

Abstract
Irreversible electroporation (IRE) is a new cancer ablation technology, but methods to improve IRE-induced therapeutic immunity are only beginning to be investigated. We developed a mouse model bearing large primary (300 mm3) and medium distant (100 mm3) EG7 lymphomas engineered to express ovalbumin (OVA) as a nominal tumor antigen. We established experimental protocols including IRE alone and IRE combined with Toll-like receptor (TLR)3/9 agonists (poly I:C/CpG) (IRE + pIC/CpG), PD-1 blockade (IRE + PD-1 blockade), or both (IRE + Combo) to investigate therapeutic effects on primary and distant EG7 tumors and conversion-promoting effects on the immunotolerant tumor microenvironment (TME). We demonstrated that IRE alone simulated very weak OVA-specific CD8+ T cell responses and did not inhibit primary tumor growth. IRE + pIC/CpG synergistically stimulated more efficient OVA-specific CD8+ T cell responses and primary tumor growth inhibition than IRE + PD-1 blockade. IRE + pIC/CpG played a major role in the modulation of immune cell profiles but a minor role in the downregulation of PD-L1 expression in the TME and vice versa for IRE + PD-1 blockade. IRE + Combo cooperatively induced potent OVA-specific CD8+ T cell immunity and rescued exhausted intratumoral CD8+ T cells, leading to eradication of not only primary tumors but also untreated concomitant distant tumors and lung metastases. IRE + Combo efficiently modulated immune cell profiles, as evidenced by reductions in immunotolerant type-2 (M2) macrophages, myeloid-derived suppressor-cells, plasmacytoid dendritic cells, and regulatory T cells and by increases in immunogenic M1 macrophages, CD169+ macrophages, type-1 conventional dendritic cells, and CD8+ T cells, leading to conversion of immunotolerance in not only primary TMEs but also untreated distant TMEs. IRE + Combo also showed effective therapeutic effects in two breast cancer models. Therefore, our results suggest that IRE + Combo is a promising strategy to improve IRE ablation therapy in cancer.
Keywords: IRE ablation, TLR3/9-agonists, PD-1-blockade, CD8+ T-cell response, antitumor immunity
Subject terms: Immunotherapy, Tumour immunology
Introduction
The tumor microenvironment (TME) plays important role in regulating tumor progression, metastases, and therapeutic responses and is composed of a variety of tumor-associated, immune, stromal, and myeloid cell subsets [1]. These tumor-associated cellular populations can be divided into two major groups with different phenotypes and distinct functional (i.e., immunogenic and immunotolerant) effects. The immunogenic group includes (i) tumoricidal CD11b+F4/80+MHCII+ type-1 (M1) macrophages that secrete inflammatory cytokines driving polarization of immunogenic CD4+ Th1 cell responses and halting tumor growth [2], (ii) CD11b+F4/80+CD169+ (M169) macrophages that dominate antitumor immunity by cross-presenting apoptotic tumor cell antigens to CD8+ T cells [3], and (iii) type-1 conventional CD8+CD103+CD11c+CD11b− dendritic cells (cDC1s), which are superior stimulators of CD8+ T cell responses in the TME [4]. The immunotolerant group includes (i) protumorigenic CD11b+F4/80+MHCII- type-2 (M2) macrophages that produce suppressive TGF-β and IL-10 and promote tumor angiogenesis; [5] (ii) CD11b+Gr1+Ly6G+ myeloid-derived suppressor cells (MDSCs), the major player or the “Queen Bee” in the immunotolerant TME [6] that activate CD4+CD25+Foxp3+ regulatory T (Treg) cells and produce inhibitory TGF-β, arginase-1, and IDO (indoleamine 2,3-dioxygenase) molecules; [7] (iii) CD317+B220+ plasmacytoid DCs (pDCs) that favor suppressive Treg cell expansion; [8] and (iv) CD4+CD25+Foxp3+ Treg cells with an inhibitory effect on CD8+ T cell responses mediated by secretion of suppressive IL-10 and TGF-β and expression of inhibitory PD-1 (programmed cell death protein-1) and CTLA-4 (cytotoxic T lymphocyte-associated protein-4) molecules [9]. The TME is profoundly immunosuppressive when the above immunotolerant cells predominate. Although CD8+ T cells play an important role in host defense against tumors [10], they are often blocked from entering tumors [11] or become dysfunctional in an immunotolerant TME [12]. The immunotolerant TME thus is a key reason why most immunotherapies based upon stimulation of tumor-specific CD8+ T cell responses against tumors consistently display limited efficacy. Therefore, developing new emergent approaches by targeting the immunotolerant TME represents a critical topic in cancer immunotherapies [1].
Irreversible electroporation (IRE) is a new nonthermal form of cancer ablation technology that delivers short bursts of current to ‘punch’ irreversible nanoholes in cell membranes, leading to massive tumor cell apoptosis [13]. Compared to radiofrequency ablation (RFA) with heat-induced collateral damage, IRE is safe for application near blood vessels, bile ducts, and nerves [14]. IRE ablation therapy has been applied to cancers in many locations, including liver, pancreas, breast, lung, and prostate tumors [15]. However, IRE ablation-induced antitumor immune responses are too weak to eradicate local primary tumors, and patients often experience local or distant tumor recurrence [16, 17]. Therefore, improving the therapeutic effects of IRE ablation is an urgent need in cancer ablation therapy. However, methods to improve IRE-induced therapeutic immunity are only beginning to be investigated.
Recently, Li’s group showed that IRE ablation alone increased vascular density and permeability and that IRE ablation combined with PD-1 blockade induced an increase in tumor-infiltrating CD8+ T cells and eradicated 33% of KRAS+ pancreatic tumors but only mildly modulated the TME by increasing the CD8+ T cell-to-Treg cell ratio [18]. In a more recent study, White’s group demonstrated that IRE ablation combined with a Toll-like receptor (TLR) 7agonist and PD-1 blockade improved the therapeutic effect by increasing cDC1s in the TME, resulting in the inhibition of primary KPC4580P pancreatic tumor growth and regression of distant tumors in ~60% of mice [19]. However, only nonspecific, not tumor-specific, CD8+ T cell responses (the critical measurement for IRE-induced antitumor immunity) were assessed in their mouse pancreatic cancer models.
TLRs are an evolutionarily ancient family of pattern recognition receptors that sense and trigger DC maturation, and most TLR agonists have been shown to significantly enhance adaptive immunity [20]. For example, the TLR3 agonist poly I:C (pIC) and TLR9 agonist CpG, which were found to enhance CD4+ Th1 and CD8+ cytotoxic T cell responses [21, 22], are used for targeting innate sensing in the TME to improve immunotherapy [23, 24]. PD-1 blockade using anti-PD-1 or anti-PD-L1 antibodies that stimulate potent antitumor immunity by blocking the inhibitory PD-1/PD-L1 pathway in CD8+PD-1+ T cells and rescuing T cell exhaustion [25] has been commonly used in clinical applications as cancer immunotherapy [26]. PD-1 blockade combined with TLR3 or TLR9 agonists was also found to enhance cancer immunotherapies [27, 28]. We recently performed an RFA study with an ovalbumin (OVA)-expressing EG7 lymphoma model and demonstrated that administration of the TLR9 agonist CpG significantly enhanced RFA-induced tumor OVA-specific CD8+ T cell responses, leading to inhibition of not only primary tumor growth but also lung metastases [29].
In the present study, we performed IRE ablation combined with PD-1 blockade as well as the TLR3 agonist pIC and TLR9 agonist CpG (i.e., IRE + PD-1 blockade+pIC/CpG or IRE + Combo) in mice bearing well-established EG7 lymphomas, followed by an investigation of OVA-specific CD8+ T cell responses and the therapeutic effects leading to eradication of primary and distant EG7 tumors and lung metastases. We systematically assessed immune cell profiles in the TME by quantitatively measuring both immunogenic and immunotolerant cell subsets. We demonstrated that IRE + pIC/CpG synergistically stimulated potent OVA-specific CD8+ T cell responses, leading to significant inhibition of primary tumor growth; the effects were stronger than those observed with IRE + PD-1 blockade. We also found that compared with PD-1 blockade, TLR3/9 agonists were more effective in promoting immunogenic cells (M1 macrophages, M169 macrophages, cDC1s, and CD4+ and CD8+ T cells) and reducing immunotolerant cells (M2 macrophages, Treg cells, MDSCs, and pDCs) but less efficient in downregulating PD-L1 (PD-ligand-1) expression in immunotolerant M2 macrophages, MDSCs and EG7 tumor cells in the TME. The combined treatment IRE + Combo cooperatively induced potent OVA-specific CD8+ T cell immunity and rescued tumor-infiltrating exhausted CD8+ T cells, leading to complete eradication of both primary tumors and distant tumors as well as lung metastases via dramatic conversion of the immunotolerant TME into an immunogenic TME in both the primary and distant tumors. Furthermore, IRE + Combo also showed effective therapeutic effects in two mouse breast cancer models (Tg1-1 and 4T1). Taken together, our results suggest that IRE + Combo is a promising strategy to improve IRE ablation therapy in cancer.
Materials and methods
Regents, cells lines and mice
A fluorescein isothiocyanate (FITC)-conjugated anti-CD8 antibody (Ab) was obtained from Bio–Rad (Hercules, CA). A phycoerythrin (PE)-conjugated H-2Kb/OVA257-264 tetramer (PE-tetramer) was obtained from the Fred Hutchinson Cancer Research Center (Seattle, WA). The following Abs and reagents were obtained from BioLegend (San Diego, CA): FITC-conjugated anti-CD8, FITC-conjugated anti-CD3, PE-conjugated anti-CD45.1, Alexa Fluor-conjugated anti-CD45.1, PE-conjugated anti-CD4, PE-conjugated anti-CD25, PE-Cy5-conjugated anti-CD11c, APC-conjugated anti-CD11b, APC/Cy7-conjugated anti-I-A/I-E (MHCII), Alexa Fluor 700-conjugated anti-Ly6G, BV421-conjugated anti-CD103, PE-Cy5-conjugated anti-F4/80, anti-Gr1, BV421-conjugated anti-CD169, PE-Cy5-conjugated anti-CD8, anti-CD317, anti-CD220, BV421-conjugated anti-PD-L1, PE-Cy5-PE-conjugated anti-IDO, PE-arginase, PE-conjugated anti-TGF-β, PE-Cy5-conjugated anti-FoxP3, PE-Cy5-conjugated anti-TNF-α, PE-Cy5-conjugated anti-IFN-γ, and the Zombie Aqua Fixable Viability Kit. CpG oligodeoxynucleotides 1826 (CpG ODN 1826) and poly:IC (pIC) were obtained from Invitrogen Inc. (San Diego, CA). Cytofix/Cytoperm kits and lysis buffer were purchased from BD Bioscience (Franklin Lakes, NJ). An anti-mouse PD-L1 antibody for the in vivo experiment was obtained from BioXCell (Lebanon, NH). The mouse lymphoma cell line EL4 and ovalbumin (OVA) transgene-transfected EL4 cell line EG7 were obtained from American Type Culture Collection (ATCC; Rockville, MD), while the OVA transgene-transfected B16 melanoma cell line BL6-10OVA was generated in our laboratory [29]. EL4 cells were maintained in RPMI medium (Life Technologies, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS), while OVA-expressing EG7 and BL6-10OVA cells were maintained in the above medium plus G418 (0.5 mg/mL; Life Technologies). Six- to 8-week-old female C57BL/6 (B6, CD45.2+), B6.1 (CD45.1+), and OVA-specific TCR transgenic OT-I mice were purchased from The Jackson Laboratory (Bar Harbor, ME). CD45.1+ OT-I mice were obtained by cross-breeding B6.1 mice with OT-I mice. All animal experiments were approved by the Animal Research Ethics Board, University of Saskatchewan (Protocol# 20160056).
