

Abstract
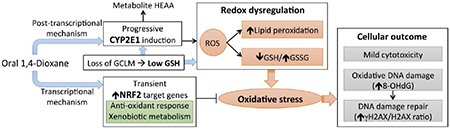
1,4-Dioxane (DX) is a synthetic chemical used as a stabilizer for industrial solvents. Recent occurrence data show widespread and significant contamination of drinking water with DX in the US. DX is classified by the International Agency for Research on Cancer as a group 2B carcinogen with the primary target organ being the liver in animal studies. Despite the exposure and cancer risk, US EPA has not established a drinking water Maximum Contaminant Level (MCL) for DX and a wide range of drinking water targets have been established across the US and by Health Canada. The DX carcinogenic mechanism remains unknown; this information gap contributes to the varied approaches to its regulation. Our recent mice study indicated alterations in oxidative stress response accompanying DNA damage as an early change by high dose DX (5,000 ppm) in drinking water. Herein, we report a follow-up study, in which we used glutathione (GSH)-deficient glutamate-cysteine ligase modifier subunit (Gclm)-null mice to investigate the role of redox homeostasis in DX-induced liver cytotoxicity and genotoxicity. Gclm-null and wild-type mice were exposed to DX for one week (1,000 mg/kg/day by oral gavage) or three months (5,000 ppm in drinking water). Subchronic exposure of high dose DX caused mild liver cytotoxicity. DX induced assorted molecular changes in the liver including: (i) a compensatory nuclear factor erythroid 2-related factor 2 (NRF2) anti-oxidative response at the early stage (one week), (ii) progressive CYP2E1 induction, (iii) development of oxidative stress, as evidenced by persistent NRF2 induction, oxidation of GSH pool, and accumulation of the lipid peroxidation by-product 4-hydroxynonenal, and (iv) elevations in oxidative DNA damage and DNA repair response. These DX-elicited changes were exaggerated in GSH-deficient mice. Collectively, the current study provides additional evidence linking redox dysregulation to DX liver genotoxicity, implying oxidative stress as a candidate mechanism of DX liver carcinogenicity.
Keywords: water contaminant, liver carcinogenicity, mechanism of action, CYP2E1, oxidative DNA damage
Graphical Abstract
1. Introduction
The synthetic chemical 1,4-dioxane (DX) was historically used as a stabilizer for industrial solvents (ATSDR, 2012; EPA, 2017). Environmental release of DX has been associated primarily with the use and improper disposal of chlorinated solvents, especially 1,1,1-trichloroethane (TCA); DX has also been found in conjunction with other chlorinated solvents such as trichloroethylene (TCE) (EPA, 2017). Owing to its high water solubility, environmental stability, and poor adsorption to solid phases and organic substrates, DX readily leaches into groundwater and contaminates surface waters that receive discharges from wastewater treatment plants (EPA, 2017; Sun et al., 2016). Based on the environmental occurrence, demonstrated toxic effects and potential for human exposures of DX, the Agency for Toxic Substances and Disease Registry (ATSDR) has listed DX as a Superfund priority substance (ATSDR, 2017). Intake of DX through drinking water, the dominant route of human exposure, is being recognized as an emerging public health threat affecting large numbers of individuals worldwide (Godri Pollitt et al., 2019). In the US, there is no federal drinking water standard for DX; a health-based reference concentration (RC) of 0.35 μg/L was provided by the Environmental Protection Agency (EPA) as part of the Unregulated Contaminant Monitoring Rule Round-3 (UCMR3) testing program (IRIS., 2013). According to the UCMR3 testing of 4,864 US public water systems (PWSs), DX ranked second in both prevalence (detected in 21% of PWSs) and RC exceedance (frequency is 6.9% of PWSs) out of the 28 contaminants tested (Adamson et al., 2017). This occurrence data highlights the widespread contamination of drinking water with DX in the US.
To our knowledge, no human studies have assessed the toxicity associated with oral DX exposure. DX-induced genotoxicity and carcinogenesis have been studied in numerous in vitro and in vivo experimental settings. While in vitro assays have shown an overall lack of mutagenic or genotoxic activity (EPA, 2017; Morita and Hayashi, 1998), in vivo studies have demonstrated that DX induces liver adenomas and carcinomas in rats and mice (of both genders) after chronic exposure (JBRC., 1998; Kano et al., 2009; Kociba et al., 1974; National Toxicology, 1978). The 50% liver tumor benchmark dose (lower confidence limit, BMDL-50) calculated by the EPA from the Kano et al. (EPA, 2013; EPA, 2020; Kano et al., 2009) female mouse data was 33 mg/kg/d, from which dose response modeling using linear low dose assumptions yielded an oral cancer slope of 0.1 per mg/kg/day (EPA, 2013; EPA, 2020; Kano et al., 2009). Based on inadequate human data and sufficient evidence of carcinogenicity in animals, the International Agency for Research on Cancer (IARC) has classified DX as a group 2B carcinogen (possibly carcinogenic to humans) (IARC, 1976; IARC, 1987).
Despite the reproducible carcinogenic effect and widespread contamination, as noted above, there is no US EPA maximal contaminant level (MCL) for DX. A number of states have established drinking water guidelines or standards ranging from 0.3 to 70 μg/L across the US (EPA, 2017), while the drinking water target set by Health Canada is 50 μg/L (HealthCanada, 2018). This wide range is, in part, due to the undetermined carcinogenic mechanisms of DX. Some studies suggest that metabolic saturation and cytotoxicity are required for DX carcinogenicity (Dourson et al., 2014; Dourson et al., 2017), which can lead to a drinking water target in the upper end of the range based upon threshold approaches (HealthCanada, 2018). Others demonstrate that long-term exposure at relatively low concentrations (i.e., 500 mg/L in drinking water for 2 years) can cause liver cancer in the absence of cytotoxicity (Kano et al., 2009). Recent in vivo mutagenicity studies show that DX is genotoxic at high doses (i.e., >1,000 mg/kg by oral gavage or >440 mg/kg/day in drinking water for 16 weeks) (Gi et al., 2018; Totsuka et al., 2020); however, whether DX induces liver carcinogenesis through a mutagenic mechanism of action (MOA) remains inconclusive. Further complicating consideration is the poor definition of the relative importance of DX metabolism. While it is widely accepted that DX is rapidly metabolized to its end-product β-hydroxyethoxyacetic acid (HEAA), the metabolic pathways by which this occurs are unclear, and it is unknown whether the metabolism of DX is a detoxifying process or results in the production of toxic intermediates (Wilbur et al., 2012). These uncertainties represent a significant knowledge gap in DX risk assessment and warrant further studies to characterize molecular details of DX-elicited changes in the liver.
We recently performed a short-term exposure study, wherein BDF-1 female mice were exposed to DX (50, 500 and 5,000 mg/L) in drinking water for 1 or 4 weeks (Charkoftaki et al., 2021). Although no gross pathological changes were observed in the liver at any exposure dose or duration, hepatocyte DNA damage was elevated at both 1 week and 4 weeks by high dose DX (5,000 ppm). Liver RNASeq and metabolomic analyses revealed DX-induced changes in genes involved in glutathione (GSH)-mediated detoxification and nuclear factor erythroid 2-related factor 2 (NRF2)-mediated oxidative stress response, and alterations of metabolites involved in metabolic pathways of nucleotide salvage synthesis, oxidative stress response and detoxification, and DNA damage (Charkoftaki et al., 2021). These results indicate that, at the early stage when hepatic cytotoxicity is absent, high dose DX induces DNA damage along with changes in cellular oxidative stress response.
Herein, we report a follow-up study targeting the high dose DX exposure, wherein we utilized a mouse model of GSH-deficiency to investigate the role of redox homeostasis in DX-induced cytotoxicity and genotoxicity in the liver. GSH attains millimolar concentrations in the liver and plays a key role in maintaining hepatic redox homeostasis and in cellular mechanisms protecting against oxidative stress (Chen et al., 2013). The glutamate-cysteine ligase modifier subunit (Gclm)-null mice exhibit only ≈15% of liver GSH levels seen in wild-type mice (Chen et al., 2005; Yang et al., 2002). While Gclm-null mice have good health when unchallenged, these mice are more sensitive to hepatotoxicity caused by acetaminophen (McConnachie et al., 2007) and TCDD (Chen et al., 2012) making them a useful model to assess oxidative stress-mediated liver damage. In the present study, following oral exposures to high dose DX for up to 3 months, wild-type (WT) and Gclm-null (KO) mice were evaluated for: (i) their susceptibility to liver cytotoxicity and genotoxicity, and (ii) biochemical and molecular components of redox dysregulation associated with DX toxic and carcinogenic mechanism.
