

Abstract
[PSI+] is a prion of Saccharomyces cerevisiae Sup35, an essential ribosome release factor. In [PSI+] cells, most Sup35 is sequestered into insoluble amyloid aggregates. Despite this depletion, [PSI+] prions typically affect viability only modestly, so [PSI+] must balance sequestering Sup35 into prions with keeping enough Sup35 functional for normal growth. Sis1 is an essential J-protein regulator of Hsp70 required for the propagation of amyloid-based yeast prions. C-terminally truncated Sis1 (Sis1JGF) supports cell growth in place of wild-type Sis1. Sis1JGF also supports [PSI+] propagation, yet [PSI+] is highly toxic to cells expressing only Sis1JGF. We searched extensively for factors that mitigate the toxicity and identified only Sis1, suggesting Sis1 is uniquely needed to protect from [PSI+] toxicity. We find the C-terminal substrate-binding domain of Sis1 has a critical and transferable activity needed for the protection. In [PSI+] cells that express Sis1JGF in place of Sis1, Sup35 was less soluble and formed visibly larger prion aggregates. Exogenous expression of a truncated Sup35 that cannot incorporate into prions relieved [PSI+] toxicity. Together our data suggest that Sis1 has separable roles in propagating Sup35 prions and in moderating Sup35 aggregation that are crucial to the balance needed for the propagation of what otherwise would be lethal [PSI+] prions.
Keywords: prion, proteostasis, J-protein, amyloid, chaperone, Sup35, Sis1
Introduction
Prions are infectious misfolded proteins associated with pathology in many organisms (McGlinchey et al. 2011; Chernova et al. 2017; Chiti and Dobson 2017; Soto and Pritzkow 2018). The [PSI+] prion of Saccharomyces cerevisiae propagates as insoluble amyloid of the essential ribosome release factor Sup35 (Stansfield et al. 1995; Zhouravleva et al. 1995; Tuite and Cox 2007; Wickner 2016). Sup35 contains a dispensable N-terminal region, separable from the functional domain, that forms the amyloid core of prion fibers (Ter-Avanesyan et al. 1994; King et al. 1997; Paushkin et al. 1997a). This prion-determining region of Sup35 is followed by a dispensable charged middle region and the C-terminal domain (CTD ) that functions in translation termination (Ter-Avanesyan et al. 1994; Liebman and Chernoff 2012). Cells propagating [PSI+] display a nonsense suppressor phenotype caused by reduced ability of Sup35 to promote release of ribosomes at termination codons of mRNAs. This obvious phenotype makes yeast an ideal system for monitoring cellular processes that promote or inhibit the ability of prions to propagate in cells.
Sup35, like other amyloid-forming proteins, can form amyloids with different physical characteristics (Toyama et al. 2007). Variants of [PSI+] prions display different strength and mitotic stability phenotypes that reflect such differences in the underlying Sup35 amyloids (Derkatch et al. 1996; Tanaka et al. 2006). A stronger nonsense suppressor phenotype reflects a greater reduction in efficiency of translation termination, which correlates with the extent Sup35 is depleted into prion aggregates (Zhou et al. 1999; Jung et al. 2000; Uptain et al. 2001). In turn, the degree of aggregation of Sup35 depends on the rates of assembly and division of Sup35 amyloid (Tanaka et al. 2004, 2006). The division rate determines the number of prion “seeds” per cell, which often correlates with strength of phenotype as it determines the number of amyloid ends available to recruit Sup35 into insoluble prion polymers. This seed number also can determine the efficiency by which the seeds are transmitted as cells divide, and therefore, the mitotic stability of a prion. Replication of prions by division depends on the same Hsp104-driven machinery that resolubilizes proteins from aggregates to help cells recover from protein-denaturing stresses (Chernoff et al. 1995; Paushkin et al. 1996; Glover and Lindquist 1998; Lum et al. 2004; Hung and Masison 2006; Tipton et al. 2008; Reidy et al. 2012). Hsp104 function requires the Hsp70 system and various components of this machinery influence prion propagation and elimination (Masison and Reidy 2015; Chernova et al. 2017).
Hsp70 is a key player in protein quality control that acts in protein translation, folding, degradation, and translocation across membranes (Sharma and Masison 2009; Craig 2018; Rosenzweig et al. 2019). Hsp70 function relies on J-proteins that recruit substrates to Hsp70 and stimulate its ATP hydrolysis (Fan et al. 2003). Altering J-proteins and many other Hsp70 co-chaperones that prions depend on can perturb prion propagation, possibly through effects on Hsp104 activity (Moriyama et al. 2000; Sondheimer et al. 2001; Kryndushkin et al. 2002; Jones et al. 2004; Kryndushkin and Wickner 2007; Reidy and Masison 2011; Reidy et al. 2012). Among J-proteins, only Sis1 is essential for both cell viability and for the propagation of the most widely studied yeast prions (Luke et al. 1991; Higurashi et al. 2008). It possesses an N-terminal J-domain that binds Hsp70 and activates Hsp70 ATPase, adjacent glycine-phenylalanine (GF) and glycine-methionine (GM) rich regions that contribute to functional specificity, and a C-terminal substrate-binding domain (CTD) that ends in a dimerization domain (DD) (Yan and Craig 1999; Li et al. 2009). Although some prions are exquisitely sensitive to alterations in Sis1 function (Reidy et al. 2014), propagation of [PSI+] and [PIN+] is supported by Sis1 lacking all but its J and GF regions (Lopez et al. 2003; Kirkland et al. 2011; Harris et al. 2014), here designated Sis1JGF. Sis1 lacking only its GF region (Sis1ΔGF) supports propagation of [PSI+], but not [PIN+] or [URE3] (Sondheimer et al. 2001; Higurashi et al. 2008; Reidy et al. 2014). Together these findings suggest that Sis1 assists propagation of different prions by cooperating with Hsp70 and Hsp104 and that [PSI+], [PIN+], and [URE3] differ in their degree of need for chaperone-mediated replication or a specific J-protein activity (Lopez et al. 2003; Harris et al. 2014; Reidy et al. 2014; Sporn and Hines 2015).
Cells propagating [PSI+] do not show obvious growth defects under routine culture conditions, so although much Sup35 is incorporated into prion aggregates, Sup35 activity remains high enough to support normal growth. We earlier showed, however, that while a strong variant of [PSI+] could propagate in cells expressing Sis1JGF in place of Sis1, the prion was highly toxic in such cells (Kirkland et al. 2011). Thus, Sis1JGF possess enough Sis1 function to propagate [PSI+], but not enough to prevent a latent toxicity of [PSI+]. Subsequently, others using a different strain and prion variants found that strong [PSI+] prions are toxic in cells with Sis1 lacking only its CTD and that such cells propagate weak [PSI+] poorly (Stein and True 2014). Still, others showed that Sis1JGF propagates strong variants of [PSI+] without apparent toxicity, but it does not support weak [PSI+] variants (Harris et al. 2014). Here, we aimed to identify the basis of our observed prion toxicity, to define more clearly the activities of Sis1 or other possible factors that are important for protecting cells from this toxicity, and to resolve the apparent discrepancies. Our extensive genetic screening indicated that no other cellular factor could compensate for Sis1 to relieve the toxicity and that the CTD of Sis1 has a critical, unique, and transferable activity needed for the protection. Our findings also suggest that the toxicity is due to over-depletion of Sup35 and that Sis1 counteracts this effect by moderating aggregation of Sup35 in [PSI+] cells.
Materials and methods
Strains, media, and culture conditions
Strains are listed in Table 1; all are [pin–]. Our base strain 779-6A is related to but not isogenic to S288C. Our strong and weak variants of [PSI+] are designated by superscripts “S” and “WSL,” respectively. For example, 970T, 970TS, and 970TWSL are the same strain, but have no prions ([psi–]) or propagate [PSI+]S or [PSI+]WSL, as indicated. The source of the strong prion variant in this strain, inherited from 779-6A, is uncertain. Weak variant [PSI+]WSL (SL designates its source as Sue Liebman) was from strain L1759, isogenic to 74-D694 (Vishveshwara et al. 2009). Strain W303α and 74-D694, each with resident strong ([PSI+]STR) and weak ([PSI+]Sc37) Sup35 prions were from Justin Hines (Harris et al. 2014). Prions were cross-infected among the three strain backgrounds by cytoduction (Jung et al. 2000). Yeast were cured of prions by growing on medium containing 3 mM guanidine (Ferreira et al. 2001; Jung and Masison 2001). Mutants of Ade1 and Ade2 produce red phenotypes due to the accumulation of a byproduct of the disrupted adenine pathway. All strains have nonsense alleles of ADE1 or ADE2 that are suppressible by [PSI+], so [psi–] cells are red and ade–, [PSI+]W cells are pink and grow slowly without adenine and [PSI+]S cells are white and ade+.
Table 1.
Yeast strains
| Strain | Genotype | Source |
|---|---|---|
| 779-6A | MATa kar1-1 ade2-1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3-52 | Jung and Masison (2001) |
| W303α | MATα ade1-14 his3-11,15 leu2-3,112 sis1::LEU2 trp1-1 ura3-1/pRS316SIS1 (URA3, SIS1) | Harris et al. (2014) |
| 74-D694a | MATα ade1-14 his3Δ-200 leu2-3,112 trp1-289 ura3-52 sis1::LEU2/pRS316SIS1 (URA3, SIS1) | Harris et al. (2014) |
| 930 | 779-6A MATα sis1::KanMX4/pYW1 7 (URA3, SIS1) [psi–] | Kirkland et al. (2011) |
| 930S | 930, [PSI+]S (endogenous prion) | |
| 970L | 930 + pRS315Sis1JGF (LEU2, sis1JGF)) [psi–] | This study |
| 970LS | 970L, [PSI+]S (endogenous prion) | This study |
| 970LWSL | 970L, [PSI+]WSL (prion donor L1759) | This study |
| 970LSTR | 970L, [PSI+]STR (prion donor 1930 via 779-6A MATa) | This study |
| 970LSc37 | 970L, [PSI+]Sc37 (prion donor 1932 via 779-6A MATa) | This study |
| 970T | 930 + pYW62 (TRP1, sis1JGF) [psi–] | This study |
| 970TS | 970T, [PSI+]S (endogenous prion) | This study |
| 1930 | W303α, [PSI+]STR (endogenous prion) | Harris et al. (2014) |
| 1932 | 1930, [PSI+]Sc37 (endogenous prion) | Harris et al. (2014) |
| 1916 | 1930, [psi–] (cured of prions) | This study |
| 1919 | 1930, [PSI+]WSL (prion donor L1759) | This study |
| 1920 | 1930, [PSI+]S (prion donor 779-6A) | This study |
| 1935 | 74-D694, [PSI+]STR (endogenous prion) | Harris et al. (2014) |
| 1937 | 1935, [PSI+]Sc37 (endogenous prion) | Harris et al. (2014) |
| 1917 | 1935, [psi–] (cured of prions) | This study |
| 1922 | 1935, [PSI+]WSL (1917, prion donor L1759) | This study |
| 1923 | 1935, [PSI+]S (1917, prion donor 779-6A) | This study |
| 1362 | MATa sis1::KanMX SUP35NGMC/pRS314Sis1::FRT, [PSI+]S | This study |
| JS131 | 1362 + pRS313 + pRS315, [PSI+]S | This study |
| JS134 | 1362 + pRS313 + pC5GAL1-FLP, [PSI+]S | This study |
| JS133 | 1362 + pJE240 (HIS3, SIS1) + pRS315, [PSI+]S | This study |
| JS132 | 1362 + pJE240 (HIS3, SIS1) + pC5GAL1-FLP, [PSI+]S | This study |
| JS124 | 1362 + pJE236 (HIS3, sis1JGF) + pRS315, [PSI+]S | This study |
| JS127 | 1362 + pJE236 (HIS3, sis1JGF) + pC5GAL1-FLP, [PSI+]S | This study |
| JS136 | 1362 + pRS313 + pRS315, [psi–] | This study |
| JS135 | 1362 + pRS313 + pC5GAL1-FLP, [psi–] | This study |
| JS139 | 1362 + pJE240 (HIS3, SIS1) + pRS315, [psi–] | This study |
| JS140 | 1362 + pJE240 (HIS3, SIS1) + pC5GAL1-FLP, [psi–] | This study |
| JS138 | 1362 + pJE236 (HIS3, sis1JGF) + pRS315, [psi–] | This study |
| JS137 | 1362 + pJE236 (HIS3, sis1JGF) + pC5GAL1-FLP, [psi–] | This study |
| 628-8Cc | MATa ade2-1 can1 kar1 leu2 lys2 SUQ5 [rho–] (cytoduction tester) | This study |
| MR1118 | 930 + HO::Sup35NM-mKate2 yTRAP sensor | This study |
All strains are [pin–] and all [psi–] strains were obtained by guanidine curing. Strains with designations 1362, 930, 970, and JS are isogenic to 779-6A. The SUP35 allele (NGMC) in strain 1362 has GFP between the N and M domains (Song et al. 2005).
