

Abstract
As chromatin accessibility data from ATAC-seq experiments continues to expand, there is continuing need for standardized analysis pipelines. Here, we present PEPATAC, an ATAC-seq pipeline that is easily applied to ATAC-seq projects of any size, from one-off experiments to large-scale sequencing projects. PEPATAC leverages unique features of ATAC-seq data to optimize for speed and accuracy, and it provides several unique analytical approaches. Output includes convenient quality control plots, summary statistics, and a variety of generally useful data formats to set the groundwork for subsequent project-specific data analysis. Downstream analysis is simplified by a standard definition format, modularity of components, and metadata APIs in R and Python. It is restartable, fault-tolerant, and can be run on local hardware, using any cluster resource manager, or in provided Linux containers. We also demonstrate the advantage of aligning to the mitochondrial genome serially, which improves the accuracy of alignment statistics and quality control metrics. PEPATAC is a robust and portable first step for any ATAC-seq project. BSD2-licensed code and documentation are available at https://pepatac.databio.org.
INTRODUCTION
Because cells package chromatin differently depending on their function and phenotype, profiling chromatin accessibility is a primary experimental approach for understanding cell states (1–3). The number of chromatin accessibility experiments has grown dramatically in recent years with the introduction of the assay for transposase-accessible chromatin (ATAC-seq) (4). With ATAC-seq now widespread, there is demand for analytical approaches (5,6), including systematic processing pipelines to facilitate the goal of reproducible research and ease cross-study comparisons (7,8).
To address this need we developed PEPATAC, a fast and effective ATAC-seq pipeline that easily generalizes across compute contexts and research environments. This pipeline has been built over years of experience analyzing chromatin accessibility experiments and implements several concepts that make it effective. These include ATAC-specific quality control outputs, both nucleotide-resolution and smoothed signal tracks, and a serial alignment strategy to deal with high mitochondrial contamination. Our serial alignment strategy, or ‘prealignments’, allows the user to configure a series of genomes to align to before the primary genome. PEPATAC provides a framework that allows a user to align serially in customized order to as many genomes as desired, which will be useful for many situations, including species contamination, dual-species experiments, repeat model alignments, decoy contamination, or spike-in controls.
While numerous ATAC-seq pipelines exist (for more in-depth coverage see: 5, 6), PEPATAC is designed with modularity and flexibility as paramount design considerations (Figure 1A). PEPATAC is compatible with the Portable Encapsulated Projects (PEP) format (9), which defines a common project metadata description, allowing projects that use PEPATAC to be easily analyzed using any PEP-compatible tool. It also provides the possibility for a single project description to be shared across pipelines, computing environments and analytical teams. PEPATAC is easily customizable, including changing individual command settings or even swapping specific software components by modifying a few lines of human readable configuration files.
Figure 1.
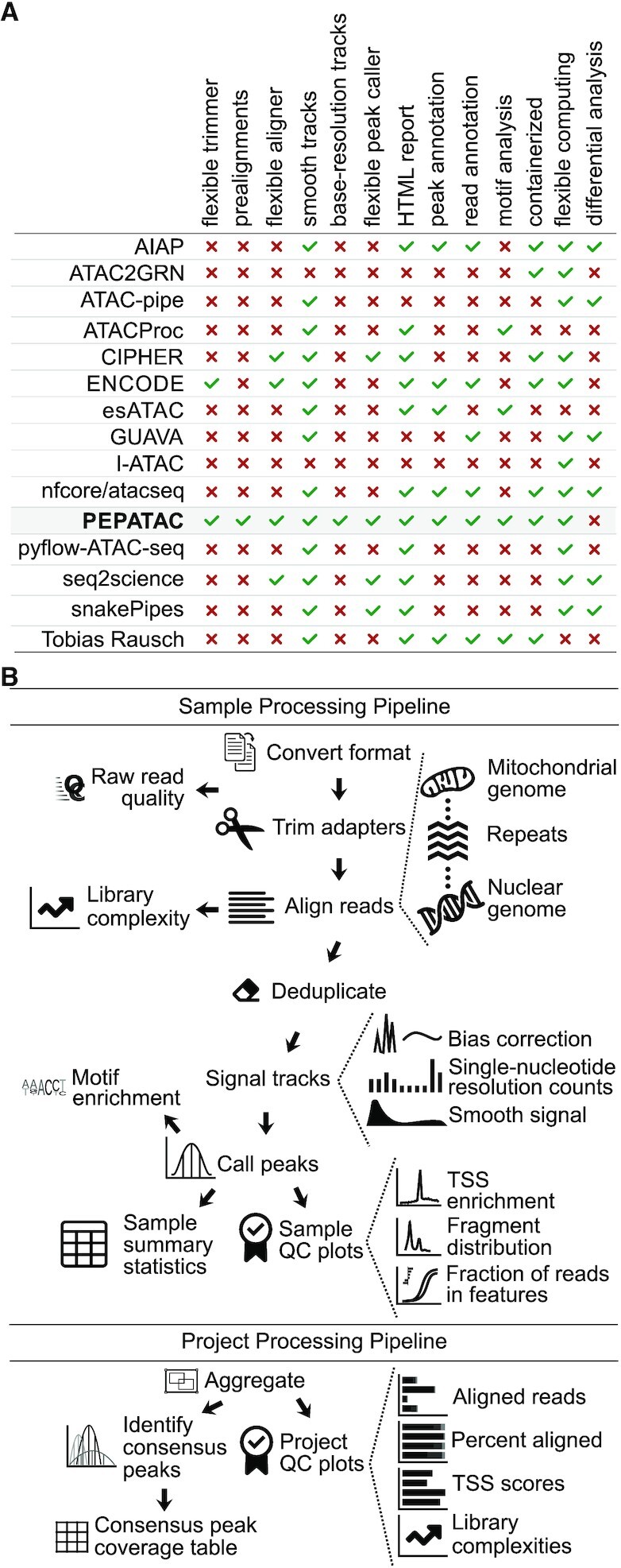
PEPATAC is feature-rich with a logical workflow. (A) We compared features across 14 ATAC-seq pipelines (AIAP (17); ATAC2GRN (18); ATAC-pipe (19); ATACProc (20); CIPHER (21); ENCODE (22); esATAC (23); GUAVA (24); I-ATAC (25); nfcore/atacseq (26); pyflow-ATAC-seq (27); seq2science (28); snakePipes (29); Tobias Rausch (30)) and PEPATAC stands out for being feature-rich . (B) Reads are preprocessed, serially aligned to the mitochondrial genome, curated repeats and then the nuclear genome. PEPATAC generates both smooth and exact signal plots, called peaks, and QC output plots and tables.