IRE ablation combined with TLR3/9 agonists and PD-1 blockade
EG7 cells were subcutaneously (s.c.) injected into the right flank of B6 mice (3 × 106 cells/mouse). Tumor growth was measured using digital calipers. Tumor volume was calculated using the formula A/2 × B2, where A and B are the long and short tumor dimensions, respectively, and once tumors reached ~300 mm3 (8–9 mm in diameter), we first assessed whether TLR3/9 agonists (pIC and CpG), PD-1 blockade (anti-PD-L1 Ab) or a combination of the above (pIC/CpG±PD-1 blockade; Combo) stimulate OVA-specific CTL responses. For CpG and/or pIC administration, mice were intratumorally (i.t.) injected with 10 μg of CpG and/or 10 μg of pIC in 30 μL of PBS for a total of three injections in three positions at peripheral areas of the tumor. Anti-PD-L1 Ab injections (200 µg/mouse) were simultaneously administered intraperitoneally (i.p.) every two days for a total of four injections. At seven days post anti-PD-L1 Ab or TLR agonist injection, blood samples were collected from the mouse tail for assessment of OVA-specific CD8± T cell responses using PE-tetramer and a FITC-conjugated anti-CD8 Ab by flow cytometry. To perform IRE ablation, mice bearing ~300 mm3 EG7 tumors were anesthetized by inhalation of isoflurane gas (5% isoflurane for anesthesia induction; 2% for maintenance) [29]. The fur covering the tumor area was removed, and two insulated custom-built pulse-delivery metal electrode needles (0.2 mm in diameter, separated by 5 mm center-to-center) of a custom-made IRE device [30] were inserted into the tumor to deliver electric pulses. The IRE parameters (voltage: 1,200 V/cm; pulse duration: 90 μs; pulse repetition frequency: 1 Hz; the number of repetition pulses: 100) were similar to a previous IRE ablation protocol for animal tumor models [18]. The needles were subsequently reoriented by 90°, and the above process was repeated once. Following all procedures, mice were given s.c. injections of buprenorphine (0.05 mg/kg of body weight) for pain control. Mice recovered post treatment on a warming blanket. To assess whether pIC, CpG, and PD-1 blockade potentiate IRE-induced CTL responses and antitumor immunity, we further developed seven different treatments by combining IRE ablation with pIC, CpG, and/or PD-1 blockade. These treatments included (i) IRE control, (ii) IRE + PD-1 blockade, (iii) IRE + pIC, (iv) IRE + CpG, (v) IRE + pIC/CpG, (vi) IRE + PD-1 blockade+pIC/CpG (IRE + Combo) and (vii) Combo alone. For CpG and/or pIC adjuvant administration, mice were similarly i.t. injected with 10 μg of CpG and/or 10 μg of pIC in 30 μL of PBS for a total of three injections in three positions at peripheral areas of the tumor post IRE ablation. Anti-PD-L1 Ab injections (200 µg/mouse) were similarly i.p. administered every two days starting one day prior to IRE for a total of four injections. At seven days post IRE, blood samples were collected from the mouse tail for assessment of OVA-specific CD8+ T cell responses by flow cytometry, followed by daily monitoring of tumor growth or regression. To examine whether IRE + Combo-induced CD8+ T cell responses affect distant tumor growth, B6 or B6.1 mice were s.c. injected with EG7 cells in the right and left flanks of the lower back (3 × 106 and 1 × 106 cells/mouse, respectively). When the right primary and left distant tumors reached ~300 and ~100 mm3, respectively, IRE + Combo, IRE or Combo alone was performed on the right primary tumors. Distant tumor growth or regression was monitored daily. To assess whether IRE + Combo-induced CD8+ T cell responses protect mice from tumor lung metastasis, B6 mice bearing primary tumors and control naïve mice were intravenously (i.v.) injected with BL6-10OVA cells (0.5 × 106 cells/mouse) to form lung tumor metastases 7 days prior to the primary tumors reaching ~300 mm3. We then performed IRE + Combo treatment to eradicate the primary tumors seven days after BL6-10OVA cell injection. Mouse lung tissues were collected 21 days after BL6-10OVA cell injection. Black tumor colonies in the lungs were counted and confirmed by histopathological examination. For ethical reasons, mice bearing tumors ~2,500 mm3 (~17 mm in diameter) were sacrificed and recorded as deaths.
CD8+ T cell depletion study
Female B6 mice (five mice/group) were i.p. injected with three doses of anti-CD8 Ab (200 µg/injection) on consecutive days prior to IRE + Combo treatment. Successful depletion was confirmed by flow cytometric analysis of mouse peripheral blood. Mice were then treated with IRE or IRE + Combo therapy one day after the last Ab injection, followed by monitoring of tumor growth or regression. The anti-CD8 Ab was additionally i.p. injected once every three days for a total of five injections to maintain depletion during the study.
Analysis of OVA-specific CD8+ T cell responses
To measure OVA-specific CD8+ T cell responses, tail nicking was used to collect peripheral blood samples from tumor-bearing mice subjected to different IRE ablation treatments into collection tubes containing the anticoagulant heparin at seven days post IRE. To assess IRE + Combo-induced long-term T cell memory, IRE + Combo-treated mice with complete primary tumor regression for 30 days were i.v. boosted with recombinant Listeria monocytogenes expressing OVA (rLmOVA) (1,000 colony-forming units/mouse) [31]. Mouse tail blood samples were collected four days post rLmOVA injection. PE-tetramer (10 µL) was added to each tube, followed by incubation for 30 min at room temperature protected from light. A FITC-conjugated anti-CD8 Ab was then added to each tube and incubated for 30 min at room temperature. Red blood cells were lysed using BD lysis buffer (BD Bioscience). Samples were analyzed for measurement of OVA-specific CD8+ T cell responses by flow cytometry.
Flow cytometric analysis of tumor-infiltrating immune cell profiles
To assess tumor-infiltrating immune cells, B6.1 (CD45.1+) mice were challenged with EG7 cells. This approach enabled us to distinguish recipient CD45.1+ mouse immune cells from CD45.2+ EG7 tumor cells when analyzing tumor-derived single-cell suspensions by flow cytometry. Two sources (untreated and IRE-treated) of tumor tissues were used for the preparation of single-cell suspensions. To compare the extent of immunotolerance in the TMEs of different-sized tumors, tumor tissues were collected when tumors grew to 4, 6, or 8–9 mm in diameter. Tumor tissue single-cell suspensions were prepared using the Tumor Dissociation Kit (Miltenyi Inc., San Diego, CA) according to the manufacturer’s instructions. Briefly, tumor tissue samples were also collected from the peripheral area of tumors treated with different IRE protocols at three days post IRE. The collected tumor tissues were first cut into 1 mm3 fragments and then incubated in 5 mL of RPMI medium containing 1 mg/mL collagenase IV and 0.2 mg/mL DNase-I at 37 °C for 30 min, followed by brief homogenization with a syringe plunger. The cell suspensions were then filtered through a 40-μm filter. Erythrocytes were finally lysed by incubating the cells with red blood cell lysis buffer (0.84% Tris-ammonium chloride) for 5 min. To exclude dead cells during analysis, live-dead cell staining with Zombie Aqua Fixable Viability dye was performed according to the manufacturer’s instructions prior to any antibody staining. Cell suspensions were then stained with a cocktail of antibodies against a combination of molecular markers used to distinguish different immune cell populations, such as immunogenic cDC1s, M1 macrophages, M19 macrophages, and CD4+ and CD8+ T cells, as well as immunotolerant M2 macrophages, MDSCs, pDCs, and Treg cells, as we and others previously described [18, 19, 29, 32]. Briefly, live tumor-infiltrating leukocyte populations were gated for initial analysis of CD45.1+ immune cell populations distinct from CD45.2+ EG7 tumor cells. Neutrophils and monocytes were later removed from the host mouse CD45.1+ cell population based on the expression of Ly6G [32]. Various immune cell populations were then progressively gated with antibodies against their cell markers for analysis. For example, the macrophage population was gated as CD11b+F4/80+ cells to further measure MHCII expression for quantification of the percentage of CD11b+F4/80+MHCII- M2 macrophages and percentage of CD11b+F4/80+MHCII+ M1 macrophages in the CD11b+F4/80+ macrophage population (Supplementary Figure S1A) or to further measure MHCII and CD169 expression for quantification of the percentage of CD11b+F4/80+MHCII+CD169+ M169 macrophages [3, 33] in the CD11b+F4/80+ macrophage population (Supplementary Figure S1B). DC populations were gated as CD11c+ cells to further analyze the expression of CD8, CD103, and MHCII for quantification of the percentage of CD8+CD103+MHCII+ cDC1s in the CD11c+ DC population (Supplementary Figure S1C). The monocyte population was gated as CD45.1+ cells to further sequentially assess the expression of CD11b and Gr1/Ly6G for quantification of the percentage of CD11b+Gr1+Ly6C+ MDSCs in the CD45.1+ cell population with the calculation formula % CD11b+CD45.1+ cells × % Gr1+Ly6G+ cells (Supplementary Figure S1D). The monocyte population was gated as CD45.1+ cells to further sequentially assess the expression of CD11b/Gr1 and CD317/B220 for quantification of the percentage of CD11b-CD317+Gr1-B220+ pDCs in the CD45.1+ cell population with the calculation formula % CD11b-Gr1- cells × % CD317+B220+ cells (Supplementary Figure S1E). The monocyte population was gated as CD45.1+ cells to further measure CD3 and CD4 or CD8 expression for quantification of the percentages of CD4+ or CD8+ T cells in the CD3+ T cell population (Supplementary Figure S1F). The monocyte population was gated as CD45.1+ cells to further sequentially measure CD3/CD4 for quantification of CD4+ T cells and CD4/Foxp3 expression for quantification of the percentage of immunotolerant CD4+Foxp3+ Treg cells in the CD4+ T cell population (Supplementary Figure S1G). In addition, the EG7 tumor cell population was gated as live CD45.1- or CD45.2+ cells (Supplementary Figure S1H). Gated cell populations such as M2 macrophages, MDSCs, and tumor cells were also analyzed for the expression of cell-surface PD-L1 by flow cytometry. For intracellular staining, cells were first stained for surface markers. Then, they were fixed and permeabilized using a Cytofix/Cytoperm kit (BD Bioscience) and stained with Abs against intracellular markers such as Foxp3, IDO, and arginase-1. Stained cells were analyzed by flow cytometry. All flow cytometry data were acquired with a CytoFLEX cytometer (Beckman Coulter Inc.) and analyzed using FlowJo (10.4.0) software (FlowJo, LLC, Ashland, OR).