74-D694 received from Harris et al. was denoted 74D-694 by them. It is the same 74-D694 first described by Chernoff et al. (1995).
Media and growth conditions were as described (Sherman 2002). Cells were grown at 30° unless indicated otherwise. YPAD rich medium contains 1% yeast extract, 2% peptone, 400 mg/L adenine, and 2% glucose. 1/2YPD (limiting adenine) is the same but contains 0.5% yeast extract and lacks supplemented adenine. Synthetic complete media (SC) contain all nutrients except those needed for plasmid or prion selection. Solid media contain 2% agar and SC plates contain limiting adenine (8–10 mg/L) when monitoring prions. Where indicated (SGal, SRaf, SGalRaf), 2% galactose and raffinose were used in place glucose. Ingredients for growth media were from Difco, Becton Dickinson, and Sunrise Science. All other chemicals were from Sigma-Aldrich.
Plasmid shuffle (counter-selection for complementation of Sis1 function)
Counter-selection was done on SC plates supplemented with uracil and 1 g/L of 5-fluoroorotic acid (FOA). FOA does not destabilize plasmids or cause plasmids to be lost or evicted. FOA is converted by Ura3 enzyme to 5-fluorouracil, which becomes incorporated into RNA and thus kills cells that express Ura3. Single-copy CEN plasmids mis-segregate in roughly 2% of cell divisions (Masison and Baker 1992), so a small proportion of cells in any population, even under selection for the plasmid, will lack the plasmid. A sis1Δ strain with SIS1 on a URA3 plasmid was transformed by test plasmids and grown as patches on medium containing uracil to relieve selection for the URA3 plasmid. If the protein expressed from the test plasmid provides Sis1 function needed for growth, then cells having lost the URA3 plasmid are not at a growth disadvantage and there is little consequence of losing it. When replica-plated as thin layers of cells onto FOA plates, there typically will be enough Ura– cells to form confluent growth in 1 day. If the gene encoded on the test plasmid provides only partial Sis1 function, then the frequency of cells having lost the URA3 plasmid encoding SIS1 will be lower because cells having lost it will divide more slowly than those that retain it. When transferred to FOA, such cells will develop less dense patches because fewer of them will be Ura– and those that are Ura– will grow slower. If the test protein does not provide enough Sis1 function to support growth, then cells that lose the URA3 plasmid will not grow, while those that retain it will die when transferred to FOA, so no cells will be recovered on FOA.
All shuffle experiments were done using freshly transformed cells. For each test plasmid, we assess 6–12 transformants on FOA and for further experiments, we select and pool 3 or 4 from the pre-FOA plate that display the consensus FOA phenotype. Because yeast are highly adaptable and there are several ways strains can bypass FOA toxicity, we consider complementation inadequate if FOA-resistant cells are not recovered consistently and evenly spread among patches of individual transformants within 3–4 days at 30°. By this time, patches of even very slowly growing cells (>3 h/cell division) show clearly detectable growth.
Plasmids
Plasmids used are listed in Table 2. All our constructs are single-copy plasmids that contain genes regulated by the Sis1 promoter. The J-domain, GF/GM region, and carboxy-terminal domain (CTD) of Sis1 and corresponding three domains of Ydj1 were swapped to create the hybrid constructs (Reidy et al. 2014). Point mutations were created using QuikChange Lightning mutagenesis kit (Agilent). Plasmid pJE236 and pJE237 were made by moving the BamHI-SalI fragment from pYW62 into pRS313 and pRS315, respectively. Plasmid pRS314Sis1FRT was constructed by moving the BamHI-SalI fragment containing SIS1 from pYW65 (Yan and Craig 1999) into pC4FMCS (Park et al. 2011). Plasmid pJE160 was made by subcloning the BamHI-PmeI fragment from pCM189 (Gari et al. 1997) into pH403 (Edskes and Wickner 2013). Plasmid pJE241 was made by cloning Sup45, amplified by PCR on a BamHI-SalI fragment, into pRS315.
Table 2.
Plasmids
| Plasmid | Description | Marker | Source |
|---|---|---|---|
| pRS313 | Empty vector | HIS3 | Sikorski and Hieter (1989) |
| pRS314 | Empty vector | TRP1 | Sikorski and Hieter (1989) |
| pRS315 | Empty vector | LEU2 | Sikorski and Hieter (1989) |
| pRS316 | Empty vector | URA3 | Sikorski and Hieter (1989) |
| pYW17 | SIS1 (wild type) on YCp50 | URA3 | Yan and Craig (1999) |
| pYW65 | SIS1 (wild type) | TRP1 | Yan and Craig (1999) |
| pJE240 | SIS1 (wild type) | HIS3 | This study |
| pYW62 | SIS1Δ122-352 (ΔGMCTD = JGF) | TRP1 | Yan and Craig (1999) |
| pYW66 | SIS1Δ171-352 (ΔCTD) | TRP1 | Yan and Craig (1999) |
| p108 | SIS1Δ70-121 (ΔGF) | TRP1 | Yan and Craig (1999) |
| pJE236 | SIS1Δ122-352 (ΔGMCTD = JGF) | HIS3 | This study |
| pJE237 | SIS1Δ122-352 (ΔGMCTD = JGF) | LEU2 | This study |
| pGCH1 | SIS1K199A | TRP1 | Kirkland et al. (2011) |
| pAK1 | SIS1Δ338-352 (ΔDD) | TRP1 | Kirkland et al. (2011) |
| pAK50 | PSIS1::HDJ1::terSIS1 | TRP1 | Kirkland et al. (2011) |
| pMR266T | SIS1cmyc | TRP1 | Reidy et al. (2014) |
| pMR266ΔDD | SIS1ΔDDcmyc | TRP1 | Reidy et al. (2014) |
| pMR267 | YDJ1cmyc | TRP1 | Reidy et al. (2014) |
| pMR274 | YSScmyc | TRP1 | Reidy et al. (2014) |
| pMR274ΔDD | YSSΔDDcmyc | TRP1 | Reidy et al. (2014) |
| pMR275 | SYYcmyc | TRP1 | Reidy et al. (2014) |
| pMR276 | SYScmyc | TRP1 | Reidy et al. (2014) |
| pMR276ΔDD | SYSΔDDcmyc | TRP1 | Reidy et al. (2014) |
| pMR277 | YSYcmyc | TRP1 | Reidy et al. (2014) |
| pMR278 | SSYcmyc | TRP1 | Reidy et al. (2014) |
| pMR279 | YYScmyc | TRP1 | Reidy et al. (2014) |
| pMR279ΔDD | YYSΔDDcmyc | TRP1 | Reidy et al. (2014) |
| pMR293 | PGPD::YYScmyc | TRP1 | Reidy et al. (2014) |
| pJE160 | SUP35 codons 254-685 | HIS3 | This study |
| pJ528 | SUP35 codons 124-685 | LEU2 | Kirkland et al. (2011) |
| pJE161 | SUP45 | LEU2 | This study |
| pC5GAL1-FLP | PGAL1::FLP | LEU2 | Park et al. (2011) |
| pRS314Sis1FRT | FRT::SIS1::FRT | TRP1 | This study |
| pJE270 | PR3-N-Sis1 | TRP1 | This study |
| pJE271 | PR3-N-Sup35 | TRP1 | This study |
| pJS272 | DHB1-Sis1 | LEU2 | This study |
| pJE273 | DHB1-Sis1JGF | LEU2 | This study |
| pJE274 | DHB1-Sis1-D36N | LEU2 | This study |
| pJE275 | DHB1-Sis1JGF-D36N | LEU2 | This study |
All plasmids are single-copy vectors. All except pYW17 and JE270-275 are derived from pRS313, pRS314, pRS315, or pRS316. All Sis1 and Sis1/Ydj1 hybrid constructs are regulated by the SIS1 promoter except where indicated. All other genes are regulated by endogenous promoters except where indicated (Pxxx).
Yeast two-hybrid plasmids were constructed according to the manufacturer’s instructions using empty bait and prey vectors provided with the DUALhunter system (Dualsystems Biotech AG). Briefly, alleles (including promoters) were cloned using the two different SfiI sites provided in the multiple cloning site of the DUALhunter bait (pDHB1) and prey (pPR3-N) plasmids. All constructs were sequence verified.
Overexpression suppressor screening
Strain 970TS, which is sis1Δ, propagates [PSI+]S and carries pYW17 (URA3, Sis1 wild type) and pYW62 (TRP1, Sis1 amino acids 1–121) was transformed by a high-copy LEU2-based tiling array library (Jones et al. 2008) that contains ∼1600 independent plasmids encoding defined segments of the yeast genome (functionally 95% of genome). Transformants were selected on plates lacking tryptophan and leucine and containing uracil. These plates were replica-plated onto FOA medium and FOA-resistant colonies were isolated. Genes present on the LEU2 plasmids were identified by sequencing. We repeated the screen using a separate, randomly assembled yeast genomic library on a TRP1-based high-copy plasmid obtained from ATCC (cat. no 77162). For the TRP1-based screen, we used strain 970LS, which is identical to 907TS except it has pJE237 (LEU2) in place of pYW62.
