

ABSTRACT
Preconditioning with a mild stressor such as fasting is a promising way to reduce severe side effects from subsequent chemo- or radiotherapy. However, the underlying mechanisms have been largely unexplored. Here, we demonstrate that the TP53/p53-FBXO22-TFEB (transcription factor EB) axis plays an essential role in this process through upregulating basal macroautophagy/autophagy. Mild stress-activated TP53 transcriptionally induced FBXO22, which in turn ubiquitinated KDM4B (lysine-specific demethylase 4B) complexed with MYC-NCOR1 suppressors for degradation, leading to transcriptional induction of TFEB. Upregulation of autophagy-related genes by increased TFEB dramatically enhanced autophagic activity and cell survival upon following a severe stressor. Mitogen-induced AKT1 activation counteracted this process through the phosphorylation of KDM4B, which inhibited FBXO22-mediated ubiquitination. Additionally, fbxo22−/− mice died within 10 h of birth, and their mouse embryonic fibroblasts (MEFs) showed a lowered basal autophagy, whereas FBXO22-overexpressing mice were resistant to chemotherapy. Taken together, these results suggest that TP53 upregulates basal autophagy through the FBXO22-TFEB axis, which governs the hormetic effect in chemotherapy.
Abbreviations: BBC3/PUMA: BCL2 binding component 3; CDKN1A/p21: cyclin dependent kinase inhibitor 1A; ChIP-seq: chromatin immunoprecipitation followed by sequencing; DDB2: damage specific DNA binding protein 2; DRAM: DNA damage regulated autophagy modulator; ESR/ER: estrogen receptor 1; FMD: fasting mimicking diet; HCQ: hydroxychloroquine; KDM4B: lysine-specific demethylase 4B; MAP1LC3/LC3: microtubule associated protein 1 light chain 3 alpha; MEFs: mouse embryonic fibroblasts; MTOR: mechanistic target of rapamycin kinase; NCOR1: nuclear receptor corepressor 1; SCF: SKP1-CUL-F-box protein; SQSTM1: sequestosome 1; TFEB: transcription factor EB
KEYWORDS: AKT1, autophagy, FBXO22, hormesis, KDM4B, MYC, TP53, ubiquitination
Introduction
More than 500 years ago, Paracelsus, the great German-Swiss physician, noted that the dose makes the poison, referencing effects related to dose dependency and lethality. This is the central idea underlying hormesis [1]. Almost all organisms whether yeasts, plants, or animals become more resilient against a high dose or severe stress when they are exposed to the same conditions or to different types of stressors of lesser potency. For example, it is well known that a dietary approach, such as fasting or a fasting mimicking diet (FMD), can increase the efficacy of chemotherapy in cancer patients [2]. This is now under evaluation in many human trials [3–6]. In one of these trials, fasting was well tolerated by all patients and was associated with a reduction in chemotherapy-induced side effects [4]. However, despite its general applicability and its potential efficacy in combination with chemo- and radiotherapy for cancer patients, the underlying molecular mechanisms are largely unknown.
Macroautophagy, hereafter referred to as autophagy, is an intracellular degradative process through which worn or damaged cytoplasmic material, including organelles and proteins, are degraded within lysosomes [7–9]. In addition to its scavenging activity inside the cell, autophagy also plays a role in supplying nutritional and other materials needed for cell survival under harmful conditions such as starvation [10]. Although the dual function of autophagy as a scavenger and a supplier might be regarded as “induced autophagy” and “basal autophagy”, respectively, its role as a physiological regulator is more complicated. For example, autophagy functions in both self-protecting and cell death-inducing mechanisms depending on the context [11]. However, the factors regulating the level of basal autophagy remain unexplored whereas those regulating induced autophagy caused by nutrient starvation have been relatively well investigated and it was found that activation is usually accompanied by suppression of MTOR (mechanistic target of rapamycin kinase) activity [12]. Nevertheless, there is an increasing awareness of the relationship between acquired stress resistance and autophagy as the latter is an adaptive and pro-survival mechanism against various forms of insult.
TP53/TRP53/p53 (tumor protein 53; the mouse protein is TRP53 but we will use TP53 for simplicity) is a well-known protector of the genome, shielding it against genotoxic stressors and playing a role in various physiological processes, such as carcinogenesis, dedifferentiation, and reprogramming [13]. It also protects against nutrient starvation [14]. These TP53-mediated responses are dependent on the cell type as well as the intensity and the nature of the signals that activate TP53. For example, when cells are sustained in the presence of intense genotoxic stressors, TP53 exerts either permanent cell cycle arrest as evidenced by senescence or apoptosis to prevent the accumulation of damaged cells. In contrast, when cells are sustained against mild and limited stress, TP53 exerts a transient cell cycle arrest through which the cell survives and is then repaired. At the molecular level, TP53 acts primarily as a transcription factor, regulating a complex network of gene expression such as that involved in cell cycle progression and apoptosis. In addition, there is an important interplay between TP53 and autophagy [15]. With respect to pro-autophagic responses, some autophagy-related genes have been identified as nuclear TP53 targets by chromatin immunoprecipitation followed by sequencing (ChIP-seq) analysis [16]. One such gene is DRAM (DNA damage regulated autophagy modulator) whose expression was reported to promote autophagy and to be essential for TP53-mediated apoptosis [17]. Other TP53-targeted autophagy-related genes including ULK1 (unc-51 like autophagy activating kinase 1) and ATG7 (autophagy related 7) were also reported to be essential for TP53-mediated apoptosis [16]. In contrast to nuclear TP53, cytoplasmic TP53 has been reported to elicit anti-autophagic responses under normal conditions [18]. The cytoplasmic TP53 inhibits the AMP-dependent kinase and in turn activate MTOR, leading to the suppression of autophagy. A molecular link of TIGAR (TP53-induced glycolysis and apoptosis regulator) with cytoplasmic TP53-mediated anti-autophagic responses has also been suggested [19]. Nevertheless, under limited stress, the mechanism for how TP53 functions as a pro-survival factor and its involvement in autophagy in this context remain elusive. Here, we demonstrate that the level of basal autophagy is determined by the transcription of TFEB (transcription factor EB) [20], which is regulated by degradation of KDM4B (lysine-specific demethylase 4B) complexed with MYC and NCOR1 (nuclear receptor corepressor 1) through TP53-FBXO22 and TP53-AKT1 axes. Mild stressors activate TP53 which in turn upregulates the level of basal autophagy without activation of MTOR-dependent-induced autophagy. Upregulation of basal autophagy by preconditioning is indispensable for hormetic protection against subsequent harmful insults.
Results
TP53-dependent hormesis upon nutrient depletion
Organisms exposed to a mild dose of stress are known to become tolerant to a lethal dose of subsequent stress. To explore this phenomenon, we first examined whether preconditioning with mild stress could increase the survival of normal human retinal pigment epithelial-1 (RPE-1) cells against subsequent harmful exposure. RPE-1 cells were subjected to preconditioning stress 24 hours after split, treated with a subsequent stress, and then subjected to analyses for 48 hours (Figure 1A). When wild-type (WT) RPE-1 cells were pre-treated with mock stress or deprived of all amino acids for 24 hours, the deprived cells became resilient against subsequent low level of glucose (2 mM) compared to the mock-treated cells, showing the reduction in the proportion of dead cells (Figure 1B). A similar resilience was also observed in TP53−/− RPE-1 cells. Interestingly, WT RPE-1 cells preconditioned with serine depletion also showed a similar resilience upon low glucose stress. Importantly, this acquired effect was not observed in RPE-1 cells lacking TP53, indicating the presence of both TP53-dependent and TP53-independent mechanisms (Figure 1C). We then examined whether depletion of other single amino acids could induce acquired resistance to low glucose. Very interestingly, depletion of arginine, glutamic acid, isoleucine, lysine, methionine, or serine from the medium resulted in an acquired resistance, showing the reduction in the proportion of dead cells, whereas that of other single amino acids did not (Figure 1D). The acquired stress resistances were completely in accord with the increased expression of CDKN1A/p21 (cyclin dependent kinase inhibitor 1A) (Figure 1D), indicating that TP53 was activated by the depletions of certain single amino acids. This TP53-dependent hormesis was also observed when RPE-1 cells were preconditioned with ionizing irradiation (IR) or heat shock (HS), both of which also activated TP53 (Figure 1E-F and S1A). A similar hormetic effect was observed in normal human fibroblast HCA2 cells (Figs. S1B-E). RPE-1 or HCA2 cells preconditioned with glucose depletion also acquired significant resilience against cysteine depletion in a TP53-dependent manner (Figure 1G and S1F). Preconditioning with serine depletion also induced hormetic effects in response to H2O2, HS, and ultraviolet (UV) treatments in RPE-1 cells (Figs. S1G-I). An impaired hormetic effect in cells preconditioned with serine depletion was almost completely recovered by ectopic expression of TP53 upon following low glucose or UV irradiation (Fig. S1J). These results suggest the presence of a mechanism underlying the induction of TP53-dependent hormesis.
Figure 1.
