1S1 transduction into LKO hepatocytes (Figure 4A). Additionally, the starvation study suggested that the AL size increases with nutrient deprivation at 10 h in the LKO hepatocytes due to the disruption in recycling (Figure 4A). To validate this, hepatocytes were loaded with LysoTracker Red and imaged under nutrient rich conditions at 48 h. Here, the size of the ALs/Lys in LKO hepatocytes was significantly larger than in the WT (Figure 4B). Together these data strongly support the disruption of the ALR in the absence of BLOC1S1.
Figure 3.
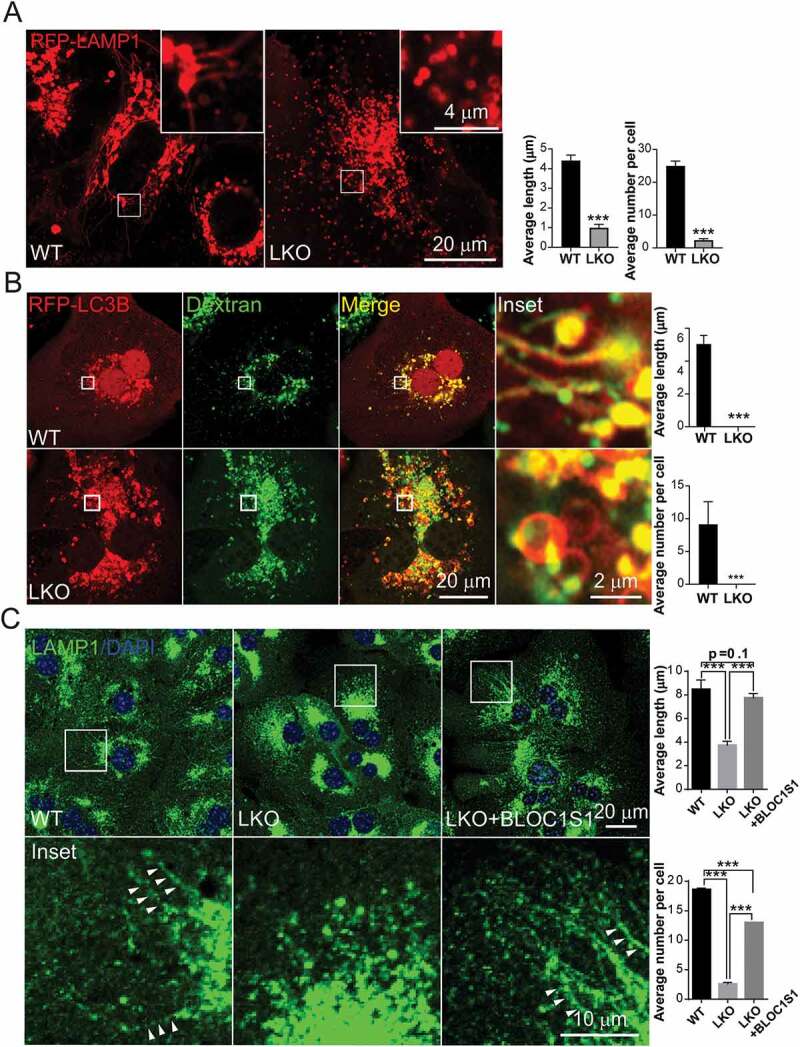
LKO hepatocytes are unable to initiate lysosomal tubulation in response to nutrient deprivation. (A) Confocal microcopy shows the greater extension of LTs from ALs in WT vs. LKO hepatocytes as assessed by live imaging of RFP-LAMP1. The average length and number of LTs per cell are quantified. n > 10 cells from each group were analyzed. (B) Confocal microcopy shows the emergence of LTs from RFP-LC3B and Dextran double-labeled autolysosomes in WT but not LKO hepatocytes as assessed by live imaging. The average length and number of LTs per cell are quantified. n > 10 cells from each group were analyzed. (C) WT and LKO hepatocytes were transduced with Ad-Ctrl or Ad-BLOC1S1 for 48 h and stained with LAMP1 antibody (green) and DAPI (blue). Arrowhead indicated the lamp1 positive tubules. The average length and number of LTs per cell are quantified. n > 5 cells from each group were analyzed
Figure 4.
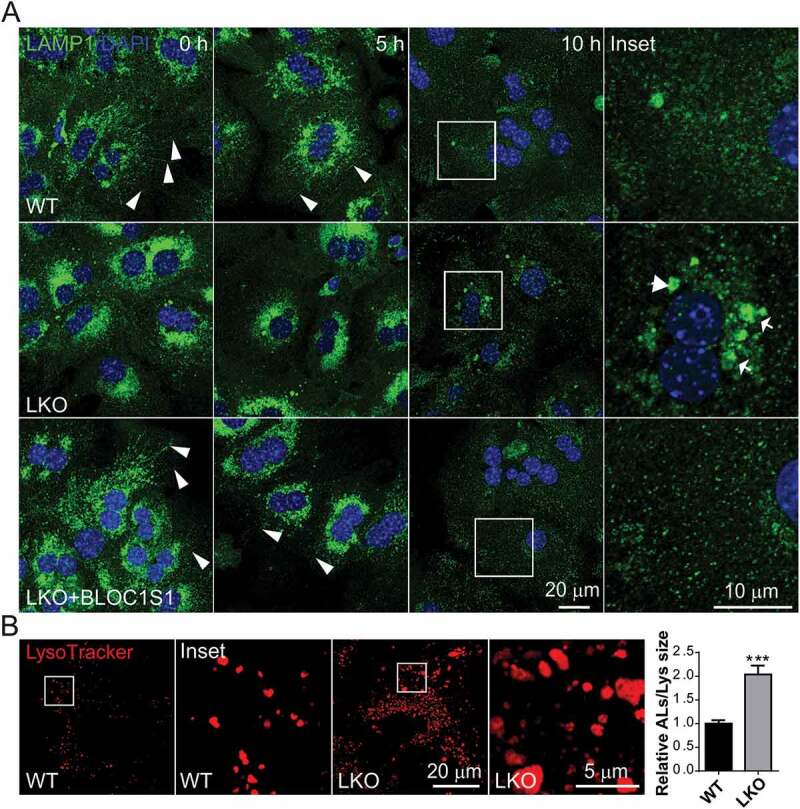
Impaired lysosomal tubulation delays ALR and results in the enlarged lysosomes in LKO. (A) WT and LKO hepatocytes were infected with Ad-Ctrl and Ad-BLOC1S1 for 48 h then starved with indicated time and stained using LAMP1 antibody (green) and DAPI (blue). Arrowhead indicated the lamp1 positive tubules. Arrow indicated the retained autolysosomes. (B) WT and LKO hepatocytes were cultured for 48 h and stained with LysoTracker (red) for 30 min. and monitored by confocal microscopy. The accompanying histograms show the relative size of the retained ALs:Lys and show the increase in LKO hepatocytes
BLOC1S1 interacts with the ARL8B-PLEKHM2-KLC1-KIF5B complex to initiate lysosomal tubulation
Although the molecular control of the LT is not completely characterized, the role of kinesin motor proteins [22] and the intracellular cytoskeleton [30] have been shown to orchestrate this process. In parallel, the BORC complex controls lysosomal mobility in conjunction with kinesin motor proteins depending on the recruitment of KIF5B to lysosomes by the ARL8B-PLEKHM2 (pleckstrin homology domain containing, family M (with RUN domain) member 2)-KLC1 (kinesin light chain 1) complex [5]. As BLOC1S1 is a subunit of BORC complex, we evaluated whether BLOC1S1 plays an integrative role in the kinesin motor for LT initiation. The motor protein KIF5B (kinesin family member 5B) plays a role in initiating LT by pulling on the autophagolysosomal membrane [22]. At the same time the interaction of KIF5B, with the KLC1, the ARF-like small GTPase ARL8B and the linker protein PLEKHM2 are essential for the centrifugal trafficking of lysosomes [5,31]. To explore the role of BLOC1S1 in LT, we first evaluated whether BLOC1S1 associated with these proteins and found by immunoprecipitation in HEK293T cells that BLOC1S1 interacted with KIF5B, KLC1, ARL8B and PLEKHM2 (Figure 5A-D). However, in-vitro pull down assays did not support a direct interaction between BLOC1S1 with KIF5B, KLC1 or PLEKHM2 (Fig S5A-C), implicating that these interactions were indirect. At the same time, as the BORC complex has been shown recruit ARL8B to lysosomes to initiate this motor complex formation [5], the interaction of ARL8B with BLOC1S1 was also assessed using in-vitro pull-down assays and here we validated that His-tagged BLOC1S1 directly bound to GFP-tagged ARL8B (Figure 5E). We further validated that the BLOC1S1-ARL8B was bound to the motor protein complex by showing that the in-vitro pull-down of His-BLOC1S1 interacted with GFP-ARL8B, HA-KLC1 and MYC-PLEKHM2 (Figure 5F). The direct interaction of BLOC1S1 with ARL8B was further supported by the finding that isolated lysosomes from WT but not from LKO hepatocytes had evidence of incorporated BLOC1S1 and ARL8B (Figure 5G and Fig S5D-E). Furthermore, confocal microscopy studies showed that GFP-tagged ARL8B colocalized with Dextran-647 in WT LTs but not in the LKO hepatocytes suggesting the association of ARL8B with LT was abolished in the absence of BLOC1S1 (Figure 5H). Interestingly, overexpression of ARL8B in the LKO cells promoted tubular formation but these were not linked to lysosomes as evident by their lack of colocalization with Dextran-647 implicating BLOC1S1 in the localization of ARL8B to lysosomes (Figure 5H). Prior studies have shown that ARL8B associate with endosomes [32] which may explain their location in the LKO hepatocytes. Consistent with Figure 3C, defective LT formation in LKO hepatocytes was restored when BLOC1S1 was overexpressed through adenoviral transduction (Figure 5I panel 3). However, this process was abolished when the motor protein KIF5B was depleted by siRNA (Figure 5I panel 4). As reported, knockdown of Kif5b in WT hepatocytes also disrupted the LT formation (Fig S6A-B) [22]. Intriguingly, the recruitment of KIF5B to lysosomes via overexpression of a lysosome targeting kinesin binding sequence (KBS) was unable to restore LT in LKO hepatocytes, implicating that the combined interactions of BLOC1S1 with KIF5B and ARL8B are necessary to initiate LT (Fig S6C).