Sis1 depletion using Flp/FRT and fluorescent microscopy
Sis1 was depleted in strain 1362 using the Flp/FRT recombination system (Park et al. 2011). Versions of strain 1362 carry pRS314Sis1FRT (TRP1, SIS1 flanked by Flp Recombinase Target sequences), pRS315Flp (LEU2, galactose inducible Flp), and pRS313 (HIS3) empty vector or carrying wild-type Sis1 or Sis1JGF. Cells grown overnight in liquid SC medium selecting for the TRP1, LEU2, and HIS3 plasmids were transferred to SRaf medium, grown overnight, and then diluted into SGalRaf medium to OD600 = 0.25. Depletion of Sis1 during subsequent outgrowth was monitored by western analysis (see below). Cultures of cells with empty pRS315 vector in place of pRS315Flp were treated similarly as nondepleted (mock) controls.
Microscopic observation of Sup35-GFP (NGMC) in log-phase cells was done immediately after the shift to galactose and 24 h later using a Nikon E-800 microscope. Images were captured using Nikon software with a Q-Imaging Retiga EXi digital camera, Plan APO VC 60X oil immersion DIC optics, and GFP filter. Images were processed for presentation using Adobe Photoshop software.
Protein expression and western analysis
Cell lysates were prepared as described (Reidy et al. 2014). Briefly, harvested cells were suspended in tris-HCl lysis buffer and broken by agitation with zirconium beads using a BioSpec bead beater. Protein concentration was measured using Bradford reagent. For western analysis, 10–15 µg of protein from cell lysates was separated on 4–20% SDS-PAGE gels and transferred to PVDF membranes. Primary antibodies were rabbit anti-Sis1 raised against an epitope in the carboxy-terminal domain of Sis1 and rabbit anti-Sup35 (a gift from Sue Liebman). Blotted membranes were subsequently stained with amido-black (Sigma # A-8181) as a loading and transfer control.
Yeast two-hybrid interaction assay
An established split-ubiquitin-based DUALhunter system was used to assess protein interactions in vivo (Mockli et al. 2007). Versions of Sis1 (bait) were fused at the N-terminus to the small membrane protein Ost4p to anchor at the membrane or at the C-terminus to a reporter module encoding the C-terminal half of ubiquitin (Cub) followed by transcription activator LexA-VP16. Sup35 and Sis1 (prey) were expressed as fusions to N-terminal half of ubiquitin (Nub). An interaction between bait and prey brings Nub and Cub close enough to reconstitute “split-ubiquitin,” which is recognized by ubiquitin-specific proteases that release LexA-VP16 to activate transcription of HIS3, ADE2, and lacZ reporter genes. Transformants of strain NMY51 [MATa, his3Δ200, trp1-901, leu2-3,112, LYS2::(lexAop)4-HIS3, ura3::(lexAop)8-lacZ, ade2::(lexAop)8-ADE2, and Gal4] were maintained on SC selecting for plasmids and then replica-plated onto similar medium lacking histidine and adenine or containing X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside). Empty bait and prey plasmids were used as a negative control. Full-length Sis1 forms dimers, so Sis1 bait and prey plasmids were used as a positive control.
Centrifugal fractionation of cell lysates
Two similar methods were used. In one, log-phase cells collected by centrifugation were lysed by agitation with zirconium beads using a BioSpec bead beater in buffer “B” (10 mM NaPO4, 250 mM NaCl, 2% SDS, 1% TritonX-100, and PMSF) containing cOmplete Mini Protease Inhibitor Cocktail (Roche). Supernatants of lysates that were cleared by centrifugation for 8–9 min at 500 ×g represent “Total” lysates. Protein in Total lysates was normalized to 12 mg/mL by diluting with buffer “B.” A portion of Total lysate was centrifuged for 45 min at 80,000 rpm in rotor TLA-120 (Beckman Coulter). The supernatant (“Soluble” fraction) was collected, and the “Pellet” was suspended in a volume of buffer “B” equal to the fractionated portion of the Total lysate. Equal volumes of Total, Soluble, and Pellet fractions representing 10 µg of Total lysate were separated by SDS-PAGE, blotted, and probed using anti-Sup35 antibody (Bagriantsev et al. 2008).
In the other fractionation method, cells were broken in 1X TBS containing 0.1% Triton X-100, 1 mM PMSF, and cOmplete protease inhibitor cocktail (Roche) and cleared by centrifugation for 3 min at 3000 ×g. The soluble (“S”) sample was the supernatant of a subsequent centrifugation for 20 min at 21,000 ×g. After removal of the supernatant, the pellet (“P”) was dissolved in an appropriate volume of 1X SDS loading dye (50 mM Tris-HCl pH 7.5, 1.5% SDS, 0.5% beta-mercaptoethanol, 5% glycerol, and bromphenol blue). All steps were performed at 4° or on ice.
FACS analysis
To monitor aggregated Sup35 in derivatives of strain 930, chromosomal integration of yTRAP mKate fluorescent reporter at the HO locus was done by transforming 930S with NotI-digested pSK260 (Addgene #129252; Newby et al. 2017). The resulting strain MR1118S was made [psi–] (strain MR1118) via passage on 3 mM guanidine-HCl. MR1118 and MR1118S were cotransformed with plasmid pJE160 (Sup35MC) and either pYW65 (full-length Sis1) or pYW62 (Sis1JGF). FOA-resistant isolates were then obtained as described above. Log-phase cultures in SC medium lacking histidine and tryptophan and containing 100 mg/L adenine were analyzed using FACS as described (Newby et al. 2017). DAPI (1 mg/L) was used as a vital stain. A BD Fortessa equipped with 407, 488, 532, and 633 laser lines was used for sample acquisition and data were analyzed using DIVA™ 6.1.2 software (BD) and FlowJo (FlowJo, LLC).
Results
No substitute for Sis1 in protecting from [PSI+] toxicity
As a first approach to understand how [PSI+] prions cause toxicity in cells expressing Sis1JGF, we used a genetic “plasmid shuffle” screen to search for factors other than Sis1 whose overexpression might mitigate the toxicity. Strain 970TS is a sis1Δ strain that has a URA3 plasmid encoding wild-type SIS1 (pYW17) to ensure viability, a TRP1 plasmid to express Sis1JGF (pYW62), and the strong prion variant [PSI+]S. If 970TS cells lose the URA3 plasmid they stop growing because Sis1JGF cannot protect them from the toxicity of [PSI+]S. We transformed 970TS using pools of a LEU2-based genomic library and selected transformants on medium containing uracil to allow loss of the URA3 plasmid. We then replica-plated the transformants to medium containing FOA, which kills cells that express URA3. Only transformants encoding proteins that alleviate the toxicity enough to allow cells to lose the URA3 plasmid and survive with Sis1JGF in place of Sis1 can grow (see Materials and Methods).
Among roughly 6000 transformants (covering roughly three to four genomes), we isolated two candidates. The library plasmids recovered from both of them carry YJL055W (LOG1). Log1 allows the growth of URA3+ cells in the presence of 5-FOA by reducing incorporation of 5-fluorouracil into RNA, so it was possible the isolated candidates still had the URA3 plasmid encoding wild-type Sis1. Indeed, both FOA-resistant isolates were Ura+. We did not isolate SIS1 because the library we chose (see Materials and Methods) did not contain an intact SIS1 gene. Using the same approach with a separate genomic library containing randomly assembled inserts averaging 8–12 kb, we recovered six FOA-resistant candidates among roughly 10,000 colonies screened. Library plasmids isolated from all six contained SIS1. These genomic screening results strongly suggest that no other yeast gene can function in place of SIS1 to prevent the toxicity caused by [PSI+]S.
CTD of Sis1 was needed to counteract [PSI+] toxicity
Much data show that altering structural regions of Sis1 and its closest human homolog DnaJB1 (here designated Hdj1), which supports the growth of yeast in place of Sis1, can influence the propagation of various yeast prions (Higurashi et al. 2008; Kirkland et al. 2011; Harris et al. 2014; Reidy et al. 2014; Stein and True 2014; Killian and Hines 2018). Yet, a systematic analysis of how different regions of Sis1 and Hdj1 contribute to protecting cells from toxicity of [PSI+] prions has not been done. Moreover, few reports resolve whether altered Sis1 or other protein quality control factors fail to support prions because they are unable to promote prion propagation or to protect from prion toxicity. Earlier we showed that transient inhibition of Hsp104 activity, which reduces seed numbers of [PSI+]S, reduces [PSI+]S toxicity enough to prevent it from killing cells expressing Sis1JGF in place of Sis1 (Kirkland et al. 2011). When the inhibition is withdrawn and Hsp104 activity is restored, [PSI+]S seed numbers recover, and toxicity reappears. These observations suggest that “weak” variants of [PSI+] prions, which have fewer seed numbers per cell and a concomitant higher amount of soluble Sup35 than strong variants (Zhou et al. 1999; Uptain et al. 2001), might not be toxic enough to kill Sis1JGF cells.
We therefore tested if Sis1JGF could support the growth of cells propagating a weak [PSI+] variant by using the same strategy as in the screen. In addition, we tested which parts of Sis1 (Figure 1A) were able to protect cells expressing only Sis1JGF from toxicity caused by strong [PSI+]. Among these is the K199A substitution of a residue involved in interaction with both substrates and the Hsp70 C-terminus (Lee et al. 2002; Li et al. 2006), and deletions of the CTD and GF regions (ΔCTD, ΔGF). We also included Hdj1. All test proteins are expressed from single-copy plasmids regulated by the SIS1 promoter to mimic expression of Sis1. Since they are expressed from a TRP1 plasmid, we used strain 970L, which is identical to 970T except it expresses Sis1JGF from a LEU2 plasmid. 970L is [psi–], 970LS propagates [PSI+]S and 970LWSL propagates a weak [PSI+] variant from Sue Liebman (see Materials and Methods).
Figure 1.

Sis1JGF supports propagation of weak [PSI+]. (A) Domain structures of Sis1, Hjd1, and mutant versions of Sis1. Numbers indicate amino acid positions. The CTD (amino acids 172–336 of Sis1) is the substrate-binding domain and the J-domain (amino acids 1–68) binds and regulates Hsp70. (B) Plasmid shuffle to assess the ability of proteins to support growth and prions in place of Sis1. Strain 970L (relevant genotype at top) lacks chromosomal SIS1, expresses Sis1JGF from a LEU2 plasmid (pJE237), and wild-type Sis1 from a URA3 plasmid (pYW17). It lacks prions ([psi–]) or propagates [PSI+]S (strong) or [PSI+]WSL (weak) as indicated. Cells were transformed by a TRP1 vector encoding the versions of Sis1 or Hdj1 (pAK50) as indicated on the left (ev is empty TRP1 vector). Transformants were grown on medium lacking only leucine and tryptophan (-LT) and replica-plated to similar medium containing FOA, which kills cells that have not lost the URA3 plasmid encoding wild-type Sis1. FOA plates, which contain adenine to allow the growth of cells without prions, were incubated 2 days at 30°. All cells growing on FOA express Sis1JGF and the proteins indicated on left; those with empty TRP1 vector express only Sis1JGF. Patches are pools of 3–4 individual transformants (see text and Supplementary Figure S1). (C) Cells of [psi–] strain 970L expressing Sis1 from a TRP1 plasmid (wt, express both Sis1 and JGF) or with empty TRP1 vector (ev, express only JGF) were recovered from FOA plate in (B), grown in YPAD and dilutions were inoculated onto YPAD plates and incubated at the indicated temperatures for 3–4 days. (D) FOA-resistant [psi–] and [PSI+]WSL cells expressing Sis1JGF in place of Sis1 were recovered from FOA and grown similarly. Generation times of these two strains are 154 and 193 min/div as indicated.