2. Materials and Methods
2.1. Chemicals and reagents
1,4-Dioxane was purchased from EMD Millipore (Burlington, MA, USA). The biochemical kits for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were purchased from Sekisui Diagnostics LLC (Burlington, MA, USA). TRIzol® Reagent was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Reagents for reverse transcription, real-time quantitative PCR, protein quantification, SDS-PAGE, coomassie blue staining, and Western immunoblotting were purchased from Bio-Rad (Hercules, CA, USA). Primary antibodies were purchased from Abcam (Waltham, MA, USA) or Cell Signaling Technology (Danvers, MA, USA) as specified below. Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The TSA Plus Biotin kit was purchased from PerkinElmer (Waltham, MA, USA). The DAB substrate kit was purchased from Vector laboratories (Burlingame, CA, USA). The reduced (GSH) and oxidized glutathione (GSSG) fluorometric assay kit (ab138881) and the Deproteinizing Sample Kit (ab204708) were purchased from Abcam (Waltham, MA, USA). The 8-OHdG competitive ELISA assay kit was purchased from Cayman Chemical (Ann Arbor, MI, USA). The DNeasy Blood & Tissue kit was purchased from Qiagen LLC (Germantown, MD). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Animals
The Gclm-null (KO) mouse line (Yang et al., 2002) has been backcrossed onto the C57BL/6J background for more than 10 generations. Experimental WT and KO littermates were generated by heterozygous breeding. All animal experiments were approved by and conducted in compliance with the Institutional Animal Care and Use Committee (IACUC) of the Yale University (Protocol #2020-11643). Mice were maintained in a temperature-controlled room (21–22°C) on a 12 hr light/dark cycle and supplied with a standard chow diet and water ad libitum.
2.3. Oral administration of DX
DX was administered to male WT and KO mice (age 12-14 weeks; N = 5-8/experimental group) following two protocols (Fig. 1A): (1) oral gavage (1,000 mg/kg/d) for one week, or (2) in drinking water (5,000 ppm) for 3 months. Daily gavage with freshly prepared DX (100 g/l) was performed between 9:00 and 9:30 AM. DX-containing drinking water was changed weekly. Mice from control (CON) groups were provided with regular drinking water. During the course of treatment, mice were housed in pairs and had free access to a standard chow diet (diet # 2018; Teklad Diets, Harlan Laboratories). The concentrations of DX in the baseline drinking water and diet are expected to be at trace levels. The drinking water is sourced from the City of New Haven municipal supply, which contained DX at an average concentration of 1.57 ppb during the UCMR3 test round (Regional Water Authority, 2016, available at https://www.rwater.com/media/1056/rwa-wqr-web.pdf). DX may occur as a trace contaminant of various food ingredients (e.g. polysorbate 60 and 80) (ATSDR, 2012); however, these emulsifying ingredients are not listed as ingredients in the chow diet used in this study (available at https://insights.envigo.com/hubfs/resources/data-sheets/2018-datasheet-0915.pdf). Mice were assessed daily for signs of pain and distress (e.g. abnormal postures, activities and breathing). Food and water consumption and body weight were recorded weekly. At the conclusion of treatment protocols, mice were euthanized by CO2 asphyxiation and blood was collected by cardiac puncture. The liver was removed and weighed on ice. One piece of liver was immediately placed in 10% neutral buffered formalin and later processed for histological examination. The remaining liver tissue was aliquoted, flash frozen in liquid nitrogen and stored at −80°C for latter analyses.
Fig. 1.
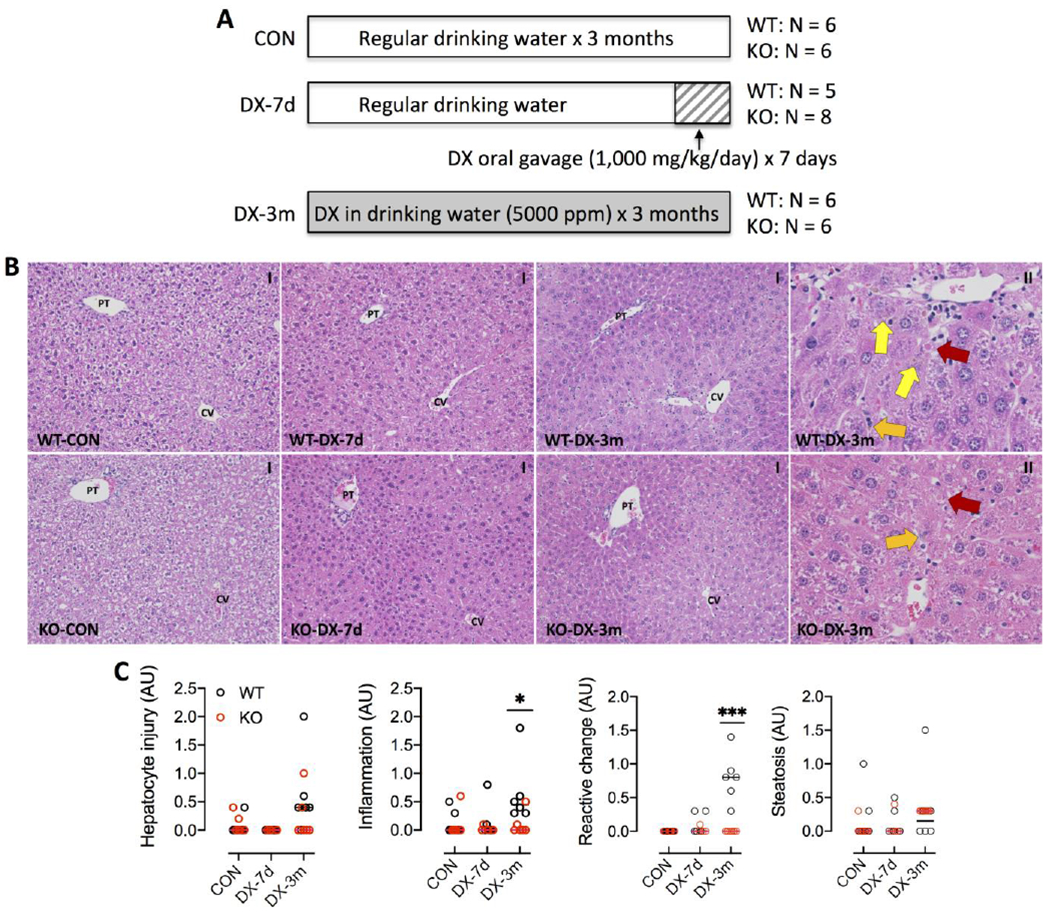
DX exposure protocol and liver histopathology. (A) Scheme of DX exposure protocols. Male wild-type (WT) and Gclm-null (KO) mice (N = 5-8/group) were provided with regular drinking water (CON) or administered with DX for 7 days (DX-7d) by oral gavage or for 3 months (DX-3m) in drinking water as described in the Materials and Methods. (B) Representative images of H&E stained liver tissues. DX exposure for 3 months caused mild pathologies, including steatosis, single cell death (red arrows), inflammatory infiltration (orange arrows), and reactive changes (yellow arrows), in the centrilobular zone in both WT and KO mice. CV, central vein; PT, portal triad. Magnifications: 200x (I) and 600x (II). (C) Liver histopathologies were scored for hepatocytes injury, inflammation, ductular reactive change, and steatosis. Results are presented in Scatter plots showing individual data points from each experimental group with the group median at the line. Differences between treatment-matched WT (black circles) to KO (red circles) mice were analyzed by Student’s unpaired t-test. *P < 0.05, ***P <0.001.
2.4. Plasma enzyme assay and liver histology
Plasma was extracted from whole blood and measured for ALT and AST activities using a biochemical kit according to the manufacturer’s protocol. For hematoxylin and eosin (H&E) staining, mouse livers were fixed in 10% neutral buffered formalin for ~24 hours at 4°C, followed by dehydration in graded ethanol solutions and then paraffin embedding. Sections (4 – 5 μm thick) were mounted onto glass slides and rendered free of paraffin by immersing in xylene, rehydrated and stained with H&E by the Pathology Core at the Yale School of Medicine using standard procedures. Liver sections were examined by a trained histologist (D.O.) blinded to the treatments and genotypes. Liver histopathology was scored for hepatocytes degeneration, inflammation, ductular reactions, steatosis, and tumor formation as described previously (Lanaspa et al., 2018); results are reported in artificial unit (AU).
2.5. Reverse transcription and real-time quantitative PCR (Q-PCR)
Total RNA was isolated from frozen liver pieces using TRIzol® Reagent according to manufacturer’s protocol. The purity of RNA was assessed by the 260/280 ratios (ranged from 1.95 to 2.0 for all samples) and 260/230 ratios (ranged from 1.97 to 2.13 for all samples) measured using a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). cDNA was synthesized using iScript™ cDNA Synthesis Kit according to manufacturer’s instructions using 1 μg total RNA in a 20 μl reaction volume. Q-PCR reaction mixtures contained 1 μl cDNA, 1x SYBR Green Supermix, and 0.15 μM gene-specific primer sets in a total volume of 10 μl. Genomic DNA and ddH2O, respectively, were used replacing cDNA as negative controls and yielded no PCR product. Sequences and amplification efficiencies of primers used for Q-PCR can be found in Table S1. Reactions were run with two technical replicates (inter-replicates CV ranged from 0.1% to 5.7% of CT values) for each biological sample using the CFX96 Touch Detection System (BioRad). Expression of housekeeping genes (18s and Rplp0) were used for normalization of CT data according to the ΔΔCT method (Livak and Schmittgen, 2001). Relative mRNA levels of individual genes were reported as fold of the control (WT-CON).