Figure 5.
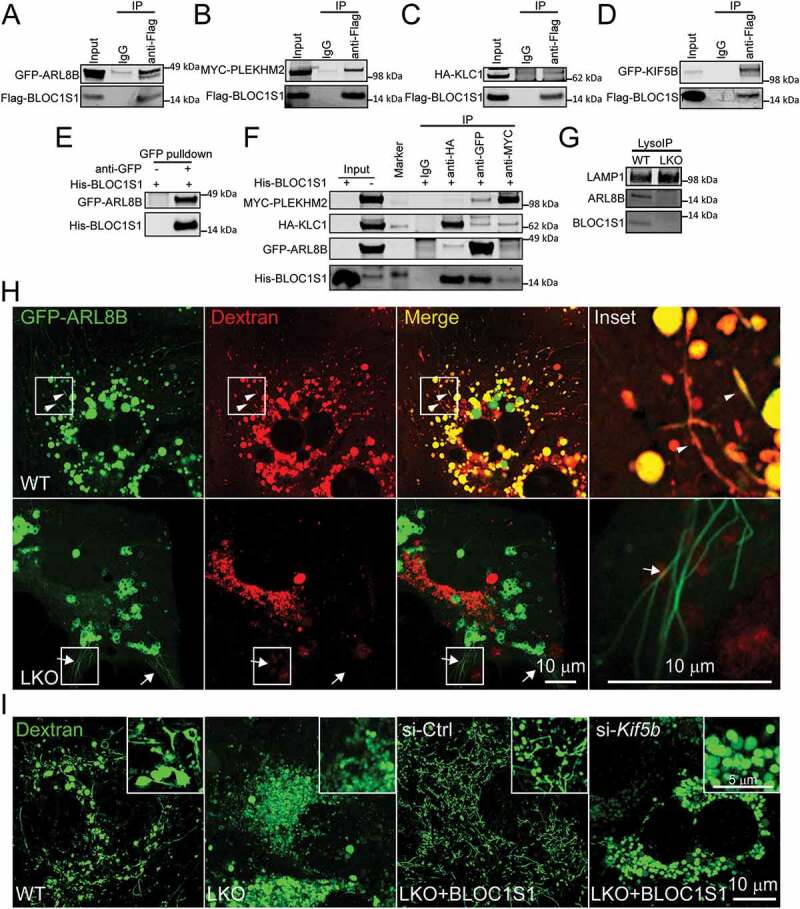
The recruitment of kinesin 1 complex to lysosome by BLOC1S1 was essential for lysosomal tubulation. (A-D) Co-immunoprecipitation studies revealed an association between BLOC1S1 with KIF5B complex subunits. 293 T cells were co-transfected Flag-BLOC1S1 with GFP-ARL8B (A), MYC-PLEKHM2 (B), HA-KLC1 (C) or GFP-KIF5B (D) for 48 h and then immunoprecipitated with either IgG or an indicated antibody and then subjected to immunoblot analysis with antibodies to Flag, MYC, HA and GFP. (E) GFP affinity isolation showed the direct interaction of His-BLOC1S1 and GFP-ARL8B protein in vitro. 293 T cells were transfected with GFP-ARL8B for 48 h and then immunoprecipitated with either IgG or an GFP antibody conjugated magnetic beads. After washing, the beads were incubated with purified His-BLOC1S1 protein and then subjected to immunoblot analysis. (F) Co-immunoprecipitation studies revealed an association between BLOC1S1 with KIF5B complex subunits. 293 T cells were co-transfected GFP-ARL8B with MYC-PLEKHM2 and HA-KLC1 for 48 h and then immunoprecipitated with either IgG or indicated antibody-conjugated magnetic beads. After washing, the beads were incubated with purified His-BLOC1S1 protein and then subjected to immunoblot analysis. (G) Lysosomes were rapidly isolated for WT and LKO hepatocytes using the LysoIP method. Immunoblot analysis shows ARL8B and BLOC1S1 are absent from LKO extracted lysosomes. LAMP1 was used as loading control. (H) WT and LKO hepatocytes were transiently transfected with GFP-ARL8B for 24 h, then incubated with fluorescence-labeled dextran (red) overnight and imaged using live cell confocal microscopy. Arrowhead indicated GFP-ARL8B and Dextran double labeled tubules. Arrows indicate GFP-ARL8B only labeled tubules indicating an absence of association with ALs. (I) WT and LKO hepatocytes were infected with AD-BLOC1S1, then transfected with si-Ctrl and si-Kif5b for 48 h and monitored using live cell confocal microscopy after loading with fluorescence-labeled dextran (green) overnight. The knockdown of Kif5b impaired BLOC1S1 rescue of LT
At the same time, clathrin and its AP2 adaptor protein are essential component for the initiation of LT and ALR and are involved in the recruitment of KIF5B to the microdomain of LT [33]. We therefore tested whether the siRNA knockdown of Cltc (clathrin, heavy polypeptide (Hc)) and/or AP2A1 (adaptor-related protein complex 2, alpha 1 subunit) could prevent the rescue of LT is LKO hepatocytes and showed that the depletion of either factor prevented BLOC1S1-mediated rescue of LT (Fig S6D-E). Furthermore, siRNA knockdown of Cltc and Ap2a1 did not disrupt the accumulation of BLOC1S1 and ARL8B within lysosomes (Fig S6F). Taken together these data show that BLOC1S1 functions in concert with these canonical ALR mediators supporting the role of BLOC1S1 in integrating the motor protein complex with ALs for LT initiation.
