

Abstract
Microglial activation is closely associated with neuroinflammatory pathologies. The nucleotide-binding and oligomerization domain-like receptor containing a pyrin domain 3 (NLRP3) inflammasomes are highly organized intracellular sensors of neuronal alarm signaling. NLRP3 inflammasomes activate nuclear factor kappa-B (NF-κB) and reactive oxygen species (ROS), which induce inflammatory responses. Moreover, NLRP3 dysfunction is a common feature of chronic inflammatory diseases. The present study investigated the effect of a novel thiazol derivative, N-cyclooctyl-5-methylthiazol-2-amine hydrobromide (KHG26700), on inflammatory responses in lipopolysaccharide (LPS)-treated BV-2 microglial cells. KHG26700 significantly attenuated the expression of several pro-inflammatory cytokines, including tumor necrosis factor-α, interleukin-1β, and interleukin-6, in these cells, as well as the LPS-induced increases in NLRP3, NF-κB, and phospho-IkBα levels. KHG26700 also suppressed the LPS-induced increases in protein levels of autophagy protein 5 (ATG5), microtubule-associated protein 1 light chain 3 (LC3), and beclin-1, as well as downregulating the LPS-enhanced levels of ROS, lipid peroxidation, and nitric oxide. These results suggest that the anti-inflammatory effects of KHG26700 may be due, at least in part, to the regulation of the NLRP3-mediated signaling pathway during microglial activation.
Keywords: Inflammasome, Microglia, N-cyclooctyl-5-methylthiazol-2-amine hydrobromide, NLRP3, Pro-inflammatory cytokines
INTRODUCTION
Microglial cells are involved in the first line of defense in the nervous system, initiating immune regulation and neuronal homeostasis (1). Microglial activation induces the production of various neurotoxic factors and pro-inflammatory cytokines, which are subsequently involved in the progression of neuroinflammatory responses, such as the microglial phagocytosis of neurons and the generation of inflammatory lesions (2, 3). Anti-inflammatory agents suppress the expression/release of pro-inflammatory mediators following traumatic injury to the brain (4). Regulation of microglial activation may therefore inhibit detrimental immune responses in the compromised central nervous system. Lipopolysaccharide (LPS) induces the production of pro-inflammatory cytokines and activates several intracellular signaling pathways associated with NF-κB (5). Although pharmacological agents that interfere with NF-κB activation may mitigate neuroinflammatory diseases, drugs that are effective against neuroinflammation are rarely developed.
The NLRP3 inflammasome complex is one of the well examined inflammasomes among NLR family members. ASC adaptor, caspase-1 enzyme, and an NLRP3 sensor are main components of this inflammasome (1, 6). The NLRP3 inflammasome complex participates in defenses against invading pathogens. In microglia, most of these NLRP3 inflammasomes regulate inflammatory responses (7). Activated NLRP3 inflammasomes may contribute significantly to the progression of neuroinflammatory diseases (8). Little is known, however, about the signaling mechanism responsible for NLRP3 inflammasome activation. Although therapeutic agents that suppress NLRP3 inflammasome activation may be effective in reducing neuroinflammation associated with neurodegenerative diseases, no such agents have been developed to date (9).
We have recently assessed the anti-neuroinflammatory activity of several newly synthesized thiazole derivatives (10-12). Several of these thiazole derivatives have shown neuroprotective effects, including the ability to scavenge reactive oxygen species (ROS), suggesting that these compounds may have therapeutic property as anti-inflammatory and anti-oxidative agents in the treatment of brain diseases (13, 14). For example, thiazol-2-amine derivatives were highly stable in liver microsomes, had high oral bioavailability, and were present in high concentrations in mouse brains, all desirable properties in the treatment of brain diseases (15, 16). To date, however, the detailed mechanisms of action of thiazoles in brain diseases remain incompletely understood.
This study investigated the anti-inflammatory function of a novel thiazol derivative, N-cyclooctyl-5-methylthiazol-2-amine hydrobromide (KHG26700), including its property to control inflammatory responses via the attenuation of inflammasome-mediated signaling pathways, in LPS-induced microglial cells. These findings showed that KHG26700 suppressed inflammatory reactions by regulating NLRP3-regulated signaling pathways for microglial activation.