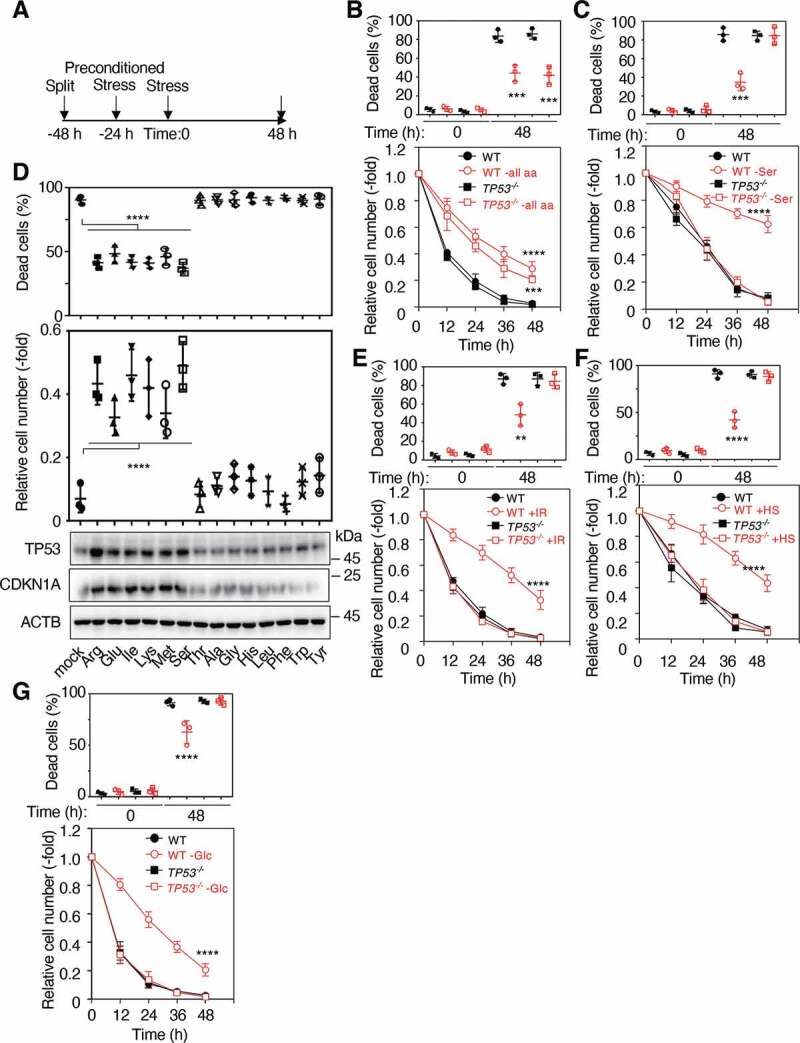
TP53 is required for an acquired stress response to various stimuli. (A) Schematic of experimental procedures for preconditioned stress. WT and TP53 KO RPE-1 cells were preconditioned with or without total amino acids depletion (B), serine depletion (C), IR (2 Gy) (E) or HS (42°C) for 4 h (F), and then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panels) and the relative cell numbers (lower panels) were determined at the indicated times as shown in (A). (D) WT RPE-1 cells were preconditioned with or without each single amino acid depletion, followed by incubation with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (middle panel) were determined at 48 h as shown in (A). Lysates from these cells at 48 h after treatment with low glucose (2 mM) media were subjected to immunoblotting using the indicated antibodies (lower panels). (G) WT and TP53 KO RPE-1 cells were preconditioned with or without glucose depletion and then treated with cysteine depletion for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were then determined at the indicated times. Data are presented as means±s.d. of three independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a WT group mean (B-G). **P < 0.01, ***P < 0.001, ****P < 0.0001
FBXO22 is a downstream effector of TP53 in hormesis
Given that depletion of all amino acids from the culture medium is known to suppress MTOR activity and subsequently activate induced autophagy, we examined the levels of the autophagy-related proteins MAP1LC3/LC3 (microtubule associated protein 1 light chain 3 alpha), SQSTM1 (sequestosome 1), and TFEB, a master transcription factor for lysosome biogenesis and autophagy [20], after preconditioning with various stressors. Preconditioning with depletion of all amino acids resulted in a downward shift of TFEB protein in both WT, TP53−/− RPE-1 and HCA2 cells (Figure 2A and S2A). In the downward shift of TFEB, corresponding to its dephosphorylated form (Figure 2A and S2A-B), this protein is reported to localize in nucleus, be in its transcriptionally active form, and mediated by the suppression of MTOR activity [21]. In fact, MTOR activity was markedly suppressed when cells were deprived of all amino acids as evaluated by phosphorylation of RPS6KB1/S6K (ribosomal protein S6 kinase B1) at Thr389, a known MTOR phosphorylation target (Figure 2A and S2A-B). Surprisingly, TP53 activation by preconditioning with mild stress such as serine depletion, glucose depletion, IR treatment or HS slightly suppressed MTOR but this suppression appeared to be insufficient for dephosphorylating TFEB (Fig. S2B), suggesting that activation of TP53 and subsequent suppression of MTOR are not involved in the regulation of nuclear translocation of TFEB under these conditions. Upon mild stress, the amount of TFEB protein was increased in WT cells whereas it was not increased in TP53−/− RPE-1 or TP53-depleted HCA2 cells (Figure 2A and S2A-B). Although a previous report showed that deletion or inhibition of TP53 promoted TFEB nuclear translocation in lung cancer cells [22], we failed to detect the downward shift and dephosphorylation of TFEB by TP53 deletion in normal cells, presumably due to the differences between normal and cancer cells. Consistent with this, preconditioning by depletion of all amino acids induced transcription of a host of TFEB target genes (Figure 2B and S2C) and increased the relative LC3-II amounts (Figure 2C) in both WT and TP53−/− RPE-1 cells. Very interestingly, even in the absence of a downward shift of TFEB and marked suppression of MTOR activity, serine depletion, glucose depletion, IR, and HS resulted in a marked increase in the level of TFEB, leading to a slight but significant increase in a transcription of TFEB target genes and the relative LC3-II amounts in WT RPE-1 cells, which was not observed in TP53−/− cells (Figure 2B,C). Taken together, the results suggest that TP53 regulates the amount of TFEB and the level of the LC3-II.
Figure 2.
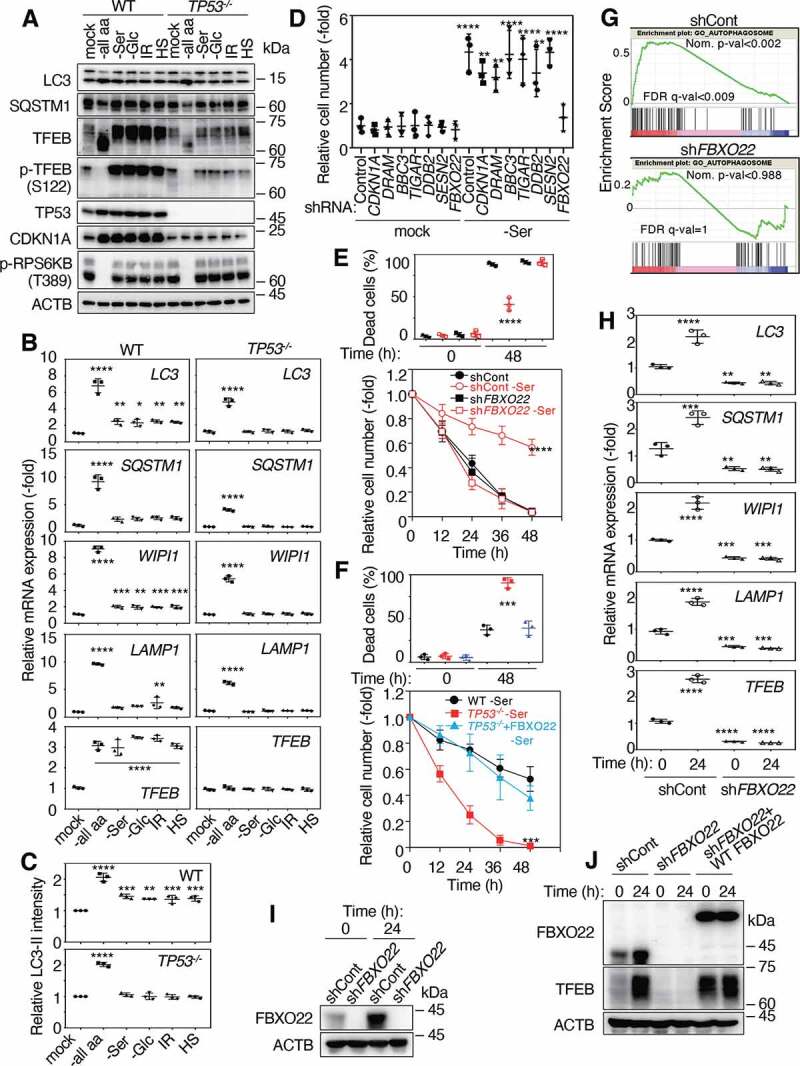
FBXO22, a TP53 target gene, is required for an acquired stress response. (A) Lysates from WT or TP53 KO RPE-1 cells preconditioned with or without total amino acid depletion (-all aa), serine depletion (-Ser), glucose depletion (-Glc), IR (2 Gy), or HS at Time 0 shown in Figure 1A were subjected to immunoblotting using the indicated antibodies. (B) QPCR analysis of cells as in (A) using the indicated primers. (C) Quantification of LC3-II intensities relative to ACTB intensities as in (A) by immunoblotting using anti-LC3 antibodies. (D) RPE-1 cells expressing the indicated Dox-inducible shRNA were preconditioned with or without serine depletion, and then treated with low glucose (2 mM) media for 48 h. The relative cell numbers were determined at 48 h as in Figure 1A. RPE-1 cells expressing the indicated Dox-inducible shRNA (E) and WT or TP53−/− RPE-1 cells expressing or not expressing FBXO22 (F), were preconditioned with or without serine-depletion and then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times as in Figure 1A. (G) GSEA plot of gene set “GO_AUTOPHAGOSOME” from the MSigDB in HCA2 cells expressing the indicated Dox-inducible shRNA in the presence or absence of IR (10 Gy) treatment. (H) QPCR analysis of cells as in (E) using the indicated primers at the indicated times after serine depletion. Lysates from RPE-1 cells expressing the indicated Dox-inducible shRNA (I) and those with or without RNA interference-resistant FLAG-FBXO22 (J) at the indicated times after serine depletion were subjected to immunoblotting using the indicated antibodies. Data are presented as means±s.d. of three independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a control (WT, mock, or shCont) group mean (B-F, H). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
TP53 is known to regulate various cellular responses essential for maintaining genome integrity, such as those involved in the cell cycle, DNA repair, apoptosis, autophagy, transcriptional activity, and metabolism [13]. We thus examined effects of the knockdown of genes representative of each of the above pathways on acquired resistance. Depletion from RPE-1 cells of known TP53 downstream effectors, such as CDKN1A/p21, DRAM, BBC3/PUMA (BCL2 binding component 3), TIGAR (TP53 induced glycolysis regulatory phosphatase), DDB2 (damage specific DNA binding protein 2), and SESN2 (sestrin 2), did not appear to affect the acquired resistance (Figure 2D and S2D-S2E). In contrast, depletion of FBXO22, another TP53-downstream target, markedly suppressed it. Previously, SCF (SKP1-CUL-F-box protein)Fbxo22 was reported to ubiquitinate KDM4A (lysine demethylase 4A), regulating histone H3 lysine 9 and lysine 36 methylation levels [23]. We also found that SCFFbxo22 targets methylated TP53 complexed with KDM4A specifically in the senescent sate and KDM4B complexed with estradiol-bound ESR/ER (estrogen receptor 1) [24,25]. Cells depleted of FBXO22 did not show any hormetic effect after serine depletion in response to subsequent incubation with low glucose (Figure 2E and S2F). Ectopic expression of FBXO22 in TP53−/− RPE-1 or TP53-depleted HCA2 cells almost completely rescued the acquired resistance, transcription, and the protein level of TFEB, but did not affect that of WT RPE-1 or HCA2 cells (Figure 2F and S2G-H), providing further the evidence for FBXO22 as a downstream effector of TP53 in terms of hormesis. Proliferation of TP53−/− RPE-1 cells, RPE-1 cells depleted of FBXO22, and TP53−/− cells expressing FBXO22 incubated in normal media after serine depletion was slightly affected as compared to that of WT RPE-1 cells (Figs. S2I-K), while that of CDKN1A/p21−/− RPE-1 cells was comparable (Fig. S2L). In order to delineate the importance of FBXO22 in TP53-dependent transcriptional activation of autophagy related genes, we performed RNA-sequencing (RNA-seq) in FBXO22-depleted normal human fibroblast HCA2 cells after TP53 activation by IR treatment. Gene set enrichment analysis revealed a significant increase in the expression of lysosomal-related and autophagosomal genes in control cells, whereas this gene ontology was not increased in cells depleted of FBXO22 (Figure 2G), indicating that TP53-dependent upregulation of the expression of these genes is dependent on the presence of FBXO22. An increase in the expression of TFEB as well as its target transcripts upon serine depletion was detected in cells expressing control short hairpin RNA (shRNA) whereas this was not found in cells expressing shFBXO22 (Figure 2H and S2M). Indeed, FBXO22 was induced by serine depletion as well as glucose depletion from the culture medium, or by IR or HS treatment, accompanied by TP53 induction (Figure 2I, S1A, and S2N) and reduction in the expression of TFEB in FBXO22-depleted cells without preconditioning was effectively rescued by the ectopic expression of FBXO22 (Figure 2J).
FBXO22 regulates TFEB transcription through MYC repressor complexes
We confirmed that TFEB is involved in hormesis after serine depletion as a preconditioning stressor from the finding that cells depleted of TFEB failed to show hormetic effects (Figure 3A and S3A-B). Proliferation of RPE-1 cells depleted of TFEB was comparable to that of WT cells (Fig. S3C). In addition, a similar impairment of hormesis was observed in RPE-1 and HCA2 cells treated with an autophagy inhibitor (hydroxychloroquine [HCQ]) or in other autophagy-deficient MEFs, such as atg7, atg9, and rb1cc1/fip200 (rb1 inducible coiled-coil 1) knockout (KO) cells (Figure 3B and S3D-G). In contrast, depletion of Rubcn, an inhibitory gene of autophagy, induced hormesis in the absence of preconditioning (Fig. S3H), further supporting the notion that the hormetic effect required autophagy. An impairment of hormesis in FBXO22-depleted cells was almost completely rescued by ectopic expression of TFEB in RPE-1 and HCA2 cells (Figure 3C and S3I-J), further supporting the epistasis of FBXO22 to TFEB in hormesis. We then tried to gain mechanistic insights into a hormetic effect by increased TFEB (Fig. S4A). Very intriguingly, preconditioning by serine depletion increased the expression of various autophagy related genes and the level of autophagy activity evaluated by LC3-II protein upon following treatment with a low level of glucose (1 mM), much more than the control (Figs. S4B-C). It should be noted that the reduction in MTOR activity upon low glucose (2 mM) treatment, evaluated by the level of p-RPS6KB, was comparable independent of the statuses of FBXO22 and TFEB (Figure 2A, S2A, and S4C). Consistent with this, ectopic expression of FBXO22 significantly increased expression of autophagy related genes as well as TFEB protein and the level of autophagy activity independent of serine-depletion (Figs. S4D-E). These effects were abrogated by the depletion of TFEB (Figs. S4D-F). In addition, although the ectopic expression of FBXO22 induced acquired resistance to low glucose independent of serine-depletion, this was also abolished by depletion of TFEB (Fig. S4F). Taken together, these results suggest that increased level of TFEB by mild stressor dramatically boosts autophagy activity upon following severe stressor, which likely governs hormesis.