Strains 970L, 970LS, and 970LWSL were transformed by the TRP1 plasmids encoding the test proteins and selected on -trp plates that lack leucine to maintain selection for the plasmid encoding Sis1JGF and contain uracil to allow loss of the URA3 plasmid encoding wild-type Sis1. Three to four individual transformants (see Materials and Methods, Supplementary Figure S1) were pooled and grown as patches of cells on similar medium and then replica-plated onto the same medium containing FOA (Figure 1B, see Materials and Methods). Any strains that grow on FOA express only Sis1JGF from the LEU2 plasmid and a test protein from a TRP1 plasmid. FOA-resistant cells with the empty TRP1 plasmid express only Sis1JGF. This configuration allows us to test which parts of Sis1 prevent the toxicity of [PSI+] independently of the degree to which the test proteins provide Sis1 activity needed for growth. The growth requirement is always supplied by Sis1JGF, but Sis1JGF cannot prevent [PSI+] toxicity.
For the [psi–] version of strain 970L (lacking prions), we recovered FOA-resistant cells (having lost the URA3 plasmid encoding wild-type Sis1) from all transformants (Figure 1B, left panels, “FOA”). Cells from the FOA plates that express only Sis1JGF (i.e., with the empty TRP1 vector) grew noticeably more slowly than those expressing both Sis1JGF and Sis1. These cells were mildly sensitive to low temperature (15°), inviable at high temperature (37°), and grew best at the suboptimal 25° (Figure 1C). These results are consistent with those found earlier showing that Sis1JGF does not provide full Sis1 activity needed to support optimal growth (Yan and Craig 1999).
For strain 970LS, we did not recover FOA-resistant cells of empty vector transformants (Figure 1B, center panels), which is in line with our earlier work showing Sis1JGF cannot protect cells from toxicity caused by [PSI+]S (Kirkland et al. 2011). Additionally, coexpressing Hdj1 with Sis1JGF failed to allow recovery of FOA-resistant cells. We also failed to recover FOA-resistant cells coexpressing Sis1ΔCTD and Sis1JGF, suggesting the substrate-binding CTD provides the protection from toxicity. In contrast, transformants expressing wild-type Sis1 from the TRP1 plasmid were recovered on FOA after 1 day. The K199A and ΔGF mutants showed wild-type Sis1 phenotypes, indicating these alterations do not hinder the ability of Sis1 to protect from the toxicity. Note that the FOA plates contain adenine, indicating that failure to grow is not due to prion loss.
For strain 970LWSL, we recovered FOA-resistant colonies of all transformants, including those with the empty TRP1 vector that express only Sis1JGF (Figure 1B, rightmost panels). Because weak [PSI+] prions are modestly unstable, we used cytoduction to confirm that all strains with weak [PSI+] variants recovered from FOA had not lost the prion (see Materials and Methods, Supplementary Figure S2). [PSI+]WSL cells expressing only Sis1JGF, and those expressing both Sis1JGF and Sis1ΔCTD, formed colonies more slowly than their isogenic [psi–] variants (Figure 1, B and D), which indicates that [PSI+]WSL had noticeable toxicity that is revealed when the Sis1 CTD is absent. Thus, as anticipated, [PSI+]WSL was toxic in a way that Sis1JGF could not counteract, but it was not toxic enough to be lethal.
These results show that Sis1JGF can propagate weak [PSI+] prions and they are consistent with our earlier finding that toxicity of [PSI+] is diminished in cells with reduced prion seed numbers, an observation seen by others (Harris et al. 2014). For [psi–] transformants, cells with Sis1JGF that coexpressed any of the test proteins grew faster than cells with Sis1JGF and the empty TRP1 vector (Figure 1B), which indicates all the test proteins possess Sis1 activity important for growth that is lacking in Sis1JGF.
Ydj1, a class A J-protein and the other major yeast cytosolic J-protein, cannot support growth in the absence of Sis1 (a class B J-protein) even when overexpressed. Using Sis1/Ydj1 hybrid proteins with swapped J, GF, and CTD domains (see Figure 2A), we earlier showed that a hybrid containing only the CTD of Sis1 (YYS) supports the growth of sis1Δ cells and propagates [PSI+]S (Reidy et al. 2014). In contrast, all Sis1/Ydj1 hybrids that contain the CTD of Ydj1 do not support the growth of sis1Δ cells, regardless of prion status. Those findings show that the CTD of Sis1 possesses an essential and transferable function unique to Sis1 and data herein suggest this CTD also has a unique ability to subdue toxic effects of [PSI+]. We used our system to test the extent that the three Sis1/Ydj1 hybrids containing the CTD of Sis1 (SYS, YSS, or YYS) might protect cells expressing Sis1JGF from prion toxicity.
Figure 2.

Sis1 CTD (see Figure 1) is enough to propagate weak [PSI+] prions. (A) Sis1 domains J (amino acids 1–68), GF/GM (69–172), and CTD (173–352) were swapped with analogous Ydj1 domains J (1–70), GF (71–106), and CTD (107–409). Numbers indicate amino acid residue positions. Ydj1 has a CaaX box prenylation site at its carboxy terminus. (B) Sis1/Ydj1 hybrid proteins with swapped J, GF, or CTD regions as indicated were assessed in strain 970L (relevant genotype at top) by plasmid shuffle as in Figure 1 for the ability to support [PSI+]S and [PSI+]WSL prions in cells expressing Sis1JGF in place of Sis1.
Coexpressing SYS, YSS, or YYS with Sis1JGF in [psi–] cells improved growth compared with cells expressing Sis1JGF alone (Figure 2B). Additionally, all full-length hybrids supported normal growth of cells with [PSI+]S or [PSI+]WSL. Thus, when appended to Ydj1 in place of the Ydj1 CTD, the Sis1 CTD was enough to confer Sis1 function needed for normal growth and for complete protection from the toxicity of both prion variants.
The same three Sis1 CTD-containing hybrids that lack the dimerization domain (SYSΔDD, YSSΔDD, or YYSΔDD) also supported normal growth of [psi–] cells and cells propagating [PSI+]WSL (Figure 2). These results show Sis1 monomers function in prion propagation and they align with the ability of Sis1JGF and Sis1ΔCTD, which also lack the dimerization region, to support a weak [PSI+] variant. However, the ΔDD hybrids with the GF region of Ydj1 (SYSΔDD and YYSΔDD) failed to support the growth of cells with [PSI+]S, suggesting they can support a [PSI+]S variant but cannot protect from [PSI+]S toxicity. Although cells expressing Sis1ΔDD also propagated [PSI+]S, they grew noticeably more slowly than [PSI+]S cells expressing full-length Sis1 (Figure 2, third row, center panel), indicating dimerization of Sis1 is needed to fully counteract the toxicity caused by [PSI+]S. Together these results suggest the CTD of Sis1 is the major contributor to the protection and they point to an interaction between the Sis CTD and GF regions as necessary for monomers to provide complete protection. Earlier findings show an interaction between CTD and GF is important for function of class B J-proteins (Yu et al. 2015; Faust et al. 2020), and an interaction between the Sis1 CTD and Ydj1 GF regions can be expected to be less functional.
All of the test proteins evaluated here that were previously shown to support growth and prions on their own also supported growth and prions for cells expressing Sis1JGF in place of Sis1. These results are more consistent with the conclusion that Sis1JGF cannot mitigate the toxicity than the interpretation that expressing Sis1JGF makes [PSI+] toxic.
Curiously, an extra copy of wild-type YDJ1 reduced the ability of Sis1JGF to support the growth of cells with or without prions (Figure 2). Even the transformants without prions grew so poorly on FOA that we had no confidence in using them. In line with these results and our data above, we failed to recover [PSI+]S cells coexpressing Sis1JGF and the extra Ydj1. Our results agree with earlier work showing elevated expression of Ydj1 fails to support growth when Sis1 is absent (Luke et al. 1991). Further work will be needed to determine how the added dosage of Ydj1 interferes with Sis1JGF function.
Sup35C alleviated [PSI+]S toxicity
We earlier found that toxicity of [PSI+]S in cells expressing only Sis1JGF was not overcome by coexpressing Sup35MC, which is a truncated Sup35 that lacks the amyloid-forming NTD and should not become incorporated into prion aggregates (Kirkland et al. 2011). We therefore concluded that [PSI+]S toxicity was not due to excessive depletion of the essential Sup35 protein into prion aggregates. Our data above, however, suggest the toxicity of [PSI+]S could be due at least in part to depletion of Sup35. We therefore re-examined the degree to which Sup35 lacking its amyloid-forming domain could suppress the toxicity. In addition to Sup35MC, we tested Sup35C, which lacks the adjacent M-region, and Sup45, an essential protein that interacts with Sup35 to promote translation termination and is found in [PSI+] prion aggregates (Stansfield et al. 1995; Paushkin et al. 1997b; Vishveshwara et al. 2009; Pezza et al. 2014).
When Sup35C or Sup35MC was coexpressed with Sis1JGF, we recovered FOA-resistant cells that propagated [PSI+]S (Figure 3). These cells grew more slowly than [psi–] cells expressing only Sis1JGF (Table 3), however, showing that rescue from the toxicity was only partial. Coexpressing Sup45 with Sis1JGF did not overcome the toxicity enough to allow growth, but when Sup45 and Sup35C were both coexpressed with Sis1JGF the FOA-resistant cells that we recovered grew as fast as the [psi–] Sis1JGF cells (Figure 3 and Table 3). Note that expression of Sup35C or Sup35MC restores translation termination, and thus ade– and red phenotypes, without affecting the propagation of prions composed of the endogenous full-length Sup35 (Figure 3 and Supplementary Figure S3). These results suggest that the toxicity of [PSI+]S is due primarily to depletion of Sup35 into prion aggregates. The ability of extra Sup45 to improve growth, but only when Sup35C is present, suggests that the residual toxicity in cells expressing Sup35C is due to partial depletion of Sup45 into the aggregates of full-length Sup35, which limits its availability to function with Sup35C.
Figure 3.
