

Summary
The Orchidaceae is of economic and ecological importance and constitutes ˜10% of all seed plant species. Here, we report a genome physical map for Cymbidium sinense, a well‐known species belonging to genus Cymbidium that has thousands of natural variation varieties of flower organs, flower and leaf colours and also referred as the King of Fragrance, which make it arose into a unique cultural symbol in China. The high‐quality chromosome‐scale genome assembly was 3.52 Gb in size, 29 638 protein‐coding genes were predicted, and evidence for whole‐genome duplication shared with other orchids was provided. Marked amplification of cytochrome‐ and photosystem‐related genes was observed, which was consistent with the shade tolerance and dark green leaves of C. sinense. Extensive duplication of MADS‐box genes, and the resulting subfunctional and expressional differentiation, was associated with regulation of species‐specific flower traits, including wild‐type and mutant‐type floral patterning, seasonal flowering and ecological adaption. CsSEP4 was originally found to positively regulate gynostemium development. The CsSVP genes and their interaction proteins CsAP1 and CsSOC1 were significantly expanded and involved in the regulation of low‐temperature‐dependent flowering. Important genetic clues to the colourful leaf traits, purple‐black flowers and volatile trait in C. sinense were also found. The results provide new insights into the molecular mechanisms of important phenotypic traits of Cymbidium and its evolution and serve as a powerful platform for future evolutionary studies and molecular breeding of orchids.
Keywords: Cymbidium sinense, genome, flower patterning, anthocyanin, fragrance
Introduction
The Orchidaceae is among the largest families of flowering plants and is distributed worldwide except in Antarctica (Chase et al., 2015; Pridgeon et al., 2014). The family comprises more than 25 000 species classified into ˜800 genera and is the largest family of monocotyledons (The plant list, 2018). Cymbidium Swartz (1799:70), a genus within the tribe Cymbidieae (Orchidaceae: Epidendroideae), contains ˜80 species. Cymbidium species are perennial herbs, combining an exotic appearance with diverse traits, including fleshy pseudobulbs, and short and thick stems bearing 4–6 leaves arranged in two rows. The raceme develops from the leaf axil of a pseudobulblet and bears unusual bilaterally symmetric and fragrant flowers (Figure 1a). Cymbidiums were among the earliest orchid species to be cultivated and have been grown for thousands of years, especially in ancient China, and became popular in Europe during the Victorian era (Hew, 2001). The traits including the diversity of floral patterning and perianth colours make Cymbidium so popular. Most importantly, the flower has its unique fragrance. Confucius referred to the Asian Cymbidium as the King of Fragrance (Kim et al., 2016; Ramya et al., 2019). More than 150 000 commercial hybrids have been registered with the Royal Horticultural Society. Thus, Cymbidium is an ideal taxon for assessment of the development of floral‐patterning, colour and fragrance and to study morphological evolution in orchids (Motomura et al., 2010).
Figure 1.
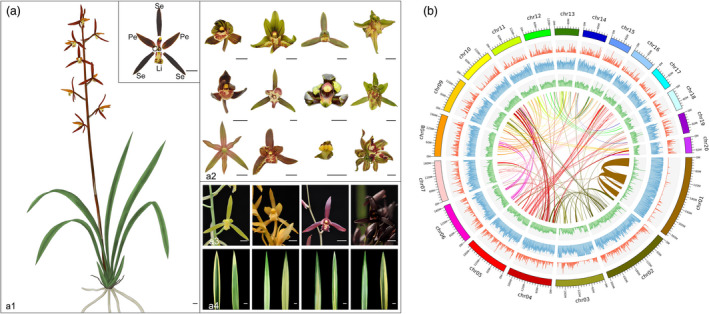
Plant morphology and genome features of Cymbidium sinense. (a) Morphological character of wild and variated types of floral organs, flower colours and leaf colours in C. sinense plants. a1. The unique floral organ includes three sepals in the first whorl, three petals in the second whorl and productive parts in the centre of the flower. The male and female productive organs are highly fused to form a gynostemium (or column), which evolved through complete fusion of the style, stigma and staminal filament and has four pollinia on a semi‐circular viscidium. The sepals and petals together are called the tepals. Among them, two of the petals are similar to each other and resemble unmodified sepals, while the third is highly modified and is called the labellum (or lip). Se, sepal, Pe, petal, Li, lip and Co, column; a2. Natural mutant (varieties) of flower types. The first row, specialization of the labellum reduced or transformed into petals, is named as lotus petals or null‐labellum varieties. The second row, the labellum expanded, sepals or petals are transformed into labellum‐like structure. The third row, the gynostemium (column) expanded, petals are transformed into Genostemium‐like structure. In the fourth row, multi‐tepal flowers develop more tepals; a3. different flower colours of C. sinense, including green, yellow, red and black; a4. Natural mutant of leaf colours. (b) High‐quality genome of C. sinense allows integration of genetic and expression data. From the inside out, Circle1. The assembled 20 chromosomes; Circle2. Gene density plotted in a 500‐kb sliding window; Circle3. Transposable element (TE) density plotted in a 500‐kb sliding window; Circle4. GC content plotted in a 500‐kb sliding window; Circle5. Intragenomic syntenic regions denoted by a single line represent a genomic syntenic region covering at least 20 paralogues.
Cymbidium sinense has a long cultural history over many centuries. It was named the black orchid in ‘Jin Zhang Lan Pu’, the earliest work on orchids published in AD1233. The species bears attractive, dark green foliage and elegant flower spikes with many strongly scented flowers. Cymbidium sinense (C. sinense) is noted for the abundant variation in flower patterning, flower colour and leaf colour (Figure 1a). More than 1000 natural variations have been derived from C. sinense, which make it an ideal species to study the evolution of phenotypic traits in orchids (Rui Chi et al., 1997; Su et al., 2018b; Zhang et al., 2013). Most previous studies on C. sinense have focused on traditional biology such as embryology, physiology, genetic diversity and population structure (Huang et al., 1998; Li and Zhang, 2016; Lu et al., 2011). Recent research involves micropropagation (Gao et al., 2014), chemical variation in essential oils (Li et al., 2017) and the unigenes associated with flower development (Su et al., 2018a, 2018b,2018a, 2018b; Zhang et al., 2013), leaf colour variation (Gao et al., 2020; Zhu et al., 2015) and the origin of pelorism (Su et al., 2018b). However, for specific traits that model plants lack, such as unique perianth development, purple‐black flower colour and fragrance (Hsu et al., 2015a; Kim et al., 2016; Pan et al., 2014), research methods for isolation of homologous genes by a reverse genetic strategy are severely restricted. More importantly, the two historical duplication events in the orchid genome led to functional redundancy or subfunctional differentiation of many genes (Cai et al., 2015; Simao et al., 2015; Zhang et al., 2017a, 2016), which makes it difficult to research the molecular regulation of species‐specific traits through model plants. The genetic basis and regulatory networks of important horticultural traits, including flower patterning, floral colour and fragrance, remain unclear. The lack of a reference genome sequence is a major obstacle to studying the basic and applied biology of Cymbidium. Here we present, to the best of our knowledge, the first chromosome‐scale assembly of a Cymbidium orchid genome. The results provide novel insights into the complete genome sequence of C. sinense that will aid in understanding the development of horticulturally important phenotypic traits, including leaf colour, floral colour, flowering time, floral patterning and fragrance. In addition, the molecular data will assist in investigation of the flowering pathway and various other biological mechanisms in other orchid species.
Results and discussion
Genome sequencing and genome characteristics
Cymbidium sinense has a karyotype of 2n = 2x = 40 with uniform chromosomes (Figure S1). To sequence the complete C. sinense genome, a total of 429.0 Gb of data were generated using Nanopore sequencing technology (Table S1). The total length of the final assembly was 3.52 Gb, comprising 8496 contigs with a corresponding contig N50 size of 1.11 Mb (Table S2). Benchmarking Universal Single‐Copy Orthologs (BUSCO) (Simao et al., 2015) assessment indicated that the completeness of the genome was 91.0%, thus suggesting that the genome assembly was relatively complete and of high quality (Table S3). We used 460.90 Gb reads from a Hi‐C library to reconstruct physical maps by recoding and clustering the assembled scaffolds into 20 chromosome‐level pseudomolecules (Table S4; Figure S2). The total length of the final assembly was 3.45 Gb distributed across 20 chromosome‐level pseudomolecules (Figure 1b) and represented ˜97.79% of the estimated genome size (Table S5). The length of the pseudochromosomes ranged from 72.95 to 376.78 Mb, suggesting the Hi‐C assembly was of high quality (Table S6; Figure S2). In addition, ˜92% of the clustered RNA sequencing (RNA‐seq) transcripts (171 transcriptomes, >6 G clean reads) was alignable to the assembly (>90% coverage and >90% identity), indicating that the assembly contained the majority of gene sequences (Table S7).
A total of 2.74 Gb repetitive elements constituting more than 77.78% of the C. sinense genome was annotated using a method combining structural and homology information (Table S8). Among transposable elements (TE), long terminal repeats (LTRs) were the most dominant type, accounting for ˜54.76% of the genome, followed by long interspersed nuclear elements (16.99%) and DNA transposons (10.84%). In addition, the percentage of de novo predicted repeats (74.02%) was notably larger than the number predicted from the Repbase database, indicating that the C. sinense genome contains many unique repeats compared with other sequenced plant genomes (Zhang et al., 2016; Tables S8 and S9). Comparison of the distribution of repeats among orchid species revealed a high percentage of repeats in both introns and intergenic regions in C. sinense compared with that of other orchid species (Phalaenopsis equestris and Dendrobium catenatum; Cai et al., 2015; Zhang et al., 2016). For regions with a repeat ratio of 50% or greater, the frequency of intergenic regions was higher than that of introns, indicating a higher repeat frequency in intergenic regions in C. sinense (Figure S3).
Gene prediction and annotation
We confidently annotated 29 638 protein‐coding genes of which more than 95% were assigned to a suite of function databases (Table S10). Of these genes, 1974 transcription factor genes were predicted and classified into 91 (sub)families. In addition, 1936 noncoding RNAs, comprising 200 conserved microRNAs, 53 novel microRNAs, 1244 transfer RNAs, 444 ribosomal RNAs and 94 small nuclear RNAs, were identified in the C. sinense genome (Table S11). These results indicated that a higher number of genes were sequenced in C. sinense compared with that of other orchid species (Cai et al., 2015; Han et al., 2020; Zhang et al., 2017a, 2016). Comparison of gene models for orchid species revealed that the length and number of exons in orchids was relatively conserved, whereas the number of introns varied substantially. However, the average length of genes was shorter in C. sinense, with a higher proportion (67.48%) of genes less than 1000 bp compared with 55.11%, 60.91% and 56.73% for Apostasia shenzhenica, P. equestris and D. catenatum, respectively. By contrast, the average length of all introns and the longest 10% of introns were relatively longer in C. sinense compared with those of other orchid species (Figure S4).
