

Abstract
Ethyl ferulate (EF) is abundant in Rhizoma Chuanxiong and grains (e.g., rice and maize) and possesses antioxidative, antiapoptotic, antirheumatic, and anti-inflammatory properties. However, its effect on lipopolysaccharide (LPS)-induced acute lung injury (ALI) is still unknown. In the present study, we found that EF significantly alleviated LPS-induced pathological damage and neutrophil infiltration and inhibited the gene expression of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) in murine lung tissues. Moreover, EF reduced the gene expression of TNF-α, IL-1β, IL-6, and iNOS and decreased the production of NO in LPS-stimulated RAW264.7 cells and BMDMs. Mechanistic experiments revealed that EF prominently activated the AMPK/Nrf2 pathway and promoted Nrf2 nuclear translocation. AMPK inhibition (Compound C) and Nrf2 inhibition (ML385) abolished the beneficial effect of EF on the inflammatory response. Furthermore, the protective effect of EF on LPS-induced ALI was not observed in Nrf2 knockout mice. Taken together, the results of our study suggest that EF ameliorates LPS-induced ALI in an AMPK/Nrf2-dependent manner. These findings provide a foundation for developing EF as a new anti-inflammatory agent for LPS-induced ALI/ARDS therapy.
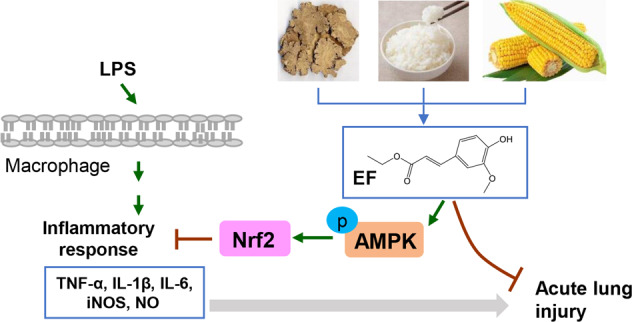
Keywords: ethyl ferulate, acute lung injury, lipopolysaccharide, inflammation, AMPK, Nrf2
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are life-threatening pulmonary disorders characterized by increased lung epithelial permeability, inflammatory cell infiltration, pulmonary edema, and diffuse alveolar damage [1, 2]. Despite decades of intensive efforts to combat this disease, morbidity and mortality rates remain very high because there is still no specific and effective treatment for ALI [3]. Therefore, it is urgent to investigate the pathogenic mechanisms of ALI and develop prevention strategies. As one of the major pathogenic factors of ALI, lipopolysaccharide (LPS) can induce a serious inflammatory response, and lead to lung damage as a result of inflammatory leukocyte infiltration. The abundant accumulation of neutrophils and an increase in proinflammatory cytokine production result in diffuse lung parenchymal damage in the alveoli [4]. Therefore, inhibiting LPS-mediated inflammation is highly effective for lessening the effects of ALI.
Macrophages in pulmonary tissues act as critical effectors to initiate host defense immunity [5]. Once activated by LPS, macrophages can release proinflammatory cytokines (TNF-α, IL-1β, and IL-6) as well as inflammatory mediators, e.g., NO [6]. Recent studies have revealed that aberrant macrophage activation is closely related to pathological changes during the development of ALI [7]. Lasting inflammation induced by macrophage activation leads to neutrophil infiltration and lung damage [5]. Thus, it is of great significance to verify the relationship between ALI and the immune response triggered by macrophages, which may provide new strategies for ALI patients.
Toll-like receptor 4 is a pattern recognition receptor that binds to LPS and subsequently triggers downstream inflammatory signaling pathways, including the mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) pathways [8]. Previous studies showed that three MAPK subfamilies, i.e., extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 MAPKs, play important roles in the production of proinflammatory mediators [4, 9, 10]. In addition, adenosine monophosphate-activated protein kinase (AMPK) has multiple functions such as regulating energy metabolism and the oxidative stress response [11, 12]. Several studies have revealed that activation of AMPK attenuates LPS-induced inflammatory responses and reduces the severity of LPS-induced ALI [11, 13]. As a downstream gene of AMPK, nuclear factor erythroid 2-related factor (Nrf2) has been demonstrated to play an important role in various inflammation-related lung diseases (e.g., chronic obstructive pulmonary disease and ALI) [14, 15]. Notably, activation of Nrf2 is dependent on AMP-activated protein kinase (AMPK) in the LPS-induced inflammatory response [16]. Notably, AMPK-mediated Nrf2 activation plays a protective role in LPS-induced murine ALI [17].
Accumulating experimental and epidemiological findings indicate that food-derived products and natural compounds extracted from plants have beneficial effects for preventing inflammation-mediated diseases [4, 18]. Ferulic acid (FA, 4-hydroxy-3-methoxycinnamic acid), a natural phenolic compound, widely exists in various plants and vegetables [19]. To date, FA has been demonstrated to possess antioxidant, antidiabetic, antihyperlipidemic, antihypertensive and anti-inflammatory functions [18, 20]. As a phenylpropanoid derivative of FA, ethyl ferulate (EF, ethyl-4-hydroxy-3-methoxycinnamate) is also widespread in plants such as Rhizoma Chuanxiong [21] and in grains such as rice and maize [22]. Many studies have shown that EF possesses several medicinal properties, such as antioxidant, antiapoptotic, antirheumatic and anti-inflammatory activities [23]. However, the role of EF in LPS-induced ALI is unclear. Based on these findings, we hypothesized that EF may have protective effects on LPS-induced inflammation and ALI. In the present study, we found that treatment with EF significantly alleviated LPS-induced ALI in mice, as demonstrated by lessened lung pathological damage, reduced neutrophil infiltration and decreased mRNA expression of proinflammatory cytokines after EF treatment. Furthermore, EF reduced LPS-induced gene expression of proinflammatory mediators (TNF-α, IL-1β, IL-6, iNOS, and NO) in RAW264.7 cells and bone marrow-derived macrophages (BMDMs) via the AMPK/Nrf2 pathway.
Materials and methods
Reagents and antibodies
Ethyl ferulate (EF, C12H14O4; Mr: 222.24; purity ≥99%) (Fig. 1a), ML385 and Compound C (Compd C) were purchased from Target Molecule Corp. (Target Mol, Shanghai, China). A stock solution of EF (for the in vitro study) was prepared at a concentration of 100 mmol/L in dimethyl sulfoxide (DMSO) (Sigma, St Louis, MO, USA) and stored at −20 °C. LPS (Escherichia coli, serotype 0111: B4) was obtained from Sigma Chemical Co.. iNOS, p-p38, p38, p-JNK, JNK, p-ERK, ERK, p-AMPK, AMPK, and β-actin antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Nrf2 antibody was purchased from Proteintech Group (Rosemont, IL, USA). Lamin B antibody was supplied by Santa Cruz Biotechnology (Dallas, TX, USA). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). CD16/32 antibody and FITC-conjugated anti-Ly-6G antibody were obtained from BD Biosciences (San Jose, CA, USA).
Fig. 1. EF alleviates LPS-induced ALI in mice.

a The chemical structure of EF. b The schematic timeline of the LPS-induced ALI model. C57BL/6 mice were subjected to an intratracheal injection of LPS (5 mg/kg) and subsequent intraperitoneal injection of vehicle (polyoxyethylene castor oil: ethanol: PBS = 1:1:8) or EF (25 and 50 mg/kg). Six hours later, the mice were sacrificed, and the lung lobes and bronchoalveolar lavage fluid (BALF) were collected. c The right lung lobes were fixed, and hematoxylin-eosin staining was performed to evaluate the lung histopathological changes (original magnification, ×100). d The lung injury score was calculated by histological analysis in different groups. e The total protein concentration in BALF supernatant was measured to determine lung permeability with a BCA kit according to the manufacturer’s instructions. The data represent the mean ± SEM, n = 5 per group. *P < 0.05; **P < 0.01; ***P < 0.001.