PEPATAC does not rely on any specific local or cloud computing infrastructure, and it has already been deployed successfully in various compute environments at multiple research institutes to yield numerous peer-reviewed studies (10–14). While all ATAC-seq pipelines use several common bioinformatic tools (Supplementary Figure S1), we simplify the creation of a computing environment with the required command-line tools using conda (15) or either docker or singularity with the bulker multi-container environment manager (16).
PEPATAC includes a well-documented code base with detailed installation instructions, tutorials, and example projects, so it is useful for both the bench biologist and bioinformatician alike. We anticipate that this pipeline will provide a useful complete analysis for basic ATAC-seq projects and serve as a unified starting point for more advanced ATAC-seq projects.
MATERIALS AND METHODS
PEPATAC configuration
The PEPATAC pipeline is divided into two major parts (Figure 1B). First, it processes each sample individually at the sample level. Once sample processing is complete, the project level part aggregates, analyzes, and summarizes the results across samples. PEPATAC is composed of two primary Python scripts that may be run from the command-line. Sample information and parameters are passed to the pipeline as command-line arguments (see pepatac.py --help), making it simple to use as a standalone pipeline for individual samples without requiring a complete project configuration. Project level output is produced using the project level pipeline (see pepatac_collator.py --help). PEPATAC is built using the Python module pypiper (31), which provides restartability, file integrity protection, copious logging, resource monitoring, and other features. Individual pipeline settings can also be configured using a pipeline configuration file (pepatac.yaml), which enables a user to specify absolute or relative paths to installed software, change adapter input files for trimming, and parameterize alignment and peak calling software tools. This configuration file comes with sensible defaults and will work out-of-the-box for research environments that include required software in the shell PATH, but it also may be configured to fit any computing environment and adapt to project-specific parameterization needs.
Refgenie reference assembly resources
Like any genome analysis, PEPATAC relies on reference genome annotations. To ensure that results are comparable across runs, it’s important to use the same reference assembly. To manage these assets in a reproducible and robust manner, PEPATAC uses refgenie. Refgenie is a reference genome assembly asset manager that simplifies access to pre-indexed genomes and annotations for common assemblies and also allows generating new standard reference genomes or annotations as needed while maintaining asset provenance (33,34). For a complete analysis, PEPATAC requires several refgenie-managed assets: fasta, chrom_sizes, bowtie2_index, blacklist, refgene_tss, and feat_annotation. These can be either downloaded automatically or built manually, which require a genome fasta file, a gene set annotation file from RefGene, and an Ensembl gene and regulatory build annotation file. Using PEPATAC with seqOutBias requires the additional refgenie tallymer_index asset built for the same read length as the data. Many of these assets may also be directly specified at the command line should a user not have refgenie-managed versions available. The TSS annotation file, region blacklist, and feature annotation file may all be specified to use a local, user-specified file. For example, while ENCODE provides a common set of regions that are aberrantly overrepresented in sequencing experiments (e.g. a blacklisted set of regions) (35), a user may create their own version of regions that should be excluded from consideration and point to this file manually.
File inputs and adapter trimming
PEPATAC sequentially trims, aligns, and analyzes sequences (Figure 1B). PEPATAC accepts sequence data input in three formats: unaligned BAM, separated FASTQ, or interleaved FASTQ format. The pipeline first converts the input format into FASTQ (if necessary) for adapter trimming. For adapter trimming, users may select between skewer (36), trimmomatic (37), or an included Python tool using command-line arguments or the PEP configuration file. The pipeline stores quality control results including the number of raw, trimmed, or duplicated reads, and runs FastQC (38) if installed.
