

Abstract
Osteosarcoma is a highly aggressive malignancy for which treatment has remained essentially unchanged for years. Our previous studies found that the F-box protein SKP2 is overexpressed in osteosarcoma, acting as a protooncogene; p27Kip1 (p27) is an inhibitor of cyclin-dependent kinases and a downstream substrate of SKP2-mediated ubiquitination. Overexpression of SKP2 and underexpression of p27 are common characteristics of cancer cells. The SCFSKP2 E3 ligase ubiquitinates Thr187-phosphorylated p27 for proteasome degradation, which can be abolished by a Thr187Ala knock-in (p27T187A KI) mutation. RB1 and TP53 are two major tumor suppressors commonly coinactivated in osteosarcoma. We generated a mouse model with a double knockout (DKO) of Rb1 and Trp53 within cells of the osteoblastic lineage, which developed osteosarcoma with full penetrance. When p27T187A KI mice were crossed on to the DKO background, p27T187A protein was found to accumulate in osteosarcoma tumor tissues. Furthermore, p27T187A promoted apoptosis in DKO tumors, slowed disease progression, and significantly prolonged overall survival. RNA sequencing analysis also linked the SCFSKP2–p27T187A axis to potentially reduced cancer stemness. Given that RB1 and TP53 loss or coinactivation is common in human osteosarcoma, our study suggests that inhibiting the SKP2–p27 axis may represent a desirable therapeutic strategy for this cancer.
Keywords: p27, phosphorylation, accumulation, SCFSKP2 inhibitors, transgenic mouse, osteosarcoma
Introduction
Osteosarcoma (OS) is the most common primary bone malignancy in children and adolescents, with a high propensity for local growth and distant dissemination. Despite comprehensive treatment including multiagent cytotoxic chemotherapy and aggressive oncologic surgery, the long-term survival of patients with localized disease remains around 70%.1 Patients who present with overt metastatic disease or who relapse attain a 5-year survival rate of less than 30%, illustrating the inadequacy of the existing treatment approach.2 Unfortunately, efforts for over three decades to modify or intensify the current three-drug regimen have failed to improve outcomes.3 Newer strategies, including immunotherapeutic approaches, have also been disappointing to date.4 At present, the treatment options for patients with either metastatic disease at presentation or for patients who relapse are extremely limited. Historically, targeted therapy for OS has been met with skepticism, in part, owing to the tumor’s high level of genomic heterogeneity and instability, coupled with its lack of a defining fusion product or gene mutation.5 However, recent in-depth next-generation sequencing studies have suggested that OS tumors share certain genetic alterations and can be classified according to their genomic landscape. While a single solution is unlikely, this finding offers new hope for a targeted, genome-informed approach.6 Given that RB1 and TP53 are the two most common inactivated tumor suppressors in OS, it is likely that the loss of function of these genes is central to the development and perseverance of OS and that the loss of RB1 and TP53 supervision is a critical failure of the antitumor mechanism in this cancer.
The F-box protein SKP2, a downstream repression target of pRB, is commonly overexpressed in OS.7 SKP2 overexpression has been correlated with a decreased p27 level, caused by increased ubiquitination and degradation.7–11 The SKP1–Cullin-1–F-box containing the SKP2 (SCFSKP2) ubiquitin complex can ubiquitinate the cyclin-dependent kinase (CDK) inhibitors p27 and p21 for proteasome degradation and, in turn, promote cell cycle progression.10,12 In the context of Rb1 and Trp53 coinactivation, a deletion of Skp2 has revealed that p27 serves a protective role by inhibiting cell division and blocking tumorigenesis in mouse models of prostate and small cell lung cancer.13,14
In OS, however, the precise mechanisms by which SKP2 promotes tumorigenesis are not fully understood. One of the most target-specific steps in SKP2-mediated ubiquitination is the interaction between SCFSKP2 and p27.15 Specifically, the phosphorylation of p27 at position Thr187 (T187) by CDK triggers p27 ubiquitination by SCFSKP2 (see Refs. 10 and 12). Replacing T187 with alanine (p27T187A) renders this position unphosphorylated and prevents SCFSKP2 from binding and ubiquitinating p27.16,17 Meanwhile, this replacement does not affect other intrinsic functions of p27. Zhao et al. recently found that p27T187A prevents Rb1-inactivated pituitary tumorigenesis.18 In addition, Rb1/Trp53 coinactivation is also a cellular context in which SKP2-mediated degradation of p27 promotes prostate tumorigenesis.15 In OS, much work is still required to elucidate the cellular context and the precise mechanisms by which SKP2 interacts with p27 to promote tumorigenesis.
OS has been characterized as a stem cell malignancy, containing a subpopulation of tumor-initiating cells that are associated with chemoresistance and clinical relapse.19,20 Therefore, targeting the stem-like features of OS may improve the response to cytotoxic chemotherapy and reduce the risk of recurrent disease. Work from our group recently showed that SKP2 acts to maintain the mesenchymal state and tumor-initiating properties of synovial sarcoma.21 Furthermore, SKP2 inhibitors have been found to restrict cancer stemness and potentiate the sensitivity to chemotherapy in other tumor models.22 At present, the involvement of the SKP2–p27 axis in OS tumorigenesis and stemness is still largely unknown.
In this study, we generated a genetically engineered mouse model (GEMM) with a double knockout (DKO) of Rb1 and Trp53 within cells of the osteoblastic lineage by using Osterix1-Cre (Osx1-Cre;Rb1lox/lox;p53lox/lox). This DKO model, first described by Walkley and colleagues,23 develops OS tumors with full penetrance and recapitulates many defining features of human OS. To test the hypothesis that SKP2-mediated degradation of p27 promotes OS tumorigenesis, we generated a new mouse strain called DKOAA (Osx1-Cre;Rb1lox/lox;Trp53lox/lox;p27T187A/T187A) by crossing the p27T187A knock-in (KI) mutation into the DKO background. The DKOAA genotype results in disruption of the interaction between SKP2 and p27T187A. The effects of p27T187A stabilization on cellular proliferation and apoptosis were assessed both in vitro and in vivo. Finally, an RNA sequencing (RNA-seq) analysis was performed to examine the transcriptomic difference between DKO and DKOAA tumors and corresponding pathway alterations.
Materials and methods
Gene expression analysis using online databases of human OS
Patient data were obtained from the cBioPortal analysis and visualization platform (https://www.cbioportal.org/). Raw expression data of 79 OS patients were retrieved from the MSK-IMPACT Clinical Sequencing Cohort (MSKCC, 2017) and the Pediatric Preclinical Testing Consortium (Maris, 2019).24,25 The tendency for the co-occurrence of paired genes was analyzed by the log odds ratio. The significance of co-occurrence tendency was analyzed by the Fisher exact test. P < 0.05 was considered statistically significant.
Animal models
Osx1-Cre mice were purchased from the Jackson Laboratory (006361) (Bar Harbor, ME). Rb1lox/lox mice,26 Trp53lox/lox mice,27 and p27T187A/T187A mice were described previously.15,28 All mice used for experiments are on the FVB, C57BL/6J, and 129Sv hybrid backgrounds. First, Rb1lox/lox mice were crossed with Trp53lox/lox mice to generate Trp53;lox/lox Rb1,lox/lox mice, which were then crossed with Osx1-Cre mice to generate Osx1-Cre; Trp53,lox/lox Rb1lox/lox mice. The p27T187A mice were crossed with the Osx1-Cre; Trp53;lox/lox Rb1;lox/lox mice to generate Osxl-Cre; Rb1lox/lox Trp5;3lox/lox p27T187A/T187A mice. The genotyping results of each crossing step are shown in Figure S2 (online only), and the correct genotypes were selected in a stepwise manner. Using RNA-seq, the deletion of exons 2–10 in Trp53, the deletion of exon 3 in Rb1, and the missense mutation of T187A in p27 (Cdkn1b) were all confirmed in our transgenic model (Fig. S4, online only). Animals were maintained under pathogen-free conditions in the Albert Einstein College of Medicine animal facility. Animal experimental protocols were reviewed and approved by the Albert Einstein Animal Care and Use Committee (#20180401), conforming to accepted standards of humane animal care. Tumor diameter was measured using a calliper every 3 days, and the relative tumor volume was calculated by the following formula: (length × width2) × 0.526. Mice were allocated into two groups based on their genotype and anatomic location of the tumors. The growth of jaw and limb tumors was assessed when the tumor volume reached approximately 400 and 700 mm3, respectively.