We compared the genomes of 18 plant species to identify gene families that are significantly expanded in C. sinense or that are unique to C. sinense. A total of 662 gene families (1723 genes) were specific to C. sinense and gene expansion was detected in 1145 families, whereas 929 families showed contraction (Table S12). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed that the significantly expanded gene families were especially enriched in the terms ‘transmembrane transport’, ‘extracellular‐glutamate‐gated ion channel activity’, ‘electron carrier activity’, ‘photosynthetic membrane’, ‘transition metal ion binding’ and ‘photosynthesis’ (Table S13). Functional exploration of these gene families indicated marked amplification of cytochrome‐ and photosystem‐related genes in the C. sinense genome (false discovery rate < 0.01), which correlated well with characters resulting from promotion of photosynthesis, namely shade tolerance and dark green leaves. In addition, 7781 genes belong to 113 gene sets (Table S14) were common to the orchid species A. shenzhenica, Gastrodia elata, P. equestris and D. catenatum, which most likely represents the ‘core’ proteome of orchids, GO and KEGG enrichment analysis revealed the genes in ‘biosynthesis of secondary metabolites’ and ‘basal transcription factors’ pathway were most representative, which may contribute to the specific characters in orchids (Table S15; Figure S5).
Genome evolution and whole‐genome duplication
Phylogenetic analyses of a concatenated sequence alignment of C. sinense and 18 other plant species indicated that C. sinense, as expected, clustered with other orchid and monocotyledonous species. The divergence time between C. sinense and the most closely related species, D. catenatum, was estimated to be 45.8 million years ago (Figure 2a). The genome comparison among different orchid plants, including genome size, assembly information and gene family evolution, are listed in detail in Figure S6. Collinearity indicated only 8.55% of the genome showed conservation of gene order and content with other regions in the genome, consistent with the small collinearity fractions in Phalaenopsis (Cai et al., 2015; Chao et al., 2018; Figure 1b). By contrast, gene collinearity between C. sinense and P. equestris accounted for 55.51% of all genes, and a higher proportion of collinear genes were observed in each collinear block (Figure S7a). The notable difference between retained homologs in collinear versus syntenic regions can probably be explained by a high degree of reshuffling of genes after duplication, fractionation (loss of either homology) and the low gene density of C. sinense (Figures 1b and S7b). The distribution of synonymous substitutions per synonymous site (K s) across all paralogous genes (regardless of gene order) and that of duplicated genes located in syntenic blocks showed two distinct peaks in K s values between 0.8–1.0 and 1.5–1.7 (Figure 2b). These results were suggestive of two whole‐genome duplication (WGD) events. Dating of the most recent WGD was similar to the K s peak of anchor pairs from the genome of A. shenzhenica and D. catenatum generated by the Orchidaceae WGD (Zhang et al., 2017a, 2016), or a single WGD event shared by all extant orchids (Zhang et al., 2017a). It is well documented that WGD events have been frequent in the evolutionary history of flowering plants and generally shaped the evolutionary trajectory of genomes and genes, in particular those genes associated with agronomic and/or plant‐specialized phenotypic traits.
Figure 2.
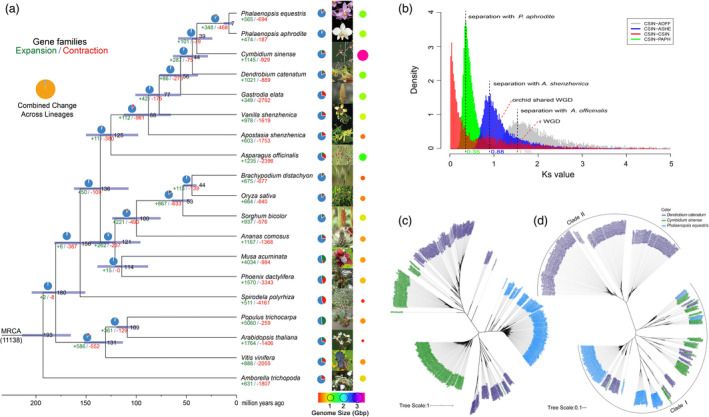
Evolution of Cymbidium sinense genome and gene families. (a) Phylogenetic tree showing divergence times and the evolution of gene family sizes. The phylogenetic tree was constructed from a concatenated alignment of 152 single‐copy gene families from 19 green plant species. Gene family expansions are indicated in orange, and gene family contractions are indicated in grey; the corresponding propotions among total changes are shown using the same colours in the pie charts. Inferred divergence dates (in millions of years) are denoted at each node in blue. MRCA, most recent common ancestor. Blue portions of the pie charts represent the conserved gene families. The corresponding genome size is shown using the different colour and size point. (b) KS distribution analysis. Distribution of KS for the one‐to‐one C. sinense–D. catenatum, C. sinense–P. equestris and C. sinense–A. officinalis. (c) Phylogenetic relationships within the Ty3/Gypsy superfamily in the three orchid species. (d) Phylogenetic relationships within the Ty1/Copia superfamily in the three orchid plants.
Evolution of LTR‐RTs in orchids
Orchid genomes contain a much higher proportion of repeated sequences than those of model plants, such as Arabidopsis (6.91%) and rice (27.39%), and of crop plants, such as kiwifruit (25.64%), coffee (33.60%) and cacao (32.69%; Xia et al., 2020). To investigate the evolution of TEs in orchids, phylogenetic trees of domains in reverse transcriptase genes were constructed for Ty1/Copia and Ty3/Gypsy retrotransposons. In the tree of the Ty3/Gypsy superfamily, long terminal repeat retrotransposons (LTR‐RTs) from C. sinense and D. catenatum were clustered in two major clades. In contrast, the fewer members observed in P. equestris were clustered in a species‐specific subclade (Figure 2c, marked in blue). The Ty1/Copia superfamily exhibited a slightly different pattern (Figure 2d), with two major clades and seven subclades resolved. Clade I included members of all three species. Clade II, the sole D. catenatum‐specific clade, showed high diversity and abundance of members, indicating an earlier split from the ancestral gene and a close relationship between C. sinense and P. equestris. It is worth noting that, although the genome was predominantly composed of repetitive sequences, and mostly LTR‐RTs (58.34% of the assembled sequences), the proportion of Ty1 and Ty3 domains with a complete structure was relatively lower (Table S16), suggesting that sequence transversions or deletions occurred after insertion of these transposon elements. Estimation of the timing of LTR insertion, in the orchid species for which genome information is available, indicated that a burst of LTR activity occurred during the last 15 million years (Figure S8). Therefore, we deduced that these LTRs were inserted into the genome after C. sinense diverged from D. catenatum but before the divergence of Phalaenopsis species.
MADS‐box genes and the evolution of floral morphology
In the orchid plants, C. sinense evolved the most natural variation types of flower organs, which are classified into lotus‐like perianth, gynostemium‐like perianth, labellum‐like petal, multi‐perianth flower and null‐labellum flower (Figure 1a), according to floral organ transformation and/or reversion. MADS‐box genes are involved in many important processes during plant development but are especially known for their roles in flower development. We thus focused on identifying and characterizing MADS‐box genes of which 94 were identified in C. sinense. Perhaps surprisingly, this number is fewer than those documented for most other sequenced angiosperm genomes, but is more than the number in other sequenced orchid genomes (Table S17; Cai et al., 2015; Han et al., 2020; Zhang et al., 2017a, 2016). Phylogenetic analysis revealed that most of the identified genes had been duplicated. However, orthologs of FLOWERING LOCUS C (FLC), AGAMOUS‐LIKE 12 (AGL12) and AGL15 were not detected in C. sinense, consistent with other sequenced orchid genomes and thus may have been specifically lost in orchids (Chao et al., 2018; He et al., 2019). FLC is an important gene for vernalization pathway and low‐temperature‐induced flowering in plants. We speculate two possibilities for the absence of FLC homologous genes in the genomes of D. catenatum, P. Aphrodite and C. sinensis. The first possibility is that it is difficult to find homologous genes by BLAST because of the divergent and short protein sequences of FLC‐like genes across the species (Ruelens et al., 2013). However, there may exist some functionally FLC‐like genes in the orchids. The second possibility is that there is a VRN‐mediated vernalization pathway independent of FLC pathway in Orchids (Shan et al., 2012; Yang et al., 2019), which may play a major role instead of FLC. Plants have two vernalization pathways, one is the FLC pathway (Michaels and Amasino, 1999) and the other is VRN pathway (Fan et al., 2021). The absence of FLC subfamily genes in orchids indicates that the regulation of genes during the flowering transition is different from Arabidopsis. However, more analyses are needed to confirm that FLC genes have been lost in the genomes of orchids and to determine the conservation and evolutionary importance of these genes.
C. sinense has 41 type II MADS‐box genes, considerably more than the number found in P. equestris (21) or A. shenzhenica (27), and D. catenatum (35) (Figure 3a). Gene expression profiling indicated the expanded gene clades included members with differential expression patterns in orchid floral organs as well as divergent coding protein domains, which supports the unique evolutionary routes of these floral organ identity genes associated with the unique labellum (lip) and gynostemium (column) innovation in orchids (Hsu et al., 2015b; Pan et al., 2014; Tsai et al., 2014). Notably, we found a greater number of paralogs with similar expression patterns to those of previously reported (Hsu et al., 2015a) AP3‐2/AGL6‐2 and AP3‐1/AGL6‐1 (Figure 3b group I and group II, respectively). The former is specifically expressed and exclusively required for labellum formation and reported as L‐code mode in orchids. The latter is reported as the P‐code of perianth development to specify the sepal/petal previously (Hsu et al., 2015a, 2021, 2015b,2015a, 2021, 2015b). Among these expanded clades, we originally found that one of the four gene copies in SEP clade, CsSEP4, was ectopically expressed in the gynostemium of the wild‐type flower and expended to all floral organs of a gynostemium‐like perianth variant. When we transformed 35S:CsSEP4 into the model plant Arabidopsis, it exhibited a severe flower phenotype. The petals were absent and only carpel‐like structures developed (Figure 3c). The floral organs failed to develop in the first and second whorls, consistent with accumulation in the gynostemium of CsSEP4, which may positively regulate gynostemium development. Parallel, transgenic plants with 35S:CsSEP3 also showed abnormal morphological features of sepals and petals, similar to the phenotype in the plant with 35S:PeSEP3. Whereas 35S:CsSEP3 had an abnormal stamen and ovule, the carpel was dehiscent, unable to develop normal seeds. The other transgenic plants 35S:CsSEP1 or 35S: CsSEP2 showed no phenotypic changes of floral organ development (Figure S9). These results indicated that there are differences in expression patterns and functional differentiation among paralogous genes or even among orthologous genes in closely related species (Malcomber and Kellogg, 2005; Morel et al., 2019; Pan et al., 2014; Zhang et al., 2017b).
Figure 3.