Prealignments and mitochondrial DNA
Because ATAC-seq data can have a high proportion of reads mapping to the mitochondrial genome (from 15 to 50% in a typical experiment up to 95% in some experiments (39)), we considered how to optimize the pipeline to deal with abundant mitochondrial DNA (mtDNA). High mtDNA exacerbates the alignment challenge caused by nuclear-mitochondrial DNA (NuMts), which are mtDNA sequences that have integrated into the nuclear genome throughout eukaryotic evolution (40). NuMts represent nonfunctional, truncated, and mutation-ridden copies of mitochondrial protein-coding genes; therefore, we assume that ATAC reads mapping to them are highly likely to be erroneous alignments. The typical strategy is to align to the mitochondrial and nuclear genomes simultaneously, and then remove nuclear-mitochondrial DNA (NuMts) post-hoc using a blacklist, but this suffers from three disadvantages: First, it is inefficient to align lots of mtDNA to the larger nuclear genome; second, reads that match both NuMt and mtDNA will be (incorrectly) split between the two; and third, this approach relies on an accurate pre-constructed annotation of NuMt locations, which may not be available for every reference genome. Furthermore, due to mitochondrial genetic diversity within and across cells, some reads derived from true mtDNA may in fact map better to the reference NuMt than to the reference mtDNA sequence. Also, reads that span the artificial breakpoint in the linear mtDNA reference may find an adequate NuMt match but would never align to the mtDNA.
We found that by separately aligning first to the mitochondrial genome, we alleviated the challenges with simultaneous alignments. To capture NuMts that span the artificial breakpoint induced by converting the circular mitochondrial DNA into a linear representation for alignment, we use a doubled mitochondrial reference sequence, which enables non-circular aligners to align reads that span the breakpoint. By default, the pipeline is configured to align reads first to the doubled mitochondrial reference genome but may be easily configured to perform any number of additional serial alignments.
Alignments, deduplication and library complexity
For prealignments and primary alignment, PEPATAC employs bowtie2 by default (41). Bowtie2 settings are configurable in the pipeline configuration file but come with sensible defaults of -k 1 -D 20 -R 3 -N 1 -L 20 -i S,1,0.50 for prealignments and --very-sensitive -X 2000 for nuclear genome alignment. Users may optionally use bwa (42) with settings similarly configurable in the pipeline configuration file (default: -M). Following alignment, reads with mapping quality scores below 10 and any residual mitochondrial reads are removed and read deduplication is carried out using samblaster (43), but picard’s MarkDuplicates (44) or samtools (45) may also be utilized based on user preference. PEPATAC utilizes preseq (46) to calculate and plot sample library complexity at the current depth and includes the number of independently calculated duplicates (Figure 2A). The pipeline also projects the unique fraction of the library at 10M total reads. These metrics provide an estimate of library complexity and allow the user to determine the value of subsequent sequencing.
Figure 2.

Example PEPATAC QC plots for reads and peaks. (A) Library complexity plots the read count versus externally calculated deduplicated read counts. Red line is library complexity curve for SRR5427743. Dashed line represents a completely unique library. Red diamond is the externally calculated duplicate read count. (B) TSS enrichment quality control plot. (C) Fragment length distribution showing characteristic peaks at mono-, di-, and tri-nucleosomes. (D) Cumulative fraction of reads in annotated genomic features (cFRiF). Inset: Fraction of reads in those features (FRiF). (E) Signal tracks including: nucleotide-resolution and smoothed signal tracks. PEPATAC default peaks are called using the default pipeline settings for MACS2 (32). (F) Distribution of peaks over the genome. (G) Distribution of peaks relative to TSS. (H) Distribution of peaks in annotated genomic partitions. Data from SRR5427743.
Library QC metrics
For quality control, PEPATAC provides a TSS enrichment plot, produced by aggregating reads present in regions 2000 bases upstream and downstream of a reference set of TSSs (Figure 2B). Enrichment is calculated as the average number of reads in a 100 bp window around the TSS divided by the average number of reads in the first 200 bases of the entire region. This yields low signals in the tails with a peak in the center, which we take to be the TSS enrichment score. PEPATAC also produces a fragment length distribution plot (Figure 2C). A standard quality ATAC-seq library is expected to yield clearly defined peaks at open chromatin (<100 bp), mononucleosomes (200 bp), and sequentially smaller peaks representing multi-nucleosomes at regular intervals. To evaluate the enrichment of all reads across genomic partitions, PEPATAC plots both the fraction and cumulative fraction of reads (FRiF and cFRiF, respectively) in genomic features (Figure 2D). A novel feature of PEPATAC includes the plotting of the fraction of reads in any feature type, not solely in peaks. This is plotted as the cumulative sum of reads in each feature divided by the total number of aligned reads against the cumulative sum of bases in each feature. The relative proportion of each feature can be then be directly compared. The standard feature annotation produced and managed by refgenie includes Ensembl defined enhancers, promoters, promoter flanking regions, 5’ UTR, 3’ UTR, exons, and introns in that order. Users can specify an alternative annotation file, either a custom one or simply a different sort order, using the --anno-name pipeline parameter. For a quality sample, the proportion of reads in peaks should be the most enriched, reflecting the specificity of the peak calls for that sample.
Signal tracks and peak calling
Alignments are used to generate two signal tracks: one that records the exact location of transposition events, and one that is smoothed (Figure 2E). These tracks may be used for different downstream analyses; the exact track is useful for analysis that requires nucleotide-resolution, while the smoothed version is often preferred for visualization and peak analysis. Reads, representing transposase cut-sites, are extracted from the deduplicated, low-quality removed, primary genome mapped BAM file into a wiggle-like track. For the exact signal track, these cut-sites are shifted +4 bases for positive strand reads and -5 bases for negative strand reads. For the smooth signal track, we extend the shifted exact sites ±25 bases to yield 50 bp smoothed windows around the exact cut-site position. seqOutBias is an optional tool that can be used to correct for enzymatic (e.g. Tn5 transposase) bias and generate tracks for visualization (47). The bias itself is corrected using a k-mer mask for the plus and minus strand Tn5 recognition sites and by taking the ratio of genome-wide observed read counts to the expected sequence based counts for each k-mer (47). The k-mer counts take into account mappability at a given read length using GenomeTools’ Tallymer program (48).