BLOC1S1 interacts with the actin cytoskeleton during LT initiation
Interestingly, the actin nucleation promoting factor WHAMM, which also associates with membranes and microtubules, has recently be identified as an essential structural protein in the initiation of LT [30]. Since our data suggests BLOC1S1 is required for LT formation, we tested whether BLOC1S1 associated with this actin scaffolding protein during LT initiation. Co-expression of His-tagged BLOC1S1 and Flag-tagged WHAMM in HEK293T cells resulted in co- immunoprecipitation of BLOC1S1 when anti-Flag antibody was used to IP WHAMM as compared to IgG control (Figure 6A). In parallel, experiments using both recombinant His-tagged BLOC1S1 and cell lysate from Flag-WHAMM transfected cells showed a direct interaction between BLOC1S1 and WHAMM (Figure 6B). Given the parallel interaction between BLOC1S1 and ARL8B, we employed the in-vitro pull-down assay to assess whether ARL8B and WHAMM directly interact. We did not find an interaction between these two proteins (data not shown), however, purified His-tagged BLOC1S1 did interact with both Flag-WHAMM and GFP-ARL8B when they were co-transfected into 293 T cells (Figure 6C). Furthermore, confocal microcopy imaging demonstrated that BLOC1S1 and WHAMM (Figure 6D) were co-localized with LTs in WT hepatocytes. Given that WHAMM has a distinct lysosome binding domain which binds to PI(4,5)P2 on lysosomes [30], it was not unexpected that BLOC1S1 was not required for WHAMM localization to lysosomes (Fig S7A). Additionally, the inhibition of actin polymerization with Latrunculin A, abolished LT in nutrient deprived WT hepatocytes indicating the actin cytoskeleton was required for LT (Fig S7B). Interestingly, inhibition of microtubule polymerization with Nocadazole similarly disrupted LT in nutrient-deprived WT hepatocytes (Fig S7B) suggested that a parallel, microtubular cytoskeleton mediated process played an important role in the LT. In support of this, siRNA depletion of both the BLOC-1 subunits (Bloc1s6 and Bloc1s5) and the BLOC-1/BORC shared subunit (snapin/BORCS3) have been shown to partially disrupt endosomal tubulation by disrupting the actin and microtubular cytoskeletons [34].
Figure 6.
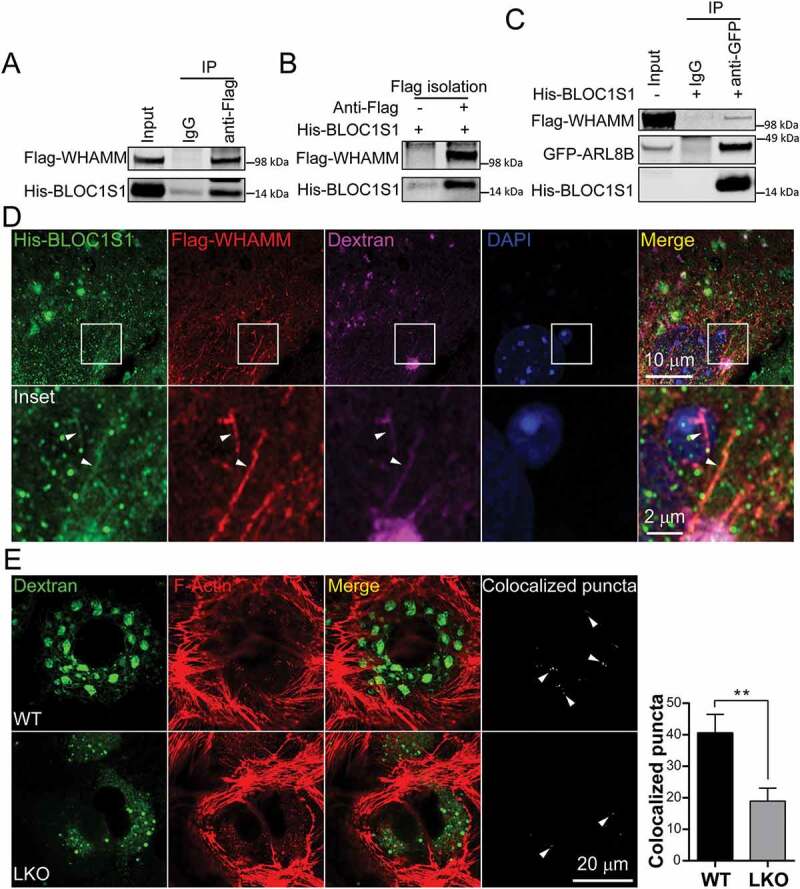
The BLOC1S1 interacted with and co-localized with WHAMM on lysosomal tubules to regulate actin nucleation on autolysosomes/lysosomes. (A) 293 T cells were co-transfected with Flag-WHAMM and His-BLOC1S1, and then immunoprecipitated with either IgG or anti-Flag antibodies and then subjected to immunoblot analysis using anti-Flag and anti-His antibodies. (B) Flag affinity isolation showed the direct interaction of and Flag-WHAMM with His-BLOC1S1 protein in vitro. 293 T cells were transfected with Flag-WHAMM for 48 h and then immunoprecipitated with either IgG or a Flag antibody conjugated magnetic beads. After washing, the beads were incubated with purified His-BLOC1S1 protein and then subjected to immunoblot analysis. (C) BLOC1S1 concurrently interacted with WHAMM and ARL8B. 293 T cells were co-transfected Flag-WHAMM with GFP-ARL8B for 48 h and then immunoprecipitated with either IgG or GFP antibody conjugated magnetic beads with or without adding His-BLOC1S1 protein and then subjected to immunoblot analysis with antibodies to Flag, His and GFP. (D) WT hepatocytes were co-transfected with His-BLOC1S1 and Flag-WHAMM for 48 h then incubated with fluorescence-labeled dextran (purple). After fixation, the cells were stained using anti-His (green) and anti-Flag (red) antibodies with the nucleus stained by DAPI (blue). The colocalization of WHAMM and BLOC1S1 is show by the arrowhead indicating His-BLOC1S1, Flag-WHAMM and Dextran triple labeled tubules. (E) WT and LKO hepatocytes were co-incubated with Dextran-488 and SiR-actin following 5 h of nutrient depletion, then observed using superresolution confocal microscopy. The colocalization of > 1 μm autolysosomes/lysosomal with actin were extracted by ImagJ and highlighted by arrowheads in far-right panels. The quantitation from n > 14 cells per group are shown in the accompanying histogram
Interestingly, the role of BLOC1S1 deficiency in the fundamental function of actin nucleation to dynamically shape and remodel membranes does not appear to be grossly impaired given that the overall similar pattern of actin distribution, as assessed by F-actin staining, in WT and LKO hepatocytes (Figure 6E). However, quantitation of the interaction points between actin and the > 1 μm autolysosome/lysosomes shows that in the absence of BLOC1S1 the colocalization of actin with autolysosome/lysosomes are diminished (Figure 6E, right panels). These data further support that the interaction of BLOC1S1 with the actin cytoskeleton play a role in lysosomal tubulation.
Bloc1s1 LKO hepatocytes exhibit constitutive MTORC1 activation
When the process of LT was initially defined, MTORC1 reactivation was implicated in playing a pivotal role in the initiation of the ALR [17]. However, we have previously shown that MTORC1 activity is constitutively active in bloc1s1 LKO hepatocytes [35], whereas in this study the LKO hepatocytes are unable to initiate LT. The prior mechanism identified driving MTORC1 activation in the LKO hepatocytes was due to the deacetylation of hepatic glutaminase and, increased enzyme activity resulting in increased α-ketoglutarate (α-KG) levels [35]. At the same time, protein degradation in ALs similarly reactivates MTORC1 signaling during prolonged autophagy [17], and MTORC1 has been shown to be associated with the lysosomal membrane [17,36,37]. These data raised the question as to whether MTORC1 activity is also increased in the LKO hepatocytes due to the accumulation of ALs during starvation. Firstly, we confirmed that MTOR binds to ALs in both the WT and LKO hepatocytes during 5 h nutrient deprivation condition (Figure 7A) and LKO hepatocytes had higher MTORC1 activity at both 5 and 10 h of starvation (Figure 7B), which was consistent with the accumulation of ALs in LKO as shown in Figure 1A-B. To further assess effects of the degradation in AL on MTORC1 activity, we prevented the digestion in AL by neutralizing the acidic AL with chloroquine (CQ). Here we found that the disruption of AL digestion blunted hyper-activity of MTORC1 in the LKO hepatocytes during starvation (Figure 7B). These data supported that the prevention of lysosomal tubular formation in the LKO hepatocytes may also contribute to increased MTORC1 activity in LKO hepatocytes through the accumulation of ALs. In addition, we found that blocking both autophagosome formation (3-MA) and lysosomal degradation (LIs) blunted the hyperactivity of MTORC1 in LKO during starvation, although the major effect on the higher MTORC1 activity in LKO hepatocytes appears to be due to lysosomal digestive activity in the enlarged AL in these cells (Figure 7C). Finally, as we had previously shown that the reconstitution of BLOC1S1 could rescue the lack of ALR in LKO hepatocytes, we explored whether this was operational when MTORC1 was inhibited in conjunction with BLOC1S1 reconstitution. Here we find that the coadministration of rapamycin and BLOC1S1 abolished the ability of BLOC1S1 to rescue ALR in LKO hepatocytes (Figure 7D). However, rapamycin pretreatment did not abolish lysosomal accumulation of BLOC1S1 and ARL8B in WT hepatocytes (Fig S8A). Taken together, these data further support that MTORC1 signaling functions in concert with the structural components afforded by BLOC1S1-ARL8B-KIF5B complex to initiate ALR in hepatocytes.