Immunohistochemistry, immunofluorescence, and TUNEL assay
Tissue sections and staining have been described previously.15 For immunohistochemistry (IHC) staining, the following antibodies were used: Ki67 (Vector Labs, SP6), p27 (BD Biosciences, #610242), cleaved caspase-3 (Cell Signaling Technology, #9661), anti-alkaline phosphatase (ALP) (Abcam, ab354), and anti-osteocalcin (Abcam, ab13420). TUNEL staining was performed with the Apoptosis Detection Kit (Millipore, S7100). Images were visualized with the EVOS® FL Auto Imaging System (AMC1000, ThermoFisher). For staining quantification, pictures were taken under 60× magnification. Approximately 300 cells in each image and three images from each animal were counted. Data are presented as means of three animals in each group.
Cell culture, cell proliferation, sphere formation, and extreme limiting dilution analysis
Primary OS cells were prepared from 0.3 cm × 0.3 cm tumor tissues, which were minced and dissociated with collagenase II in EMEM for 30–60 min at 37 °C. All cells were cultured at 37 °C in a humidified incubator with 5% CO2. Proliferation and sphere formation assays were performed as previously described.7,21 C1 and flavokawain A (FKA) were prepared as previously described.21 C25 was purchased from Millipore-Sigma (#5063050001), and pevonedistat was from MedChemExpress (#HY-70062). Cells were plated overnight and treated with each compound for the indicated duration and concentration. Extreme limiting dilution analysis (ELDA) was performed and analyzed based on the protocol from Hu et al.29 Cells were harvested, counted, seeded in ultra-low attachment 96-well plates at decreasing densities (at 20, 10, 5, and 1 cells/well, 24 duplicates per density), and cultured for 12 days. Each well was monitored for signs of spheroid formation from day 3, with the criterion of containing one or more spheres greater than~50 μm diameter.
Western blot analysis, cycloheximide and MG132 treatments, and qRT-PCR
Western blot, cycloheximide (CHX), MG132, and qRT-PCR experiments were performed as described.7 Antibodies for western blots included p27 (BD Bioscience, #610242), cleaved caspase-3 (Cell Signaling Technology, #9661), Skp2 (Protein-tech, #15010–1-AP), p21 (Santa Cruz, #SC-397), and α-tubulin (Cell Signaling Technology, #2125). PCR primers are shown in Table S2 (online only).
Flow cytometry for apoptosis and ALDH activity assay
The Annexin V-APC/7-AAD Apoptosis Kit (Abnova, #KA3808) was used for the apoptosis analysis by flow cytometry. Cells were harvested, counted, and washed in the binding buffer. Cells were then stained with annexin V-APC and 7-ADD, and analyzed by a flow cytometer (BD Biosciences, San Jose, CA) as recommended by the manufacturer. Results were analyzed by the FlowJo software (version 10.1, Ashland, OR). ALDH assays were performed as described previously.21 All experiments were performed in triplicate.
RNA sequencing
Mouse tumor tissues from DKO or DKOAA animals were harvested and processed for RNA-seq. Approximately 100 mg of fresh tumor tissue was collected and processed using the RNeasy Mini Kit (#74104 Qiagen) for RNA extraction following the manufacturer’s protocol. Samples were first quantified and then evaluated for RNA integrity, degradation, and contamination. Next, the rRNA-depleted RNA sample was enriched using oligo(dT) beads and randomly fragmented. This was then followed by cDNA synthesis using random hexamers and reverse transcriptase. After first-strand synthesis, a custom second-strand synthesis buffer (Illumina) was added with dNTPs, RNase H, and E. coli polymerase I to generate the second strand by nick translation. The final cDNA library was completed following a round of purification, terminal repair, A-tailing, ligation of sequencing adapters, size selection, and PCR enrichment. The library concentration was quantified using a Qubit 2.0 fluorometer (Life Technologies). The insert size was checked by an Agilent 2100 Bioanalyzer and then further quantified by qPCR. Libraries were then sequenced using an Illumina HiSeq platform. Raw data were processed by the Illumina pipeline, and the resulting RNA-seq reads were mapped to the mouse reference genome (mm10) using the STAR software (v2.6.1).30 Downstream analysis was performed using R (v3.6.3). Exploratory data analysis revealed intragenotype variance driven by some genes highly expressed in only one of the three (DKO) or four (DKOAA) replicates. To limit this, we excluded extreme outlier genes with 5× higher expression in one sample than all the other replicates in each genotype, many of which were noncoding. Read counts for the filtered protein-coding genes were then used for differential expression analysis by the DESeq2 R package (v1.26.0),31 with adjusted P (padj) <0.05 for statistical significance. Genes ranked by –log10 (P values) multiplied by the sign of the log2(fold-change) computed by DESeq2 were used for gene set enrichment analysis using the FGSEA package (1.12.0). Enrichment plots were made with the GSEA Java app in PreRanked mode (v4.1.0).
Statistical analyses
Survival curves were estimated by the Kaplan–Meier method and compared between groups by log-rank tests. Differences between groups were assessed using the Student’s t-test for continuous variables and the Fisher’s exact test for categorical variables. Differences in proliferation, viability, and drug inhibition assays were analyzed by two-way ANOVA. All statistical analyses were performed using SPSS version 22 (IBM, Armonk, NY). All tests were two-sided. P values of 0.05 or less were considered statistically significant.
Results
The co-occurrence of RB1 and TP53 inactivation is statistically significant in human OS
To determine the frequency of RB1 and TP53 coinactivation in human OS, we queried these tumor suppressors’ genetic status in accessible databases from cBioPortal (Table 1). Two datasets were identified, with available information from 79 OS patients. The rate of inactivation of RB1 and TP53 is 14.89% and 40.42%, respectively, in the MSKCC dataset and 37.56% and 68.75%, respectively, in the Pediatric Preclinical Testing Consortium dataset. To determine the tendency for pairwise co-occurrence of genetic alterations, the statuses of five additional OS drivers (PTEN, ATRX, NF1, FLCN, and MYC) were queried. Overall, the inactivation of RB1 and TP53 was the only pair of genetic alterations that co-occurred with statistical significance in both datasets (P = 0.011 and P = 0.012). The alterations of RB1 and TP53 occur by deletion, followed by gene fusion and missense mutation (Fig. S1, online only). When a literature review was performed and studies with RB1 and TP53 expression data in OS were included, the rate of RB1 and TP53 coinactivation was approximately 20–64%, suggesting that the loss of function of these tumor suppressors is a common event in OS (Table S1, online only).
Table 1.
Pairwise co-occurrence relationships among RB1, TP53, PTEN, NF1, ATRX, FLCN, and MYC in osteosarcoma in two cBioPortal datasets
| cBioPortal dataset |
||
|---|---|---|
| Msk_impact_2017 | Maris_2019 | |
| Number of specimens | 47 | 32 |
| RB1 inactivation (R) | 14.89% | 37.56% |
| TP53 inactivation (T) | 40.42% | 68.75% |
| PTEN inactivation (P) | 0% | 12.50% |
| NF1 inactivation (N) | 6.38% | 9.38% |
| ATRX inactivation (A) | 12.77% | 9.38% |
| FLCN activation (F) | 12.77% | 6.25% |
| MYC activation (M) | 4.26% | 6.25% |
| Co-occurrence (log odds ratio, statistical significance) | RT (0.051, 0.011) | RT (0.208, 0.012) |
| RN (0.489, 0.916) | RP (0.802, 0.041) | |
| RA (0.954, 0.765) | RN (0.580, 0.015) | |
| RF (0.054, 0.765) | RM (0.237, 0.005) | |
| FM (0.892, 0.267) | TP (0.153, <0.001) | |
| TN (0.528, <0.001) TM (0.201, <0.001) NA (1.290, 0.504) |
NM (1.146, 0.823) | |
p27T187A KI delays the progression of OS tumorigenesis
DKO tumorigenesis is invasive, metastatic, and lethal within 20–35 weeks. DKOAA mice were fertile without evidence of delayed or stunted growth. DKO and DKOAA mice developed OS tumors with full penetrance at an average of 154.65 ± 29.08 and 169 ± 43.69 days, respectively (P = 0.052; Fig. 1A). The histological features of primary tumors (Fig. 1A) and metastatic lung nodules (Fig. S3A, online only) from each genotype were determined by IHC staining for ALP and osteocalcin. Using Kaplan–Meier analysis, the overall survival of the DKOAA mice was significantly longer than that of the DKO mice (P < 0.0001) (Fig. 1B). The anatomic distribution of DKO and DKOAA tumors was very similar, including the jaw (42.9% versus 40.7%), the limb (38.8% versus 39%), the spine and sacrum (8.2% versus 10.2%), and the trunk (10.2% versus 10.2%) (Fig. S3D, online only). To examine whether the anatomic site affects survival, we compared the survival of DKO and DKOAA cohorts separately for animals with jaw and limb tumors. We found that DKOAA animals survived longer than DKO animals, regardless of whether tumors were located in the jaw (P = 0.0014) or the limb (P < 0.001) (Fig. S3B and C, online only).