An earlier study found multiple peak callers worked well with chromatin accessibility data (49), and PEPATAC provides the option to use F-Seq (50), MACS2 (32), Genrich (51), HOMER (52), or HMMRATAC (53) for peak calling, with parameters customizable in the pipeline configuration file. MACS2 is used by default (--shift -75 --extsize 150 --nomodel --call-summits --nolambda --keep-dup all -p 0.01). The default settings are intended to maximize recall and sensitivity. More stringent settings can be easily adopted by modifying the pipeline configuration file. Called peaks are standardized by extending up and down 250 bases (a tunable parameter, --extend) from the summit of each peak to establish peaks 500 bases in width. Any peaks which then extend beyond chromosome boundaries are trimmed. Utilizing fixed-width peaks reduces bias toward larger peaks in both count-based and motif analyses while simultaneously improving the identification of consensus peak sets by reducing the likelihood of extraordinarily large peaks created through the union and merging of multiple peak sets. Finally, peak scores are normalized to score per million by dividing by the sum of scores over 1M.
PEPATAC also produces several plots detailing enrichment of reads in peaks including: the distribution of peaks across the genome by chromosomal location (Figure 2F), the distribution of peaks relative to TSSs (Figure 2G), and the distribution of peaks within genomic partitions (Figure 2H). The TSS distance distribution shows the distance of called peaks with respect to TSSs grouped in log-scale bins. Finally, users may optionally employ HOMER to calculate motif enrichments in called peaks (54).
Running multiple samples with PEPATAC
To run the pipeline across multiple samples in a larger project, the pipeline uses the job submission engine looper (55), which employs the Portable Encapsulated Project standardized definition of project metadata (9) (Supplementary Figure S2). This standard project format enables a pipeline to be run on any project that follows the format, which is simple, standardized, and well-documented. Looper enables the PEPATAC pipeline to be run in any compute environment, including locally (the default) on a single laptop or desktop, or with any cluster resource manager. It also can be used with containers. Additionally, looper’s project format gives pipeline users access to APIs written in Python and R for downstream analysis of pipeline results.
For the user whose environment is set up to run containers, we enable container use with either Docker or Singularity via a single image file or through the multi-container environment manager, bulker (16). Using bulker, PEPATAC may be run in containers across samples and compute environments, simplifying deployment by requiring only bulker and the PEPATAC pipeline itself, eliminating the need to install each required package independently.
Aggregating results from multiple samples
To summarize and incorporate data across samples, the second step in a PEPATAC analysis is to run a project-level pipeline (pepatac_collator.py) that identifies consensus peaks across a project and calculates sample coverage of those consensus peaks in a convenient table for easy downstream analysis. To establish consensus peaks, PEPATAC identifies overlapping (1 bp, a tunable parameter: --min-olap) peaks between every sample in a project and defines the consensus peak’s coordinates based on the overlapping peak with the highest score. Peaks present in at least 2 (parameter: --cutoff) samples with a minimum score per million ≥5 (parameter: --min-score) are retained. A peak count table is then provided where every sample peak set is overlapped against the consensus peak set. Individual peak counts for an overlapping peak are weighted by multiplying by the percent overlap of the sample peak with the consensus peak.
For navigating results, PEPATAC provides both sample and project level reports in a convenient, easy-to-navigate HTML report with project-level summary table and plots, job status page and individual sample pages with sample statistics and QC plots all at your fingertips. In addition, looper will produce summary plots from individual sample statistics including the number of aligned reads, percent aligned reads, TSS scores and library complexities. A user can produce the HTML report during a run or after completion, with the job status page providing information on whether a sample has failed, is still running, or has already completed.
RESULTS
To demonstrate PEPATAC’s default workflow and output, we analyzed samples from the original standard ATAC (4), fast ATAC (56), and omni ATAC (57) protocol papers. This dataset includes human ATAC-seq reads from 33 standard ATAC, 152 fast ATAC, and 139 omni ATAC samples (Supplementary File S1). PEPATAC provides output and quality control results both for individual samples and for the project as a whole. For each sample, PEPATAC produces narrowPeak and bigWig files to visualize nucleotide-resolution alignments, smoothed alignments, and peak calls. PEPATAC also produces summary statistics files that report the number of reads, duplicates, genome alignment rates, transcription start site (TSS) enrichment score, number of called peaks, fraction of reads in peaks (FRiP), and job runtime, among others, for every sample in a project.
Performance
PEPATAC is designed to be computationally efficient. To evaluate how PEPATAC scales with increasing numbers of reads, we ran 430 ATAC-seq samples of varying input size through PEPATAC (Supplementary File S4). We then placed samples in 500 MB input file size bins and compared runtimes and peak memory usage (Supplementary Figure S3). Runtime scales linearly with increasing file size, but importantly, even samples with >150 million reads completed in <8 h (Supplementary Figure S3a). We also show that PEPATAC, with default settings, only utilizes between 5 and 9 GB at peak memory use (Supplementary Figure S3b).