Figure 3.
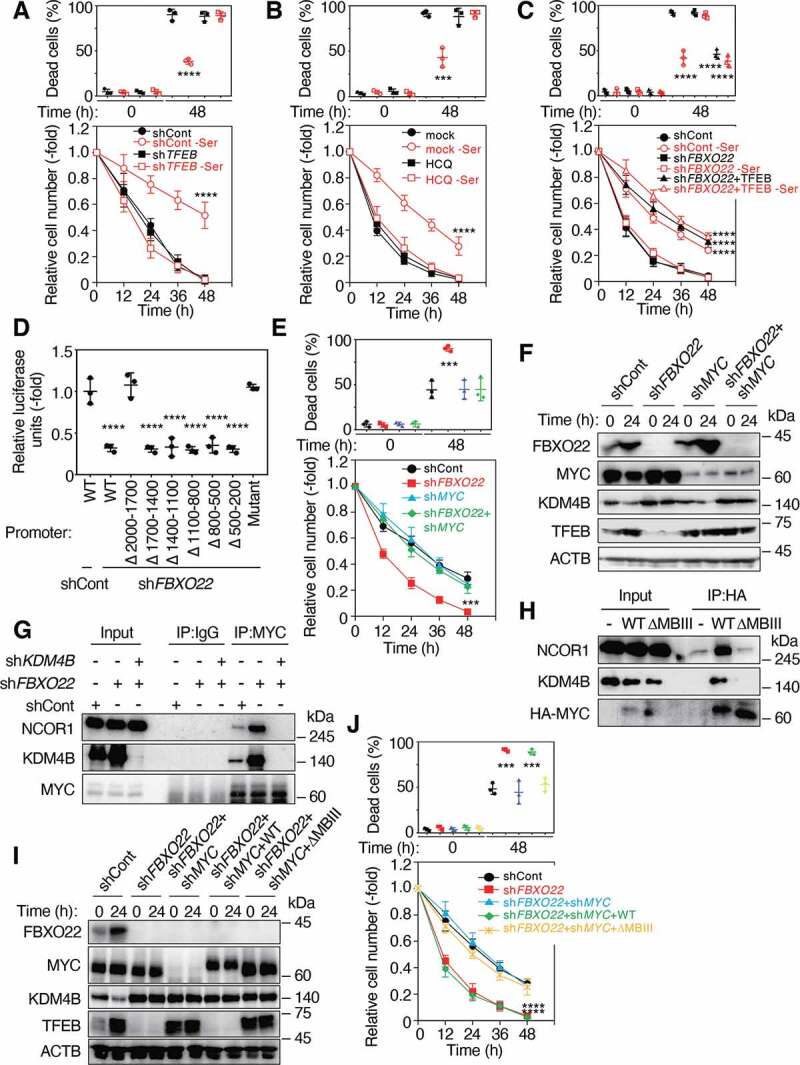
FBXO22 is required for proper TFEB transcription through the inhibition of MYC-KDM4B-NCOR1 transcription suppressor. (A) RPE-1 cells expressing the indicated Dox-inducible shRNA, preconditioned with or without serine depletion and then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times as in Figure 1A. (B) RPE-1 cells were preconditioned as in (A) and then treated with low glucose (2 mM) media in the presence or absence of HCQ (10 μM). The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) are determined at the indicated times. (C) RPE-1 cells expressing the indicated Dox-inducible shRNA and/or TFEB were preconditioned and treated as in (A). The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) are determined at the indicated times. (D) HeLa cells expressing the indicated Dox-inducible shRNAs were transfected with the WT or the indicated deletion mutants of the TFEB promoter fused with the luciferase gene, and treated with doxycycline (1 μg/mL) for 48 h. The lysates were subjected to a luciferase assay. (E) RPE-1 cells expressing the indicated Dox-inducible shRNA, were preconditioned with serine depletion and then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times as in (A). (F) Lysates from RPE-1 cells as in (E) at the indicated times after incubation with low glucose media were subjected to immunoblotting using the indicated antibodies. (G) Lysates from HeLa cells expressing the indicated Dox-inducible shRNAs were immunoprecipitated and then subjected to immunoblotting using the indicated antibodies. (H) Lysates from RPE-1 cells expressing WT or a MYC Box III deletion mutant (ΔMBIII) MYC were immunoprecipitated and then subjected to immunoblotting using the indicated antibodies. (I) Lysates from RPE-1 cells expressing the indicated Dox-inducible shRNAs and/or WT MYC or ΔMBIII at the indicated times after serine depletion were subjected to immunoblotting using the indicated antibodies. (J) Cells as in (I) were preconditioned with serine depletion and then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined. Data are presented as means±s.d. of three independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a control (shCont or mock) (A-E, J). ***P < 0.001, ****P < 0.0001
In order to gain mechanistic insight into FBXO22-mediated regulation of TFEB expression, we identified repressor elements on the TFEB promoter. Reporter analysis using the WT and its truncated TFEB promoters revealed that the repressor element(s) is located between 1700 bp and 2000 bp upstream from the transcription start site (Figure 3D). Given that this upstream region contains a consensus MYC binding motif and MYC was reported to act as a transcriptional repressor as well as activator [26,27], we first examined whether MYC actually binds to this binding motif. ChIP analysis revealed that MYC bound to this region (Fig. S5A). We then found that FBXO22-depletion-dependent suppression of reporter activity was markedly compromised when a mutant promoter in which a consensus MYC response element (−1777 CACGTG −1772) was substituted with CCCGGG was used (Figure 3D). Importantly, co-depletion of MYC in FBXO22-depleted cells effectively recovered the ability of cells to acquire stress resistance upon serine depletion (Figure 3E and S5B) and rescued the reduced expression of TFEB (Figure 3F and S5C), indicating that FBXO22 upregulated TFEB transcription through inhibiting MYC function. Interestingly, the sole depletion of MYC per se induced acquired resistance and increased the level of TFEB without preconditioning in RPE-1 cells and TP53-deficient cancer cells (Figs. S5B-G).
MYC repressor complexes were reported to contain NCOR1 co-repressor [28]. Moreover, KDM4A was also reported to form a complex with NCOR1 [29]. Thus, we speculated that TFEB transcription was suppressed by MYC-KDM4B-NCOR1 repressors because we previously found that SCFFbxo22 preferentially targets KDM4B complexed with transcription factors or their cofactors [24,25]. Formation of endogenous MYC-KDM4B-NCOR1 ternary complex was dramatically facilitated in the absence of FBXO22 (Figure 3G). Complex formation between MYC and NCOR1 appeared to be dependent on the presence of KDM4B, suggesting that KDM4B serves as a scaffold in the complex. In addition, the sole depletion of KDM4B itself induced acquired resistance and increased the level of TFEB without preconditioning in TP53-deficient cancer cells (Figs. S5D-G).Transcriptional repression by MYC was reported to require a conserved motif within MYC Box III (MBIII) [30]. Indeed, KDM4B formed a complex with WT MYC and NCOR1, but failed with a mutant lacking MBIII (ΔMBIII) (Figure 3H). Reintroduction of the WT MYC greatly suppressed TFEB expression in FBXO22 and MYC double-depleted RPE-1 and HCA2 cells (Figure 3I and S5H). Consistent with this observation, reintroduction of the ΔMBIII effectively rescued the ability of cells to acquire stress resistance upon serine depletion in RPE-1 and HCA2 cells (Figure 3J and S5I). Thus, these results indicate that MYC-KDM4B-NCOR1 complex represses TFEB transcription.
SCFFbxo22 ubiquitinates KDM4B in a complex with NCOR1 for degradation
To elucidate the mechanism by which FBXO22 regulates MYC-KDM4B-NCOR1 repressors, we examined the involvement of KDM4B degradation because we previously found that SCFFbxo22 ubiquitinates KDM4B for degradation [24]. The reduced expression of TFEB in FBXO22-depleted cells was rescued by the co-depletion of KDM4B (Figure 4A and S5J), indicating the direct involvement of KDM4B in FBXO22 function. Both catalytic and non-catalytic activities of KDM family proteins in gene regulation have been reported [31]. Thus, we examined whether the role of KDM4B in the suppression of TFEB transcription was dependent on its catalytic activity. Reintroduction of the WT and its catalytic mutant KDM4BH189A in FBXO22 and KDM4B double-depleted cells severely suppressed TFEB expression. In contrast, that of the Tudor domain mutant (D959R) failed. Thus, the results indicate that KDM4B suppresses TFEB transcription independent of its demethylase activity, but in a manner dependent on its Tudor domain. Consistent with this, co-depletion of KDM4B and FBXO22 rescued the ability of cells to acquire hormesis after serine depletion as a preconditioning stressor. Reintroduction of WT and KDM4BH189A but not KDM4BD959R mutant strongly suppressed the hormetic effect in RPE-1 and HCA2 cells (Figure 4B and S5K).
Figure 4.