Figure 7.
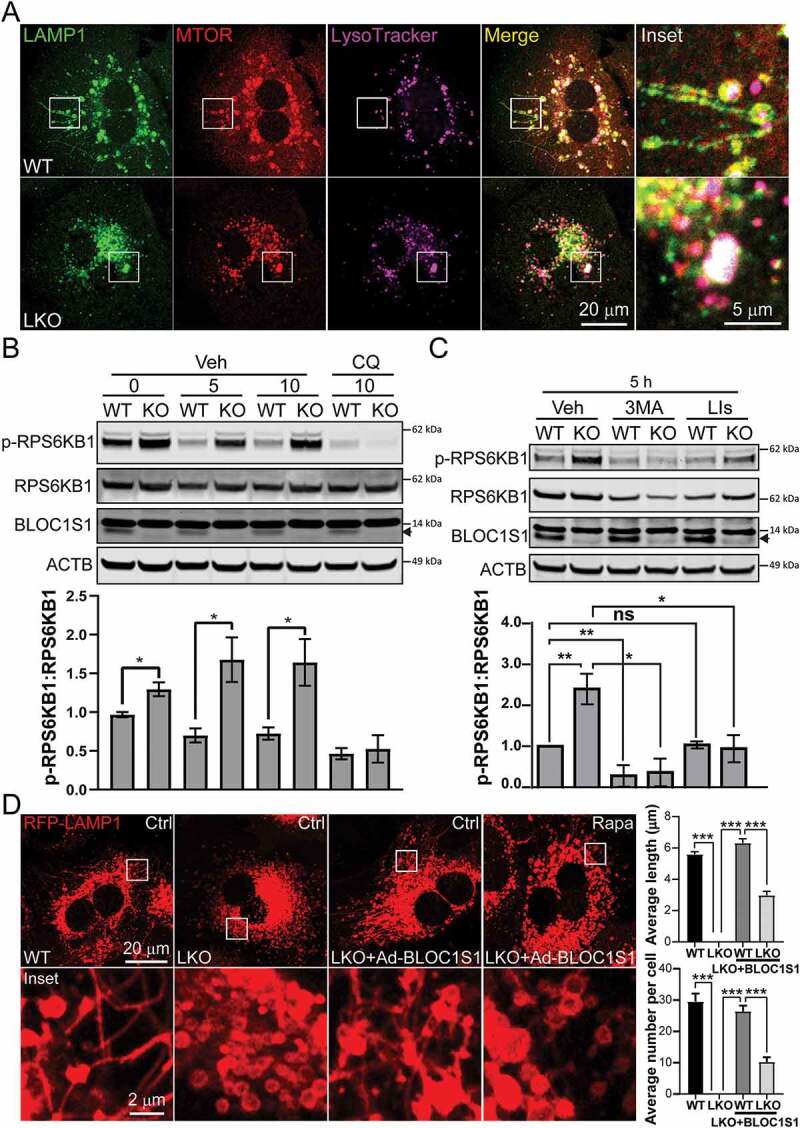
Impaired autolysosome clearance resulted in hyperactivity of MTORC1 in LKO hepatocytes. (A) WT and LKO hepatocytes were starved for 5 h and then stained with LysoTracker (purple). After fixation, the cells were stained by anti-LAMP1 (green) and anti-MTOR (red) antibodies. The representative confocal microscopy images show the enhanced MTOR (yellow and whiteoverlaid images) in LKO hepatocytes. (B) WT and LKO hepatocytes were starved for indicated time and treated with DMSO (Veh) or chloroquine (CQ; 50 nM). The cell lysates were immunoblotted with antibodies recognizing phospho-RPS6KB1 (p-RPS6KB1), RPS6KB1, BLOC1S1, and ACTB and show the dissipation of RPS6KB1 phosphorylation by the inhibition of autophagosome lysosome fusion. The ratio of phospho-RPS6KB1 to RPS6KB1 were shown on the accompanying histogram. Endogenous BLOC1S1 immunoblot expression is depicted with a solid arrowhead. (C) WT and LKO hepatocytes were starved for 5 h and treated with DMSO (Veh), 3-MA (5 mM) or LIs (leupeptin and pepstatin A (20 µM each)). The cell lysates were immunoblotted with antibodies against phospho-RPS6KB1, RPS6KB1, BLOC1S1, and ACTB. These data support that blocking both autophagosome formation (3-MA) and lysosomal degradation (LIs) blunted the hyperactivity of MTORC1 in LKO during starvation. The ratio of phospho-RPS6KB1 to RPS6KB1 were shown on the accompanying histogram. (D) WT and LKO hepatocytes were transduced Ad-RFP-LAMP1 with Ad-Ctrl or Ad-BLOC1S1 for 48 h and stained in parallel with the administration of DMSO (Ctrl) or rapamycin (100 nM) in conjunction with nutrient deprivation (5 h). Live imaging confocal microscopy was then performed and the average length and number of LTs per cell were quantified. n > 10 cells from each group were analyzed
Discussion
The results presented here extend our understanding of the role of BLOC1S1/GCN5L1/BORCS1 in autophagosome-lysosome biology by showing its requirement for the initiation of lysosomal tubulation to enable lysosomal reformation. BLOC1S1 is shown to bind to and promote the accumulation of KIF5B, KLC1, ARL8B and PLEKHM2 on lysosomes as a putative structural complex to initiate tubulation. In parallel, BLOC1S1 also interacts with the actin nucleation promoting factor WHAMM, a required cytoskeletal factor for LT initiation. Moreover, the data from this study shows that the colocalization of BLOC1S1 and its motor and structural protein cofactors to ALs function in concert with MTORC1 mediated signaling to initiate LT. A schematic depicting these roles of BLOC1S1 is shown in Figure 8.
Figure 8.
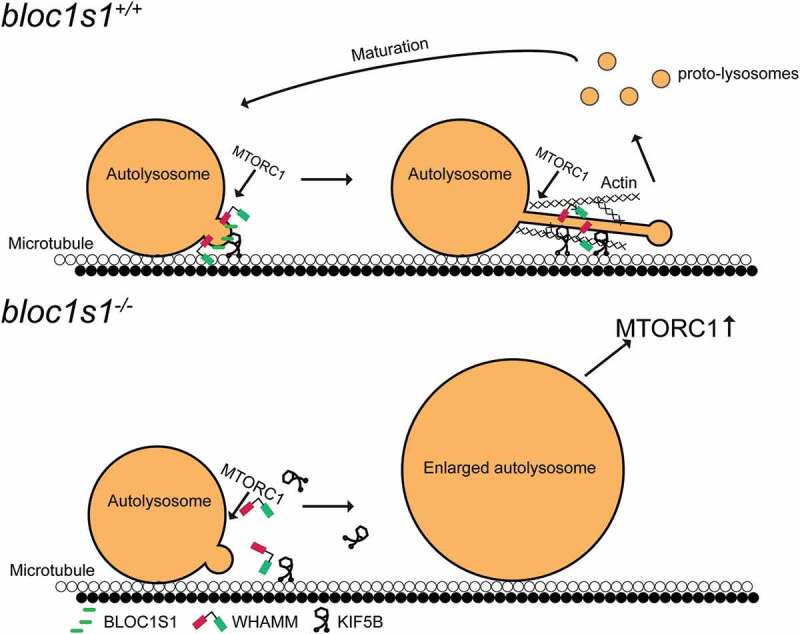
Schematic representation of BLOC1S1 coordinating with KIF5B and WHAMM to initiate LT. In WT hepatocytes, BLOC1S1 recruits KIF5B and WHAMM to the lysosome membrane to initiate lysosomal tubulation for subsequent ALR. In LKO hepatocytes, KIF5B and WHAMM are unable to be recruited to ALs. This abolishes the initiation or extension of lysosomal tubules, thereby impairing the ALR. The consequences of this blockade resulted in accumulation of enlarged autolysosomes which induced hyperactive MTORC1
The expanding role of BLOC1S1 in endosomal, autophagosome, lysosome and lysosome-related organelle biology
The initial indication that BLOC1S1 was linked to vacuolar organelle homeostasis was its identification as a subunit of the BLOC-1 complex linked to the biogenesis of lysosome related organelles [11]. A drosophila BLOC1S1 deficiency model subsequently showed impaired intracellular protein trafficking and reduced eye pigmentation [38]. Interestingly, although the murine germline bloc1s1 knockout was embryonic lethal [8,20], E14.5 embryos similarly showed defects in eye pigmentation, supporting the role of BLOC1S1 in melanocyte biogenesis [8]. The role of BLOC1S1 in vesicular trafficking was further confirmed where BLOC1S1 depletion delayed epidermal growth factor receptor (EGFR) degradation [8]. In that context BLOC1S1 interacted with sorting nexin 2 (SNX2) and the endosomal sorting complex required for transport 1 component, TSG101 to mediate the sorting of EGFR into endosomal compartments [8].