Prealignments
To evaluate the advantage of serially aligning to the mitochondrial genome (Figure 3A), we measured the total alignment runtime of synthetic mixtures of mitochondrial-aligning (mtDNA) and whole human-aligning (hg38) sequences with and without prealignments. We constructed libraries of mixed mtDNA:hg38 mapping ATAC-seq reads from 0% to 100% mtDNA in increments of 10%, at 10 million, 20 million, and up to 200 million total reads in increments of 20 million reads, resulting in 121 different library combinations. We recorded the alignment time for each input file with and without prealignments (Figure 3B). To determine for which scenarios using prealignments is beneficial, we calculated the log ratio of run times with prealignments versus without prealignments and found that using prealignments reduces the total time of alignment even when mtDNA alignment rates are under 10% (Figure 3C). In addition to speed and efficiency gains, PEPATAC with prealignment compared to without prealignment to mtDNA yields higher alignment rates to mitochondrial sequence than aligning to a combined human and mitochondrial genome as is commonly performed (Figure 3D). This is true for every sample tested no matter the library preparation protocol nor percent mitochondrial contamination (Supplementary Figure S4). This result indicates that the common approach of simultaneously aligning to the nuclear and mitochondrial genomes systematically underestimates the fraction of mitochondrial reads in an experiment. We therefore propose that mitochondrial alignment rates are generally underestimated by about 1–5% in published reports.
Figure 3.

PEPATAC prealignments increase mapped mtDNA reads, improve computational efficiency and positively influences the fraction of reads in peaks (FRiP) metric. (A) NuMTs represent a significant complication of simultaneous alignment. (B) At mtDNA percentages from 10 to 100% at total read numbers ranging from 10 to 200 M, using prealignments dramatically reduces run time. (C) Log ratio of prealignments runtimes versus no prealignment runtimes yields significant savings. (D) There is a significant increase in the percent of reads mapped to mitochondrial sequence when using prealignments versus not across standard, fast and omni-ATAC protocols. (E) As reported for ChIP-seq (58), FRiP is positively correlated with the number of called peaks. (F) With prealignments, the positive correlation between FRiP and the number of called peaks tends to increase ((D) **P < 0.001; t-test (mu = 0) with Benjamini–Hochberg correction. (E and F):*P < 0.0001; Kendall rank correlation coefficient).
To show how prealignments successfully depletes reads aligning to NuMTs, we ran a standard ATAC (SRR5427804), fast ATAC (SRR2920492) ,and omni ATAC (SRR5427806) sample through PEPATAC with no prealignments, prealignment to mitochondrial sequence, and prealignment to mitochondrial, ribosomal, and known repeat sequences. We then compared the highest signal peaks between each prealignment strategy across each ATAC-seq protocol. We used BLAST (59) to annotate the highest signal peaks and then intersected called peaks under each strategy with the ENCODE blacklist (35), which normally is used to filter results in PEPATAC by default. The omni ATAC sample had the least number of aberrant high signal peaks with only a single NuMT peak identified in the top 10 highest signal peaks and only present when analyzed without prealignments. Significantly, as soon as mitochondrial prealignment is included, this peak is excluded (Supplementary File S3, Supplementary Figure S5a). Of the top 100 omni ATAC peaks, there are fewer overlaps with blacklisted regions, both overall, and as we increase the number of prealignments. With no prealignments there are 4 blacklisted regions in the top 100 and only 2 with prealignments (Supplementary File S3). As omni ATAC is reported to reduce mitochondrial reads, this result is expected. Furthermore, this difference is highlighted as we compare both fast ATAC and standard ATAC. Three of the top 10 peaks from the fast ATAC sample without prealignments aligned to mitochondrial sequence (Supplementary File S3). These are eliminated with prealignments. Additionally, without prealignments, 22 of the top 100 peaks intersect blacklisted regions. Only 18 overlap with mitochondrial prealignment, and significantly, only 3 of the top 100 overlap blacklisted regions when prealigning includes ribosomal and repeat regions (i.e. satellite DNA). This suggests that a number of regularly identified peaks should typically be excluded in the absence of prealignments. While a blacklist does an excellent job at removing these regions, prealignment achieves similar results while also removing additional non-blacklisted regions that are likely spurious (mapping to unmapped regions or to different species, see Supplementary File S3). These results are even more obvious with standard ATAC. Standard ATAC without prealignment to mitochondria mapped 8 of the top 10 peaks to NuMTs (Supplementary File S3). These are removed with prealignment to mitochondria. Furthermore, the number of blacklisted regions drops from 17 without prealignments to 7 with mitochondrial prealignment and only 2 with mitochondrial, ribosomal and repeat region prealignment. Because prealignment reduces spurious peak assignment (Supplementary File S3 and Supplementary Figure S5b) and it reduces total runtime in nearly every scenario (Figure 3C), prealignment is an effective strategy to include in every pipeline run.