2.6. Western immunoblotting (WB) analyses
Frozen liver pieces were homogenized in RIPA buffer (150 mM NaCl, 1% TritonX-100, 0.25% sodium deoxycholate, 0.1% SDS, 50 mM Tris, 1 mM EDTA, 1 mM PMSF, protease inhibitor cocktail, pH 7.4) on ice with the TissueLyser II (Qiagen). Whole cell liver homogenates were centrifuged at 12,000 rpm at 4°C for 20 min and the supernatant was collected. Protein concentrations were determined using the Bio-Rad protein assay kit according to the manufacturer’s protocol. Proteins in total homogenates (20 - 40 μg) were resolved by 4-15% SDS-PAGE, followed by parallel gel Coomassie blue staining (for total protein loading) and immunoblotting using following primary antibodies directed against: (i) (from Abcam) GCLM (ab126704, 1:5000), GCLC (ab190685, 1:5000), CYP2E1 (ab28146, 1:2000), GST-pi (ab138491, 1:5000) and CK7 (ab181598, 1:1000), and (ii) (from Cell Signaling Technology) NRF2 (#12721, 1:1000), HMOX1 (#43966, 1:1000), NQO1 (#62262, 1:1000), H2AX (#2595, 1:1000), γH2AX (#9718, 1:500), TRX1 (#2429, 1:1000) and TRX2 (#14907, 1:1000), according to the manufacturers’ instructions. Corresponding HRP-conjugated secondary antibodies were used at 1:2000 dilution. A quality control (QC) sample was prepared by pooling a small aliquot from each biological sample and loaded on every single gel to control for SDS-PAGE variations. Coomassie blue-stained gels and immunoblots were imaged on the ChemiDoc Touch imaging system (BioRad) and quantified using the Image Lab software (BioRad). Protein levels were quantified by densitometry analysis of band intensity and normalized to total protein loading per lane (Fig. S1). Relative protein abundance was reported as fold of the control (WT-CON).
2.7. Immunohistochemical (IHC) analyses
Paraffin-embedded liver sections (4 - 5 μm) were prepared as described for H&E staining and processed for IHC staining using a TSA Plus Biotin kit according to the manufacturer’s protocol. Briefly, deparaffinized and rehydrated liver sections were heated in 0.01 M citrate buffer (pH 6.6) at boiling for 20 min for antigen retrieval. After cooling down to room temperature (RT), tissue sections were immersed in 3% hydrogen peroxide in 30% methanol for 10 min to deplete endogenous peroxidase activity. After 4 washes with distilled water, the sections were incubated in TNB blocking solution in a humidified chamber for 1 h at RT to block nonspecific staining. The sections were then incubated with primary antibodies diluted in TNB blocking solution overnight at 4°C. One section from each biological sample was incubated with blank blocking solution to serve as the negative control in every group of slides stained, which showed no immunopositivity (Fig. S5C). All subsequent procedures in the next day were carried out at RT. The sections were washed 3 times with PBST (PBS + 0.5% Tween-20) and incubated with HRP-conjugated secondary antibodies diluted in TNB blocking buffer for 1 h. The sections were then incubated for 30 min in streptavidin HRP, washed in PBST, incubated for 10 min with biotinyl tyramide, and washed in PBST. The Sections were then visualized by incubating with DAB substrate solution for 10 min and counterstained with hematoxylin (1:10 dilution in distilled water) for 2 min. Lastly, the sections were dehydrated in ascending concentrations of ethanol and mounted with Permount for microscopic examination. Primary antibodies were used at dilutions of 1:300 for CYP2E1 (ab28146, Abcam), 1:100 for 4-HNE (ab46545, Abcam), 1:400 for γH2AX (#2595, Cell Signaling Technology), 1:2000 for GST-pi (ab138491, Abcam), and 1:8000 for CK7 (ab181598, Abcam) for overnight incubation at 4°C. Corresponding HRP-conjugated secondary antibodies were used at 1:500 for 60 min incubation at room temperature. IHC staining was developed using the DAB substrate kit (Vector Laboratories), followed by counterstaining with Hematoxylin.
2.8. CYP2E1 activity
Whole cell liver homogenates prepared from frozen liver tissues were used in the following assays. CYP2E1 activity was assessed by measuring the rate of oxidation of p-nitrophenol (PNP) to p-nitrocatechol as described (Chang et al., 2006) with some modifications. Briefly, frozen liver tissues were homogenized in the assay buffer (50 mM potassium phosphate, pH 7.4) on ice with TissueLyser II (Qiagen). Whole cell liver homogenates were centrifuged at 12,000 rpm at 4°C for 20 min and the supernatant was collected. Protein concentrations were determined using the Bio-Rad protein assay kit (BioRad) according to the manufacturer’s protocol and adjusted to a final concentration of 5 μg/μl with the assay buffer. For each reaction, 180 μl reaction mixture (50 mM potassium phosphate, pH 7.4, 5 mM PNP, 100 mM NADPH) was pre-warmed to 37°C, followed by the addition of 20 μl liver homogenate (100 μg protein), or serial diluted p-nitrocatechol standards, or negative control samples (including blank assay buffer, reaction mixture with no substrate, and heat inactivated lysate) to start the reaction. Reaction was allowed to carry on at 37°C for 1 h and stopped by the addition of 50 μl trichloroacetic acid (20%, w/v). Mixtures were then centrifuged at 13,000 rpm for 5 min at 4°C and 200 μl clear supernatant were transferred to each well of a 96-well spec-reader plate. Neutralization was made by adding 20 μl 10N NaOH to each well and the absorbance at 535 nm was immediately read on a Spectramax M3 microplate reader (Molecular Devices). Assays were run with two technical replicates (inter-replicates CV ranged from 1.2% to 7.4%) for each biological sample. The amount of product p-nitrocatechol formed in each sample was calculated by plotting against the p-nitrocatechol standard curve and normalized to total protein levels (mg). The CYP2E1 activity was reported as pmol/min/mg protein.
2.9. Liver GSH and GSSG levels
The concentrations of GSH and GSSG were measured using a fluorometric assay kit according to the manufacturer’s protocol. Briefly, frozen liver tissues were weighed and homogenized in the lysis buffer (phosphate-buffered saline, 0.5% NP-40) on ice with TissueLyser II (Qiagen). Whole cell liver homogenates were centrifuged at 14,000 rpm at 4°C for 20 min. The supernatants were then deproteinized by using the Deproteinizing Sample Kit and the resultant clear lysates were immediately used for measurements of GSH and GSSG in the same sample by parallel fluorometric reactions with respective assay buffers and probes. GSH and GSSG standards, and negative controls containing no tissue lysates were processed and assayed in the same manner. Fluorescence intensity was read at Ex490/Em520 nm on a Spectramax M3 microplate reader (Molecular Devices). Assays were run with two technical replicates (inter-replicates CV ranged from 0.6% to 8.3%) for each biological sample. Concentrations of liver GSH and GSSG were calculated by plotting against GSH and GSSG standard curves, respectively, and normalized to liver wet weight (gram). GSH and GSSG concentrations were reported as μmol/g liver. Their values were used to calculate the GSH/GSSG ratio for each biological sample.
2.10. HEAA measurement in the plasma
Circulating levels of HEAA were derived from untargeted metabolomic analysis of plasma using the HILIC-MS method as described previously (Charkoftaki et al., 2021). Briefly, 50 μl frozen plasma samples were thawed overnight at 4°C and mixed with 400 μl acetonitrile:methanol (1:1, %v/v), followed by centrifugation at 13,000 rpm at 4°C. Supernatants were transferred to LC-MS glass vials for HILIC-MS analysis on a quadrupole time-of flight (Q-ToF) mass spectrometer (Xevo G2-XS Q-ToF, Waters Corporation) equipped with an ultra-performance liquid chromatography (UPLC) Acquity I Class (Waters Corporation) unit. The electrospray ionization source (ESI) was operated in the negative mode. A quality control (QC) sample was prepared by pooling a small aliquot from each biological sample and was analyzed every five injections. Raw MS data were acquired using MassLynx 4.1 software (Waters Corporation) and processed using MZmine 2.52 (https://github.com/mzmine/mzmine2/releases/tag/v2.52) to generate a data matrix consisting of m/z value, retention time (RT), and peak abundances. HEAA was annotated based on the standard run using the same LC-MS conditions as the samples (Fig. S2). Results are reported as HEAA peak area generated by MZmine.
2.11. 8-Hydroxy-2’-deoxyguanosine (8-OHdG) ELISA assay
Oxidative DNA damage was assessed by measuring the oxidized derivative of deoxyguanosine (8-OHdG) using a competitive ELISA assay kit. Briefly, genomic DNA was isolated from frozen liver tissues using the DNeasy Blood & Tissue kit (Qiagen Inc.) following the manufacturer’s instruction. The purity of DNA was assessed by the 260/280 ratios (ranged from 1.84 to 2.02 for all samples) and 260/230 ratios (ranged from 2.06 to 2.2 for all samples) measured using a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). The quality of genomic DNA was assessed by nucleic acid electrophoresis (Fig. S3). Genomic DNA (30 – 50 μg) was digested with 50U DNase I in 40 μl reaction buffer (pH 7.5) containing 10 mM Tris-HCl, 2.5 mM MgCl2 and 0.1 mM CaCl2, at 37°C for 30 min. The reaction mixture was added with 1U nuclease P1, 5 μl 10x reaction buffer (0.2 M sodium acetate, 50 mM ZnSO4, and 0.5 M NaCl, pH 5.2,) and ultrapure H2O to a final volume of 50 μl and incubated at 37°C for 30 min. The pH of this reaction mixture was then readjusted to 7.5 by the addition of 6 μl 1M Tris (pH 8.0), and incubated with 1U alkaline phosphatase at 37°C for 30 min in a final reaction mixture of 60 μl, followed by boiling for 10 min and cooling on ice. Fifty μl of the resultant DNA hydrolysate was used in the 8-OHdG ELISA assay according to manufacturer’s protocol. Ultrapure H2O replacing genomic DNA was processed in the same manner to serve as the control sample. 8-OHdG levels are reported as pg/μg DNA.
