

Summary
Azalea belongs to Rhododendron, which is one of the largest genera of flowering plants and is well known for the diversity and beauty in its more than 1000 woody species. Rhododendron contains two distinct groups: the most high‐altitude and a few low‐altitude species; however, the former group is difficult to be domesticated for urban landscaping, and their evolution and adaptation are little known. Rhododendron ovatum has broad adaptation in low‐altitude regions but possesses evergreen characteristics like high‐altitude species, and it has floral fragrance that is deficient in most cultivars. Here we report the chromosome‐level genome assembly of R. ovatum, which has a total length of 549 Mb with scaffold N50 of 41 Mb and contains 41 264 predicted genes. Genomic micro‐evolutionary analysis of R. ovatum in comparison with two high‐altitude Rhododendron species indicated that the expansion genes in R. ovatum were significantly enriched in defence responses, which may account for its adaptability in low altitudes. The R. ovatum genome contains much more terpene synthase genes (TPSs) compared with the species that lost floral fragrance. The subfamily b members of TPS are involved in the synthesis of sesquiterpenes as well as monoterpenes and play a major role in flora scent biosynthesis and defence responses. Tandem duplication is the primary force driving expansion of defence‐responsive genes for extensive adaptability to the low‐altitude environments. The R. ovatum genome provides insights into low‐altitude adaptation and gain or loss of floral fragrance for Rhododendron species, which are valuable for alpine plant domestication and floral scent breeding.
Keywords: Azalea, Rhododendron ovatum, altitude, adaptability, floral scent, terpene synthase (TPS), tandem duplication, defence response
Introduction
The genus Rhododendron comprises one of the largest and most diverse groups of woody ornamental plants in cultivation, ranging from spectacular trees to evergreen or deciduous shrubs and alpine cushions (Cameron, 1993; De Riek et al., 2018). Rhododendron plants have attractive foliage and flowers, which in some species are sweetly fragrant, and are economically important as ornamental landscapes or pot plants (De Riek et al., 2018; Norton and Norton, 1989). The genus Rhododendron contains more than 1,000 species. Because of the large number of the species in this genus, taxonomists have made several classifications based on morphology, now the most universally accepted classification system subdivides the genus Rhododendron into eight subgenera, including the five most important subgenera: Rhododendron, Hymenanthes, Azaleastrum, Tsutsusi, and Pentanthera (Chamberlain et al., 1996). The former two subgenera comprise what gardeners loosely refer to as ‘rhododendrons’, whereas the latter three subgenera comprise the ‘azaleas’ (Figure 1a) (De Riek et al., 2018). The evolution and adaptation of the two different groups, high‐altitude rhododendron and low‐altitude azalea of this genus, remain largely unexplored.
Figure 1.
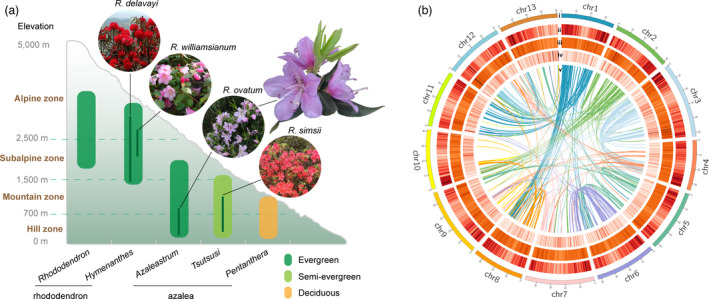
Characteristics of Rhododendron ovatum and its genome assembly. (a) Low‐altitude and evergreen characteristics of R. ovatum. The genus Rhododendron contains five main subgenera, of which two subgenera (Rhododendron and Hymenanthes) commonly called ‘rhododendron’ are mainly distributed in alpine and subalpine zone, and the other three (Azaleastrum, Tsutsusi, and Pentanthera) commonly called ‘azalea’ are distributed in mountain and hill zone. The subgenera of Rhododendron, Hymenanthes, and Azaleastrum are mostly evergreens, while Tsutsusi are mainly semi‐evergreens and Pentanthera are deciduous. Genome sequences of R. delavayi and R. williamsianum in subgenus Hymenanthes, and R. simsii in subgenus Tsutsusi were previously reported (Soza et al., 2019; Yang et al., 2020; Zhang et al., 2017a). (b) Overview of the R. ovatum genome assembly. (i) The 13 pseudochromosomes, (ii) gene density, (iii) transposon elements, (iv) tandem repeats, and (v) collinear blocks of the R. ovatum genome.
The species of rhododendron group account for a large portion of the genus and possess diverse fascinating flowers. However, evergreen rhododendrons prefer to grow in cool and humid forest environments with acidic soil in the alpine or subalpine zone, which shows a temperate climate even in the tropics or subtropics. Thus, high‐altitude rhododendrons can hardly adapt to the hot and dry urban summer environment during domestication. Another group azaleas distribute in low altitudes and are widely cultivated and applied in urban landscapes. The azalea group contains evergreen, semi‐evergreen, and deciduous species, of which the subgenus Azaleastrum species all have evergreen characteristics like high‐altitude rhododendrons but mainly distributed at low altitudes of approximately 300–2000 m (Figure 1a). Moreover, the evolutionary position of Azaleastrum in the genus Rhododendron has been proposed with different points. According to morphological classification, the evolutionary process of the genus Rhododendron was deduced from evergreen to semi‐evergreen and deciduous plants, and the subgenus Azaleastrum has a transitional evolutionary position that links rhododendrons and azaleas (Ming and Fang, 1990). In another report, Azaleastrum was supported to be the early subgenus that diverged from Rhododendron ancestor based on the dated molecular phylogeny using nine chloroplast genes and seven nuclear regions (Shrestha et al., 2018). Therefore, the subgenus Azaleastrum has important position for evolution and adaptation in the genus Rhododendron. To date, there are two draft genomes for R. delavayi (Zhang et al., 2017a) and R. williamsianum (Soza et al., 2019) in subgenus Hymenanthes, and a genome assembly for the semi‐evergreen R. simsii in subgenus Tsutsusi was newly published (Yang et al., 2020). However, no genome sequencing was reported for the important subgenus Azaleastrum. Genome‐wide analysis of the species in Azaleastrum will contribute to elucidating its evolutionary position and adaptive mechanisms between high‐altitude rhododendrons and low‐altitude azaleas.
Floral scent is one of the most important trait for ornamental plant and has been always the breeding objective for Rhododendron cultivars (Kobayashi et al., 2008). In the large genus Rhododendron, only a small amount of species has floral scents, and the fragrant flowers generally exhibit light or faded colours like white, cream, pink or pale yellow (Cameron, 1993). Floral scents comprise complex compound with over 1700 volatile compounds have been identified from nearly 1000 species of flowering plants, which mainly belong to three compound classes of terpenes, benzenoids and fatty acid derivatives (Knudsen et al., 2006). Floral scent constitutes an ancient and important channel of communication between flowering plants and their pollinators or enemies, and important for the understanding of adaptations and evolutionary processes of angiosperms (Raguso, 2008). Orchidaceae are by far the best‐investigated family for floral scent, followed by several families, such as Araceae, Arecaceae, Magnoliaceae and Rosaceae (Knudsen et al., 2006). However, the compounds and evolution of floral scents of Rhododendron plants are still poorly investigated.
To fill the gaps in adaptation and floral scents of Rhododendron plants, we present a high‐quality reference genome of Rhododendron ovatum (Lindl.) Maxim., the representative species of subgenus Azaleastrum, using PacBio sequencing and Hi‐C technology. R. ovatum is an evergreen shrub or a small tree that is widely distributed in subtropical zones with altitudes <1000 m (Figure 1a) (Zhang et al., 2017b). In addition to its extensive adaptability, R. ovatum possesses white to pink or pinkish purple corolla, often with dark purple spots in the upper part, and has pleasant fragrance, which shows a fine combination of flower coloration and aroma. We performed comparative genomic analysis between R. ovatum and two high‐altitude rhododendrons to elucidate their micro‐evolutionary differences in low‐altitude adaptation. Analyses of biosynthesis pathways and key genes of the flora scent compounds elucidated evolution path of floral fragrance in Rhododendron species and provided candidate genes for breeding. This genomic study of R. ovatum will provide a better understanding of the evolution and adaptation of Rhododendron and make more contributions to evolutionary research on Ericales.
Results
Genome sequencing, assembly and annotation
The genome size of the R. ovatum was estimated to be 529 Mb with an extremely high heterozygosity of 1.55% based on k‐mer analysis of 76 Gb of clean short reads (˜144‐fold coverage) (Figure S1A, Table S1). For genome sequencing, we obtained a total of 56.7 Gb of PacBio long‐read sequencing data, which represents ˜107‐fold coverage of the estimated genome (Table S2), using single‐molecule real‐time (SMRT) sequencing technology. We initially assembled the PacBio reads into contig sequences of 712 Mb using Falcon/Falcon‐Unzip (Chin et al., 2016) and polished them using Arrow (Table S3). The redundant sequences from heterozygous genomic regions were filtered out using purge_haplotigs (Figure S1B, C; Table S4) (Roach et al., 2018), and the contig sequences were further polished using short reads. This yielded a genome assembly of 549 Mb with a contig N50 length of 1.24 Mb, and the longest contig is 7.45 Mb (Table 1, Table S3). A total of 64.5 Gb of clean reads (Table S5) were generated during construction of the Hi‐C sequencing library. This enabled 99.05% of the assembled sequences to be anchored onto 13 pseudochromosomes (2n = 26) (Figure 1b), which could be well distinguished in the chromatin interaction heatmap (Figure S2). The final chromosome‐level genome assembly of R. ovatum was 549 Mb with a scaffold N50 of 41 Mb, which has higher integrity and continuity than that of the other three Rhododendron species (scaffold N50: 36, 0.6, and 29 Mb, separately) (Table 1).
Table 1.
Comparisons of genome assemblies and annotations among species in the Ericales
| Species | Assembled genome size (Mb) | Contig N50 (kb) | Scaffold N50 (kb) | Complete BUSCOs of assembly (%) | Annotated gene numbers | Complete BUSCOs of annotation (%) | Repetitive sequences ratio (%) | Reference |
|---|---|---|---|---|---|---|---|---|
| Rhododendron ovatum | 549 | 1241 | 41 000 | 95.30 | 41 264 | 96.10 | 44.70 | This study |
| Rhododendron simsii | 529 | 2235 | 36 351 | 93.68 | 32 999 | 89.91 | 47.48 | Yang et al. (2020) |
| Rhododendron delavayi | 695 | 62 | 638 | 92.80 | 32 938 | 87.40 | 51.77 | Zhang et al. (2017a) |
| Rhododendron williamsianum | 532 | 219 | 29 011 | 89.00 | 23 559 | 77.20 | 58.80 | Soza et al. (2019) |
| Vaccinium corymbosum | 1680 | 15 | 186 | 95.50 | 32 140 | 97.21 | 44.31 | Colle et al. (2019) |
| Actinidia chinensis | 654 | 1430 | 20 000 | 90.80 | 38 202 | 86.33 | 43.42 | Wu et al. (2019) |
| Primula vulgaris | 411 | 1 | 295 | 89.92 | 24 000 | 86.23 | 37.03 | Cocker et al. (2018) |
| Diospyros lotus | 746 | 1060 | 29 749 | 78.97 | 40 532 | 74.07 | 66.60 | Akagi et al. (2020) |
| Camellia sinensis | 2940 | 600 | 167 113 | 90.60 | 50 525 | 90.24 | 86.87 | Xia et al. (2020) |
To provide RNA‐level evidence for genome annotation, we generated 147.8 Gb of PacBio ISO‐seq subreads (Table S6) obtained from three tissue types (stem, leaf and flower) and finally got 269 Mb of high‐quality, full‐length and consistent isoform sequences after processing. In combination with ab initio‐, homologous‐ and ISO‐seq‐based predictions, we identified 41,264 protein‐coding genes in the R. ovatum genome, which is greater than other sequenced species in Ericales except in tea plant (Camellia sinensis) (Table 1, Table S7). Of these protein‐coding genes, 39,405 were annotated with known proteins. We identified 245.48 Mb (44.71%) of repetitive sequences, and long terminal repeat (LTR) retrotransposons accounted for 28.42% of the entire genome (Table S8). A total of ˜158 037 simple sequence repeats were annotated (Table S8), which will provide valuable molecular markers to assist azalea breeding. Genome comparison between R. ovatum and R. williamsianum indicated that more genes and tandem repeats were identified in the former (Figure S3), indicating a higher quality of annotation for the R. ovatum genome.