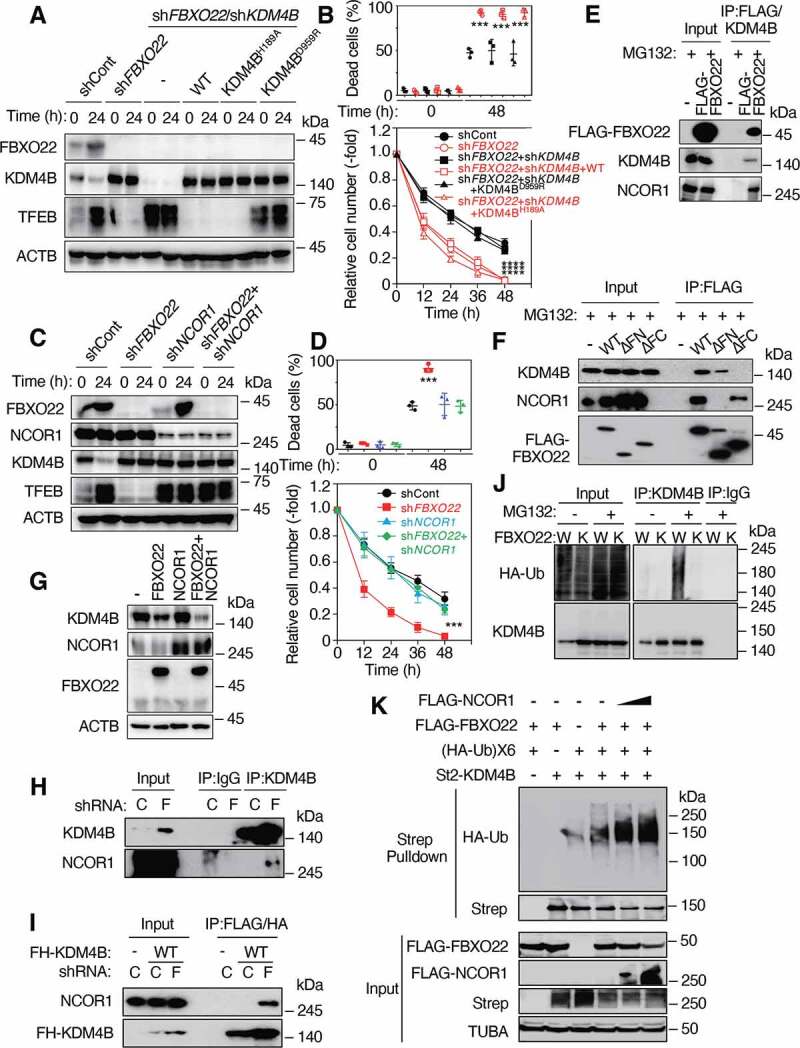
SCFFbxo22 ubiquitinates KDM4B complexed with NCOR1 for degradation, which is critical for induction of TFEB transcription and an acquired stress resistance. (A) Lysates from RPE-1 cells expressing the indicated Dox-inducible shRNA and/or WT KDM4B or its mutants after (24 h) or before (0 h) preconditioning with serine depletion, were subjected to immunoblotting using the indicated antibodies. (B) Cells treated as in (A) were then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times as in Figure 1A. (C) Lysates from RPE-1 cells expressing the indicated Dox-inducible shRNA after (24 h) or before (0 h) preconditioning with serine depletion were subjected to immunoblotting using the indicated antibodies. (D) Cells treated as in (C) were then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times as in (A). (E) Lysates from HeLa cells expressing the indicated genes were sequentially immunoprecipitated with anti-FLAG M2 affinity gel and anti-KDM4B antibodies. The resultant precipitates were subjected to immunoblotting using the indicated antibodies. (F) Lysates from HeLa cells expressing WT FBXO22 or its deletion mutants were immunoprecipitated with an anti-FLAG M2 affinity gel and then subjected to immunoblotting using the indicated antibodies. ΔFC: FIST-C domain deletion, ΔFN: FIST-N deletion (G) Lysates from RPE-1 cells expressing the indicated genes were subjected to immunoblotting using the indicated antibodies. (H) Lysates from HeLa cells expressing the Dox-inducible shControl (C) or shFBXO22 (F) were immunoprecipitated and then subjected to immunoblotting using the indicated antibodies. (I) Lysates from HeLa cells expressing FLAG-HA-KDM4B (FH-KDM4B) with the Dox-inducible shControl or shFBXO22 were sequentially immunoprecipitated using anti-FLAG M2 affinity gel and anti-HA affinity gel. The resultant immunoprecipitates were subjected to immunoblotting. (J) WT (W) or FBXO22−/− (K) HeLa cells were transfected with the HA-Ubiquitin (HA-Ub), treated with or without MG132, lysed under denaturing conditions, and subjected to immunoprecipitation with the indicated antibodies, followed by immunoblotting. (K) FBXO22−/− HeLa cells were transfected with the indicated genes, treated with MG132, lysed under denaturing conditions, and subjected to StrepTactin pulldown, followed by immunoblotting. Data are presented as means±s.d. of three independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a control (shCont, WT, or C) group mean (B, D). ***P < 0.001, ****P < 0.0001
The reduced expression of TFEB and the loss of hormetic response to low glucose media in FBXO22-depleted cells were rescued by co-depletion of NCOR1 (Figure 4C-D and S5L-M). Importantly, NCOR1 depletion per se increased the expression of TFEB and induced the acquisition of a hormetic response independent of preconditioning and FBXO22 status, indicating the epistasis of FBXO22 to NCOR1 and KDM4B in TFEB transcription and hormesis. In the presence of proteasome inhibitor MG132, FBXO22, KDM4B and NCOR1 formed a ternary complex (Figure 4E). Using truncated mutants of FBXO22, we found that KDM4B bound to FBXO22 through the F-box and intracellular signal transduction, C-terminal (FIST-C) domain and NCOR1 through the F-box and intracellular signal transduction, N-terminal (FIST-N) domain (Figure 4F). Intriguingly, ectopic expression of both FBXO22 and NCOR1 synergistically reduced the level of endogenous KDM4B, whereas expression of NCOR1 failed to do so, suggesting that FBXO22 might specifically target KDM4B-NCOR1 complexes (Figure 4G). Indeed, endogenous KDM4B bound to NCOR1 specifically in the absence of FBXO22 (Figure 4H). An interaction between KDM4B and NCOR1 in the absence of FBXO22 was also confirmed when FLAG-HA-tagged KDM4B was overexpressed (Figure 4I). Ubiquitination of endogenous KDM4B was detectable in WT cells, but not FBXO22−/− cells in the presence of MG132 (Figure 4J). To further confirm the possibility that SCFFbxo22 ubiquitinates KDM4B complexed with NCOR1, we performed an in vivo ubiquitination assay with ectopically expressed proteins. Using denatured extracts, an enhanced ubiquitination signal was detected on KDM4B in the presence of FBXO22. This signal was further enhanced by co-expression of NCOR1 (Figure 4K). Thus, the results suggest that FBXO22 preferentially targets KDM4B in the complex with NCOR1 for ubiquitination-dependent degradation.
AKT1-mediated phosphorylation of KDM4B suppress its ubiquitination by SCFFbxo22
We next examined whether mitogen signals could affect TP53-FBXO22-mediated acquired resistance and upregulation of TFEB transcription. Surprisingly, serum starvation further enhanced the survival of cells preconditioned with serine depletion in RPE-1 and HCA2 cells, showing the reduction in the proportion of dead cells (Figure 5A and S6A). Serum starvation per se increased a hormetic response of cells to incubation with low glucose media without preconditioning and the effect of serum starvation on hormesis appeared to be dependent on the presence of FBXO22. Interestingly, serum starvation and serine depletion synergistically reduced and increased the levels of KDM4B and TFEB in RPE-1 and HCA2 cells, respectively (Figure 5B and S6B). As expected, serum starvation resulted in a marked reduction in the phosphorylation of AKT1S1/PRAS40 (AKT1 substrate 1), a known target of AKT1. We then tried to uncover the underlying mechanism. Given that AKT1 is a hub kinase in mitogen signaling [32], we examined the effect of AKT1 inhibitor on acquired stress resistance. In the presence of MK-2206, a specific AKT1 inhibitor, the level of KDM4B was markedly reduced in the control, but not in FBXO22-deleted cells (Figure 5C). The level of p-AKT1S1 was markedly reduced. In addition, the stability of KDM4B was drastically reduced in the presence of the AKT1 inhibitor (Figure 5D). Taken together, the results suggest that AKT1 suppresses SCFFbxo22-mediated ubiquitination of KDM4B. Indeed, a signal of phospho-AKT1 substrate was detected in WT KDM4B immunoprecipitates from cells expressing ERBB2/HER2 (human epidermal growth factor receptor 2) as an AKT1 activator (Figure 5E). A recent comprehensive proteogenomics identified S666 within a consensus AKT1 phosphorylation site as a phosphorylation site of KDM4B [33]. Consistent with this, the signal was not detected in the presence of AKT1 inhibitor or when its KDM4BS666A mutant was immunoprecipiated (Figure 5E). In addition, the signal was detectable in WT, but not KDM4BS666A precipitates, when constitutively active AKT1 (AKT1[CA]) was expressed (Figure 5F). KDM4BS666A and a phospho-mimic KDM4BS666D mutant were less stable and more stable than the WT, respectively (Figure 5G). These results suggest that AKT1 directly phosphorylates KDM4B on S666, which inhibited its FBXO22-mediated ubiquitination and degradation. Immunoprecipitation (IP)-immunoblotting analysis revealed that the WT as well as KDM4BS666A bound to FBXO22 whereas KDM4BS666D failed to do so (Figure 5H). An in vivo ubiquitination assay showed that SCFFbxo22 ubiquitinated KDM4B complexed with NCOR1, which was markedly inhibited by co-expression of WT AKT1 and AKT1[CA] in RPE-1 and HCA2 cells, respectively (Figure 5I and S6C-E). This inhibition was effectively reversed in the presence of AKT1 inhibitor. When AKT1[CA] was expressed, the acquired resistance upon serine depletion (Figure 5J and S6F) and stabilization of TFEB (Figure 5K and S6G) were strongly suppressed without affecting TP53 activation and induction of FBXO22 in RPE-1 and HCA2 cells. Taken together, these results suggest that cancer cells, even those with WT TP53, are defective in terms of a hormetic response to chemotherapy through AKT1-mediated KDM4B phosphorylation because AKT1 signals are highly activated in most of cancer cells [34].
Figure 5.
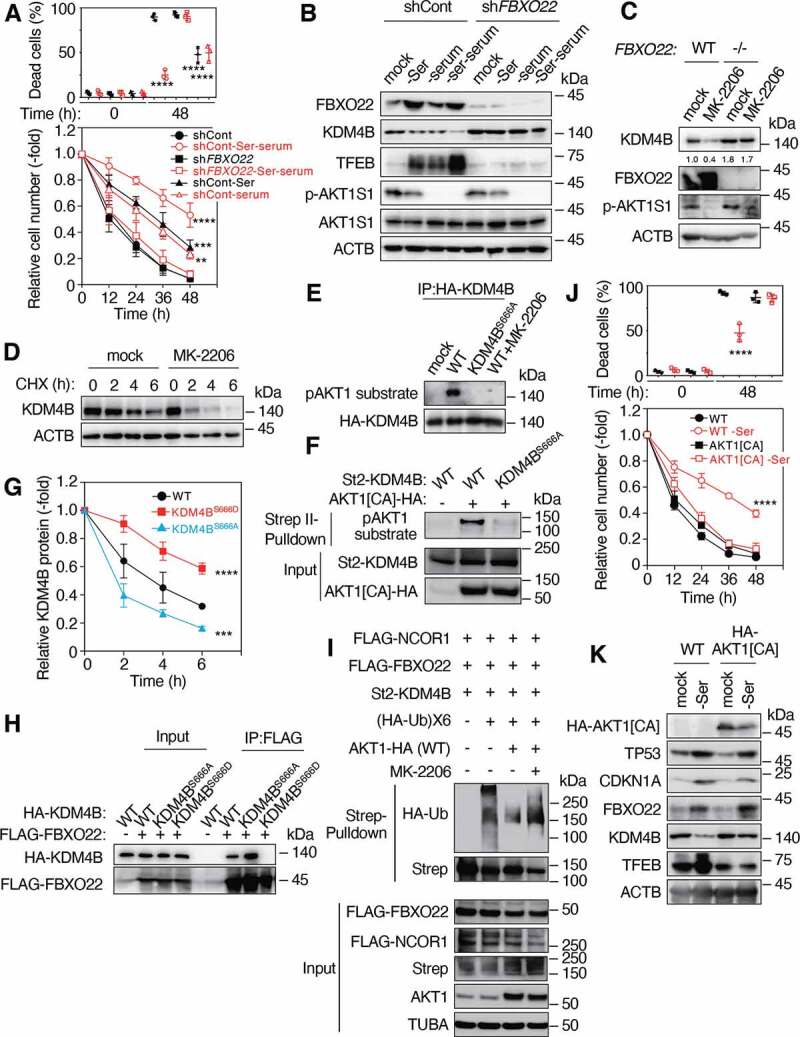
Mitogen-activated AKT1 phosphorylates KDM4B and inhibits its FBXO22-mediated degradation. (A) RPE-1 cells expressing the indicated Dox-inducible shControl or shFBXO22, preconditioned with or without serine depletion plus serum starvation, followed by incubation with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times as in Figure 1A. (B) Lysates from cells as in (A) were subjected to immunoblotting using the indicated antibodies. (C) WT or FBXO22−/− HeLa cells were treated with or without MK-2206 for 4 h and the lysates were subjected to immunoblotting using the indicated antibodies. (D) FBXO22−/− HeLa cells were treated with or without 20 μM MK-2206 together with 50 μg/mL CHX. The lysates were collected at the indicated times and subjected to immunoblotting using the indicated antibodies. (E) Lysates from 293 T cells expressing HA-KDM4B WT or KDM4BS666A treated with or without 20 μM MK-2206 were immunoprecipitated using anti-HA affinity gel. The resultant immunoprecipitates were subjected to immunoblotting. (F) 293 T cells were transfected with or without WT St2-KDM4B or its KDM4BS666A mutant and/or AKT1[CA] lysed under denaturing conditions and subjected to StrepTactin pulldown, followed by immunoblotting. (G) HeLa cells expressing the WT KDM4B or its mutants were treated with 50 μg/mL CHX. The lysates were collected at the indicated times and subjected to immunoblotting, and the relative KDM4B intensities were determined using ImageJ. (H) Lysates from HeLa cells expressing the indicated genes were immunoprecipitated using anti-FLAG M2 affinity gel. The resultant immunoprecipitates were subjected to immunoblotting. (I) FBXO22−/− HeLa cells were transfected with the indicated genes, treated with MG132, lysed under denaturing conditions and subjected to StrepTactin pulldown, followed by immunoblotting. (J) WT RPE-1 cells or cells expressing AKT1[CA], preconditioned with or without serine depletion and then treated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times. (K) Lysates from cells preconditioned as in (J) were subjected to immunoblotting using the indicated antibodies. Data are presented as means±s.d. of three independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a control (shCont or WT) group mean (A, G, J). **P < 0.01, ***P < 0.001, ****P < 0.0001
FBXO22 is necessary and its induction is sufficient for hormesis in chemotherapy in vivo
We then analyzed fbxo22−/− mice to examine whether FBXO22 is essential for hormesis in vivo. We have previously demonstrated that fbxo22−/− mice could survive but were a significantly smaller in size [25]. However, after several generations of backcross with C57BL/6 N to obtain mice with a uniform genetic background, we found that all fbxo22−/− mice died within 10 hours of birth without any obvious developmental and histological abnormalities (Figure 6A-C and S7A), and milk suckling appeared to be normal in fbxo22−/− mice (Figure 6B). These observations are interesting because various mouse models lacking autophagy-related genes showed a similar neonatal lethality [35]. Consistent with this finding, the levels of Lc3, Sqstm1, Wipi1 (WD repeat domain, phosphoinositide interacting 1), and Lamp1 (lysosomal-associated membrane protein 1) as well as Tfeb transcripts were markedly reduced (Figure 6D). A reduction in TFEB, LC3-II, and an increase in KDM4B were also observed (Figure 6E). Very intriguingly, although the level of induced autophagy was comparable between WT and fbxo22−/− mice, the level of basal autophagy in fbxo22−/− mice was significantly lower than that of WT mice (Figure 6F). In addition, the level of L-amino acids in brains from fbxo22−/− mice was lower than that from Fbxo22+/− mice (Figure 6G). These results suggest that FBXO22 is essential for maintaining a basal level of autophagy and that its loss results in neonatal lethality through a reduction in the level of L-amino acids. Importantly, mouse embryonic fibroblasts (MEFs) from fbxo22−/− mice failed to show a hormetic effect after preconditioning with serine depletion (Figure 6H) and other stressors such as UV, butylated hydroxytoluene (BHT), MG132 treatments, and glucose depletion (Figure 6I).