2.12. Statistical analyses
Statistical analysis was performed using the GraphPad Prism 8.0 software (San Diego, CA). For liver histopathology score and blood HEAA levels, differences between treatment-matched WT and KO mice were analyzed by Student’s unpaired t-test. For GCLM mRNA and protein abundance in WT mice, and GSH levels, GSSG levels and GSH/GSSG ratios in WT and KO mice, respectively, differences between treatment groups were analyzed by one-way ANOVA with post-hoc Dunnett’s test correction. For liver 8-OHdG levels, differences between groups were analyzed by one-way ANOVA with post-hoc Dunnett’s test correction. For all other measurements, multiple group differences were analyzed by two-way ANOVA, where the two factors were genotype (WT vs KO) and DX treatments (CON vs DX-7d vs DX-3m), with post-hoc Bonferroni test correction. Tests for normality (Kolmogorov-Smirnov test) and homoscedasticity (Brown-Forsythe test) were performed with residuals of the dependent variables from one-way or two-way ANOVA; the data did not deviate from the normality and equal variance assumptions. P < 0.05 was considered significant.
3. Results
3.1. Administration of DX in drinking water (5,000 ppm) for 3 months caused mild liver cytotoxicity in WT and GSH-deficient (Gclm-null) mice
To investigate the role of redox homeostasis in DX-induced liver damage, we utilized the Gclm-null (KO) mouse model of chronic oxidative stress due to compromised de no biosynthesis of GSH (Yang et al., 2002). Two DX exposure regimes (Fig. 1A) were applied to WT and KO mice (male, age 12-14 weeks) to evaluate hepatic response to short-term (1000 mg/kg by daily oral gavage for 7 days) and subchronic (5,000 ppm in drinking water for 3 months) exposure. Oral gavage at 1000 mg/kg daily is estimated to be equivalent to dosing 5,000 ppm in drinking water based on an average daily water intake of 5 ml in an adult male mouse weighing 25 g. This administration route is meant to deliver the chemical with a consistent and accurate dose over a short period of time. DX exposure for up to 3 months caused no change in food consumption or water intake; however, there was a significant interaction between the genotype and 1.4-DX exposure (Table 1). We calculated the DX intake by drinking water in WT and KO mice to be 651 ± 131 and 647 ± 106 mg/kg/day, respectively (P = 0.95). Exposure to DX for 3 months had a slight effect on body weight (BW) gain (Table 1). The liver weights measured as % of BW were not altered by DX treatments, but were higher in KO mice than in WT mice due to a lower BW in KO mice (Table 1); this is consistent with the lean phenotype of the KO mice previously reported (Kendig et al., 2011). Plasma ALT and AST activities were unaltered in mice exposed for 7 days, but were elevated ~2-fold in mice exposed for 3 months (Table 1). Histological examination of the liver revealed mild pathologies, manifesting as single cell death (apoptosis and necrosis) and associated inflammatory infiltration, reactive changes, and steatosis, in the centrilobular zone in mice from the 3-month exposure cohort (Fig. 1B). Histopathological scoring revealed no significant difference in direct cytotoxic effects between the WT and KO mice; however, WT mice appeared to experience increased inflammation and had more reactive changes than KO mice at the 3 months time point (Fig. 1C). Overall, our data show that short-term or sub-chronic oral exposure to high dose DX does not cause significant liver cytotoxicity in either WT or KO mice.
Table 1.
Food and water intake and clinical parameters
|
Gclm wild-type (WT) (N = 5-6/group) |
Gclm-null (KO) (N = 6-8/group) |
P value (2W-ANOVA) |
|
|---|---|---|---|
| Food intake (gram/mouse/day) | |||
| CON | 4.2 ± 0.7 | 4.8 ± 1.1 | PI = 0.03 |
| DX-3m | 4.9 ± 0.5 | 4.1 ± 0.7 | |
|
| |||
| Water intake (ml/mouse/day) | |||
| CON | 3.8 ± 0.4 | 4.4 ± 0.3 | PI = 0.01 |
| DX-3m | 4.1 ± 0.5 | 3.7 ± 0.7 | |
|
| |||
| Body weight (gram) | |||
| CON | 29.8 ± 2.3 | 26.8 ± 1.4 |
PT = 0.04 PG < 0.001 |
| DX-7d | 28.2 ± 1.4 | 26.5 ± 2.2 | |
| DX-3m | 32.0 ± 2.9 | 27.5 ± 1.9 | |
|
| |||
| Liver weight (% of Body weight) | |||
| CON | 4.6 ± 0.3 | 5.4 ± 0.4 |
PG = 0.04 PI = 0.001 |
| DX-7d | 5.1 ± 0.6 | 4.8 ± 0.4 | |
| DX-3m | 4.6 ± 0.3 | 5.0 ± 0.2 | |
|
| |||
| Plasma ALT (U/dL) | |||
| CON | 20.0 ± 5.7 | 19.0 ± 1.9 | PT < 0.001 |
| DX-7d | 23.7 ± 6.2 | 15.7 ± 2.7 | |
| DX-3m | 41.6 ± 7.7 | 43.8 ± 14.2 | |
|
| |||
| Plasma AST (U/dL) | |||
| CON | 29.1 ± 3.3 | 35.1 ± 5.5 | PT < 0.001 |
| DX-7d | 34.7 ± 4.2 | 38.1 ± 3.4 | |
| DX-3m | 62.1 ± 4.6 | 73.6 ± 19.6 | |
Male mice (age 12-14 weeks) were provided with regular drinking water (CON) or administered with 1,4-dioxane (DX) for 7 days (DX-7d) by oral gavage (1,000 mg/kg/day) or for 3 months (DX-3m) in drinking water (5,000 ppm) as described in the Materials and Methods. Food and water consumptions and body weights were recorded weekly. Liver weights and plasma ALT and AST levels were measured at the conclusion of treatment protocols. Group differences were analyzed using two-way ANOVA for the main effects of DX exposures (PT), genotypes (PG), or the interaction between these two factors (PI). Post-hoc Bonferroni test was performed for multiple comparisons test. Results are presented as mean ± S.D.. P < 0.05 was considered significant.
3.2. High dose DX upregulated NRF2 and its target proteins involved in GSH metabolism and anti-oxidative response
To assess whether DX alters cellular redox status, we first measured the expression of a panel of NRF2 target genes involved in GSH biosynthesis (Gclm and Gclc) and recycling (Gpx4 and Gsr), antioxidant defense (Hmox1 and Nqo1), and xenobiotic metabolism (Gstm1, Ugt1a1, and Abcc3) (Figs. 2A & 2B). As expected, most of these genes were constitutively upregulated in KO livers (PG < 0.05). They were also induced by DX (PT < 0.05), which displayed a trend of peaking at 7 days and declining to nearly basal levels at 3 months; in contrast, Nqo1 exhibited a unique pattern of persistent induction (Fig. 2B). Given that some of these genes are also members of the AHR gene battery (Fig. 2B), we examined the expression of the signature AHR target genes Cyp1a1 and Cyp1a2, both of which were only slightly induced by DX in KO livers (Fig. 2C). Examination of the protein abundance of GCLM, GCLC, HMOX1 and NQO1 revealed a pattern of temporal changes (Figs. 3A & 3B) that were similar to those observed for gene expression (Figs. 2A & 2B). Notably, NRF2 protein was persistently upregulated by DX exposure at 3 months (Figs. 3A & 3B). DX-induced redox disturbance was also evidenced by changes in the hepatic GSH pool (Fig. 3C). We observed a transient increase in GSH (glutathione in its reduced form) in both WT and KO livers at 7 days when NRF2 response was most robust. At 3 months of DX exposure, liver GSSG (glutathione in its oxidized form) were elevated in both WT and KO mice leading to the decrease in GSH/GSSG ratios. This observation is consistent with more oxidation of the hepatic GSH pool in WT mice and by a larger extent in KO mice. Serving as the primary redox backup system, thioredoxin 1 gene and protein were found induced by DX, whereas thioredoxin 2 protein appeared to be upregulated only in KO mice and was not altered by DX exposure (Fig. S4). Collectively, our data show that high dose DX disrupts hepatic redox homeostasis prompting NRF2 anti-oxidative response and leading to oxidative stress by 3 months, a process enhanced by the low GSH status in KO mice.
Fig. 2. Q-PCR analysis of gene expression in liver tissues.