The completeness of the R. ovatum genome assembly was evaluated using the Benchmarking Universal Single‐Copy Orthologs (BUSCO) gene sets and available ISO‐seq data. BUSCO analysis showed that 96.4% of the core eudicotyledon genes are present in the R. ovatum genome, of which 95.3% had complete coverage (Table S9), higher than that of R. simsii (93.7%), R. delavayi (92.8%), R. williamsianum (89.0%) and most other sequenced genomes in Ericales (Table 1). In addition, 269 Mb of polished PacBio ISO‐seq data were mapped onto the R. ovatum genome assembly and showed a high mapping rate of 99.66%. LTR annotation showed an LTR assembly index (LAI) score of 18.32, which meets the reference quality (10 ≤ LAI < 20) and is higher than that of tea plant (Camellia sinensis) (12.45) and R. simsii (18.10) (Xia et al., 2020; Yang et al., 2020). In summary, the extensive coverage of core eudicotyledon genes, high mapping rate of ISO‐seq reads and the high LAI score that represents reference genome quality indicated the high completeness and accuracy of the assembled genome. Moreover, our gene predictions covered 96.10% of highly conserved core proteins in the eudicot lineage, which is much higher than that of the other three Rhododendron genomes (89.91%, 87.40% and 77.20%, separately) and most other plants in Ericales (Table 1, Table S10). Overall, the evaluations demonstrated that the R. ovatum genome has superior quality in assembly and annotation among the reported plants in Rhododendron and Ericales.
Whole‐genome duplication of R. ovatum
Whole‐genome duplication (WGD) has been considered as an important factor for genome evolution (Moriyama and Koshiba‐Takeuchi, 2018). To investigate the WGD events during the evolution of R. ovatum, gene duplications were searched and classified into four types, among which WGD/segmental duplication covered 8027 genes (18.4%) (Figure S4). We characterized the distribution of the synonymous substitution rate (Ks) based on the collinear gene pairs of R. ovatum and other species, including kiwifruit (Actinidia chinensis), C. sinensis and R. williamsianum from Ericales, as well as tomato (Solanum lycopersicum) and grape (Vitis vinifera) from eudicots (Figure 2a). Our results confirmed that an ancient whole‐genome triplication (WGT, At‐γ, Ks peak value = 1.43), which is shared by core eudicots (Badouin et al., 2017), and a recent WGD (Ad‐β, Ks peak value = 0.63), which is shared by some families within Ericales (Zhang et al., 2020a), had occurred during the evolutionary history of R. ovatum. The occurrence time of Ad‐β is close to that of the triplication event (Solanaceae‐T) of tomato. Moreover, kiwifruit had experienced another recent WGD (Ad‐α) in addition to Ad‐β, which is consistent with previous reports (Huang et al., 2013; Shi et al., 2010).
Figure 2.
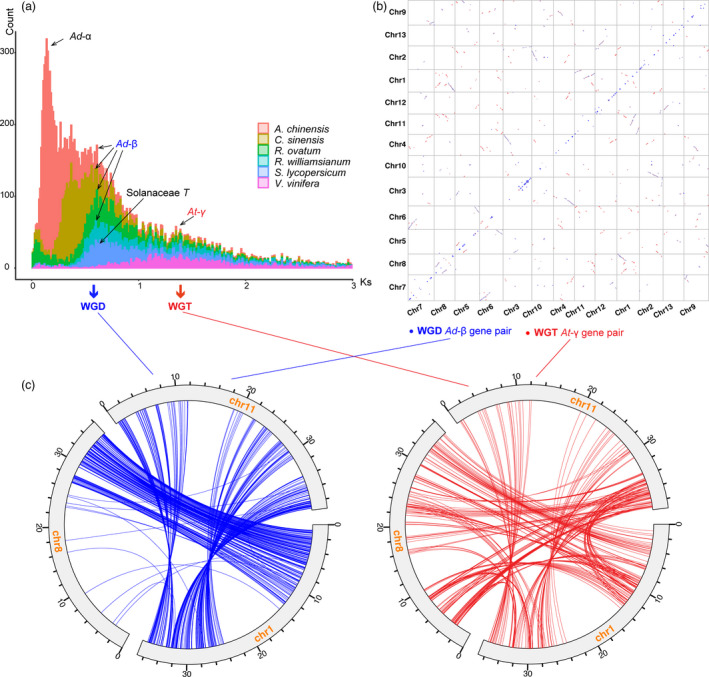
Whole‐genome duplication (WGD) events of R. ovatum. (a) The synonymous substitution rates (Ks) distributions of paralogous genes in A. chinensis, C. sinensis, R. ovatum, and R. williamsianum of the Ericales, and the other two core eudicot plant S. lycopersicum and V. vinifera. (b) Ks dot plot based on Ks value of R. ovatum syntenic gene pairs. (c) Intergenomic synteny blocks among chromosomes 1, 8, and 11 of R. ovatum. The blue and red lines indicate the retained genes that were derived from Ad‐β and At‐γ events, respectively.
To further elucidate the WGD of R. ovatum genome, we performed Ks dot plot (Figure 2b) based on the Ks value of paralogues inherited from Ad‐β (blue dots) and At‐γ (red dots) using TBtools (v 1.074) (Chen et al., 2020). The Ad‐β paralogues shows obvious 1:1 diagonal relationships between Chr1:Chr11, Chr2:Chr10, Chr3:Chr4, Chr6:Chr7, etc. Furthermore, Ks peak boundaries corresponding to Ad‐β (0.20–1.05) and At‐γ (1.05–2.46) were derived from Gaussian mixture modelling (Teh et al., 2017). We illustrated the intra‐genomic synteny blocks among chromosomes 1, 8 and 11 of R. ovatum, and the syntenic gene pairs based on Ks peak boundaries showed the retained genes that were derived from Ad‐β (blue lines, Ks mean = 0.71) and At‐γ (red lines, Ks mean = 1.59) (Figure 2c). In addition, Ks dot plot of retained orthologues of R. ovatum‐V. vinifera (Figure S5) and R. ovatum‐A. chinensis (Figure S6) supported the recent Ad‐β and Ad‐α events, respectively. These data further supported the occurrence of the recent WGD event (Ad‐β) in R. ovatum followed by extensive gene loss.
Phylogenetic placement of R. ovatum
To explore the genome evolution of R. ovatum, genes of eight species from Ericales with tomato were clustered into 28 190 orthologous groups (OGs) (Data S1). Of these, 380 single‐copy orthologues were identified and used to reconstruct a phylogenetic tree (Figure 3a). According to the phylogenetic tree, R. ovatum (subgenus Azaleastrum) diverged from Rhododendron ancestor around 11.85 million years ago (MYA), and R. simsii (subgenus Tsutsusi) diverged from ancestor of R. delavayi and R. williamsianum (subgenus Hymenanthes) 10.59 MYA. The divergence time between the R. delavayi and R. williamsianum was estimated to be about 6.95 MYA. Thus, subgenus Azaleastrum diverged earlier than Tsutsusi, followed by Hymenanthes. However, the evolutionary distance between R. ovatum and Hymenanthes is less than that between R. simsii and Hymenanthes, indicating that R. ovatum, compared with R. simsii, has less genetic variance with Hymenanthes. Moreover, R. ovatum has least genetic variance from Rhododendron ancestor among these four Rhododendron species. These genetic variance relationships are in accord with morphological characteristic differences among Azaleastrum, Tsutsusi and Hymenanthes. Taken together, R. ovatum had early divergence but conservative evolution, which probably make it most similar to the Rhododendron ancestor.
Figure 3.
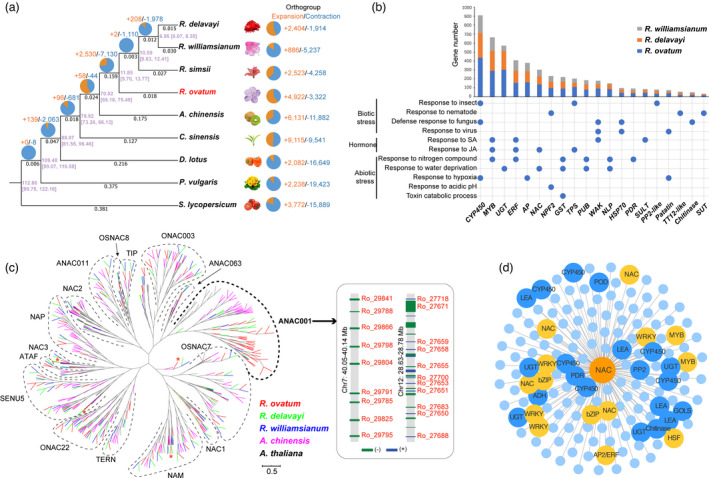
Phylogenetic relationship and comparative genomics analyses. (a) Phylogenetic tree based on single‐copy genes from 8 plant species in the Ericales, and tomato (S. lycopers) was used as the outgroup. The numbers in purple beside each node shows the estimated divergent time of each node (myr), and those in brackets are 95% confidence intervals for the time of divergence between different clades. The pie diagram on each branch of the tree represents the proportion of orthogroups undergoing gain (orange) or loss (blue) events, and the numbers beside the pie diagram denote the total number of expansion and contraction orthogroups. The numbers under each branch indicate evolutionary distance that represent genetic variance. (b) Gene expansions in R. ovatum compared to R. delavayi and R. williamsianum. Left panel shows enriched GO terms of expansive genes related to stress responses. Bottom and up panels show the names and gene numbers of the expansive gene families involved in the GO terms. The dots indicate corresponding relations between GO terms and gene families. The full names of gene families are listed in Data S3. (c) The phylogenetic tree of NAC genes from R. ovatum and representative plants showing the extremely expanded clade of ANAC001 in R. ovatum. Branches are colour‐coded according to species. Asterisks (*) indicate the genes that have undergone positive selection (Ka/Ks > 1). Right penal shows dense tandem arrays of ANAC001 members that are distributed on chromosomes 7 and 12 of R. ovatum. (d) Co‐expression network analysis based on a highly expressed NAC gene member (Ro_40853). Orange circles represent transcription factors and blue circles represent structural proteins.
Expansion of stress‐responsive genes related to low‐altitude adaptability
The four Rhododendron species have various genetic expansion and contraction patterns (Figure 3a). To identify the differential evolution and adaptation between low‐altitude evergreen R. ovatum and the two high‐altitude evergreen rhododendrons, a total of 394 expanded OGs were determined in R. ovatum compared with R. delavayi and R. williamsianum. Gene Ontology (GO) term enrichment analyses of the expanded genes demonstrated that a large number of genes were involved in responses to biotic stresses (insect, nematode, fungus and virus), abiotic stresses (nitrogen compound, water deprivation, hypoxia, acidic pH and toxin) and hormones (salicylic acid and jasmonic acid) (Figure 3b, Data S2). Some of these gene families, such as cytochrome P450 (CYP450), MYB, ethylene‐responsive factor (ERF), NAC and terpene synthase (TPS), are related to more than one GO term, indicating their multiple functions in defence responses. Gene members of the transcription factors MYB, ERF and NAC were identified and used to construct phylogenetic trees to explore their evolutionary significance of lineage‐specific duplications in R. ovatum (Figure 3c, Figures S7 and S8). Most of the expanded genes in the three gene families of R. ovatum show uniformly dispersed individual duplication, except one of the NAC subfamily ANAC001, which was densely expanded with 39 genes in R. ovatum, much higher than that in R. delavayi and R. williamsianum (12 and 4 genes, respectively) (Figure 3c, Figure S9). Chromosome localization of NAC genes demonstrated that most of the members in the ANAC001 clade show tandem repeats on chromosomes 7, 9, 11, 12 and 13 (Figure S10). In particular, 9 and 11 copies are densely distributed on chromosomes 7 and 12 across stretches of 90 kb and 152 kb, respectively (Figure 3c). In addition, 2, 9 and 4 genes experienced positive selection (Ka/Ks>1) of MYB, ERF and NAC, respectively, in R. ovatum compared to either R. delavayi or R. williamsianum (Figure 3c, Figures S7 and S8). Based on the weighted gene co‐expression network analysis (WGCNA) of the transcriptomes (Figure S11), the transcription factors NAC, MYB, WRKY, HSF and bZIP, and the structural proteins CYP450, UGT, PP2, PDR and chitinase were found in the co‐expression network of an NAC member (Ro_40853), which is highly expressed in leaf, stem and flower tissues of R. ovatum (Figure 3d). We revealed that NAC genes are likely critical transcriptional factors in regulating massive stress‐responsive genes involved in extensive adaptability of R. ovatum in the complex environments of low‐altitude areas.
In consideration of temperature as a major factor in low‐altitude environments compared with high altitude, the responses of the expanded gene families mentioned above to medium (37°C) and severe (42°C) heat stress were investigated. Among these gene families, some members of HSP70s and TPSs had elevated transcript abundance after medium or severe heat stress (Figure S12), which indicates their involvement in the temperature response besides the GO‐enriched responses above. In addition, HSFs, which serve as the master transcriptional regulators in response to heat stress, were identified in R. ovatum and compared with the two rhododendrons. Phylogenetic analysis showed that the HSF family of R. ovatum has duplications in subfamilies of A2, A4, A6 and A9. Moreover, HSFA2, A4, A7 and B1 exhibited significant responses to heat stress (Figure S13).