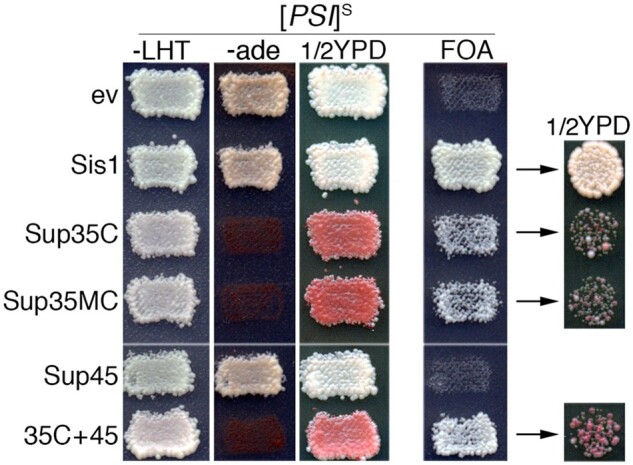
Sup35C restores growth of [PSI+]S cells expressing Sis1JGF. Strain 970TS (sis1Δ expressing Sis1JGF from TRP1 plasmid pYW62 and Sis1 from URA3 plasmid pYW17) was transformed by empty vector (ev) or plasmids encoding Sis1 (pJE240, HIS3), Sup35C (pJE160, HIS3), Sup35MC (pJ528, LEU2), or Sup45 (pJE161, LEU2) together with appropriate empty vectors to maintain all strains on the same selection plate. Transformants were assessed for the ability of Sis1JGF to support growth in place of Sis1 (i.e., on FOA) as in Figure 1. -LHT contains uracil, but lacks leucine, histidine, and tryptophan. YPD and -ade plates show relevant prion phenotypes of transformants. All FOA-resistant strains lack pYW17 and express Sis1JGF from plasmid pYW62. The rightmost column shows cells recovered from FOA, diluted to the same cell density, and grown on rich medium for 2 days to show color and growth phenotypes.
Table 3.
Growth rates (min/cell div)
| Proteins, plasmids | [psi–] | [PSI+]S |
|---|---|---|
| JGF+ev+ev | 155 | na |
| JGF+Sis1+ev | 110 | 117 |
| JGF+Sup35C+ev | 160 | 175 |
| JGF+Sup35MC+ev | 150 | 168 |
| JGF+Sup35C+Sup45 | nd | 149 |
Strain used is 970T (sis1Δ expressing Sis1JGF from a TRP1 plasmid) with combinations of indicated empty vectors (ev) or plasmids encoding indicated proteins (see Figure 3). Cells were grown in YPAD at 30° with constant shaking at 220 rpm. OD600 was measured at 10-min intervals for 24 h. na, not applicable as Sis1JGF [PSI+]S cells are not viable; nd, not determined.
Sis1 maintains Sup35 solubility in [PSI+]S cells
Differences in the strength of [PSI+] nonsense suppression phenotypes correlate with differences in the extent that Sup35 is depleted into insoluble prion aggregates (Jung et al. 2000; Uptain et al. 2001). Excessive depletion of Sup35 into [PSI+] prion aggregates can be lethal (McGlinchey 2011, #43). Accordingly, although there is evidence that Sup35 in prion aggregates might retain some function (Baxa et al. 2011; Pezza et al. 2014), it is generally accepted that the Sup35 in prion aggregates has little release factor activity. The viability of [PSI+] cells therefore depends mostly on the residual soluble Sup35.
To test the importance of Sis1 in maintaining the solubility of Sup35 in [PSI+] cells, we depleted Sis1 and then visually monitored the aggregation status of a functional GFP-tagged version of Sup35 (designated NGMC) expressed in place of Sup35 from the endogenous chromosomal locus (Song et al. 2005). Fluorescent prion aggregates of NGMC are seen as many small, disperse foci (Figure 4) that are highly mobile (Song et al. 2005).
Figure 4.
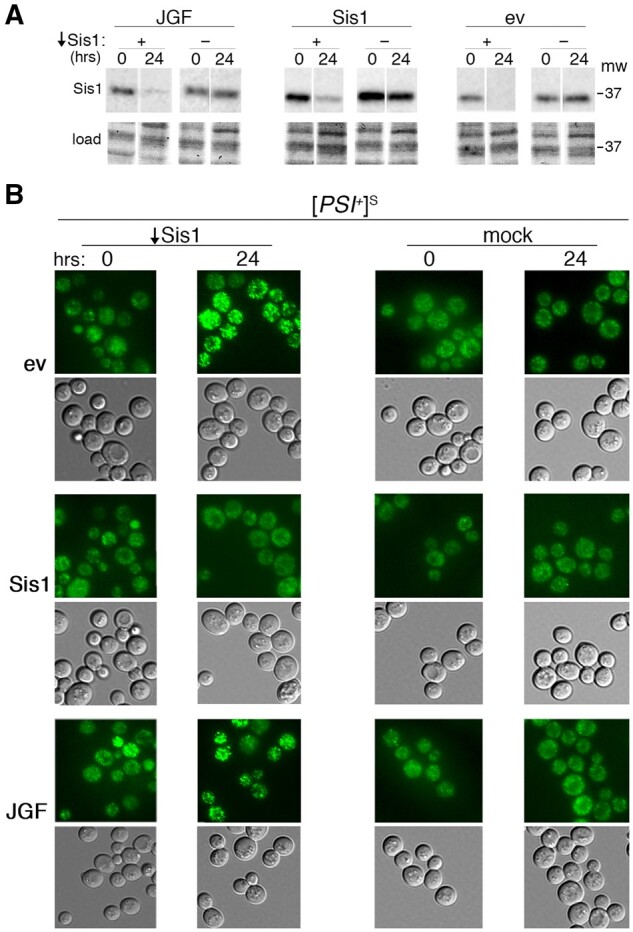
Sup35GFP forms brighter foci in cells depleted of Sis1. Flp-FRT-based recombination was used to deplete Sis1 by excising a plasmid-borne SIS1 gene in sis1Δ strain 1362, which propagates [PSI+]S and expresses NGMC (Sup35GFP) in place of Sup35 from the chromosomal SUP35 locus. (A) Western analysis showing abundance of Sis1 in cells immediately after (0) and 24 h after inducing depletion. (B) Fluorescent images of Sup35GFP were obtained at the same time points, as indicated above. Cells also carry an empty vector (ev) or the same vector encoding wild-type Sis1 or Sis1JGF (JGF) as indicated on the left. “Mock” columns are identical except cells carry the empty vector in place of the one encoding the inducible Flp recombinase.
We used [PSI+]S versions of NGMC strain 1362, which is sis1Δ and carries a TRP1 plasmid encoding a SIS1 gene flanked by FRT sites. This SIS1 gene can be excised by galactose-induced expression of Flp recombinase encoded on a separate LEU2 plasmid (see Materials and Methods; Park et al. 2011). Sis1 protein then becomes depleted by normal turnover and dilution among cells of the growing population. We excised the FRT-flanked SIS1 gene in cells that also carry an HIS3 plasmid encoding wild-type Sis1 or Sis1JGF, or the empty HIS3 vector.
As anticipated, the amount of Sis1 remaining 24 h after inducing excision of the SIS1 gene was essentially undetectable for cells with the empty HIS3 vector and was reduced to a one-gene amount in cells with the extra copy of wild-type SIS1 (Figure 4A). Cells expressing Sis1JGF had a detectable, but considerably reduced amount of full-length Sis1.
After depleting Sis1 in cells carrying the empty HIS3 vector, we observed a visually obvious change in the fluorescence of prion aggregates. The aggregates became strikingly brighter in 70% of cells (Figure 4B, top rows, Table 4). Thus, in a large proportion of cells the size of [PSI+]S aggregates increased when abundance of Sis1 decreased. In contrast, after similarly depleting Sis1 in cells that also express wild-type Sis1 from the HIS3 plasmid, we saw no increase in the proportion of cells with brighter foci (Figure 4B, middle rows, Table 4). After depleting Sis1 in cultures that express Sis1JGF from the HIS3 plasmid, however, foci in half of the cells had the brighter phenotype. This high proportion of Sis1JGF cells with the brighter aggregates is nine times greater than that of cells that retained a copy of wild-type SIS1, and it was similar to cultures with the empty HIS3 vector (Figure 4B, bottom rows, Table 4). These results provide clear visual evidence suggesting that Sis1JGF is much less capable than Sis1 at limiting incorporation of Sup35 into prion aggregates of [PSI+]S cells.
Table 4.
Cells with large Sup35 foci (%) after Sis1 depletion
| Sis1 Depleted? | 2nd copy of Sis1 | Hours Sis1 depleted |
|
|---|---|---|---|
| 0 | 24 | ||
| Yes | Wild type | 14 ± 2 | 6 ± 3 |
| No | Wild type | 8 ± 3 | 2 ± 1 |
| Yes | None | 14 ± 1 | 75 ± 8 |
| No | None | 5 ± 2 | 7 ± 2 |
| Yes | JGF | 14 ± 3 | 56 ± 7 |
| No | JGF | 12 ± 3 | 20 ± 5 |
Values are average percentages of cells (±SD) with visually obvious bright foci of fluorescent Sup35NGMC from cultures shown in Figure 4. Different pools of 200–420 cells were scored blindly by two people and the data were combined to derive each value. Cells carry pRS314Sis1FRT, the indicated second copy of Sis1, and either pRS315Flp (yes) or pRS315 empty vector (no). Sis1 was depleted by galactose-induced Flp-mediated excision of SIS1 from RS314Sis1FRT. Cells were scored immediately after the shift to galactose (0) and 24 h later.
In matching [PSI+]S cultures that were treated similarly but had an empty LEU2 vector in place of the one encoding Flp recombinase (Figure 4B, “mock”), or in [psi–] cells with or without Sis1 depletion (Supplementary Figure S4), there were no noticeable fluorescence changes in any of the strains after switching to excision-inducing medium. Thus, the observed differences in NGMC aggregation seen in Figure 4 required the presence of the prion and were not due to notable differences in extent of Sis1 depletion or to unrelated physiological effects caused by the shift to galactose medium.
It is widely reported that most Sup35 in [PSI+]S cells is incorporated into insoluble prion aggregates that are readily separated by centrifugation. We used an established centrifugation assay (see Materials and Methods) as another way to compare the solubility of Sup35 in [PSI+]S cells expressing Sis1JGF or wild-type Sis1. Because Sup35C is needed to maintain the viability of [PSI+]S cells expressing Sis1JGF in place of Sis1, we coexpressed it in wild type and Sis1JGF strains.
In lysates of Sis1JGF cells, we observed a noticeable reduction in soluble Sup35 and a corresponding increase in the relative amount of insoluble Sup35 in pellet fractions (Figure 5A). For [psi–] cells, we saw very little difference in amounts or proportions of Sup35 in the different lysate fractions from cells expressing Sis1JGF or wild-type Sis1. Thus, coexpressing Sup35C had little effect on abundance or solubility of full-length Sup35.
Figure 5.