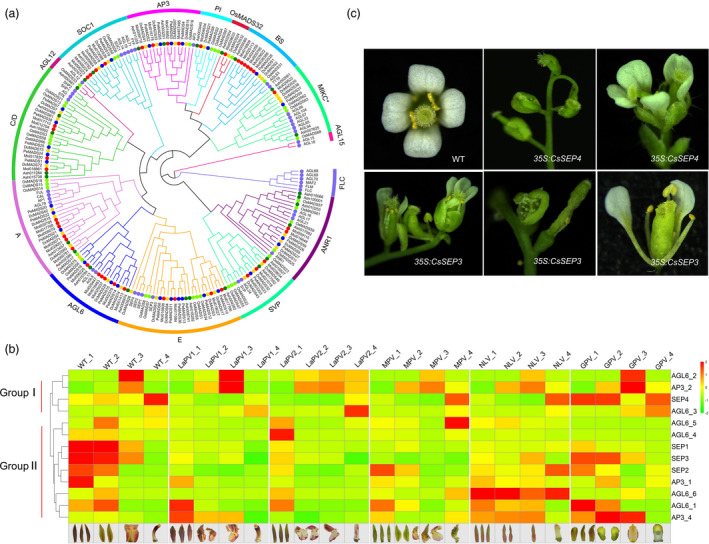
MADS‐box genes and floral morphology evolution. (a) The phylogenetic tree of type Ⅱ MADS‐box genes among C. sinense, D. catenatum, A. shenzhenica, P. equestris, O. sativa and A. Thaliana. The MADS‐box proteins in C. sinense are marked by red solid cycle. (b) Gene expression patterns of AP3‐/SEP‐/AGL genes in individual floral organs from different flower varieties, including labellum‐like perianth variety (LaPV), multi‐perianth variety (MPV), null‐lip variety (NLV), Genostemium‐like perianth variety (GPV), 1 to 4 represent the individual floral organ sepal, petal, labellum and Genostemium, respectively. (c) Floral organ morphology of transgenic Arabidopsis with ectopic expression of CsSEP genes.
In addition, we detected extensive expansion of type I MADS‐box genes of which 53 members were identified in C. sinense. Of these genes, 40% were generated by tandem duplication, indicating that these genes have mainly arisen by recent, small‐scale duplication (Figure S10). We observed strong differences among plant species, even among closely related orchid species, but a large number of lineage‐specific expansions of type I genes were resolved (Figure S10b,c). The present results corroborate previous findings that type I genes exhibit a faster evolutionary rate than type II genes and show that the number of duplications of type I genes is high even in short time frames (Ng and Yanofsky, 2001; Smaczniak et al., 2012; Theißen et al., 2018). Compared with type II genes, type I genes are less well studied, but a number of recent reports indicate a key regulatory role for them in plant reproduction, in particular in specifying female gametophyte, embryo and endosperm development, and are decisive for imposing reproductive boundaries between species (Masiero et al., 2011). In this work, the expression dynamics of type I genes indicated highly correlation with floral patterning. Generally, most of the genes showed strict spatial expression patterns in the wild type (WT), which restricted their expression in specific individual floral organs. However, these specific expression patterns disappeared in mutants showing abnormal proliferation of floral organs or severe inhibition of floral organ differentiation. Some other genes were expressed homogeneously in all floral organs of the WT, but the expression patterns differed entirely in individual floral organs of the mutant (Figure S11), which suggested these genes played a regulatory role in normal floral differentiation and development.
Low‐temperature‐dependent flowering and adaptive evolution of flowering time genes
Despite the probable tropical origins of orchids, species are now distributed in temperate ecosystems and evolved cold tolerance. Indeed, seasonal cues and vernalization responsiveness coupled with flowering time are commonly observed in many orchid species. For most Cymbidium orchids, floral development from initiation to flower opening progresses over 6–7 months (Figure 4a). Cold conditions are needed to avoid bud abortion and promote flower development during the period of semi‐endodormancy. Bud dormancy is common among perennial plants as an adaptive process to survive the winter in temperate climates. The SHORT VEGETATIVE PHASE (SVP) genes, especially the DORMANCY ASSOCIATED MADS‐BOX (DAM) clade, evolved as crucial regulators of this process (Falavigna et al., 2018; Liu et al., 2020; Wu et al., 2017). In the present study, we observed three SVP genes in C. sinense compared with two in P. equestris and A. shenzhenica. However, we detected no orthologs of DAM genes, which are considered to regulate growth cessation and terminal bud formation in the endodormancy cycle in perennial species. This result indicated that orchids did not follow the trend for expansion of SVP/StMADS11 genes observed in other perennial plants, such as Rosaceae species, kiwifruit and Populus trichocarpa (Falavigna et al., 2018; Liu et al., 2020, 2018; Wu et al., 2012). However, sites with a high probability of having been under positive Darwinian selection were detected in the K‐box domain of orchid SVP genes (Table S18). Expression analysis confirmed that the SVP genes were expressed during floral bud developmental stages 0–5 (Figure 4b) and were responsive to prolonged cold exposure, but the nature of the response differs qualitatively and quantitatively among paralogs (Figure S12). When we overexpressed CsSVP genes in Cymbidium protoplasts using a transient expression system. The expression of CsFT, CsAP1 and CsSOC1 was significantly suppressed after CsSVP3 was overexpressed 24–36 h post‐treatment (Figure S13a). By comparison, CsSVP1 had no direct effect on CsFT expression, but markedly repressed expression of CsSVP1, CsAP1 and CsSOC1 (Figure S13b), which was indicative of functional differentiation among the proteins.
Figure 4.
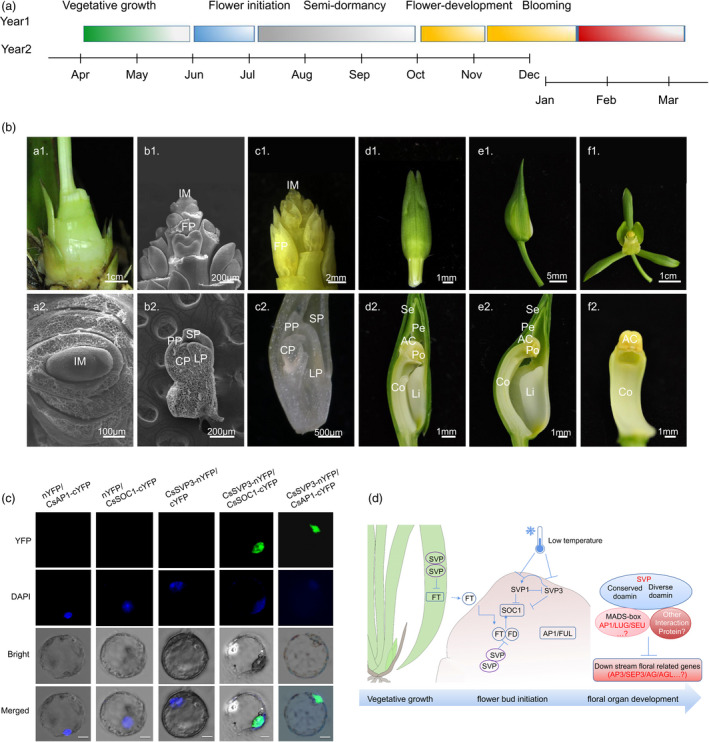
Seasonal flowering and the analysis of floral organ identity and flowering‐time‐related genes. (a) Schematic diagram of C. sinense seasonal growth and development in 12‐month cycle. (b) Floral development of C. sinense. (a–c) Scanning electron micrograph (SEM) of early floral developmental stages. (a) Dormant lateral buds (S0). (b) potential floral bud initiation in the lateral buds of developing shoots (S1). (c) Developing floret. d‐f. developing flowers, Bar = 1 cm. The developing flower of stage 3 (d), stage 4 (e) and mature flowers (f). The first line: plant morphology, the second line: flower bud or floret form, the third line: microstructure of floral bud and floral organ. Im, inflorescence meristem; Br, Bract; FP, floret primordium; SP, sepal primordium; PP, petal primordium; LP, labellum primordium; CP, column primordium. Se, sepal, Pe, petal, Li, lip and Co, column, AC, anther cap, Lo, locule. Po, pollinium. (c) Biomolecular fluorescence complementation visualization. Fusion proteins CsSVP3‐YFPn and CsAP1‐YFPc or CsSOC1‐YFPc were co‐expressed in C. sinense protoplasts and YFP signals were detected in nuclei where the DAPI signal presented, while negative controls did not produce any BiFC fluorescence. (d) Control of flowering in C. sinense. Arrows with solid line indicated positive interaction, and right angle indicated negative interaction.
Additionally, when focus on flowering key regulators, expansion of six AP1 and four SOC1 genes was detected in the C. sinense genome compared with P. equestris and A. shenzhenica. Bayesian inference and maximum likelihood analyses revealed that orchid AP1/FUL and SOC1 subfamilies were clustered distinct from Arabidopsis and rice and supported two major clades among orchids, consistent with WGD and gene duplication (Figure 3a). Moreover, paralogous genes were distributed in different subclades, which indicated a subset of paralogs may have evolved after divergence of the orchid ancestor, and additional duplications occurred during the evolution of these lineages. Interestingly, SOC1 and AP1 were also detected when we screened SVP‐interacting proteins using a yeast two‐hybrid system. This result prompted us to explore the flowering regulatory roles of the strong expansion of AP1/SOC1 genes in ecological adaptation of orchids. Yeast two‐hybrid and bimolecular fluorescence complementation (BiFC) assays confirmed that CsSVP proteins formed homodimers and could interact with AP1 and SOC1 (Figures 4c and S14), suggesting more extensive protein‐binding activity as previously reported for Arabidopsis and Brassica juncea (Jang et al., 2009; Lee et al., 2007; Li et al., 2019; Mendez‐Vigo et al., 2013). These results suggested that C. sinense, unlike other perennial plants, did not harbour DAM genes responsive to low‐temperature dormancy, but the crucial genes SVP, AP1 and SOC1 were significantly expanded during environmental adaptation (Figure 4d). Interestingly, analysis of the interaction motif indicated that SVP 137k was conserved for protein interaction, whereas SOC1 137C was not conserved. For example, in B. juncea, mutation of SOC1 C137K led to loss of protein interaction between BjuSOC1 and BjuSVP in regulating flowering time (Li et al., 2019). However, we observed that the 137th amino acid of CsSOC1 that interacted with CsSVP was K (Figure S15). Thus, regulatory mechanisms for SOC1/SVP interaction may differ between the Cruciferae and Orchidaceae.
Metabolic regulation and leaf‐colour variation
Cymbidium sinense includes many cultivars with highly ornamental variations in leaf colour, such as yellow or white plaques and stripes on the leaves (Figure 1a). Previously, we observed that the chloroplast ultrastructure of leaf‐colour mutants of C. sinense showed abnormal starch granule enlargement (Gao et al., 2020), which indicated that sugar metabolism was abnormal in the chloroplast. Sugars are the primary end product of CO2 assimilation and act as a retrograde signal to modulate expression of nuclear‐encoded PS genes (Rolland et al., 2006).