Tandem duplication had critical effects on the expansion of NAC genes. In addition, it has been reported that expanded genes generated by tandem duplication exhibited important roles in responses to various environmental stimuli (Dassanayake et al., 2011; Hanada et al., 2008). Thus, we performed analyses of relationships between tandem duplication and gene functions. First of all, the ratios of tandem duplication in all types of duplication of the four Rhododendron species were investigated, and we found that R. ovatum has a higher duplication ratio (20.21%) compared with that of R. simsii (14.39%), R. delavayi (12.37%) and R. williamsianum (11.96%) (Figure S14A). All the tandem‐duplicated genes of R. ovatum were extracted, and their Pfam domains were annotated and ranked with target gene numbers. The top 20 Pfam domains contained in the hundreds of tandem‐duplicated genes include protein tyrosine kinase (489 genes), leucine‐rich repeat (325), F‐box domain (210), cytochrome P450 (184), UDP‐glucoronosyl and UDP‐glucosyltransferase (182), NB‐ARC domain (178), ankyrin repeats (117) and PPR repeat family (104) (Figure S14B and Data S5).
To deeply understand the roles of tandem duplication in the evolutionary process of R. ovatum, GO annotation and enrichment of all the tandem‐duplicated genes were also performed. For the biological processes, more downstream GO terms were significantly enriched (corrected P‐value < 0.05) compared with upstream GO terms in the hierarchical structure of the network (Figure S15 and Data S6). The most enriched group is related to stimulus responses, including biotic stresses (virus, insect and bacterium), chemical stimulus, singlet oxygen, organic nitrogen, growth hormone and indolebutyric acid. In addition, immune response, glutathione and xyloglucan metabolic processes, toxin catabolic process, terpene and steroid metabolic processes, which were involved in defence responses, were also significantly enriched. Other enrichments are small molecule (organic acid and carboxylic acid) biosynthetic process, auxin transport, pollination and pollen tube growth, regulation and negative regulation of peptidase activity. These results indicated that most functions of the tandem‐duplicated genes are related to defence responses. Furthermore, we investigated the proportions of tandem duplication of the above‐mentioned expansive genes in Figure 3b. The results showed that 15 of 20 gene families exhibited higher tandem duplication ratio than the average level (20.21%) of the whole genome (Figure S16), indicating a primary contribution of tandem duplication for expansion of these stress‐responsive genes.
Flora scent and biosynthesis pathways of the volatiles in R. ovatum
Floral fragrance is one of the most important objectives in evergreen azalea breeding (Kobayashi et al., 2008). However, in the genus Rhododendron, only a few species have flower fragrance, and most of the cultivated azaleas are short of the attractive trait. R. ovatum flowers have pleasant aroma, and the flora volatile organic compounds (VOCs) were analysed using headspace collection combined with GC‐MS (Figure 4a, Table S11). Terpenes and fatty acids are the two main classes of VOCs that were detected. The terpenes include three monoterpenes (3‐carene, linalool and L‐α‐terpineol), one sesquiterpene (α‐farnesene) and one triterpene (squalene), among which α‐farnesene had the highest content. The fatty acids include tetradecanoic acid (14:0), palmitoleic acid (16:1), n‐hexadecanoic acid (16:0), 6‐octadecenoic acid (18:1) and octadecanoic acid (18:0). Other components with lower contents, such as methyl cinnamate, elemicin and isopropyl myristate, were also detected. With the identification of scent compounds from R. ovatum flowers, next we analysed the genes in the respective biosynthetic pathways.
Figure 4.
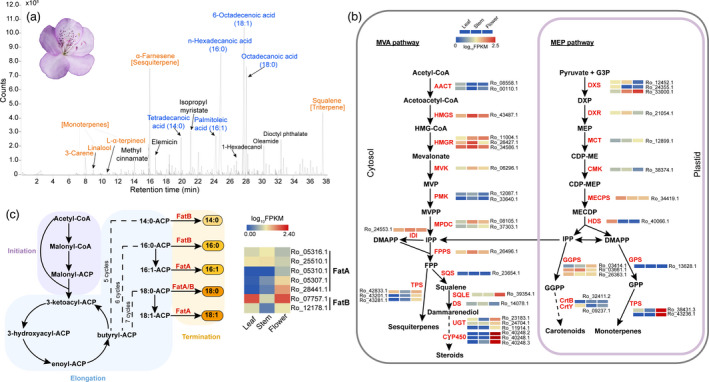
Flora volatiles compounds and biosynthesis pathways. (a) Gas chromatogram of floral volatiles from the flower of R. ovatum. Compounds shown in orange are terpenes and that in blue are fatty acids. (b) Expression profiles of genes encoding enzymes involved in canonical terpenoid biosynthesis pathways. Abbreviations for enzymes in each catalytic step are shown in red and their full names are listed in Data S3. (c) The fatty acid biosynthesis pathway and expression profiles of the genes coding the key enzymes FatA and FatB in leaf, stem, and flower tissues.
In green plants, two canonical pathways are involved in the biosynthesis of terpenes: the mevalonic acid (MVA) pathway in the cytosol for sesquiterpene and triterpene biosynthesis and the methylerythritol phosphate (MEP or non‐mevalonate) pathway in plastids for monoterpene, diterpene and tetraterpene biosynthesis (Chen et al., 2011; Gershenzon and Kreis, 1999). In the present study, the genes involved in both pathways were isolated, and their expression levels were evaluated and compared between the tissues of leaves, stems and flowers (Figure 4b). TPSs are the key enzymes responsible for biosynthesis of terpenoid compounds, which are also involved in significantly expanded genes in R. ovatum compared to the two rhododendrons (Figure 3b). The TPSs (Ro_38431 and Ro_43236) with plastid transit peptides and high gene expression in flowers probably play important roles in monoterpene synthesis in plastids related to flower fragrance and those without plastid transit peptides (Ro_42833, Ro_43201, Ro_43281) probably contribute to sesquiterpene biosynthesis in the cytosol. Key genes encoding the enzymes in the pathway for biosynthesis of steroids, a subclass of terpenoids, are also enriched in the GO terms of expanded genes (Data S2). High expression of squalene monooxygenase (SQLE) and UDP‐glycosyltransferase (UGT) in flowers may facilitate steroid synthesis derived from squalene. It is noteworthy that abundant CYP450 could result in the structural diversity of terpene derivatives, especially triterpenoid saponins (Seki et al., 2015). Thus, given that its three alternative transcripts all exhibited significant expression in flowers, the CYP450 gene member Ro_40248 is probably a key player in the metabolism of triterpenes (Figure 4b, Figure S17).
Fatty acid derivatives in flower VOCs are mainly esters, alcohols, aldehydes and ketones (Knudsen et al., 1993). It is rare to observe free fatty acids (Knudsen et al., 1993), despite their occurrence in abundance in some flowers such as Hydnora Africana (Burger et al., 1988). It is very interesting that five fatty acids, including saturated 14:0, 16:0 and 18:0, and unsaturated 16:1 and 18:1, were detected in the flower VOCs of R. ovatum (Figure 4a). In the fatty acid biosynthesis pathway, fatty acyl‐acyl carrier protein (ACP) thioesterases play an essential role in chain termination during de novo fatty acid synthesis, hence determining the amount and type of free fatty acids that are exported from the plastids for regulating lipid metabolism (Bonaventure et al., 2003; Jones et al., 1995). There are two different classes of fatty acyl‐ACP thioesterases described in plants: FatA and FatB (Salas and Ohlrogge, 2002). FatAs exhibit the highest activity for 18:1‐ACP and much lower activity for saturated acyl‐ACPs, whereas FatBs prefer saturated acyl‐ACP substrates but also have activity for unsaturated acyl groups (Bonaventure et al., 2003; Salas and Ohlrogge, 2002). There are eight FatAs and two FatBs in R. ovatum, which is similar to the two rhododendrons (Figure S18). Of the five expressed FatAs, Ro_05316 and Ro_25510 had higher mRNA levels in leaves and stems compared with flowers, whereas Ro_05310, Ro_05307 and Ro_28441 were mainly expressed in flowers (Figure 4c). The two FatBs had similar expression in leaves and flowers, and the levels were considerably increased compared with that in stems.
TPSs evolution reveals loss of flora fragrance in the other Rhododendron species
The terpene products of TPSs are extremely diverse and constitute the largest class of plant secondary metabolites (Dudareva et al., 2005). Besides their role in floral scents biosynthesis and plant response to abiotic environmental stress, such as heat stress previously mentioned, terpenes are involved in plant defences against various biotic stresses (Pichersky and Gershenzon, 2002). In addition, terpenes may function as chemical signals mediating beneficial interactions between plants and other organisms (Pichersky and Gershenzon, 2002). Therefore, the TPS family plays an important role in plant adaptation. One of the most striking features of the R. ovatum genome is the extremely expanded large number of TPS genes (RoTPS). There are 111 RoTPS genes in R. ovatum genome (Figure S19 and Data S4), which is significantly larger than that of other Rhododendron species [R. simsii (65), R. delavayi (49) and R. williamsianum (43)] without floral scents. Moreover, the TPS number of R. ovatum is also larger than A. chinensis (32), C. sinensis (72), S. lycopersicum (52), wintersweet (52) (Shang et al., 2020) and most of the other plants (Chen et al., 2011). Phylogenetic analyses of TPS from seven species showed that RoTPSs are ascribed to six previously recognized TPS subfamilies, TPS‐a, TPS‐b, TPS‐c, TPS‐e, TPS‐f and TPS‐g and showed 1–3 Rhododendron‐specific clades within the TPS subfamilies (Figure 5a, Figures S20–S26). The majority of RoTPSs were classified into the TPS‐a and TPS‐b subfamilies (Figures S19 and S20). Comparative genomic analysis showed that RoTPSs have been extremely expanded, especially in the TPS‐b subfamily (Figure 5a, Figures S19–S26). Chromosome localization showed that numerous RoTPS genes occur in tandem arrays especially on chromosomes 6 and 7, where 27 (TPS025˜TPS051) and 16 (TPS055˜TPS070) TPS genes are organized as extremely dense gene clusters across stretches of 921 and 378 kb, respectively (Figure S27 and Data S4).
Figure 5.
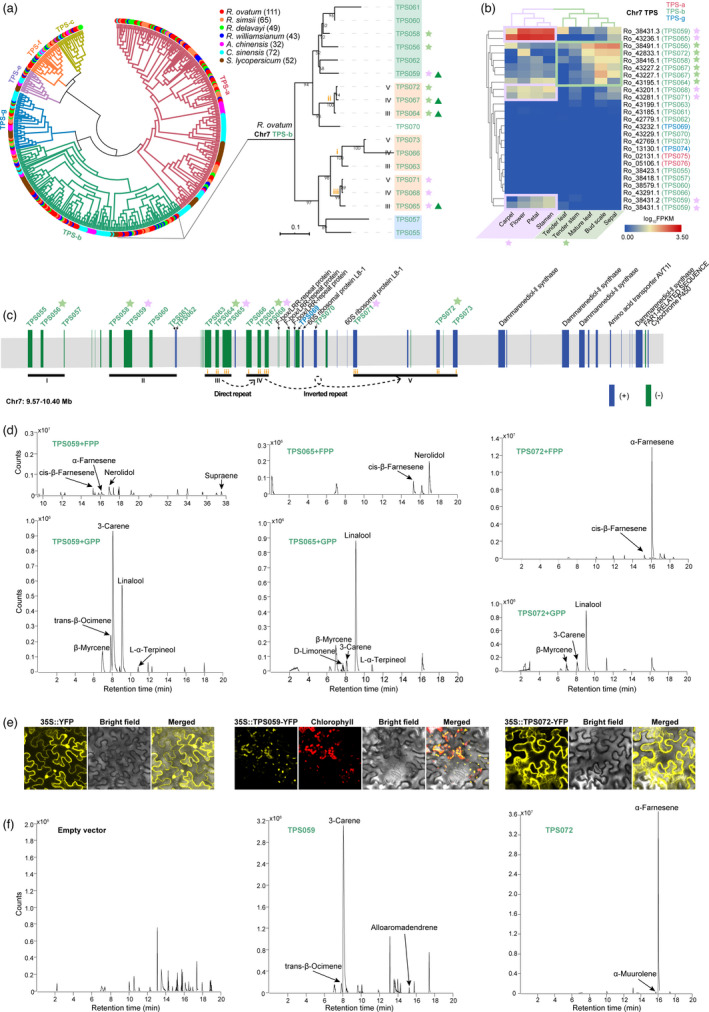
Characterization of terpene synthase‐encoding genes in the R. ovatum genome. (a) Phylogeny of TPSs identified in R. ovatum and 6 other sequenced plant genomes. The numbers after species names indicate TPS gene numbers. Right panel shows details of the TPS‐b clade on Chromosome 7 (Chr7) of R. ovatum. (b) Expression profiles of the TPS transcripts on Chr7. (c) Arrangement and chromosomal position as indicated of the 830‐kb TPS gene cluster and adjacent genes on Chr7. The black lines under the genes indicate tandem repeat modules (I–V). The gene order of the genes in modules III˜V are marked as i–iii. Stars indicate highly expressed genes, with purple indicating the members mainly expressed in flower tissues and green indicating that in vegetative tissues and sepal. Triangles indicate the genes that have plastid transit peptides coding sequence. (d) In vitro enzymatic products of recombinant TPS proteins incubated with farnesyl diphosphate (FPP)/geranyl diphosphate (GPP). (e) Subcellular localization of TPS‐YFP fusion proteins in tobacco mesophyll cells. (f) In vivo enzymatic products of TPS059 and 072 that transiently expressed in tobacco leaves. The volatile terpenes were analysed by GC‐MS analysis.