Sup35 is less soluble in cells expressing Sis1JGF in place of Sis1. (A) Shown is an immunoblot of whole lysates (T) of cells expressing Sis1JGF (JGF) or wild-type Sis1 (wt) separated by centrifugation into supernatant (S) and pellet (P) fractions, probed with Sup35 antibody. Cells coexpress Sup35C to ensure viability of Sis1JGF [PSI+]S cells. [psi–] cells, with and without Sup35C, are shown for comparison. The upper band (FL) is full-length Sup35, lower band (C) is Sup35C (amino acids 254–685). The lower panel shows relevant portion of the blotted membrane stained by amido-black as a loading and transfer control. (B) Relevant sections of blots from three additional replicates using [PSI+]S cells and a slightly modified version of the fractionation assay (see Materials and Methods) and yTRAP sensor strain MR1118S. (C) Relative proportions of soluble Sup35 in [PSI+]S cells expressing wild-type Sis1 (19.5 ± 5.8) or Sis1JGF (3.4 ± 2.2) were measured using Image-J. Values are averages of the four [PSI+]S replicates shown; error bars indicate SD (**P < 0.01; t-test). (D) Sup35NM-mKate2 yTRAP sensor was used to monitor Sup35 aggregation in MR1118S (105 live cells) expressing wild-type Sis1 or Sis1JGF. Profile of Sis1JGF [psi–] cells is shown; wild-type Sis1 [psi–] cells were essentially indistinguishable.
As an additional approach to compare amounts of soluble Sup35 in [PSI+] cells, we used the yTRAP system (Khalil et al. 2012; Newby et al. 2017). yTRAP is a fluorescence-based tool to monitor and quantify aggregation states of proteins in vivo. It was developed using Sup35 and [PSI+] to demonstrate its usefulness. In the yTRAP system, a Sup35NM-transcription factor fusion protein activates the expression of the mKate2 reporter. This fusion protein can incorporate into Sup35 prion aggregates, and when it does it loses its ability to activate mKate expression. The extent to which mKate fluorescence becomes diminished reports on the extent that Sup35 is aggregated. We integrated the Sup35NM-mKate2 (red) sensor at the HO locus of our base 930S strain to create strain MR1118S and we obtained MR1118 ([psi–]) by guanidine curing. We then isolated cells with plasmids that express Sis1 or Sis1JGF and the Sup35C plasmid using the plasmid shuffle.
We first repeated the fractionation assay with these cells and got similar results showing reduced solubility of Sup35 in Sis1JGF cells (Figure 5B). Quantitation of the combined data from experiments in Figure 5, A and B showed that cells expressing Sis1JGF in place of Sis1 had a significant 5.7-fold reduction in amount of soluble Sup35 (Figure 5C). These results add to the confidence in concluding that Sup35 is less soluble in [PSI+] cells expressing Sis1JGF and show that the Sup35NM-mKate sensor system does not affect aggregation of Sup35 differently in [PSI+]S cells expressing Sis1 or Sis1JGF.
The fractionation experiments in Figure 5A used denaturing lysis buffer (containing SDS) while those in Figure 5B used buffer without SDS. If Sis1 was limiting incorporation of Sup35 into SDS-sensitive aggregates such as Sup35 “gels” or “biocondensates” (Franzmann et al. 2018), then the amount of Sup35 in the soluble fraction of Sis1JGF lysates made using nondenaturing conditions would be less than in lysates using denaturing conditions. In fact, the differences in solubility we observed using both conditions were so similar that the data from the different experiments were combined to calculate the 5.7-fold difference in solubility between wild type and Sis1JGF strains. These results are in line with Sis1 limiting incorporation of Sup35 into toxic amyloid rather than into condensates from which functional Sup35 could be recovered.
Using the yTRAP system to monitor Sup35 aggregation by fluorescence, we found that [PSI+]S cells expressing Sis1JGF in place of Sis1 produced less fluorescence than cells expressing wild-type Sis1 (Figure 5D). Thus, more Sup35 is aggregated in the [PSI+]S cells expressing Sis1JGF. These results were anticipated on the basis of the fluorescence and fractionation assays, and they further support the conclusion that Sis1JGF is less able than wild-type Sis1 to moderate aggregation of Sup35 in [PSI+]S cells.
Sis1 interacts with Sup35 in vivo; Sis1JGF does not
Sis1 is detected in aggregates of Sup35 from lysates of [PSI+] cells, suggesting it binds Sup35 prions in vivo, and it binds the soluble and amyloid forms of Sup35NM in vitro (Bagriantsev et al. 2008; Barbitoff et al. 2020). We considered that Sis1JGF might bind less well to Sup35, which could underlie its reduced ability to moderate Sup35 aggregation in [PSI+] cells. To assess binding of Sis1JGF to Sup35 in live cells, we used an established two-hybrid system with yeast strain NMY51 that depends on physical interactions between two proteins to produce a transcriptional readout. Strain NMY51 is [psi–], so any interaction detected is presumed to be with soluble Sup35. An interaction between proteins being tested results in activation of reporter genes that include HIS3, ADE2, and bacterial lacZ, which encodes β-galactosidase.
We coexpressed bait and prey-tagged versions of Sup35 with wild-type Sis1 or Sis1JGF and tested for interactions by monitoring growth on medium lacking both histidine and adenine or containing X-gal, a chromogenic substrate of β-galactosidase (Figure 6A). We included versions of Sis1 with the D36N mutation in the J-domain that disrupts binding to Hsp70 to determine if any Sis1 interaction with Sup35 could be direct or mediated by an interaction with Hsp70. We quantified the strengths of interactions using a standard β-galactosidase assay (Miller 1972; Figure 6B).
Figure 6.

Sis1 interacts with Sup35 in vivo. (A) Example of interactions of wild Sis1 (wt) with Sup35 expressed from LEU2 (bait, top) plasmids or TRP1 (prey, left) plasmids that were assessed by growing transformants on medium lacking leucine and tryptophan (-LT) and then replica-plating onto medium also lacking histidine and adenine (-LTHA) or containing x-gal (-LT + x-gal), where amount of blue color reflects degree of β-galactosidase activity. (B) Quantitation of strength of interactions of Sis1 with Sup35 was done by measuring lacZ expression as Miller units of β-galactosidase activity (Miller 1972). In addition to wild-type Sis1, we included empty vector (ev) controls and versions of Sis1 with the D36N mutation that disrupts functional interaction with Hsp70. ev, empty vector; wt, Sis1 wild type; S35, Sup35; JGF, Sis1JGF; wt-36N, Sis1D36N; and JGF-36N, Sis1JGFD36N. By one-way ANOVA test: ****P < 0.0001; ***P < 0.001; ns, not significant. Data for both panels were from [psi–] strain NMY51.
Combining wild-type Sis1 with itself, which served as a positive control because Sis1 acts as a dimer, produced 1000 Miller units of β-galactosidase activity. Combining Sis1 with empty vector produced less than 12 units of background activity, while combining it with Sup35 produced about 90 units, an increase over sevenfold. In contrast, combining Sis1JGF with Sup35 produced a similar amount of β-galactosidase (27 units) as when combining it with empty vector (33 units). The D36N mutation in Sis1 affected β-galactosidase values only modestly while including D36N in Sis1JGF only reduced the apparent background. Growth on plates without histidine and adenine correlated with these measured activities, as did the development of color on the plates with X-gal. These results suggest that Sis1 can interact with Sup35 in vivo independently of Hsp70 and that Sis1JGF was unable to do so with or without Hsp70.
Phenotypes of Sis1JGF are consistent across strain backgrounds and prion variants
Harris et al. (2014) found that Sis1JGF (there designated Sis1-121) and Hdj1 support strong [PSI+] variants without apparent toxicity, and that neither supported weak variants .Those findings differ from our results here and earlier (Kirkland et al. 2011). Although we both use a similar Sis1JGF plasmid, they expressed Hdj1 to a much higher level. They also used different genetic strain backgrounds and prion variants. They kindly provided their yeast strains W303 and 74-D694, each carrying their plasmids and propagating strong ([PSI+]STR) and weak ([PSI+]Sc37) prions. Using cytoduction, we cross-infected prions among strains W303, 74-D694, and 930 to obtain each of the three strains propagating [PSI+]S, [PSI+]WSL, [PSI+]STR, [PSI+]Sc37, or no prions. All of these 15 combinations are sis1Δ, carry a URA3 plasmid encoding wild-type Sis1, and express the test proteins wild-type Sis1, Sis1JGF, or Hdj1 from a TRP1 plasmid.
For each of the 930, W303, or 74-D694 strains, we performed the plasmid shuffle to compare the ability of Sis1JGF or the two expression levels of Hdj1 to support both cell growth and propagation of the four prions in the absence of Sis1. As expected, regardless of prion status the control cells in all three strain backgrounds that express wild-type Sis1 from the TRP1 plasmid showed heavy confluent growth on FOA after 1 day. This efficient recovery of cells on FOA reflects lack of dependence on the URA3 plasmid encoding wild-type SIS1 and thus maximal loss of the URA3 plasmid. Controls with the empty TRP1 vector, with or without prions, showed no growth on FOA after 4 days, reflecting the dependence of SIS1 on the URA3 plasmid for growth.
Also as expected, all [psi–] transformants expressing Sis1JGF or Hdj1 similarly showed obvious growth on FOA after 1–2 days. In line with the reduced Sis1 activity of Sis1JGF, the transformants expressing Sis1JGF showed noticeably lighter density on FOA in all strain backgrounds. For all transformants, the presence or absence of growth on FOA was unambiguous after 4 days.
As we saw with our strain 930, Sis1JGF did not support growth of strains W303 or 74-D694 propagating either of the two strong [PSI+] variants, but it did support growth of 930, W303, and 74-D694 propagating either of the two weak variants. The growth of all three strains that propagate weak [PSI+] and express only Sis1JGF lagged somewhat behind their isogenic [psi–] counterparts, which is consistent with both of the weak prion variants causing toxicity in W303 and 74-D694. Using cytoduction, we confirmed these FOA-resistant cells did not lose the weak prions (Supplementary Figure S2C). Therefore, all conclusions we make here regarding the ability of Sis1JGF to propagate weak and strong variants of [PSI+] prions, and the degree of toxicity of the prion variants, holds for two strong prions and two weak prions in three different strain backgrounds.
To confirm the prions were responsible for any observed reduction of growth, we replica-plated the same master plates of cells onto plates containing 3 mM guanidine and grew the cells overnight before replica-plating to FOA. The guanidine inactivates Hsp104, which arrests replication of prions (Eaglestone et al. 2000; Ferreira et al. 2001; Jung and Masison 2001). The nondividing prions are diluted among dividing cells and eventually lost as they become outnumbered by the cells. For all combinations of transformants and prions, except those with the empty vector, we recovered cells on FOA after incubating only 1 day (Figure 7, rightmost panels). Thus, aside from the empty vector controls, any reduced growth or failure to recover cells on FOA was due solely to the presence of toxic prions. Note that these results imply that proteins tested here that do not support growth of cells with prions are able to support propagation of the prion that is preventing growth.
Figure 7.