CD8+ T cell proliferation and cytotoxicity assays
To assess CD8+ T cell proliferation and cytotoxic effects, we first enzymatically prepared tumor-derived single-cell suspensions as described above, followed by purification of live cells with Ficoll-based density gradient centrifugation using Ficoll-Paque PREMIUM 1.084 Solution (GE Healthcare Bio-sciences Inc., Uppsala, Sweden) according to the manufacturer’s instructions. We then further purified tumor-infiltrating CD8+ T cells from the tumor-derived single-cell suspensions using the CD8+ T Cell Isolation Kit (StemCell Tech Inc., Vancouver, BC, Canada) according to the manufacturer’s instructions. To measure T cell proliferation, CD8+ T cells purified from IRE-, IRE ± Combo-, and Combo-treated tumor tissues (0.5 × 106 cells/well in a U-bottomed 96-well plate) were incubated at 37 °C in RPMI medium containing 10% FCS, IL-2 (40 U/ml) and the OVAI (SIINFEKL) peptide or control (unrelated) Mut1 (FEQNTAQP) peptide (0.1 nM) for 48 h [34], followed by cell counting. To assess T cell cytotoxicity, purified CD8+ T cells derived from IRE-, IRE+ Combo-, and Combo-treated tumor tissues were first incubated in RPMI medium containing 10% FCS, phosphomolybdic acid (PMA, 0.081 µM) and ionomycin (1.34 µM) (Sigma–Aldrich, St. Louis, MO) at 37 °C for 1 h [35, 36] and then used as effector cells in a T cell cytotoxicity assay [31]. Briefly, EG7 and EL4 tumor cells labeled with GranToxiLux (a cell-permeable fluorogenic granzyme-B substrate) using the GranToxiLux®–PLUS Kit (OncoImmunin Inc., Gaithersburg, MD) according to the manufacturer’s instructions were used as target cells and control target cells, respectively. Mixtures containing effector CD8± T cells and GranToxiLux-labeled target cells (10:1 and 2:1) in culture medium were incubated at 37 °C for 1 h, followed by flow cytometric analysis to detect the fluorescence emitted due to fluorogenic granzyme-B substrate cleavage (GBSC) in the target cells undergoing cell apoptosis [31].
Tumor-draining lymph node cell analysis
Tumor-draining lymph nodes (TDLNs) were collected from IRE + Combo- and control IRE-treated mice on day 7 post IRE and homogenized by forcing the tissues through 40-µm nylon mesh with a syringe plunger. Single-cell suspensions were stained with Abs against CD8, CD11c, and CD103 for detection of cDC1s by flow cytometry. For analysis of endogenous cytokine production by CD8+ T cells, single-cell suspensions were first stimulated with PMA (0.081 µM), ionomycin (1.34 µM), 3 μg/mL brefeldin A (BD Biosciences), and 2 μM monensin (BD Biosciences) for 5 h in complete RPMI and then fixed and permeabilized using Cytofix/Cytoperm kits (BD Bioscience), followed by staining with Abs against intracellular IFN-γ and TNF-α molecules. Cells were analyzed by flow cytometry. Data were acquired with a CytoFLEX cytometer and analyzed using FlowJo software.
Cytokine ELISAs
Sera were collected from tumor-bearing mice on day 3 post IRE + Combo treatment, IRE ablation, or Combo treatment alone. The concentrations of cytokines (IL-2, IFN-γ, and TGF-β) in mouse sera were measured using Mouse IL-2, IFN-γ, and TGF-β ELISA Kits (Abcam Inc.) according to the manufacturer’s instructions.
Immunohistochemistry
Frozen tumors were completely embedded in optimal cutting temperature (OCT) compound, and then 6-μm cryostat sections were cut and used for immunohistochemical detection. Briefly, slides were fixed with cold acetone for 20 min and then rinsed with PBS two times for 5 min each time. The slides were incubated in 3% hydrogen peroxide for 10 min to block endogenous peroxidases and then rinsed with PBS two times for 5 min each time. Then, the tissue sections were blocked with 1% BSA at room temperature for 30 min and incubated with polyclonal rabbit anti-CD8 or anti-Ly6G Abs in PBS with 1% BSA at 4 °C overnight in a humidified chamber for the detection of tumor-infiltrating CD8+ T cells and MDSCs, respectively [37]. After washing with PBS three times, the sections were incubated with an HRP-labeled anti-rabbit IgG Ab for 30 min at room temperature, followed by incubation with a DAB (3,3-diaminobenzidine) developing solution. Hematoxylin was used for counterstaining. The sections were dehydrated using an increasing ethanol gradient (75, 90, and 100%) and xylene and then mounted with a coverslip using a permanent mounting medium. The slides were imaged using a microscope at 50× and 200× magnification.
Statistical analysis
Data were analyzed using GraphPad Prism version 8 (GraphPad Software Inc.). Tumor growth curves were first analyzed with two-way ANOVA, and groups were compared with Tukey’s test. Kaplan–Meier survival curves were analyzed with the log-rank test. A two-tailed Student’s t test was applied to compare two experimental groups. Multiple comparisons were conducted using one-way ANOVA followed by Tukey’s test. A value of p < 0.05 was considered statistically significant. Results are presented as the mean ± standard error of the mean (SEM).
Results
TME immunotolerance increases with tumor stage
To investigate whether tumor stage affects TME development, different sizes (4, 6, and 8–9 mm in diameter or ~30, ~100, and ~300 mm3 in volume) of subcutaneous (s.c.) EG7 lymphomas (Fig. 1A) grown in B6.1 mice were sectioned for histopathological examination. We found some areas of focal tumor necrosis in the center of large tumors (Fig. 1B), possibly due to lack of blood supply, but not in small or medium-sized tumors. To assess TME immunotolerance, we performed flow cytometric analysis of single-cell suspensions prepared from tumor tissues (Supplementary Fig. S1). With increasing tumor size (small to medium to large, respectively), we observed trends toward increases in immunotolerant CD11b+F4/80+MHCII- M2 macrophages (ranging from 13.9 to 19.4 to 42.4% of total CD11b+F4/80+ macrophages), CD4+CD25+Foxp3+ Treg cells (ranging from 6.4 to 10.5 to 17.8% of total CD4+ T cells), CD11b+Gr1+Ly6C+ MDSCs (ranging from 7.9 to 17.1 to 36.1% of total CD45.1+ cells), and CD317+B220+ pDCs (ranging from 0.4 to 0.7 to 1.4% of total CD45.1+ cells) (Fig. 1C) and toward upregulation of immunosuppressive PD-L1 expression in MDSCs, M2 macrophages, and EG7 tumor cells in tumors (Fig. 1D). Together, our data indicate that TME immunotolerance increases with tumor progression (i.e., the later the stage, the more immunotolerant the TME is). Therefore, we chose to study a well-established EG7 tumor (~300 mm3 in volume) model with a more immunotolerant TME, mimicking late stages of disease in the clinic. In addition, we also found that tumor cells and tumor-infiltrating immune cells comprised ~55% and ~45% of live tumor-derived single-cell suspensions, respectively (Fig. 1E), and that MDSCs comprised ~35% of total CD45.1+ tumor-infiltrating immune cells in the TME of large tumors, while M2 macrophages, pDCs and Treg cells comprised ~4%, ~1.4% and ~1.1%, respectively (Fig. 1F), indicating that MDSCs are the major tolerant immune cells in the TME of large tumors.
Fig. 1. An enhanced immunotolerant TME is associated with tumor progression.

A Representative image of EG7 tumors of different sizes. B Representative hematoxylin and eosin (H&E) staining of tissue sections of small, medium, and large EG7 tumors. Black arrows, areas of focal necrosis in the center of the large tumor; red arrow, amplified tumor necrosis. C Tumor-derived single-cell suspensions (TSCSs) were enzymatically prepared from tissues of EG7 tumors of different sizes. Cell samples were stained with a cocktail of antibodies against a combination of molecular markers and then analyzed by flow cytometry with progressive gating strategies. The last sets of representative flow cytometry plots show quantitative measurements of various immune cell subsets. The relative quantification of (i) M1/M2 macrophage ratio calculated as % MHCII+ M1 macrophages/% MHCII- M2 macrophages in total CD11b+F4/80+ macrophages; (ii) % Treg cells calculated as CD4+Foxp3+ Treg cells/total CD4+ T cells; (iii) % MDSCs and (iv) % pDCs in tumor-infiltrating host CD45.1+ cells calculated as % CD11b+CD45.1+ cells in upper square × % Gr1+Ly6G+ cells in lower square) and % CD11b-CD11c- cells in upper square × % B220+CD137+ cells in lower square, respectively, are described in the Methods and Supplementary Figure S1. D Flow cytometric analysis of PD-L1 expression in M2 macrophages, MDSCs, and tumor cells. The gray line represents control isotype antibody staining. MFI, mean fluorescence intensity. Flow cytometry plots representing one of two independent experiments (4-5 replicates each) are presented as the mean ± SEM. E The average percentages of CD45.2+ EG7 tumor cells and CD45.1+ tumor-infiltrating immune cells in the TME of large tumors and (F) the average percentages of CD11b+F4/80+MHCII- M2 macrophages, CD4+Foxp+ Treg cells, Gr1+Ly6G+ MDSCs and B220+CD317+ pDCs in CD45.1+ tumor-infiltrating immune cells were measured based upon Fig. 1C and Supplementary Figure S1 (n = 5/group). *P < 0.05, **P < 0.01 by one-way ANOVA with Tukey’s test. ns, not significant
IRE ablation induces massive tumor cell apoptosis and weak OVA-specific CD8+ T cell responses but does not induce any significant inhibition of tumor growth in large tumors
To assess IRE-induced tumor cell death, B6 mice were s.c. injected in the right thigh with EG7 cells followed by IRE ablation and histopathologic and flow cytometric analyses (Fig. 2A). When tumors reached a large size (8–9 mm in diameter or ~300 mm3 in volume) (Fig. 2B), we performed IRE ablation (voltage: 1,200 V/cm; pulse duration: 90 μs; pulse repetition frequency: 1 Hz; number of repetition pulses: 100) using our newly constructed custom-made IRE device with two needle array electrodes (5 mm apart) (Fig. 2C) [30]. To assess IRE-induced tumor cell death, we collected tumors at 3 days post IRE for histological examination. IRE caused a large area of tumor cell apoptosis in the central part of tumors in association with large surrounding inflamed areas (Fig. 2D). To assess IRE-induced CD8+ T cell responses and tumor growth inhibition, we conducted flow cytometry to measure CD8+ T cell responses in mouse peripheral blood samples collected 7 days post IRE and closely monitored tumor growth. IRE stimulated weak OVA-specific CD8+ T cell responses (0.33%) (Fig. 2E) but did not induce any significant growth inhibition of treated tumors compared to untreated tumors (Fig. 2F).
Fig. 2. IRE ablation induces tumor cell apoptosis but weak OVA-specific CD8+ T cell responses and is ineffective in inhibiting tumor growth.