Expression analysis of the 111 RoTPS genes by RNA‐seq of leaf, stem, bud scale and floral tissues (whole flower, petal, sepal, stamen and carpel) demonstrated that most of the genes had no or low expression, whereas the highly expressed genes had a concentrated distribution on chromosome 7 (Chr7) (Figure S28). Expression profiles of TPS genes in sepal were clustered with that in vegetative green tissues instead of other floral tissues (Figure 5b). There are eighteen TPS‐b, two TPS‐a and two TPS‐g genes on Chr7, and only transcripts of TPS‐b genes were expressed in the tissues, of which some were mainly expressed in the green tissues and some in flora tissues. Among the TPS transcripts, Ro_38431.3 (TPS059) and Ro_43236.1 (TPS065) exhibited high abundance in floral tissues. The other two alternative splicing transcripts (Ro_38431.1 and Ro_38431.2) of TPS059 exhibited much lower expression levels than Ro_38431.3. Sequence alignment of the three transcripts of TPS059 to genomic DNA showed that Ro_38431.3 lacks an exon that encodes 32 and 22 amino acids for Ro_38431.1 and Ro_38431.2, respectively (Figure S29). The TPS‐b of Chr7 were clustered into two major clades in phylogenetic analysis, and only four TPSs (TPS059, 064, 065 and 067) contain predicted plastid transit peptides, indicating that other expressed TPS‐b proteins (TPS058, 056, 071, 068 and 072) probably work in the cytosol (Figure 5a, Figure S30).
The TPS gene clusters located within the 9.57–10.40 Mb region on Chr7 showed more detailed relationships among these genes (Figure 5c). According to the tandem repeat identification, there are five tandem repeat modules (I–V) of the 17 TPS genes with 4 on the plus strand and 13 on the minus strand. TPS055 and TPS057 of module I are sister gene pairs, while TPS056 are in the clade of module II genes (Figure 5a), which indicates that TPS056 and TPS058 may result from a duplication event because of their close relationship and similar expression patterns. TPS genes of module III–V are evenly clustered to three phylogenetic clades (i, ii and iii) with the same gene relationships of the three modules. In combination with the phylogenetic relationship, chromosome location and gene expression profiles, we inferred that module IV duplicated from module III by direct repeat, and module V duplicated from module IV by inverted repeat. The differences in gene structure and expression profiles between the duplicated gene pairs, such as TPS058 and TPS059, TPS065 and TPS068, demonstrate that one of the gene copies may have occurred neofunctionalization. The adjacent genes of the TPS tandem arrays on Chr7 encoding F‐box/LRR‐repeat proteins, 60S ribosomal proteins L8‐1 and Dammarenediol‐II synthases also show the characteristic of tandem repeat, but they had very low or no expression in the samples detected with the exception of one L8‐1 gene (Figure S31). This finding reveals high transcriptional activity for TPSs and low transcriptional activity for the TPS‐adjacent genes in the TPS densely distributed region on Chr7.
To validate whether the representative TPSs that have high expressions are crucial for flower cent biosynthesis, enzyme activities of recombinant TPS059, 065 and 072 were performed in vitro. The three TPSs can catalyse biosynthesis of sesquiterpenes (cis‐β‐farnesene, α‐farnesene and nerolidol) and triterpene (supraene) when with FPP as substrate and of monoterpenes (β‐myrcene, trans‐β‐ocimene, 3‐carene, linalool and L‐α‐terpineol) when with GPP as substrate (Figure 5d). The subcellular localization showed that TPS059 distributes in plastid and TPS072 in the cytosol (Figure 5e). The in vivo expression of TPS059 in tobacco leaves exhibited the main product of 3‐carene, as well as little trans‐β‐ocimene and alloaromadendrene, and TPS072 catalysed the main product of α‐farnesene and little α‐Muurolene (Figure 5f). These results demonstrate that the representative TPSs are crucial for flower cent biosynthesis in R. ovatum.
To investigate whether floral fragrance was gained in R. ovatum after its divergence from other species or floral fragrance was lost in other Rhododendron species, the evolutionary history of TPS‐b, the mainly expressed subfamily related to floral scents, was further analysed. Phylogenetic analyses of TPS‐b from seven species showed that Rhododendron TPS‐b members were ascribed to four groups (b1–4) (Figure 6a). Most of the R. ovatum TPS‐b genes are distributed on chromosomes 2, 7 and 12 as tandem arrays (Figure 6b). The b1 (TPS001 and TPS002, with their sister gene pairs TPS007 and TPS008, respectively) and b3 groups (TPS003˜006, with their sister gene pairs TPS009˜TPS012, respectively) are mainly distributed on Chr2, and gene cluster TPS009˜TPS012 are likely duplicated from TPS001˜TPS006. The b2 and b4 groups are mainly distributed on Chr12 and Chr7, respectively. Collinearity analysis of TPS‐b‐located regions between R. ovatum and R. simsii showed that many chromosomal variations (including insertion, deletion or translocation or inversion) as well as tandem duplication have occurred during their evolutionary history, which resulted in more TPS‐b genes in R. ovatum than R. simsii (Figure 6b).
Figure 6.
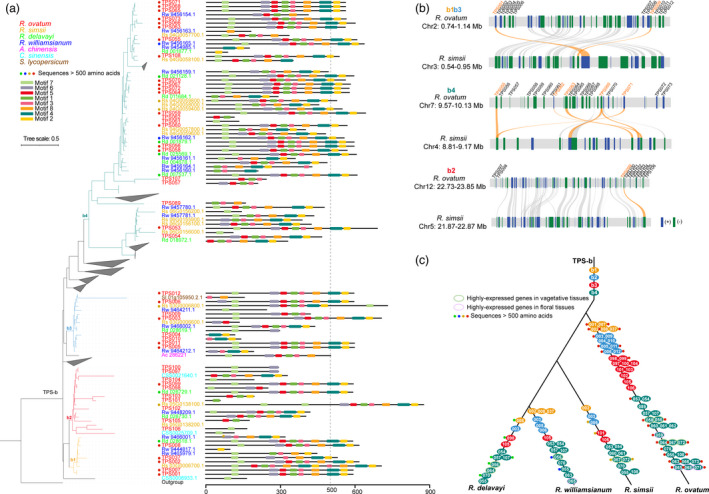
Evolution of TPS‐b genes in Rhododendron species. (a) Phylogeny tree of TPS‐b subfamily identified in R. ovatum and 6 other sequenced plant genomes. The b1˜4 indicate four groups of Rhododendron TPS‐b. Motifs were predicted using MEME tool (http://meme‐suite.org/tools/meme). The length of 500 amino acids was used to define relatively complete sequences. (b) Intergenomic synteny blocks of the TPS‐b‐located chromosome regions of R. ovatum and R. simsii. TPS‐b gene names of R. ovatum were marked above the gene blocks and the lines link the syntenic TPS‐b genes are highlighted in orange. (c) Schematic drawing of the evolution history of TPS‐b aligned with the lineage phylogeny of R. ovatum, R. simsii, R. williamsianum, and R. delavayi. The black lines indicate the lineage phylogeny of the four Rhododendron species. The TPS‐b orthologues are aligned in the branches according to the phylogeny relationships. The complete sequences and the highly expressed genes in vegetative or floral tissues were marked.
As the expressed TPS proteins all have more than 500 amino acids and complete motifs, we use this length to define the relatively complete sequences. R. ovatum has 28 complete TPS‐b sequences, which is much more than that in R. simsii (4), R. williamsianum (2) and R. delavayi (6) (Figure 6a). According to the phylogenetic relationship of TPS‐b in four Rhododendron species, we identified and named orthologous members based on the TPS‐b names of R. ovatum and aligned them with the species lineage phylogeny (Figure 6c). Four Rhododendron species all possess b1–4 groups of TPS‐b subfamily, which indicated the existing of the four TPS‐b groups in Rhododendron ancestor. However, R. ovatum has more members of each group compared with the other three species. For the highly expressed TPS genes in R. ovatum, the other three species have some of the orthologues, such as TPS056, 064, 065, 067 and 072, but only the orthologues that are highly expressed in vegetative tissues have complete sequences. R. williamsianum has the TPS065 orthologues that are highly expressed in R. ovatum floral tissues but with incomplete sequence (Rw 9456154.1). TPS065 of R. ovatum also generated two more copies (TPS068 and 071), and they all have high transcript levels in floral tissues, which may enhance the content or diversity of terpenes. Another highly expressed gene in R. ovatum floral tissues, TPS059, generated earlier than TPS064, 070, 055, 063, 065 and their copies of the other three species (Figure 6a) but still have high sequence similarity with them, which indicates that the other three species may lost TPS059 orthologues. From these results, we inferred that the other three Rhododendron species likely lost floral fragrance in evolutionary history, which has tight relationships with the loss and fragmentization of the essential genes for floral scents biosynthesis.
Discussion
In this study, we present a high‐quality reference genome assembly for the evergreen azalea species R. ovatum, which has important ornamental value and evolutionary position in the genus Rhododendron. The high heterozygosity of 1.55% presents a significant challenge for genome assembly. We applied PacBio sequencing technology and assembled the genome by Falcon using purge_haplotigs to filter redundant sequences from heterozygous genomic regions. The R. ovatum genome has a higher quality of assembly and annotation compared with other sequenced species in Ericales. Thus, the genome assembly of R. ovatum improved the quantity and quality of genome information not only for Rhododendron but also for Ericales. In addition, the genome analyses provide more evidence that the WGD of Ad‐β, which was previously identified in the analysis of the kiwifruit genome (Huang et al., 2013; Shi et al., 2010; Tang et al., 2019; Wu et al., 2019), was shared by some families within Ericales (Zhang et al., 2020a). Importantly, phylogenetic analysis indicated that R. ovatum (subgenus Azaleastrum) diverged from Rhododendron ancestor earlier than R. simsii (subgenus Tsutsusi), R. delavayi and R. williamsianum (subgenus Hymenanthes). However, the genetic variance between R. ovatum and Hymenanthes is less than that between R. simsii and Hymenanthes, which explains why Azaleastrum, compared with Tsutsusi, has more similar morphological characteristics with Hymenanthes. Thus, the early but conservative evolution of R. ovatum probably makes it similar to the Rhododendron ancestor, and its genome will contribute to resolving the origin of Rhododendron.
Habitat heterogeneity was proved to have the highest effects on species diversity and net diversification rate in the genus Rhododendron (Shrestha et al., 2018). The availability of the R. ovatum genome sequence facilitates in‐depth comparative genomic analysis to elucidate the evolution and adaptation divergences between low‐altitude azalea and high‐altitude rhododendron. Numerous studies have reported cold and ultraviolet tolerance of alpine plants (Klatt et al., 2018; Larson et al., 1990; Peng et al., 2011; Zhang et al., 2019a, 2019b), but the adaptation advantages of low‐altitude plants have rarely been investigated. It is commonly considered that high temperatures may be the primary factor of low‐altitude environments compared to high altitudes (Wang et al., 2020; Zhang et al., 2019a). However, a comparison of R. ovatum with R. delavayi and R. williamsianum showed significant expansion of genes related to diverse biotic and abiotic responses, although high‐temperature‐responsive genes (HSP70s and TPSs) are also included. The enriched GO terms, such as response to nematode, nitrogen, water deprivation, hypoxia and acidic pH, provided probable evidence that points to the rhizosphere environment. These findings indicated that low‐altitude azaleas have encountered more complex stresses in addition to higher temperatures compared to alpine rhododendrons. A species living in a specific environment has the tendency of preferential amplification of gene responses to environmental factors. The significantly enriched expansive hormone signals in R. ovatum are SA and JA, which play important roles in various physiological processes, especially in biotic stress responses (Clarke et al., 2000; Kanno et al., 2012). This finding is consistent with the fact that more pathogens are present in low‐altitude environments compared with high‐altitude environments (Zhang et al., 2019a). The transcription factors MYB, ERF and NAC are important regulators in defence responses (Riechmann and Ratcliffe, 2000). One ANAC001 subfamily member of the NAC family in Chrysanthemum lavandulifolium named ClNAC4 was induced by salt, drought, cold or ABA treatment (Huang et al., 2012). The expression of the ANAC001 subfamily member of Chinese cabbage increased under both cold and heat stress (Ma et al., 2014). These reports imply that the significantly expanded ANAC001 subfamily of R. ovatum may have important roles in abiotic stress responses. The expansive genes involved in the stress responses of R. ovatum, we expect, lie the unique solutions to understanding the particular adaptation to its low‐altitude ecological niche.