RESULTS AND DISCUSSION
KHG26700 attenuated cytokine levels in LPS-treated BV-2 cells
The progression of neurodegenerative diseases frequently involves the activation of microglia. For example, microglia in neuron-glia cultures secrete pro-inflammatory cytokines and cytotoxic factors when treated with N-methyl-D-aspartate, glutamate, β-amyloid, and LPS (11). Abnormally activated microglia were found to produce a variety of pro-inflammatory cytokines and other mediators, including TNF-α, IL-1β, and IL-6. To examine the effects of KHG26700 on pro-inflammatory cytokine level, cells were treated with KHG26700 (0, 1, 5, 10, or 20 μM) and LPS (1 μg/ml) for 24 h in culture media. The concentrations of cytokines (IL-6, IL-1β, and TNF-α) were determined by ELISA. Although LPS treatment increased the concentrations of all cytokines determined in culture media, these concentrations were markedly decreased when BV-2 cells were treated with KHG26700 (Fig. 1). KHG26700 itself did not show any cytotoxicity at concentrations used in this study. These results suggested that KHG26700 effectively attenuates the production of pro-inflammatory cytokines in LPS-induced BV-2 cells.
Fig. 1.
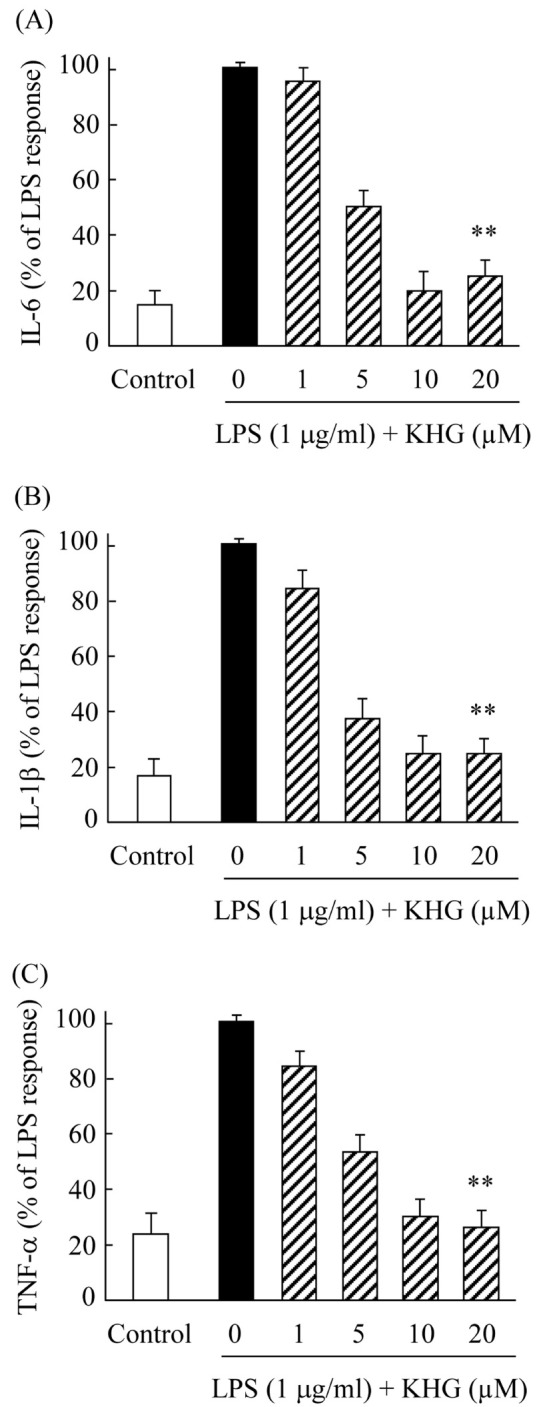
Effects of KHG26700 on the production of IL-1b (A), IL-6 (B), and TNF-α (C) in LPS-treated BV-2 cells. BV-2 cells were treated with KHG26700 for 30 min followed by treatment with LPS (1 μg/ml) for 24 h. Data are presented as means ± S.D. (n = 3). *P < 0.01 for comparisons between cells treated with LPS and LPS plus KHG26700.