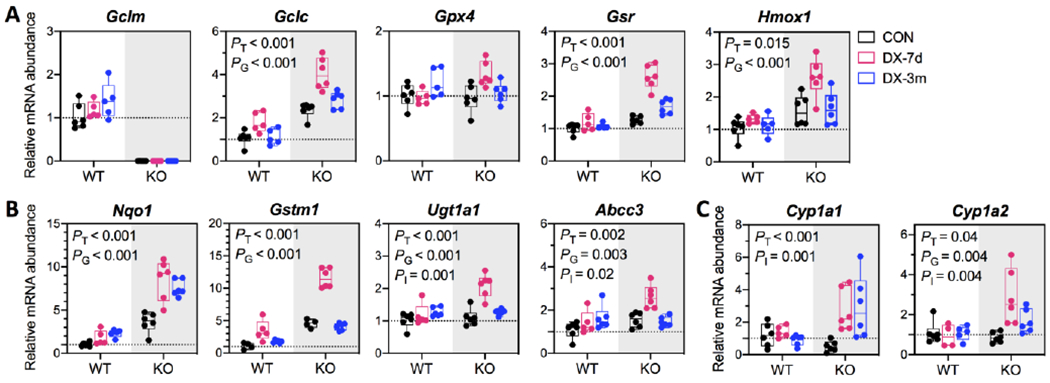
Examined genes are known to be regulated by (A) NRF2, (B) NRF2 and AHR, or (C) AHR. Expressions of housekeeping genes 18s and Rplp0 were used for normalization of CT data. Relative mRNA abundance of individual gene is reported as fold of the control (WT-CON). Results are presented in Box plots showing individual data points from each experimental group (N = 5-6/group). Group differences were analyzed using one-way ANOVA with post-hoc Dunnett’s test correction for Gclm gene in WT mice or using two-way ANOVA with post-hoc Bonferroni test correction for all other genes for the main effects of DX exposures (PT), genotypes (PG), or the interaction between these two factors (PI). P < 0.05 was considered significant. WT, wild-type mice; KO, Gclm-null mice. CON (black), provided regular water; DX-7d (pink), exposed to DX by gavage for 7 days; DX-3m (blue), exposed to DX in drinking water for 3 months.
Fig. 3. Expressions of redox sensitive proteins and glutathione concentrations in liver tissues.
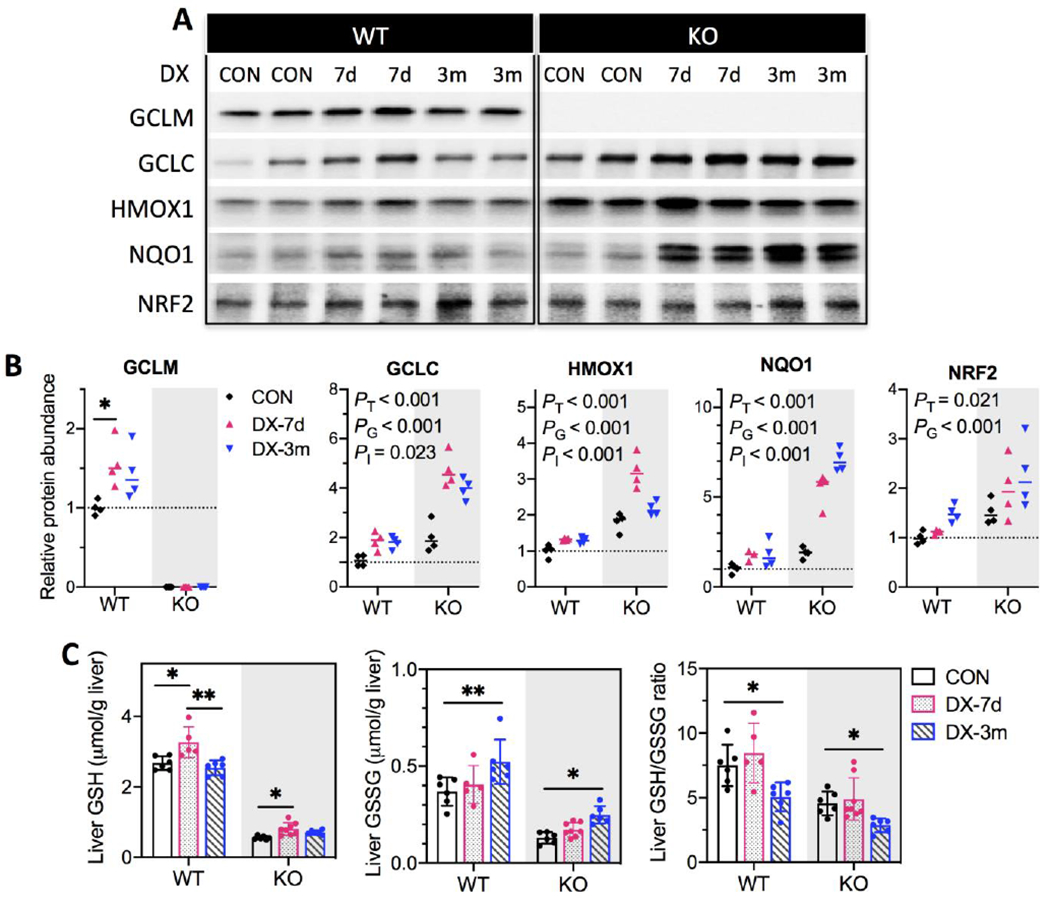
(A) Representative Western blotting images for protein expressions using liver whole-cell lysates. (B) proteins levels were quantified by densitometry analysis and normalized to total protein loading per sample. Relative protein abundance was reported as fold of control (WT-CON). Results are presented in Scatter plots showing individual data points from each experimental group with the group median at the line (N = 4/group). Group differences were analyzed using one-way ANOVA with post-hoc Dunnett’s test correction for GCLM protein in WT mice or using two-way ANOVA with post-hoc Bonferroni test correction for all other proteins for the main effects of DX exposures (PT), genotypes (PG), or the interaction between these two factors (PI). P < 0.05 was considered significant. (C) Liver concentrations of reduced (GSH) and oxidized glutathione (GSSG) were measured in whole-cell lysates using a fluorometric assay kit and normalized to liver weight (g). Differences between treatment groups of WT and KO mice, respectively, were analyzed by one-way ANOVA with post-hoc Dunnett’s test correction. Results are presented as mean ± S.D. in Bar plots showing individual data points from each experimental group (N = 5-8/group). P < 0.05 was considered significant. *P < 0.05, **P < 0.01. WT, wild-type mice; KO, Gclm-null mice. CON (black), provided regular water; DX-7d (pink), exposed to DX by gavage for 7 days; DX-3m (blue), exposed to DX in drinking water for 3 months.
3.3. DX caused time-dependent induction of CYP2E1 and centrilobular accumulation of the lipid peroxidation by-product 4-hydroxynonenal (4-HNE)
CYP2E1 is postulated to be involved in mammalian DX metabolism (Nannelli et al., 2005). This is consistent with a progressive induction of CYP2E1 protein (Figs. 4A & 4B) and corresponding increases in CYP2E1 activity (Fig. 4C) with time by DX treatment in WT livers and a more robust response in KO livers; however, Cyp2e1 mRNA abundance was unaffected by DX exposure (Fig. 4D). IHC examination of liver tissues revealed time-dependent expansion of CYP2E1-positive hepatocytes in the centrilobular zone (Fig. 4E), which is characteristic of xenobiotic activation of hepatic CYP2E1 (Lindros, 1997). We also observed a progressive increase in immunopositivity for 4-HNE, the by-product of lipid peroxidation. Changes in 4HNE occurred predominantly in centrilobular hepatocytes and was most dramatic in 3-month exposed KO livers (Fig. 4E). This agrees with the known role of CYP2E1 in generating ROS and thereby promoting lipid peroxidation (Caro and Cederbaum, 2004). In line with a plausible role of CYP2E1 in metabolizing DX, we detected higher plasma levels of HEAA in KO mice relative to WT mice after 3 months DX exposure (Fig. 4F). Collectively, our data show that high dose DX induces CYP2E1 expression in a time-dependent manner, which likely contributes to the oxidative stress and lipid peroxidation observed in the liver following 3 months exposure.
Fig. 4. CYP2E1 activity, lobular distribution of CYP2E1 and 4-HNE in liver tissues, and plasma HEAA.
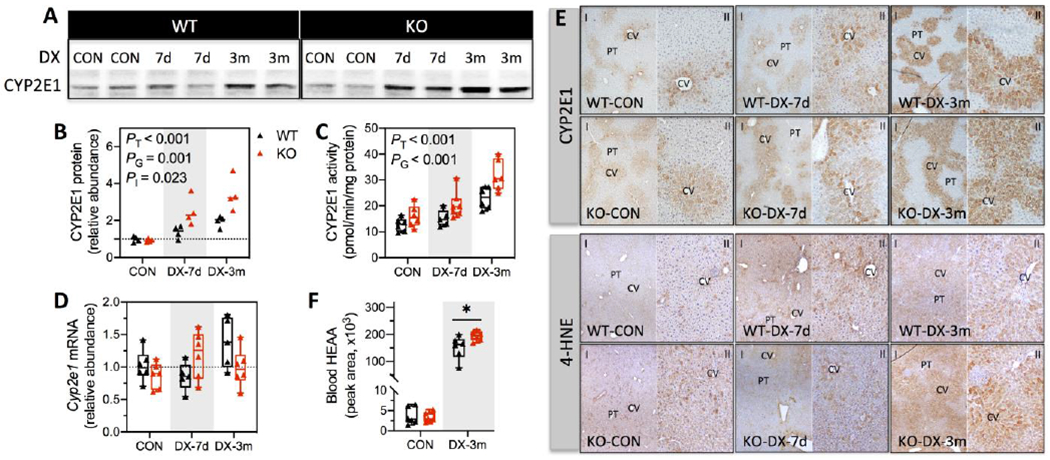
(A) Representative Western blotting images of CYP2E1 using liver whole-cell lysates. (B) CYP2E1 protein levels were quantified by densitometry analysis and normalized to total protein loading per sample (N = 4/group). (C) CYP2E1 activity was determined by measuring the rate of p-nitrophenol oxidation using liver whole-cell lysates and was normalized to total protein levels (mg) per sample (N = 5-8/group). (D) Cyp2e1 mRNA level was measured by Q-PCR (N = 5-8/group). Relative abundance of CYP2E1 protein and mRNA were reported as fold of control (WT-CON). (E) IHC detection of CYP2E1 and 4-HNE in liver tissues. CV, central vein; PT, portal triad. Magnifications, 100x (I) and 200x (II). (F) Plasma levels of HEAA were estimated by untargeted metabolomics analysis as described in the Materials and Methods (N = 6/group). Results are presented in Scatter plots (B) or Box plots (C, D, & F) showing individual data points from each experimental group. Differences between groups for CYP2E1 protein, mRNA and activity were analyzed using two-way ANOVA with post-hoc Bonferroni test correction for the main effects of DX exposures (PT), genotypes (PG), or the interaction between these two factors (PI). Differences between treatment-matched WT and KO mice were analyzed using unpaired student’s t-test. *P < 0.05 was considered significant. WT (black), wild-type mice; KO (red), Gclm-null mice. CON, provided regular water; DX-7d, exposed to DX by gavage for 7 days; DX-3m, exposed to DX in drinking water for 3 months.