A Diagram illustrating the design of the IRE ablation experiment. B Experimental setup for IRE treatment. C Schematic diagram showing the placement of the IRE device electrode in a tumor (8–9 mm in diameter) during IRE ablation. D Representative H&E staining of tumor tissue sections collected at 3 days post IRE ablation. Arrows indicate areas of massive apoptosis in IRE-treated tumors. Arrowheads indicate the surrounding tumor tissues. E Blood cells collected from the tail vein of IRE-treated or naïve control mice were stained with OVA-specific PE-Tetramer and a FITC-conjugated anti-CD8 antibody and analyzed by flow cytometry. OVA-specific CD8+ T cells were defined as CD8 and tetramer double-positive cells. The value in each panel represents the percentage of OVA-specific CD8+ T cells in the total CD8+ T cell population. **P < 0.01 by two-tailed Student t test. F Tumor-bearing mice were monitored for tumor growth post IRE ablation. ns, not significant by two-way ANOVA with Tukey’s test. Flow cytometry or tumor growth plots representing one of two independent experiments are presented as the mean ± SEM (n = 5/group)
Combo treatment alone induces only very weak OVA-specific CD8+ T cell responses
In this study, we selected an anti-PD-L1 antibody for PD-1 blockade since PD-L1 expression in both the host and tumor compartments contributes to immune suppression in a nonredundant fashion [38] and anti-PD-L1 antibodies have been shown to be more effective than anti-PD-1 antibodies in blocking PD-1/PD-L1 signaling [39, 40]. In addition, we chose to intraperitoneally (i.p.) inject the anti-PD-L1 antibody into mice for PD-1 blockade to make our data more comparable to other cancer ablation reports since i.p. administration of the PD-1 blockade agent is the most common route used in animal tumor models of RFA and IRE ablation therapy [18, 19, 41–43]. To assess whether pIC, CpG, PD-1 blockade and Combo treatments stimulate OVA-specific CTL responses, we administered the various treatments to mice bearing large EG7 tumors and measured OVA-specific CD8+ T cell responses by flow cytometry (Supplementary Fig. S2). We found that Combo treatment alone induced weak OVA-specific CD8+ T cell responses (0.69%) in large EG7 tumors (Fig. 3C), while the CD8+ T cell responses in the other groups treated with PD-1 blockade, the TLR3 or TLR9 agonist or the TLR3/9 agonists were negligible (Supplementary Fig. S2), which is consistent with previous reports using a TLR agonist and PD-1 blockade [18, 41, 44]. Therefore, we selected the Combo group as another control group for comparison with the IRE + Combo group in subsequent studies. In addition, Combo treatment did not show any inhibition of tumor growth (Fig. 3D).
Fig. 3. IRE combined with PD-L1 blockade and TLR3/9 agonists results in potent OVA-specific CD8+ T cell responses and antitumor immunity.

A Schematic diagram of IRE ablation combined with PD-1 blockade (anti-PD-L1 Ab) and/or TLR3/9 agonists (pIC/CpG) in six protocols: (i) IRE, (ii) IRE + anti-PD-L1, (iii) IRE + pIC, (iv) IRE + CpG, (v) IRE + pIC/CpG, (vi) IRE + Combo and (vii) Combo alone (treatment schedules described in the Methods). B Schematic illustrating IRE device electrode placement and pIC/CpG injection points (red color) in the tumor (8–9 cm in diameter) during IRE ablation. C Blood cells collected from the tail vein of mice treated with each of the above six different protocols (n = 4/group) were stained with OVA-specific PE-Tetramer and a FITC-conjugated anti-CD8 antibody and analyzed by flow cytometry. The value in each panel represents the percentage of OVA-specific CD8+ T cells in the total CD8+ T cell population. **P < 0.01 by one-way ANOVA with Tukey’s test. D Tumor-bearing mice were monitored for tumor growth or regression. *P < 0.05, **P < 0.01 by two-way ANOVA with a post hoc Tukey’s test. Tumor-bearing mice were also monitored for mouse survival post IRE ablation. Kaplan–Meier survival analysis for the same experiments (n = 12/group) with the log-rank test. **P < 0.01. E Tumor growth curves of IRE + Combo-treated tumors with and without depletion of CD8+ cells using anti-CD8 and control antibodies. Tumor growth or regression was monitored. Tumor-bearing mice were monitored for tumor growth post IRE + Combo ablation. **P < 0.01 by two-way ANOVA with Tukey’s test. Tumor growth plots representing one of two independent experiments are presented as the mean ± SEM (n = 4/group)
PD-1 blockade enhances OVA-specific CD8+ T cell responses and antitumor immunity in IRE-treated tumors
To improve IRE-induced CD8+ T cell responses, we incorporated PD-1 immune checkpoint blockade (anti-PD-L1 antibody) into our IRE ablation protocol. When tumors reached ~300 mm3 in volume, mice were first intraperitoneally (i.p.) injected with the anti-PD-L1 antibody (Ab) 1 day prior to IRE and then every 2 days for a total of four times (Fig. 3A). One day after the first anti-PD-L1 Ab administration, mice were subjected to IRE ablation (Fig. 3A, B). Eight days post IRE + PD-1 blockade, we performed flow cytometry to measure CD8+ T cell responses and monitored tumor growth. We demonstrated that PD-1 blockade (1.79%) significantly enhanced OVA-specific CD8+ T cell responses compared to control IRE ablation alone (0.32%) (Fig. 3C). We also showed that PD-1 blockade significantly inhibited tumor growth and prolonged mouse survival compared to control IRE ablation (Fig. 3D).
TLR3/9 agonists synergistically stimulate potent OVA-specific CD8+ T cell responses and strong antitumor immunity in IRE-treated tumors
To improve IRE-induced CD8+ T cell immunity, we also incorporated pIC and CpG administrations into the IRE ablation protocol. Tumor-bearing mice were subjected to IRE immediately followed by intratumoral (i.t.) injection of pIC (IRE + pIC), CpG (IRE + CpG) or both (IRE + pIC/CpG) into peripheral tumor areas (Fig. 3B). At seven days post IRE, we performed flow cytometry to measure CD8+ T cell responses and monitored tumor growth. Although IRE ± pIC and IRE ± CpG promoted OVA-specific and CD8± T cell responses (1.04% and 1.45%) and inhibited primary tumor growth compared to control IRE treatment, the latter was more efficient than the former (Fig. 3C, D). Interestingly, IRE combined with the TLR3/9 agonists (IRE ± pIC/CpG) synergistically stimulated potent OVA-specific CD8± T cell responses (3.80%), significantly inhibited tumor growth and prolonged mouse survival post IRE compared to IRE ± pIC or IRE ± CpG (Fig. 3C, 3D), indicating that the TLR3/9 agonists synergistically stimulated potent OVA-specific CD8± T cell responses and strong antitumor immunity in IRE-treated tumors. In addition, IRE + pIC/CpG-induced CD8+ T cell immunity was more efficient than that induced by IRE + PD-1 blockade (Fig. 3C, D).
TLR3/9 agonists play a major role in modulating immune cell profiles and a minor role in reducing PD-L1 expression in the TME and vice versa for PD-1 blockade in IRE-treated tumors
To assess the modulatory effect of the TLR3/9 agonists on the immunotolerant TME, single-cell suspensions prepared from peripheral areas of tumors collected at 3 days posttreatment were analyzed by flow cytometry (Supplementary Fig. S1). We found that compared with pIC, CpG more efficiently modulated immune cell profiles by increasing the M1/M2 macrophage ratio and M169 macrophage and cDC1 levels and reducing Treg cell, MDSC, and pDC levels (Fig. 4A). Interestingly, we demonstrated that IRE + pIC/CpG significantly increased the M1 (71.2%)/M2 (27.5%) macrophage ratio (2.6) and frequencies of immunogenic M169 macrophages (9.9% of total macrophages) and cDC1s (16.4% of total DCs) and reduced the frequencies of immunotolerant Treg cells (7.6% of CD4+ T cells), MDSCs (16.9% of CD45.1+ cells), and pDCs (0.3% of CD45.1+ cells) compared to IRE + pIC and IRE + CpG. Interestingly, the modulation of immune cell profiles by IRE + pIC/CpG was also more efficient than that achieved with IRE + PD-1 blockade, as illustrated by the M1 (53.9%)/M2 (43.7%) macrophage ratio (1.2) and M169 macrophage (3.1% of total macrophages), cDC1 (10.8% of total DCs), Treg cell (10.3% of CD4+ T cells), MDSC (29.8% of CD45.1+ cells), and pDC (0.5% of CD45.1+ cells) frequencies (Fig. 4B). In contrast, we found that IRE + PD-1 blockade more efficiently downregulated cell-surface PD-L1 expression in M2 macrophages, MDSCs, and tumor cells than did IRE + pIC/CpG (Fig. 4C). Taken together, our data indicate that the TLR3/9 agonists play a major role in modulating immune cell profiles but a minor role in downregulating PD-L1 expression in the TME and vice versa for PD-1 blockade.
Fig. 4. IRE combined with PD-L1 blockade and TLR3/9 agonists modulates immune cell profiles in the TME.

A Diagram illustrating the experimental setup for analyzing intratumoral immune cell subsets of B6.1 mice bearing primary tumors (8–9 mm in diameter) at 3 days post IRE ablation. Single-cell suspensions were enzymatically prepared from primary tumor tissues at 3 days post IRE ablation. Cell samples were stained with a cocktail of antibodies and then analyzed by flow cytometry with progressive gating strategies. B The last sets of representative flow cytometry plots show quantitative measurements of various immune cell subsets. The relative quantitation of (i) the M1/M2 macrophage ratio calculated as % MHCII+ M1 macrophages/% MHCII- M2 macrophages in total CD11b+F4/80+ macrophages; (ii) % M169 macrophages calculated as CD169+CD11b+F4/80+ M169 macrophages/total CD11b+F4/80+ macrophages; (iii) % cDC1s calculated as CD8+CD103+CD11c+ cDC1s/total CD11c+ DCs; (iv) % Treg cells calculated as CD4+Foxp3+ Treg cells/total CD4+ T cells; and (v) % MDSCs and (vi) % pDCs in tumor-infiltrating host CD45.1+ cells calculated as % CD11b+CD45.1+ cells in upper square × % Gr1+Ly6G+ cells in lower square and % CD11b-CD11c- cells in upper square × % B220+CD137+ cells in lower square, respectively, is described in the Methods and Supplementary Figure S1. * p < 0.05, **P < 0.01 by one-way ANOVA with Tukey’s test. C Cell samples were also stained with a cocktail of antibodies and then analyzed by flow cytometry. PD-L1 expression in gated M2 macrophages, MDSCs, and tumor cells were analyzed by flow cytometry. * p < 0.05, **P < 0.01 by one-way ANOVA with Tukey’s test. D Immunohistochemistry (IHC) analysis of frozen primary tumor tissue sections. Representative micrographs of IHC staining for MDSCs. The above data representing one of two independent experiments are presented as the mean ± SEM (n = 5/group)
IRE + Combo treatment cooperatively stimulates potent OVA-specific CD8+ T cell responses, leading to complete eradication of primary tumors
Although an incremental improvement in OVA-specific CD8+ T cell responses was observed with both IRE + PD-1 blockade and IRE + pIC/CpG compared to IRE alone, each of these combination approaches was still insufficient to overcome the aggressive nature of EG7 tumors (Fig. 3D), suggesting that a combined IRE treatment protocol incorporating both PD-1 blockade and CpG/pIC is worth assessing for potentially improved therapeutic effects. Therefore, we combined IRE ablation with coadministration of the PD-1 blockade agent and CpG/pIC (IRE + Combo) to treat our EG7 tumor model (Fig. 3A). At seven days post IRE, we performed flow cytometry to measure OVA-specific CD8+ T cell responses and monitored tumor growth. Our experiments revealed enhanced OVA-specific CD8+ T cell responses (5.81%) (Fig. 3C), which were more efficient than the CD8+ T cell responses observed in the other groups, leading to complete eradication of primary tumors with no tumor recurrence for one month (Fig. 3D). To confirm that IRE + Combo-induced CD8+ T cell responses contribute to primary tumor eradication, we performed a CD8+ T cell depletion assay using an anti-CD8 Ab to deplete CD8+ T cells 1 day prior to and once every three days during IRE + Combo treatment (total of four injections). This resulted in a complete loss of the ability of IRE + Combo to induce a therapeutic effect that eradicated primary tumors (Fig. 3E), which is consistent with previous studies on tumor ablation therapy [18, 19, 29] and indicates that CD8+ T cells are the major effectors in IRE + Combo-induced tumor eradication.