Figure 6.
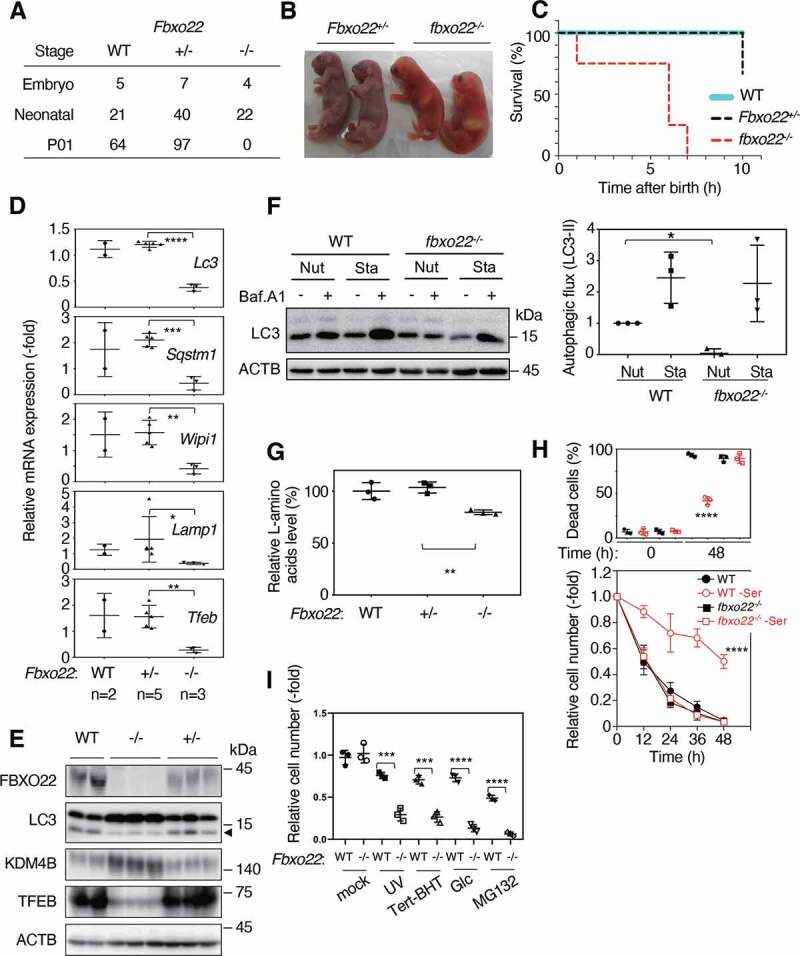
The deletion of FBXO22 in mice results in early neonatal death, accompanied by low basal autophagy levels. (A) The surviving numbers of mice with the indicated genotypes at the embryonic, neonatal, or at postnatal day 1 (P01). (B) Representative images of Fbxo22+/− (or +/-) and fbxo22−/− (or -/-) pups immediately after birth. (C) Survival curves of mice with the indicated genotypes which were nutrient starved immediately after birth. (D) QPCR analysis using total RNA from brains of mice with the indicated genotypes at 8 h after birth using the indicated primers. (E) Lysates from brains as in (C) were subjected to immunoblotting using the indicated antibodies. Arrowhead indicates LC3-II. (F) Autophagic flux of MEFs with the indicated genotypes under nutrient-rich (Nut) or starvation (Sta) conditions (right). The representative images of immunoblotting were shown (left). (G) Lysates from brains as in (C) were assessed for L-amino acid levels. (H) MEFs from WT or fbxo22−/− mice, preconditioned with or without serine depletion, followed by incubation with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) were determined at the indicated times. (I) MEFs from WT or fbxo22−/− mice preconditioned with the indicated stressors, followed by incubation with low glucose (2 mM) media. The relative cell numbers were determined at 48 h as in Figure 1A. Data are presented as means±s.d. of three independent experiments. Unpaired two-tailed Student’s t-test (D, G, I), one-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a WT group mean (F, H). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
We then examined whether increased expression of FBXO22 is sufficient for becoming resilient in vivo. We first generated a Fbxo22 transgenic (Fbxo22-Tg) mice in which the Fbxo22 gene is driven by the CAG promoter. In these mice, a high level of TFEB as well as FBXO22 expression was detected in all tissues tested (Fig. S7B) and any histological abnormalities were not observed (Fig. S7C). MEFs from Fbxo22-Tg mice showed a high level of FBXO22 that was similar to that of WT MEFs depleted of serine (Figure 7A). They also showed a low level of KDM4B and a high level of TFEB independent of serine depletion. Consistent with this, although the levels of induced autophagy were comparable between WT and Fbxo22-Tg mice, the level of basal autophagy was much higher in Fbxo22-Tg mice as compared to WT mice (Figure 7B). MEFs from Fbxo22-Tg mice were highly resilient to low glucose without preconditioning with serine depletion at a level similar to that of WT MEFs with preconditioning (Figure 7C). Both preclinical and clinical studies have shown that fasting can protect a host against toxicity from high-dose chemotherapy [2] although the molecular basis of this phenomenon remains largely unknown. Similarly, short-term fasting in mice prevented lethality from a high dose of etoposide, a treatment that was lethal for mice fed ad libitum [36–39]. The principle underlying these observations is that fasting induces hormesis in normal cells, but not in cancer cells, most of which possess a loss of function due to mutated TP53. Five-week-old WT B6J mice were allowed to feed ad libitum or were fasted for 24 hours. These mice were then injected with etoposide via the tail vein. As reported previously, mice fed ad libitum died at day 6 after injection whereas the fasted mice survived (Figure 7D) [39]. Most intriguingly, 80% of 5-weeks-old non-fasting Fbxo22-Tg mice survived and treatment of these mice with HCQ, an autophagy inhibitor, resulted in the death of all mice at day 6 or day 7. Taken together, these results suggest that a strong expression of FBXO22 upregulates the level of basal autophagy that ensures hormesis in vivo.
Figure 7.
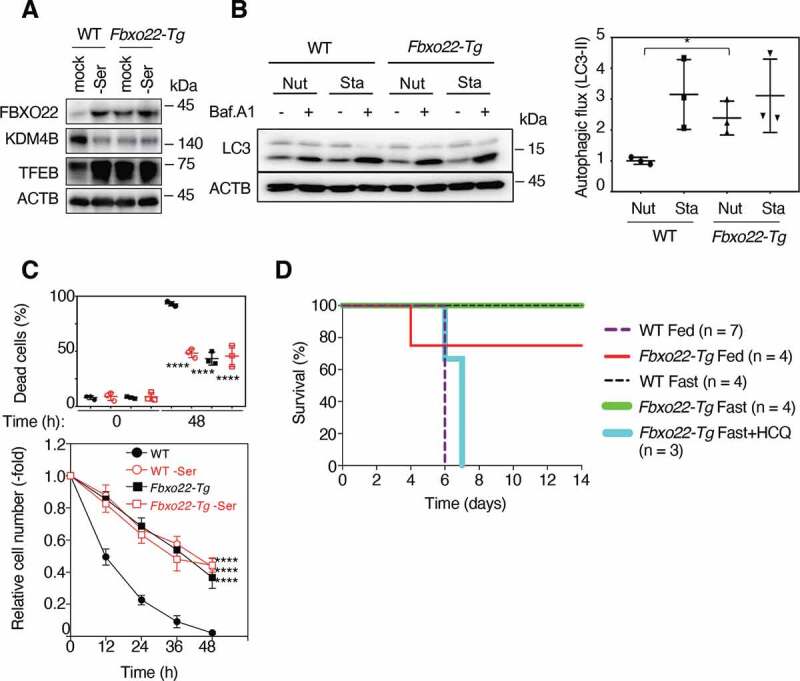
Fbxo22-Tg mice are highly resistance to chemotherapy even without fasting. (A) Lysates from MEFs of WT or Fbxo22-Tg mice, preconditioned with or without serine depletion, were subjected to immunoblotting using the indicated antibodies. (B) Autophagic flux of MEFs from WT or Fbxo22-Tg under nutrient-rich (Nut) or starvation (Sta) conditions (right). The representative images of immunoblotting were shown (left). (C) Cells treated as in (A), were then incubated with low glucose (2 mM) media for 48 h. The proportions of dead cells (upper panel) and the relative cell numbers (lower panel) was determined at the indicated times. (D) WT or Fbxo22-Tg mice were preconditioned under fasting (Fast) or feeding (Fed) conditions for 1 day and then injected with etoposide and/or HCQ. Their survival curves were determined at the indicated times. Data are presented as means±s.d. of three independent experiments. Unpaired two-tailed Student’s t-test (B), one-way ANOVA with Dunnett’s multiple comparisons post hoc test was performed against a WT group mean (C). *P < 0.05, ****P < 0.0001
Discussion
Organisms frequently experience stressful conditions that can threaten their lives. Therefore, they have evolved intricate defense systems such as hormesis to protect themselves against such stressors. This phenomenon has been observed in many organisms including bacteria, plants, and mammals, and is regulated by a variety of mechanisms depending on the species. As applied to human patients, a hormetic approach in disease treatment appears promising. For example, use of fasting and FMDs as preconditioning tools to reduce severe and sometimes fatal side effects of subsequent chemotherapy is currently being evaluated in several clinical trials [2]. Using fasting or FMDs, some studies demonstrated a potential reduction in a variety of side effects as well as in leukocyte DNA damage [3,4]. Therefore, fasting and FMDs have been proposed as promising strategies for reducing side effects of chemotherapy through selectively inducing acquired stress resistance in normal cells and for enhancing the cytotoxicity for cancer cells. In the present study, we provide clues to understanding the mechanisms underlying TP53-mediated regulation of basal autophagy in hormesis through the FBXO22-KDM4B-TFEB axis. The TP53-mediated regulation of basal autophagy is summarized as a schematic (Fig. S7D). The essential role of FBXO22 in hormesis was confirmed using in vivo models, in one, fbxo22−/− mice died as a result of neonatal stress as did autophagy-deficient mice, and in the other, Fbxo22-Tg mice were highly resistance to chemotherapy without fasting as a precondition. This resistance was efficiently reversed by treatment with an autophagy inhibitor. Previously, we reported that SCFFbxo22 preferentially ubiquitinates methylated TP53 complexed with KDM4A for degradation specifically in the senescent state [25], and that KDM4B complexed with estradiol-unbound ER [24], regulating their transcriptional activity. In the latter case, KDM4B degradation determined the antagonistic activity of selective ER modulators (SERMs) for it is required for the release of NCOA1 (nuclear receptor coactivator 1) from SERMs-bound ER. In the current study, SCFFbxo22 ubiquitinates KDM4B complexed with NCOR1, leading to the inhibition of MYC suppressor complexes. The degradation of NCOR1-bound KDM4B also facilitates antagonistic activity of SERMs because transcriptional suppression of ER requires its binding to KDM4B-unbound NCOR1. Taken together, SCFFbxo22 is likely to be a specific regulator of KDM4 family-related transcriptional complexes.