In the starch and sucrose metabolism pathway, β‐glucosidases (EC 3.2.1.21) are glycosyl hydrolases that hydrolyse the β‐O‐glycosidic bond at the anomeric carbon of a glucose moiety at the nonreducing end of a carbohydrate or glycoside molecule (Opassiri et al., 2006). Compared with other species, the gene number of CH1 gene subfamily which belongs to β‐xylosidase gene family in Orchidaceae (C. sinense, A. shenzhenica, Vanilla shenzhenica, P. equestris, P. Aphrodite and D. catenatum) showed significant contraction. There are only 24 CH1 genes in C. sinense (Figure 5a), compared with 48 and 40 CH1 genes in Arabidopsis and rice, respectively. Gene distribution indicated that AtCH1s in Arabidopsis are distributed on all five chromosomes and the number of genes on each chromosome is more than four. OsCH1 genes were located on eight of 12 chromosomes in rice (Chr1, 3, 4, 5, 6, 8, 9 and 11). By comparison, 17 of the 24 CsCH1 genes are located on five of 20 chromosomes in C. sinense (Chr2, 5, 8, 10 and 18; Figure 5b), the remaining seven genes were individually distributed on seven chromosomes, suggesting that there may be gene loss or pseudogenization in the process of evolution. Gene expression profiles indicated that 15 CsCH1 genes showed significant down‐regulation in a leaf variegation mutant compared with WT (Figure 5c). We thus speculated the significant contraction of CH1 gene subfamily may be an important reason for the decrease of starch content, abnormal sugar metabolism in chloroplast act as a sugar signal to trigger the response of nuclear‐encoded PS genes which leads to the impair of chloroplast in C. sinense (Figure 5d).
Figure 5.
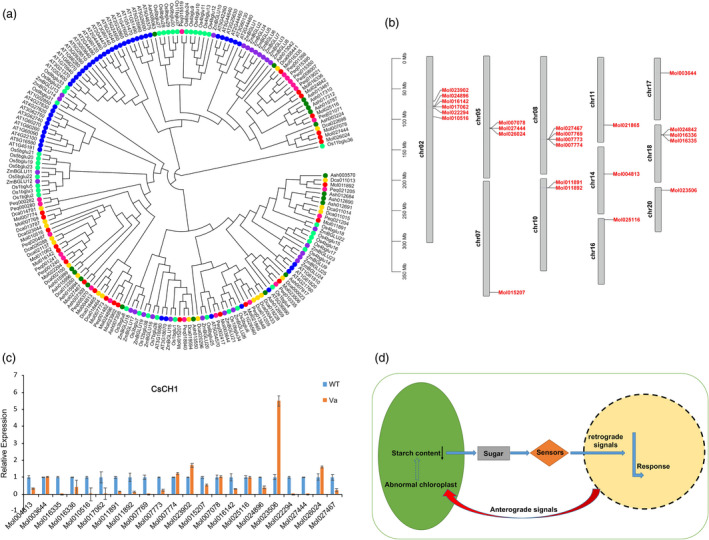
Gene contraction of β‐xylosidase gene family related to sucrose metabolism in Cymbidium sinense. (a) The phylogenetic tree of CH1 genes among C. sinense, D. catenatum, A. shenzhenica, P. equestris, O. sativa and Arabidopsis. (b) Chromosomal localization of CsCH1 genes. (c) Gene expression patterns of CsCH1 genes between WT and leaf variegation mutant. (d) Control of leaf colour in C. sinense.
Activated F3′5H and combined cyanidin‐ and delphinidin‐based pigmentation in Cymbidium sinense
Cyanidin‐based (cyanidins and peonidins) and delphinidin‐based (petunidin) anthocyanins were detected in C. sinense by high‐performance liquid chromatography (HPLC). Hybrids differed only in the contents of the pigments (Table S17). Pigmentation‐related homologs, including CHS, CHI, FLS, F3H, F3′H, F3′5′H, DFR, ANS and UFGT, involved in anthocyanin synthesis were identified genome‐wide, and 56 genes showed differential expression among the flower‐colour variants including green, yellow, pink and purple‐black (Figures 6a and S16). Among them, FLS, ANS, F3H belong to the 2‐oxoglutarate‐dependent oxygenase family. Based on phylogenetic analysis, we identified two CsFLS, two CsANS and four CsF3H genes in the C. sinense genome (Figures 6b and S16b). The number of these genes showed little difference from that reported for other orchid species, but the expression patterns of different collateral homologous genes in different flower‐colour variants differed significantly. These results indicated that these homologs may show subfunctional differentiation in substrate recognition and regulation of flower colour.
Figure 6.
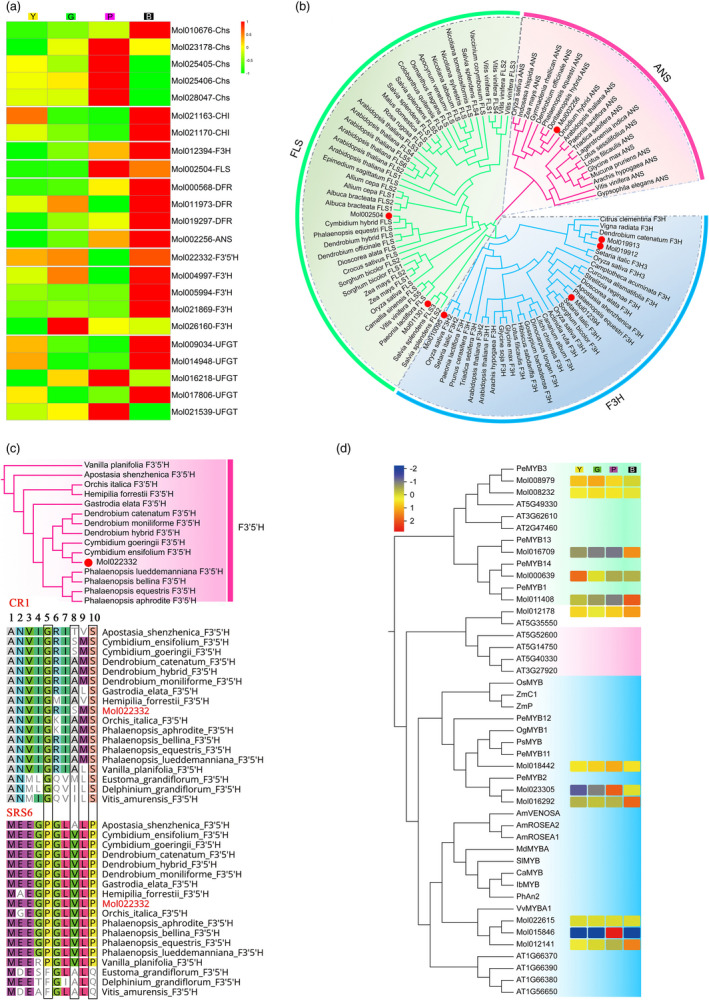
Evolution and expression of key genes involved in anthocyanin biosynthesis in Cymbidium sinense. (a) The differential expression patterns of genes involved in anthocyanin biosynthesis in four different colour flowers of C. sinense. (b) The phylogenetic tree of 2ODD genes among Cymbidium sinense, Epimedium sagittatum, Albuca bracteata, Dioscorea alata, Dendrobium officinale, Citrus clementine, Camptotheca acuminate, Actinidia rufa, Curcuma alismatifolia, Strelitzia reginae, Apostasia shenzhenica, Phalaenopsis equestri, Setaria italic, Oryza sativa, Zea mays, Vigna radiate, Dendrobium catenatum, Gymnadenia rhellican, Dendrobium hybrid, Doritaenopsis hybrid, Cymbidium hybrid, Arabidopsis thaliana, Oncidium hybrid, Indosasa hispida, Triadica sebifera, Lotus sessilifolius, Lotus filicaulis, Vitis vinifera, Gypsophila elegans, Glycine max, Lagerstroemia indica, Mucuna pruriens, Arachis hypogaea, Paeonia lactiflora, Canarium album, Litchi chinensis, Gossypium barbadense, Hibiscus sabdariffa, Prunus cerasifera, Dimocarpus longan, Glycine soja, Rosa rugosa, Malus domestica, Sorghum bicolor, Osmanthus fragrans, Colobanthus quitensis, Apocynum venetumv, Salvia splendens, Crocus sativus, Allium cepa, Vaccinium corymbosum, Camellia sinensis, Nicotiana tabacum, Nicotiana tomentosiformis, Nicotiana sylvestris. (c) The phylogenetic tree and multiple alignment analysis of F3'5'H from Cymbidium sinense (highlighted in red) and other members of Orchidaceae. Positions 5, 8 and 10 of CR1 and SRS6 amino acids are shown in black squares. (d) The phylogenetic tree of MYB genes among Cymbidium sinense, Apostasia shenzhenica, Oryza sativa, Zea mays, Petunia hybrida, Dendrobium spp, Capsicum annuum, Solanum lycopersicum, Antirrhinum majus, Oncidium spp. Gower Ramsey, Malus domestica, Ipomoea batatas, Vitis vinifera, Phalaenopsis equestris, Phalaenopsis spp and gene expression patterns of MYB genes in four different colour flowers of Cymbidium sinense.
At the branch node of the anthocyanin pathway, F3′H and F3′5′H are needed for synthesis of cyanidins and delphinidins for red and blue pigmentation, respectively. Four to five duplicated F3′H genes were detected in different orchid species (Figure 6b) and underwent volatile lineage‐specific gene duplication (Jia et al., 2019; Li et al., 2020). Significant differences in expression of F3′H genes were observed in different flower‐colour variants and were especially highly expressed in pink and purple flowers, concomitant with higher cyanidin‐based anthocyanin accumulation. In contrast, F3′5′H was present as a single copy in orchid genomes (Figure 6c). Interestingly, the substrate recognition domain of F3′5′H differs notably from that of other plants but is conserved among orchids. For example, CR1 and SRS6, which are considered to be crucial to the functional activity of F3′5′H, are highly consistent among orchids, whether delphinidins are synthesized or not (Figure S17). With regard to crucial amino acids that determine functional divergence between F3′H and F3′5′H, we observed that Orchidaceae F3′5′Hs have conserved A, V and S residues at positions 1, 3 and 10 of CR1, and P, V and P residues at positions 5, 8 and 10 of SRS6, respectively (Figure 6e). Although the amino acids and domains of F3'5'H in orchids are conserved, significant differences in their expression are observed among species and cultivars, which have a functional impact on anthocyanin biosynthesis. We detected accumulation of delphinin and strong expression of F3'5'H in C. sinense, whereas delphinin accumulation and F3'5'H activity were not detected in Phalaenopsis (Liang et al., 2020; Whang et al., 2011). It was speculated that the existence of F3′5′H activity in orchids may depend not only on the amino acids of the substrate recognition domain.
We thus analysed the 2 kb promoter region of orchid F3′5′H genes to search for DNA‐binding motifs. In the five orchid species that have been sequenced, a large gap was detected in the 300 bp region upstream of the ATG start codon in Apostasia and Phalaenopsis ‘Aphrodite’. For the remaining orchid F3′5′H genes, six MYB binding sites (MBS) were detected in the CsF3′5′H promoter, five of which were concentrated within 1 kb of the promoter. Four MBS were detected in the DcF3'5'H promoter of which only one was within 1 kb of the promoter. Similarly, only one MBS was located within 1 kb of the PeF3'5'H promoter, with two additional MBS located beyond 1 kb of the promoter (Figure S18). Considering that the MYB binding domain is important for activation and expression of F3'5'H, we speculated that differences in the MYB binding motif in the promoter region may account for differences in F3'5'H expression among orchid variants.