IRE + Combo treatment potently modulates immune cell profiles and significantly downregulates PD-L1 expression in the TME in IRE-treated tumors
Having shown the effective therapeutic efficacy of IRE + Combo with respect to the eradication of primary tumors, we then focused on investigating its modulatory effect on immune cell profiles in the TME. We performed flow cytometry with progressive gating strategies to analyze immune cell profiles and PD-L1 expression in single-cell suspensions enzymatically prepared from IRE + Combo-treated tumors at 3 days post IRE (Fig. 4A). We demonstrated that IRE + Combo cooperatively promoted the M1 (76.0%)/M2 (22.7%) macrophage ratio (3.3), increased the frequencies of immunogenic M169 macrophages (14.8% of the macrophage population) and cDC1s (25.3% of the DC population), and reduced the frequencies of immunotolerant Treg cells (4.5% of CD4+ T cells), MDSCs (11.9% of CD45.1+ cells), and pDCs (0.1% of CD45.1+ cells) compared to IRE + PD-1 blockade, IRE + pIC/CpG or Combo alone (Fig. 4B). The decreased frequency of MDSCs in IRE + Combo-treated tumors compared to control IRE- or Combo alone-treated tumors was confirmed by immunohistochemical analysis (Fig. 4D). In addition, IRE + Combo more significantly downregulated PD-L1 expression in M2 macrophages, MDSCs, and tumor cells in the TME than did IRE + pIC/CpG, IRE + PD-1 blockade, or Combo alone (Fig. 4C).
IRE + Combo modulates tolerant immune and tumor cells to reduce suppression and induces a systemic decrease in immune tolerance
Because we observed the ability of IRE + Combo to reduce immunotolerant cell populations, such as M2 macrophages and MDSCs, in the TME, we next wanted to assess whether IRE + Combo modulates immunotolerant M2 macrophages and MDSCs in IRE + Combo-treated tumors or affects systemic immune tolerance. To this end, we performed various analyses at three or seven days post IRE to evaluate (i) the expression of immunosuppressive IDO and arginase-1 in immunotolerant M2 macrophages, MDSCs, and tumor cells; (ii) the frequencies of MDSCs and M1 and M2 macrophages in blood cell samples; (iii) cytokine concentrations in serum; (iv) the frequencies of CD4+ and CD8+ T cells in the TME; and (v) the frequencies of cDC1s and CD8+ T cells in the tumor-draining lymph nodes (TDLNs) (Fig. 5A). We demonstrated that compared to IRE or Combo alone, IRE + Combo significantly downregulated suppressive IDO and arginase-1 in immunotolerant M2 macrophages, MDSCs, and EG7 tumor cells at three days post IRE (Fig. 5B), indicating that IRE + Combo alters tolerant immune and tumor cells to favor less suppression. In addition, the abundance of immunotolerant MDSCs and the ratio of M1 versus M2 macrophages were significantly reduced and increased, respectively, in the blood of IRE + Combo-treated mice at 3 days post IRE compared to that of control IRE-ablated or Combo-treated mice (Fig. 5C). Finally, we tested cytokine expression in sera collected from IRE + Combo-, IRE- or Combo-treated mice at three days post IRE using colorimetric cytokine ELISAs. We showed increased concentrations of IL-2 and IFN-γ (~700 pg/ml and ~1,100 pg/ml) and a reduced concentration of TGF-β (~8 ng/ml) in IRE + Combo-treated mouse sera compared to control IRE- and Combo-treated mouse sera (Fig. 5D). Taken together, our data indicate that IRE + Combo treatment induces a systemic decrease in immune tolerance.
Fig. 5. IRE + Combo treatment modulates immune cells and cytokines in the blood and promotes CD8+ T cells in IRE + Combo-treated tumor tissues and the tumor-draining lymph nodes.

A Diagram illustrating the experimental setup for different analyses. B TSCSs were enzymatically prepared from primary tumor tissues collected from mice treated with IRE, Combo or IRE + Combo at 3 days post IRE. Cell samples were stained with a cocktail of antibodies and analyzed by flow cytometry. The expression of IDO and arginase-1 in gated M2 macrophages, MDSCs and tumor cells was analyzed by flow cytometry, as described in the Methods and Supplementary Figure S1. The gray line represents control isotype antibody staining. MFI, mean fluorescence intensity. C Mouse blood was collected at 3 days post IRE ablation, and blood monocytes purified by Ficoll-Hypaque density gradient centrifugation were stained with a cocktail of antibodies. The relative quantitation of MDSCs calculated as % CD11b+Gr1+Ly6G+ MDSCs in total monocytes and the ratio of M1/M2 macrophages calculated as the amount of MHCII+CD11b+F4/80+ M1 macrophages/the amount of MHCII-CD11b+F4/80+ M2 macrophages were determined by flow cytometry. D Quantification of TGF-β, IL-2, and IFN-γ in mouse sera collected on day 3 post IRE ablation. E TSCSs were enzymatically prepared from primary tumor tissues collected at 3 days post IRE + Combo or IRE ablation. Cell samples were stained with a cocktail of antibodies and analyzed by flow cytometry with progressive gating strategies as described in the Methods and Supplementary Figure S1. The last sets of representative flow cytometry plots show quantitative measurement of CD4+ and CD8+ T cells in total tumor-infiltrating host CD45.1+ cells by gating CD3+CD45.1+ T cells for measurement of CD4+ and CD8+ T cells, respectively. F IHC analysis of frozen primary tumor tissue sections. Representative micrographs of IHC staining for CD8+ T cells. G Purified CD8+ T cells from TSCSs were cultured in medium containing IL-2 and OVAI or the unrelated Mut1 peptide for three days and then counted in a T cell proliferation assay. H Purified CD8+ T cells from TSCSs were measured for their cytotoxic effect in a cell cytotoxicity assay, in which activated CD8+ T cells and GranToxiLux-labeled EG7 or EL4 tumor cells were used as effector (E) and target (T) cells, respectively. The percentage of positive fluorogenic granzyme-B substrate cleavage (GBSC+) cells was measured at the indicated E:T (10:1 and 2:1) ratios. I Single-cell suspensions prepared from the tumor-draining lymph nodes (TDLNs) at 7 days post primary tumor treatment with IRE + Combo or IRE ablation. Cell samples were stained with a cocktail of antibodies and analyzed by flow cytometry with progressive gating strategies as described in the Methods and Supplementary Figure S2. The last sets of representative flow cytometry plots show quantitative measurement of the percentage of CD8+CD103+ cDC1s in total DCs by analysis of gated CD11c+MHCII+ DCs and quantitative measurement of the percentage of IFN-γ+TNF-α+ (double-positive) CD8+ effector T cells in total CD3+ T cells by analysis of gated CD3+CD8+ T cells. J T cell memory recall responses. Mice with complete eradication of IRE + Combo-treated primary tumors for 30 days or naïve mice as a control were i.v. boosted with recombinant rLmOVA bacteria. Blood cell samples were collected from the tail vein and stained with OVA-specific PE-Tetramer and a FITC-conjugated anti-CD8 antibody, and OVA-specific CD8+ T cell responses were then analyzed by flow cytometry on day 4 after the booster immunization. Each bar represents an average of 4 mice/group. Error bars indicate the mean ± SEM. *P < 0.05, **P < 0.01 by two-tailed Student t test. One representative experiment out of two independent experiments is shown
IRE + Combo promotes tumor-infiltrating CD4+ and CD8+ T cells in the TME in IRE-treated tumors
Tumor-infiltrating T cells play an important role in tumor eradication [10, 45]. To test whether the IRE + Combo-treated TME favors T cell tumor infiltration, we first assessed the number of tumor-infiltrating T cells in IRE + Combo-treated tumors at 3 days after IRE ablation by flow cytometric and immunohistochemical analyses (Fig. 5A). We demonstrated that the frequencies of both CD4+ T cells and CD8+ T cells, assessed as the percentages of CD4+ and CD8+ T cells in the total live CD45.1+ cell population, were significantly elevated by 2- and 3-fold, respectively, in IRE + Combo-treated tumors compared to control IRE-ablated or Combo-treated tumors (Fig. 5E). The increased frequency of CD8+ T cells in IRE + Combo-treated tumors was also confirmed by immunohistochemical analysis (Fig. 5F).
IRE + Combo-promoted tumor-infiltrating CD8+ T cells are functional effectors
CD8+ T cell dysfunction in the TME is functionally characterized by a reduced proliferative capacity and diminished cytotoxic effects, in part because of the upregulation of immune checkpoint molecules [46]. To assess whether IRE + Combo-promoted tumor-infiltrating CD8+ T cells are functional effectors, we purified CD8+ T cells from single-cell suspensions derived from IRE + Combo-, IRE- or Combo-treated tumors using a CD8+ T Cell Isolation Kit and assessed their in vitro proliferative and cytotoxic effects in T cell proliferation and cytotoxicity assays, respectively. We found that compared with CD8+ T cells derived from control IRE- or Combo-treated tumors, CD8+ T cells derived from IRE + Combo-treated tumors had a much more efficient OVA-specific proliferative potential and greater cytolytic effects against EG7 but not EL4 target cells (Fig. 5G, H).
IRE + Combo promotes cDC1s and effector CD8+ T cells in the tumor-draining lymph nodes and long-term CD8+ T cell memory
To confirm the promotive effect of IRE + Combo on functional immune cells, single-cell suspensions were prepared from the TDLNs on day 7 post IRE + Combo treatment of primary tumors and analyzed by flow cytometry (Supplementary Fig. S3). We demonstrated that IRE + Combo significantly promoted CD8+CD103+ cDC1s (10.6% in total CD11c+ DCs) and IFN-γ/TNF-α double-positive effector CD8+ T cells (26.5% in total CD3+ T cells) in the TDLNs, while the control IRE-ablated and Combo groups showed frequencies of 3.0% and 3.5% for cDC1s and 6.7% and 8.9% for effector CD8+ T cells, respectively (Fig. 5I). To assess whether IRE + Combo ablation induces long-term CD8+ T cell memory, we i.v. immunized IRE + Combo-treated mice with recombinant Listeria expressing OVA (rLmOVA) one month after achieving complete regression of treated primary tumors. We then measured recall responses on day 4 post rLmOVA boost. We demonstrated significant recall CD8+ T cell responses (11.6%) in IRE + Combo-treated mice but not in untreated control mice (Fig. 5J), indicating that IRE + Combo induces long-term CD8+ T cell memory in mice.