KHG26700 inhibits the activation of NF-κB, NLRP3 inflammasomes, and p-IκB-α in LPS-treated BV-2 cells
NLRP3 inflammasomes are highly organized intracellular sensors of neuronal alarm signals, with activation of NLRP3 inflammasome complexes being involved in regulating cytokine production in microglia (12). We previously reported that NLRP3 inflammasomes expressed in LPS-induced BV-2 cells was regulated by anti-inflammatory agents (13). In addition, NLRP3 inflammasomes were reported to activate the pro-inflammatory mediators NF-κB and ROS (14, 15), with NLRP3 inflammasome activation mediated through NF-κB translocation into the nucleus (14). We therefore investigated the effects of KHG26700 on the relationship between NF-κB activation and the NLRP3 inflammasome pathway. Western blotting and immunofluorescence study showed that the levels of NF-κB protein and NLRP3 inflammasomes were similarly upregulated in LPS-treated BV-2 cells, with LPS stimulation inducing NF-κB translocation into the nucleus (Fig. 2A) and increasing the levels of expression of NLRP3 inflammasomes (Fig. 2B). By contrast, KHG26700 treatment blocked these processes, indicating that KHG26700 suppresses inflammatory reactions by blocking the NF-κB and NLRP3 inflammasome signaling pathways. KHG26700 reduces both immune responses associated with NF-κB signaling and NLRP3 inflammasome activation.
Fig. 2.
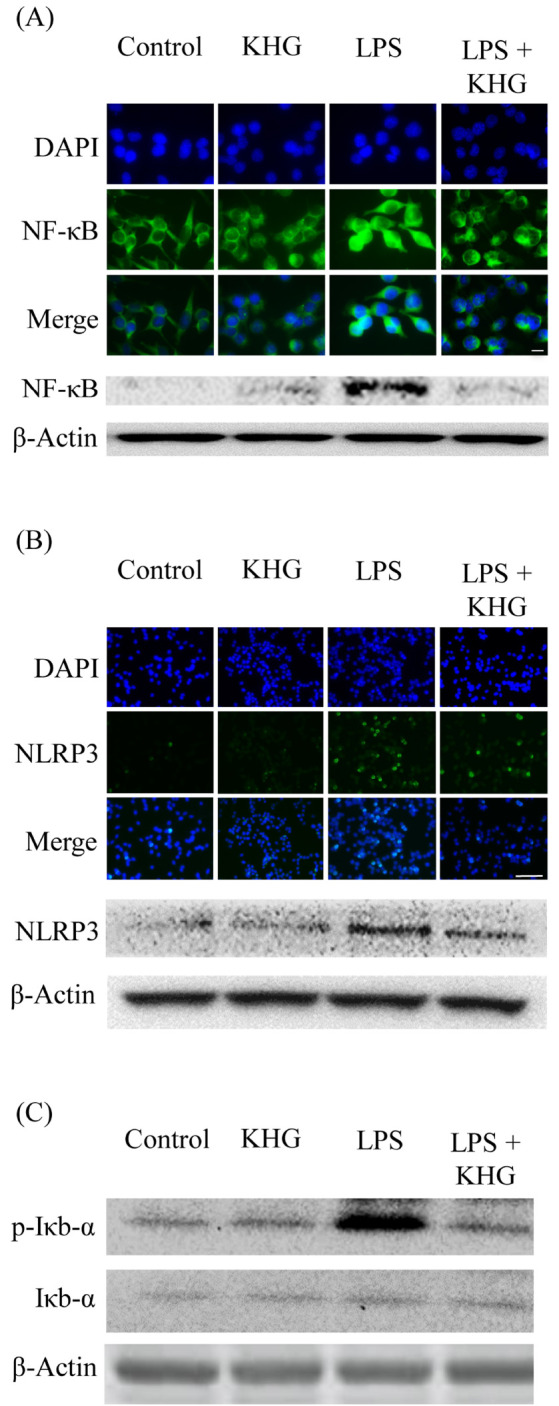
Effects of KHG26700 on NF-κB (A), NLRP3 (B), and Iκb-α (C) in LPS-treated BV-2 cells, as determined by immunofluorescence assays and western blot analysis. BV-2 cells were treated with KHG26700 for 30 min followed by treatment with LPS (1 μg/ml) for 24 h. Images presented are from a single experiment and are representative of three independent experiments. β-Actin was used as a loading control. Scale bars were 10 μm in (A) and 50 μm in (B).