Animals
Male C57BL/6 mice (8–10 weeks old, specific pathogen free) were purchased from Slac Laboratory Animal Co., Ltd. (Shanghai, China). Mice were housed five per cage with free access to food and water in pathogen-free and climate-controlled rooms with 12 h light/dark cycles. All animal experimental protocols described in this study were approved by the Instructional Animal Care and Use Committee of Jiangnan University.
LPS-induced ALI mouse model
Part I (Protective study): Wild-type C57BL/6 mice were randomly divided into four groups, namely, the PBS group, LPS + vehicle group, LPS + EF (25 mg/kg) group, and LPS + EF (50 mg/kg) group (n = 5). Part II (Therapeutic study): C57BL/6 mice were randomly divided into three groups, namely the PBS group, LPS + vehicle group and LPS + EF (25 mg/kg) group. Mice were subjected to an intratracheal injection of LPS (5 mg/kg) for 1 h, followed by intraperitoneal injection of vehicle or EF (n = 4). Part III (Mechanistic study): Nrf2 knockout (KO) (Nrf2−/−) mice were randomly divided into three groups, namely, the PBS group, LPS + vehicle group and LPS + EF (50 mg/kg) group (n = 4–5). EF was dissolved in vehicle (polyoxyethylene castor oil: ethanol: PBS = 1:1:8). Mice were anesthetized by pentobarbital and then treated intratracheally (i.t.) with LPS (5 mg/kg) or PBS. In the LPS + EF group, EF (25 or 50 mg/kg) was intraperitoneally (i.p.) injected subsequently after LPS challenge [24]. At 6 h (Part I and III) or 24 h (Part II) after treatment with LPS, the mice were euthanized. Serum, bronchoalveolar lavage and lung tissue samples were collected for subsequent experiments.
Collection of blood and bronchoalveolar lavage fluid
After anesthesia, peripheral blood was collected and centrifuged. Serum was obtained for the measurement of MDA. The trachea was exposed and cannulated with a tracheal catheter, and then the lungs were lavaged three times with 0.6 mL ice-cold PBS. BALF was centrifuged at 500 × g for 5 min [25], and cell-free supernatants were collected for total protein concentration quantification using a BCA protein assay kit (Beyotime, Shanghai, China).
Histopathological analysis
Lung tissues were fixed with 4% paraformaldehyde, paraffin embedded and cut into 4 μm thick sections. Tissue sections were stained with hematoxylin and eosin, and images were obtained using a Pannoramic MIDI (3D, HISTECH, Budapest, Hungary). The degree of lung pathological injury was scored as described in a previous study [26]. Briefly, hemorrhage, lung edema, inflammatory cell infiltration, atelectasis and hyaline membrane were assayed. Each variable was scored as 0–4 according to the severity of damage as follows: no injury, 0; injury to 25% of the field, 1; injury to 50% of the field, 2; injury to 75% of the field, 3; and diffuse injury, 4.
Immunohistochemical analysis
The lung lobe was fixed, embedded in paraffin, cut into 4 μm thick sections, and then immunostained with antibodies against MPO (Abcam, Cambridge, MA, USA) at a dilution of 1:200. Subsequently, biotinylated secondary antibodies were used followed by the addition of 3′3-diaminobenzidine (DAB) solution to detect the avidin-biotin complex signal [27]. Images of the lung tissue sections subject to immunohistochemical staining were captured by Pannoramic MIDI (3D HISTECH, Budapest, Hungary) and analyzed using Pannoramic Viewer software (3D HISTECH).
Flow cytometry assay
BALF cells were incubated with an Fc-blocking anti-mouse CD16/32 antibody; the population of neutrophils (Ly6G+) in each BALF sample was analyzed using a flow cytometer (BD Accuri C6, BD Biosciences, San Jose, CA, USA) and graphed using FlowJo software [26, 28].
Isolation of lung macrophages from ALI mice
After 6 h of challenge with LPS, the C57BL/6 mice were euthanized, and lung tissues were collected for the isolation of lung macrophages as previously described [29]. Briefly, the whole lungs of the mice were perfused with ice-cold PBS, and the lung lobes were excised. Then, macrophages were isolated via collagenase digestion (collagenase [1.0 mg/ml], DNase [25–50 U/ml]; Sigma-Aldrich). Cells were adherence-purified for 1 h in serum-free media and nonadherent cells (such as lymphocytes) were removed before complete medium was replaced for overnight incubation. Lung macrophages were collected for subsequent analysis.
Cell culture
RAW264.7 macrophages (ATCC, Manassas, VA, USA) were maintained in RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum (Gibco-BRL, Grand Island, NY, USA) and 1% penicillin–streptomycin (Gibco-BRL, Grand Island, NY, USA) in an incubator (37 °C, 5% CO2). When cells reached 80% confluence, they were seeded into 6-well or 12-well plates and treated for subsequent experiments.
Preparation of BMDMs and treatment
BMDMs from C57BL/6 wild-type mice were flushed with PBS and cultured in Dulbecco’s modified Eagle’s medium (Gibco-BRL, Grand Island, NY, USA) supplemented with 10% FBS, 1% penicillin–streptomycin and macrophage colony-stimulating factor (M-CSF, 10 ng/mL) (PeproTech, Rocky Hill, NJ, USA). At the day 3, the medium was replenished once. At day 6, cells were harvested and seeded in plates [9]. BMDMs were pretreated with EF for 0.5 h and stimulated with LPS for the indicated times.
Cell viability assay
Cells were seeded in 96-well plates at a density of 1 × 104 cells/well in the presence or absence of EF (0–100 μM) for 24 h, and 0.1% DMSO was used as a vehicle control. Cell viability was measured using the Cell Counting Kit-8 (CCK-8, Biosharp, Hefei, China) according to the manufacturer’s instructions. The optical density (OD) was measured at 450 nm using a microplate reader (Synergy H4, BioTek, Vermont, USA). Cell viability is presented as a percentage of the control.
Determination of nitrite levels
Macrophages were seeded in 6-well plates and incubated overnight. Cells were treated with various doses of EF (0, 3, 10, 30, and 100 μM) for 0.5 h, followed by challenge with LPS (100 ng/mL) for 24 h. The supernatant was collected, and nitrite levels were measured by Griess reagents (Beyotime, Shanghai, China) to reflect NO production [26, 28].
Measurement of malondialdehyde (MDA) content
MDA content in serum was measured using an MDA assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
RNA isolation, reverse transcription, and quantitative PCR
Total RNA from macrophages and lung tissues was extracted using TRIzol Reagent (Life Technologies, Waltham, MA, USA). Isolated RNA was reverse-transcribed into cDNA with a PrimeScript RT Reagent Kit (TaKaRa, Otsu, Shiga, Japan). Target mRNA quantification was measured by real-time PCR using SYBR Premix Ex TaqTM (TaKaRa, Otsu, Shiga, Japan) and a LightCycler® 480 detection PCR system (Roche, Foster City, CA, USA). Quantification was performed using the 2(−∆∆Ct) method. The primer sequences are shown in Table 1.
Table 1.