Figure 1.
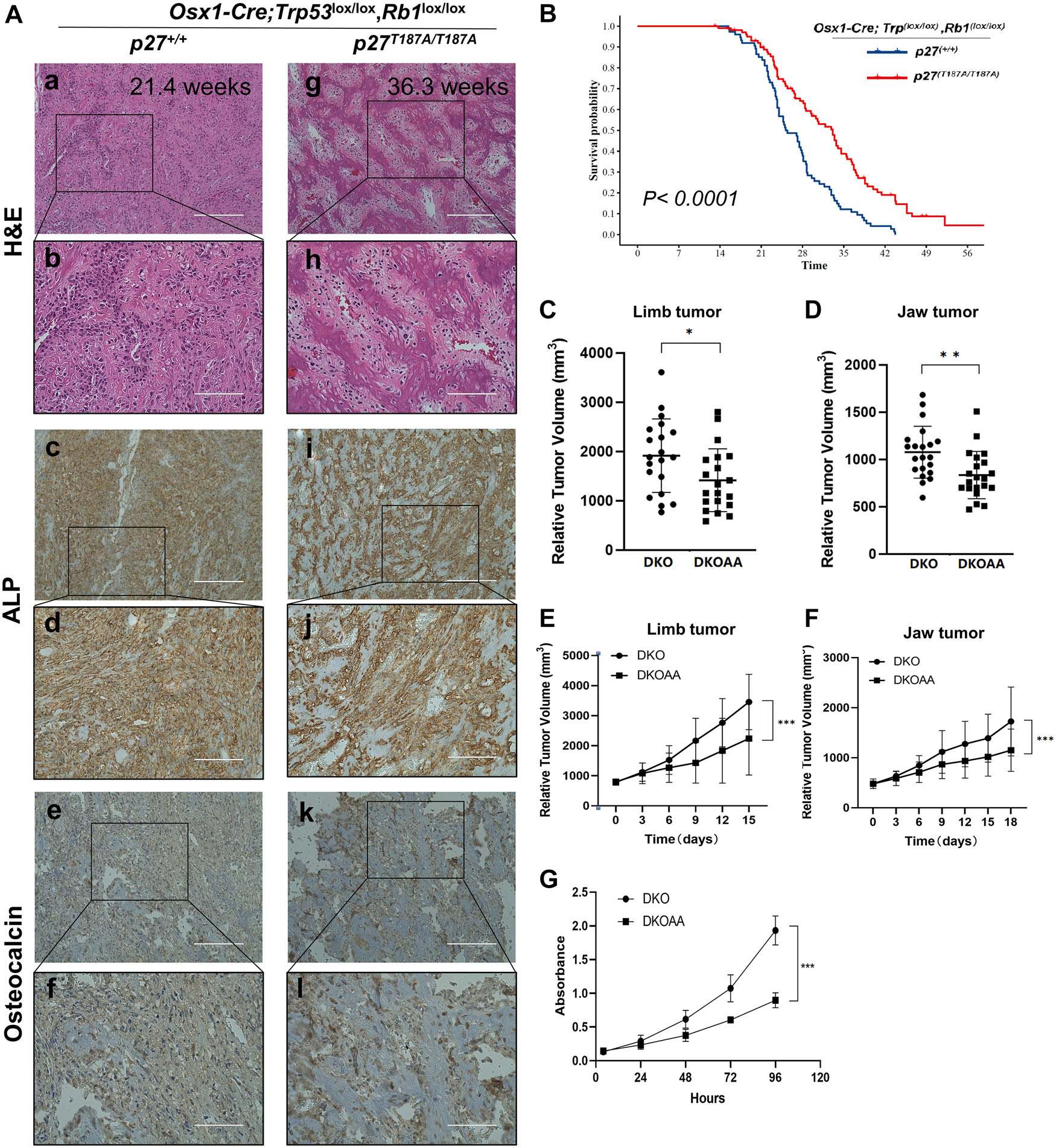
The progression of DKO osteosarcoma tumorigenesis is inhibited in p27T187A/T187A mice. (A) Representative H&E, alkaline phosphatase (ALP), and osteocalcin staining of OS tissue of the indicated genotype and age. Scale bars = 200 μ m (a, c, e, g, i, and k) and 100 μ m (b, d, f, h, j, and l). (B) Kaplan–Meier survival analysis comparing the DKO and DKOAA cohorts of mice undergoing tumorigenesis. P value is by a log-rank test. Mice at the same age with measurable limb tumors (n = 20 per group) (C) and jaw tumors (n = 21 per group) (D) were randomly selected from each group, and tumor volume was measured and compared among two groups at the same age (P = 0.039 and P = 0.003). Mice with limb (E) (n = 8 per group) or jaw (F) (n = 8 per group) tumors were randomly selected from each group once the tumor reached a similar volume, and tumor measurement was carried out every 3 days. The tumor growth rate was plotted and compared among the two groups (P < 0.001 and P < 0.001). (G) Graphs showing the proliferation of DKO and DKOAA tumor cells in monolayer cultures. Error bars are SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
In vivo tumor growth was also compared between the DKO and DKOAA groups and analyzed separately by anatomic sites (jaw and limb). Mice with measurable jaw tumors (n = 20) and limb tumors (n = 21) were randomly selected from each group. Tumor volume was measured in animals of the same age and compared between groups Tumors from the DKO group were significantly larger compared with those from the DKOAA group in both the limb (1915.01 ± 728.62 mm3 versus 1316.14 ± 623.16 mm3, P = 0.039) (Fig. 1C) and jaw (1096.86 ± 267.04 mm3 versus 804.11 ± 203.36 mm3, P = 0.003) (Fig. 1D). In addition, the growth rate of DKOAA tumors was significantly slower than that of the DKO cohort (P < 0.0001), regardless of the limb and jaw anatomic site (Fig. 1E and F). In vitro, DKOAA primary culture cells proliferated significantly less than DKO cells (Fig. 1G), consistent with the in vivo results. These findings collectively support the notion that p27T187A KI delays the progression and proliferation of DKO tumors and significantly extends the survival of DKO mice.
p27T187A accumulates in DKO tumors
Previously, p27T187A KI has been shown to stabilize p27 in a transgenic prostate cancer model.15 To determine whether p27T187A leads to an accumulation of p27 in Rb1/Trp53 double-knockout OS, IHC staining was used, and it showed that p27T187A levels were markedly elevated in DKOAA compared with those in DKO tumors (Fig. 2A). Similarly, western blots of tumor lysates from three individual animals showed elevated p27 protein levels, with similar SKP2 expression (Fig. 2B). To assess p27 protein stability, we treated early passage cells from DKO and DKOAA tumors with the protein synthesis inhibitor CHX and the proteasome inhibitor MG132. After MG132 treatment, p27T187A protein levels were upregulated significantly in DKO (6.22-fold) compared with a more moderate change in DKOAA (1.38-fold), suggesting that p27 in DKOAA has been stabilized previously and was less affected by proteasome degradation compared with that in DKO (Fig. 2C). After CHX treatment, p27 protein levels decreased markedly in DKO in the time-course analysis compared with a modest change in DKOAA, suggesting that p27 protein levels in DKOAA are more durable after protein synthesis inhibition than DKO (Fig. 2D). Together, the MG132 and CHX treatment experiments showed that the stability of p27T187A protein was enhanced in DKOAA over DKO cells. Quantitative RT-PCR experiments demonstrated no significant difference in p27 mRNA levels between DKO and DKOAA cells (Fig. 2E), suggesting that p27T187A accumulation in DKOAA tumors was not due to increased transcription.