Sis1JGF phenotypes are consistent across yeast strains and prion variants. Plasmid shuffle as in Figure 1B. The left column shows strains 930, W303, and 74-D694 (all sis1Δ) each with two weak [PSI+] variants (W is WSL; 37 is Sc37), two strong variants (STR, S), or no prions (−) indicated above. Each expresses the proteins indicated on the left from a TRP1 plasmid (ev is empty TRP1 vector) and all express wild-type Sis1 from a URA3 plasmid. Hdj-lo is expressed from a single-copy plasmid, while Hdj-hi is overexpressed from a high-copy plasmid. Patches of cells that each contain three pooled transformants were grown on -trp medium containing uracil and limiting adenine (leftmost column) and then replica-plated onto FOA. FOA plates were imaged after incubating 2 days at 30° (2 days) and again after two additional days at 23° (4 days) as indicated. Panels in rightmost column (via Gdn) are from the same -trp lim. ade plates (leftmost column) that were first replica-plated onto YPAD containing 3 mM guanidine and grown overnight. The YPAD plates were then replica-plated onto the FOA plates shown, which were incubated 1 day at 30°. Centers of some patches of 74-D694 cells did not transfer well when replica-plated.
Recovery of guanidine-treated W303 and 74D strains with STR prions was least efficient. This reduced efficiency could be explained most simply if the average numbers of prion seeds per cell of the STR prion variant were highest in these strains. More cell divisions would be required to dilute the seeds enough to result in a similar extent of prion loss.
For strains W303 and 74-D694, high-level expression of Hdj1 was required to recover FOA-resistant cells propagating strong [PSI+] variants (Figure 7, compare Hdj1-hi with Hdj1-lo). Even so, these cells were less confluent on FOA than wild-type cells after 1–2 days. Thus, Hdj1 possesses a Sis1-like activity that can protect cells from [PSI+] toxicity, but it was very much less effective. We suspect this reduced effectiveness is related to a reduced ability of the human Hdj1 to cooperate with the yeast Hsp70 system.
Even with high expression of Hdj1, we did not recover cells of strain 930 propagating either strong prion. Thus, ability of Hdj1 to overcome toxicity of strong [PSI+] was strain-dependent.
Discussion
It is widely observed that in [PSI+] cells most of the essential Sup35 is depleted into prion aggregates, yet the presence of the prion causes only a modest stress (Jung et al. 2000) and has little effect on growth under optimal conditions. Thus, enough Sup35 must remain soluble in [PSI+] cells to support near-normal cell growth or Sup35 retains some of its function even when it is within prion aggregates (Baxa et al. 2011; Pezza et al. 2014). Abundant data show that the range of strengths of [PSI+] phenotypes, whether due to prion variation, altered protein quality control or environmental factors such as temperature, correlate with the degree that Sup35 is depleted into prion aggregates. Therefore, despite any residual function Sup35 might have in prion aggregates, the degree of nonsense suppression that underlies the strength of phenotype primarily reflects function of the nonaggregated Sup35.
There is an upper limit to how much Sup35 can be depleted into prion aggregates for cells to remain viable. This limit was shown by the finding that many spontaneously arising variants of [PSI+] fail to become established because they kill cells by depleting too much Sup35 or they are supplanted by less toxic variants (McGlinchey et al. 2011). Persistent [PSI+] prions must therefore attain a balance of propagating efficiently enough to be stable without depleting too much Sup35.
Our findings point to Sis1 as having an important role in determining this balance by moderating incorporation of Sup35 into prion aggregates. The requirement of the CTD in this role separates it from the role of Sis1 in [PSI+] propagation, in which the CTD is dispensable (Kirkland et al. 2011; Harris et al. 2014; Stein and True 2014). Whereas Sis1JGF might act in the disaggregation reaction needed for [PSI+] propagation, the Sis1 CTD is required to maintain solubility of Sup35 in [PSI+] cells. Harris et al. (2014) observed that after depleting Sis1 in otherwise wild-type cells, strong and weak [PSI+] prions inhibited growth to an extent that correlated with strength of prion phenotype. Those findings are in complete agreement with our conclusions here and earlier about [PSI+] toxicity and the unique role of Sis1 to protect cells from it.
The CTD is recognized as the substrate-binding region of Sis1 and other Hsp40 J-proteins (Lu and Cyr 1998; Yan and Craig 1999; Johnson and Craig 2001; Li et al. 2009). Therefore, the ability of Sis1JGF to support growth in place of Sis1, while no other J-protein can do so (Sahi and Craig 2007), suggests that the essential function of Sis1 can be provided by a specific regulation of Hsp70 rather than by specific or general substrate binding by the CTD (Yan and Craig 1999). Our findings and those of others that show Sis1 lacking its CTD can support prion propagation suggest a similarly specific Hsp70-regulating activity can be enough to provide Sis1 function needed for the Hsp104 machinery to promote replication of prions (Sondheimer et al. 2001; Lopez et al. 2003; Higurashi et al. 2008; Hines et al. 2011; Harris et al. 2014; Stein and True 2014). However, the possibility that Sis1JGF supports growth in place of Sis1 while another Hsp40 provides the J-protein function needed for [PSI+] replication, has not been ruled out.
Like other Hsp40 family J-proteins, Sis1 combines delivering its CTD-bound substrates to Hsp70 with activating the Hsp70 ATPase cycle to help proteins fold to their native state. Each extraction cycle of the Hsp104 machinery that divides a [PSI+] prion fiber should release a Sup35 monomer, which then would be expected to require assistance from Hsp70 to refold to its native state. Our findings here support our earlier proposal that the roles of Sis1 in helping the Hsp104 machinery promote prion replication and in helping non-native proteins fold properly are separable. While Sis1JGF might still be able to regulate Hsp70 in a way that helps it activate Hsp104 for protein disaggregation (Seyffer et al. 2012; Chamera et al. 2019), it could fail to coordinate regulation of Hsp70 ATPase with capture of its clients, which is needed to help Hsp70 refold them. Misfolded Sup35 extracted from prion aggregates that is not captured properly by Hsp70 might aggregate in a nonprion manner or rejoin the aggregates more readily.
When considering that Sis1JGF propagates [PSI+], questions arise about an earlier proposal that the role of Sis1 in [PSI+] propagation is to recruit the Hsp104 machinery to prion aggregates (Tipton et al. 2008), which requires Sis1 to bind Sup35 prion aggregates before Hsp70 and Hsp104. By that scenario, the GF region of Sis1JGF would have to be the part that binds Sup35 prions, and this interaction must be specific since Sis1JGF does not propagate [URE3] prions (Reidy et al. 2014). Another interpretation consistent with the data of that study is that a necessary action of Sis1 can occur after engagement of the machinery with prion aggregates. Accordingly, Hsp104 can bind Sup35 without help from Sis1 and Hsp70 can compete with Hsp104 for binding to Sup35 prions (Inoue et al. 2004; Helsen and Glover 2012; Winkler et al. 2012).
Although coexpressing YYS restored viability of [PSI+]S cells expressing Sis1JGF, coexpressing additional Ydj1 did not, which is consistent with substrate binding specifically by the CTD of Sis1 being necessary to keep Sup35 soluble enough for viability. Beyond influencing the fate of Sup35 monomers extracted from prion aggregates, this requirement might be met in different ways. We present evidence indicating Sis1 can bind soluble Sup35 in vivo, while Sis1JGF cannot. Such Sis1 activity might limit the ability of Sup35 to adopt a conformation that facilitates its joining to prion fibers. Sis1 also binds [PSI+] prion aggregates (Bagriantsev et al. 2008) and might reduce the ability of fibers to sequester Sup35 from the soluble pool by binding at fiber ends. It is also possible that Sis1JGF, unhindered by binding to misfolded protein or prion aggregates, is freer to promote higher than normal activity of the Hsp104 machinery to replicate prions, which would result in more fiber ends that can soak up Sup35.
Despite the dispensability of the CTD for many Sis1 functions, YYS supports cell viability and promotes Hsp104-driven [PSI+] replication in sis1Δ cells (Yan and Craig 1999; Reidy et al. 2014). So the CTD of Sis1 is enough to provide Sis1 functions needed for growth and prion propagation. In line with work showing the CTD must act in cis with a JGF region (Johnson and Craig 2001), we did not observe protection from prion toxicity by expressing the Sis1 CTD alone (unpublished observation). When appended to the heterologous JGF regions, the CTDs of Sis1 and Ydj1 also determine specific J-protein requirements for certain Hsp90 functions and for efficiency of Hsp104 functions needed for prion replication and for disaggregating heat-denatured protein aggregates (Reidy et al. 2014). Altogether, the data suggest that the role of Sis1 in prion replication can be accomplished either by Sis1JGF regulating a specific functional interaction between Hsp70 and Hsp104 or by the CTD of Sis1 modifying function of a related JGF to confer a similar activity to that JGF.
Sup35 binds to Sup45 to promote release of ribosomes from mRNA and Sup45 is found in [PSI+] aggregates (Stansfield et al. 1995; Paushkin et al. 1997b). Our data suggest that reduced availability of Sup45 due to its binding to prion aggregates can explain why Sup35C rescued cells from the [PSI+] toxicity only partially. Therefore, if Sis1 protects cells from [PSI+]S by moderating depletion of Sup35 into prion aggregates, then there would be less aggregated Sup35 to deplete Sup45. Others found that overexpressing intact Sup35 is toxic in wild-type [PSI+] cells and that this toxicity can be overcome by expressing Sup45 (Chernoff et al. 1992; Vishveshwara et al. 2009; Pezza et al. 2014). Depletion of Sup45 is therefore primarily responsible for that toxicity. Under those conditions, the excess Sup35 apparently increases the mass of aggregated Sup35, which causes lethal depletion of Sup45, without limiting the functional Sup35 needed for growth. The toxicity caused by artificially overproducing Sup35 is qualitatively different than that caused by deleting the CTD of Sis1.
Sis1JGF clearly supported propagation of several weak and strong variants of [PSI+] and the relative toxicity of all the different variants was very similar across three different yeast strain backgrounds. Sup35 aggregated more extensively in cells expressing Sis1JGF in place of Sis1 and, consistently, the degree of [PSI+] toxicity was proportional to the strength of prion phenotype, and thus the degree to which Sup35 is depleted by the individual prion variants. The ability of Sis1 to protect from [PSI+] toxicity by moderating Sup35 aggregation is thus a general property that we expect to be found with other strain backgrounds and possibly other prions.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplementary material is available at GENETICS online.
Supplementary Material
Acknowledgments
The authors thank Justin Hines for generously providing many yeast strains, Sue Liebman for Sup35 antibody, and NIH colleagues for critical review of the manuscript.
Funding
This work was supported by the Intramural Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney diseases (ZIA DK024946-23).
Conflicts of interest
The authors declare that there is no conflict of interest.