3.4. Subchronic DX exposure elevated DNA damage marker γH2AX and 8-OHdG in the liver
Acting as part of the cellular response to direct genotoxic insults, phosphorylation of histone H2A variant (H2AX) at Ser139 residue (γH2AX) occurs rapidly at DNA damage foci. As such, γH2AX has been used as a sensitive molecular markers to track DNA damage and subsequent damage repair (Sharma et al., 2012). The changes of H2AX and γH2AX in our study cohorts were twofold. First, hepatic content of H2AX was ~30% lower in KO-CON mice relative to WT-CON mice, and DX exposure for 3 months further reduced H2AX levels by ~35% in both WT and KO livers (Figs. 5A & 5B). This observation agrees with a previous study reporting accelerated H2AX degradation under cellular oxidative stress (Gruosso et al., 2016), and provides an additional line of evidence supporting DX-elicited oxidative stress in the liver. Second, Ser139 phosphorylation of H2AX, reflected by the relative abundance of γH2AX to the total H2AX pool (measured by the γH2AX/H2AX ratio), was elevated by approximately 1.3- and 2.1-fold, respectively, in the livers of WT and KO mice exposed to DX for 3 months (Figs. 5A & 5B). Notably, IHC analysis using the same primary antibody detected minimal γH2AX-positive hepatocytes in liver tissues of CON mice, but revealed scattered foci of γH2AX-positive hepatocytes in the centrilobular zone of liver tissues from DX-exposed WT and KO mice (Fig. 5C). To further assess if this DNA damage response was directly associated with DX-induced oxidative stress at 3 months, we measured 8-OHdG (an index of oxidative DNA damage) in the liver. Indeed, subchronic DX exposure caused a significant increase in liver 8-OHdG levels in KO mice (~89% increase, P = 0.01) (Fig. 5D), whereas this effect was marginal in WT mice (~33% increase, P = 0.08). Collectively, our data show that subchronic exposure to high dose DX leads to genotoxicity in the liver, which is in part mediated by oxidative stress. To evaluate the presence of preneoplastic lesions in the liver, we examined the expression and distribution of GST-pi and CK7, both of which have been used as markers for preneoplastic changes (Sakai and Muramatsu, 2007; Santos et al., 2014). In our study cohorts, neither protein was altered following 3 months of DX exposure (Fig. S5).
Fig. 5. Detection of DNA damage marker (γH2AX) and oxidative DNA damage (8-OHdG) in the liver.
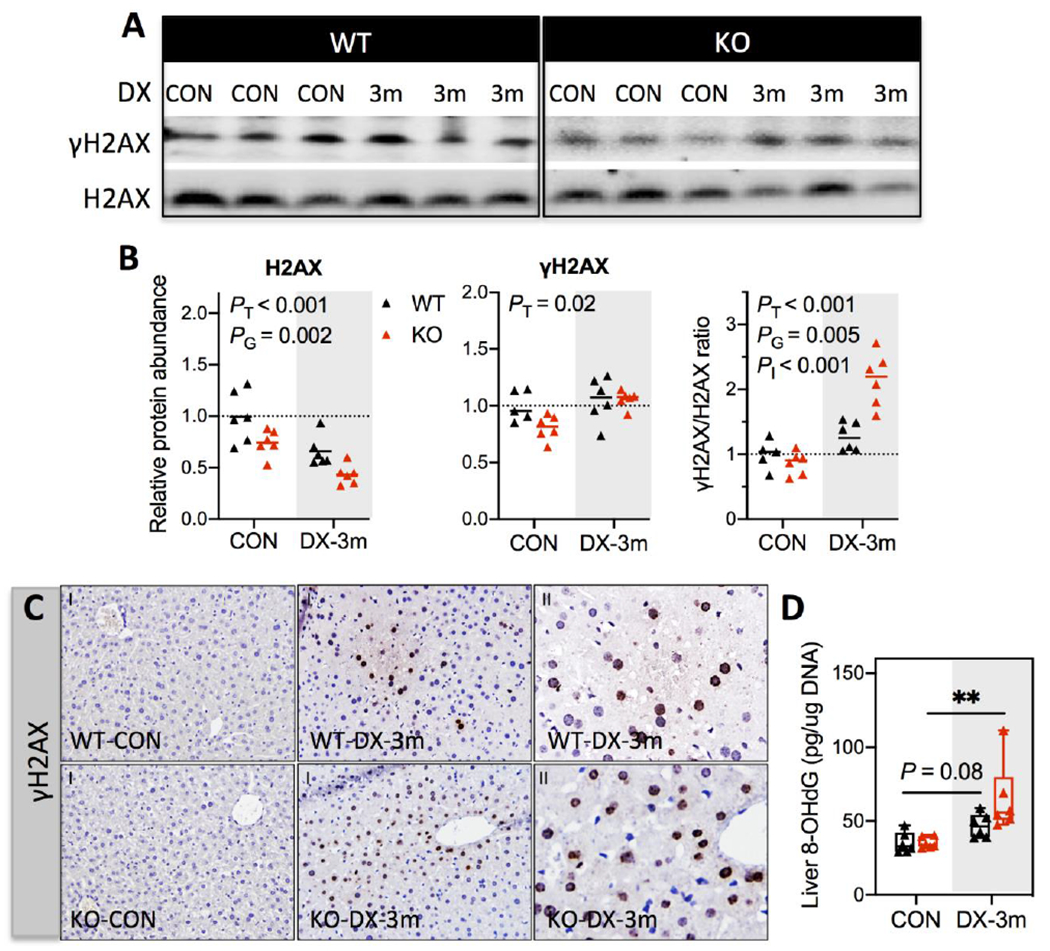
(A) Representative Western blotting images of H2AX and γH2AX using liver whole-cell lysates. (B) Protein levels were quantified by densitometry analysis and normalized to total protein loading per sample (N = 6/group). Relative abundance was reported as fold of control (WT-CON). Results are presented in Scatter plots showing individual data points from each experimental group with the group median at the line (N = 6/group). Differences between groups were analyzed using two-way ANOVA with post-hoc Bonferroni test correction for the main effects of DX exposures (PT), genotypes (PG), or the interaction between these two factors (PI). P < 0.05 was considered significant. (C) IHC detection of γH2AX in liver tissues. Magnifications, 100x (I) and 400x (II). (D) Liver 8-OHdG Levels were measured by a competitive ELISA assay. Results are reported as pg/μg genomic DNA and presented in Box plots showing individual data points from each experimental group (N = 6/group). Group differences were analyzed using one-way ANOVA with post-hoc Dunnett’s test correction. *P < 0.05 was considered significant. CON, provided regular water; DX-3m, exposed to DX in drinking water for 3 months.
4. Discussion
Defining the mechanisms of DX toxicity and carcinogenicity require a comprehensive understanding at the molecular level. Our recent multi-omics study in BDF-1 mice indicated alterations in oxidative stress defense systems accompanying DNA damage as an early change by high dose DX (Charkoftaki et al., 2021). These changes were investigated in more molecular detail in the current mechanistic study, wherein we exposed the C57BL/6J GSH-deficient Gclm-null mice (a model of chronic oxidative stress) and their WT littermates to high dose DX orally for one week or three months. Subchronic DX exposure at 5,000 ppm in drinking water caused mild cytotoxicity in the livers of WT or KO mice. This lack of overt hepatotoxicity from high dose DX exposure is consistent with other subchronic studies (Gi et al., 2018; Kano et al., 2008; Lafranconi et al., 2021). It should be noted that the development of preneoplastic lesions (characterized by GST-pi positive foci) in the liver was observed mostly in rats in these studies. The absence of this phenotype in mice, however, was noted in the current study and has been reported previously (Kano et al., 2008). Such a discrepancy may suggest species differences in the biological process of DX-induced preneoplasia. At the molecular level, we identified a series of changes in the liver by high dose DX, including: (i) a compensatory NRF2 anti-oxidative response in the acute phase (i.e. at 7 days), (ii) progressive induction of CYP2E1 protein expression and enzymatic activity, (iii) development of oxidative stress following subchronic exposure, as evidenced by persistent NRF2 induction, a more oxidized GSH pool, and accumulation of the lipid peroxidation by-product 4-HNE, and (iv) elevations in oxidative DNA damage and DNA damage repair. Importantly, these DX-elicited molecular changes were amplified in mice deficient of GSH. These results provide molecular evidence linking redox dysregulation to DX-induced liver genotoxicity and potentially carcinogenesis.