Induced autophagy is known to regulate the response to such cellular stressors as hypoxia, genomic instability, and nutrient depletion [40]. Therefore, induced autophagy per se would comprise a part of hormesis. However, the clinical response to fasting or an FMD as a precondition is different in normal and cancer cells; namely upon preconditioning, normal cells acquire stress resistance whereas cancer cells fail to do so, suggesting the presence of mechanisms that are specific to normal cells. Furthermore, short-term fasting or an FMD might not be sufficient for activation of induced autophagy in vivo. Consistent with this, we found that single amino acid depletion, low glucose, IR, and HS failed to activate induced autophagy (suppression of MTOR activity), although all still induced hormetic effects (Figure 1C-G, 2A, S2A, and S1C-F). Most intriguingly, this hormesis requires functional TP53 in which most cancer cells are defective, which supports the clinical relevance of fasting or FMDs.
TP53 has been implicated previously in activation of autophagy through several mechanisms including transcriptional induction of autophagy-related genes [16]. Such genes include DRAM [17] and AEN (apoptosis enhancing nuclease) [41]. DRAM appears to be essential for apoptosis induction by TP53 and AEN to promote cell death when it is overexpressed, although the molecular basis in both cases is unclear. A more systemic approach using ChIP-seq analysis has identified many autophagy-related genes, such as ULK1 and ATG7, as a TP53-bound gene [16]. Although the activation of TP53 does indeed induce transcription of these genes, it remains unclear whether the induction is due to direct binding of TP53 to their promoters or is an indirect consequence of the activities of other transcription factors. In addition, the role of TP53-mediated autophagy in cellular function under physiological conditions in vivo is largely unknown. In addressing this, the current study identified TFEB, a master modulator of autophagy and lysosomal biogenesis, as an indirect downstream transcription factor of TP53 through activation of the FBXO22-KDM4B axis. The finding that FBXO22-depletion almost completely eliminated the ability to induce lysosomal-related and autophagosomal genes in response to TP53 activation (Figure 2G), suggests that transcriptional induction of autophagy related genes requires activation of the TP53-FBXO22-KDM4B-TFEB axis.
Surprisingly, the amount of TFEB appeared to determine the level of basal autophagy, whose upregulation was essential for hormesis. In the presence of nutrients, TFEB is phosphorylated by MTOR and tethered in cytoplasm [21]. Under conditions of stress, such as nutrient starvation, inhibition of MTOR and concomitant activation of the phosphatase calcineurin induces TFEB dephosphorylation, leading to its nuclear localization. Even under nutrient-rich conditions, an increased amount of phosphorylated TFEB due to the expression of FBXO22 upregulated basal autophagy. This might be achieved by an indirect increase in nuclear TFEB during continuous nuclear-cytoplasmic shuttling. Although basal autophagy is thought to contribute to the routine turnover of cellular wastes, it also determines cellular resistance to various stressors.
In vivo, most normal cells exist in a quiescent state during which mitogen signals are inactivated whereas cancer cells continuously proliferate with ongoing activation of mitogen signaling. AKT1 comprises a hub-node in the network of mitogen signals and is reported to be highly activated in many cancer cells [32,34]. AKT1 phosphorylated KDM4B at serine 666, which in turn inhibited its binding to FBXO22. Therefore, KDM4B appears to be continuously phosphorylated and stabilized in many cancer cells, which ultimately suppresses upregulation of basal autophagy and hormesis independent of the TP53 status. This mechanism is also responsible for the difference in responses to preconditioning between normal and cancer cells.
In conclusion, the current study provides clues to understanding the clinical relevance of fasting or FMDs as a preconditioning therapeutic strategy for reducing severe and fatal side effects resulting from subsequent chemotherapy. Our results clearly demonstrated that single amino acid depletion, HS, or IR as well as low glucose by fasting or by an FMD are also effective for inducing hormesis through activation of TP53. Thus, these treatments could act as alternative forms of preconditioning before a patient undergoes high-dose exposure to chemotherapy or radiotherapy.
Materials and methods
Antibodies
Mouse anti-ACTB (Abcam, 6276), rabbit anti-AKT (Cell Signaling Technology, 9272), rabbit anti-pAKT substrate (Cell Signaling Technology, 100B7E), rabbit anti-DDB2 (Cell Signaling Technology, D4C4), rabbit anti-DRAM (Abcam, ab64739), rabbit anti-FBXO22 (GeneTex, N3C3), mouse anti-FBXO22 (Santa Cruz Biotechnology, FF-7), mouse anti-FLAG (Sigma Aldrich, M2), mouse anti-HA (Boehringer Ingelheim, 12 CA5), rabbit anti-KDM4B (Abcam, 191434), rabbit anti-KDM4B (Cell Signaling Technology, D7E6), rabbit anti-LC3 (Cell Signaling Technology, 2775), rabbit anti-MYC (Santa Cruz Biotechnology, N262), mouse anti-MYC (Abcam, 17355), rabbit anti-MYC (Cell Signaling Technology, D3N8F), rabbit anti-MYC (Cell Signaling Technology, D84 C12), rabbit anti-NCOR1 (Cell Signaling Technology, 5948), rabbit anti-AKT1S1/PRAS40 (Cell Signaling Technology, D23 C7), rabbit anti-p-AKT1S1/PRAS40 (Cell Signaling Technology, C77D7), mouse anti-BBC3/PUMA (Santa Cruz Biotechnology, G3), mouse anti-CDKN1A/p21 (Santa Cruz Biotechnology, F-5), mouse anti-TP53 (Santa Cruz Biotechnology, DO-1), rabbit anti-SQSTM1/p62 (Cell Signaling Technology, D5E2), rabbit anti-SESN2 (Cell Signaling Technology, D1B6), rabbit anti-StrepII (Abnova, PAB16603), rabbit anti-p-T389-RPS6KB/S6K (Cell Signaling Technology, 108D2), rabbit anti-TFEB (Cell Signaling Technology, 4240), rabbit anti-TFEB (Cell Signaling Technology, D2O7D), rabbit anti-TFEB (Proteintech, 13372), rabbit anti-phospho-TFEB (Ser122; Cell Signaling Technology, 86843), rabbit anti-TIGAR (Cell Signaling Technology, D3F4A), rabbit anti-TUBA/tubulin (Cell Signaling Technology, 2144).
Cell culture
Cell cultures, cell cycle synchronization, and treatment with various drugs were performed as described previously [42]. Briefly, early passage HCA2 [42], RPE-1 (ATCC, CRL-4000), HeLa (ATCC, CCL-2), HCT116-TP53−/− (a gift from Dr. Vogelstein, Johns Hopkins Medicine), Saos-2 (ATCC, HTB-85), 293 T (ATCC, ACS-4500) cells, or MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, 08459–35) supplemented with 10% fetal bovine serum (FBS) at 37°C under 5% CO2. For any amino acid deletion or low-glucose experiments, dialyzed FBS (Thermo Fisher Scientific, 16000036) was used. Bafilomycin A1 (Baf.A1; Sigma Aldrich, B1793), cycloheximide (CHX; Sigma Aldrich, C7698), or MG132 (Sigma Aldrich, M8699) was used at a concentration of 125 nM, 50 μg/mL, or 10 μg/mL, respectively, for the specified intervals. For amino acid or glucose starvation, cells were washed with phosphate-buffered saline (Nacalai Tesque, 14249–24), and then incubated with glucose or the indicated amino acid-free DMEM (customized by IFP for this experiment) supplemented with 10% dialyzed FBS (Thermo Fisher Scientific, 16000036). For IR, UV, or HS, cells were treated with either IR or UVC at the indicated doses, or cultured at 42°C under 5% CO2 for 4 h (preconditioning) or more (stress). Viable and dead cell numbers were determined by staining cells with 0.4% trypan blue dye and counting viable cells with a hemocytometer.
Plasmid construction
Lentivirus-based shRNA constructs and Tet-on-inducible lentivirus constructs were generated as described previously [42]. The following lentiviral shRNA target sequences were used with shLuciferase as the control (shCont).
shLuciferase (shCont): CGTACGCGGAATACTTCGA
shDRAM: CCACGATGTATACAAGATA
shDDB2: GAGCGAGATCCGAGTTTAC
shFBXO22: GGAATTGTAGTGACTCCAATG
shKDM4B: GGAAGGACATGGTCAAGAT
shMYC: GATGAGGAAGAAATCGATG
shNCOR1: GCATTCAGCACAGAACAAAGT
shCDKN1A/p21: GAGATGGCAGAGCACCTCCGC
shBBC3/PUMA: CCTGGAGGGTCATGTACAATC
shSESN2: GGTCCACGTGAACTTGCTGC
shTFEB: GAGACGAAGGTTCAACATCAA
shTIGAR: GATTAGCAGCCAGTGTCTTAG
To generate lentivirus-based shRNA constructs, a 19–21 base shRNA-coding fragment with a 5ʹ-ACGTGTGCTGTCCGT-3ʹ loop was introduced into pENTR4-H1 (RIKEN, RDB04395) digested with AgeI/EcoRI. To insert the H1tetOx1-shRNA into the lentivirus vector, we mixed the resulting pENTR4-H1-shRNA vector and CS-RfA-ETBsd (RIKEN, RDB07917) or CS-RfA-ETPuro [42] vector with Gateway LR clonase (Invitrogen, 11791100). To construct Tet-on-inducible lentivirus constructs, the PCR-generated BamHI/NotI fragments containing cDNA for human/mouse FBXO22-WT or the respective mutants, human KDM4B-WT, KDM4BS666A, KDM4BS666D, KDM4BH189A, or KDM4BD959R, human MYC WT or MBIII- deleted [27], or TFEB were inserted into a pENTR-1A vector (Invitrogen, A10462) containing the FLAG, HA, FLAG-HA epitope or EGFP digested with BamHI/NotI. The resultant plasmid was mixed with CS-IV-TRE-RfA-UbC-Puro [42] or CS-IV-TRE-RfA-UbC-Hygro [42] vector, and reacted with Gateway LR clonase to generate the lentivirus plasmid. pcDNA3-(HA-Ub)x6 containing six tandem repeats of HA-tagged Ub was described previously [24]. pcDNA3-St2-KDM4B was generated with KDM4B cDNA subcloned into pcDNA3 (Invitrogen, V79020) with the following oligonucleotide corresponding to two Strep II epitopes with a linker peptide:
5ʹTGGAGCCATCCTCAGTTCGAGAAAGGTGGCGGTTCTGGCGGAGGGTCGGGCGGCTCCGCCTGGAGTCACCCTCAGTTTGAGAAA-3ʹ.