Primer sequences.
| Target gene | Primer | Primer sequences (5′-3′) |
|---|---|---|
| GAPDH | Forward | TGGCCTTCCGTGTTCCTAC |
| Reverse | GAGTTGCTGTTGAAGTCGCA | |
| TNF-α | Forward | CCTGTAGCCCACGTCGTAG |
| Reverse | GGGAGTAGACAAGGTACAACCC | |
| IL-1β | Forward | GAAATGCCACCTT TTGACAGTG |
| Reverse | TGGATGCTCTCATCAG GACAG | |
| IL-6 | Forward | CTGCAAGAGACTTCCATCCAG |
| Reverse | AGTGGTATAGACAGGTCTG TTGG | |
| tGPX4 | Forward | CGCAGCCGTTCTTATCAATG |
| Reverse | CACTGTGGAAATGGATGAAAGTC |
Immunoblotting
Total protein extracted from cells and lung tissues was homogenized in RIPA buffer containing protease inhibitor cocktail and phosphatase inhibitor (MedChem Express, Monmouth Junction, NJ, USA). A BCA protein assay kit (Beyotime, Shanghai, China) was used to measure the total protein in the samples. For the analysis of nuclear Nrf2 expression, the nuclear fraction was obtained using nuclear extraction reagents (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of proteins were loaded into 10% SDS/PAGE gels and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk and then incubated with primary antibodies (p-p38, p38, p-JNK, JNK, p-ERK, ERK, Nrf2, p-AMPK, AMPK, and β-actin, diluted 1:1000), followed by horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibodies (diluted 1:10,000). Subsequently, data were collected with a ChemiDoc MP Imaging System (Bio-Rad Laboratories, Hercules, CA, USA), and quantification was performed by ImageJ software (National Institute of Mental Health, Bethesda, MD, USA) [4, 25].
Statistical analysis
All data are presented as the mean ± SEM obtained from at least three independent experiments. Differences between two groups were compared by Student’s t test, and comparisons among three or more groups were performed by one-way ANOVA (Tukey’s post hoc test). Statistical analyses were performed using Prism 7 (GraphPad, San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
EF ameliorates LPS-induced pathological changes in the lungs
An LPS-induced ALI mouse model was established to determine the protective role of EF (Fig. 1b). Pathological alterations in the lungs were detected, and the severity of the lung injury was scored in each mouse. As shown in Fig. 1c, d, serious pathological damage was observed in the LPS group, such as inflammatory cell infiltration, hemorrhage and destruction of the alveolar histological structure. EF treatment at both 25 and 50 mg/kg remarkably lessened LPS-induced pulmonary histopathological changes. Moreover, we evaluated the total protein concentration of the BALF to assess the protective role of EF. The results showed that the BALF concentration was markedly increased in LPS-challenged mice, whereas mice treated with EF at a 50 mg/kg dose exhibited a significant BALF concentration decrease (Fig. 1e). Based on the above results, we proposed that EF at 25 and 50 mg/kg exerted protective effects on mice with LPS-induced ALI. Meanwhile, EF at a 50 mg/kg dose showed better benefits than 25 mg/kg in ALI mice. All these results demonstrate that EF significantly attenuates LPS-induced ALI.
In addition, to determine the therapeutic effect of EF on LPS-induced ALI, we intratracheally injected LPS (5 mg/kg) into C57BL/6 mice. After 1 h, mice were treated with EF (25 mg/kg) by i.p. injection (Supplementary Fig. 1a). Lung injury was evaluated 24 h after LPS challenge. As shown in Supplementary Fig. 1, EF treatment significantly lessened the destruction of alveolar histological structures (Supplementary Fig. 1b, c) and decreased the total protein levels in BALF (Supplementary Fig. 1d). These data further demonstrate that EF alleviates LPS-induced ALI.
EF decreases LPS-induced pulmonary inflammatory cell infiltration and the mRNA expression of proinflammatory cytokines
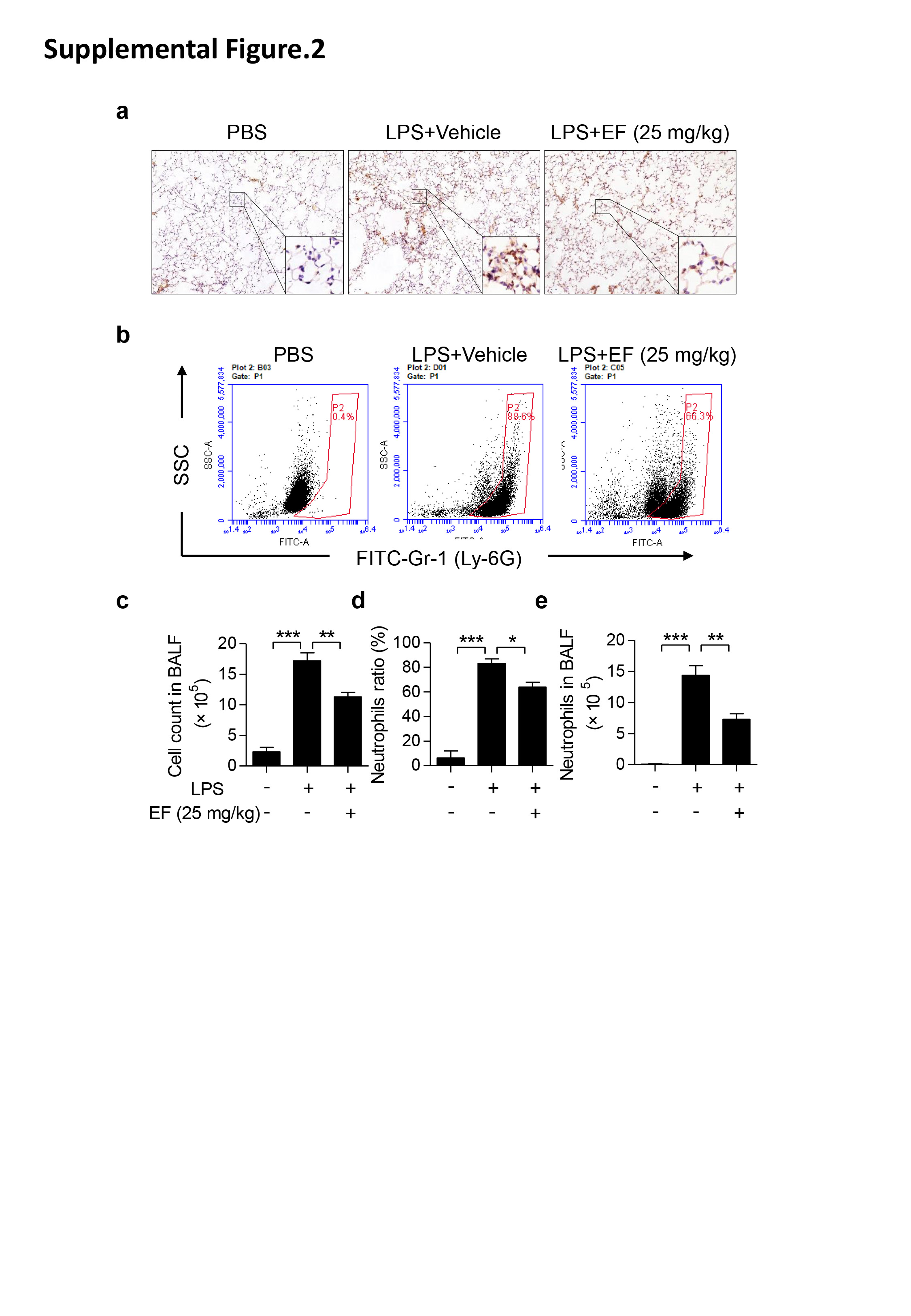
Since MPO in lung tissues is a specific marker of neutrophil infiltration, MPO-positive content was determined using immunohistochemical staining. As displayed in Fig. 2a, MPO was rarely expressed in the lungs of the PBS group and increased dramatically in the LPS group. This increase was significantly attenuated by the EF treatment. These results indicate that EF reduces neutrophil infiltration in ALI mice. Moreover, total cells in BALF were counted, and neutrophils were classified by staining with anti-Ly6G-FITC (Fig. 2b). As shown in Fig. 2c, EF decreased inflammatory cell infiltration induced by LPS. In addition, LPS resulted in 83.14% ± 1.233% neutrophil (Ly-6G+) infiltration into BALF, whereas the infiltration level was 0.8167% ± 0.2482% in PBS group. Meanwhile, EF treatment reduced the neutrophil ratio to 71.92% ± 3.590% and 58.34% ± 7.637% (Fig. 2d) at the doses of 25 and 50 mg/kg, respectively. Based on the total cells in BALF and the ratio of neutrophils, neutrophil infiltration levels were calculated, and we found that both doses (25 and 50 mg/kg) of EF substantially reduced LPS-induced neutrophil infiltration (Fig. 2e). Moreover, to confirm the protective effect of EF on inflammatory cell infiltration in LPS-induced ALI, C57BL/6 mice were intraperitoneally injected with EF 1 h before LPS challenge, and neutrophil infiltration was assayed as described above. The results demonstrated that injection of EF after LPS also decreased LPS-induced neutrophil infiltration (Supplementary Fig. 2). To further clarify the anti-inflammatory effects of EF in vivo, the mRNA levels of proinflammatory cytokines in lung tissues were measured. The results demonstrated that the mRNA levels of TNF-α (Fig. 2f), IL-1β (Fig. 2g), and IL-6 (Fig. 2h) were significantly reduced by EF treatment. Notably, to further identify the role of macrophages in the protective effects of EF on LPS-induced ALI in mice, we isolated lung macrophages from ALI mice with or without EF treatment and tested the mRNA expression levels of proinflammatory cytokines. As displayed in Fig. 2i, k, EF (50 mg/kg) decreased the mRNA level of proinflammatory cytokines (TNF-α, IL-β, and IL-6) in LPS-induced ALI lung macrophages from mice. Together, these results suggest that EF inhibits LPS-induced pulmonary inflammation.