Terpenoids are the largest class of secondary metabolites in plants (Dudareva et al., 2005). Terpenes can be released from vegetative and floral tissues to attract pollinators or protect plants by attracting predators of attacking herbivores, and they can also serve directly as toxic agents against herbivores or pathogens (Gershenzon and Kreis, 1999; Pichersky and Gershenzon, 2002). Regarding the terpenes in flower VOCs of R. ovatum, three monoterpenes, one sesquiterpene and one triterpene were detected, demonstrating significant diversity in functions. Among the terpenes in R. ovatum, a sesquiterpene (α‐farnesene) exhibited the highest content. In general, sesquiterpene synthases belong to the TPS‐a group (Chen et al., 2011). However, farnesene synthase has been reported to be coded by TPS‐b members in apple (Green et al., 2007) and soybean (Lin et al., 2017). (E)‐β‐farnesene was detected in floral VOCs of water lily but no gene was found in the TPS‐a clade, and a member of TPS‐b, which is highly expressed in flowers, was deduced to be a candidate gene for sesquiterpene synthase (Zhang et al., 2020b). Moreover, two tandem‐duplicated TPS‐b genes TPS02 and TPS03 showed (E)‐β‐ocimene and (E,E)‐α‐farnesene synthase activities in two ecotypes of Arabidopsis, respectively, and their differential subcellular compartmentalization in plastids and the cytosol was responsible for the ecotype‐specific difference (Huang et al., 2010). In addition, it was also found that sesquiterpenes could be catalysed by TPS‐e (Sallaud et al., 2009) or TPS‐g (Aharoni et al., 2004). Overall, in recent years, numerous studies have demonstrated that plants have evolved complex routes for terpene biosynthesis, including new enzymes, alternative substrates and localization (Sun et al., 2016). Hence, the functions or species specificity of TPS may be more complex than previously thought. In the present study, the in vitro enzyme activities showed that TPSs can use both FPP and GPP as substrate to synthesis sesquiterpene and monoterpene, respectively. However, the in vivo enzyme activities showed that plastid‐localized TPS059 mainly catalyse monoterpene synthesis while cytosol‐localized TPS072 mainly catalyse sesquiterpene synthesis. Therefore, our study demonstrated that differential subcellular compartmentalization is the dominant factor for TPSs function, like in above‐mentioned Arabidopsis (Huang et al., 2010). TPS‐b genes without plastid transit peptide coding sequences can participate in sesquiterpene synthesis in the cytosol. Moreover, TPS‐b members expressed not only in flower but also vegetative tissue of R. ovatum, which indicates that they probably play major roles in floral fragrance as well as defence responses.
Flower colour and fragrance are equally important in terms of attracting consumers of ornamentals, and these two traits are essential for the attraction of pollinators and hence for the evolutionary success of plants (Parachnowitsch et al., 2012; Zuker et al., 2002). Large amounts of Rhododendron species have bright and beautiful flowers but no floral fragrance. It is common for the flowering plant that bright coloured flowers have no or less fragrance compared with light coloured flowers, which is a balance for pay (energy distribution on flower colour or fragrance) and gain (attraction of pollinators). The carnation plants with colour modification resulted in lighter colour and more fragrant than control plants, which was due to diversion of metabolic flow from anthocyanin biosynthesis for colour to benzoic acid production for fragrance, both originating from the phenylpropanoid pathway (Zuker et al., 2002). Moreover, the anthocyanin biosynthetic enzyme chalcone isomerase could modulate terpenoid production in glandular trichomes of tomato, although the mechanisms were not clarified (Kang et al., 2014). There may be complementary relationships between flower colour and fragrance of Rhododendron plants, as R. simsii and R. delavayi have bright red flowers without fragrance, whereas R. ovatum has light pinkish purple flowers with delightful aroma, and some white flowers generally exhibit strong fragrance (Cameron, 1993). The less TPS genes of other Rhododendron species may be related to the loss of floral fragrance, and the expansion of the TPS genes in R. ovatum not only promotes the retention of the floral fragrance but also is related to the greater diversity of the floral fragrant compounds.
An important type of duplication common in genome is tandem duplication, where identical copies appear next to each other (Hanada et al., 2008). The tandem duplication occurred not only from single gene but also from gene clusters with mode of direct repeat or inverted repeat. In addition, the gene retention following tandem duplication shows a bias in comparison to segmental and whole‐genome duplication and often exhibits a lineage‐specific fashion (Freeling, 2009), which can be confirmed in the subfamilies a and b of TPS and ANAC001 of NAC in present study. The characteristics of tandem duplication of TPS genes are commonly found in other plants (Chaw et al., 2019; Shang et al., 2020; Xia et al., 2020). However, in this study, tandem duplication has more contribution to the diversification of TPS genes associated with the production of terpenoids for floral scents and defence responses of R. ovatum compared with the other Rhododendron species. In a previous report, the genes responsive to abiotic or biotic stimuli were most multiplied by tandem duplication in the comparison of the extremophile crucifer Thellungiella parvula with Arabidopsis thaliana (Dassanayake et al., 2011). Other studies also supported the notion that expanded genes via tandem duplication tend to be involved in responses to various stresses, which is important for adaptive evolution to dynamically changing environments (Hanada et al., 2008; Myburg et al., 2014). Our analyses of tandem‐duplicated genes showed that most of these genes contain stress‐related domains and are involved in defence‐responsive biological processes. The GO enrichments for tandem‐duplicated genes and the expansive genes of R. ovatum compared with the high‐altitude rhododendrons exhibited high similarity, which indicates that tandem duplication has tight relationships with the genes mediating extensive adaptability of R. ovatum in the complex environments of low‐altitude areas. Despite the same genus undergoing same WGD events, azaleas and rhododendrons have different habitat preferences, which are probably due to the subsequent high rate of tandem duplications of stress‐responsive genes. Therefore, tandem duplication is the primary evolutionary force driving the expansion of genes related to stress responses, especially for the divergent evolution of the closely related species that experienced the same WGD events.
In conclusion, we report a high‐quality chromosome‐level reference genome of the evergreen azalea R. ovatum and provide novel insights into tandem duplication‐facilitated low‐altitude adaptation and flower fragrance. We identified the biological processes and candidate genes associated with biotic and abiotic stress responses, which can be taken into consideration in domestication and breeding of alpine rhododendrons for urban landscaping. In addition, the genome sequence will provide valuable resources for evolutionary research of Rhododendron and Ericales.
Materials and methods
Plant materials and sequencing
An individual plant of R. ovatum growing at the Ornamental Germplasm Resource Nursery of Zhejiang University, Hangzhou, China, was used for reference genome construction. R. ovatum is an azalea species with several excellent ornamental traits, such as evergreen features, graceful architecture, elegant and fragrant flowers, and resistance to complex environmental stresses. Fresh tissues, including mature leaves, tender leaves, tender stems, bud scale, whole flower and different tissues of flower (sepal, petal, stamen and carpel), were harvested and immediately frozen in liquid nitrogen. Samples were stored at −80 °C prior to DNA or RNA extraction.
High‐quality genomic DNA was extracted from the tender leaves of R. ovatum using a modified CTAB method (Porebski et al., 1997). Approximately 20‐kb SMRTbell libraries were constructed for PacBio sequencing on the PacBio Sequel platform. Hi‐C libraries were also constructed from tender leaves and sequenced using the MGISEQ‐2000 system. Samples collected from three tissues (leaves, stems and flowers) were used to construct individual SMRTbell libraries, and the libraries were pooled with equimolar ratios before ISO‐seq. All of the raw sequencing data were filtered using SOAPnuke (v 1.6.5) (Chen et al., 2018) to remove the reads that exhibit low quality, adapter contamination and PCR duplication before subsequent analyses.
Genome assembly and quality assessment
Genome size and heterozygosity were evaluated by k‐mer analysis using Illumina sequence data. The long reads generated from PacBio sequencing were initially assembled into contigs using Falcon/Falcon‐Unzip with parameters ‘length_cutoff = −1, seed_coverage = 40’ (PacBio Assembly Tool Suite) (https://github.com/PacificBiosciences/pb‐assembly) (Chin et al., 2016), and the contigs were self‐corrected using Arrow (https://github.com/PacificBiosciences/GenomicConsensus). The resultant sequences from heterozygous regions were then removed using purge_haplotigs (Roach et al., 2018) (https://bitbucket.org/mroachawri/purge_haplotigs) with parameters ‘‐a 75 ‐m 500’. To increase the accuracy of the assembly, the short reads used in the genome survey were recruited for further polishing with the Pilon program (v1.23) (https://github.com/broadinstitute/pilon). The paired‐end reads from Hi‐C were used to cluster, order and orient the draft contigs onto chromosomes using the ALLHIC (v 0.8.11) pipeline (https://github.com/tangerzhang/ALLHiC) (Zhang et al., 2019b).
Two assessment strategies, BUSCO alignment and transcript alignment, were performed to evaluate the assembly quality of the R. ovatum genome. The Eudicotyledons_odb10 dataset (busco.ezlab.org) was employed to evaluate the completeness of the genome assembly using BUSCO (v 3.1.0) (Simao et al., 2015). The polished transcripts generated from PacBio ISO‐seq were aligned to the genome assembly using Minimap2 (v 2.6) (Li, 2018) to assess the accuracy of the R. ovatum genome. We evaluated the assembly continuity by calculating the LAI score with LTR‐RTs using LTR_retriever (v 2.6) (https://github.com/oushujun/LTR_retriever) (Ou and Jiang, 2018).
Gene prediction and functional annotation
De novo and homologous predictions were integrated to identify repetitive sequences. LTR_Finder (v 1.07) (parameters: ‐D 15000 ‐d 1000 ‐L 7000 ‐l 100 ‐p 20 ‐C ‐M 0.9) and LTRharvest (GenomeTools suite v1.5.9) (parameters: ‐similar 90 ‐vic 10 ‐seed 20 ‐seqids yes ‐minlenltr 100 ‐maxlenltr 7000 ‐mintsd 4 ‐maxtsd 6 ‐motif TGCA ‐motifmis 1) were first used to identify candidate LTR‐RTs, and the outputs were feeded to LTR_retriever (https://github.com/oushujun/LTR_retriever) (Ou and Jiang, 2018) to generate non‐redundant LTR‐RT library. This library was used in de novo prediction to obtain LTR elements. Other repeats were identified using RepeatModeler (v 1.0.11) (https://github.com/Dfam‐consortium/RepeatModeler) (Flynn et al., 2020). The LTR elements and other repeats were merged to the de novo repeats database. Homologous predictions were performed using Repeatmasker (v 4.0.5) (http://www.repeatmasker.org/) (Tarailo‐Graovac and Chen, 2009) based on the de novo repeats database, Repbase and TE protein database. Repetitive sequences in the R. ovatum genome were masked before gene prediction. The ab initio prediction was performed by using the BRAKER2 pipeline (v 2.1.5) (https://github.com/Gaius‐Augustus/BRAKER) (Bruna et al., 2021). In addition, GlimmerHMM and SNAP were used for ab initio gene predictions with gene model parameters trained from R. delavayi and V. corymbosum. We aligned the protein sequences from R. delavayi, V. corymbosum, A. chinensis, P. vulgaris and C. sinensis to the R. ovatum genome using GeMoMa (v 1.4.2) (http://www.repeatmasker.org/) (Keilwagen et al., 2019) in homologue predictions. For transcriptome‐based predictions, the full‐length transcripts obtained by PacBio sequencing were aligned to the assembled genome by PASA (https://github.com/PASApipeline/PASApipeline) (Haas et al., 2008). Finally, all evidence from ab initio, homology‐based and RNA‐seq‐based predictions were combined into the consensus gene sets using the EVidenceModeler (v1.1.1) (http://evidencemodeler.github.io/) (Haas et al., 2008) and then optimized with PASA (Haas et al., 2008) to obtain the integrated gene model.