Peak caller comparison
To evaluate the difference in called peaks when using different peak callers, we compared both the PEPATAC determined consensus peaks and the peaks from a single sample (SRR5210416) produced when using different peak callers (Fseq, Genrich, HOMER, HMMRATAC, MACS2 with variable peaks and MACS2 with fixed peaks). Similarity between the intervals was evaluated with a modified Jaccard statistic (60) implemented in the bedtools (61) package. At the single sample level MACS2 with variable peak width is the most similar in output to MACS2 with fixed peaks and Fseq (Supplementary Figure S6a and see Supplementary File S2). Interestingly, the least similar peak results are from Genrich and HMMRATAC, which possibly reflects the goal of both tools being designed to evaluate ATAC-seq data as opposed to originally being developed for ChIP-seq (Supplementary Figure S6a). These differences become more pronounced at the consensus peak level, with HMMRATAC becoming more dissimilar (average jaccard statistic = 0.31, Supplementary File S2) to the other peak callers (Supplementary Figure S6b).
We also asked whether this difference was due to an improvement in reduced peak calling at nuclear mitochondrial sequences (NuMTs), repeat regions, or high signal regions. One way to evaluate this is to determine the number of intersections of the individual peak caller called regions against a known blacklist (35) and to BLAST (59) the highest signal peaks. Indeed, HMMRATAC overlaps the least number of blacklisted regions (231 versus the maximum of 756 with HOMER; see Supplementary File S2) and it turns out a number of both the blacklisted regions and the highest signal peaks are NuMTs or repeat regions (Supplementary File S3). While MACS2 remains the most commonly employed peak caller across ATAC-seq pipelines, further comparative studies may better illustrate the utility of some of the more recently developed peak callers.
Library QC comparison
Several of the QC metrics (e.g. TSS enrichment score, the fragment distributions, non-redundant fractions, and the PCR bottlenecking coefficients 1 and 2) employed by PEPATAC are near-universal in the field, and as such are calculated in the same manner. To evaluate how different annotations may affect the TSS score, we also compared TSS annotations from Ensembl, Gencode, and Refgene (PEPATAC default). Refgene produces higher TSS scores (Supplementary Figure S7), which reflects the fact that Refgene contains only the most commonly employed transcription start sites for each gene whereas both Ensembl and Gencode include all known sites, diluting the aggregated signal.
Fraction of reads in peaks
It has also been reported that in ChIP-seq experiments, but not specifically in ATAC-seq, that FRiP correlates positively with the number of identified peaks (58) (Figure 3E). In libraries with significant mitochondrial contamination, for example, from libraries produced using standard-ATAC library preparation protocols, this correlation is emphasized when using prealignments (Figure 3F). We next sought to understand how the serial alignment strategy affects calculation of Fraction of Reads in Peaks (FRiP). FRiP is a common qualitative measure of enrichment and sample quality. However, FRiP calculations are poorly defined, making it dangerous to compare FRiP scores among different protocols and approaches. ENCODE defines the denominator of the FRiP score to be total mapped reads (ENCODE Terms). If only one genome is used for alignment, then the calculation is clear, but for a serial alignment pipeline, the FRiP score depends on whether the denominator includes reads mapped to the nuclear genome only, or to all genomes (Supplementary Figure S8c,d). By default, PEPATAC uses the deduplicated, low-quality removed, primary genome mapped BAM file to calculate the fraction of reads in the final called peak output file, which by default utilizes fixed width peaks and has removed any blacklisted regions. This has the consequence of changing the FRiP calculation based on whether prealignments were used (Supplementary Figure S8c,d). When using prealignments, the default FRiP calculation will significantly increase because the number of reads mapped to the primary genome is reduced due to reads mapping more accurately to the mitochondrial genome and thus being excluded from downstream analysis. When FRiP is calculated using the total mapped reads (prealignments and primary alignment), these relationships are inversed (Supplementary Figure S8c,d). In any scenario, prealignments lead to more total mapped reads, due to more efficient mitochondrial alignment. As more recent ATAC-seq sample preparation protocols intentionally reduce mitochondrial contamination, these differences are most pronounced when using the original, standard ATAC-seq protocol. Therefore, reliance on a specific cutoff (e.g. 0.3 or greater) as indicative of a quality sample must be relative to protocol and method.
DISCUSSION
PEPATAC is an efficient, user-friendly ATAC-seq pipeline that produces helpful quality control plots and signal tracks that provide a comprehensive starting point for further downstream analysis. Two key benefits of the PEPATAC pipeline over existing pipelines are its flexibility and modularity. PEPATAC is uniquely flexible, for example, by allowing pipeline users to serially align to multiple genomes, to select from multiple aligners, peak callers, and adapter trimmers, while providing a convenient, configurable interface so a user can adjust parameters for individual pipeline tasks. Furthermore, PEPATAC reads projects in PEP format, a standardized, well-described project definition format, providing a reproducible interface with Python and R APIs to simplify downstream analysis.
Because PEPATAC is built on looper, it is easily deployable on any compute infrastructure, including a laptop, a compute cluster, or the cloud. It is thereby inherently expandable from single to multi-sample analyses with both project level and individual sample level quality control reporting. This means that a user may submit any number of samples using a single looper command and corresponding PEP metadata file. Its design allows for simple restarts at any step in the process should the pipeline be interrupted. Due to its modular construction multiple software options for primary pipeline steps are available, creating a swappable pipeline flow path with individual steps adaptable to future changes in the field. PEPATAC is a rapid, flexible and portable ATAC-seq project analysis pipeline providing a standardized foundation for more advanced inquiries.
Documentation and links
PEPATAC v0.9.16: pepatac.databio.org.
PEP metadata standards: pep.databio.org.
Looper job submission engine: looper.databio.org.
Refgenie reference genomes: refgenie.databio.org.