Fig. 2. EF suppresses the inflammatory response in LPS-induced ALI mice.

a Sections of mouse lung tissue immunostained for MPO content. b Cell pellets in BALF were suspended and stained for a marker of neutrophils (Ly-6G) and analyzed by FACS. c Total infiltrated cells in BALF were counted by a counting chamber. d Statistical analysis of the neutrophil percentage was performed. e Total infiltrated neutrophils in BALF were calculated based on the total BALF cell number and percentage of neutrophils. f–h TNF-α (f), IL-1β (g), and IL-6 (h) mRNA in the lungs was quantified by real-time PCR. i–k TNF-α (i), IL-1β (j), and IL-6 (k) mRNA levels in the lung macrophages of LPS-induced ALI mice were quantified by real-time PCR. Values represent the mean ± SEM, n = 5 mice each group. *P < 0.05; **P < 0.01; ***P < 0.001.
EF inhibits LPS-induced iNOS expression and NO production in macrophages
Since macrophage activation often produces several inflammatory mediators, such as iNOS and NO, we investigated whether EF could inhibit the expression of iNOS and the production of NO. RAW264.7 cells and primary BMDMs were chosen in this study to identify the anti-inflammatory effect of EF on macrophages. First, the potential cytotoxicity of EF on macrophages was detected after incubation with EF for 24 h by CCK-8 assay. The results showed that, even at 100 μM, EF exhibited no cellular toxicity against RAW264.7 cells and BMDMs (Fig. 3a, e). Macrophages were incubated with EF (3, 10, 30, and 100 μM) for 0.5 h, followed by stimulation with LPS (100 ng/mL) for 24 h. Then, the cells were harvested for Western blot assay, and the supernatant was collected to detect nitrite levels. The results revealed that EF suppressed iNOS mRNA expression and NO production in RAW264.7 cells (Fig. 3b–d) and BMDMs (Fig. 3f–h) in a dose-dependent manner.
Fig. 3. EF inhibits iNOS expression and nitrite generation in LPS-stimulated RAW 264.7 cells and BMDMs.

RAW264.7 cells (a–d) and BMDMs (e–h) were treated with EF at 0, 3, 10, 30, and 100 μM for 24 h. a, e The cytotoxicity of EF in RAW264.7 cells (a) and BMDMs (e) was assayed using CCK-8. RAW264.7 cells (b–d) and BMDMs (f–h) were preincubated with EF at 0, 3, 10, 30, and 100 μM for 0.5 h before stimulation with or without LPS (100 ng/mL) for 24 h. Cell lysates were collected for Western blot analysis, and supernatants were collected to detect the level of nitrite. The protein expression of iNOS (b, f) was measured, GAPDH was used as a loading control, and iNOS/GAPDH (c, g) was analyzed by ImageJ software. d, h The amount of nitrite in the supernatant was detected by the Griess Reagent System. Data shown as the mean ± SEM, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001.
EF reduces the expression of proinflammatory cytokines in LPS-challenged macrophages
To investigate the anti-inflammatory effects of EF in LPS-stimulated macrophages, the mRNA levels of TNF-α, IL-1β, and IL-6 were measured by real-time PCR. As shown in Fig. 4, LPS significantly increased the mRNA levels of TNF-α, IL-1β, and IL-6 in RAW264.7 cells (Fig. 4a–c) and BMDMs (Fig. 4d–f). EF treatment remarkably decreased the expression of these cytokines in a dose-dependent manner.
Fig. 4. EF decreases proinflammatory cytokine mRNA expression in LPS-stimulated macrophages.

a–c RAW264.7 cells were treated with EF at the indicated concentrations for 0.5 h, followed by challenge with LPS (100 ng/mL) for 4 h. Total RNA was isolated, and the mRNA levels of TNF-α (a), IL-1β (b), and IL-6 (c) were then quantified by real-time PCR. d–f BMDMs were also pretreated with EF at 0, 3, 10, 30, and 100 μM for 0.5 h and then challenged with LPS (100 ng/mL) for 4 h, and the mRNA levels of TNF-α (d), IL-1β (e), and IL-6 (f) were quantified by real-time PCR. The experiments were independently repeated at least three times. Values represent the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
EF activates the AMPK-mediated pathway but fails to inhibit the activation of the MAPK pathway in macrophages
The MAPK pathway plays an important role in regulating the expression of proinflammatory mediators. Therefore, we investigated whether the anti-inflammatory role of EF on LPS-stimulated macrophages involved the MAPK pathway. As shown in Fig. 5a, b, LPS stimulation increased p38, JNK and ERK phosphorylation. However, EF failed to inhibit p38, JNK, and ERK phosphorylation.
Fig. 5. EF activates the AMPK-mediated signaling pathway but fails to deactivate the MAPK pathway.

RAW264.7 cells (a) and BMDMs (b) were pretreated with EF (0, 3, 10, 30, and 100 μM) for 0.5 h, followed by stimulation with LPS (100 ng/mL) for another 0.5 h. a, b The total protein of cells was extracted, and the expression levels of p-p38, p38, p-JNK, JNK, p-ERK, and ERK were analyzed by Western blot with β-actin as the loading control. RAW264.7 cells (c, e) and BMDMs (d, f) were plated in 6-well plates, pretreated with EF (0, 3, 10, 30, and 100 μM) for 0.5 h, and then challenged with LPS (100 ng/mL) for another 0.5 h. c, d The protein expression levels of p-AMPK and AMPK were measured using Western blot analysis. e, f The ratio of p-AMPK/AMPK was quantified using ImageJ software. g–i RAW264.7 cells pretreated with 5 μM Compd C were treated with EF (100 μM) for 0.5 h and then challenged with LPS (100 ng/mL) for another 24 h. The protein expression levels of iNOS and Nrf2 were measured, β-actin was used as a loading control (g), iNOS/β-actin (h), and Nrf2/β-actin (i) were analyzed by ImageJ software. Data are shown as the mean ± SEM, n = 3, *P < 0.05; **P < 0.01; ***P < 0.001.