IRE + Combo ablation eradicates distant tumors by modulating the immunotolerant TME and promoting tumor-infiltrating CD4+ and CD8+ T cells
The term “abscopal” effect originally indicated a local therapy, such as radiation therapy, that not only shrank the targeted tumor but also led to shrinkage of untreated distant tumors [47]. To assess the potential abscopal effect of IRE + Combo ablation, we monitored the growth or regression of distant (left flank-implanted) untreated tumors following IRE + Combo therapy of primary (right flank-implanted) tumors (Fig. 6A). Remarkably, we found that distant tumors were also completely eradicated in IRE + Combo-treated mice, while those in in IRE- or Combo-treated control mice grew aggressively (Fig. 6A). To investigate whether IRE + Combo ablation modulates the TME of distant tumors, we analyzed immune cell profiles and PD-L1 expression by flow cytometric analysis of single-cell suspensions prepared from distant tumors collected at 3 days post primary tumor IRE ablation (Fig. 6B). We demonstrated significant increases in the M1/M2 macrophage ratio and immunogenic cDC1s and significant reductions in immunotolerant Treg cells, MDSCs, and pDCs (Fig. 6C) in distant tumors from IRE + Combo-treated mice compared to those from control IRE- or Combo-treated mice. In addition, we assessed tumor-infiltrating CD4+ and CD8+ T cells in distant tumors by flow cytometric and immunohistochemical analyses. We demonstrated that more CD4+ and CD8+ T cells were detected in distant tumors in mice with IRE + Combo-treated primary tumors than in those in mice with control IRE- or Combo-treated primary tumors (Fig. 6C). The increased frequencies of CD8+ T cells in the distant tumors of IRE + Combo-treated mice in comparison to those of IRE- and Combo-treated mice were confirmed by immunohistochemistry (Fig. 6D). Next, we assessed the modulatory effect on PD-L1 expression by flow cytometry and found downregulation of inhibitory PD-L1 expression in M2 macrophages, MDSCs, and EG7 tumor cells in the distant tumors of IRE + Combo-treated mice (Fig. 6E). Collectively, our data indicate that IRE + Combo ablation in primary tumors also eradicates distant tumors via conversion of the immunotolerant TME into an immunogenic TME, leading to increased CD4+ and CD8+ T cell infiltration in the distant tumors.
Fig. 6. IRE + Combo induces an “abscopal” effect that eradicates distant tumors by converting the immunotolerant TME of the distant tumors.

A Schematic diagram illustrating the experimental design for the measurement of the “abscopal” effect. B6.1 mice bearing both primary (8–9 mm in diameter) and distant (6 mm in diameter) tumors were monitored for distant tumor regression post IRE + Combo, IRE ablation or Combo alone treatment. **P < 0.01 by two-way ANOVA with Tukey’s test. B Diagram displaying the experimental design for immune cell profiling, PD-L1 expression evaluation, and IHC analyses. C TSCSs were enzymatically prepared from distant tumor tissues on day 3 post IRE ablation of primary tumors. Cell samples were stained with a cocktail of antibodies and analyzed by flow cytometry. The last sets of representative flow cytometry plots show quantitative measurements of various immune cell subsets. The relative quantitation of (i) the M1/M2 macrophage ratio calculated as % MHCII+ M1 macrophages/% MHCII- M2 macrophages in total CD11b+F4/80+ macrophages; (ii) % cDC1s calculated as CD8+CD103+CD11c+ cDC1s/total CD11c+ DCs; (iii) % Treg cells calculated as CD4+Foxp3+ Treg cells/total CD4+ T cells; (iv) % MDSCs, (v) % pDCs and (vi) % CD4+ or CD8+ T cells in total tumor-infiltrating host CD45.1+ cells calculated as % CD11b+CD45.1+ cells in upper square × % Gr1+Ly6G+ cells in lower square % CD11b-CD11c- cells in upper square × % B220+CD137+ cells in lower square and % CD3+CD45.1+ cells in upper square × % CD4+ or CD8+ cells in lower square, respectively, is described in the Methods and Supplementary Figure S1. *P < 0.05, **P < 0.01 by two-tailed Student t test. D IHC analysis of frozen distant tumor tissue sections after IRE-Combo or IRE + Combo treatment of primary tumors. Representative micrographs of IHC staining for CD8+ T cells. E Flow cytometric analysis of PD-L1 expression in M2 macrophages, MDSCs, and tumor cells. The gray line represents control isotype antibody staining. MFI, mean fluorescence intensity. Tumor growth and flow cytometry plots representing one of two independent experiments are presented as the mean ± SEM (n = 5/group). *P < 0.05, **P < 0.01 by two-tailed Student t test
IRE + Combo treatment of primary tumors eradicates tumor lung metastases
To assess whether IRE + Combo ablation in primary tumors affects existing lung metastases, we i.v. injected highly lung metastatic B16 melanoma-derived BL6-10OVA cells engineered to express OVA into mice bearing small s.c. EG7 tumors and control naïve mice (Fig. 7A). One week after melanoma cell injection, when the s.c. EG7 tumors reached ~300 mm3 in volume, we treated the EG7 tumor-bearing mice with IRE + Combo. Two weeks after IRE + Combo treatment, we collected mouse lungs to measure visible metastatic black melanoma colonies, followed by histopathological examination (Fig. 7A). We observed numerous black BL6-10OVA melanoma lung metastases present in untreated control mice but none in IRE + Combo-treated mice (Figs. 7B, C), indicating that IRE + Combo treatment of primary tumors is able to eradicate lung tumor metastases.
Fig. 7. IRE + Combo treatment of primary tumors inhibits lung tumor metastasis.

A Schematic diagram of the experimental design for assessing the antimetastatic activity of IRE + Combo treatment of primary tumors. Mice bearing small 7-day EG7 tumors or untreated control mice (n = 4 mice/group) were i.v. injected with BL6-10OVA cells. Seven days later, IRE + Combo treatment was performed on the mice bearing primary EG7 tumors (8–9 mm in diameter). Mice were sacrificed 14 days after treatment, and lung tissues were collected. B Black metastatic BL6-10OVA melanoma metastases in the lungs were counted. C Representative micrographs of H&E-stained tissue sections from lungs collected from control (untreated) and IRE + Combo-treated mice. One representative experiment out of two independent experiments is shown
Potent therapeutic effects of IRE + Combo in two mouse breast cancer models
Based on the effectiveness of IRE + Combo in the mouse lymphoma EG7 model, we also sought to determine its therapeutic effect in two additional mouse tumor models. We s.c. injected Tg1-1 or 4T1 breast cancer cells into FVB/NJ and BALB/c mice, respectively. When tumors reached ~300 mm3 in volume, we then performed IRE + Combo treatment of the tumor-bearing mice, followed by monitoring tumor growth or regression, and tumor-bearing mice treated with IRE or Combo were used as control groups. We found that IRE + Combo completely eradicated Tg1-1 breast cancer tumors and significantly inhibited 4T1 breast tumor growth (Fig. 8), thus indicating the effective therapeutic effects of IRE + Combo in two mouse breast cancer models.
Fig. 8. IRE + Combo treatment effectively eradicates tumors or significantly inhibits tumor growth in two mouse breast cancer models.
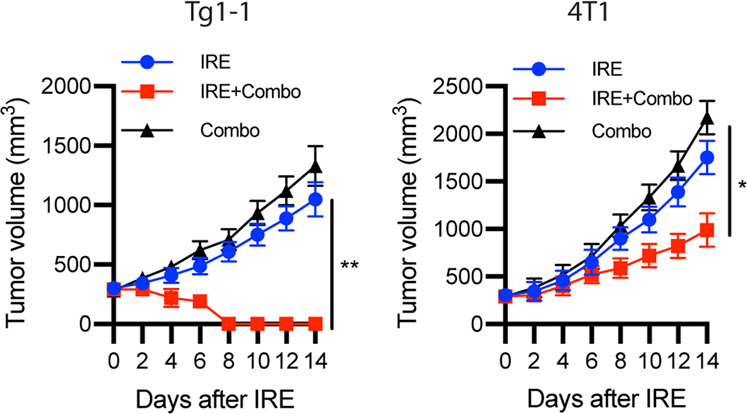
Mice bearing Tg1-1 or 4T1 breast cancer tumors (8–9 mm in diameter) were treated with IRE, Combo or IRE + Combo. Tumor-bearing mice treated with IRE or Combo alone were used as controls. Tumor-bearing mice were monitored for tumor growth or regression post IRE + Combo ablation. Tumor growth plots representing one of two independent experiments are presented as the mean ± SEM (n = 4/group). *P < 0.05, **P < 0.01 by two-way ANOVA with Tukey’s test
Discussion
Tumor cells often evade immunosurveillance by downregulating immunogenic MHC-I molecules while upregulating the expression of inhibitory molecules, such as PD-L1, IDO, arginase-1, and TGF-β [48]. Tumor growth is associated with remodeling of the TME, which often becomes more suppressive as tumors increase in size [49]. By quantitatively measuring immune cell profiles and qualitatively evaluating myeloid and tumor cells, we demonstrated that CD45.2+ tumor cells and CD45.1+ tumor-infiltrating immune cells comprised approximately 55% and 45% of the total tumor cell composition, respectively. Among tumor-infiltrating immune cells, MDSCs comprising ~35% of the total CD45.1+ tumor-infiltrating immune cells in the TME of large tumors represented the major population of immunotolerant cells, while M2 macrophages, pDCs and Treg cells comprised ~4%, ~1.4%, and ~1.1%, respectively. Importantly, our study provides the first evidence that more immunotolerant M2 macrophages, Treg cells, MDSCs, and pDCs and more inhibitory PD-L1 expression on immunotolerant M2 macrophages, MDSCs, and tumor cells are found in larger tumors, indicating a trend toward a more immunotolerant TME in larger tumors. This is possibly due to alterations in tumor cell metabolism derived from the hypoxic and oxidative conditions in larger tumors [49–51].
In this study, we performed IRE with two needle array electrodes (5 mm apart) in mice bearing large primary tumors (8–9 mm in diameter or ~300 mm3 in volume) with an immunotolerant TME, mimicking the situation in clinical cancer patients. This is in contrast to two recent reports conducting IRE in mice bearing small (5–6 mm in diameter or ~80 mm3 in volume) [19] or medium-sized (7 mm in diameter or ~180 mm3 in volume) primary tumors [18] with a less immunotolerant TME. Using an OVA transgene-engineered EG7 tumor cell line in our animal model, we were able to quantitatively measure OVA-specific CD8+ T cell responses, in contrast to the two previous studies that measured only nonspecific CD8+ T cell responses [19]. To improve the IRE-induced therapeutic effect, we incorporated PD-1 blockade, a TLR3 agonist (pIC), and a TLR9 agonist (CpG) into IRE ablation to form various combination therapies including IRE + PD-1 blockade, IRE + pIC, IRE + CpG, IRE + pIC/CpG, IRE + Combo and Combo alone. We then assessed OVA-specific CD8+ T cell responses and antitumor immunity limiting tumor growth. We demonstrated that the TLR3/9 agonists (pIC/CpG) synergistically stimulated stronger IRE-induced OVA-specific CD8+ T cell responses, leading to more efficient inhibition of primary tumor growth and prolonged mouse survival, than did PD-1 blockade. The synergistic promotive effect of the TLR3/9 agonists on IRE-induced CD8+ T cell responses might be derived from their synergistic abilities to drive gene expression and cytokine release [52, 53].