Further analysis of anthocyanin‐regulating MYB genes revealed that C. sinense harboured 125 MYB genes. This number is higher than that of other sequenced orchids (Figure S19). Specific expansion and contraction of the MYB subfamily in C. sinense was detected, probably contributing to species‐specific traits, given the important roles of MYB genes in plant growth and development. For example, accompanying the smooth leaves and roots phenotype of C. sinense, the subgroup of homologous genes associated with trichome and root hair development in Arabidopsis is absent (Figure 6d, marked in pink). In contrast, gene numbers in the subgroup reported to positively or negatively regulate anthocyanin synthesis (Chaves‐Silva et al., 2018; Gonzalez et al., 2008; Hsu et al., 2015a) were increased (Figures 6e and S19). We observed that MYB genes regulating the early biosynthesis stages were generally up‐regulated in C. sinense with purple‐black flowers, which was positively correlated with overall anthocyanin accumulation. In contrast, the paralogous genes regulating the late biosynthesis stages showed significantly different expression profiles. For example, three of the six homologous genes of subgroup 7 were highly expressed in black flowers (Figure 6d, marked in green); two of the six homologous genes of subgroup 6 were highly expressed in black flowers, three were highly expressed in pink flowers, and expression of one did not differ significantly (Figure 6d, marked in blue). Thus, different genes may be associated with different anthocyanin contents in different branches of red and purple flowers. These observations indicated that gene duplications and the expression dynamics of these anthocyanin‐regulating MYB genes may be responsible for the uniquely high contents of pigments in dark‐coloured flowers.
Enzymology specially evolved in the biosynthesis of floral fragrance
The floral scent of C. sinense is unique and is useful for industrial development. Fifty‐eight volatile compounds were detected in C. sinense flowers. Compared with the bud stage, the contents of four monoterpenes (linalool, L‐α‐terpineol, 1‐cyclohexene‐1‐carboxaldehyde, 2,6,6‐trimethyl‐ and 2,4‐decadienal, (E,E)‐) and five sesquiterpenes ((Z)‐β‐farnesene, α‐farnesene, β‐bisabolene, 2,6,10‐dodecatrien‐1‐ol,3,7,11‐trimethyl‐ and 2,6,10‐dodecatrienal,3,7,11‐trimethyl‐, (E,E)‐) were significantly increased at the full‐bloom stage (8‐1). Based on these results, we concluded that terpenes play a crucial role in the floral scent of C. sinense. The terpene synthase (TPS) gene family, a critical gene involved in terpenes synthesis (Chen et al., 2011), was significantly expanded in C. sinense compared with that of other orchid species: 59 members in C. sinense, 48 members in D. officinale, 24 members in P. equestris and 15 members in Apostasia (Figure 7a). Compared with a non‐scented variant, TPS expression in a fragrant variant of C. sinense was significantly up‐regulated and the highest expression levels were detected in the labellum (Figure 7b). The 59 putative TPS genes in C. sinense were ascribed to five previously recognized TPS subfamilies in angiosperms: TPS‐a, TPS‐b, TPS‐c, TPS‐d and TPS‐e/f, but no member of TPS‐g was detected (Figure 7a). The TPS genes were predominantly located on chromosomes 2, 3, 11 and 15 (Figure S20). Notably, the TPS‐b subfamily, which is responsible for monoterpene biosynthesis, contained 26 putative TPS genes of which 19 encoded putative α‐terpineol synthases. The content of putative α‐terpineol synthase at the full‐bloom stage was six‐times higher than that at the bud stage, and six α‐terpineol synthase genes showed significant up‐regulation at the full‐bloom stage compared with that at the bud stage (Figure 7c). These results suggested that monoterpenes and sesquiterpenes may be crucial components of the floral scent in C. sinense. Expansion of α‐terpineol synthase genes in the TPS‐b subfamily of C. sinense may play an important role in the formation of its specific fragrance.
Figure 7.
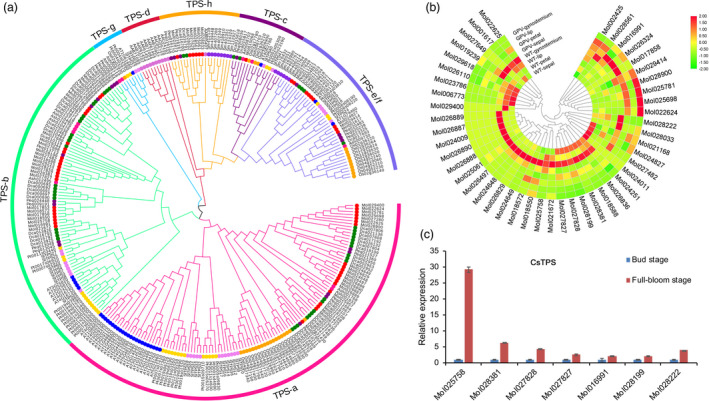
Evolution and expression of TPS genes in Cymbidium sinense. (a) The phylogenetic tree of TPS genes among C. sinense, D. catenatum, A. Shenzhenica and P. equestris. (b) Gene expression patterns of TPS genes between WT and non‐scented variety. (c) Differentially expressed TPS‐b genes in full‐bloom stage flowers compared with bud stage flowers.
In conclusion, we compiled a high‐quality assembly of the genome of C. sinense. The assembled genome sequence is 3.52 Gb, distributed across 20 chromosome‐level pseudomolecules ranging from 72.95 to 376.78 Mb. Using a combination of genomic and genetic approaches, we demonstrated that the reference sequence can be used to analyse important ornamental traits, such as orchid‐specific floral patterning, seasonal flowering, flower colour, leaf colour and floral scent. Cymbidium is, after Phalaenopsis, the most economically important group of orchids, especially in Asian countries. Cymbidium holds a pre‐eminent position in the Orchidaceae on account of its high economic value and abundant natural variants, which are ideally suited to study biologically relevant characters. Therefore, the availability of a complete reference genome for C. sinense provides a valuable resource for comparative genomics studies on the diversity and the evolutionary mechanisms of ornamental traits (e.g. floral scent and colour) at the genome level. The genome sequence will ultimately facilitate modernization of traditional orchids through molecular breeding in the future.
Methods
Plant materials, library construction and sequencing
Wild‐type and natural variants of Cymbidium sinense used in this study. For genome sequencing, we collected leaves and flowers from several individuals of wild‐type C. sinense and extracted genomic DNA using the modified cetyltrimethylammonium bromide (CTAB) protocol (Murray and Thompson, 1980). Quantitative detection of the constructed DNA library was conducted accurately using a Qubit instrument. After library construction, a certain concentration and volume of DNA library was added to a flow cell, which was transferred to the Nanopore GridION X5 sequencer for real‐time single‐molecule sequencing.
We performed RNA‐seq for transcriptome analysis of fresh flowers from different floral‐patterning variants, individual plant organs, and different development stages (the materials used for transcriptomes are listed in Table S20). Total RNA was isolated using the modified CTAB protocol (Tel‐zur et al., 1999) and used for cDNA library preparation. RNA‐seq was performed on an Illumina HiSeq 2000 platform. For small RNA sequencing, a total amount of 3 μg total RNA from the wild‐type and variant were used as input material to construct small RNA libraries, following the procedures described by Yang et al. (2017). The PCR products were size‐selected by PAGE using the TruSeq® Small RNA Sample Prep Kit (Illumina, San Diego, CA) in accordance with the manufacturer’s instructions. The purified library products were evaluated using the Agilent 2200 TapeStation and diluted to 10 pM for cluster generation in situ on the HiSeq 2500 single‐end flow cell followed by sequencing (1 × 50 bp) on the HiSeq 2500 platform.
Genome assembly and assessment of assembly quality
Using the raw reads generated by third‐generation sequencing, NextDenovo (https://github.com/Nextomics/NextDenovo) was used for correction of the raw data. The default parameters of BWA (0.7.12‐R1039) were used to match the second‐generation sequencing data to the assembled genome. Pilon (v1.22) was used for continuous iteration and four rounds of correction. Busco (v3.0.1) prediction was performed on the assembled sequences. Single‐copy embryophyta_odb10 homologous genes were used to predict the gene status of the existing sequences in the genome. Detailed statistics for the results are shown in Table S3. BWA (0.7.12‐r1039) was used to compare the second‐generation sequencing data with the corrected genome to evaluate the coverage of the second‐generation data and genome and to judge the integrity of the assembly. GATK (GATK4.0.4.0) CallSNP was used. iTools (0.23) samtools stat was used to review the genome coverage and the coverage of each sequence.
Analysis of genome synteny and WGD
The C. sinense genome was compared with three other plant genomes by pairwise alignment using the Large‐Scale Genome Alignment Tool (LAST; http://last.cbrc.jp/). We defined syntenic blocks using LAST hits with a distance cut‐off of 20 genes apart from the two retained homologous pairs, in which at least four consecutive retained homologous pairs were required. We then obtained the one‐to‐one blocks to exclude ancient duplication blocks with QUOTA‐ALIGN (Tang et al., 2011).
We detected and compared WGD events in the C. sinense genome and three other plant genomes (P. equestris, Asparagus officinalis and A. shenzhenica). We identified paralogous gene pairs using BLAST‐based methods and determined syntenic paralogs using MCScanX (http://chibba.pgml.uga.edu/mcscan2/MCScanX.zip). We also identified orthologs between the four species. We calculated the number of synonymous substitutions per synonymous site (K s) for gene pairs based on the NG method of Yang implemented in the PAML program (v4.8). The synonymous substitution rate of 8.25 × 10−9 mutations per site per year for asterids was applied to calculate the ages of the WGDs (Badouin et al., 2017).
Hi‐C data analysis
The data generated by sequencing is the original off‐machine sequences, which will include sequences containing joints and low‐quality sequences. To ensure the quality of the information analysis data, the original sequences were filtered with FASTP (software version: 0.20.0) using the default parameters to obtain high‐quality clean reads and duplicate reads were removed. Subsequent analysis was performed on the clean reads. A HI‐C library creates two sets of data, called Invalid Interaction Pairs and Valid Interaction Pairs. Invalid Interaction Pairs are mainly composed of Self Circle, Dangling End and Dumped Pairs. Statistical analysis of effective paired‐end reads showed that 327 450 535 Valid Interaction Pairs were obtained, which accounted for 46.99% of Unique Map Reads and 14.21% of clean data.
Repeat and noncoding RNA annotation
Tandem repeats and TEs in the genome were identified separately. The repeat annotation process was similar to that applied and described in a previous study (Zhang et al., 2012). Tandem repeats were identified using TRF63 and RepeatMasker (v3.2.7; Tarailo‐Graovac and Chen, 2009). All repeat sequences identified by the different methods were combined into the final repeat annotation. The repeat elements were categorized in a hierarchical manner as described previously (Zhang et al., 2012). To study the divergence of LTRs, we identified LTRs with a complete structure using LTR_STRUC (McCarthy and McDonald, 2003) with default parameters. Divergence was then estimated as described previously (Zhang et al., 2012). The LTRs with a complete structure were aligned using MUSCLE (Edgar, 2004).