Due to the pivotal role of activated AMPK in regulating LPS-induced inflammatory responses [12], the phosphorylation of AMPK was measured to further illustrate the mechanism by which EF exerts anti-inflammatory activity. As shown in Fig. 5c, d, EF pretreatment in RAW264.7 cells and primary BMDMs significantly increased AMPK phosphorylation. Furthermore, an AMPK inhibitor (Compound C) rescued the decreased iNOS expression suppressed by EF (Fig. 5e, f) in LPS-stimulated RAW264.7 cells. Moreover, we also measured the downstream gene expression of AMPK (Nrf2) after EF treatment. The results revealed that EF promoted Nrf2 expression, while Compound C inhibited Nrf2 expression induced by LPS (Fig. 5e, g). Taken together, these results suggest that the anti-inflammatory activities of EF might be dependent on the AMPK/Nrf2 signaling pathway.
EF exhibits an anti-inflammatory effect on the LPS-induced inflammatory response in a Nrf2-dependent manner
To determine the role of Nrf2 in the inhibitory effect of EF on inflammation, we confirmed the effect of EF on the expression of Nrf2 in LPS-induced macrophage activation. As shown in Fig. 6a–d, EF prominently accelerated Nrf2 expression in both RAW264.7 cells and BMDMs. Furthermore, we found that EF promoted the expression of Nrf2 in the lung tissues of ALI mice (Fig. 6e). These results suggested that EF might exert an anti-inflammatory effect by promoting Nrf2 signaling. Additionally, GPX4 reduction and MDA augmentation also contributed to lung damage, and Nrf2 was reported to regulate GPX4 activity and reduce MDA content [30, 31]. Therefore, the expression of GPX4 in lung tissues and the level of MDA in serum were measured to further clarify the role of Nrf2 in ALI. As expected, EF reversed the GPX4 decrease (Fig. 6f) and decreased the MDA concentration (Fig. 6g) induced by LPS in vivo. The above results demonstrated that EF markedly accelerated Nrf2 expression in vitro and in vivo. Therefore, we speculated that EF may regulate LPS-induced macrophage activation and ALI through Nrf2 signaling. To verify this hypothesis, ML385, a specific Nrf2 inhibitor, was applied in this study and incubated with EF in RAW264.7 cells under LPS stimulation. The results revealed that ML385 increased iNOS expression (Fig. 7a, b) and NO production (Fig. 7c) upon LPS stimulation. In addition, we demonstrated that EF facilitated Nrf2 translocation in a dose-dependent manner by immunoblotting (Fig. 7d). We also detected the nuclear translocation of Nrf2 by an immunofluorescence assay. The results revealed that EF promotes the nuclear translocation of Nrf2 and enhances its expression in the cytoplasm (Fig. 7e). In summary, these results suggest that EF inhibits the LPS-induced inflammatory response through AMPK-mediated Nrf2 signaling.
Fig. 6. EF accelerates the expression of Nrf2 in vitro and in vivo.

RAW264.7 cells (a, c) and BMDMs (b, d) were pretreated with EF (0, 3, 10, 30, and 100 μM) for 0.5 h, followed by stimulation with LPS (100 ng/mL) for another 0.5 h. a, b The total cell protein was extracted to evaluate the expression of Nrf2 by Western blot, and β-actin was used as a loading control. c, d The ratios of Nrf2/β-actin were semiquantitatively analyzed by ImageJ software. e–g LPS-induced ALI mice were generated by intratracheal injection of LPS (5 mg/kg) with or without EF (25 mg/kg) administration for 6 h. e Lung tissues were homogenized, and the expression of Nrf2 was measured by Western blot analysis. Then, the ratio of Nrf2/β-actin was calculated by densitometry. f–g The levels of tGPX4 (f) in the lung tissue and MDA (g) in the serum were measured by real-time PCR and MDA assay kits, respectively. The results represent the mean ± SEM, n = 3 for in vitro tests, and n = 5 mice per group for in vivo experiments. *P < 0.05; **P < 0.01.
Fig. 7. EF reduces LPS-induced inflammatory mediator production in a manner dependent on the Nrf2 pathway.

a–c RAW264.7 cells were pretreated with or without ML385 (10 μM) for 0.5 h before EF (100 μM) treatment, and 0.5 h later, the cells were stimulated with LPS (100 ng/mL) for another 24 h. a Cell lysates were analyzed by Western blotting to measure the expression of iNOS, and β-actin was used as a loading control. b The ratios of iNOS/β-actin were calculated by ImageJ software. c Culture supernatant was collected for NO detection. d RAW264.7 cells were treated with EF (0, 3, 10, 30, and 100 μM) for 0.5 h before coincubation with LPS (100 ng/mL) for 0.5 h. Nuclear fractions were extracted and subjected to immunoblot analysis using Nrf2 antibody and Lamin B antibody. e RAW264.7 cells were treated with EF (100 μM) for 0.5 h before coincubation with LPS (100 ng/mL) for 0.5 h. Then, the cells were used for immunofluorescence analysis and stained with anti-Nrf2 (red) and DAPI (blue). Images were obtained by confocal microscopy. Data are shown as the mean ± SEM, n = 3, *P < 0.05; **P < 0.01.
Nrf2 knockout abolishes the beneficial effect of EF on LPS-challenged ALI mice
Nrf2−/− mice were used to confirm the role of Nrf2 in the protective effect of EF against LPS-induced ALI. As displayed in Fig. 8, the results showed that after intratracheal treatment with LPS, Nrf2−/− mice experienced severe lung injury, which was characterized by the altered lung histology (Fig. 8a, b), increased BALF protein concentration (Fig. 8c), increased MPO content (Fig. 8d), and greatly enhanced expression of proinflammatory cytokines, including TNF-α (Fig. 8e), IL-1β (Fig. 8f), and IL-6 (Fig. 8g). However, EF (even 50 mg/kg) failed to exert a protective effect on Nrf2−/− ALI mice. Altogether, these results indicate that EF inhibits macrophage activation and alleviates ALI through the AMPK/Nrf2 signaling pathway.
Fig. 8. EF protecteds against LPS-induced ALI in mice is dependent on the Nrf2 pathway.

Nrf2−/− mice were subjected to an intratracheal injection of LPS (5 mg/kg) and subsequent intraperitoneal injection of vehicle (polyoxyethylene castor oil: ethanol: PBS = 1:1:8) or EF (50 mg/kg). Six hours later, the mice were sacrificed, and the lung lobes and bronchoalveolar lavage fluid (BALF) were collected. a The lung lobes were fixed, and hematoxylin-eosin staining was performed to evaluate the lung histopathological changes (original magnification, ×100). b The lung injury score was calculated by histological analysis in different groups. c The total protein concentration in the BALF supernatant was measured to determine lung permeability with a BCA kit according to the manufacturer’s instructions. d MPO expression in lung tissue was detected by immunohistochemical localization. e–g TNF-α (e), IL-1β (f), and IL-6 (g) mRNA in the lungs was quantified by real-time PCR. The data represent the mean ± SEM, for (a–d), n = 5 per group; for (e–g), n = 4. **P < 0.01; ***P < 0.001, ns means no significant difference.
Discussion
As a major cause of ALI, pulmonary inflammation still has high morbidity and mortality [32]. No effective therapeutic strategies for ALI have been developed to date. Therefore, novel pharmacological agents for ALI treatment are urgently needed. LPS is a component of the gram-negative bacterial cell membrane and is considered to be a mainstay for ALI model induction. The LPS-induced ALI model is a well-accepted model of human ALI and is widely used for the study of ALI/ARDS [33]. In this study, we established an LPS-induced ALI model to assess the therapeutic potential of EF. We report here for the first time that EF protects against LPS-induced ALI in mice.
During LPS-induced ALI, increased pulmonary microvascular permeability accompanied by massive inflammatory cell (especially neutrophil) infiltration are the main characteristics of the alteration of lung tissue [34]. Along with these alterations, an increased MPO content, elevated BALF protein concentration and alveolar structure destruction were also reported during this process [4]. In addition, Gr-1-positive cells (neutrophils) were significantly increased, as determined by flow cytometry analysis [35]. In the present study, the effects of intratracheal LPS treatment were consistent with the above alterations, suggesting that we successfully established an LPS-induced ALI model in mice. Meaningfully, these alterations were reduced by treatment with EF, indicating that EF possesses the capacity to alleviate ALI induced by LPS.