Figure 2.
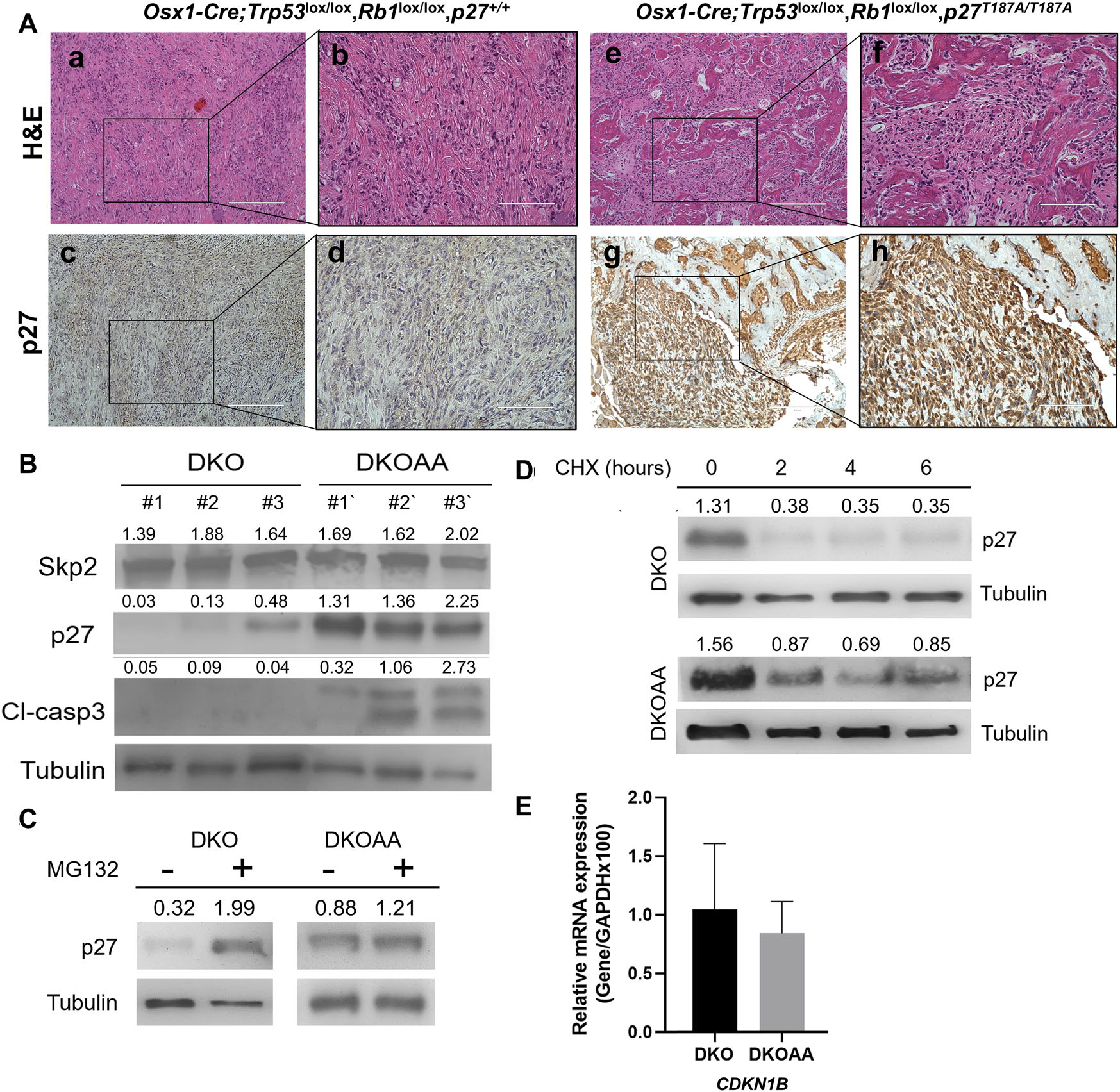
p27T187A/T187A mice accumulate p27T187A protein in DKO osteosarcoma. (A) Representative photographs (10 mice of each genotype) of H&E and p27 immunostaining of tissue sections of each genotype as marked. Scale bars = 200 μ m (a, c, e,and g) and 100 μ m (b, d, f, and h). (B) Protein levels were determined by western blotting of OS tissues from the indicated genotypes. (C and D) DKO and DKOAA cells were treated with MG132 and CHX, and western blots were used to determine p27 protein stability compared with α-tubulin. (E) DKO and DKOAA cells were subject to qRT-PCR to determine levels of p27 mRNA relative to GAPDH. Error bars indicate SEM of the means of three samples. CDKN1B is the official gene name for p27.
p27T187A promotes apoptosis in DKO tumors
As previously reported, both genetic deletion and pharmacological inhibition of SKP2 led to elevated levels of p27 and a concomitant increase in apoptosis.7 To assess whether p27T187A contributes to apoptosis, IHC analyses were performed in DKO and DKOAA tumors. TUNEL and cleaved caspase-3 immunostaining suggested that DKOAA tumors underwent increased apoptosis compared with DKO tumors (Fig. 3A), which was also confirmed by western blotting that showed a variable increase of cleaved caspase-3 (Fig. 2B). The quantification of TUNEL-positive and -negative staining showed that the DKOAA cohort exhibited increased apoptosis (P < 0.001) (Fig. 3B). Furthermore, flow cytometry using annexin V/7-AAD staining showed a significant increase in the early apoptotic population (P = 0.023) (Fig. 3C). Both DKO and DKOAA tumors were further analyzed by RNA-seq. To better compare molecular pathway changes in the two groups of tumors, we carried out gene set enrichment analysis (GSEA) using the Molecular Signatures Database hallmark gene set collection.32 This demonstrated that genes involved in cell cycling and immune response were significantly enriched among the genes expressed higher in DKOAA, while gene sets associated with myogenesis and oxidative phosphorylation were more active in the DKO (Fig. 3D). One example was that E2F target genes were found enriched after p27T187A stabilization in the DKOAA group. As an important transcription regulation factor, E2F1 is responsible for the transcription initiation of many apoptosis-related genes and was found to be activated in our previous transgenic model of p27 stabilization.15 We further examined genes involved in apoptosis and found that they were more active in the DKOAA in an apoptosis-related gene set (Fig. 3E). Additionally, we found that genes associated with cell cycle arrest and the G2/M checkpoint were significantly upregulated in DKOAA (Fig. 3F). Together, these data suggest that p27T187A promotes a proapoptotic and less proliferative environment in OS tumors in the absence of p53 and pRb.
Figure 3.