Four types of noncoding RNA genes (tRNAs, rRNAs, miRNAs and snRNAs) were predicted in the C. sinense genome. The tRNA genes were predicted using tRNAscan‐SE (v1.3.1; Lowe and Eddy, 1997) with eukaryote parameters. Identification of the rRNA genes was conducted by aligning known Arabidopsis and rice rRNAs.
Structural and functional annotation of genes
Putative protein‐coding genes in the C. sinense genome were predicted using the Maker package (v2.31.8). We also included de novo predictions of gene structures obtained using Augustus software (v3.0.3; Stanke et al., 2006). Functional annotation of the protein‐coding genes was conducted by performing BlastP (e‐value cut‐off 1e−05) searches against entries in the NCBI nr and SwissProt databases. Searches for gene motifs and domains were performed using InterProScan (v5.28). The GO terms for genes were obtained from the corresponding InterPro or Pfam entry. Pathway reconstruction was performed using KOBAS (v2.0) (Xie et al., 2011) and the KEGG database.
Gene family and phylogenomic analysis
Orthologous gene clusters in the genomes of C. sinense and 18 other representative plants were identified using the OrthoMCL program (Li et al., 2003). We determined gene family expansion or contraction using CAFE (v3.0) (De Bie et al., 2006). The gene families with >200 members per species were selected based on this analysis. Alignments from MUSCLE were converted to coding sequences, and RAxML (v8.2.10) (Stamatakis, 2014) was used to construct the phylogenetic trees. The Bayesian Relaxed Molecular Clock method was used to estimate species divergence times using the program MCMCTREE (v4.0) within the PAML package (v4.8; Yang, 2007). PAML was used to calculate the value under evolutionary pressure based on 500 single‐copy gene families.
HPLC for flower colour
Five individual flowers were selected from each plant, and all petals of the five flowers of each individual plant were removed and stored in liquid nitrogen. The samples were ground into powder and dried at low temperature for later use.
The chromatographic analysis system comprised an Agilent 1200 HPLC (Agilent Technologies, Santa Clara, CA). The structure of anthocyanin and flavonoid glycosides was inferred from HPLC‐DAD‐HRMS mass spectrometry data. The comprehensive analysis was conducted, and the UV‐Vis absorption spectrum characteristics, molecular weight, molecular formula and secondary mass spectrometry fragments were combined, as well as the relevant literature reports (Mikanagi et al., 1995; Veberic et al., 2015) to determine the structure of the anthocyanins and flavonols. The anthocyanin and flavonoid glycosides were quantitatively analysed according to the HPLC‐DAD‐HRMS data. The content of similar compounds was determined using the external standard method for similar structural compounds (Mikanagi et al., 1995; Veberic et al., 2015).
GC‐MS for floral fragrance
The samples were stored in a refrigerator at −80 °C. On use, the samples were ground in liquid nitrogen. The raw data obtained by GC‐MS was processed using Unknowns Analysis (v10.0, Agilent) in the Masshunter's Worksite. The compounds were characterized based on the MS data (match score > 70.0) and linear retention index (deviation within 10 units), both of which were compared with the data in the NIST14 library.
BiFC assay in orchid protoplasts
The vectors used for BiFC assay were prepared accordingly. Protoplast isolation and transfection from the leaf bases of Cymbidium sinense was conducted based on protocols established by Ren et al. (2021). Empty vectors pSPYNE‐35S (harbouring the nitrogen terminal of the yellow fluorescent protein [YFP]) and pSPYCE‐35S (harbouring the carbon terminal of the YFP) were both driven by the CaMV 35S promoter. Full‐length coding sequences of CsSVP1, CsSVP3, CsSOC1 and CsAP1 were cloned into pSPYNE‐35S and pSPYCE‐35S resulting in the recombinant plasmids pSPYNE‐35S‐CsSVP3‐YFPn, pSPYNE‐35S‐CsSOC1‐YFPc and CsAP1‐YFPc, respectively. For the BiFC assay, pSPYNE‐35S‐CsSVP3‐YFPn and pSPYNE‐35S‐CsSOC1‐YFPc or pSPYNE‐35S‐CsAP1‐YFPc were cotransfected into protoplasts. As negative controls, the vector combinations pSPYNE‐35S‐CsAP1‐YFPc + pSPYNE‐35S, pSPYNE‐35S‐CsSOC1‐YFPc + pSPYNE‐35S and pSPYCE‐35S + pSPYNE‐35S‐CsSVP3‐YFPn were also cotransfected into the protoplasts.
Funding information
This research was funded by the Natural Science Foundation of Guangdong Province (2017A030312004), grants from National Key R&D Program (2018YFD1000400, 2019YFD1001003), Science and Technology Project of Guangdong Province (2019B030316033) and Guangzhou (201707010307), Innovation Team of Modern Agricultural Industry Technology System in Guangdong Province (2021KJ121), and the Guangdong Academy of Agricultural Sciences Discipline Team Construction Project (202127 TD).
Conflict of interest
No conflict of interest was declared.
Author contributions
G.F.Z., Z.J.L., Y.V.P. and W.C.T. designed the experiments and edited the manuscript; F.X.Y., J.G. and R.R. executed the experiments and assembled the figures; Y.L.W. and G.Q.Z. performed genome analyses; C.Q.L. and J.P.J. conducted the qRT‐PCR and protein–protein interaction; Y.A., D.Y.Z. and Y.Q.W. performed the transcriptome analysis. W.H.S. and L.J.C. prepared the DNA and RNA samples and libraries; carried out the experiments P.S., V.R., A.P., and B.V.; K.M.G., B.C., and F.E.R. B.V., V.R., and P.S. F.X.Y., J.G., and Y.L.W. wrote the paper with inputs from other authors. All authors read and approved the final manuscript.
Supporting information
Figure S1 Karyotype analysis of C. sinense.
Figure S2 Genome‐wide all‐by‐all Hi‐C interaction.
Figure S3 Comparison of the distribution of repeat elements between intergenic and intron regions in orchid plants.
Figure S4 Comparison of gene models in orchid plants.
Figure S5 Venn diagram represents the shared and unique gene families among five closely related orchid plants. Each number represents the number of gene families.
Figure S6 Evolution of orchids genome and gene families.
Figure S7 Genome features of Cymbidium sinense.
Figure S8 Comparison of the timing of LTR‐RTs insertions among three orchid plants.
Figure S9 Floral organ morphology of transgenic Arabidopsis with overexpression of CsSEP1 and CsSEP2.
Figure S10 Chromosomal localization and phylogenetic analysis of Type I MADS‐box genes.
Figure S11 Gene expression patterns of Type I MADS‐box genes in individual floral organs from different flower varieties.
Figure S12 The expression patterns of SVP, SOC1, AP1/FUL genes at different flower developmental stages (a) and cold condition (b).
Figure S13 The gene expression analysis when overexpressing CsSVP1 (a) and CsSVP3 (b) in Cymbidium sinense protoplasts using a protoplast‐based transient expression system.
Figure S14 Yeast two‐hybrid assay (Y2H) of proteins encoded by CsSVP, CsAP1 and CsSOC1.
Figure S15 Amino acid sequence alignment of SOC1 and SVP proteins in C. sinense. The 137th amino acid of CsSOC1 is marked with red square frame.
Figure S16 Expression analysis of UFGT genes in four different colour flowers of C. sinense.
Figure S17 Substrate recognition domains of orchids F3'5'H. Positions 5 and 10 of SRS6 amino acids are shown in black squares, and position 8 of SRS6 is shown in a black box.
Figure S18 Analysis of MYB Binding Sites in F3'5'H gene promoter in Orchidaceae.
Figure S19 The phylogenetic tree of MYB genes among C. sinense, O. sativa and Arabidopsis.
Figure S20 Chromosomal localization of TPS genes in Cymbidium sinense.
Table S1 Summary of Nanopore sequencing
Table S2 Statistics of final assembly results
Table S3 Evaluation of the genome completeness and coverage
Table S4 Summary of Hi‐c data
Table S5 Summary of Hi‐c assembly
Table S6 Statistics of the 20 pseudochromosomes length
Table S7 Evaluation of the genome completeness using dataset of RNA sequencing
Table S8 Statistics of repeated sequence classification
Table S9 Statistics of transposable elements classification
Table S10 Statistics of gene annotation
Table S11 Statistics of noncoding gene annotation
Table S12 Comparison of gene family clusters between C. sinense and 17 other land plants
Table S13 Functional enrichment of expanded gene families in C. sinense. (P<0.01)
Table S14 Statistics of specific gene of Orchids
Table S15 Functional enrichment of core proteome in Orchids. (P < 0.01)
Table S16 Ty3/Gypsy and Ty1/Copia reverse transcriptase domains and LTR TE numbers
Table S17 MADS‐box genes in the A. shenzhenica, P. equestris, D. catenatum, C. sinense, O. sativa and A. thaliana genomes
Table S18 Positive Darwinian selection (PDS) were found in K domain of orchid SVP genes
Table S19 HPLC analysis of C. sinense pigmentation
Table S20 Information about samples used for transcriptome sequencing
Table S21 Primer used in this work
Acknowledgements
We thank the engineers of Nextomics Biosciences Co., Ltd and PubBio Co. Ltd for technical assistance in data processing and bioinformatics analysis, and the Yuandong Chinese Orchid Corporation for plant cultivation. We are grateful to Prof. Da Luo of Sun Yat‐sen University and Prof. Di‐Qiu Yu of Yunnan University for their help during the preparation of the article.
Yang, F.‐X. , Gao, J. , Wei, Y.‐L. , Ren, R. , Zhang, G.‐Q. , Lu, C.‐Q. , Jin, J.‐P. , Ai, Y. , Wang, Y.‐Q. , Chen, L.‐J. , Ahmad, S. , Zhang, D.‐Y. , Sun, W.‐H. , Tsai, W.‐C. , Liu, Z.‐J. and Zhu, G.‐F. (2021) The genome of Cymbidium sinense revealed the evolution of orchid traits. Plant Biotechnol. J., 10.1111/pbi.13676
†These authors are contributed equally to this work.
Contributor Information
Wen‐Chieh Tsai, Email: tsaiwc@mail.ncku.edu.tw.
Zhong‐Jian Liu, Email: zjliu@fafu.edu.cn.
Gen‐Fa Zhu, Email: zhugenfa@gdaas.cn.
Data availability statement
Genome dataset and expression data are available at the GenBank with accession number SAMN20059972. Genome sequencing data of Illumina, Nanopore, HI‐C and RNA‐Seq reads are available in the NCBI Sequence Read Archive under accessions SRR15070575, SRR15127750, SRR15127751, SRR15127752, SRR15170672–SRR15170679, SRR15174943–SRR15174947 and SRR15194893–SRR15194902.