BLOC1S1/BORCS1 has also been implicated in modulating lysosome and synaptic vesicular positioning within cells. Here, the genetic knockdown of bloc1s1 phenocopied depletion of other BORC subunits and resulted in reduced dissemination of lysosomes to the cell periphery [5]. This perturbation in the trafficking of lysosomes resulted from BORC defect-mediated impairment of the recruitment of the GTPase ARL8B to the lysosome membrane [5]. An addition role of BLOC1S1 in lysosome positioning was identified by its role in microtubular acetylation and stability [3]. Furthermore, the axonal transport of synaptic vesicle precursors (SVPs), an additional ARL8B-dependent trafficking event, was similarly disrupted following the genetic depletion of BLOC1S1 [6]. However, biochemical studies suggested that a different BORC subunit termed SAM-4, rather than BLOC1S1/BORCS1, functions as the ARL8 guanine nucleotide exchange factor in SVPs [6].
In this study we expand the understanding of the role of BLOC1S1 showing that it, in association with the motor proteins KIF5B and the GTPase ARL8B, are necessary for the initiation of lysosomal tubulation as the initiating event of ALR. Concurrently, despite the known close physical proximity of BLOC1S1 [3] to additional LT initiation proteins clathrin and AP2 [33,39] we have not proven a direct interaction of these proteins with BLOC1S1. However, the inability of BLOC1S1 to rescue LT in LKO hepatocytes following the depletion of clathrin and AP2, and the role of clathrin and AP2 in the recruitment of KIF5B to the LT initiation complex [33], support a synergistic role of BLOC1S1 with clathrin and AP2 in the composition of the motor protein complex for LT initiation.
At the same time, a paradox has emerged, given that autophagic flux appears to be maintained in LKO hepatocytes despite the persistent activation of MTORC1 in the liver [35]. Whether this reflects redundancy in hepatic autophagy, to circumvent MTORC1 activation, due to the liver’s central role in systemic fuel substrate recycling under nutrient deprivation conditions will need to be investigated.
Furthermore, our data shows that the number of lysosomes in the LKO hepatocytes are increased, despite disruption in lysosome recycling. This does not appear to be solely due to TFEB activity, although TFEB activation has been shown to be induced in BLOC1S1 deficient in primary mouse embryonic fibroblast cells [40]. In contradiction to this, MTORC1 activity is increased in LKO hepatocytes and the activation of TFEB is known to be blunted by MTORC1 activity [41]. Taken together these data suggest that the mechanism/s orchestrating increased lysosomal content in the bloc1s1 LKO hepatocytes require additional study.
Overlapping and distinct roles of the BORC/BLOC-1 complex-subunits in organelle tubulation
A major limitation to our understanding of BLOC1S1 and the other BLOC-1 and BORC multi-subunit complexes, is that together, these two complexes are composed of 13 core subunits, including 3 shared subunits and a myriad of associated motor, cytoskeletal, adaptor/linker and catalytic proteins and enzymes. The combination of genetic manipulations required, using reductionist approaches, to dissect the specific function of each subunits, its interaction with all other subunits and the concomitant cellular adaptations to the disruption of individual subunits is technically feasible, but biologically more challenging. Nevertheless, insight can be gained from evaluating the overlapping phenotypes described by the individual disruption of BLOC-1 and BORC subunits to compare them to the knockout of BLOC1S1 in the processes of vacuolar organelle tubulation.
Snapin/BORCS3, which like BLOC1S1, is a shared subunit between BLOC-1 and BORC, has also been linked to lysosome tubulation in neurons. It has been shown that the overexpression of Snapin increases LT length whereas a dominant negative Snapin mutant, blunts LT length [27]. Although, the assessment of LT initiation was not assessed in snapin KO neurons, these cells did phenocopy our finding with evidence of AL accumulation and a modest reduction in CTSD maturation [27]. Although, the disruption of Snapin appears to have a more robust effect on modulating lysosomal acidification than the disruption of BLOC1S1 [42]. These data suggest that Snapin/BORCS3 and BORCS1/GCN5L1 may both play important and potentially interrelated roles in AL and ALR biology. The role of other BLOC-1 or BORC complexes in LT initiation do not appear to have been extensively evaluated. However, the combined reduction of BLOC-1 exclusive subunits (BLOC1S6, BLOC1S5 and DTNBP1) impair endosomal tubulation and recycling [34]. In that study endosomal tubulation is dependent of KIF13A-dependent mobility of endosomal tubules along the actin and microtubule cytoskeletons [34].
The initial description of lysosomal tubulation showed that the reactivation of MTORC1 after a period of prolonged starvation was a necessary signaling event for LT initiation [17]. Here we show that the structural association of BLOC1S1 and its interacting partners work in concert with MTORC1 signaling to initiate LT. The synergistic effects of MTORC1 activation and the role of BLOC1S1 is further evident in that LKO hepatocytes exhibit persistent activation of MTORC1 [35], but are unable to initiate LT in the absence of BLOC1S1. Interestingly, the genetic knockout of the exclusive BORC subunits BORCS5 through BORCS8 in HeLa cells did not result in MTORC1 activation [12]. This again, supports distinct roles of the different subunits. Alternatively, as the liver is one of primary organs for fuel substrate synthesis during starvation, an additional possibility would be that the higher autophagic capacity in the liver, with AL accumulation would result in greater MTORC1 activity in primary hepatocytes compared to in HeLa cells.
Evaluation of the role of individual BORC subunits in the control of MTORC1 activity further illuminates differences in their function roles. BORCS4 was specifically knocked out in HeLa cells to explore whether the disruption of lysosomal positioning affected MTORC1 activity. This BORCS4 study confirmed the prior observation that the disruption of any of the BORC subunits retained the perinuclear clustering of lysosomes [5,43]. In BORCS4 KO cells, the temporal activation of MTORC1 in response to serum reconstitution was delayed compared to the wildtype controls43. In contrast to this, the hepatic KO of bloc1s1, had previously been shown to elicit constitutively increased MTORC1 activity via the induction of mitochondrial glutaminolysis [35]. Whether the accumulation of ALs as we found in this study, also contribute to increased MTORC1 activity is possible, although difficult to distinguish from the BLOC1S1 effects on glutamine production. Given that snapin KO neurons phenocopy the AL accumulation we find in LKO hepatocytes, and it would be of interest to evaluate MTORC1 signaling in those cells [27].
The requirement of BLOC1S1 in the initiation of LT lysosomal tubulation, is further supported where the knockout of Kif5b, results in foreshortened, rather than absent lysosomal tubules [44]. This difference in LT phenotype may be due to the multifaceted role of BLOC1S1 in recruiting both kinesin and components of the actin cytoskeleton organization required to tubular extension. This latter concept parallels, in part, the roles played by the BLOC-1 subunits, BLOC1S6, BLOC1S5 and DTNBP1, in endosomal tubulation [34]. Although not fully characterized these data suggest that the different BORC subunits may play distinct and complementary roles in the broader vacuolar organelle tubulation biology within cells.