The pcDNA3-HA-KDM4B, FLAG-NCOR1, HA-WT MYC and MBIII-deleted mutant, or pLNCX1-myr-AKT1-delta4-129 (Addgene, 15989) were kindly provided by Dr. Kristian Helin (Sloan Kettering Institute, New York, USA), Dr. Shunsuke Ishii (RIKEN, Tsukuba, Japan), Dr. William P. Tansey (Vanderbilt University, Nashville, USA), and Dr. Richard Roth (Stanford University, Stanford, USA), respectively. The pCS4-HA-AKT1 WT was kindly provided by Dr. Yukiko Goto (University of Tokyo, Bunkyo, Japan) and a constitutive active mutant (AKT1[CA]) was generated by introducing the substitutions of T308 and S473 to aspartic acid.
CRISPR-Cas9-mediated gene KO
A single guide RNA (sgRNA) for human TP53 or CDKN1A/p21 was obtained as oligonucleotides, annealed, and cloned into the dual Cas9 and sgRNA expression vector pX330 (kindly provided by Dr. Feng Zhang, University of Cambridge, Cambridge, United Kingdom) with a BbsI site as reported previously [43]. The plasmid was transfected into RPE-1 cells using Lipofectamine 3000 (Thermo Fisher Scientific, LC3000001) according to the manufacturer’s protocol. After 48-h incubation, the cells were split individually to construct a clonal cell line. The ~500 bp genomic fragments containing the target in the center were PCR-amplified and sequenced to confirm the gene disruption.
SgRNA sequence: TP53 (5ʹ-TCGACGCTAGGATCTGACTG-3ʹ), CDKN1A/p21 (5ʹ-CCGCGACTGTGATGCGCTAA-3ʹ).
Virus generation and infection
Generation of lentiviruses and their infection of cells were performed as described previously [42]. Lentiviruses expressing the respective shRNAs or genes were generated by co-transfection of 293 T cells with pCMV-VSV-G-RSV-RevB (RIKEN, RDB04393), pCAG-HIVgp (RIKEN, RDB04394), and the respective CS-RfA-ETBsd (RIKEN, RDB07917), CS-RfA-ETPuro, CS-IV-TRE-RfA-UbC-Puro, CS-IV-TRE-RfA-UbC-Hygro, or CSII-CMV-MCS (RIKEN, RDB04377) using the calcium phosphate co-precipitation method. Cells infected with the indicated viruses were treated with 10 μg/mL of blasticidin (Thermo Fisher Scientific, A1113903), and/or 2 μg/mL of puromycin (Sigma Aldrich, P8833) for 2–3 days. Doxycycline (Dox; Sigma Aldrich, D9891) was added to the medium at a concentration of 1 μg/mL for inducible expression of the respective shRNAs or genes.
IP and immunoblotting analyses
IP and immunoblotting were performed as described previously [42]. Cells were lysed in Tris buffered saline containing NP-40 (TBSN) buffer (20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 0.5% NP-40 [Thermo Fisher Scientific, FNN0021], 5 mM ethylene glycol-bis[2-aminoethylether]-N,N,N′,N′-tetraacetic acid [EGTA], 1.5 mM ethylenediamine-tetraacetic acid [EDTA], 0.5 mM Na3VO4). The resulting lysates were clarified by centrifugation at 15,000 × g for 20 min at 4°C before IP with the specified antibody. For immunoprecipitation against KDM4B and MYC, nuclear extracts were prepared as reported previously [44]. Cell pellets were suspended in a 5 × packed cell volume of hypotonic buffer A (10 mmol/L 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid [HEPES]-KOH, pH 7.9, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 0.5 mmol/L dithiothreitol [DTT], and 0.5 mmol/L phenylmethylsulfonyl fluoride [Sigma Aldrich, P7626; PSMF]) supplemented with a cocktail of protease inhibitors (Nacalai Tesque, 25955–11) and incubated on ice for 5 min. Cells were then centrifuged at 500 × g for 5 min at 4°C, suspended in a 2 × packed cell volume of buffer A and lysed by Dounce homogenization using a tight-fitting pestle (Wheaton, 357538). Nuclei were collected as a pellet by centrifugation at 4,000 × g for 5 min at 4°C and extracted in an equal volume of buffer C (20 mmol/L HEPES-KOH, pH 7.9, 600 mmol/L KCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, 25% glycerol, 0.5 mmol/L DTT, 0.5 mmol/L PMSF) supplemented with a protease inhibitor cocktail, and mixed on a rotator at 4°C for 30 min. Nuclear extracts (supernatants) were recovered by centrifugation (16,000 × g for 15 min at 4°C) and dialyzed using Slide-A-Lyzer Dialysis Cassettes (Thermo Fisher Scientific, 66330; 3,500-D protein molecular weight) against buffer D (20 mmol/L HEPES-KOH, pH 7.9, 100 mmol/L KCl, 0.2 mmol/L EDTA, 20% glycerol [Avantar, E-550], 0.5 mmol/L DTT, and 0.5 mmol/L PMSF). Dialyzed nuclear extracts were centrifuged (16,000 × g for 30 min at 4°C) to eliminate residual precipitates. For whole lysates, cells were directly lysed with Laemmli-buffer (2% sodium dodecyl sulfate [SDS], 10% glycerol, 5% 2-mercaptoethanol, 0.002% bromophenol blue, 62.5 mM Tris HCl, pH 6.8). The whole lysates (20–50 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene difluoride (PVDF) (Millipore, IPVH00010; Immobilon-P) membrane, and then subjected to immunoblotting with the indicated antibodies using the enhanced chemiluminescence (ECL) detection system. Quantification of bands was performed using ImageJ. Band detection was within the linear range.
RNA-Seq analysis
Total RNA was isolated from HCA2 cells expressing Dox-inducible shControl or shFBXO22 with IR, and RNA integrity was assessed using a 2100 Bioanalyzer (Agilent Technologies). Samples with an RNA integrity number (RIN) greater than 8.0 were further processed for sequencing. Library preparation was carried out with an Ion Total RNA-Seq kit v2 (Thermo Fisher Scientific, 4479789) according to manufacturer’s instructions. The prepared libraries were loaded onto Ion PI chips v2 (Thermo Fisher Scientific, A26770), and single-end sequencing was performed using an Ion Proton System from Thermo Fisher Scientific. The sequencing reads were aligned to the hg19/GRCh37 reference human genome using Bowtie2 (v2.0.0) [45] and STAR (v2.3.0) [46]. Data normalization and differential expression analysis were performed using a DESeq2 package (v1.14.1) in the R language (v3.2.2). The biological significance of the sequencing data was assessed using Gene Set Enrichment Analysis (GSEA) v2-2.3 [47,48]. The C5 collection (GO gene sets) was retrieved from Molecular Signature Database v5.2 (MSigDB). A description of each gene set can be found at the MSigDB website (http://software.broadinstitute.org/gsea/msigdb/).
Quantitative-PCR (qPCR)
QPCR was performed as described previously [42]. Total RNA was extracted using ISOGEN II (FUJIFILM Wako Pure Chemical Corporations, 317–07363) according to the manufacturer’s instructions. For qPCR analysis, cDNAs were synthesized using the SuperScript II cDNA synthesis kit (Invitrogen, 18064). Real-time PCR amplifications were performed in 96-well optical reaction plates with Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659). The relative expression values of each gene were determined by normalization to ACTB/beta-actin expression for each sample. Primer sequences are available upon request.
Luciferase assay
The human TFEB promoter region (2000 bp upstream of the transcription start site) was cloned into pGL4-luciferase reporter vector (Promega, E6651). Its truncated mutants and an MYC response element mutant (from CACGTG to CCCGGG) at the TFEB promoter were constructed by site-directed mutagenesis. HeLa cells were transfected with the pGL4-luciferase reporter plasmid and pCMV-NanoLuc (Promega, N1001) for normalization, and were measured 48 h after transfection following the Dual-Luciferase Reporter Assay System (Promega, E1910) protocol.
ChIP-qPCR
Chromatin immunoprecipitation was performed with a SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling Technology, 9003) according to the manufacturer’s instructions. Briefly, about 4 × 106 cells were fixed with 1% formaldehyde for 10 min at room temperature, followed by quenching with 125 mM glycine. Chromatin was prepared from cell pellets and was digested with micrococcal nuclease for 15 min at room temperature. The digested chromatin was incubated with about 2 μg of anti-MYC antibody (Abcam, ab9106) or normal rabbit IgG (Cell Signaling Technology, 2729) at 4°C overnight, followed by incubation with 20 μL of magnetic beads (Cell Signaling Technology, 9003) at 4°C for 2 h. Beads were thoroughly washed 4 times with the wash buffer, and the chromatin was eluted with the ChIP elution buffer, followed by the reversal of cross-linking with proteinase K at 65°C for 4 h. DNA was then purified on the DNA purification column. Real-time PCR amplifications were performed in 96-well optical reaction plates with Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659). The relative values using anti-MYC antibody were determined by normalization to the control values using normal rabbit IgG for each sample. Primer sequences are available upon request.
Ubiquitination assay
Using previously described methods [24] with some modifications, plasmids were transiently transfected into cells using Lipofectamine 3000. After 48 h, cells were treated for 12 h with 10 μM MG132, and subsequently lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.5% Triton X-100 [Sigma Aldrich, X100]) containing protease inhibitors (Roche, 5056489001) and a deubiquitinase inhibitor (Sigma Aldrich, SML0430). An equal volume of 2× denaturing IP buffer (100 mM Tris-HCl, pH 7.5, 2% SDS, 10 mM DTT) was added to the cell lysates which were then incubated at 100°C for 10 min and centrifuged at 15,000 x g at room temperature for 10 min. The supernatants were diluted with five volumes of lysis buffer and were immunoprecipitated with the indicated antibodies at 4°C followed by immunoblotting. For the detection of in vivo ubiquitinated KDM4B, cells were transfected with the indicated plasmids including 2 x Strep II (WSHPQFEKGGGSGGGSGGSAWSHPQFEK)-tagged KDM4B, treated with 20 μM MG132 for 16 h, and harvested 48 h after the transfection. Cells were lysed under denaturing conditions with 1% SDS-containing buffer, centrifuged, and the supernatant was diluted as described previously [49,50]. Affinity isolation with 10 μL of 50% StrepTactin resin was performed according to the manufacturer’s instructions (Nacalai Tesque, 2–1201-010), using washing buffer containing a high salt concentration (2 M NaCl, 50 mM Tris-HCl, pH 7.5, 0.5% Nonidet P-40 [NP-40; Sigma Aldrich, 21–3277], 150 mM NaCl, 50 mM NaF, 1 mM DTT). The resin was boiled in sample buffer and subjected to immunoblotting.
Autophagy analysis
Cells were plated on 6-well plates at a density of 200,000 cells 1 day before further analysis, washed once with phosphate-buffered saline and cultured in serum-free DMEM media (Nutrients) or Earle’s balanced buffered solution (Starvation) with or without 125 nM Baf.A1 for 2 h. The cells were directly lysed with Laemmli buffer, and the lysates were subjected to immunoblotting using anti-LC3 and anti-ACTB antibodies. The differences in the relative LC3-II intensities (LC3-II:ACTB) between cells treated with or without Baf.A1 were calculated.
Preparation of MEFs
Mouse embryos were dissected from the uterus and extraembryonic membranes at E13.5-E14.5 post-coitus. After the dissection of head and visceral organs for genotyping, embryos were minced into pieces, trypsinized for 15 min at 37°C, and then rinsed with DMEM supplemented with 10% FBS. Cell aggregates were resuspended by pipetting, placed on culture dishes, and maintained in DMEM with 10% FBS for further experiments.