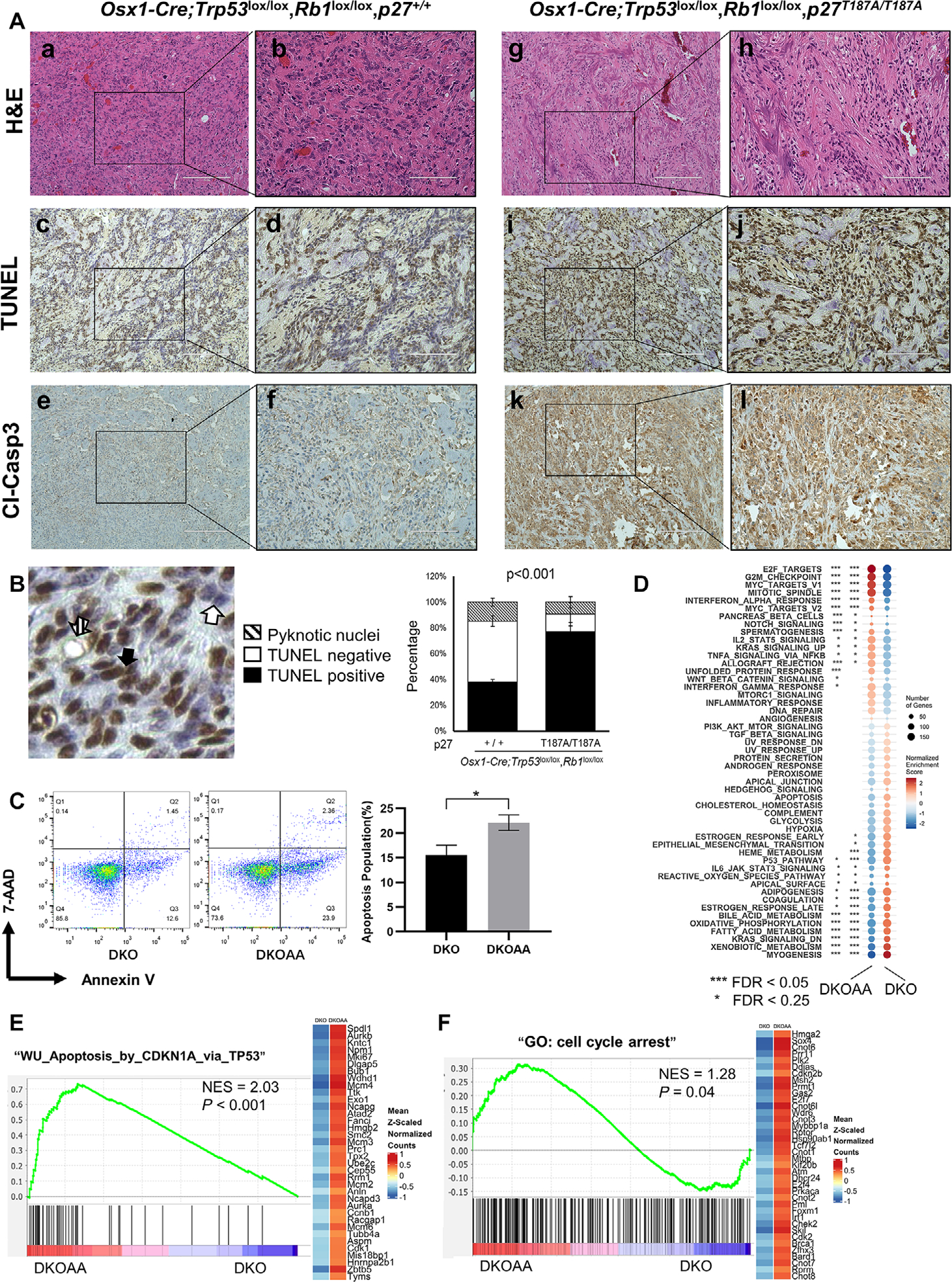
p27T187A increases apoptosis in DKO osteosarcoma. (A) Consecutive OS tumor sections of each genotype, as indicated, stained with H&E, TUNEL, and cleaved caspase-3. Scale bars = 200 μ m (a, c, e, g, i, and k) and 100 μ m (b, d, f, h, j, and l). (B) Quantification of apoptosis by TUNEL staining. Arrows demonstrate how total apoptosis was quantified. The bar graph was based on the average quantification of apoptosis from three mice. (C) 7-AAD and annexin V staining of tumor cells of the indicated genotypes. Flow cytometry was used to detect and quantify apoptosis in the sub-G1 population. Error bars are SEM *P < 0.05. (D) GSEA for the hallmark gene sets. Circle sizes indicate gene numbers, while orange and blue colors represent enrichment or depletion of a set, respectively, in the DKOAA (left) or DKO (right) group. (E) GSEA plot showing enrichment of apoptosis genes in DKOAA. The y-axis is the enrichment score, and the lines in the x-axis indicate genes ranked by their expression difference between DKOAA and DKO. The right heatmap displays average expression of the leading edge genes in DKO (left) and DKOAA (right), with orange for high and blue for low expression and cleaved caspase. NES, normalized enrichment score. (F) GSEA plot showing the enrichment of cell cycle arrest genes in DKOAA.
p27T187A reduces tumor-initiating properties in DKO tumors
Differential expression analysis with RNA-seq comparing DKOAA versus DKO tumors found that 376 and 498 genes were up- and downregulated, respectively, at a multiple test-adjusted P < 0.05. Aldh3a1 and multiple genes in the Hox loci were among the most significant ones (Fig. 4A). The altered genes were enriched for functions related to cell cycling, DNA damage repair, development, and various metabolic processes, including oxidative phosphorylation (Fig. S5, online only). Interestingly, when we applied GSEA to a previously annotated set of stemness genes, we found that they were enriched among the downregulated genes in DKOAA (Fig. 4B). Among them were known OS stemness markers, including KIT, ALDH7A1, ALDH1A1, ALDH2, and CD133. This was further validated with more RNA samples from independent DKO and DKOAA tumors by quantitative RT-PCR (Fig. 4C). In order to determine whether overexpressed stemness markers correlated with a stemness phenotype, an ALDH activity assay was performed and revealed a much lower portion of cells expressing a high level of ALDH (ALDHBR) in DKOAA (P < 0.001; Fig. 4E). DKOAA tumor cells also exhibited less sphere-forming capacity (P = 0.009; Fig. 4D). Furthermore, ELDA demonstrated lower stem cell frequency and less self-renewal ability (P < 0.001) of DKOAA compared with DKO cells (Fig. 4F). Overall, these data indicate that p27T187A reduces the tumor-initiating properties of OS and provides insights into the role of the SKP2–p27 axis in cancer stemness.
Figure 4.
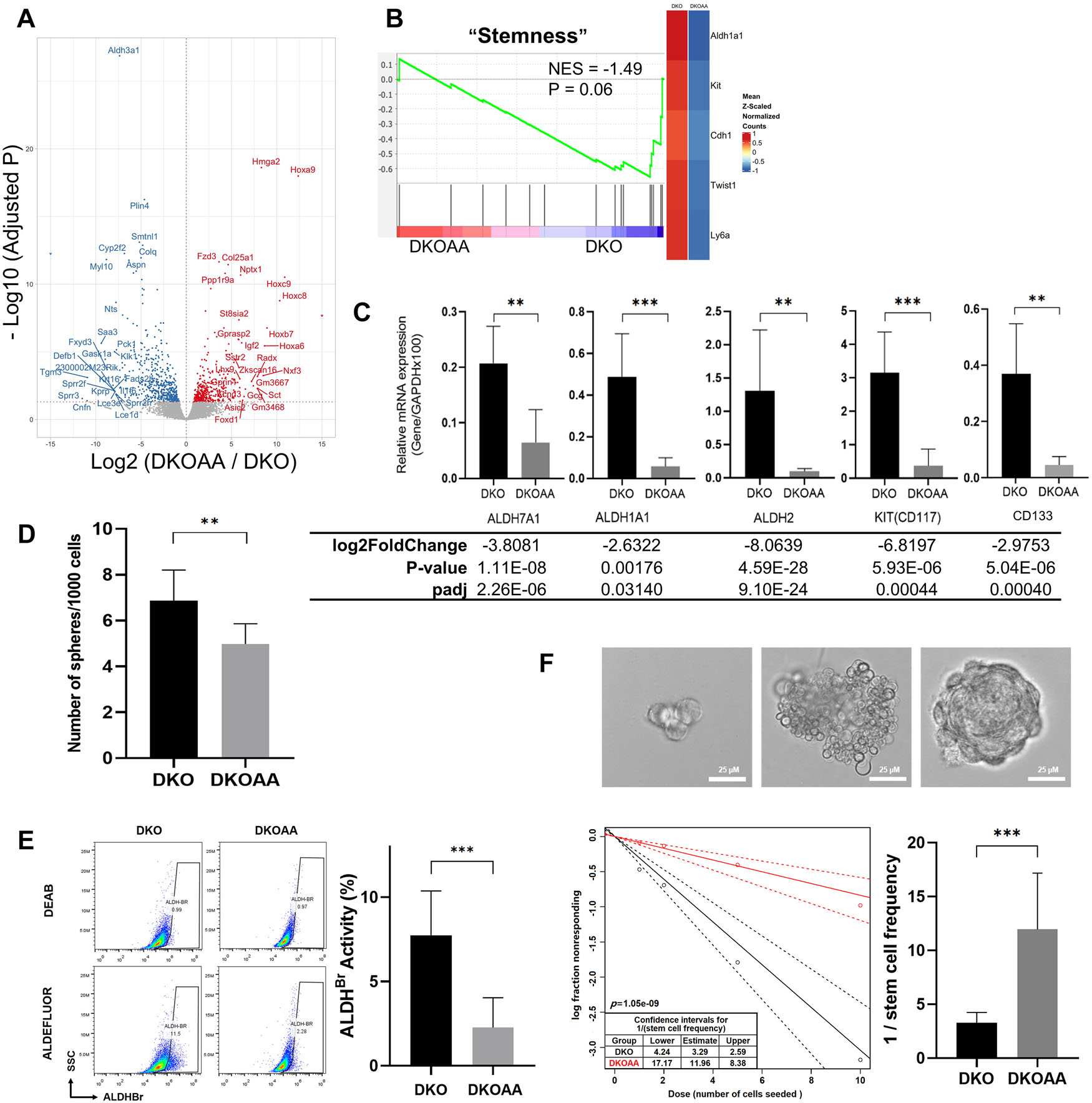
p27T187A inhibits stemness in DKO osteosarcoma. (A) Volcano plot for genes differentially expressed between the DKOAA and DKO tumors. (B) GSEA plot showing the enrichment of stemness genes in DKO. The y-axis is the enrichment score, and the lines on the x-axis indicate genes ranked by their expression difference between DKOAA and DKO. The right heatmap displays average expression of the leading edge genes in DKO (left) and DKOAA (right), with orange for high and blue for low expression. NES, normalized enrichment score. (C) Cells of each indicated genotype were subject to RT-qPCR validation to determine levels of CD133, CD117, ALDH7A1, ALDH1A1, and ALDH2 mRNA relative to GAPDH mRNA. Differential expression changes, including fold changes and adjusted P value from