LPS stimulation of BV-2 microglial cells was also found to increase the phosphorylation of IκBα, an increase suppressed by treatment with KHG26700 (Fig. 2C). In the resting state, NF-κB is present in the cytoplasm as a complex with IκBα complex, whereas phosphorylation of IκBα leads to its ubiquitination and subsequent degradation (16). The inhibitory protein IκBα has been found to control inflammatory processes and immune responses by regulating NF-κB activity (17). Our results suggested that KHG26700 may suppress NF-κB signaling by downregulating LPS-induced IκBα phosphorylation, and so decreasing IκBα degradation and retaining NF-κB in the cytoplasm (Fig. 2C). The results in the present study suggest that NLRP3 inflammasomes could be regulated via the activation of NF-κB in BV-2 cells.
Effects of KHG26700 on protein levels of ATG5, beclin-1, and LC3 in LPS-treated BV-2 cells
Although initial responses in microglia-mediated inflammation can be protective, excessive pro-inflammatory responses may contribute to the pathogenesis of neurodiseases in microglial cells. Autophagy is known to be related with inflammatory responses, and the dysregulation of autophagy may result in the induction of autophagic cell death (13, 14). Autophagy is modulated cooperatively by several marker proteins, such as ATG5, LC3, and beclin 1 (15), and has been reported to regulate inflammatory responses by downregulating NF-κB and pro-inflammatory cytokines (16, 17). Glutamate-induced cortical neuronal cell injury was reported related to the expression of LC3, ATG5, and beclin-1 proteins (18, 19). Nevertheless, the role and mechanism of action of autophagy in microglia remain unclear and the cross-regulation of LPS-induced inflammation in microglia and autophagy has not been intensively investigated.
The involvement of neuronal autophagy in LPS-treated BV-2 cells was assessed by measuring the expression levels of LC3, ATG5, and beclin-1 proteins 24 h after LPS (1 μg/ml) treatment in the presence or absence of KHG26700. Compared with control, untreated cells, LPS treatment dramatically enhanced the levels of ATG5, LC3, and beclin-1 proteins (Fig. 3A-C), suggesting that LPS treatment enhanced the activation of autophagy. However, KHG26700 effectively attenuated the LPS-induced autophagic activation almost to the level observed in control cells (Fig. 3A-C). These findings suggested that the suppression of autophagy activity coincided with the neuroprotective function of KHG26700, and that KHG26700 may play an important role in protecting microglial cells from LPS-induced neuronal damage through inhibition of autophagy.
Fig. 3.

Western blot analysis of the effects of KHG26700 on protein levels of ATG5, beclin-1, and LC3 in LPS-treated BV-2 cells. (A) Western blot analysis was performed using antibodies against ATG5, beclin-1, and LC3, with β-actin used as a loading control. (B-D) Relative protein levels were quantified by densitometry and normalized relative to the expression of β-actin. Data are presented as means ± S.D. (n = 3). *P < 0.01 for comparisons between cells treated with LPS and LPS plus KHG26700.
Effects of KHG26700 on the formation of ROS and NO as well as on lipid peroxidation in LPS-treated BV-2 cells
LPS can promote oxidative damage to neighboring neurons by stimulating the level of ROS and nitric oxide (NO) through intracellular signaling pathways in microglia (20). Moreover, suppression of iNOS has been reported to regulate LPS-induced microglial activation (20). Regulating oxidative stress can therefore be a promising strategy to regulate LPS-induced inflammation in microglial cells. To further determine the relation between antioxidant activities of KHG26700 and its anti-inflammatory property in LPS-induced microglial activation, the concentrations of indicators of oxidative stress, such as ROS, NO, and lipid peroxidation, were measured. Nitric oxide is one of the most reactive nitrogen intermediates, which is regarded as an active messenger that mediates various cellular signaling pathways, and as a mediator in the immune systems (21). The induction of NO in activated microglia results in NO-associated stress and direct injury to neuronal cells (22, 23).
Protective activity of KHG26700 on ROS production in LPS-treated BV-2 cells was also examined. Immunofluorescence analysis demonstrated that the levels of expression of ROS were ∼5-fold higher in LPS-treated than in untreated BV-2 microglial cells, an increase that was dramatically suppressed by treatment with KHG26700 (Fig. 4A). To investigated the anti-inflammatory property of KHG26700 on BV-2 cells, the concentration of nitric oxide was measured in cell culture supernatants. Treatment with LPS markedly enhanced the formation of nitric oxide, whereas KHG26700 treatment efficiently protected its production (Fig. 4B). LPS can increase lipid peroxidation and attack polyunsaturated fatty acids, resulting in mem-brane damage (24). Increased malondialdehyde (MDA) levels are thought to be markers of lipid peroxidation (24, 25). Cells exposed to LPS had higher levels of MDA than control cells, whereas KHG26700 treatment efficiently suppressed the LPS-associated increase in MDA (Fig. 4C). Our results suggested that KHG26700 might attenuate LPS-induced oxidative stress by reducing ROS generation and lipid peroxidation.