In addition, we further uncovered distinct roles played by the TLR3/9 agonists and PD-1 blockade in the modulation of immune cell profiles and downregulation of PD-L1 expression in the immunotolerant TME. The TLR3/9 agonists were more efficient in modulating immune cell profiles by promoting immunogenic M1 macrophages, M169 macrophages, and cDC1s and reducing immunotolerant M2 macrophages, pDCs, Treg cells, and MDSCs but less potent in downregulating PD-L1 expression on M2 macrophages, MDSCs, and tumor cells and vice versa for PD-1 blockade. This is possibly because the TLR3/9 agonists directly bind to TLR3/9 on the membrane of endosomes within macrophages and DCs, leading to activation of M1 macrophages, M169 macrophages, and cDC1s and promotion of M2 macrophage and MDSC differentiation into M1 macrophages [54] via TLR-mediated metabolic reprogramming [55], while the anti-PD-L1 Ab (PD-1 blockade) directly binds to and blocks inhibitory PD-L1 on M2 macrophages, MDSCs, and tumor cells via Ab binding-mediated cellular internalization of anti-PD-L1 Ab/PD-L1 complexes [56, 57], leading to downregulation of PD-L1 expression in the TME. Heterogeneous TMEs containing different degrees of tolerant immune cells and PD-L1 expression have been found in different types of tumors or different tumors of the same tumor type, and these features are distinct tumor cell intrinsic factors and indicative of different genetic and/or phenotypic traits [50, 58, 59]. Understanding the distinct roles of TLR agonists and PD-L1 blockade in combating an immunotolerant TME helps in the design of better protocols to improve the therapeutic efficacy of IRE ablation based upon genetic and phenotypic characterization of individual TMEs for personalized medicine.
IRE + Combo therapy, which combines IRE ablation-induced massive destruction in tumors with PD-1 blockade and TLR3/9 agonists, cooperatively stimulated potent peripheral OVA-specific CD8+ T cell responses compared to either IRE + PD-1 blockade or IRE + pIC/CpG, leading to complete eradication of large (~300 mm3) primary tumors and long-term OVA-specific CD8+ T cell memory, suggesting that IRE + Combo treatment is a potent therapeutic protocol for cancer therapy. Our mechanistic studies further revealed that IRE + Combo treatment cooperatively promoted immunogenic cDC1s and M169 macrophages and increased the M1/M2 macrophage ratio but reduced immunotolerant Treg cells, MDSCs, and pDCs in the TME compared to either IRE + PD-1 blockade or IRE + pIC/CpG. In addition, IRE + Combo treatment downregulated immunosuppressive PD-L1, IDO and arginase-1 expression in M2 macrophages, MDSCs, and EG7 tumor cells, as measured by flow cytometric analysis, indicating that IRE + Combo therapy significantly converts an immunotolerant TME into an immunogenic TME. Furthermore, IRE + Combo also increased the M1/M2 macrophage ratio but reduced immunotolerant MDSCs in IRE + Combo-treated mouse blood. Various cytokines and chemokines contribute to the modulation of the immunotolerant TME [60]. We measured the concentrations of three major cytokines including two representative immunogenic cytokines, IL-2 and IFN-γ, and one representative immunosuppressive cytokine, TGF-β, in mouse sera. Our data demonstrated that IRE + Combo increased IL-2 and IFN-γ cytokine levels but reduced the level of the immunotolerant cytokine TGF-β in mouse sera, indicating a systemic reduction in immunotolerance and an elevation in immunogenic CD4+ Th1 responses post IRE + Combo treatment. To gain better insight into the modulatory effects of IRE + Combo on cytokines and chemokines as well as immune cell subsets, cytokine/chemokine array analyses, including measurement of the important cytokines IL-12 and IFN-α, will be carried out in the future, and some other important immune cell subsets that could be considered novel immune targets in the TME, such as Th17 cells [61, 62], should also be included in immune cell profiling analyses in the future.
It is worth noting that our data demonstrated that IRE ablation alone induced very weak CTL responses in large EG7 tumors, possibly due to remaining tumor tissues becoming more immunotolerant and accelerating tumor progression post ablation [63], and that Combo treatment alone also failed to induce efficient CTL responses, possibly due to the strong immunotolerance within the large tumor TME. Overall, IRE + Combo therapy was shown to synergistically induce potent CTL responses and antitumor immunity, leading to eradication of primary and distant tumors and lung tumor metastases, possibly due to its conversion of not only the local tolerant TME in the primary tumor but also systemic immunotolerance. Therefore, our IRE + Combo protocol may represent another good example in support of the newly emerging concept that efficiently reducing the tumor burden (i.e., by IRE ablation) and increasing the immunogenicity of the TME (i.e., by PD-1 blockade and TLR3/9 agonist administration) are two key factors for improving cancer immunotherapy [64], leading to a significantly synergistic therapeutic effect on cancer mediated by IRE + Combo.
Tumor-infiltrating CD8+ T cells play an important role in eradicating malignant tumors [10, 45] and are a key factor predicting clinical outcome in cancer patients [65]. In addition to the above peripheral CD8+ T cell responses, CD8+ T cell responses in the TDLNs were also promoted by IRE + Combo. More importantly, IRE + Combo promoted tumor-infiltrating CD8+ T cells, as indicated by flow cytometric and immunohistochemistry analyses, and converted exhausted T cells, as shown by T cell proliferation and cytotoxicity analyses, indicating the generation of an immunogenic TME favorable for CD8+ T cell recruitment, expansion, and effector function post IRE + Combo treatment. The increase in tumor-infiltrating CD8+ T cells may result from a combination of factors, including IRE + Combo-induced elevations in immunogenic (i) M1 macrophages that polarize CD4+ Th1 cell differentiation for enhancement of CD8+ T cell survival and tumor infiltration; [2, 66] (ii) cDC1s, superior stimulators of CD8+ T cell responses [4, 5] leading to tumor-infiltrating CD8+ T cell clonal expansion and efficient T cell killing of tumor cells; [67] and (iii) M169 macrophages, which are capable of not only directly priming CD8+ T cell responses [68] but also transferring tumor antigens derived from apoptotic tumor cells to cDC1s for further CD8+ T cell cross-priming [3, 69]. Finally, enhanced recruitment of CD8+ T cells into IRE + Combo-treated tumors may also be supported by the IRE-modulated tumor stroma with increased microvessel density and permeability, which is expected to favor T cell tumor infiltration and tumor destruction [18].
The “abscopal” effect observed in our IRE + Combo treatment was also demonstrated in a recent report showing elimination of 3-day “palpable” distant tumors post treatment combining IRE ablation with a TLR7 agonist and PD-1 blockade [19]. In comparison, our IRE + Combo approach, which combined IRE ablation with both PD-1 blockade and TLR-3/9 agonists, completely eradicated not only primary tumors but also concomitant distant ~100 mm3 EG7 tumors and BL6-10OVA lung metastases. We further conducted a systemic analysis of immune cell profiles in distant tumors post IRE + Combo treatment of primary tumors. Our data demonstrated that IRE + Combo dramatically modulated the TME of distant tumors by reducing the frequencies of immunotolerant M2 macrophages, Treg cells, MDSCs, and pDCs, which is consistent with previous reports using RFA + PD-1 blockade or RFA + CpG/PD-1 blockade therapeutic protocols [41, 42], and downregulating PD-L1 expression in M2 macrophages, MDSCs, and tumor cells, leading to increased frequencies of tumor-infiltrating CD4+ and CD8+ T cells in the distant tumors. The potential molecular mechanisms underlying the conversion of an immunotolerant TME to an immunogenic TME, as observed in untreated distant tumors, are currently unclear. The IRE + Combo-induced downregulation of systemic immunotolerance could partially contribute to the conversion of the immunotolerant TME in distant tumors. PD-1 blockade has been reported to promote the frequencies of cDC1s and CD8+ T cells and reduce the frequencies of Treg cells in distant tumors post IRE or RFA ablation [19, 41]. Our administration (i.v.) of the anti-PD-L1 Ab in the IRE + Combo protocol could specifically contribute to its conversion-promoting effect. Further elucidation of other factors responsible for the IRE + Combo-induced conversion-promoting effect on the distant tumor TME is now underway in our laboratory.
Finally, we extended our IRE + Combo therapeutic findings obtained from the mouse EG7 lymphoma model to two mouse breast cancer models. We demonstrated that IRE + Combo completely eradicated Tg1-1 breast cancer tumors and significantly inhibited triple-negative 4T1 breast cancer growth, indicating that IRE + Combo is an effective protocol for cancer ablation therapy. The varied therapeutic effects of IRE + Combo observed in the two breast tumor models are possibly due to the heterogeneous TMEs of these two breast cancers [50, 58, 59].
Taken together, our data demonstrate that IRE + Combo induces potent CD8+ T cell responses, leading to complete eradication of both primary and distant tumors as well as lung metastases by converting the immunotolerant TME into an immunogenic TME in both the primary and distant tumors. These findings warrant further study in other mouse solid tumor models and in human trials for IRE ablation therapy in cancer.
Supplementary information
Acknowledgements
This work was supported by grants to J.X. from the Saskatchewan Cancer Agency (SCA), Saskatchewan Health Research Foundation (SHRF), College of Medicine Research Awards (CoMRAD), Royal University Hospital Foundation (RUHF) and Prostate Cancer Fight Foundation (PCFF). F.B. and A.X. were supported by the Lisa Rendall Breast Cancer Graduate Student Scholarship and Postdoctoral Fellowship, respectively, from the SCA.
Author contributions
JX conceived the project and designed the experiments. JX and AX analyzed data and wrote the manuscript. FB, JW, and AX performed experiments and organized data. ZW provided technical help. WZ and BZ provided the custom-made IRE device. R.C. and Y.W. provided technical help related to histopathology and immunohistochemistry. SA, AF, MM and GG interpreted results and reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Fatma Babikr, Jiangbo Wan, Aizhang Xu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-021-00796-4.