Oxidative stress due to the overproduction of reactive oxygen species (ROS) is an important mutagenic mechanism for numerous chemical carcinogens (Checa and Aran, 2020; Kirtonia et al., 2020). The involvement of oxidative stress in DX toxicity and carcinogenicity has been investigated in a limited number of studies. Mnaa et al reported the inhibition of catalase activity and increase in malondialdehyde (a lipid peroxidation product) content in the livers of rats exposed to a modest dose of DX in drinking water (100 mg/kg/day) for 6 weeks (Mnaa et al., 2016). Totsuka et al analyzed the spectrum of DNA mutations and adducts in livers from DX-exposed male gpt delta transgenic F344 rats; 8-OHdG was identified as one of the three characteristic DNA adducts formed following 16 weeks exposure to medium (200 ppm) or high (5,000 ppm) doses of DX (Totsuka et al., 2020). DX-elicited redox disturbance has also been reported in kidney tissues following DX exposure for 12 weeks (Qiu et al., 2019). Using ELISA in the current study, we show that exposure to high dose DX produced 8-OHdG in mouse liver tissues. Taken together, results from this study and others support a causal role of oxidative stress in DX-induced liver toxicity and genotoxicity.
In the current study, the hepatic oxidative stress resulting from subchronic DX exposure is likely associated with the observed progressive induction of CYP2E1 enzyme. CYP2E1 activation is an important cellular source of ROS formation, including superoxide anion, hydrogen peroxide and the lipid peroxidation by-products MDA and 4-HNE, some of which are highly reactive and can form mutagenic DNA-adducts (Guengerich, 2020; Linhart et al., 2014; Peter Guengerich and Avadhani, 2018). Our present results also show that CYP2E1 induction by DX in the liver is likely mediated by a post-transcriptional mechanism. It is an interesting finding that low GSH appears to escalate this process as seen in the Gclm-null liver, the underlying mechanism of which is unknown. When comparing WT and Gclm-null exposed mice, we noted a correlation between the CYP2E1 activity in the liver and the HEAA concentration in the plasma, suggesting that liver CYP2E1 is likely an important contributor to DX metabolism in vivo. In addition, recent studies have reported both histopathologic and transcriptomic results that are consistent with a mitogenic response, independent of cytotoxicity, to high dose DX exposure in mouse livers; these effects were found at 6,000 ppm in drinking water for 90 days, but not at lower doses (e.g. ≤ 2000 ppm) (Chappell et al., 2021; Lafranconi et al., 2021). Our current data showing CYP2E1 induction and oxidative stress at 5,000 ppm for 90 days is compatible with the mitogenic findings because CYP2E1 and oxidative stress are both capable of activating mitogenic pathways and inducing a proliferative response in the liver (Cederbaum et al., 2012; Schattenberg and Czaja, 2014). Whether CYP2E1 induction is essential to mitogenesis, genotoxicity and carcinogenesis induced by DX warrants further investigation.
The gap in our mechanistic understanding of DX liver carcinogenicity is a major factor underlying the wide range in DX drinking water targets and the lack of a federal MCL in the US. The focus of the current study was to elucidate redox changes associated with DX cytotoxicity and genotoxicity, a potential MOA that has not been carefully investigated before. We used a high dose of DX that has been shown to cause tumor development in laboratory animals (> 66 mg/kg/day) (Kano et al., 2009). Of note, the cancer effect level demonstrated in Kano et al. (Kano et al., 2009) of 500 ppm in drinking water is 10 fold below the dose level used in this study; in neither case was there remarkable evidence of hepatocellular damage. It will thus be important to track the dose response for effects seen in this study to the lower doses which remain associated with DX carcinogenesis to determine which transcriptomic and biochemical markers are most related to the carcinogenic response. Further, the cancer effect level in animal studies is approximately 10,000 fold above levels to which humans are exposed in contaminated drinking water (Adamson et al., 2017). While toxicological studies using environmentally relevant concentrations of DX are essential, high dose studies are important and necessary to initially explore the adverse biological effects possible from a given chemical in a limited number of animals and thereby to identify mechanistically-relevant events. As such, mechanistic data derived from the current and other high dose studies (Gi et al., 2018; Lafranconi et al., 2021; Totsuka et al., 2020) are expected to serve as important references to refine the assessment of carcinogenic pathways that may be triggered at lower doses, not only in the general population but also in sub-populations of greater susceptibility.
In the current study, we identified the NRF2 stress response, GSH homeostasis, and CYP2E1 induction as being among the cellular targets of high dose DX exposure that can lead to the observed oxidative stress and genotoxicity in mouse livers. This evidence comports with the generally negative findings of DX genotoxicity in vitro, but is consistent with evidence of liver genotoxicity in vivo (EPA, 2020; NJDWQI, 2020). The liver may be vulnerable to the induction of CYP2E1 and other oxidative stress pathways triggered by DX leading to genotoxicity via mechanisms that are less likely used to screen for genotoxicity in the in vitro systems. It is critical to determine how the molecular changes identified in the current study progress at environmentally relevant doses in future longer-term studies. Utilizing cells or animal model systems that are deficient in key steps in these redox and metabolic pathways (e.g. Nrf2-null or Cyp2e1-null mice) will help to delineate their functional significance in DX metabolism and carcinogenicity.
Supplementary Material
Highlights.
1,4-Dioxane evoked transient compensatory NRF2 antioxidant response in mouse livers
CYP2E1 was induced by 1,4-Dioxane, accompanied by oxidative stress in mouse livers
GSH deficiency escalated 1,4-dioxane-induced oxidative DNA damage in mouse livers
Redox dysregulation may play a causal role in 1,4-dioxane liver genotoxicity
Funding
This work was supported in part by the National Institutes of Health grants K01AA025093, R24AA022057, and P30DK034989.
Abbreviations:
- DX
1,4-dioxane
- 4-HNE
4-hydroxynonenal
- 8-OHdG
8-Hydroxy-2’deoxyguanosine
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATSDR
Agency for Toxic Substances and Disease Registry
- BMDL-50
50% liver tumor benchmark dose (lower confidence limit)
- ESI
electrospray ionization source
- GCL
glutamate-cysteine ligase
- Gclc
glutamate-cysteine ligase catalytic subunit gene
- Gclm
glutamate-cysteine ligase modifier subunit gene
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- H2AX
histone H2A variant
- γH2AX
phosphorylation of H2AX at Ser139 residue
- H&E
hematoxylin and eosin
- HEAA
β-hydroxyethoxyacetic acid
- IARC
International Agency for Research on Cancer
- IHC
immunohistochemical
- LOAEL
lowest-observed-adverse-effect level
- MCL
maximal contaminant level
- MOA
mechanism of action
- NOAEL
no-observed-adverse-effect level
- NRF2
nuclear factor erythroid 2-related factor 2
- TCA
1,1,1-trichloroethane
- TCE
trichloroethylene
- PWSs
public water systems
- QC
quality control
- Q-ToF
quadrupole time-of flight
- RC
reference concentration
- ROS
reactive oxygen species
- UCMR3
Unregulated Contaminant Monitoring Rule Round-3
- UPLC
ultra-performance liquid chromatography
- WB
western immunoblotting
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no conflict of interest with the contents of the article.