Source code to reproduce output for this paper: github.com/databio/pepatac_paper_data.
DECLARATION
H.Y.C. is a co-founder of Accent Therapeutics and Boundless Bio, a consultant for Arsenal Biosciences and Spring Discovery, and an advisor of 10x Genomics. Stanford University holds a patent on ATAC-seq on which H.Y.C. is a named inventor.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge helpful discussions and contributions from Yuning Wei, Ying Shen and Jason Buenrostro on early versions of the pipeline.
Contributor Information
Jason P Smith, Center for Public Health Genomics, University of Virginia, VA,22908, USA; Department of Biochemistry and Molecular Genetics, University of Virginia, VA 22908 USA.
M Ryan Corces, Center for Personal Dynamic Regulomes, Stanford University, Stanford, CA 94304, USA.
Jin Xu, Center for Personal Dynamic Regulomes, Stanford University, Stanford, CA 94304, USA.
Vincent P Reuter, Genomics and Computational Biology Graduate Group, University of Pennsylvania, PA 19087, USA.
Howard Y Chang, Center for Personal Dynamic Regulomes, Stanford University, Stanford, CA 94304, USA.
Nathan C Sheffield, Center for Public Health Genomics, University of Virginia, VA,22908, USA; Department of Biochemistry and Molecular Genetics, University of Virginia, VA 22908 USA; Department of Public Health Sciences, University of Virginia, VA 22908, USA; Department of Biomedical Engineering, University of Virginia, VA 22908, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
NIH [RM1-HG007735 to H.Y.C., R35-GM128636 to N.C.S.]; Howard Hughes Medical Institute (to H.Y.C.); American Society of Hematology [K99-AG059918 to M.R.C].
Conflict of interest statement. None declared.
REFERENCES
- 1. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. et al. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sheffield N.C., Thurman R.E., Song L., Safi A., Stamatoyannopoulos J.A., Lenhard B., Crawford G.E., Furey T.S. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 2013; 23:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sheffield N., Furey T. Identifying and characterizing regulatory sequences in the human genome with chromatin accessibility assays. Genes. 2012; 3:651–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics. Nat. Methods. 2013; 10:1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan F., Powell D.R., Curtis D.J., Wong N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020; 21:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith J.P., Sheffield N.C. Analytical approaches for ATAC-seq data analysis. Curr. Protoc. Human Genet. 2020; 106:e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins F.S., Tabak L.A. Policy: NIH plans to enhance reproducibility. Nature. 2014; 505:612–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lauer M., Tabak L., Collins F. Opinion: The next generation researchers initiative at NIH. Proc. Natl. Acad. Sci. USA. 2017; 114:11801–11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheffield N.C., Stolarczyk M., Reuter V.P., Rendeiro A. Linking big biomedical datasets to modular analysis with portable encapsulated projects. GigaScience. 2021; 10.1093/gigascience/giab077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W. et al. The chromatin accessibility landscape of primary human cancers. Science (New York, N.Y.). 2018; 362:eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ram-Mohan N., Thair S.A., Litzenburger U.M., Cogill S., Andini N., Yang X., Chang H.Y., Yang S. Integrative profiling of early host chromatin accessibility responses in human neutrophils with sensitive pathogen detection. Life Sci. Alliance. 2020; 4: 10.1101/2020.04.28.066829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR: An integrative and scalable software package for single-cell chromatin accessibility analysis. Nature Genetics. 2020; 53:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou J., Li X., Chen J., Li T., Zhan W., Zhao J., Li M., Yu Z., Yu R., Zou H. et al. CATA: A comprehensive chromatin accessibility database for cancer. 2020; bioRxiv doi:17 May 2020, preprint: not peer reviewed. 10.1101/2020.05.16.099325. [DOI] [PMC free article] [PubMed]
- 14. Fan H., Atiya H., Wang Y., Pisanic T.R., Wang T.-H., Shih I.-M., Foy K.K., Frisbie L., Chandler C., Shen H. et al. Epigenetic reprogramming towards mesenchymal-epithelial transition in ovarian cancer-associated mesenchymal stem cells drives metastasis. Cell Reports. 2020; 33:108473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anaconda software distribution Anaconda Documentation. 2021; https://docs.anaconda.com/. [Google Scholar]
- 16. Sheffield N.C. Bulker: A multi-container environment manager. 2019; OSF Preprints doi:06 October 2019, preprint: not peer reviewed 10.31219/osf.io/natsj. [DOI] [Google Scholar]
- 17. Liu S., Li D., Lyu C., Gontarz P., Miao B., Madden P., Wang T., Zhang B. Improving ATAC-seq data analysis with AIAP, a quality control and integrative analysis package. 2019; bioRxiv doi:28 June 2019, preprint: not peer reviewed 10.1101/686808. [DOI] [PMC free article] [PubMed]
- 18. Pranzatelli T.J., Michael D.G., Chiorini J.A. ATAC2GRN: optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference. BMC Genomics. 2018; 19:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuo Z., Jin Y., Zhang W., Lu Y., Li B., Qu K. ATAC-pipe: general analysis of genome-wide chromatin accessibility, 1934–1943. Bioinformatics. 2019; 20:1934–1943. [DOI] [PubMed] [Google Scholar]
- 20. Sourya Bhattacharyya P.V., Ferhat A. Y. ATACProc - a pipeline for processing ATAC-seq data. 2019; https://github.com/ay-lab/ATACProc.