Functional annotation of protein‐coding genes was performed using BLASTP (E‐value cut‐off 1e−5) homologue search against the NCBI non‐redundant (Nr) protein database (2020‐7‐10) and Swiss‐Prot database (2020‐8‐12). InterProScan package (v5.29‐68.0) was used to run the scanning algorithms from the InterPro database to perform a comprehensive annotation including TIGRFAM, Phobius, SignalP, SUPERFAMILY, PANTHER, Gene3D, ProSite, Coils, PRINTS, SMART, Pfam, PIRSF, TMHMM and GO. eggNOG‐mapper (v1.0.3, https://github.com/eggnogdb/eggnog‐mapper) was used for functional annotation based on fast orthology assignments using precomputed clusters and phylogenies from the eggNOG database. GO and KO (KEGG Orthology) information was retrieved from InterPro and eggNOG annotation. In addition, KAAS (KEGG Automatic Annotation Server) (https://www.genome.jp/tools/kaas/) was used to provide functional annotation of genes by BLAST against the KEGG GENES database to obtain KO assignments and automatically generated KEGG pathways.
Construction of species phylogenetic tree
Protein sequences of R. ovatum, R. simsii, R. delavayi, R. williamsianum, Actinidia chinensis, Camellia sinensis, Diospyros lotus, Primula vulgaris and Solanum lycopersicum were collected for the construction of the species phylogenetic tree. Orthofinder (v 2.4.0) (Emms and Kelly, 2015), an integrated tool for gene family clustering, was adopted for the analysis of gene families. The protein datasets for the chosen species were clustered into orthologous groups and paralogous groups at first, and then, genes of single‐copy orthologous groups were selected for the alignment and construction of the species phylogenetic tree. After the single‐copy orthologues were obtained, the longest protein sequences of each single‐copy gene were extracted according to the Orthofinder results. ClustalO (v 1.2.4) (Madeira et al., 2019) was used for the multiple sequence alignment (MSA) of protein sequences, and then trimAl (v 1.2rev59) (Capella‐Gutierrez et al., 2009) was adopted for the trimming of MSA results. We chose RAxML (v 8.2.12) (Stamatakis, 2014) finally for the construction of a species tree exhibiting the phylogenetic relationship of selected species using trimmed MSA results with the concatenation method.
Divergence time estimation and gene family expansion
After construction of the species phylogenetic tree, the calibration time of divergence of these species was obtained from TimeTree (http://www.timetree.org/) (Hedges et al., 2015), a database for calibration time among different species supported by integrated studies, as the benchmark of the following analysis. Single‐copy orthologous genes are required for the analysis of fourfold degenerate sites. We applied MCMCtree, a tool comprised in the PAML software package (Yang, 2007), for the construction of the ultrametric tree, which contains not only phylogenetic relationships but also 95% confidence intervals of divergence time. The CAFE (v 4.2.1) (Han et al., 2013) workflow with a probabilistic graphical model was then chosen for the analysis of gene family expansion and contraction with P‐value of 0.05.
Polyploidization events analysis
The longest protein sequences of each gene within the genomes of R. ovatum, A. chinensis, C. sinensis, D. lotus, R. delavayi, R. williamsianum, S. lycopersicum and V. vinifera were selected for synteny and collinearity detection within themselves using mcscan, a tool contained in JCVI (Goll et al., 2010), to shed light on polyploidization events. PAML (v 4.9i) (Yang, 2007) was chosen to compute the Ks value between gene pairs of synteny gene blocks. For R. ovatum, we modelled the distribution of Ks rates as a mixture model and identified syntenic gene pairs falling within the Ks pea ± 1 SD as paralogs likely derived from the Ad‐β and At‐γ event (Teh et al., 2017). Ks Dot plots of orthologues between genomes and intergenomic synteny blocks were visualized using TBtools (v 1.074) (Chen et al., 2020). Types of gene duplication and tandem repeats were analysed using MCScanX (Wang et al., 2012) and TBtools (v 1.045) (Chen et al., 2020).
RNA sequencing and gene expression analysis
All tissue samples mentioned above and leaves with different temperature treatments (25, 37 and 42 °C) were used to perform RNA sequencing on the Illumina nova‐seq 6000 platform. After obtaining reads data of transcriptomes, Trimmomatic (v 0.39) (Bolger et al., 2014) was used for the trimming of the adapter sequences. Then, HISAT2 (v 2.2.1) (Wen, 2017) was chosen for the mapping of trimmed sequences to genome assemblies, followed by the analysis for FPKM (Fragments Per Kilobase per Million) using StringTie (v 2.1.4) (Pertea et al., 2015). Gene co‐expression networks were analysed by weighted correlation network analysis (WGCNA) (Langfelder and Horvath, 2008) and visualized by using Cytoscape (v 3.8.0) (Smoot et al., 2011). Gene expression profiles of different tissues were presented as heatmaps using TBtools (v 1.045) (Chen et al., 2020).
Determination of floral volatiles by GC‐MS analysis
The floral volatiles of R. ovatum were detected using a headspace solid‐phase microextraction system combined with gas chromatography–mass spectrometry (GC–MS). One gram of fully opened flower petals (approximately five flowers) was detached and immediately put into sample vials, and the volatiles were extracted with solid‐phase microextraction (SPME, Supelco) for 30 min in a water bath at 50 °C. Then, the SPME was immediately transferred to the injection port (250 °C) of the GC‐MS system (Agilent 7890B‐7000C) to be desorbed for 5 min. Separation was performed on an HP‐5MS capillary column (30 m × 0.25 mm) with helium as the carrier at a flow rate of 1 mL/min under splitless injection conditions. The temperature programming started from an initial oven temperature at 50 °C (2‐min hold) and a temperature gradient of 5 °C per min increase to 180 °C (5‐min hold) followed by a temperature gradient of 10 °C per min increase to 250 °C (8‐min hold). Other settings included electron impact ionization (EI) at 70 eV, a 230 °C ion source temperature, and a 280 °C interface temperature. The mass spectrum was analysed in the range of 20–350 atom mass units. National Institute of Standards and Technology mass spectral database (NIST17.L) was used to identify the mass spectra of the compounds.
Enzyme activities and subcellular localization of TPSs
For in vitro enzyme activity characterization, coding sequences of TPS059, 065 and 072 were amplified from cDNA of flower tissue of R. ovatum and cloned into the prokaryotic expression vector pCold TF containing a His tag and expressed in the Escherichia coli strain Rosetta (DE3). The recombinant His‐TPSs proteins were induced by 0.3mM IPTG at 16 °C overnight and purified using the ProteinIso® Ni‐NTA Resin (Transgen, Beijing, China) according to the manufacturer’s instructions. Assays for TPS enzyme activity were performed in a 1 mL assay buffer (30 mm HEPES, pH 7.5, 5 mm DTT, 25 mm MgCl2) containing 10 μg purified TPS proteins and 60 μμ GPP/FPP (Shang et al., 2020). The mixture was incubated at 30 °C for 1 h and then 45 °C for 15 min before the synthesized volatiles were collected and analysed using the same methods with flower scents measurement.
For subcellular localization and enzyme activity characterization, coding sequences of TPS059 and 072 were cloned into pHB vector to obtained constructs of 35S::TPSs‐YFP. Then, the plasmids were transformed into Agrobacterium tumefaciens strain GV3101 and injected into tobacco (Nicotiana benthamiana) leaves. After 2 days of incubation, the injected parts were sampled for subcellular localization observation by laser scanning confocal microscope and for volatile detection by headspace solid‐phase microextraction with GC‐MS.
Gene family identification and characterization
HMM search (Finn et al., 2011) and BLASTp (Altschul et al., 1990) were integrated to identify gene family members. The predicted proteins of the R. ovatum genome were screened by HMM search (Finn et al., 2011) with Pfam motifs (http://pfam.xfam.org) of the target gene family. The hits with E‐values greater than 1e‐5 were individually evaluated. In addition, gene homologs were obtained by running a local BLASTp search (Altschul et al., 1990) using previously characterized proteins from Swiss‐Prot as queries against all protein sequences with an E‐value cut‐off of 1e‐5. The obtained sequences from the two methods were integrated and manually checked to correct erroneous automatic annotation. Multiple sequence alignment was performed by MAFFT (v 7.467) (Katoh and Standley, 2013) with default parameters, and the maximum likelihood tree was constructed using FastTree (v 2.1.11) (Price et al., 2009). Tree visualization and labelling were performed on MAGA (v 7.0.26) (Kumar et al., 2016) or iTOL (https://itol.embl.de/) (Letunic and Bork, 2019). Plastid transit peptide was predicted using ChloroP 1.1 Server (Emanuelsson et al., 1999). Chromosome localization and gene structure were visualized using TBtools (v 1.045) (Chen et al., 2020).
Tandem duplication analysis
The genes generated by tandem duplication were identified using MCScanX (Wang et al., 2012) inserted in TBtools (v 1.045) (Chen et al., 2020). Tandem duplication ratio was calculated as number of genes generated by tandem duplication/total number of genes. Pfam domains were annotated using HMMER (v 3.1b2) (http://hmmer.org/) (Finn et al., 2011) search against Pfam database (http://pfam.xfam.org/). The file of GO annotation for network analysis was prepared using TBtools (v 1.045) (Chen et al., 2020) and visualized using BiNGO in Cytoscape (v 3.8.0) (Smoot et al., 2011).
Competing interests
The authors declare that they have no competing interests.
Author contributions
Y.X., L.Z., and C.Z. conceived, designed the research, and managed the project. X.Y.W., H.Z., Z.L., B.L., and J.W. contributed to sample preparation and sequencing. X.P.W. and Y.G. performed the assembly and annotation. X.Y.W. and Y.G. analysed data and wrote the manuscript. X.H.W. and D.L. worked on WGCNA analyses. Fei C., Feng C., X.P.W., C.Z., L.Z., and Y.X. revised the manuscript. All authors read and approved the manuscript.
Data availability statement
The raw data files of genome sequencing and RNA sequencing have been deposited at Sequence Read Archive (SRA) under the accession PRJNA671625. This whole‐genome project also has been deposited at DDBJ/ENA/GenBank under the accession JADHZH000000000. Genome assembly and gene annotations could be downloaded from Rhododendron Plant Genome Database (http://bioinfor.kib.ac.cn/RPGD/download_genome.html).
Supporting information
Figure S1 Evaluation of Rhododendron ovatum genome by k‐mer analysis and read‐depth.
Figure S2 Genome‐wide analysis of chromatin interactions in the R. ovatum genome based on Hi‐C data.
Figure S3 Genome assemblies of R. ovatum (ov) and R. williamsianum (wi).
Figure S4 Types of gene duplication in the R. ovatum genome.
Figure S5 Ks dot plot between R. ovatum and grape (V. vinifera).
Figure S6 Ks dot plot between R. ovatum and kiwifruit (A. chinensis).
Figure S7 Phylogenetic tree of ERF gene family.
Figure S8 Phylogenetic tree of MYB gene family.
Figure S9 Phylogenetic tree of ANAC001 clade of NAC gene family.
Figure S10 Chromosome localization of NAC genes of R. ovatum.
Figure S11 Weighted gene co‐expression network analysis (WGCNA) of the transcriptomes.
Figure S12 Expression profiles of HSP70s and TPSs in response to different temperature treatments (25, 37, and 42 °C).
Figure S13 Phylogenetic analysis and heat‐induced expression profiles of HSFs in R. ovatum.
Figure S14 Characteristics of tandem duplication in R. ovatum genome.
Figure S15 GO functional enrichment network of tandem‐duplicated genes.
Figure S16 Tandem duplication ratio of the stress responsive genes.
Figure S17 Expression profiles of CYP450s in different tissues.
Figure S18 Phylogenetic tree of fatty acyl‐ACP thioesterases coding genes.
Figure S19 Statistic of TPS gene subfamilies.
Figure S20 Phylogenetic tree of TPSs in R. ovatum.
Figure S21 Phylogeny of the TPS‐a subfamily.
Figure S22 Phylogeny of the TPS‐b subfamily.
Figure S23 Phylogeny of the TPS‐c subfamily.
Figure S24 Phylogeny of the TPS‐e subfamily.
Figure S25 Phylogeny of the TPS‐f subfamily.
Figure S26 Phylogeny of the TPS‐g subfamily.
Figure S27 Chromosome localization of RoTPS genes.
Figure S28 Expression profiles of TPS genes of R. ovatum.
Figure S29 Structure features of the three alternative splicing transcripts of Ro_38431.
Figure S30 Amino sequence alignment of Ro_38431.3 and Ro_43236.1.
Figure S31 Expression profiles of adjacent genes of TPS clusters on chromosome 7.
Table S1 Statistics for Illumina sequencing data of R. ovatum genome.