References
- Adamson DT, Pina EA, Cartwright AE, Rauch SR, Hunter Anderson R, Mohr T, et al. 1,4-Dioxane drinking water occurrence data from the third unregulated contaminant monitoring rule. Sci Total Environ 2017; 596-597: 236–245. [DOI] [PubMed] [Google Scholar]
- ATSDR. Toxicological Profile for 1,4 Dioxane. Agency for Toxic Substances and Disease Registry, Division of Toxicology and Human Health Sciences, Atlanta, GA, 2012. [Google Scholar]
- ATSDR. Support Document to the 2017 Substance Priority List (Candidates for Toxicological Profiles). Agency for Toxic Substances and Disease Registry, Division of Toxicology and Human Health Sciences, Atlanta, GA, 2017. [Google Scholar]
- Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol 2004; 44: 27–42. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI, Yang L, Wang X, Wu D. CYP2E1 Sensitizes the Liver to LPS- and TNF alpha-Induced Toxicity via Elevated Oxidative and Nitrosative Stress and Activation of ASK-1 and JNK Mitogen-Activated Kinases. Int J Hepatol 2012; 2012: 582790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TK, Crespi CL, Waxman DJ. Spectrophotometric analysis of human CYP2E1-catalyzed p-nitrophenol hydroxylation. Methods Mol Biol 2006; 320: 127–31. [DOI] [PubMed] [Google Scholar]
- Chappell GA, Heintz MM, Haws LC. Transcriptomic analyses of livers from mice exposed to 1,4-dioxane for up to 90 days to assess potential mode(s) of action underlying liver tumor development. Current Research in Toxicology 2021; 2: 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charkoftaki G, Golla JP, Santos-Neto A, Orlicky DJ, Garcia-Milian R, Chen Y, et al. Identification of dose-dependent DNA damage and repair responses from subchronic exposure to 1,4-dioxane in mice using a systems analysis approach. Toxicol Sci 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checa J, Aran JM. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J Inflamm Res 2020; 13: 1057–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dong H, Thompson DC, Shertzer HG, Nebert DW, Vasiliou V. Glutathione defense mechanism in liver injury: insights from animal models. Food Chem Toxicol 2013; 60: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Krishan M, Nebert DW, Shertzer HG. Glutathione-deficient mice are susceptible to TCDD-Induced hepatocellular toxicity but resistant to steatosis. Chemical Research in Toxicology 2012; 25: 94–100. [DOI] [PubMed] [Google Scholar]
- Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. The Journal of biological chemistry 2005; 280: 33766–74. [DOI] [PubMed] [Google Scholar]
- Dourson M, Reichard J, Nance P, Burleigh-Flayer H, Parker A, Vincent M, et al. Mode of action analysis for liver tumors from oral 1,4-dioxane exposures and evidence-based dose response assessment. Regul Toxicol Pharmacol 2014; 68: 387–401. [DOI] [PubMed] [Google Scholar]
- Dourson ML, Higginbotham J, Crum J, Burleigh-Flayer H, Nance P, Forsberg ND, et al. Update: Mode of action (MOA) for liver tumors induced by oral exposure to 1,4-dioxane. Regul Toxicol Pharmacol 2017; 88: 45–55. [DOI] [PubMed] [Google Scholar]
- EPA. Toxicological Review of 1,4- Dioxane (With Inhalation Update) (CAS No. 123-91-1) in Support of Summary Information on the Integrated Risk Information System (IRIS) [EPA Report]. U.S. Environmental Protection Agency, Washington DC, 2013. [Google Scholar]
- EPA. United States Environmental Protection Agency. Technical Fact Sheet–1,4-Dioxane In: EPA, editor. EPA 505-F-17-011, 2017. [Google Scholar]
- EPA. Final Risk Evaluation for 1,4-Dioxane. U.S. Environmental Protection Agency, Washington DC, 2020. [Google Scholar]
- Gi M, Fujioka M, Kakehashi A, Okuno T, Masumura K, Nohmi T, et al. In vivo positive mutagenicity of 1,4-dioxane and quantitative analysis of its mutagenicity and carcinogenicity in rats. Arch Toxicol 2018; 92: 3207–3221. [DOI] [PubMed] [Google Scholar]
- Godri Pollitt KJ, Kim JH, Peccia J, Elimelech M, Zhang Y, Charkoftaki G, et al. 1,4-Dioxane as an emerging water contaminant: State of the science and evaluation of research needs. Sci Total Environ 2019; 690: 853–866. [DOI] [PubMed] [Google Scholar]
- Gruosso T, Mieulet V, Cardon M, Bourachot B, Kieffer Y, Devun F, et al. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol Med 2016; 8: 527–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP. Cytochrome P450 2E1 and its roles in disease. Chem Biol Interact 2020; 322: 109056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HealthCanada. Health Canada. 1,4-Dioxane in Drinking Water -Guideline Technical Document for Public Consultation, http://www.canada.ca/content/dam/hc-sc/documents/programs/consultation-1-4-dioxane-drinking-water/pub-eng.pdf. 2018; Accessed April 10, 2019.
- IARC. Cadmium, Nickel, Some Epoxides, Miscellaneous Industrial Chemicals andGeneral Considerations on Volatile Anaesthetics. IARC Monogr Eval Carcinog Risk Chem Man 1976; 11: 247–56. [PubMed] [Google Scholar]
- IARC. Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42. IARC Monogr Eval Carcinog Risk Chem Man 1987; Suppl. 7: 201. [PubMed] [Google Scholar]
- IRIS. 1,4-Dioxane (CASRN 123-91-1). U.S. Environmental Protection Agency, Washington DC, 2013. [Google Scholar]
- JBRC. Two-year studies of 1,4-dioxane in F344 rats and BDF1 mice (drinking water). Japan Bioassay Research Center, Kanagawa, Japan, 1998. [Google Scholar]
- Kano H, Umeda Y, Kasai T, Sasaki T, Matsumoto M, Yamazaki K, et al. Carcinogenicity studies of 1,4-dioxane administered in drinking-water to rats and mice for 2 years. Food Chem Toxicol 2009; 47: 2776–84. [DOI] [PubMed] [Google Scholar]
- Kano H, Umeda Y, Saito M, Senoh H, Ohbayashi H, Aiso S, et al. Thirteen-week oral toxicity of 1,4-dioxane in rats and mice. J Toxicol Sci 2008; 33: 141–53. [DOI] [PubMed] [Google Scholar]
- Kendig EL, Chen Y, Krishan M, Johansson E, Schneider SN, Genter MB, et al. Lipid metabolism and body composition in Gclm(−/−) mice. Toxicol Appl Pharmacol 2011; 257: 338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci 2020; 77: 4459–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kociba RJ, Mccollister SB, Park C, Torkelson TR, Gehring PJ. 1,4-Dioxane Toxicity as Determined by a 2-Year Dose-Response Study in Rats. Toxicology and Applied Pharmacology 1974; 29: 86–86. [Google Scholar]
- Lafranconi M, Budinsky R, Corey L, Klapacz J, Crissman J, LeBaron M, et al. A 90-day drinking water study in mice to characterize early events in the cancer mode of action of 1,4-dioxane. Regul Toxicol Pharmacol 2021; 119: 104819. [DOI] [PubMed] [Google Scholar]
- Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J Clin Invest 2018; 128: 2226–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindros KO. Zonation of cytochrome P450 expression, drug metabolism and toxicity in liver. Gen Pharmacol 1997; 28: 191–6. [DOI] [PubMed] [Google Scholar]
- Linhart K, Bartsch H, Seitz HK. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol 2014; 3: 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- McConnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, Fernandez C, et al. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol Sci 2007; 99: 628–36. [DOI] [PubMed] [Google Scholar]
- Mnaa S, Shaker ES, Mahmoud HI. Inhibitory Activity of Protected Edible Plants on Oxidative Stress Induced by Oral 1,4-Dioxane. J Egypt Soc Parasitol 2016; 46: 135–43. [DOI] [PubMed] [Google Scholar]
- Morita T, Hayashi M. 1,4-Dioxane is not mutagenic in five in vitro assays and mouse peripheral blood micronucleus assay, but is in mouse liver micronucleus assay. Environ Mol Mutagen 1998; 32: 269–80. [DOI] [PubMed] [Google Scholar]
- Nannelli A, De Rubertis A, Longo V, Gervasi PG. Effects of dioxane on cytochrome P450 enzymes in liver, kidney, lung and nasal mucosa of rat. Archives of Toxicology 2005; 79: 74–82. [DOI] [PubMed] [Google Scholar]
- National Toxicology P. Bioassay of 1,4-dioxane for possible carcinogenicity. Natl Cancer Inst Carcinog Tech Rep Ser 1978; 80: 1–123. [PubMed] [Google Scholar]
- NJDWQI. Health-Based Maximum Contaminant Level Support Document, 1,4-dioxane. http://www.state.nj.us/dep/watersupply/pdf/14-dioxane-pub-rev-health-sub.pdf. 2020.
- Peter Guengerich F, Avadhani NG. Roles of Cytochrome P450 in Metabolism of Ethanol and Carcinogens. Adv Exp Med Biol 2018; 1032: 15–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Cheng J, Xie Y, Jiang L, Shi P, Li X, et al. 1,4-Dioxane exposure induces kidney damage in mice by perturbing specific renal metabolic pathways: An integrated omics insight into the underlying mechanisms. Chemosphere 2019; 228: 149–158. [DOI] [PubMed] [Google Scholar]
- Sakai M, Muramatsu M. Regulation of glutathione transferase P: a tumor marker of hepatocarcinogenesis. Biochem Biophys Res Commun 2007; 357: 575–8. [DOI] [PubMed] [Google Scholar]
- Santos NP, Oliveira PA, Arantes-Rodrigues R, Faustino-Rocha AI, Colaco A, Lopes C, et al. Cytokeratin 7/19 expression in N-diethylnitrosamine-induced mouse hepatocellular lesions: implications for histogenesis. Int J Exp Pathol 2014; 95: 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattenberg JM, Czaja MJ. Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol 2014; 3: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 2012; 920: 613–26. [DOI] [PubMed] [Google Scholar]
- Sun M, Lopez-Velandia C, Knappe DR. Determination of 1,4-Dioxane in the Cape Fear River Watershed by Heated Purge-and-Trap Preconcentration and Gas Chromatography-Mass Spectrometry. Environ Sci Technol 2016; 50: 2246–54. [DOI] [PubMed] [Google Scholar]
- Totsuka Y, Maesako Y, Ono H, Nagai M, Kato M, Gi M, et al. Comprehensive analysis of DNA adducts (DNA adductome analysis) in the liver of rats treated with 1,4-dioxane. Proc Jpn Acad Ser B Phys Biol Sci 2020; 96: 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbur S, Jones D, Risher JF, Crawford J, Tencza B, Llados F, et al. Toxicological Profile for 1,4-Dioxane, Atlanta (GA), 2012. [PubMed] [Google Scholar]
- Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. Journal of Biological Chemistry 2002; 277: 49446–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.