The inflammatory response is frequently accompanied by excessive inflammatory cell infiltration and is thought to play a pivotal role in the process of ALI [4, 17, 35]. Upon LPS stimulation, inflammatory cells release large levels of inflammatory cytokines and mediators, including TNF-α, IL-1β, IL-6, iNOS, and NO [4, 28]. These inflammatory mediators have been reported to play a crucial role in the initiation and expansion of inflammation in LPS-induced ALI [36]. For example, TNF-α and IL-1β are early inflammatory cytokines in ALI that can stimulate the activation of endothelial cells, epithelial cells, and alveolar macrophages to release cytokines containing IL-8, TNF-α, IL-1β and IL-6 and recruit and activate neutrophils and macrophages [28, 37]. Meanwhile, high levels of NO produced from iNOS can lead to aggravated cytokine production and mitochondrial dysfunction, which in turn accelerate inflammation and tissue injury [38]. Increased levels of proinflammatory mediators have been observed in ALI patients and are associated with major inflammatory disorders [39]. Previous studies showed that the expression of TNF-α, IL-1β, and IL-6 was significantly upregulated in the lung tissues of LPS-induced ALI mice [4, 28], which was consistent with the findings of our study. Similarly, the decreased inflammatory cytokines paralleled the reductions in lung pathological injuries [4, 28], which indicated that EF alleviated LPS-induced ALI partly through the inhibition of inflammatory cytokines. In addition, the reduction in the mRNA levels of TNF-α, IL-1β, and IL-6 in the lung macrophages of EF-treated ALI mice suggested that EF may improve ALI by regulating macrophage function.
As a specific receptor of LPS on the surface of monocytes/macrophages, TLR4 can be recognized by LPS and initiate downstream signal transduction and the activation of NF-κB and MAPKs [28]. As two classical inflammatory signaling pathways, the activated NF-κB and MAPK pathways are reported to promote the production of proinflammatory mediators. Therefore, suppression of the NF-κB and MAPK pathways contributes to the inhibition of proinflammatory cytokine production [28]. PSN, a Stemona alkaloid isolated from S. sessilifolia, was demonstrated to reduce proinflammatory cytokine expression via the NF-κB and MAPK pathways in our previous study [9]. Nevertheless, Lio et al. found that Nardosinanone N, a natural compound, exhibits anti-inflammatory effects through neither the NF-κB nor MAPK pathway [40]. Since a previous study revealed that EF could inhibit the translocation of NF-κB p65 in LPS-stimulated RAW 264.7 cells, we initially focused on the activation of MAPKs in the present study. Interestingly, our findings showed that EF did not inhibit the phosphorylation of p38, JNK, or ERK.
Growing evidence suggests that pulmonary inflammation and injury are modulated by AMPK activation [41]. Treatment with a specific activator could significantly reduce proinflammatory cytokine release, indicating that AMPK activation is beneficial for inhibiting the development of lung inflammation [42]. This effect is probably regulated by a master transcription factor downstream of Nrf2 [43]. AMPK/Nrf2 axis activation also exhibits anti-inflammatory activities in an LPS-challenged inflammatory response model [44]. Previous studies have shown that natural compounds, such as triterpene acids and neochlorogenic acid, show potent anti-inflammatory effects by activating the AMPK/Nrf2 pathway [45, 46]. In the present study, we also verified that EF exhibits anti-inflammatory effects via the AMPK/Nrf2 signaling pathway by using a specific AMPK inhibitor (Compd C) and a Nrf2 inhibitor (ML385) in vitro and an in vivo Nrf2 KO mouse model. As EF reduced the mRNA levels of TNF-α, IL-1β and IL-6 in lung macrophages of ALI mice and macrophages were applied for mechanistic analysis, one limitation of this study is that Nrf2 global KO mice, not macrophage-specific Nrf2 KO mice, were used in this study.
EF, a naturally occurring ester derivative of FA, is present in many plants, such as Rhizoma Chuanxiong [21] and grains [22]. In fact, FA is a phenolic acid that has multiple bioactivities and was approved in Japan as a food additive [47]. It is worth mentioning that FA causes unpleasant bitter and astringent tastes in alcoholic beverages, while its derivative, EF, which is found in sake and sake mash [48], has a more pleasant taste than FA, making it more palatable for the food industry and drug development. EF has multiple bioactivities, including antioxidant [49], antiapoptotic, neuroprotective [50], vasculoprotective [51] and anti-inflammatory [24] activities. EF displays an inhibitory effect on the production of inflammatory mediators, including TNF-α, IL-1β, IL-6, and NO [24]. Our results were consistent with this finding, as EF inhibited the expression of these inflammatory mediators in LPS-stimulated RAW264.7 cells and BMDMs.
Many ingredients found in plants have been listed as Nrf2 activators [52]. Given that EF exerts an anti-inflammatory effect via the AMPK/Nrf2 signaling axis in the current study, we believe that EF is a novel Nrf2 activator. As HO-1 is a major cytoprotective protein regulated by Nrf2 activation [53], combined with Scapagnini et al.’s finding that EF is able to induce HO-1 protein expression and protect rat neurons [54], we speculated that EF may exhibit an anti-inflammatory response and attenuate ALI through AMPK/Nrf2-mediated HO-1 upregulation, but this hypothesis needs further investigation.
In summary, the results of our study suggested that EF has a potential therapeutic effect on LPS-induced ALI. Furthermore, we demonstrated that EF suppresses macrophage inflammation by activating AMPK/Nrf2 signaling. KO of Nrf2 abolishes the protective role of EF in LPS-induced ALI mice. Taken together, our results show that EF exerts anti-inflammatory effects in vivo and in vitro. Because Nrf2 and the inflammatory response are implicated in various diseases, including colitis, arthritis, asthma, and fibrosis, we believe that EF may be a new agent for the treatment of ALI/ARDS and other Nrf2 inflammation-related diseases.
Supplementary Information
{kind=link}
{kind=link}
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81871518, 81901522); China Postdoctoral Science Foundation (2020M671347); Fundamental Research Funds for the Central Universities (JUSRP11955); Natural Science Foundation of Jiangsu Province (BK20200602); the Wuxi Health and Family Planning Commission (Z201810); and the Public Health Research Center at Jiangnan University (JUPH201805).
Author contributions
QFP and YXW designed the study and drafted the paper; FQ participated in the design and helped draft the paper; YXW and YYW performed the experiments; ZQG, DC, GL, and BBW participated in the animal experiments; FJJ, MXW, and Jing Zuo performed the data analysis; and Jun Zhu, YQC were involved in the discussion of the experiments. All authors read and approved the final paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Feng Qian, Email: fengqian@sjtu.edu.cn.
Qing-feng Pang, Email: qfpang@jiangnan.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00742-0.