References
- Badouin, H. , Gouzy, J. , Grassa, C.J. , Murat, F. , Staton, S.E. , Cottret, L. , Lelandais‐Brière, C. et al. (2017) The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature, 546, 148–152. [DOI] [PubMed] [Google Scholar]
- Cai, J. , Liu, X. , Vanneste, K. , Proost, S. , Tsai, W.‐C. , Liu, K.‐W. , Chen, L.‐J. et al. (2015) The genome sequence of the orchid Phalaenopsis equestris . Nat. Genet. 47, 65–72. [DOI] [PubMed] [Google Scholar]
- Chao, Y.T. , Chen, W.C. , Chen, C.Y. , Ho, H.Y. , Yeh, C.H. , Kuo, Y.T. , Su, C.L. , Yen, S.H. , Hsueh, H.Y. , Yeh, J.H. , Hsu, H.L. , Tsai, Y.H. , Kuo, T.Y. , Chang, S.B. , Chen, K.Y. and Shih, M.C. (2018) Chromosome‐level assembly, genetic and physical mapping of Phalaenopsis aphrodite genome provides new insights into species adaptation and resources for orchid breeding. Plant biotechnology journal, 16(12), 2027–2041. 10.1111/pbi.12936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, M.W. , Cameron, K.M. , Freudenstein, J.V. , Pridgeon, A.M. , Salazar, G. , van den Berg, C. and Schuiteman, A. (2015) An updated classification of Orchidaceae. Bot. J. Linn. Soc. 177, 151–174. [Google Scholar]
- Chaves‐Silva, S. , Santos, A. , Chalfun‐Júnior, A. , Zhao, J. , Peres, L. and Benedito, V.A. (2018) Understanding the genetic regulation of anthocyanin biosynthesis in plants ‐ Tools for breeding purple varieties of fruits and vegetables. Phytochemistry, 153, 11–27. 10.1016/j.phytochem.2018.05.013 [DOI] [PubMed] [Google Scholar]
- Chen, F. , Tholl, D. , Bohlmann, J. and Pichersky, E. (2011) The family of terpene synthases in plants: a mid‐size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. [DOI] [PubMed] [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falavigna, V.D.S. , Guitton, B. , Costes, E. and Andres, F. (2018) I want to (Bud) break free: the potential role of DAM and SVP‐Like genes in regulating dormancy cycle in temperate fruit trees. Front. Plant Sci. 9, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, M. , Miao, F. , Jia, H. , Li, G. , Powers, C. , Nagarajan, R. , Alderman, P.D. et al. (2021) O‐linked n‐acetylglucosamine transferase is involved in fine regulation of flowering time in winter wheat. Nat. Commun. 12(1), 2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J. , Liang, D. , Xu, Q. , Yang, F. and Zhu, G. (2020) Involvement of CsERF2 in leaf variegation of Cymbidium sinense 'Dharma'. Planta, 252, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, R. , Wu, S.Q. , Piao, X.C. , Park, S.Y. and Lian, M.L. (2014) Micropropagation of Cymbidium sinense using continuous and temporary airlift bioreactor systems. Acta Physiol. Plant. 36, 117–124. [Google Scholar]
- Gonzalez, A. , Zhao, M. , Leavitt, J.M. and Lloyd, A.M. (2008) Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. The Plant journal : for cell and molecular biology, 53(5), 814–827. 10.1111/j.1365-313X.2007.03373.x [DOI] [PubMed] [Google Scholar]
- Han, B. , Jing, Y. , Dai, J. , Zheng, T. , Gu, F. , Zhao, Q. , Zhu, F. et al. (2020) A chromosome‐level genome assembly of dendrobium huoshanense using long reads and Hi‐C data. Genome Biol. Evol. 12, 2486–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, C. , Si, C. , Teixeira da Silva, J.A. , Li, M. and Duan, J. (2019) Genome‐wide identification and classification of MIKC‐type MADS‐box genes in Streptophyte lineages and expression analyses to reveal their role in seed germination of orchid. BMC plant biology, 19(1), 223. 10.1186/s12870-019-1836-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hew, C.S. (2001). Ancient Chinese orchid cultivation: a fresh look at an age‐old practice. Sci. Hortic. 87, 1–10. [Google Scholar]
- Hsu, C.C. , Chen, Y.Y. , Tsai, W.C. , Chen, W.H. and Chen, H.H. (2015a) Three R2R3‐MYB transcription factors regulate distinct floral pigmentation patterning in Phalaenopsis spp. Plant Physiol. 168, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, H. , Chen, W. , Shen, Y. , Hsu, W. , Mao, W. and Yang, C. (2021) Multifunctional evolution of B and AGL6 MADS box genes in orchids. Nat. Commun. 12, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, H. , Hsu, W. , Lee, Y.I. , Mao, W. , Yang, J. , Li, J. and Yang, C. (2015b) Model for perianth formation in orchids. Nat. Plants, 1, 15046. [Google Scholar]
- Huang, B.Q. , Ye, X.L. , Yeung, E.C. and Zee, S.Y. (1998) Embryology of Cymbidium sinense: the microtubule organization of early embryos. Ann. Bot. 81, 741–750. [Google Scholar]
- Jang, S. , Torti, S. and Coupland, G. (2009) Genetic and spatial interactions between FT, TSF and SVP during the early stages of floral induction in Arabidopsis. Plant J. 60, 614–625. [DOI] [PubMed] [Google Scholar]
- Jia, Y. , Li, B. , Zhang, Y. , Zhang, X. , Xu, Y. and Li, C. (2019) Evolutionary dynamic analyses on monocot flavonoid 3'‐hydroxylase gene family reveal evidence of plant‐environment interaction. BMC Plant Biol. 19, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.M. , Jang, E.J. , Hong, J.W. , Song, S.H. and Pak, C.H. (2016) A comparison of functional fragrant components of Cymbidium (Oriental Orchid) species. Horticult. Sci. Technol. 34, 331–341. [Google Scholar]
- Lee, J.H. , Yoo, S.J. , Park, S.H. , Hwang, I. , Lee, J.S. and Ahn, J.H. (2007) Role of SVP in the control of flowering time by ambient temperature in Arabidopsis . Genes Dev. 21, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B.‐J. , Zheng, B.‐Q. , Wang, J.‐Y. , Tsai, W.‐C. , Lu, H.‐C. , Zou, L.‐H. , Wan, X. et al. (2020) New insight into the molecular mechanism of colour differentiation among floral segments in orchids. Commun. Biol. 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Gu, H. , Jiang, W. , Zou, C. , Wei, D. , Wang, Z. and Tang, Q. (2019) Protein interactions of SOC1 with SVP are regulated by a few crucial amino acids in flowering pathways of Brassica juncea. Acta Physiol. Plant. 41, 43. [Google Scholar]
- Li, J.W. and Zhang, S.B. (2016). Differences in the responses of photosystems I and II in Cymbidium sinense and C. tracyanum to long‐term chilling stress. Front. Plant Sci. 6, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Zhu, G. and Wang, Z. (2017) Chemical variation in essential oil of Cymbidium sinense flowers from six cultivars. J. Essential Oil Bear. Plants 20, 385–394. [Google Scholar]
- Li, L. , Stoeckert, C.J. Jr. and Roos, D.S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, C.Y. , Rengasamy, K.P. , Huang, L.M. , Hsu, C.C. , Jeng, M.F. , Chen, W.H. and Chen, H.H. (2020) Assessment of violet‐blue color formation in Phalaenopsis orchids. BMC Plant Biol. 20, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Ren, M. , Chen, H. , Wu, S. , Yan, H. , Jalal, A. and Wang, C. (2020) Evolution of SHORT VEGETATIVE PHASE (SVP) genes in Rosaceae: implications of lineage‐specific gene duplication events and function diversifications with respect to their roles in processes other than bud dormancy. Plant Genome, 13, e20053. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Sun, Z. , Dong, W. , Wang, Z. and Zhang, L. (2018) Expansion and functional divergence of the SHORT VEGETATIVE PHASE (SVP) genes in eudicots. Genome Biol. Evol. 10, 3026–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. (1997) tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Hu, X. , Liu, J. and Wang, H. (2011) Genetic diversity and population structure of 151 Cymbidium sinense cultivars. J. Horticul. Forest. 3, 104–114. [Google Scholar]
- Malcomber, S.T. and Kellogg, E.A. (2005) SEPALLATA gene diversification: brave new whorls. Trends Plant Sci. 10, 427–435. [DOI] [PubMed] [Google Scholar]
- Masiero, S. , Colombo, L. , Grini, P.E. , Schnittger, A. and Kater, M.M. (2011) The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell, 23, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, E.M. and McDonald, J.F. (2003) LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics, 19, 362–367. [DOI] [PubMed] [Google Scholar]
- Mendez‐Vigo, B. , Martinez‐Zapater, J.M. and Alonso‐Blanco, C. (2013) The flowering repressor SVP underlies a novel Arabidopsis thaliana QTL interacting with the genetic background. PLoS Genet. 9, e1003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels, S.D. and Amasino, R.M. (1999) FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell, 11(5), 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikanagi, Y. , Yokoi, M. , Ueda, Y. and Saito, N. (1995) Flower flavonol and anthocyanin distribution in subgenus Rosa. Biochem. Syst. Ecol. 23, 183–200. [Google Scholar]
- Morel, P. , Chambrier, P. , Boltz, V. , Chamot, S. , Rozier, F. , Rodrigues Bento, S. , Trehin, C. et al. (2019) Divergent functional diversification patterns in the SEP/AGL6/AP1 MADS‐box transcription factor superclade. Plant Cell, 31, 3033–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motomura, H. , Selosse, M.A. , Martos, F. , Kagawa, A. and Yukawa, T. (2010) Mycoheterotrophy evolved from mixotrophic ancestors: evidence in Cymbidium (Orchidaceae). Ann. Bot. 106, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M.G. and Thompson, W.F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, M. and Yanofsky, M.F. (2001) Function and evolution of the plant MADS‐box gene family. Nat. Rev. Genet. 2, 186–195. [DOI] [PubMed] [Google Scholar]
- Opassiri, R. , Pomthong, B. , Onkoksoong, T. , Akiyama, T. , Esen, A. and Ketudat Cairns, J.R. (2006) Analysis of rice glycosyl hydrolase family 1 and expression of Os4bglu12 beta‐glucosidase. BMC Plant Biol. 6, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Z.J. , Chen, Y.Y. , Du, J.S. , Chen, Y.Y. , Chung, M.C. , Tsai, W.C. , Wang, C.N. et al. (2014) Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA‐like genes. New Phytol. 202, 1024–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon, A.M. , Cribb, P.J. , Chase, M.W. and Rasmussen, F. (2014) Genera Orchidacearum. Oxford: Oxford University Press. [Google Scholar]