Table S2 Statistics for PacBio sequencing data of R. ovatum genome.
Table S3 Summary of contig‐level genome assembly for R. ovatum.
Table S4 BUSCO evaluation of the genomic completeness before and after elimination of redundancy.
Table S5 Statistics for Hi‐C sequencing data of R. ovatum genome.
Table S6 Statistics for PacBio Iso‐seq data of R. ovatum.
Table S7 Gene prediction of R. ovatum genome.
Table S8 Summary of repeat annotation of R. ovatum.
Table S9 BUSCO evaluation of the genomic completeness of R. ovatum.
Table S10 BUSCO evaluation of the gene prediction of R. ovatum.
Table S11 Main flora volatiles compounds of R. ovatum.
Data S1 Orthologous groups identified in these species.
Data S2 Biological_process GO enrichment of expansive genes in R. ovatum compared to R. delavayi and R. williamsianum.
Data S3 Full name list of the gene abbreviations.
Data S4 TPS gene family of R. ovatum.
Data S5 Pfam annotation of tandem‐duplicated genes in R. ovatum.
Data S6 Biological_process GO enrichment of tandem‐duplicated genes in R. ovatum.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 31901356 and 31800597), the Fundamental Research Funds for the Central Universities (grant number 2021QNA6021), and Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (grant number 2016C02056‐12).
Wang, X. , Gao, Y. , Wu, X. , Wen, X. , Li, D. , Zhou, H. , Li, Z. , Liu, B. , Wei, J. , Chen, F. , Chen, F. , Zhang, C. , Zhang, L. and Xia, Y. (2021) High‐quality evergreen azalea genome reveals tandem duplication‐facilitated low‐altitude adaptability and floral scent evolution. Plant Biotechnol. J., 10.1111/pbi.13680
Contributor Information
Chengjun Zhang, Email: zhangchengjun@mail.kib.ac.cn.
Liangsheng Zhang, Email: zls83@zju.edu.cn.
Yiping Xia, Email: ypxia@zju.edu.cn.
References
- Aharoni, A. , Giri, A.P. , Verstappen, F.W.A. , Bertea, C.M. , Sevenier, R. , Sun, Z.K. , Jongsma, M.A. et al. (2004) Gain and loss of fruit flavor compounds produced by wild and cultivated strawberry species. Plant Cell, 16, 3110–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi, T. , Shirasawa, K. , Nagasaki, H. , Hirakawa, H. , Tao, R. , Comai, L. and Henry, I.M. (2020) The persimmon genome reveals clues to the evolution of a lineage‐specific sex determination system in plants. Plos Genet. 16, e1008566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Badouin, H. , Gouzy, J. , Grassa, C.J. , Murat, F. , Staton, S.E. , Cottret, L. , Lelandais‐Briere, C. et al. (2017) The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature, 546, 148–152. [DOI] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventure, G. , Salas, J.J. , Pollard, M.R. and Ohlrogge, J.B. (2003) Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell, 15, 1020–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna, T. , Hoff, K. , Lomsadze, A. , Stanke, M. and Borodovsky, M. (2021) BRAKER2: automatic eukaryotic genome annotation with GeneMark‐EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinform. 3, lqaa108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger, B.V. , Munro, Z.M. and Visser, J.H. (1988) Determination of plant volatiles.1. Analysis of the insect‐attracting allomone of the parasitic plant Hydnora‐africana using grob‐habich activated‐charcoal traps. J. High Resolut. Chromatogr. Chromatogr. Commun. 11, 496–499. [Google Scholar]
- Cameron, P. (1993) Fragrance in Rhododendron species. J. Am. Rhodo. Soc. 47, 128–130. [Google Scholar]
- Capella‐Gutierrez, S. , Silla‐Martinez, J.M. and Gabaldon, T. (2009) TrimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain, D.F. , Hyam, G. , Argent, G. , Fairweather, G. and Walter, K. (1996) The Genus Rhododendron: Its Classification and Synonymy. Edinburgh: Royal Botanical Garden. [Google Scholar]
- Chaw, S.M. , Liu, Y.C. , Wu, Y.W. , Wang, H.Y. , Lin, C.Y.I. , Wu, C.S. , Ke, H.M. et al. (2019) Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants, 5, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.J. , Chen, H. , Zhang, Y. , Thomas, H.R. , Frank, M.H. , He, Y.H. and Xia, R. (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant, 13, 1194–1202. [DOI] [PubMed] [Google Scholar]
- Chen, F. , Tholl, D. , Bohlmann, J. and Pichersky, E. (2011) The family of terpene synthases in plants: a mid‐size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Chen, Y. , Shi, C. , Huang, Z. , Zhang, Y. , Li, S. , Li, Y. et al. (2018) SOAPnuke: a MapReduce acceleration‐supported software for integrated quality control and preprocessing of high‐throughput sequencing data. Gigascience, 7, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.S. , Peluso, P. , Sedlazeck, F.J. , Nattestad, M. , Concepcion, G.T. , Clum, A. , Dunn, C. et al. (2016) Phased diploid genome assembly with single‐molecule real‐time sequencing. Nat. Methods, 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, J.D. , Volko, S.M. , Ledford, H. , Ausubel, F.M. and Dong, X.N. (2000) Roles of salicylic acid, jasmonic acid, and ethylene in cpr‐induced resistance in Arabidopsis. Plant Cell, 12, 2175–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocker, J.M. , Wright, J. , Li, J.H. , Swarbreck, D. , Dyer, S. , Caccamo, M. and Gilmartin, P.M. (2018) Primula vulgaris (primrose) genome assembly, annotation and gene expression, with comparative genomics on the heterostyly supergene. Sci. Rep. 8, 17942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colle, M. , Leisner, C.P. , Wai, C.M. , Ou, S. , Bird, K.A. , Wang, J. , Wisecaver, J.H. et al. (2019) Haplotype‐phased genome and evolution of phytonutrient pathways of tetraploid blueberry. Gigascience, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassanayake, M. , Oh, D.H. , Haas, J.S. , Hernandez, A. , Hong, H. , Ali, S. , Yun, D.J. et al. (2011) The genome of the extremophile crucifer Thellungiella parvula . Nat. Genet. 43, 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Riek, J. , De Keyser, E. , Calsyn, E. , Eeckhaut, T. , Van Huylenbroeck, J. and Kobayashi, N. (2018) Azalea. In Ornamental Crops, ( Van Huylenbroeck, J. , ed.), pp. 237–271. Switzerland: Springer. [Google Scholar]
- Dudareva, N. , Andersson, S. , Orlova, I. , Gatto, N. , Reichelt, M. , Rhodes, D. , Boland, W. et al. (2005) The nonmevalonate pathway supports both monoterpene and sesquiterpene formation in snapdragon flowers. Proc. Natl. Acad. Sci. USA, 102, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson, O. , Nielsen, H. and Heijne, G.V. (1999) ChloroP, a neural network‐based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 8, 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D.M. and Kelly, S. (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Clements, J. and Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, J.M. , Hubley, R. , Goubert, C. , Rosen, J. , Clark, A.G. , Feschotte, C. and Smit, A.F. (2020) RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA, 117, 9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling, M. (2009) Bias in plant gene content following different sorts of duplication: tandem, whole‐genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453. [DOI] [PubMed] [Google Scholar]
- Gershenzon, J. and Kreis, W. (1999) Biochemistry of terpenoids: monoterpenes, sesquiterpenes, diterpenes, sterols, cardiac glycosides and steroid saponins. In Biochemistry of Plant Secondary Metabolism, ( Wink, M. , ed.), pp. 222–299. Sheffield: Sheffield Academic Press. [Google Scholar]
- Goll, J. , Rusch, D.B. , Tanenbaum, D.M. , Thiagarajan, M. , Li, K. , Methe, B.A. and Yooseph, S. (2010) METAREP: JCVI metagenomics reports‐an open source tool for high‐performance comparative metagenomics. Bioinformatics, 26, 2631–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, S. , Friel, E.N. , Matich, A. , Beuning, L.L. , Cooney, J.M. , Rowan, D.D. and MacRae, E. (2007) Unusual features of a recombinant apple alpha‐farnesene synthase. Phytochemistry, 68, 176–188. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, M.V. , Thomas, G.W.C. , Lugo‐Martinez, J. and Hahn, M.W. (2013) Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. [DOI] [PubMed] [Google Scholar]
- Hanada, K. , Zou, C. , Lehti‐Shiu, M.D. , Shinozaki, K. and Shiu, S.H. (2008) Importance of lineage‐specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges, S.B. , Marin, J. , Suleski, M. , Paymer, M. and Kumar, S. (2015) Tree of life reveals clock‐like speciation and diversification. Mol. Biol. Evol. 32, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H. , Wang, Y. , Wang, S.L. , Wu, X. , Yang, K. , Niu, Y.J. and Dai, S.L. (2012) Transcriptome‐wide survey and expression analysis of stress‐responsive NAC genes in Chrysanthemum lavandulifolium . Plant Sci. 193, 18–27. [DOI] [PubMed] [Google Scholar]
- Huang, M.S. , Abel, C. , Sohrabi, R. , Petri, J. , Haupt, I. , Cosimano, J. , Gershenzon, J. et al. (2010) Variation of herbivore‐induced volatile terpenes among Arabidopsis Ecotypes depends on allelic differences and subcellular targeting of two terpene synthases, TPS02 and TPS03. Plant Physiol. 153, 1293–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S.X. , Ding, J. , Deng, D.J. , Tang, W. , Sun, H.H. , Liu, D.Y. , Zhang, L. et al. (2013) Draft genome of the kiwifruit Actinidia chinensis . Nat. Commun. 4, 2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, A. , Davies, H.M. and Voelker, T.A. (1995) Palmitoyl‐acyl carrier protein (ACP) thioesterase and the evolutionary origin of plant acyl‐ACP thioesterases. Plant Cell, 7, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, J.H. , McRoberts, J. , Shi, F. , Moreno, J.E. , Jones, A.D. and Howe, G.A. (2014) The flavonoid biosynthetic enzyme chalcone isomerase modulates terpenoid production in glandular trichomes of tomato. Plant Physiol. 164, 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno, H. , Hasegawa, M. and Kodama, O. (2012) Accumulation of salicylic acid, jasmonic acid and phytoalexins in rice, Oryza sativa, infested by the white‐backed planthopper, Sogatella furcifera (Hemiptera: Delphacidae). Appl. Entomol. Zool. 47, 27–34. [Google Scholar]
- Katoh, K. and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen, J. , Hartung, F. and Grau, J. (2019) GeMoMa: homology‐based gene prediction utilizing intron position conservation and RNA‐seq data. Methods Mol. Biol. 1962, 161–177. [DOI] [PubMed] [Google Scholar]
- Klatt, S. , Schinkel, C.C.F. , Kirchheimer, B. , Dullinger, S. and Horandl, E. (2018) Effects of cold treatments on fitness and mode of reproduction in the diploid and polyploid alpine plant Ranunculus kuepferi (Ranunculaceae). Ann. Bot. 121, 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, J.T. , Eriksson, R. , Gershenzon, J. and Stahl, B. (2006) Diversity and distribution of floral scent. Bot. Rev. 72, 1–120. [Google Scholar]
- Knudsen, J.T. , Tollsten, L. and Bergstrom, L.G. (1993) Floral scents‐a checklist of volatile compounds isolated by head‐space techniques. Phytochemistry, 33, 253–280. [Google Scholar]
- Kobayashi, N. , Mizuta, D. , Nakatsuka, A. and Akabane, M. (2008) Attaining inter‐subgeneric hybrids in fragrant azalea breeding and the inheritance of organelle DNA. Euphytica, 159, 67–72. [Google Scholar]
- Kumar, S. , Stecher, G. and Tamura, K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, R.A. , Garrison, W.J. and Carlson, R.W. (1990) Differential responses of alpine and non‐alpine Aquilegia species to increased ultraviolet‐B radiation. Plant Cell Environ. 13, 983–987. [Google Scholar]
- Letunic, I. and Bork, P. (2019) Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J. , Wang, D. , Chen, X. , Köllner, T.G. , Mazarei, M. , Guo, H. , Pantalone, V.R. et al. (2017) An (E, E)‐α‐farnesene synthase gene of soybean has a role in defence against nematodes and is involved in synthesizing insect‐induced volatiles. Plant Biotech. J. 15, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. , Wang, F. , Li, M.Y. , Jiang, Q. , Tan, G.F. and Xiong, A.S. (2014) Genome wide analysis of the NAC transcription factor family in Chinese cabbage to elucidate responses to temperature stress. Sci. Hortic. 165, 82–90. [Google Scholar]