References
- 1.Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 2018;19:50. doi: 10.1186/s12931-018-0756-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jordan S, Mitchell JA, Quinlan GJ, Goldstraw P, Evans TW. The pathogenesis of lung injury following pulmonary resection. Eur Respir J. 2000;15:790–9. doi: 10.1034/j.1399-3003.2000.15d26.x. [DOI] [PubMed] [Google Scholar]
- 3.Han J, Li Y, Li Y. Strategies to enhance mesenchymal stem cell-based therapies for acute respiratory distress syndrome. Stem Cells Int. 2019;2019:5432134. doi: 10.1155/2019/5432134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu YX, He HQ, Nie YJ, Ding YH, Sun L, Qian F. Protostemonine effectively attenuates lipopolysaccharide-induced acute lung injury in mice. Acta Pharmacol Sin. 2018;39:85–96. doi: 10.1038/aps.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rayees S, Rochford I, Joshi JC, Joshi B, Banerjee S, Mehta D. Macrophage TLR4 and PAR2 signaling: role in regulating vascular inflammatory injury and repair. Front Immunol. 2020;11:2091. doi: 10.3389/fimmu.2020.02091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsieh YH, Deng JS, Chang YS, Huang GJ. Ginsenoside Rh2 ameliorates lipopolysaccharide-induced acute lung injury by regulating the TLR4/PI3K/Akt/mTOR, Raf-1/MEK/ERK, and Keap1/Nrf2/HO-1 signaling pathways in mice. Nutrients. 2018;10:1208. [DOI] [PMC free article] [PubMed]
- 7.Li Q, Ran Q, Sun L, Yin J, Luo T, Liu L, et al. Lian Hua Qing Wen Capsules, a potent epithelial protector in acute lung injury model, block proapoptotic communication between macrophages, and alveolar epithelial cells. Front Pharmacol. 2020;11:522729. doi: 10.3389/fphar.2020.522729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin WC, Deng JS, Huang SS, Wu SH, Chen CC, Lin WR, et al. Anti-Inflammatory activity of sanghuangporus sanghuang mycelium. Int J Mol Sci. 2017;18:347. [DOI] [PMC free article] [PubMed]
- 9.Wu Y, Nie Y, Huang J, Qiu Y, Wan B, Liu G, et al. Protostemonine alleviates heat-killed methicillin-resistant Staphylococcus aureus-induced acute lung injury through MAPK and NF-kappaB signaling pathways. Int Immunopharmacol. 2019;77:105964. doi: 10.1016/j.intimp.2019.105964. [DOI] [PubMed] [Google Scholar]
- 10.Ko IG, Hwang JJ, Chang BS, Kim SH, Jin JJ, Hwang L, et al. Polydeoxyribonucleotide ameliorates lipopolysaccharide-induced acute lung injury via modulation of the MAPK/NF-kappaB signaling pathway in rats. Int Immunopharmacol. 2020;83:106444. doi: 10.1016/j.intimp.2020.106444. [DOI] [PubMed] [Google Scholar]
- 11.Tao L, Cao F, Xu G, Xie H, Zhang M, Zhang C. Mogroside IIIE attenuates LPS-Induced acute lung injury in mice partly through regulation of the TLR4/MAPK/NF-kappaB axis via AMPK activation. Phytother Res. 2017;31:1097–106. doi: 10.1002/ptr.5833. [DOI] [PubMed] [Google Scholar]
- 12.Badamjav R, Sonom D, Wu Y, Zhang Y, Kou J, Yu B, et al. The protective effects of Thalictrum minus L. on lipopolysaccharide-induced acute lung injury. J Ethnopharmacol. 2020;248:112355. doi: 10.1016/j.jep.2019.112355. [DOI] [PubMed] [Google Scholar]
- 13.Chen JJ, Deng JS, Huang CC, Li PY, Liang YC, Chou CY, et al. p-coumaric-acid-containing adenostemma lavenia ameliorates acute lung injury by activating AMPK/Nrf2/HO-1 signaling and improving the anti-oxidant response. Am J Chin Med. 2019;47:1483–506. doi: 10.1142/S0192415X19500769. [DOI] [PubMed] [Google Scholar]
- 14.Yu D, Liu X, Zhang G, Ming Z, Wang T. Isoliquiritigenin inhibits cigarette smoke-induced COPD by attenuating inflammation and oxidative stress via the regulation of the Nrf2 and NF-kappaB signaling pathways. Front Pharmacol. 2018;9:1001. doi: 10.3389/fphar.2018.01001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q, Lv H, Wen Z, Ci X, Peng L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-like receptor protein 3 inflammasome and inhibits the NF-kappaB pathway in macrophages and in acute lung injury. Front Immunol. 2017;8:1518. doi: 10.3389/fimmu.2017.01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park SY, Jin ML, Chae SY, Ko MJ, Choi YH, Park G, et al. Novel compound from Polygonum multiflorum inhibits inflammatory response in LPS-stimulated microglia by upregulating AMPK/Nrf2 pathways. Neurochem Int. 2016;100:21–9. doi: 10.1016/j.neuint.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Huang XT, Liu W, Zhou Y, Sun M, Yang HH, Zhang CY, et al. Galectin-1 ameliorates lipopolysaccharide-induced acute lung injury via AMPK-Nrf2 pathway in mice. Free Radic Biol Med. 2020;146:222–33. doi: 10.1016/j.freeradbiomed.2019.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Lin L, Wu H. Ferulic acid alleviates lipopolysaccharide-induced acute lung injury through inhibiting TLR4/NF-kappaB signaling pathway. J Biochem Mol Toxicol. 2021;35:e22664. [DOI] [PubMed]
- 19.Lin TY, Lu CW, Huang SK, Wang SJ. Ferulic acid suppresses glutamate release through inhibition of voltage-dependent calcium entry in rat cerebrocortical nerve terminals. J Med Food. 2013;16:112–9. doi: 10.1089/jmf.2012.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mir SM, Ravuri HG, Pradhan RK, Narra S, Kumar JM, Kuncha M, et al. Ferulic acid protects lipopolysaccharide-induced acute kidney injury by suppressing inflammatory events and upregulating antioxidant defenses in Balb/c mice. Biomed Pharmacother. 2018;100:304–15. doi: 10.1016/j.biopha.2018.01.169. [DOI] [PubMed] [Google Scholar]
- 21.Kong L, Yu Z, Bao Y, Su X, Zou H, Li X. Screening and analysis of an antineoplastic compound in Rhizoma Chuanxiong by means of in vitro metabolism and HPLC-MS. Anal Bioanal Chem. 2006;386:264–74. doi: 10.1007/s00216-006-0621-0. [DOI] [PubMed] [Google Scholar]
- 22.Zhang LW, Al-Suwayeh SA, Hsieh PW, Fang JY. A comparison of skin delivery of ferulic acid and its derivatives: evaluation of their efficacy and safety. Int J Pharm. 2010;399:44–51. doi: 10.1016/j.ijpharm.2010.07.054. [DOI] [PubMed] [Google Scholar]
- 23.Cunha FVM, do Nascimento Caldas Trindade G, da Silva Azevedo PS, Coelho AG, Braz EM, Pereira de Sousa Neto B, et al. Ethyl ferulate/beta-cyclodextrin inclusion complex inhibits edema formation. Mater Sci Eng C Mater Biol Appl. 2020;115:111057. doi: 10.1016/j.msec.2020.111057. [DOI] [PubMed] [Google Scholar]
- 24.Cunha FV, Gomes Bde S, Neto Bde S, Ferreira AR, de Sousa DP, de Carvalho e Martins Mdo C, et al. Ferulic acid ethyl ester diminished complete Freund’s adjuvant-induced incapacitation through antioxidant and anti-inflammatory activity. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:117–30. doi: 10.1007/s00210-015-1180-8. [DOI] [PubMed] [Google Scholar]
- 25.Song Y, Wu Y, Li X, Shen Y, Ding Y, Zhu H, et al. Protostemonine attenuates alternatively activated macrophage and DRA-induced asthmatic inflammation. Biochem Pharmacol. 2018;155:198–206. doi: 10.1016/j.bcp.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Z, Sun L, Hao R, Jiang H, Qian F, Ye RD. Nedd8 modification of Cullin-5 regulates lipopolysaccharide-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2017;313:L104–14. doi: 10.1152/ajplung.00410.2016. [DOI] [PubMed] [Google Scholar]