- Ramya, M. , Park, P.H. , Chuang, Y.‐C. , Kwon, O.K. , An, H.R. , Park, P.M. , Baek, Y.S. et al. (2019) RNA sequencing analysis of Cymbidium goeringii identifies floral scent biosynthesis related genes. BMC Plant Biol. 19, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, R. , Gao, J. , Yin, D. , Li, K. , Lu, C. , Ahmad, S. , Wei, Y. , Jin, J. , Zhu, G. and Yang, F. (2021) Highly Efficient Leaf Base Protoplast Isolation and Transient Expression Systems for Orchids and Other Important Monocot Crops. Frontiers in plant science, 12, 626015. 10.3389/fpls.2021.626015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland, F. , Baena‐Gonzalez, E. and Sheen, J. (2006) Sugar sensing and signaling in plants: conserved and novel mechanisms. Annu. Rev. Plant Biol. 57, 675–709. [DOI] [PubMed] [Google Scholar]
- Ruelens, P. , de Maagd, R.A. , Proost, S. , Theißen, G. , Geuten, K. and Kaufmann, K. (2013). Flowering locus c in monocots and the tandem origin of angiosperm‐specific mads‐box genes. Nat. Commun. 4, 2280. [DOI] [PubMed] [Google Scholar]
- Rui Chi, P. , Qing Sheng, Y. and Choy Sin, H. (1997) Physiology of Cymbidium sinense: a review. Sci. Hortic. 70, 123–129. [Google Scholar]
- Shan, L. , Ye, Q.S. , Li, R.H. , Leng, J.Y. and Li, H.Q. (2012) Transcriptional regulations on the low‐temperature‐induced floral transition in an orchidaceae species, dendrobium nobile: an expressed sequence tags analysis. Comp. Funct. Genomics, 3, 757801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smaczniak, C. , Immink, R.G. , Angenent, G.C. and Kaufmann, K. (2012) Developmental and evolutionary diversity of plant MADS‐domain factors: insights from recent studies. Development, 139, 3081–3098. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. and Morgenstern, B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, S. , Shao, X. , Zhu, C. , Xu, J. , Tang, Y. , Luo, D. and Huang, X. (2018a) An AGAMOUS‐like factor is associated with the origin of two domesticated varieties in Cymbidium sinense (Orchidaceae). Horticul. Res. 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, S. , Shao, X. , Zhu, C. , Xu, J. , Lu, H. , Tang, Y. , Jiao, K. et al. (2018b) Transcriptome‐wide analysis reveals the origin of peloria in Chinese cymbidium (Cymbidium sinense). Plant Cell Physiol. 59, 2064–2074. [DOI] [PubMed] [Google Scholar]
- Tang, H. , Lyons, E. , Pedersen, B. , Schnable, J.C. , Paterson, A.H. and Freeling, M. (2011) Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinform. 12(1), 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics, Chapter, 4, 4–10. 10.1002/0471250953.bi0410s25 [DOI] [PubMed] [Google Scholar]
- Tel‐zur, N. , Abbo, S. , Myslabodski, D. and Mizrahi, Y. (1999) Modified CTAB procedure for DNA Isolation from epiphytic cacti of the genera hylocereus and selenicereus (Cactaceae). Plant Mol. Biol. Rep. 17, 249–254. [Google Scholar]
- Theißen, G. , Rümpler, F. and Gramzow, L. (2018) Array of MADS‐box genes: facilitator for rapid adaptation? Trends Plant Sci. 23, 563–576. [DOI] [PubMed] [Google Scholar]
- Tsai, W.C. , Pan, Z.J. , Hsiao, Y.Y. , Chen, L.J. and Liu, Z.J. (2014). Evolution and function of MADS‐box genes involved in orchid floral development. J. Syst. Evol. 52, 397–410. [Google Scholar]
- Veberic, R. , Slatnar, A. , Bizjak, J. , Stampar, F. and Mikulic‐Petkovsek, M. (2015). Anthocyanin composition of different wild and cultivated berry species. LWT Food Sci. Technol. 60, 509–517. [Google Scholar]
- Whang, S.S. , Um, W.S. , Song, I.J. , Lim, P.O. , Choi, K. , Park, K.W. , Kang, K.‐W. et al. (2011) Molecular analysis of anthocyanin biosynthetic genes and control of flower coloration by flavonoid 3′,5′‐hydroxylase (F3′5′H) in Dendrobium moniliforme . J. Plant Biol. 54, 209–218. [Google Scholar]
- Wu, R. , Tomes, S. , Karunairetnam, S. , Tustin, S.D. , Hellens, R.P. , Allan, A.C. , Macknight, R.C. et al. (2017) SVP‐like MADS box genes control dormancy and budbreak in apple. Front. Plant Sci. 8, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, R.M. , Walton, E.F. , Richardson, A.C. , Wood, M. , Hellens, R.P. and Varkonyi‐Gasic, E. (2012) Conservation and divergence of four kiwifruit SVP‐like MADS‐box genes suggest distinct roles in kiwifruit bud dormancy and flowering. J. Exp. Bot. 63, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, E. , Tong, W. , Hou, Y. , An, Y. , Chen, L. , Wu, Q. , Liu, Y. et al. (2020) The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant, 13, 1013–1026. [DOI] [PubMed] [Google Scholar]
- Xie, C. , Mao, X. , Huang, J. , Ding, Y. , Wu, J. , Dong, S. , Kong, L. , Gao, G. , Li, C.Y. and Wei, L. (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic acids research, 39, W316–W322. 10.1093/nar/gkr483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. , Zhu, G. , Wang, Z. , Liu, H. , Xu, Q. , Huang, D. and Zhao, C. (2017) Integrated mRNA and microRNA transcriptome variations in the multi‐tepal mutant provide insights into the floral patterning of the orchid Cymbidium goeringii . BMC Genom. 18, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. , Zhu, G. , Wei, Y. , Gao, J. , Liang, G. , Peng, L. , Lu, Q. et al. (2019) Low‐temperature‐induced changes in the transcriptome reveal a major role of cgsvp genes in regulating flowering of cymbidium goeringii . BMC Genom. 20, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol., 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhang, G.‐Q. , Liu, K.‐W. , Li, Z. , Lohaus, R. , Hsiao, Y.‐Y. , Niu, S.‐C. , Wang, J.‐Y. et al. (2017a) The Apostasia genome and the evolution of orchids. Nature, 549, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Liu, X. , Quan, Z. , Cheng, S. , Xu, X. , Pan, S. , Xie, M. et al. (2012) Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554. [DOI] [PubMed] [Google Scholar]
- Zhang, G.Q. , Xu, Q. , Bian, C. , Tsai, W.C. , Yeh, C.M. , Liu, K.W. , Yoshida, K. et al. (2016) The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 19029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Wu, K. , Zeng, S. , Teixeira da Silva, J.A. , Zhao, X. , Tian, C.E. , Xia, H. et al. (2013) Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genom. 14, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Zhao, Y. , Juntheikki, I. , Mouhu, K. , Broholm, S.K. , Rijpkema, A.S. , Kins, L. et al. (2017b) Dissecting functions of SEPALLATA‐like MADS box genes in patterning of the pseudanthial inflorescence of Gerbera hybrida . New Phytol. 216, 939–954. [DOI] [PubMed] [Google Scholar]
- Zhu, G. , Yang, F. , Shi, S. , Li, D. , Wang, Z. , Liu, H. , Huang, D. et al. (2015) Transcriptome characterization of Cymbidium sinense 'Dharma' using 454 pyrosequencing and its application in the identification of genes associated with leaf color variation. PLoS One, 10, e0128592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Karyotype analysis of C. sinense.
Figure S2 Genome‐wide all‐by‐all Hi‐C interaction.
Figure S3 Comparison of the distribution of repeat elements between intergenic and intron regions in orchid plants.
Figure S4 Comparison of gene models in orchid plants.
Figure S5 Venn diagram represents the shared and unique gene families among five closely related orchid plants. Each number represents the number of gene families.
Figure S6 Evolution of orchids genome and gene families.
Figure S7 Genome features of Cymbidium sinense.
Figure S8 Comparison of the timing of LTR‐RTs insertions among three orchid plants.
Figure S9 Floral organ morphology of transgenic Arabidopsis with overexpression of CsSEP1 and CsSEP2.
Figure S10 Chromosomal localization and phylogenetic analysis of Type I MADS‐box genes.
Figure S11 Gene expression patterns of Type I MADS‐box genes in individual floral organs from different flower varieties.
Figure S12 The expression patterns of SVP, SOC1, AP1/FUL genes at different flower developmental stages (a) and cold condition (b).
Figure S13 The gene expression analysis when overexpressing CsSVP1 (a) and CsSVP3 (b) in Cymbidium sinense protoplasts using a protoplast‐based transient expression system.
Figure S14 Yeast two‐hybrid assay (Y2H) of proteins encoded by CsSVP, CsAP1 and CsSOC1.
Figure S15 Amino acid sequence alignment of SOC1 and SVP proteins in C. sinense. The 137th amino acid of CsSOC1 is marked with red square frame.
Figure S16 Expression analysis of UFGT genes in four different colour flowers of C. sinense.
Figure S17 Substrate recognition domains of orchids F3'5'H. Positions 5 and 10 of SRS6 amino acids are shown in black squares, and position 8 of SRS6 is shown in a black box.
Figure S18 Analysis of MYB Binding Sites in F3'5'H gene promoter in Orchidaceae.
Figure S19 The phylogenetic tree of MYB genes among C. sinense, O. sativa and Arabidopsis.
Figure S20 Chromosomal localization of TPS genes in Cymbidium sinense.
Table S1 Summary of Nanopore sequencing
Table S2 Statistics of final assembly results
Table S3 Evaluation of the genome completeness and coverage
Table S4 Summary of Hi‐c data
Table S5 Summary of Hi‐c assembly
Table S6 Statistics of the 20 pseudochromosomes length
Table S7 Evaluation of the genome completeness using dataset of RNA sequencing
Table S8 Statistics of repeated sequence classification
Table S9 Statistics of transposable elements classification
Table S10 Statistics of gene annotation
Table S11 Statistics of noncoding gene annotation
Table S12 Comparison of gene family clusters between C. sinense and 17 other land plants
Table S13 Functional enrichment of expanded gene families in C. sinense. (P<0.01)
Table S14 Statistics of specific gene of Orchids
Table S15 Functional enrichment of core proteome in Orchids. (P < 0.01)
Table S16 Ty3/Gypsy and Ty1/Copia reverse transcriptase domains and LTR TE numbers
Table S17 MADS‐box genes in the A. shenzhenica, P. equestris, D. catenatum, C. sinense, O. sativa and A. thaliana genomes
Table S18 Positive Darwinian selection (PDS) were found in K domain of orchid SVP genes
Table S19 HPLC analysis of C. sinense pigmentation
Table S20 Information about samples used for transcriptome sequencing
Table S21 Primer used in this work
Data Availability Statement
Genome dataset and expression data are available at the GenBank with accession number SAMN20059972. Genome sequencing data of Illumina, Nanopore, HI‐C and RNA‐Seq reads are available in the NCBI Sequence Read Archive under accessions SRR15070575, SRR15127750, SRR15127751, SRR15127752, SRR15170672–SRR15170679, SRR15174943–SRR15174947 and SRR15194893–SRR15194902.