RNA-seq, are listed below. (D) Sphere formation assay of tumor cells of the indicated genotypes. (E) ALDH activity staining of tumor cells of indicated genotypes. FACS (representative of three experiments) was used to detect and quantify the subpopulation that expresses a high level of ALDH (ALDHBR). (F) Extreme limiting dilution analysis (ELDA) for the assessment of stem-like cell self-renewal ability. Representative figures show a cell cluster but not sphere formation (upper left), a cluster of differentiated and apoptotic cells showing no indication of sphere formation (upper middle), and a single sphere that was scored as “positive” (upper right). Scale bars = 25 μ m. The lower left figure shows sphere-forming capacity output using the previously described algorithm. The graph in the lower right shows the reciprocal of the calculated stem cell frequency. Error bars are SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
SCFSKP2 inhibitors reduce the proliferation of DKO cells
To investigate the efficacy of SCFSKP2 inhibitors on OS proliferation, we performed MTT assays on DKO cells using the following known SKP2 inhibitors: C1; pevonedistat and FKA, which are neddylation inhibitors;33,34 and C25, which is a SKP1-SKP2 pocket inhibitor.22 Our results indicated that all four drugs exerted antiproliferative effects on DKO cells, among which pevonedistat and C1 exerted more potent effects than FKA or C25 (Fig. 5A). Confirmed by in silico screening and in vitro assays, the compound C1 has been shown to bind to the SKP2/CKS1 pocket, thereby preventing the binding of p27 to the SCFSKP2 complex.35 Next, we tested the specificity of C1 and pevonedistat by using DKOAA cells, in which the SCFSKP2–p27 interaction has already been disrupted and, therefore, should be less responsive to C1. As expected, C1 (at 2 μM) reduced the cell proliferation of DKO and DKOAA by 45.9% and 20.3%, respectively, relative to DMSO (vehicle control). The selectivity of C1 for DKO cells continued to be observed at both low and high concentrations (Fig. 5B). Pevonedistat also exerted a similar selective antiproliferative effect on DKO over DKOAA cells (Fig. 5C). By immunoblotting, we found that both C1 and pevonedistat increased p27 (Fig. 5D and E). C1 treatment also stabilized p27 more efficiently in DKO than DKOAA cells, corresponding to the selectivity in the previous drug proliferation assay (Fig. 5D). Additionally, C1 treatment also increased p21 levels, which is consistent with the structural design and specificity of C1. Together, these findings suggest that anti-SKP2 drugs have efficacy in pRB/p53 double-deficient OS, and that the SCFSKP2–p27 axis is susceptible to selective inhibitors. The difference in drug activity of C1 and pevonedistat in DKO over DKOAA cells suggests that these two agents have a degree of selectivity as SKP2 inhibitors.
Figure 5.
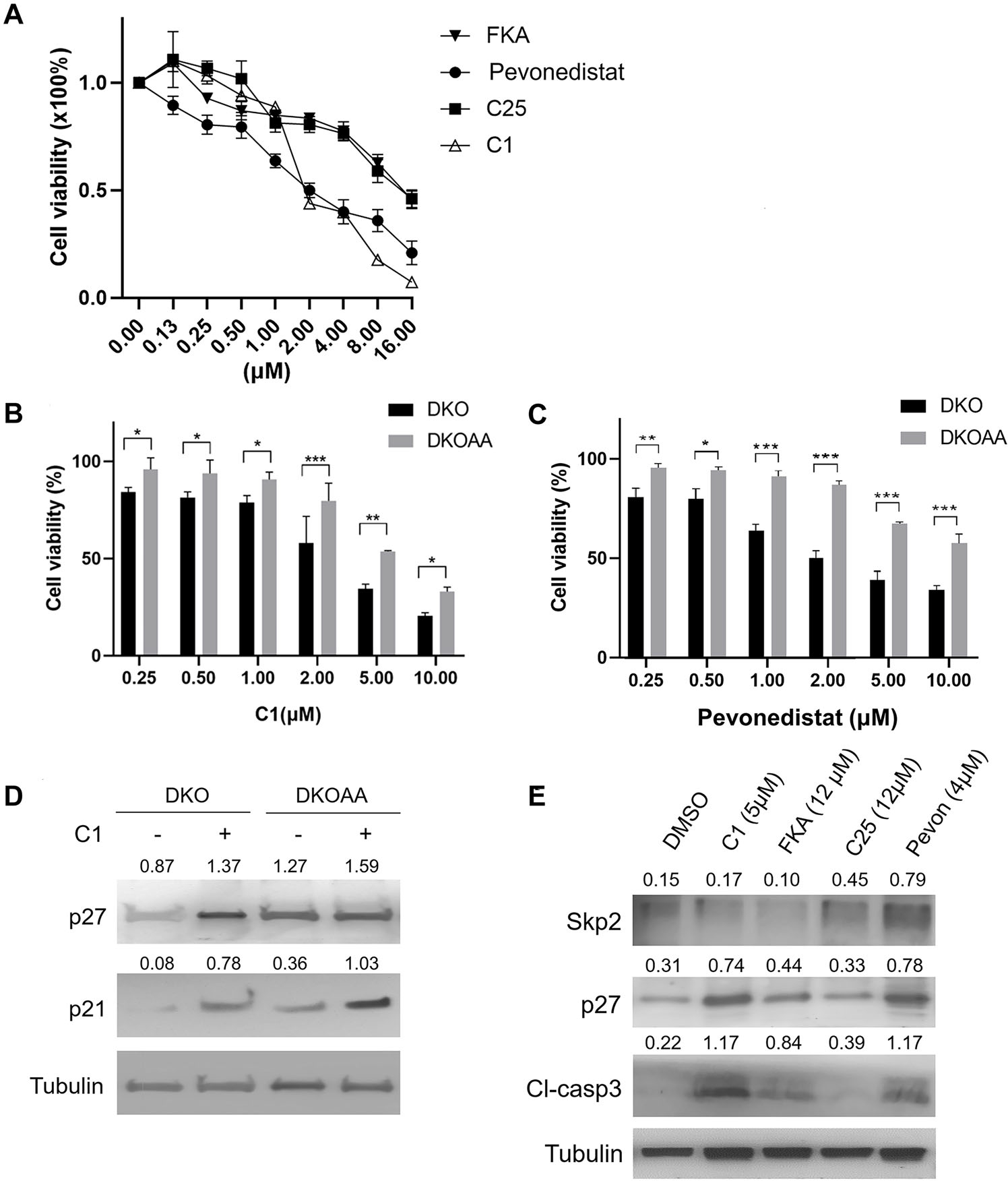
Inhibitors of SCFSKP2 selectively inhibit DKO tumor cells. (A) Graphs showing DKO cell viability following treatment with serial dilutions of SCFSKP2 inhibitors. (B and C) Graphs showing the cell viability of DKO and DKOAA cells treated with C1 (B) or pevonedistat (C) at indicated concentrations. The cell viability is calculated relative to the treatment with DMSO (vehicle control). Cells were treated for 72 h, and a representative of three independent experiments is shown. Cell proliferation was determined by the MTT method. Error bars are SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. (D) Western blots of DKO and DKOAA cells following treatment with C1 (5 μ M) for 72 hours. (E) Western blots of DKO cells following treatment with several SCFSKP2 inhibitors at indicated concentrations for 72 hours.