- 21. Guzman C., D’Orso I. CIPHER: a flexible and extensive workflow platform for integrative next-generation sequencing data analysis and genomic regulatory element prediction. BMC Bioinformatics. 2017; 18:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J. ENCODE ATAC-seq pipeline. 2020; https://github.com/ENCODE-DCC/atac-seq-pipeline.
- 23. Wei Z., Zhang W., Fang H., Li Y., Wang X. esATAC: an easy-to-use systematic pipeline for ATAC-seq data analysis. Bioinformatics (Oxford, England). 2018; 34:2664–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Divate M., Cheung E. GUAVA: A graphical user interface for the analysis and visualization of ATAC-seq data. Front. Genet. 2018; 9:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahmed Z., Ucar D. I-ATAC: Interactive pipeline for the management and pre-processing of ATAC-seq samples. PeerJ. 2017; 5:e4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ewels P.A., Peltzer A., Fillinger S., Alneberg J., Patel H., Wilm A., Garcia M.U., Di Tommaso P., Nahnsen S. Nf-core: community curated bioinformatics pipelines. Nat Biotechnol. 2019; 38:276–278. [DOI] [PubMed] [Google Scholar]
- 27. Tang M. pyflow-ATACseq: a snakemake based ATAC-seq pipeline Zenodo. 2017; https://zenodo.org/record/1043588#.YYyxBbrhWUk.
- 28. Maarten van der Sande J.S., Siebren F. seq2science Zenodo. 2021; https://zenodo.org/record/5579087#.YYyxWbrhWUk.
- 29. Bhardwaj V., Heyne S., Sikora K., Rabbani L., Rauer M., Kilpert F., Richter A.S., Ryan D.P., Manke T. snakePipes: facilitating flexible, scalable and integrative epigenomic analysis. Bioinformatics. 2019; 35:4757–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rausch T., Hsi-Yang Fritz M., Korbel J.O., Benes V. Alfred: interactive multi-sample BAM alignment statistics, feature counting and feature annotation for long-and short-read sequencing. Bioinformatics. 2019; 35:2489–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rendeiro A.F., Stolarczyk M., Reuter V.P., Smith J.P., Klughammer J., Schoenegger A., Sheffield N.C. Pypiper: a python toolkit for building restartable pipelines. 2020; https://github.com/databio/pypiper.
- 32. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stolarczyk M., Reuter V.P., Magee N.E., Sheffield N.C. Refgenie: a reference genome resource manager. Gigascience. 2020; 9:giz149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stolarczyk M., Xue B., Sheffield N.C. Identity and compatibility of reference genome resources. NAR Genom, Bioinform. 2021; 3:lqab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep.-UK. 2019; 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang H., Lei R., Ding S.-W., Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014; 15:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics (Oxford, England). 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andrews S. FastQC: a quality control tool for high throughput sequence data. 2017; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 39. Wu J., Huang B., Chen H., Yin Q., Liu Y., Xiang Y., Zhang B., Liu B., Wang Q., Xia W. et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016; 534:652–657. [DOI] [PubMed] [Google Scholar]
- 40. Lopez J.V., Yuhki N., Masuda R., Modi W., O’Brien S.J. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 1994; 39:174–90. [DOI] [PubMed] [Google Scholar]
- 41. Langmead B., Salzberg S.L. Fast gapped-read alignment with bowtie 2. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H., Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics (Oxford, England). 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Faust G.G., Hall I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics (Oxford, England). 2014; 30:2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Institute B. Picard toolkit. Broad Institute, GitHub Repository. 2019; http://broadinstitute.github.io/picard/. [Google Scholar]
- 45. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., R.D. The sequence alignment/map format and SAMtools. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Daley T., Smith A.D. Modeling genome coverage in single-cell sequencing. Bioinformatics. 2014; 30:3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martins A. fqdedup: remove PCR duplicates from FASTQ files. 2018; https://github.com/guertinlab/fqdedup.
- 48. Kurtz S., Narechania A., Stein J.C., Ware D. A new method to compute k-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics. 2008; 9:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koohy H., Down T.A., Spivakov M., Hubbard T. A comparison of peak callers used for DNase-seq data. PLoS One. 2014; 9:e96303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boyle A.P., Guinney J., Crawford G.E., Furey T.S. F-seq: a feature density estimator for high-throughput sequence tags. Bioinformatics (Oxford, England). 2008; 24:2537–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gaspar J.M. Genrich: Detecting sites of genomic enrichment. 2018; https://github.com/jsh58/Genrich.
- 52. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tarbell E.D., Liu T. HMMRATAC: a hidden markov ModeleR for ATAC-seq. 2019; 47:e91–e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell. 2010; 38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stolarczyk M., Reuter V.P., Rendeiro A.F., Smith J.P., Gu A., Sheffield N.C. Looper: a python-based pipeline submission engine and project manager. GitHub repository. 2020; http://looper.databio.org. [Google Scholar]
- 56. Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016; 48:1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Sci. Rep.-UK. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012; 22:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 60. Favorov A., Mularoni L., Cope L.M., Medvedeva Y., Mironov A.A., Makeev V.J., Wheelan S.J. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012; 8:e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinlan A.R. BEDTools: The swiss-army tool for genome feature analysis: BEDTools: the swiss-army tool for genome feature analysis. 2014; 47:11.12.1–11.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.