In this study we also demonstrate that BLOC1S1 interacts with the actin nucleation promoting factor WHAMM and that they colocalize with lysosomal tubules. Interestingly the KO of whamm in rat kidney epithelial cells has significant overlap with the phenotype of the LKO hepatocytes. The KO of whamm results in the presence of increased LC3B-II puncta, increased LAMP1 positive structures, retention of autophagosome maturation, an accumulation of starvation-induced enlarges ALs and the disruption of LT formation [30]. Together the robust phenotypic parallels following the genetic depletion of BLOC1S1/GCN5L1 and WHAMM suggest that the interaction between BLOC1S1 and WHAMM may play a pivotal role in LT initiation. Alternatively, or in parallel, the overlap of phenotypes between the KO of bloc1s1 and whamm could suggest that disruption of the cytoskeletal contribution to LT constipation of all upstream events, with consequent increase in ALs and signatures of autophagy etc. An additional role of the interaction between BLOC1S1 and WHAMM may play a role in the interaction of this LT initiating complex with the AL membrane. This concept is possible given that WHAMM which binds with PI(4,5)P2 to localize to membranes, preferentially binds to AL membranes and not to other PI(4,5)P2 enriched membranes [30]. Hence, the interaction of BLOC1S1 and WHAMM may direct WHAMM to ALs to facilitate the initiation of ALR.
Interestingly, BLOC-1 brings together the actin and microtubule cytoskeletons to generate recycling endosomes [34]. Based on this a hypothetical model of lysosomal tubulation, could entail BLOC1S1 with BORC and or BLOC-1 complex subunits playing a similar role to coalesce the microtubules (ARL8B-KIF5B-dependent) and actin (WHAMM) cytoskeletons, where KIF5B supplies the pulling force for tubular extension and microtubule functions as the track for the KIF5B motor protein, with the actin cytoskeleton enabling stabilization of the structure.
Conclusions
This study has explored the role of BLOC1S1/GCN5L1/BORCS1 in autolysosomal reformation in primary hepatocytes and has identified that it is a necessary component of the initiation of lysosomal tubulation. Moreover, BLOC1S1 has been identified as a possible link between the kinesin motor proteins and the actin cytoskeleton in initiating this lysosomal recycling program. Additional insight gleaned from this study strongly supports that the structural accumulation of LT initiation factors work in parallel with MTORC1 signaling to inaugurate autolysosomal reformation. Finally, this study expands our understanding of the complex interactions of proteins linked to the BLOC-1 and BORC subunits that play pivotal roles in the overall control of positioning, formation and recycling of numerous vacuolar organelles within the cell.
Materials and methods
Animal studies
The NHLBI Animal Care and Use Committee approved all animal studies used in this protocol. The mice were maintained on a 12-h light/dark cycle and housed 3–5 mice per cage with free access to water and normal chow diet (LabDiet, 5001). bloc1s1 liver knockout mice were generated by crossing bloc1s1flox/flox mice with albumin-cre mice [15]. All mice were generated in the C56BL/6b background. For immunoblot analysis, mice were anesthetized, then livers were removed and homogenized in RIPA buffer (Boston BioProducts, BP-115), 40 μg total protein was used for each sample [45].
Cell Culture and Transfection
Primary hepatocytes were isolated from normal chow fed mice at age of 8–12 weeks as previously described [45]. Briefly, each mouse was anesthetized with isoflurane, and the liver was perfused with Krebs Ringer buffer with glucose (135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.4 mM K2HPO4, 20 mM HEPES, 20 mM glucose, pH 7.4) and EGTA (100 mM) for 3 min, followed by continuous perfusion with the same buffer containing CaCl2 (1.4 mM) and collagenase (7000 IU/mouse, type I; Worthington, LS004196) for 8 min. Hepatocytes were isolated by disrupting and agitating the perfused liver in Krebs-ringer buffer and filtering through a 100 µm cell strainer. This hepatocyte rich fraction was then purified using a 43.2% Percoll (Sigma, P1644) density gradient (250 x g for 6 min). Pelleted liver hepatocytes were collected and plated (5 × 105 cells per well in 6-well plates) into collagen (Sigma, C3867)-coated plates and cultured in DMEM medium (Gibco, 11,995–065) containing 10% fetal bovine serum (FBS; Gibco, 16,000,044) and 1% penicillin-streptomycin (P/S; Gibco, 15,140,122). HeLa cells (ATCC, CCL-2), 293A cells (Agilent, 240,085), 293 T cells (ATCC, CRL-11,268) and COS7 cells (ATCC, CRL-1651) were cultured in DMEM containing 10% FBS, 1% P/S and 2 mM glutamine (Gibco, 25,030,081) at 37°C and 5% CO2.
For overexpression studies, primary hepatocytes, HeLa cells and COS7 cells were transfected with Lipofectamine 2000 (Invitrogen, 11,668,019) following the manufacturer’s instructions. 293A and 293 T cells were transfected with PolyJet In Vitro DNA Transfection Reagent (SignaGen Laboratories, SL100688). Cells were analyzed 48 h after transfection. For knockdown experiments, cells were transfected with indicated 25 nM siRNAs (GE Dharmacon, si-Kif5b: L-040710-01-0005; si-Ap2a1: L-043307-01-0005; si-Cltc: L-063954-00-0005) or ON-TARGETplus Non-Targeting SMARTpool (GE Dharmacon, D-001810-10-50) with Lipofectamine RNAiMAX Reagent (Invitrogen, 13,778,150) and analyzed 48 h later.
Adenovirus Production and Transfection
RFP-LAMP1, GFP-LC3B, BLOC1S1 overexpression adenoviruses were produced using Adeasy Adenoviral System (Agilent, 240,009) as described [15]. For primary hepatocytes infection, adenovirus overexpressing either empty vector (control), BLOC1S1, GFP-LC3B or RFP-LAMP1 were added to hepatocytes at a dose of 20 plaque-forming units/cell, and analyzed 48 h later.
Primary hepatocytes or COS7 cells were plated on 12 mm diameter, # 1 thickness coverslips coated with rat tail type I collagen (Neuvitro, GG-12-Collagen) and grown to 80% confluency before transfection. Twenty-four or 48 h after transfection, coverslips were washed in PBS (Gibco, 10,010–023) and fixed with 4% paraformaldehyde (Electron Microscopy Sciences, 157–8) in PBS for 10 min. Coverslips were washed twice for 5 min in PBS and permeabilized for 15 min in 0.2% Triton X-100 (Sigma, T8787). Cells were blocked for 60 min in 1% BSA (Sigma, A6003), 10% goat normal serum (Abcam, ab7481) and 0.3 M glycine in PBS and stained for 60 min with primary antibodies in 10% goat normal serum in PBS at room temperature. The secondary antibody was an Alexa Fluor conjugated antibody (Invirogen, A32723, A32731, A32754, A32744, A32733 and A32728) used at a 1:1000 or 1:250 dilution in PBS for 1 h. Coverslips were incubated with 300 nM DAPI (Cell Signaling Technology, 4083) for 5 min then washed three times with PBS and mounted on slides using Fluoroshield with 1,4-diazabicyclo[2.2.2]octane (Sigma, F6182). Images were obtained on an inverted confocal laser-scanning microscope (LSM 880 or LSM 880 with Airyscan; CarlZeiss) fitted with a 63×, 1.4 numerical aperture (NA) objective. For colocalization analysis, one cell was selected each time as ROI based on DAPI staining. Background fluorescence was subtracted in all channels, and fluorescence intensity thresholds were used to discriminate between signal and noise. For colocalized particle counting, colocalization plugin was employed using Fiji ImageJ (NIH). Pixel intensity was measured by ImageJ between images of the same particle structures in two channels. Correlation coefficients of pixel intensity correlation analysis were calculated by linear fitting using GraphPad Prism. The length of lysosomal tubules analyzed by ImageJ. For analyzing lysosomal size, the LysoTracker Red (Invitrogen, L7528) stained lysosomes were segmented using trainable Weka segmentation plugin [46], then lysosomal sizes were measured by ImageJ. DNA constructs used in this study are shown in Table 1 and Antibodies used in this study are shown in Table 2.