Literature cited
- Bagriantsev SN, Gracheva EO, Richmond JE, Liebman SW.. 2008. Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol Biol Cell. 19:2433–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbitoff YA, Matveenko AG, Bondarev SA, Maksiutenko EM, Kulikova AV, et al. 2020. Quantitative assessment of chaperone binding to amyloid aggregates identifies specificity of Hsp40 interaction with yeast prion fibrils. FEMS Yeast Res. 20.foaa025. doi: 10.1093/femsyr/foaa025. [DOI] [PubMed] [Google Scholar]
- Baxa U, Keller PW, Cheng N, Wall JS, Steven AC.. 2011. In Sup35p filaments (the [PSI+] prion), the globular C-terminal domains are widely offset from the amyloid fibril backbone. Mol Microbiol. 79:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamera T, Kłosowska A, Janta A, Wyszkowski H, Obuchowski I, et al. 2019. Selective Hsp70-dependent docking of Hsp104 to protein aggregates protects the cell from the toxicity of the disaggregase. J Mol Biol. 431:2180–2196. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Inge-Vechtomov SG, Derkach IL, Ptyushkina MV, Tarunina OV, et al. 1992. Dosage-dependent translational suppression in yeast Saccharomyces cerevisiae. Yeast. 8:489–499. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW.. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science. 268:880–884. [DOI] [PubMed] [Google Scholar]
- Chernova TA, Wilkinson KD, Chernoff YO.. 2017. Prions, chaperones, and proteostasis in yeast. Cold Spring Harb Perspect Biol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM.. 2017. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. 86:27–68. [DOI] [PubMed] [Google Scholar]
- Craig EA. 2018. Hsp70 at the membrane: driving protein translocation. BMC Biol. 16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW.. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 144:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaglestone SS, Ruddock LW, Cox BS, Tuite MF.. 2000. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI(+)] of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 97:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Wickner RB.. 2013. The [URE3] prion in Candida. Eukaryot Cell. 12:551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CY, Lee S, Cyr DM.. 2003. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 8:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust O, Abayev-Avraham M, Wentink AS, Maurer M, Nillegoda NB, et al. 2020. HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature. 587:489–494. [DOI] [PubMed] [Google Scholar]
- Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF.. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 40:1357–1369. [DOI] [PubMed] [Google Scholar]
- Franzmann TM, Jahnel M, Pozniakovsky A, Mahamid J, Holehouse AS, et al. 2018. Phase separation of a yeast prion protein promotes cellular fitness. Science. 359.eaao5654. doi: 10.1126/science.aao5654 10.1093/femsyr/foaa025. [DOI] [PubMed] [Google Scholar]
- Gari E, Piedrafita L, Aldea M, Herrero E.. 1997. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 13:837–848. [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S.. 1998. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 94:73–82. [DOI] [PubMed] [Google Scholar]
- Harris JM, Nguyen PP, Patel MJ, Sporn ZA, Hines JK.. 2014. Functional diversification of hsp40: distinct j-protein functional requirements for two prions allow for chaperone-dependent prion selection. PLoS Genet. 10:e1004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsen CW, Glover JR.. 2012. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J Biol Chem. 287:542–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi T, Hines JK, Sahi C, Aron R, Craig EA.. 2008. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc Natl Acad Sci U S A. 105:16596–16601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines JK, Higurashi T, Srinivasan M, Craig EA.. 2011. Influence of prion variant and yeast strain variation on prion-molecular chaperone requirements. Prion. 5:238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung GC, Masison DC.. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics. 173:611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Taguchi H, Kishimoto A, Yoshida M.. 2004. Hsp104 binds to yeast Sup35 prion fiber but needs other factor(s) to sever it. J Biol Chem. 279:52319–52323. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Craig EA.. 2001. An essential role for the substrate-binding region of Hsp40s in Saccharomyces cerevisiae. J Cell Biol. 152:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G, Song Y, Chung S, Masison DC.. 2004. Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol Cell Biol. 24:3928–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GM, Stalker J, Humphray S, West A, Cox T, et al. 2008. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat Methods. 5:239–241. [DOI] [PubMed] [Google Scholar]
- Jung G, Jones G, Wegrzyn RD, Masison DC.. 2000. A role for cytosolic hsp70 in yeast [PSI(+)] prion propagation and [PSI(+)] as a cellular stress. Genetics. 156:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Masison DC.. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol. 43:7–10. [DOI] [PubMed] [Google Scholar]
- Khalil AS, Lu TK, Bashor CJ, Ramirez CL, Pyenson NC, et al. 2012. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian AN, Hines JK.. 2018. Chaperone functional specificity promotes yeast prion diversity. PLoS Pathog. 14:e1006695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CY, Tittmann P, Gross H, Gebert R, Aebi M, et al. 1997. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci U S A. 94:6618–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland PA, Reidy M, Masison DC.. 2011. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics. 188:565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin D, Wickner RB.. 2007. Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol Biol Cell. 18:2149–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin DS, Smirnov VN, Ter-Avanesyan MD, Kushnirov VV.. 2002. Increased expression of Hsp40 chaperones, transcriptional factors, and ribosomal protein Rpp0 can cure yeast prions. J Biol Chem. 277:23702–23708. [DOI] [PubMed] [Google Scholar]
- Lee S, Fan CY, Younger JM, Ren H, Cyr DM.. 2002. Identification of essential residues in the type II Hsp40 Sis1 that function in polypeptide binding. J Biol Chem. 277:21675–21682. [DOI] [PubMed] [Google Scholar]
- Li J, Qian X, Sha B.. 2009. Heat shock protein 40: structural studies and their functional implications. Protein Pept Lett. 16:606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wu Y, Qian X, Sha B.. 2006. Crystal structure of yeast Sis1 peptide-binding fragment and Hsp70 Ssa1 C-terminal complex. Biochem J. 398:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebman SW, Chernoff YO.. 2012. Prions in yeast. Genetics. 191:1041–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez N, Aron R, Craig EA.. 2003. Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. Mol Biol Cell. 14:1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Cyr DM.. 1998. Protein folding activity of Hsp70 is modified differentially by the hsp40 co-chaperones Sis1 and Ydj1. J Biol Chem. 273:27824–27830. [DOI] [PubMed] [Google Scholar]
- Luke MM, Sutton A, Arndt KT.. 1991. Characterization of SIS1, a Saccharomyces cerevisiae homologue of bacterial dnaJ proteins. J Cell Biol. 114:623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum R, Tkach JM, Vierling E, Glover JR.. 2004. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 279:29139–29146. [DOI] [PubMed] [Google Scholar]
- Masison DC, Baker RE.. 1992. Meiosis in Saccharomyces cerevisiae mutants lacking the centromere-binding protein CP1. Genetics. 131:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masison DC, Reidy M.. 2015. Yeast prions are useful for studying protein chaperones and protein quality control. Prion. 9:174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey RP, Kryndushkin D, Wickner RB.. 2011. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A. 108:5337–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. 1972. Assays of lac operon enzymes: β-galactosidase permease, repressor, transacetylase, a complementation. In: Miller J, editor. Experiments in Molecular Genetics. Cold Spring Harbor, NY: CSHL Press. p. 352–355. [Google Scholar]
- Mockli N, Deplazes A, Hassa PO, Zhang Z, Peter M, et al. 2007. Yeast split-ubiquitin-based cytosolic screening system to detect interactions between transcriptionally active proteins. Biotechniques. 42:725–730. [DOI] [PubMed] [Google Scholar]
- Moriyama H, Edskes HK, Wickner RB.. 2000. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 20:8916–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby GA, Kiriakov S, Hallacli E, Kayatekin C, Tsvetkov P, et al. 2017. A genetic tool to track protein aggregates and control prion inheritance. Cell. 171:966–979.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YN, Masison D, Eisenberg E, Greene LE.. 2011. Application of the FLP/FRT system for conditional gene deletion in yeast Saccharomyces cerevisiae. Yeast. 28:673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD.. 1996. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD.. 1997a. In vitro propagation of the prion-like state of yeast Sup35 protein. Science. 277:381–383. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD.. 1997b. Interaction between yeast Sup45p (eRF1) and Sup35p (eRF3) polypeptide chain release factors: implications for prion-dependent regulation. Mol Cell Biol. 17:2798–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezza JA, Villali J, Sindi SS, Serio TR.. 2014. Amyloid-associated activity contributes to the severity and toxicity of a prion phenotype. Nat Commun. 5:4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy M, Masison DC.. 2011. Modulation and elimination of yeast prions by protein chaperones and co-chaperones. Prion. 5:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy M, Miot M, Masison DC.. 2012. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics. 192:185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy M, Sharma R, Shastry S, Roberts BL, Albino-Flores I, et al. 2014. Hsp40s specify functions of Hsp104 and Hsp90 protein chaperone machines. PLoS Genet. 10:e1004720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B.. 2019. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 20:665–680. [DOI] [PubMed] [Google Scholar]
- Sahi C, Craig EA.. 2007. Network of general and specialty J protein chaperones of the yeast cytosol. Proc Natl Acad Sci U S A. 104:7163–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, et al. 2012. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol. 19:1347–1355. [DOI] [PubMed] [Google Scholar]
- Sharma D, Masison DC.. 2009. Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept Lett. 16:571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. 2002. Getting started with yeast. Methods Enzymol. 350:3–41. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P.. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N, Lopez N, Craig EA, Lindquist S.. 2001. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 20:2435–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Wu YX, Jung G, Tutar Y, Eisenberg E, et al. 2005. Role for Hsp70 chaperone in Saccharomyces cerevisiae prion seed replication. Eukaryot Cell. 4:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Pritzkow S.. 2018. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci. 21:1332–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn ZA, Hines JK.. 2015. Hsp40 function in yeast prion propagation: Amyloid diversity necessitates chaperone functional complexity. Prion. 9:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, et al. 1995. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 14:4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein KC, True HL.. 2014. Structural variants of yeast prions show conformer-specific requirements for chaperone activity. Mol Microbiol. 93:1156–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS.. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature. 428:323–328. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Collins SR, Toyama BH, Weissman JS.. 2006. The physical basis of how prion conformations determine strain phenotypes. Nature. 442:585–589. [DOI] [PubMed] [Google Scholar]
- Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN.. 1994. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics. 137:671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton KA, Verges KJ, Weissman JS.. 2008. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol Cell. 32:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Kelly MJ, Gross JD, Weissman JS.. 2007. The structural basis of yeast prion strain variants. Nature. 449:233–237. [DOI] [PubMed] [Google Scholar]
- Tuite MF, Cox BS.. 2007. The genetic control of the formation and propagation of the [PSI+] prion of yeast. Prion. 1:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain SM, Sawicki GJ, Caughey B, Lindquist S.. 2001. Strains of [PSI(+)] are distinguished by their efficiencies of prion-mediated conformational conversion. EMBO J. 20:6236–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishveshwara N, Bradley ME, Liebman SW.. 2009. Sequestration of essential proteins causes prion associated toxicity in yeast. Mol Microbiol. 73:1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB. 2016. Yeast and fungal prions. Cold Spring Harb Perspect Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Tyedmers J, Bukau B, Mogk A.. 2012. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol. 198:387–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Craig EA.. 1999. The glycine-phenylalanine-rich region determines the specificity of the yeast Hsp40 Sis1. Mol Cell Biol. 19:7751–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HY, Ziegelhoffer T, Osipiuk J, Ciesielski SJ, Baranowski M, et al. 2015. Roles of intramolecular and intermolecular interactions in functional regulation of the Hsp70 J-protein co-chaperone Sis1. J Mol Biol. 427:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Derkatch IL, Uptain SM, Patino MM, Lindquist S, et al. 1999. The yeast non-Mendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J. 18:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhouravleva G, Frolova L, Goff XL, Guellec RL, Inge-Vechtomov S, et al. 1995. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 14:4065–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplementary material is available at GENETICS online.