References
- 1.Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27:1482–92. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- 2.Soncin I, Sheng J, Chen Q, Foo S, Duan K, Lum J, et al. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat Commun. 2018;9:582. doi: 10.1038/s41467-018-02834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Li L, Kim S, Herndon JM, Goedegebuure P, Belt BA, Satpathy AT, et al. Cross-dressed CD8alpha+/CD103+ dendritic cells prime CD8+ T cells following vaccination. Proc Natl Acad Sci USA. 2012;109:12716–21. doi: 10.1073/pnas.1203468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6:1670–90. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. 2018;9:398. doi: 10.3389/fimmu.2018.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tesi R. MDSC; the most important cell you have never heard of. Trends Pharmacol Sci. 2019;40:4–7. doi: 10.1016/j.tips.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012;72:5240–9. doi: 10.1158/0008-5472.CAN-12-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110:2080–9. doi: 10.1111/cas.14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 11.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia A, Zhang Y, Xu J, Yin T, Lu XJ. T cell dysfunction in cancer immunity and immunotherapy. Front Immunol. 2019;10:1719. doi: 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tekle E, Wolfe MD, Oubrahim H, Chock PB. Phagocytic clearance of electric field induced ‘apoptosis-mimetic’ cells. Biochem Biophys Res Commun. 2008;376:256–60. doi: 10.1016/j.bbrc.2008.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubinsky B. Irreversible electroporation in medicine. Technol Cancer Res Treat. 2007;6:255–60. doi: 10.1177/153303460700600401. [DOI] [PubMed] [Google Scholar]
- 15.Lyu T, Wang X, Su Z, Shangguan J, Sun C, Figini M, et al. Irreversible electroporation in primary and metastatic hepatic malignancies: A review. Med (Baltim) 2017;96:e6386. doi: 10.1097/MD.0000000000006386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin RC, 2nd, McFarland K, Ellis S, Velanovich V. Irreversible electroporation in locally advanced pancreatic cancer: potential improved overall survival. Ann Surg Oncol. 2013;20:S443–449. doi: 10.1245/s10434-012-2736-1. [DOI] [PubMed] [Google Scholar]
- 17.Huang KW, Yang PC, Pua U, Kim MD, Li SP, Qiu YD, et al. The efficacy of combination of induction chemotherapy and irreversible electroporation ablation for patients with locally advanced pancreatic adenocarcinoma. J Surg Oncol. 2018;118:31–36. doi: 10.1002/jso.25110. [DOI] [PubMed] [Google Scholar]
- 18.Zhao J, Wen X, Tian L, Li T, Xu C, Wen X, et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat Commun. 2019;10:899. doi: 10.1038/s41467-019-08782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narayanan JSS, Ray P, Hayashi T, Whisenant TC, Vicente D, Carson DA, et al. Irreversible electroporation combined with checkpoint blockade and TLR7 stimulation induces antitumor immunity in a murine pancreatic cancer model. Cancer Immunol Res. 2019;7:1714–26. doi: 10.1158/2326-6066.CIR-19-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fitzgeraid KA, Kagan JC. Toll-like receptor and the control of immunity. Cell. 2020;180:1044–66. doi: 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu M, Xu W, Su H, Huang Q, Wang B. Addition of CpG ODN and Poly (I:C) to a standard maturation cocktail generates monocyte-derived dendritic cells and induces a potent Th1 polarization with migratory capacity. Hum Vaccin Immunother. 2015;11:1596–605. doi: 10.1080/21645515.2015.1046659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee BR, Jeong SK, Ahn BC, Lee BJ, Shin SJ, Yum JS, et al. Combination of TLR1/2 and TLR3 ligands enhances CD4(+) T cell longevity and antibody responses by modulating type I IFN production. Sci Rep. 2016;6:32526. doi: 10.1038/srep32526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amos SM, Pegram HJ, Westwood JA, John LB, Devaud C, Clarke CJ, et al. Adoptive immunotherapy combined with intratumoral TLR agonist delivery eradicates established melanoma in mice. Cancer Immunol Immunother. 2011;60:671–83. doi: 10.1007/s00262-011-0984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Han C, Fu Y. Targeting innate sensing in the tumor microenvironment to improve immunotherapy. Cell Mol Immunol. 2020;17:13–26. doi: 10.1038/s41423-019-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson CE, Mills LJ, McCurtain JL, Thompson EA, Seelig DM, Bhela S, et al. Reprogramming responsiveness to checkpoint blockade in dysfunctional CD8 T cells. Proc Natl Acad Sci USA. 2019;116:2640–5. doi: 10.1073/pnas.1810326116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y, Chen M, Nie H, Yuan Y. PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccin Immunother. 2019;15:1111–22. doi: 10.1080/21645515.2019.1571892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. 2017;19:1874–87. doi: 10.1016/j.celrep.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Reilley MJ, Morrow B, Ager CR, Liu A, Hong DS, Curran MA. TLR9 activation cooperates with T cell checkpoint blockade to regress poorly immunogenic melanoma. J Immunother Cancer. 2019;7:323. doi: 10.1186/s40425-019-0811-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu A, Zhang L, Yuan J, Babikr F, Freywald A, Chibbar R, et al. TLR9 agonist enhances radiofrequency ablation-induced CTL responses, leading to the potent inhibition of primary tumor growth and lung metastasis. Cell Mol Immunol. 2019;16:820–32. doi: 10.1038/s41423-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, Moser MA, Zhang EM, Xiang J, Zhang W. An in vitro experimental study of the pulse delivery method in irreversible electroporation. J Eng Sci Med Diagnostics Ther. 2018;1:014501. [Google Scholar]
- 31.Ahmed KA, Xiang J. mTORC1 regulates mannose-6-phosphate receptor transport and T-cell vulnerability to regulatory T cells by controlling kinesin KIF13A. Cell Disco. 2017;3:17011. doi: 10.1038/celldisc.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–52. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chavez M, Silvestrini MT, Ingham ES, Fite BZ, Mahakian LM, Tam SM, et al. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics. 2018;8:3611–28. doi: 10.7150/thno.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umeshappa CS, Huang H, Xie Y, Wei Y, Mulligan SJ, Deng Y, et al. CD4+ Th-APC with acquired peptide/MHC class I and II complexes stimulate type i helper CD4+ and central memory CD8+ T cell responses. J Immunol. 2009;182:193–206. doi: 10.4049/jimmunol.182.1.193. [DOI] [PubMed] [Google Scholar]
- 35.Spear S, McNeish IA, Capasso M. Generation of orthotopic pancreatic tumors and ex vivo characterization of tumor-infiltrating T cell cytotoxicity. J Visualized Exp. 2019;154:e60622. doi: 10.3791/60622. [DOI] [PubMed] [Google Scholar]
- 36.Severson JJ, Serracino HS, Mateescu V, Raeburn CD, McIntyre RC, Jr, Sams SB, et al. PD-1+Tim+ CD8+ T lymphocytes display varied degrees of functional exhaustion in patients with regional metastatic differentiated thyroid cancer. Cancer Immunol Res. 2015;3:620–30. doi: 10.1158/2326-6066.CIR-14-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu C, Tan X, Hu X, Zhou M, Yan J, Ding C. Tumor Microenvironment following gemcitabine treatment favors differentiation of immunosuppressive Ly6C(high) myeloid cells. J Immunol. 2020;204:212–23. doi: 10.4049/jimmunol.1900930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, et al. Tumor and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun. 2017;8:14572. doi: 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 40.Linhares ADS, et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci Rep. 2019;9:11472. doi: 10.1038/s41598-019-47910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi L, Chen L, Wu C, Zhu Y, Xu B, Zheng X, et al. PD-1 blockade boosts radiofrequency ablation-elicited adaptive immune responses against tumor. Clin Cancer Res. 2016;22:1173–84. doi: 10.1158/1078-0432.CCR-15-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silvestrini MT, Ingham ES, Mahakian LM, Kheirolomoom A, Liu Y, Fite BZ, et al. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight. 2017;2:e90521. doi: 10.1172/jci.insight.90521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eranki A, Srinivasan P, Ries M, Kim A, Lazarski CA, Rossi CT, et al. High-intensity focused ultrasound (HIFU) triggers immune sensitization of refractory murine neuroblastoma to checkpoint inhibitory therapy. Clin Cancer Res. 2020;26:1152–61. doi: 10.1158/1078-0432.CCR-19-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.den Brok MH, Sutmuller RP, Nierkens S, Bennink EJ, Toonen LW, Figdor CG, et al. Synergy between in situ cryoablation and TLR9 stimulation results in a highly effective in vivo dendritic cell vaccine. Cancer Res. 2006;66:7285–92. doi: 10.1158/0008-5472.CAN-06-0206. [DOI] [PubMed] [Google Scholar]
- 45.Nolz JC. Molecular mechanisms of CD8(+) T cell trafficking and localization. Cell Mol Life Sci. 2015;72:2461–73. doi: 10.1007/s00018-015-1835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thormmen D, Schumacher T. T cell dysfunctionin cancer. Cancer Cell. 2018;33:547–62. doi: 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Dong Y, Kong L, Shi F, Zhu H, Yu J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J Hematol Oncol. 2018;11:104. doi: 10.1186/s13045-018-0647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 49.Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20:840. doi: 10.3390/ijms20040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sormendi S, Wielockx B. Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front Immunol. 2018;9:40. doi: 10.3389/fimmu.2018.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sooriakumaran P, Kapa R. Angiogenesis and tumor hypoxia response in prostate cancer: a review. Int J Surg. 2005;3:61–67. doi: 10.1016/j.ijsu.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 52.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergisticlly trigger a T helper type-1 polarizing program in dendritic cells. Nat Immunol. 2005;6:769–76. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tross D, PPetrenko L, Klaschik S, Zhu Q, Klinman DM. Global changes in gene expression and synergistic interactions induced by TLR9 and TLR3. Mol Immunol. 2009;46:2557–64. doi: 10.1016/j.molimm.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Noci V, Tortoreto M, Gulino A, Storti C, Bianchi F, Zaffaroni N, et al. Poly(I:C) and CpG-ODN combined aerosolization to treat lung metastases and counter the immunosuppressive microenvironment. Oncoimmunology. 2015;4:e1040214. doi: 10.1080/2162402X.2015.1040214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prodam F, Chiocchetti A, Dianzani U. Diet as a strategy for type 1 diabetes prevention. Cell Mol Immunol. 2018;15:1–4. doi: 10.1038/cmi.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contreras-Sandoval AM, Merino M, Vasquez M, Trocóniz IF, Berraondo P, Garrido MJ. Correlation between anti-PD-L1 tumor concentrations and tumor-specific and nonspecific biomarkers in a melanoma mouse model. Oncotarget. 2016;7:76891–901. doi: 10.18632/oncotarget.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rudkouskaya A, Barroso M. Internalization and trafficking of PD‐L1 in MDAMB231 breast cancer cells. FASEB J. 2017;31:809.807–809.807. [Google Scholar]
- 58.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. 2018;49:178–93 e177. doi: 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landskron G, Fuente MDI, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi: 10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zou W, Restifo NP. Th17 cells in tumor immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–56. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bilska M, Pawłowska A, Zakrzewska E, Chudzik A, Suszczyk D, Gogacz M, et al. Th17 cells and IL-17 as novel immune targets in ovarian cancer therapy. J Oncol. 2020;2020:8797683–15. doi: 10.1155/2020/8797683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi L, Wang J, Ding N, Zhang Y, Zhu Y, Dong S, et al. Inflammation induced by incomplete radiofrequency ablation accelerates tumor progression and hinders PD-1 immunotherapy. Nat Commun. 2019;10:5421. doi: 10.1038/s41467-019-13204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33:581–98. doi: 10.1016/j.ccell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 66.Huang H, Hao S, Li F, Ye Z, Yang J, Xiang J. CD4+ Th1 cells promote CD8+ Tc1 cell survival, memory response, tumor localization and therapy by targeted delivery of interleukin 2 via acquired pMHC I complexes. Immunology. 2007;120:148–59. doi: 10.1111/j.1365-2567.2006.02452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fearon DT. Immune-suppressing cellular elements of the tumor microenvironment. Annu Rev Cancer Biol. 2017;1:241–55. [Google Scholar]
- 68.Bernhard CA, Ried C, Kochanek S, Brocker T. CD169+ macrophages are sufficient for priming of CTLs with specificities left out by cross-priming dendritic cells. Proc Natl Acad Sci USA. 2015;112:5461–6. doi: 10.1073/pnas.1423356112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Dinther D, Veninga H, Iborra S, Borg E, Hoogterp L, Olesek K, et al. Functional CD169 on macrophages mediates interaction with dendritic cells for CD8(+) T cell cross-priming. Cell Rep. 2018;22:1484–95. doi: 10.1016/j.celrep.2018.01.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.