- Madeira, F. , Park, Y.M. , Lee, J. , Buso, N. , Gur, T. , Madhusoodanan, N. , Basutkar, P. et al. (2019) The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming, T.L. and Fang, R.Z. (1990) The phylogeny and evolution of genus Rhododendron . Acta Bot. Yunnanica, 12, 353–365. [Google Scholar]
- Moriyama, Y. and Koshiba‐Takeuchi, K. (2018) Significance of whole‐genome duplications on the emergence of evolutionary novelties. Brief. Funct. Genomics, 17, 329–338. [DOI] [PubMed] [Google Scholar]
- Myburg, A.A. , Grattapaglia, D. , Tuskan, G.A. , Hellsten, U. , Hayes, R.D. , Grimwood, J. , Jenkins, J. et al. (2014) The genome of Eucalyptus grandis . Nature, 510, 356–362. [DOI] [PubMed] [Google Scholar]
- Norton, C.R. and Norton, M.E. (1989) Rhododendrons. In Trees II ( Bajaj, Y.P.S. , ed.), pp. 428–451. Berlin, Heidelberg: Springer Berlin Heidelberg. [Google Scholar]
- Ou, S. and Jiang, N. (2018) LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parachnowitsch, A.L. , Raguso, R.A. and Kessler, A. (2012) Phenotypic selection to increase floral scent emission, but not flower size or colour in bee‐pollinated Penstemon digitalis . New Phytol. 195, 667–675. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Yang, Z.H. , Zhang, H. , Cui, C.Y. , Qi, X.B. , Luo, X.J. , Tao, X.A. et al. (2011) Genetic variations in Tibetan populations and high‐altitude adaptation at the Himalayas. Mol. Biol. Evol. 28, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G.M. , Antonescu, C.M. , Chang, T.C. , Mendell, J.T. and Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichersky, E. and Gershenzon, J. (2002) The formation and function of plant volatiles: perfumes for pollinator attraction and defense. Curr. Opin. Plant Biol. 5, 237–243. [DOI] [PubMed] [Google Scholar]
- Porebski, S. , Bailey, L.G. and Baum, B.R. (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. [Google Scholar]
- Price, M.N. , Dehal, P.S. and Arkin, A.P. (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raguso, R.A. (2008) Wake up and smell the roses: The ecology and evolution of floral scent. Annu. Rev. Ecol. Evol. Syst. 39, 549–569. [Google Scholar]
- Riechmann, J.L. and Ratcliffe, O.J. (2000) A genomic perspective on plant transcription factors. Curr. Opin. Plant Biol. 3, 423–434. [DOI] [PubMed] [Google Scholar]
- Roach, M.J. , Schmidt, S.A. and Borneman, A.R. (2018) Purge Haplotigs: allelic contig reassignment for third‐gen diploid genome assemblies. BMC Bioinform. 19, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas, J.J. and Ohlrogge, J.B. (2002) Characterization of substrate specificity of plant FatA and FatB acyl‐ACP thioesterases. Arch. Biochem. Biophys. 403, 25–34. [DOI] [PubMed] [Google Scholar]
- Sallaud, C. , Rontein, D. , Onillon, S. , Jabes, F. , Duffe, P. , Giacalone, C. , Thoraval, S. et al. (2009) A novel pathway for sesquiterpene biosynthesis from Z, Z‐farnesyl pyrophosphate in the wild tomato Solanum habrochaites. Plant Cell, 21, 301–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki, H. , Tamura, K. and Muranaka, T. (2015) P450s and UGTs: key players in the structural diversity of triterpenoid saponins. Plant Cell Physiol. 56, 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J.Z. , Tian, J.P. , Cheng, H.H. , Yan, Q.M. , Li, L. , Jamal, A. , Xu, Z.P. et al. (2020) The chromosome‐level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 21, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, T. , Huang, H.W. and Barker, M.S. (2010) Ancient genome duplications during the evolution of kiwifruit (Actinidia) and related Ericales. Ann. Bot. 106, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha, N. , Wang, Z.H. , Su, X.Y. , Xu, X.T. , Lyu, L.S. , Liu, Y.P. , Dimitrov, D. et al. (2018) Global patterns of Rhododendron diversity: the role of evolutionary time and diversification rates. Glob. Ecol. Biogeogr. 27, 913–924. [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smoot, M.E. , Ono, K. , Ruscheinski, J. , Wang, P.L. and Ideker, T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics, 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soza, V.L. , Lindsley, D. , Waalkes, A. , Ramage, E. , Patwardhan, R.P. , Burton, J.N. , Adey, A. et al. (2019) The Rhododendron genome and chromosomal organization provide insight into shared whole‐genome duplications across the heath family (Ericaceae). Genome Biol. Evol. 11, 3353–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, P. , Schuurink, R.C. , Caissard, J.C. , Hugueney, P. and Baudino, S. (2016) My way: noncanonical biosynthesis pathways for plant volatiles. Trends Plant Sci. 21, 884–894. [DOI] [PubMed] [Google Scholar]
- Tang, W. , Sun, X.P. , Yue, J.Y. , Tang, X.F. , Jiao, C. , Yang, Y. , Niu, X.L. et al. (2019) Chromosome‐scale genome assembly of kiwifruit Actinidia eriantha with single‐molecule sequencing and chromatin interaction mapping. Gigascience, 8, giz027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N.S. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, 4, unit 4.10. [DOI] [PubMed] [Google Scholar]
- Teh, B.T. , Lim, K. , Yong, C.H. , Ng, C.C.Y. , Rao, S.R. , Rajasegaran, V. , Lim, W.K. et al. (2017) The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 49, 1633–1641. [DOI] [PubMed] [Google Scholar]
- Wang, X.Y. , Li, Z. , Liu, B. , Zhou, H. , Elmongy, M.S. and Xia, Y.P. (2020) Combined proteome and transcriptome analysis of heat‐primed azalea reveals new insights into plant heat acclimation memory. Front. Plant Sci. 11, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.P. , Tang, H.B. , DeBarry, J.D. , Tan, X. , Li, J.P. , Wang, X.Y. , Lee, T.H. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, G.Z. (2017) A simple process of RNA‐sequence analyses by Hisat2, Htseq and DESeq2. Proceedings of the 2017 International Conference on Biomedical Engineering and Bioinformatics, 11–15. [Google Scholar]
- Wu, H. , Ma, T. , Kang, M. , Ai, F. , Zhang, J. , Dong, G. and Liu, J. (2019) A high‐quality Actinidia chinensis (kiwifruit) genome. Hortic. Res., 6, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, E.H. , Tong, W. , Hou, Y. , An, Y.L. , Chen, L.B. , Wu, Q. , Liu, Y.L. et al. (2020) The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant, 13, 1013–1026. [DOI] [PubMed] [Google Scholar]
- Yang, F.S. , Nie, S. , Liu, H. , Shi, T.L. , Tian, X.C. , Zhou, S.S. , Bao, Y.T. et al. (2020) Chromosome‐level genome assembly of a parent species of widely cultivated azaleas. Nat. Commun. 11, 5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.H. (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhang, C.F. , Zhang, T.K. , Luebert, F. , Xiang, Y.Z. , Huang, C.H. , Hu, Y. , Rees, M. et al. (2020a) Asterid phylogenomics/phylotranscriptomics uncover morphological evolutionary histories and support phylogenetic placement for numerous whole genome duplications. Mol. Biol. Evol. 37, 3188–3210. [DOI] [PubMed] [Google Scholar]
- Zhang, L.S. , Chen, F. , Zhang, X.T. , Li, Z. , Zhao, Y.Y. , Lohaus, R. , Chang, X.J. et al. (2020b) The water lily genome and the early evolution of flowering plants. Nature, 577, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Xu, P.W. , Cai, Y.F. , Ma, L.L. , Li, S.F. , Li, S.F. , Xie, W.J. et al. (2017a) The draft genome assembly of Rhododendron delavayi Franch. var. delavayi. Gigascience, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T.C. , Qiao, Q. , Novikova, P.Y. , Wang, Q. , Yue, J.P. , Guan, Y.L. , Ming, S.P. et al. (2019a) Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. USA, 116, 7137–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Zhang, S. , Zhao, Q. , Ming, R. and Tang, H. (2019b) Assembly of allele‐aware, chromosomal‐scale autopolyploid genomes based on Hi‐C data. Nat. Plants, 5, 833–845. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.H. , Ni, J. , Tang, F.P. , Jiang, L.F. , Guo, T.R. , Pei, K.Q. , Sun, L.F. et al. (2017b) The effects of different human disturbance regimes on root fungal diversity of Rhododendron ovatum in subtropical forests of China. Canadian J. Forest Res. 47, 659–666. [Google Scholar]
- Zuker, A. , Tzfira, T. , Ben‐Meir, H. , Ovadis, M. , Shklarman, E. , Itzhaki, H. , Forkmann, G. et al. (2002) Modification of flower color and fragrance by antisense suppression of the flavanone 3‐hydroxylase gene. Mol. Breeding, 9, 33–41. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Evaluation of Rhododendron ovatum genome by k‐mer analysis and read‐depth.
Figure S2 Genome‐wide analysis of chromatin interactions in the R. ovatum genome based on Hi‐C data.
Figure S3 Genome assemblies of R. ovatum (ov) and R. williamsianum (wi).
Figure S4 Types of gene duplication in the R. ovatum genome.
Figure S5 Ks dot plot between R. ovatum and grape (V. vinifera).
Figure S6 Ks dot plot between R. ovatum and kiwifruit (A. chinensis).
Figure S7 Phylogenetic tree of ERF gene family.
Figure S8 Phylogenetic tree of MYB gene family.
Figure S9 Phylogenetic tree of ANAC001 clade of NAC gene family.
Figure S10 Chromosome localization of NAC genes of R. ovatum.
Figure S11 Weighted gene co‐expression network analysis (WGCNA) of the transcriptomes.
Figure S12 Expression profiles of HSP70s and TPSs in response to different temperature treatments (25, 37, and 42 °C).
Figure S13 Phylogenetic analysis and heat‐induced expression profiles of HSFs in R. ovatum.
Figure S14 Characteristics of tandem duplication in R. ovatum genome.
Figure S15 GO functional enrichment network of tandem‐duplicated genes.
Figure S16 Tandem duplication ratio of the stress responsive genes.
Figure S17 Expression profiles of CYP450s in different tissues.
Figure S18 Phylogenetic tree of fatty acyl‐ACP thioesterases coding genes.
Figure S19 Statistic of TPS gene subfamilies.
Figure S20 Phylogenetic tree of TPSs in R. ovatum.
Figure S21 Phylogeny of the TPS‐a subfamily.
Figure S22 Phylogeny of the TPS‐b subfamily.
Figure S23 Phylogeny of the TPS‐c subfamily.
Figure S24 Phylogeny of the TPS‐e subfamily.
Figure S25 Phylogeny of the TPS‐f subfamily.
Figure S26 Phylogeny of the TPS‐g subfamily.
Figure S27 Chromosome localization of RoTPS genes.
Figure S28 Expression profiles of TPS genes of R. ovatum.
Figure S29 Structure features of the three alternative splicing transcripts of Ro_38431.
Figure S30 Amino sequence alignment of Ro_38431.3 and Ro_43236.1.
Figure S31 Expression profiles of adjacent genes of TPS clusters on chromosome 7.
Table S1 Statistics for Illumina sequencing data of R. ovatum genome.
Table S2 Statistics for PacBio sequencing data of R. ovatum genome.
Table S3 Summary of contig‐level genome assembly for R. ovatum.
Table S4 BUSCO evaluation of the genomic completeness before and after elimination of redundancy.
Table S5 Statistics for Hi‐C sequencing data of R. ovatum genome.
Table S6 Statistics for PacBio Iso‐seq data of R. ovatum.
Table S7 Gene prediction of R. ovatum genome.
Table S8 Summary of repeat annotation of R. ovatum.
Table S9 BUSCO evaluation of the genomic completeness of R. ovatum.
Table S10 BUSCO evaluation of the gene prediction of R. ovatum.
Table S11 Main flora volatiles compounds of R. ovatum.
Data S1 Orthologous groups identified in these species.
Data S2 Biological_process GO enrichment of expansive genes in R. ovatum compared to R. delavayi and R. williamsianum.
Data S3 Full name list of the gene abbreviations.
Data S4 TPS gene family of R. ovatum.
Data S5 Pfam annotation of tandem‐duplicated genes in R. ovatum.
Data S6 Biological_process GO enrichment of tandem‐duplicated genes in R. ovatum.
Data Availability Statement
The raw data files of genome sequencing and RNA sequencing have been deposited at Sequence Read Archive (SRA) under the accession PRJNA671625. This whole‐genome project also has been deposited at DDBJ/ENA/GenBank under the accession JADHZH000000000. Genome assembly and gene annotations could be downloaded from Rhododendron Plant Genome Database (http://bioinfor.kib.ac.cn/RPGD/download_genome.html).