Discussion
OS development is increasingly linked to the inactivation of TP53, often potentiated by the loss of function of RB1. 6,23 Although the exact cellular origin of OS has remained elusive, Nakashima et al. have suggested that OS arises from cells that are at or beyond the Osterix expression stage of osteoblast differentiation.36 By recreating the loss of Rb1 and Trp53 within the osteoblast lineage, the DKO GEMM serves to recapitulate common genetic and clinical features of human OS and permits an in-depth inquiry into the role of the SCFSKP2–p27 axis in OS tumorigenesis. In this study, when we queried a cohort of human OS specimens from cBioPortal in which the inactivation of both RB1 and TP53 is frequent, they co-occurred with statistical significance. In our mouse models, we have shown that p27T187A promotes apoptosis and significantly delays the progression of Rb1/Trp53 double-knockout OS to lethality. Our results indicate that p27 accumulates to a higher level in DKOAA tumors compared with DKO tumors. Furthermore, small-molecule inhibitors of the SCFSKP2 complex, including C1 and pevonedistat, exert a dose-dependent antiproliferative effect on DKO tumor cells. Finally, RNA sequencing and subsequent analyses revealed that p27T187A reduces biomarkers of cancer stem cells (CSCs), suggesting that inhibitors of the SKP2–p27 axis may have a role in targeting stemness plasticity and chemoresistance in OS.
In the current study, reduced levels of p27 were found in the setting of Trp53/Rb1 codeletion. SCFSKP2-mediated ubiquitination and degradation of p27 have been shown as the principal mechanism that impedes the function of p27 as a cell cycle regulator. As a tumor suppressor, pRb suppresses SKP2 through multiple pathways, involving both transcriptional and posttranslational mechanisms.37,38 pRb has been shown to bind directly to the N-terminus of SKP2 and stabilize p27 by repressing SKP2 ligase activity.37 In addition, pRb is known to promote SKP2 protein degradation through CDH1 and repress SKP2 mRNA expression via E2F.38,39 Therefore, the loss of pRb in our DKO model leads to overactivated SKP2, thus promoting the degradation of p27. The p27T187A KI mutation is designed to abolish T187 phosphorylation, which is indispensable for the binding of SCFSKP2 to p27 and for the latter’s subsequent ubiquitination and degradation. Consistent with this mechanism, we observed that p27 is stabilized and accumulates in DKOAA tumor cells, resulting in increased apoptosis and decreased cellular proliferation. Interestingly, although multiple lines of evidence confirmed the apoptotic status in DKOAA, an increased cleaved caspase-3 level was not precisely correlated with an increased level of p27. RNA-seq revealed that DKOAA induces enriched expression of genes related to cell cycle arrest, the G2/M checkpoint, and the mitotic spindle. It is possible that DKOAA may cause aberrant mitotic regulation, leading to apoptosis that is independent from caspase; further studies are, therefore, warranted to test this hypothesis.40,41
Genetic and pharmacological inactivation of SKP2 has been shown to reduce the CSC population in prostate cancer.22 In OS, the stemness marker CD133 has been linked to an increase in metastatic potential and a poor prognosis.20,42,43 Similarly, the CSC marker CD117 has been shown to be preferentially expressed in tumorspheres and chemoresistant OS cells.44 In adenoid carcinoma, the subpopulation of CD133+ cells has been shown to express reduced levels of p27.45 Recently, Meyer et al. found that restoring p27 suppresses a stem cell signature and induces cell death by a miRNA-mediated pathway in leukemia.46 In our study, RNA sequencing and subsequent analyses revealed that CD133, CD117, and other stemness markers are markedly reduced in DKOAA tumors compared with those in the DKO cohort. Similarly, other stem-like properties, such as ALDH activity, stem-cell frequency, and self-renewal ability, are also suppressed in DKOAA tumor cells. Together, these data suggest that blocking SCFSKP2-mediated degradation of p27 may reduce tumor-initiating properties and should be explored further as a therapeutic strategy to reduce chemoresistance in OS. Although bulk RNA sequencing has uncovered a potential role for p27 in regulating OS stemness, a more in-depth analysis using single-cell transcriptome analysis may elucidate the complex cell–cell interaction responsible for this regulation.
Although p27T187A extended the survival of DKO animals, this mutation delays disease progression but does not block tumorigenesis entirely. In addition to p27, it is conceivable that the ubiquitination of p21 by SCFSKP2 may also play a role in tumor progression. However, given that p53 is inactivated in our models, and p21 is a known downstream effector of p53, one may argue that the contribution of p21 may be less consequential in DKO tumors. In the future, generating a transgenic model that disrupts the binding of SKP2 to p21 may be desirable for examining the role of p21 ubiquitination in the pathobiology of OS.47 Another interesting finding from RNA-seq was that genes from the Hox locus, such as HoxA9, were overexpressed in DKOAA samples. Hox family transcription factors have been reported as either oncogenes or tumor suppressor genes in different cancer types.48 Since there is minimal data regarding Hox genes in sarcoma, it would be meaningful to proceed with further studies focusing on the role of Hox family genes in OS tumorigenesis.
In our study, the clear survival advantage of DKOAA mice suggests that targeting the SKP2–p27 axis may have clinical relevance. As such, drugs that block the SCFSKP2-mediated degradation of p27 should be investigated as a complementary strategy to the current standard-of-care chemotherapy for OS. SKP2 gene knockout has been shown to lastingly block tumorigenesis in pRb/p53 double-knockout prostate cancer,13 suggesting that inhibiting SKP2 may act as a synthetic lethal and exert a stronger tumor-suppressive effect than p27T187A. Interestingly, ongoing experiments in our laboratory also show that crossing mice harboring an SKP2 deletion into the Osx1-Cre;Rb1lox/lox; Trp53lox/lox background leads to a significant delay in tumorigenesis, consistent with the notion that SCFSKP2 is essential for OS progression. Our findings that pevonedistat and C1 are more effective in DKO than DKOAA cells suggest that these compounds have a degree of selectivity for the SKP2–p27 axis. Among the tested compounds, pevonedistat is known to block the neddylation of SCFSKP2 and is the only anti-SKP2 drug currently in human clinical trials.33,49 In the future, preclinical studies using OS transgenic models and patient-derived xenografts are needed to determine the efficacy of SKP2 inhibitors, either alone or in combination with standard chemotherapy, to reduce OS progression.
Although our GEMM OS model largely mimics the genetic and phenotypical features of human OS, a few limitations exist. One of them is that the model may not reflect the entire range of human OS heterogeneity. Although loss of Rb1 and Trp53 has been widely found, other driver mutations, including in PTEN, ATRX, and BCRA, have also been shown to play a role in OS tumorigenesis.6 Hence, secondary mutations in addition to Rb1/Trp53 loss of function may also contribute to resistant oncogenic features. Transgenic models designed for specific oncogenic drivers combined with genetic sequencing techniques would be one way to elucidate this question and warrant further research.
Conclusion
Our study identifies the SCFSKP2–p27 axis as a potential therapeutic target for OS, which frequently harbors gene loss or mutation of two major tumor suppressors, RB1 and TP53. Our findings revealed that blocking the interaction of SKP2 and p27 promotes apoptosis and cell cycle arrest, and inhibits cancer stemness. These data support the notion that conventional chemotherapies may not prevent relapses in OS if the CSC subpopulation is not appropriately targeted. Specifically, disrupting the interaction between SKP2 and its substrate p27 may lead to strategies to overcome the stemness plasticity and chemoresistance of OS. As such, combination trials of SCFSKP2 inhibitors and conventional chemotherapeutic drugs in OS may be desirable. In the future, efforts to decipher the interaction of different components within the SCF complex will undoubtedly lead to better-targeted approaches to improve OS survival.
Supplementary Material
Table S1. The mutated status of TP53 and RB1 in human osteosarcoma.
Table S2. qPCR primers used in this study.
Figure S1. Oncoprints of two studies acquired from cBioPortal, showing TP53 and RB1 mutations and deletions of human osteosarcoma.
Figure S2. Genotyping results using agarose gel electrophoresis.
Figure S3. H&E, alkaline phosphatase, and osteocalcin staining of OS nodules; Kaplan–Meier curves comparing the survival of DKO with DKOAA mice with tumors of the jaw and limb.