Table 1.
Recombinant DNA Constructs used in this study
| Plasmids | Protein | Notes |
|---|---|---|
| Flag-BLOC1S1 | BLOC1S1 | [3] |
| His-BLOC1S1 | BLOC1S1 | This work |
| pEX-PK-hLC3 (pHluorin-mKate2-LC3) | HsLC3B | pEX-PK-hLC3 was a gift from Isei Tanida (Addgene, 61,458) |
| mEos2-Lysosomes-20 (GFP-LAMP1) | LAMP1 | mEos2-Lysosomes-20 was a gift from Michael Davidson (Addgene, 57,397) |
| LAMP1-RFP | LAMP1 | LAMP1-RFP was a gift from Walther Mothes (Addgene, 1817) |
| pmRFP-LC3 | LC3B | pmRFP-LC3 was a gift from Tamotsu Yoshimori (Addgene, 21,075) |
| HA-KLC1 | KLC1 | [5] |
| GFP-ARL8B | ARL8B | [5] |
| GFP-KIF5B | KIF5B | [5] |
| MYC-PLEKHM2 | PLEKHM2 | [5] |
| KBS-LAMP1-GFP | KBS-LAMP1 | [5] |
| Flag-WHAMM | WHAMM | OriGene (CAT: MR223421) |
| pLJC5-TMEM192-3xHA (LysoIP) | TMEM192 | pLJC5-TMEM192-3xHA was a gift from David Sabatini (Addgene, 102,930) |
Table 2.
Antibodies used in this study
| Antibody | Cat. number | Working dilution | Source |
|---|---|---|---|
| Mouse anti-ACTB/β-Actin | 3700 | 1:1000(IB) | Cell Signaling Technology |
| Rabbit anti-ARL8B | LS-C456372 | 1:500(IB) | LSBio |
| Rabbit anti-VDAC1 | 4661 | 1:1000(IB) | Cell Signaling Technology |
| Mouse anti-TUBA1B/α-tubulin | sc-8035 | 1:1000(IB) | Santa Cruz Biotechnology |
| Rabbit anti-TOMM20 | 42,406 | 1:1000(IB) | Cell Signaling Technology |
| Mouse anti-CANX | MAB3126 | 1:1000(IB) | Millipore |
| Mouse anti-CTSD | AF1029 | 1:1000(IB) | R&D Systems |
| Mouse anti-LC3B | L8918 | 1:1000(IB) | Sigma |
| Rabbit anti-SQSTM1/p62 | 5114 | 1:1000(IB) | Cell Signaling Technology |
| Rabbit anti-TFEB | A303-673A | 1:1000(IB) | Bethyl Laboratories |
| Rabbit anti-TFE3 | ABE1400 | 1:1000(IB) | Millipore |
| Rabbit anti-RPS6KB1 | 9202 | 1:1000(IB) | Cell Signaling Technology |
| Rabbit anti-phospho-RPS6KB1 | 9204 | 1:1000(IB) | Cell Signaling Technology |
| Rabbit anti-MTOR | 2972 | 1:1000(IB) 1:100(IP) |
Cell Signaling Technology |
| Rabbit anti-H4C1 | 13,919 | 1:1000(IB) | Cell Signaling Technology |
| Rabbit anti-BLOC1S1 | Home made | 1:1000(IB) | [1] |
| Mouse anti-Flag | F3165 | 1:1000(IB) 1:400(IF) 1:250(IP) |
Sigma |
| Mouse anti-HA | 11,583,816,001 | 1:1000(IB) 1:100(IP) 1:100(IF) |
Roche |
| Rabbit anti-HA | sc-7392 | 1:1000(IB) | Santa Cruz Biotechnology |
| Rabbit anti-GFP | sc-8334 | 1:1000(IB) | Santa Cruz Biotechnology |
| Rabbit anti-LAMP1 | ab24170 | 1:1000(IB) 1:800(IF) |
Abcam |
| Rat anti-LAMP1 | 1D4B | 1:1000(IB) 1:200(IF) |
Developmental Studies Hybridoma Bank |
| Rat anti-LAMP2 | sc-19,991 | 1:1000(IB) | Santa Cruz Biotechnology |
| Anti-HA Magnetic Beads | 88,837 | 20 μL per sample | Thermo ScientificThermo Fisher |
| GFP-Trap Magnetic Agarose | gtma-20 | 20 μL per sample | Chromotek |
| Myc-Trap Magnetic Agarose | ytma-20 | 20 μL per sample | Chromotek |
| IRDye® 800CW Goat anti-Mouse IgG (H + L) | 926–32,210 | 1:10,000 (IB) | LI-COR |
| IRDye® 800CW Goat anti-Rabbit IgG (H + L) | 926–32,211 | 1:10,000 (IB) | LI-COR |
| IRDye® 680RD Goat anti-Mouse IgG (H + L) | 926–68,070 | 1:10,000 (IB) | LI-COR |
| IRDye® 680RD Goat anti-Rabbit IgG (H + L) | 926–68,071 | 1:10,000 (IB) | LI-COR |
Immunofluorescence Microscopy.
Live Cell Imaging
Primary hepatocytes were seeded on µ-Slide 4 well glass (ibidi, 80,426) bottom coated with rat tail type I collagen (Sigma, C3867). Twenty-four or 48 h after transfection, cells were imaged using confocal laser-scanning microscope (LSM 880 or LSM 880 with Airyscan; CarlZeiss) fitted with a 63×, 1.4 numerical aperture (NA) objective in an environmental control system at 37°C in 5% CO2, and images were acquired continually. Labeling of lysosomes with internalized dextran was performed by loading the cells with Dextran, Alexa Fluor 647 (Invitrogen, D22914) or Dextran, Alexa Fluor 488 (Invitrogen, D22910) for 16 h before imaging. For lysosomal pH, Cathepsin D assay or Cathepsin L assay, 1 µM LysoSensor Green (Invitrogen, L7535), 1 µM BODIPY FL-pepstatin A (Invitrogen, P12271) or 20 μl Magic Red (ImmunoChemistry Technologies, 941) were incubated with hepatocytes at 37°C for 30 min. For F-actin staining, 100 nM SiR-Actin (Cytoskeleton Inc, CY-SC001) were incubated in starvation medium for 6 h. After live imaging, fluorescent intensity of cells and colocalization were analyzed by Fiji ImageJ (NIH).
Purification of Lysosomes by LysoIP
TMEM192-3xHA were inserted to pShuttle-CMV vector, then TMEM192-3xHA overexpression adenovirus were produced using Adeasy Adenoviral System (Agilent, 240,009) as described.15 Primary hepatocytes were infected by these adenovirus for 48 h, then lysosomes were isolated using anti-HA magnetic beads (Thermo Scientific, 88,837) as described [47].
Transmission Electron Microscopy (TEM)
Mice liver tissue was extracted and post fixed for 90 min in 2% glutaraldehyde, 2% formaldehyde (Electron Microscopy Sciences, 157–8), in 0.1 phosphate buffer (pH 7.2), post-fixed in aqueous 1% osmium tetroxide, block stained with 1% uranyl acetate, dehydrated in graded ethanol solutions, and embedded in EMbed-812 (Electron Microscopy Sciences, 14,120). Thin sections were stained with uranyl acetate, and lead citrate then examined on a JEM-1200EX (JEOL, USA) transmission electron microscope (accelerating voltage 80 keV) equipped with an AMT 6 megapixel digital camera (Advanced Microscopy Techniques Corp).
Immunoprecipitation
For immunoprecipitation, transfected 293 T cells were treated with dithio-bis-succinimidyl propionate (2 mM; Thermo Scientific, 22,585) then lysed in 50 mM Tris, pH7.4, 137 mM NaCl, 10 mM MgCl2, 10% glycerol, 1%Triton X-100 containing protease inhibitors (Thermo Scientific, 78,438) and centrifuged at 12,000 x g for 10 min at 4°C. Supernatants (1–2 mg of total protein in 1 ml) were incubated with 2 µg normal IgG (EMD-Millipore, 12–371) or 2 µg primary antibody overnight at 4°C. 20 µl Sera-Mag Protein A/G particles (GE healthcare life sciences, 17,152,104,010,150) were added to each sample and incubated at room temperature for 1 h. For GFP-tagged protein immunoprecipitation, 20 µl GFP-Trap Magnetic Agarose (Chromotek, gtma-20) were incubated with cell lysates overnight. These beads were separated using a magnet (GE healthcare life sciences, 28,948,964) and washed four times in TBS with 0.1% Triton X-100, then the bound proteins were eluted in 1xLDS sample buffer (Invitrogen, NP0007) and analyzed by SDS–PAGE and immunoblotting.