- 27.Chen D, Qiu YB, Gao ZQ, Wu YX, Wan BB, Liu G, et al. Sodium propionate attenuates the lipopolysaccharide-induced epithelial-mesenchymal transition via the PI3K/Akt/mTOR signaling pathway. J Agric Food Chem. 2020;68:6554–63. doi: 10.1021/acs.jafc.0c01302. [DOI] [PubMed] [Google Scholar]
- 28.Zhang D, Li X, Hu Y, Jiang H, Wu Y, Ding Y, et al. Tabersonine attenuates lipopolysaccharide-induced acute lung injury via suppressing TRAF6 ubiquitination. Biochem Pharmacol. 2018;154:183–92. doi: 10.1016/j.bcp.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Nie Y, Sun L, Wu Y, Yang Y, Wang J, He H, et al. AKT2 regulates pulmonary inflammation and fibrosis via modulating macrophage activation. J Immunol. 2017;198:4470–80. doi: 10.4049/jimmunol.1601503. [DOI] [PubMed] [Google Scholar]
- 30.Kim YS, Podder B, Song HY. Cytoprotective effect of alpha-lipoic acid on paraquat-exposed human bronchial epithelial cells via activation of nuclear factor erythroid related factor-2 pathway. Biol Pharm Bull. 2013;36:802–11. doi: 10.1248/bpb.b12-00977. [DOI] [PubMed] [Google Scholar]
- 31.Xie YZ, Zhang XJ, Zhang C, Yang Y, He JN, Chen YX. Protective effects of leonurine against ischemic stroke in mice by activating nuclear factor erythroid 2-related factor 2 pathway. CNS Neurosci Ther. 2019;25:1006–17. doi: 10.1111/cns.13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen R, Xie F, Zhao J, Yue B. Suppressed nuclear factor-kappa B alleviates lipopolysaccharide-induced acute lung injury through downregulation of CXCR4 mediated by microRNA-194. Respir Res. 2020;21:144. doi: 10.1186/s12931-020-01391-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Z, Zhong R, Yang Y, Xia T, Wang W, Wang Y, et al. Systems pharmacology reveals the mechanism of activity of Ge-Gen-Qin-Lian decoction against LPS-induced acute lung injury: a novel strategy for exploring active components and effective mechanism of TCM formulae. Pharmacol Res. 2020;156:104759. doi: 10.1016/j.phrs.2020.104759. [DOI] [PubMed] [Google Scholar]
- 34.Jing W, Chunhua M, Shumin W. Effects of acteoside on lipopolysaccharide-induced inflammation in acute lung injury via regulation of NF-kappaB pathway in vivo and in vitro. Toxicol Appl Pharmacol. 2015;285:128–35. doi: 10.1016/j.taap.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Wu Y, He H, Ding Y, Liu S, Zhang D, Wang J, et al. MK2 mediates macrophage activation and acute lung injury by regulating let-7e miRNA. Am J Physiol Lung Cell Mol Physiol. 2018;315:L371–81. doi: 10.1152/ajplung.00019.2018. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Chang G, Huang J, Wang Y, Ma N, Roy AC, et al. Sodium butyrate inhibits the inflammation of lipopolysaccharide-induced acute lung injury in mice by regulating the toll-like receptor 4/nuclear factor kappaB signaling pathway. J Agric Food Chem. 2019;67:1674–82. doi: 10.1021/acs.jafc.8b06359. [DOI] [PubMed] [Google Scholar]
- 37.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin M, Suh SJ, Yang JH, Lu Y, Kim SJ, Kwon S, et al. Anti-inflammatory activity of bark of Dioscorea batatas DECNE through the inhibition of iNOS and COX-2 expressions in RAW264.7 cells via NF-kappaB and ERK1/2 inactivation. Food Chem Toxicol. 2010;48:3073–9. doi: 10.1016/j.fct.2010.07.048. [DOI] [PubMed] [Google Scholar]
- 39.Huang GJ, Huang SS, Deng JS. Anti-inflammatory activities of inotilone from Phellinus linteus through the inhibition of MMP-9, NF-kappaB, and MAPK activation in vitro and in vivo. PLoS One. 2012;7:e35922. doi: 10.1371/journal.pone.0035922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lio CK, Luo JF, Shen XY, Dai Y, Machado J, Xie Y, et al. Nardosinanone N suppresses LPS-induced macrophage activation by modulating the Nrf2 pathway and mPGES-1. Biochem Pharmacol. 2020;173:113639. doi: 10.1016/j.bcp.2019.113639. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, Cheng X, Yue L, Cui W, Zhou W, Gao J, et al. Molecular pathogenesis in chronic obstructive pulmonary disease and therapeutic potential by targeting AMP-activated protein kinase. J Cell Physiol. 2018;233:1999–2006. doi: 10.1002/jcp.25844. [DOI] [PubMed] [Google Scholar]
- 42.Cheng XY, Li YY, Huang C, Li J, Yao HW. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget. 2017;8:22513–23. doi: 10.18632/oncotarget.15116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui W, Zhang Z, Zhang P, Qu J, Zheng C, Mo X, et al. Nrf2 attenuates inflammatory response in COPD/emphysema: Crosstalk with Wnt3a/beta-catenin and AMPK pathways. J Cell Mol Med. 2018;22:3514–25. doi: 10.1111/jcmm.13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. 2017;12:311–24. doi: 10.1016/j.redox.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jian T, Ding X, Li J, Wu Y, Ren B, Li J, et al. Triterpene acids of loquat leaf improve inflammation in cigarette smoking induced COPD by regulating AMPK/Nrf2 and NFkappaB pathways. Nutrients. 2020;12:657. [DOI] [PMC free article] [PubMed]
- 46.Gao XH, Zhang SD, Wang LT, Yu L, Zhao XL, Ni HY, et al. Anti-Inflammatory effects of neochlorogenic acid extract from Mulberry leaf (Morus alba L.) against LPS-stimulated inflammatory response through mediating the AMPK/Nrf2 signaling pathway in A549 cells. Molecules. 2020;25:1385. [DOI] [PMC free article] [PubMed]
- 47.Rychlicka M, Maciejewska G, Niezgoda N, Gliszczynska A. Production of feruloylated lysophospholipids via a one-step enzymatic interesterification. Food Chem. 2020;323:126802. doi: 10.1016/j.foodchem.2020.126802. [DOI] [PubMed] [Google Scholar]
- 48.Hashizume K, Ito T, Shimohashi M, Ishizuka T, Okuda M. Ferulic acid and ethyl ferulate in sake: comparison of levels between Sake and Mirin and analysis of their sensory properties. Food Sci Technol Res. 2013;19:705–9. [Google Scholar]
- 49.Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta-peptide(1-42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem. 2005;92:749–58. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- 50.Cunha FVM, Coelho AG, Azevedo P, da Silva AAJ, Oliveira FA, Nunes LCC. Systematic review and technological prospection: ethyl ferulate, a phenylpropanoid with antioxidant and neuroprotective actions. Expert Opin Ther Pat. 2019;29:73–83. doi: 10.1080/13543776.2019.1568410. [DOI] [PubMed] [Google Scholar]
- 51.Tsai YC, Lee YM, Hsu CH, Leu SY, Chiang HY, Yen MH, et al. The effect of ferulic acid ethyl ester on leptin-induced proliferation and migration of aortic smooth muscle cells. Exp Mol Med. 2015;47:e180. doi: 10.1038/emm.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scapagnini G, Vasto S, Abraham NG, Caruso C, Zella D, Fabio G. Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol Neurobiol. 2011;44:192–201. doi: 10.1007/s12035-011-8181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lei L, Chai Y, Lin H, Chen C, Zhao M, Xiong W, et al. Dihydroquercetin activates AMPK/Nrf2/HO-1 signaling in macrophages and attenuates inflammation in LPS-induced endotoxemic mice. Front Pharmacol. 2020;11:662. doi: 10.3389/fphar.2020.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scapagnini G, Butterfield DA, Colombrita C, Sultana R, Pascale A, Calabrese V. Ethyl ferulate, a lipophilic polyphenol, induces HO-1 and protects rat neurons against oxidative stress. Antioxid Redox Signal. 2004;6:811–8. doi: 10.1089/ars.2004.6.811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.