Figure S4. Sashimi plots showing splicing RNA-seq reads mapped to the Trp53 and RB1 loci; summary of RNA-seq reads mapped to the T187A mutation sites.
Figure S5. GO enrichment analysis of differentially expressed genes.
Acknowledgments
Funding was provided by grants from the Live for Others Foundation, Sarcoma Strong, Montefiore Medical Center (to B.H.), and the National Institutes of Health (R01CA193967 to X.Z.; R01CA230032 and R01201458 to H.Z.).
Footnotes
Competing interests
The authors declare no competing interests.
Supporting information
Additional supporting information may be found in the online version of this article.
References
- 1.Harrison DJ, Geller DS, Gill JD, et al. 2018. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer Ther. 18: 39–50. [DOI] [PubMed] [Google Scholar]
- 2.Botter SM, Neri D & Fuchs B. 2014. Recent advances in osteosarcoma. Curr. Opin. Pharmacol. 16: 15–23. [DOI] [PubMed] [Google Scholar]
- 3.Gill J, Ahluwalia MK, Geller D & Gorlick R. 2013. New targets and approaches in osteosarcoma. Pharmacol. Ther. 137: 89–99. [DOI] [PubMed] [Google Scholar]
- 4.Saraf AJ, Fenger JM & Roberts RD. 2018. Osteosarcoma: accelerating progress makes for a hopeful future. Front. Oncol. 8. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luetke A, Meyers PA, Lewis I & Juergens H. 2014. Osteosarcoma treatment — where do we stand? A state of the art review. Cancer Treat. Rev. 40: 523–532. [DOI] [PubMed] [Google Scholar]
- 6.Sayles LC, Breese MR, Koehne AL, et al. 2019. Genomeinformed targeted therapy for osteosarcoma. Cancer Discov. 9: 46–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Zvi YS, Batko B, et al. 2018. Down-regulation of Skp2 expression inhibits invasion and lung metastasis in osteosarcoma. Sci. Rep. 8. 14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao QD, Zhong D & Chen Q. 2008. Protein expression of Skp2 in osteosarcoma and its relation with prognosis. J. Cent. South Univ. (Med. Sci.) 33: 606–611. [PubMed] [Google Scholar]
- 9.Ding L, Li R, Han X, et al. 2017. Inhibition of Skp2 suppresses the proliferation and invasion of osteosarcoma cells. Oncol. Rep. 38: 933–940. [DOI] [PubMed] [Google Scholar]
- 10.Hershko DD 2008. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 112: 1415–1424. [DOI] [PubMed] [Google Scholar]
- 11.Ding L, Li R, Sun R, et al. 2017. S-phase kinase-associated protein 2 promotes cell growth and motility in osteosarcoma cells. Cell Cycle 16: 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bornstein G, Bloom J, Sitry-Shevah D, et al. 2003. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cipl in S phase. J. Biol. Chem. 278: 25752–25757. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Bauzon F, Fu H, et al. 2013. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 24: 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao H, Iqbal NJ, Sukrithan V, et al. 2020. Targeted inhibition of the E3 ligase SCF(Skp2/Cksl) has antitumor activity in RB1-deficient human and mouse small-cell lung cancer. Cancer Res. 80: 2355–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao H, Lu Z, Bauzon F, et al. 2017. P27tl87a knockin identifies Skp2/Cksl pocket inhibitors for advanced prostate cancer. Oncogene 36: 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vlach J, Hennecke S & Amati B. 1997. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 16: 5334–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montagnoli A, Fiore F, Eytan E, et al. 1999. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 13: 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao H, Bauzon F, Bi E, et al. 2015. Substituting threonine 187 with alanine in p27Kip1 prevents pituitary tumorigenesis by two-hit loss of Rb1 and enhances humoral immunity in old age. J. Biol. Chem. 290: 5797–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang N, Song WX, Luo J, et al. 2008. Osteosarcoma development and stem cell differentiation. Clin. Orthop. Relat. Res. 466: 2114–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ying M, Liu G, Shimada H, et al. 2013. Human osteosarcoma CD49f(–)CD133(+)cells: impaired in osteogenic fate while gain of tumorigenicity. Oncogene 32: 4252–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Sato K, O’Donnell E, et al. 2020. Skp2 depletion reduces tumor-initiating properties and promotes apoptosis in synovial sarcoma. Transl. Oncol. 13. 100809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan CH, Morrow JK, Li CF, et al. 2013. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 154: 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walkley CR, Qudsi R, Sankaran VG, et al. 2008. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 22: 1662–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zehir A, Benayed R, Shah RH, et al. 2017. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23: 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rokita JL, Rathi KS, Cardenas MF, et al. 2019. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep. 29: 1675–1689.e1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sage J, Miller AL, Perez-Mancera PA, et al. 2003. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424: 223–228. [DOI] [PubMed] [Google Scholar]
- 27.Jonkers J, Meuwissen R, van der Gulden H, et al. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 29: 418–425. [DOI] [PubMed] [Google Scholar]
- 28.Malek NP, Sundberg H, McGrew S, et al. 2001. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 413: 323–327. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y & Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347: 70–78. [DOI] [PubMed] [Google Scholar]
- 30.Dobin A, Davis CA, Schlesinger F, et al. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders S & Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11. R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liberzon A, Birger C, Thorvaldsdóttir H, et al. 2015. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockhart AC, Bauer TM, Aggarwal C, et al. 2019. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest. New Drugs 37: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Yokoyama NN, Zhang S, et al. 2015. Flavokawain A induces deNEDDylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget 6: 41809–41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu L, Grigoryan AV, Li Y, et al. 2012. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol. 19: 1515–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakashima K, Zhou X, Kunkel G, et al. 2002. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108: 17–29. [DOI] [PubMed] [Google Scholar]
- 37.Ji P, Jiang H, Rekhtman K, et al. 2004. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 16: 47–58. [DOI] [PubMed] [Google Scholar]
- 38.Rinne UK, Classon MK, Dick FA, et al. 2007. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat. Cell Biol. 9: 225–232. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L & Wang C. 2006. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene 25: 2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruan W, Lim HH & Surana U. 2018. Mapping mitotic death: functional integration of mitochondria, spindle assembly checkpoint and apoptosis. Front. Cell Dev. Biol. 6. 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinha D, Duijf PHG & Khanna KK. 2019. Mitotic slippage: an old tale with a new twist. Cell Cycle 18: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tirino V, Desiderio V, Paino F, et al. 2011. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J. 25: 2022–2030. [DOI] [PubMed] [Google Scholar]
- 43.He A, Qi W, Huang Y, et al. 2012. CD 133 expression predicts lung metastasis and poor prognosis in osteosarcoma patients: a clinical and experimental study. Exp. Ther. Med. 4: 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adhikari AS, Agarwal N, Wood BM, et al. 2010. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 70: 4602–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panaccione A, Chang MT, Carbone BE, et al. 2016. NOTCH1 and SOX10 are essential for proliferation and radiation resistance of cancer stem-like cells in adenoid cystic carcinoma. Clin. Cancer Res. 22: 2083–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer SE, Muench DE, Rogers AM, et al. 2018. miR-196b target screen reveals mechanisms maintaining leukemia sternness with therapeutic potential. J. Exp. Med. 215: 2115–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Child ES & Mann DJ. 2006. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle 5: 1313–1319. [DOI] [PubMed] [Google Scholar]
- 48.Shah N & Sukumar S. 2010. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 10: 361–371. [DOI] [PubMed] [Google Scholar]
- 49.Swords RT, Coutre S, Maris MB, et al. 2018. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 131: 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The mutated status of TP53 and RB1 in human osteosarcoma.
Table S2. qPCR primers used in this study.
Figure S1. Oncoprints of two studies acquired from cBioPortal, showing TP53 and RB1 mutations and deletions of human osteosarcoma.
Figure S2. Genotyping results using agarose gel electrophoresis.
Figure S3. H&E, alkaline phosphatase, and osteocalcin staining of OS nodules; Kaplan–Meier curves comparing the survival of DKO with DKOAA mice with tumors of the jaw and limb.
Figure S4. Sashimi plots showing splicing RNA-seq reads mapped to the Trp53 and RB1 loci; summary of RNA-seq reads mapped to the T187A mutation sites.
Figure S5. GO enrichment analysis of differentially expressed genes.