In-vitro affinity-isolation Assay
293 T cells were transfected with indicated plasmids for 48 h and then immunoprecipitated with either 2 µg normal IgG (EMD-Millipore, 12–371) or an indicated tag antibody conjugated magnetic beads. After washing 3 times, these beads were incubated with purified His-BLOC1S1 protein overnight and then washed four times in TBS with 0.1% Triton X-100, then the bound proteins were eluted in 1xLDS sample buffer and analyzed by SDS–PAGE and immunoblotting.
Statistical Analysis
Results are displayed as the mean ± standard error of the mean. A comparison of groups was performed using two-tailed unpaired Students t-test. A value of P < 0.05 was considered to indicate statistical significance.
Supplementary Material
Acknowledgments
We thank Dr. Juan S. Bonifacino (NICHD) for the kind gifts of Kinesin complex related plasmids, Dr. Joseph Hill (UT Southwestern) for GFP-LC3B overexpression adenovirus, Dr. Campellone (University of Connecticut) for WHAMM plasmids; We thank the US National Heart Lung and Blood Institute (NHLBI) Electron Microscopy core; specifically, E. Stempinski for assistance during the EM acquisition and analyses; We thank NHLBI Light Microscopy core for assistance on imaging acquisition and analysis and animal facility for animal maintenance.
Funding Statement
This work was supported by the National Heart, Lung, and Blood Institute [ZIA-HL005199-10].
Disclosure statement
No conflicts to declare.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Scott I, Webster BR, Li JH, et al. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J. 2012;443(3):655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Scott I, Wang L, Wu K, et al. GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell Biol. 2018;28(5):346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wu K, Wang L, Chen Y, et al. GCN5L1 interacts with alphaTAT1 and RanBP2 to regulate hepatic alpha-tubulin acetylation and lysosome trafficking. J Cell Sci. 2018;131(22):pii: jcs221036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Donato V, Bonora M, Simoneschi D, et al. The TDH-GCN5L1-Fbxo15-KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat Cell Biol. 2017;19(4):341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pu J, Schindler C, Jia R, et al. BORC, a multisubunit complex that regulates lysosome positioning. Dev Cell. 2015;33(2):176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Niwa S, Tao L, Lu SY, et al. BORC regulates the axonal transport of synaptic vesicle precursors by activating ARL-8. Curr Biol. 2017;27(17):2569–78 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bae D, Moore KA, Mella JM, et al. Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J Cell Biol. 2019;218(4):1118–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang A, He X, Zhang L, et al. Biogenesis of lysosome-related organelles complex-1 subunit 1 (BLOS1) interacts with sorting nexin 2 and the endosomal sorting complex required for transport-I (ESCRT-I) component TSG101 to mediate the sorting of epidermal growth factor receptor into endosomal compartments. J Biol Chem. 2014;289(42):29180–29194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].John Peter AT, Lachmann J, Rana M, et al. The BLOC-1 complex promotes endosomal maturation by recruiting the Rab5 GTPase-activating protein Msb3. J Cell Biol. 2013;201(1):97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Thapa D, Wu K, Stoner MW, et al. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial beta-oxidation enzyme HADHA. J Biol Chem. 2018;293(46):17676–17684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Starcevic M, Dell’Angelica EC.. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J Biol Chem. 2004;279(27):28393–28401. [DOI] [PubMed] [Google Scholar]
- [12].Jia R, Guardia CM, Pu J, et al. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy. 2017;13(10):1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Webster BR, Scott I, Han K, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci. 2013;126(21):4843–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alrob OA, Sankaralingam S, Ma C, et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res. 2014;103(4):485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang L, Scott I, Zhu L, et al. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat Commun. 2017;8(1):523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu H, Ren D.. Lysosomal physiology. Annu Rev Physiol. 2015;77(1):57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen Y, Yu L. Autophagic lysosome reformation. Exp Cell Res. 2013;319(2):142–146. [DOI] [PubMed] [Google Scholar]
- [19].Munson MJ, Ganley IG. MTOR, PIK3C3, and autophagy: signaling the beginning from the end. Autophagy. 2015;11(12):2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Webster BR, Scott I, Traba J, et al. Regulation of autophagy and mitophagy by nutrient availability and acetylation. Biochim Biophys Acta. 2014;1841(4):525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li X, Rydzewski N, Hider A, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016;18(4):404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Du W, Su QP, Chen Y, et al. Kinesin 1 drives autolysosome tubulation. Dev Cell. 2016;37(4):326–336. [DOI] [PubMed] [Google Scholar]
- [23].Chen Y, Yu L. Recent progress in autophagic lysosome reformation. Traffic. 2017;18(6):358–361. [DOI] [PubMed] [Google Scholar]
- [24].Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31(21):7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen CS, Chen WN, Zhou M, et al. Probing the cathepsin D using a BODIPY FL-pepstatin A: applications in fluorescence polarization and microscopy. J Biochem Biophys Methods. 2000;42(3):137–151. [DOI] [PubMed] [Google Scholar]
- [27].Cai Q, Lu L, Tian JH, et al. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68(1):73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tanida I, Ueno T, Uchiyama Y. A super-ecliptic, pHluorin-mKate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PloS One. 2014;9(10):e110600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Johnson DE, Ostrowski P, Jaumouille V, et al. The position of lysosomes within the cell determines their luminal pH. J Cell Biol. 2016;212(6):677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dai A, Yu L, Wang HW. WHAMM initiates autolysosome tubulation by promoting actin polymerization on autolysosomes. Nat Commun. 2019;10(1):3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Guardia CM, Farias GG, Jia R, et al. BORC functions upstream of kinesins 1 and 3 to coordinate regional movement of lysosomes along different microtubule tracks. Cell Rep. 2016;17(8):1950–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jongsma ML, Bakker J, Cabukusta B, et al. SKIPHOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. Embo J. 2020;39(6):e102301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14(9):924–934. [DOI] [PubMed] [Google Scholar]
- [34].Delevoye C, Heiligenstein X, Ripoll L, et al. BLOC-1 brings together the actin and microtubule cytoskeletons to generate recycling endosomes. Curr Biol. 2016;26(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang L, Zhu L, Wu K, et al. Mitochondrial general control of amino acid synthesis 5 like 1 regulates glutaminolysis, mammalian target of rapamycin complex 1 activity, and murine liver regeneration. Hepatology. 2020;71(2):643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rebsamen M, Pochini L, Stasyk T, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519(7544):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-rag complex targets mtorc1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cheli VT, Daniels RW, Godoy R, et al. Genetic modifiers of abnormal organelle biogenesis in a Drosophila model of BLOC-1 deficiency. Hum Mol Genet. 2010;19(5):861–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu K, Wang L, Chen Y, et al. GCN5L1 interacts with alphaTAT1 and RanBP2 to regulate hepatic alpha-tubulin acetylation and lysosome trafficking. J Cell Sci. 2018;131(22):jcs221036. doi: 10.1242/jcs.221036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Scott I, Webster BR, Chan CK, et al. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem. 2014;289(5):2864–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shi B, Huang QQ, Birkett R, et al. SNAPIN is critical for lysosomal acidification and autophagosome maturation in macrophages. Autophagy. 2017;13(2):285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jia R, Bonifacino JS. Lysosome positioning influences mTORC2 and AKT signaling. Mol Cell. 2019;75(1):26–38 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chiricozzi A, Suarez-Farinas M, Fuentes-Duculan J, et al. Increased expression of interleukin-17 pathway genes in nonlesional skin of moderate-to-severe psoriasis vulgaris. Br J Dermatol. 2016;174(1):136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu H, Fergusson MM, Wu JJ, et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011;4(158):ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Arganda-Carreras I, Kaynig V, Rueden C, et al. Trainable Weka segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics. 2017;33(15):2424–2426. [DOI] [PubMed] [Google Scholar]
- [47].Abu-Remaileh M, Wyant GA, Kim C, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358(6364):807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.