Mouse experiments
All animals were maintained under specific pathogen-free conditions and were handled in accordance with the Guidelines for Animal Experiments of the Institute of Medical Science, the University of Tokyo and the Institutional Laboratory Animal Care and Use Committee of Nagoya City University. Thefbxo22−/−mice were generated as previously reported [25]. Briefly, 4-week-old C57BL/6 N female mice were superovulated and mated with 7-week-old C57BL/6 N males, and fertilized eggs were collected from the oviducts. The pronuclear stage eggs were injected with pX330-mFbxo22-4 plasmid at 5 ng/uL. The eggs were cultivated in KSOM (Merck, MR-121-D) overnight, then transferred into the oviducts of 5-week-old pseudopregnant ICR females. Potential off-target sites were found using optimized CRISPR design software (crispr.mit.edu/). The ~500 bp genomic fragments containing the target or off-target in the center were PCR-amplified and sequenced. Total RNA was extracted from organs using a RNeasy Mini kit (Qiagen, 74104) and subjected to qPCR analysis. A small portion of tissues (approximately 100 mg) was lysed with radioimmunoprecipitation lysis buffer (Thermo Fisher Scientific, 89900) and subjected to immunoblotting or testing with an L-Amino Acid Assay kit (Abcam, ab65347) to measure L-amino acid levels according to the manufacturer’s instructions. For the generation of Fbxo22-Tg,mouseFbxo22open reading frame was subcloned into theEcoRI site of a pCAGGS1 expression vector (kindly provided by Dr. Jyunichi Miyazaki, Osaka University, Ibaraki, Japan). The fragment containing the promoter, open reading frame and poly-adenylation signal was excised withSalI andBamHI, gel-purified, and used as the transgene. C57BL6/N female mice were superovulated and mated with C57BL6/N males, and fertilized eggs were collected from the oviduct. The pronuclear stage eggs were injected with the transgene. The eggs were cultivated in KSOM overnight, then transferred into the oviducts of pseudopregnant ICR females. The founder and transgenic descendants were bred onto a C57BL6/N background for at least five generations. The F6-8 transgenic mice and their littermates were used in experiments. For etoposide treatments, 8-week-old mice were allowed to feed ad libitum or were fasted for 24 h, followed by a 75 mg/kg body weight tail vein injection of etoposide and/or intraperitoneal injection of 60 mg/kg body weight hydroxychloroquine (HCQ; Sigma Aldrich, H0915). After the etoposide injection, moistened food pellets were placed at the bottom of all cages. Survival was monitored daily for 2 weeks post-injection, unless otherwise noted. Ethical endpoints were determined by ~25% body weight loss, low body temperature, and/or impaired mobility.
Histological analysis
The hematoxylin and eosin (H&E) assay was used to analyze the morphological characteristics of the hearts, lungs, kidneys, and intestines in the control WT C57BL/6 J (JAX stock. 0006664), fbxo22−/−, and Fbxo22-Tg. For each group, 2–3 male littermates were chosen for sampling. For the fbxo22−/− mice, neonatal mice (8–10 hours after birth) were sacrificed by hypothermia anesthesia, followed by freezing. The entire body was fixed in 10% formalin. For the Fbxo22-Tg mice, 25–28-week-old mice were sacrificed by avertin anesthesia, followed by cervical dislocation. Each organ was excised and fixed in 10% formalin. In both cases, the samples were embedded in paraffin to create 5 μm sections, stained with H&E, then observed using the Keyence fluorescent microscope (KEYENCE, BZ-9000).
Statistical analysis and reproducibility
Data are presented as means ±s.d. unless otherwise noted. Comparisons between two groups were performed by an unpaired two-tailed Student’s t-test, and analysis of variance (ANOVA) with Dunnett’s multiple comparisons post hoc test. For consistency in comparisons, significance in all figures is denoted as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. For all representative findings, the results of triplicate experiments were similar.
Supplementary Material
Acknowledgments
We thank Dr. M. Pagano (NYU Cancer Institute) and Z.Q. Pan (The Icahn School of Medicine at Mount Sinai) for critical reading of the manuscript, suggestions, and discussion. We are grateful to Dr. M. Komatsu (Juntendo University) for atg7 KO MEFs, Dr. J.L. Guan (University of Cincinnati) for rb1cc1/fip200 KO MEFs, Dr. S. Akira (Osaka University) for atg9 KO MEFs. We also thank Dr. T. Miyamoto (Nagoya City University) for generating fbxo22−/− and Fbxo22-Tg mice. This study was supported by MEXT/JSPS KAKENHI under Grant Numbers JP26250027, JP22118003, and JP16K15239, and by AMED under Grant Numbers JP17cm0106122, JP17fk0310111, and JP17gm5010001, as well as by Ono Medical Research Foundation, Princess Takamatsu Cancer Research Fund, and RELAY FOR LIFE JAPAN CANCER SOCIETY to M.N. Y.J. was supported by MEXT/JSPS KAKENHI JP18H05026m, JP16H06148, and JP16K15238. TO was supported by MEXT/JSPS KAKENHI under Grant Numbers 17H03585 and JP24112005, and by AMED under Grant Number 16ck0106085h0003. WW was by MEXT/JSPS KAKENHI under Grant Number 17K08676.
Funding Statement
This work was supported by the Japan Agency for Medical Research and Development [16ck0106085h0003]; Japan Agency for Medical Research and Development [JP17gm5010001]; Japan Agency for Medical Research and Development [JP17fk0310111]; Japan Agency for Medical Research and Development [JP17cm0106122]; Japan Society for the Promotion of Science [JP16K15239]; Japan Society for the Promotion of Science [17K08676]; Japan Society for the Promotion of Science [JP26250027]; Japan Society for the Promotion of Science [JP22118003]; Japan Society for the Promotion of Science [JP24112005]; Japan Society for the Promotion of Science [17H03585]; Japan Society for the Promotion of Science [JP18H05026m]; Japan Society for the Promotion of Science [JP16H06148]; Japan Society for the Promotion of Science [JP16K15238]; Ono Medical Research Foundation; RELAY FOR LIFE JAPAN CANCER SOCIETY; Princess Takamatsu Cancer Research Fund.
Disclosure statement
All authors declare no conflict of interest.
Data availability
The RNA-seq data from this publication have been deposited in the NCBI GEO database (GSE163178).
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Mattson MP, Calabrese EJ.. Hormesis: What it is and why it matters. in Hormesis: A Revolution in Biology, Toxicology, and Medicine. Humana Press Inc. (Totowa NJ) [Internet]. 2010. [cited 2020 Jan 26];1–13. Available from:http://link.springer.com/ 10.1007/978-1-60761-495-1_1 [DOI] [Google Scholar]
- [2].Nencioni A, Caffa I, Cortellino S, et al. Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer. 2018;18:707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bauersfeld SP, Kessler CS, Wischnewsky M, et al. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: a randomized cross-over pilot study. BMC Cancer. 2018;18:476–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Safdie FM, Dorff T, Quinn D, et al. Fasting and cancer treatment in humans: a case series report. Aging (Albany NY). 2009;1:988–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dorff TB, Groshen S, Garcia A, et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer. [Internet]. 2016. cited 2020 Jan 26;16:360. Available from: http://bmccancer.biomedcentral.com/articles/ 10.1186/s12885-016-2370-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].De Groot S, Vreeswijk MP, Welters MJ, et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: a randomized pilot study. BMC Cancer. [Internet]. 2015. [cited 2020 Jan 26];15:652. Available from: http://bmccancer.biomedcentral.com/articles/ 10.1186/s12885-015-1663-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yoshimori T, Noda T.. Toward unraveling membrane biogenesis in mammalian autophagy. Curr Opin Cell Biol. 2008;20:401–407. [DOI] [PubMed] [Google Scholar]
- [8].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [9].Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol. 2019;35:453–475. [DOI] [PubMed] [Google Scholar]
- [10].Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. [DOI] [PubMed] [Google Scholar]
- [11].Yan X, Zhou R, Ma Z. Autophagy-cell survival and death. Adv Exp Med Biol. 2019;1206:667–696. [DOI] [PubMed] [Google Scholar]
- [12].Wang Y, Zhang H. Regulation of autophagy by mTOR signaling pathway. Adv Exp Med Biol. 2019;1206:67–83. [DOI] [PubMed] [Google Scholar]
- [13].Laptenko O, Prives C. p53: master of life, death, and the epigenome. Genes Dev. 2017;31:955–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. [DOI] [PubMed] [Google Scholar]
- [15].White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6:a026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kenzelmann Broz D, Mello SS, Bieging KT, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. [DOI] [PubMed] [Google Scholar]
- [18].Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. [Internet]. 2008. [cited 2020 Dec 21];10:676–687. Available from: https://pubmed.ncbi.nlm.nih.gov/18454141/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. [DOI] [PubMed] [Google Scholar]
- [20].Slade L, Pulinilkunnil T. The MiTF/TFE family of transcription factors: master regulators of organelle signaling, metabolism, and stress adaptation. Mol Cancer Res. 2017;15:1637–1643. [DOI] [PubMed] [Google Scholar]
- [21].Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018;37:e98804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Z, Wang H, Ding Q, et al. The tumor suppressor p53 regulates autophagosomal and lysosomal biogenesis in lung cancer cells by targeting transcription factor EB. Biomed Pharmacother. [Internet]. 2017. [cited 2020 Dec 22];89:1055–1060. Available from: https://pubmed.ncbi.nlm.nih.gov/28292013/ [DOI] [PubMed] [Google Scholar]
- [23].Tan M-KM, Lim H-J, Harper JW. SCFFBXO22 regulates histone H3 Lysine 9 and 36 Methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol Cell Biol. 2011;31:3687–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Johmura Y, Maeda I, Suzuki N, et al. Fbxo22-mediated KDM4B degradation determines selective estrogen receptor modulator activity in breast cancer. J Clin Invest. 2018;128:5603–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Johmura Y, Sun J, Kitagawa K, et al. SCF Fbxo22-KDM4A targets methylated p53 for degradation and regulates senescence. Nat Commun. 2016;7:10574-10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4:a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008;68:3624–3629. [DOI] [PubMed] [Google Scholar]
- [28].Alland L, Muhle R, Hou H, et al. Role for NCOR1 and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. [DOI] [PubMed] [Google Scholar]
- [29].Zhang D, Yoon H-G, Wong J. JMJD2A is a novel NCOR1-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2). Mol Cell Biol. 2005;25:6404–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Herbst A, Hemann MT, Tworkowski KA, et al. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Rep. 2005;6:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marte BM, Downward J. PKB/AKT1: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–358. [DOI] [PubMed] [Google Scholar]
- [33].Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. [Internet]. 2016. [cited 2020 Apr 1];534:55–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27251275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Alzahrani AS. PI3K/AKT1/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125–132. [DOI] [PubMed] [Google Scholar]
- [35].Kuma A, Komatsu M, Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017;13:1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pietrocola F, Pol J, Vacchelli E, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Caffa I, D’Agostino V, Damonte P, et al. Fasting potentiates the anticancer activity of tyrosine kinase inhibitors by strengthening MAPK signaling inhibition. Oncotarget. 2015;6:11820–11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee C, Raffaghello L, Brandhorst S, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med. 2012;4:124ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Raffaghello L, Lee C, Safdie FM, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. [Internet]. 2008. [cited 2020 Jan 26];105:8215–8220. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18378900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Mol Cell. 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Eby KG, Rosenbluth JM, Mays DJ, et al. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer. 2010;9:95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Johmura Y, Shimada M, Misaki T, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell. 2014;55:73–84. [DOI] [PubMed] [Google Scholar]
- [43].Shimada M, Goshima T, Matsuo H, et al. Essential role of autoactivation circuitry on Aurora B-mediated H2AX-pS121 in mitosis. Nat Commun. 2016;7:12059–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hirokawa T, Shiotani B, Shimada M, et al. CBP-93872 inhibits NBS1-mediated ATR activation, abrogating maintenance of the DNA double-strand break-specific G2 Checkpoint. Cancer Res. 2014;74:3880–3889. [DOI] [PubMed] [Google Scholar]
- [45].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. [DOI] [PubMed] [Google Scholar]
- [49].Sato K, Hayami R, Wu W, et al. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279:30919–30922. [DOI] [PubMed] [Google Scholar]
- [50].Nishikawa H, Ooka S, Sato K, et al. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem. [Internet]. 2004. [cited 2020 Jan 26];279:3916–3924. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1463869 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data from this publication have been deposited in the NCBI GEO database (GSE163178).