Fig. 4.

Effects of KHG26700 on ROS (A), NO (B), and MDA (C) production in LPS-treated BV-2 cells. BV-2 cells were treated with KHG26700 for 30 min followed by treatment with LPS (1 μg/ml) for 24 h. In (A), images presented are from a single experiment and are representative of three independent experiments. Data are presented as means ± S.D. (n = 3). *P < 0.01 for comparisons between cells treated with LPS and LPS plus KHG26700.
Taken together, the results in the present study may indicate that the anti-inflammatory property of KHG26700 in microglial activation is due to its activity for controlling the NLRP3-mediated signaling pathway at least in part. Further in vivo studies may be necessary to understand the mechanism by which KHG26700 protects microglial cells from LPS-induced inflammatory responses.
MATERIALS AND METHODS
Materials
LPS, fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco (Grand Island, NY). Antibodies against NF-κB, NLRP3, Iκb-α, ATG5, beclin-1, LC3, and β-actin were purchased from Cell Signaling Technology (Beverly, MA). N-cyclooctyl-5-methylthiazol-2-amine hydrobromide (KHG26700) was kindly provided by Dr. Hyo-Kyu Hahn (KIST, Seoul, Korea). All other commercial reagents were of the highest available purity.
Culture and drug treatment of microglial BV2 cells
BV-2 microglial cells were maintained in DMEM containing 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in a humidified incubator in an atmosphere containing 5% CO2. The medium was changed every day, and plated cells were grown at an appropriate density on an experiment-by-experiment basis. KHG26700 was freshly prepared in DMSO and diluted to the desired final concentrations in culture medium. For all experiments, equivalent amounts of DMSO were added to control and LPS-treated cells. Cell viability was determined by MTT reduction assay as described elsewhere (26) and expressed as a percentage of the control.
Measurement of IL-6, IL-1β, and TNF-α
IL-6, IL-1β, and TNF-α, levels in the extracellular medium were determined using ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions, as described (27, 28). Optical density was measured using a microplate reader. The concentration of each sample was calculated from the linear equation derived from a standard curve of known concentrations of each cytokine.
Immunofluorescent staining
BV-2 cells were fixed with 3.7% paraformaldehyde in phosphate buffered saline (PBS), permeabilized in 0.2% Triton X-100 in PBS, and blocked by incubation in 2% bovine serum albumin in PBS. The samples were subsequently incubated with rabbit polyclonal anti-NF-κB p65 primary antibodies, rabbit monoclonal NLRP3 primary antibodies, and Alexa Fluor 488-conjugated goat anti-rabbit IgG. The cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Images were captured under a laser scanning confocal microscope (Carl Zeiss AG, Oberkochen, Germany).
Western blotting
Total protein extracts were subjected to 10% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with antibodies to NF-κB, NLRP3, Iκb-α, ATG5, beclin-1, LC3, and β-actin. Proteins were detected by enhanced chemiluminescence according to the manufacturer’s instructions (Amersham Bioscience, Piscataway, NJ, USA) and analyzed using a Molecular Imager ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA) as described (29). Detected protein bands were normalized against those of β-actin in the same samples to confirm equal protein loading.
Measurement of ROS
The intracellular formation of ROS was measured using DCF-DA (2’,7’-dichlorofluorescin diacetate), as described (30). Briefly, BV-2 cells were plated at a density of 1 × 105 cells/well in a 96-well plate and treated with various concentrations of KHG26700 for 30 min, followed by treatment with LPS (1 μg/ml) for an additional 24 h. The culture medium was removed and the cells were washed with PBS three times. DCFH-DA, diluted to a final concentration of 10 μM with DMEM/F12, was added to the culture medium and incubated at 37°C for 20 min in the dark. After washing the cells with serum-free medium, the cells were observed using an inverted fluorescence microscope at an excitation wavelength of 352 nm and an emission wavelength of 461 nm (Olympus Opticals, Tokyo, Japan). Four continual fields in each group were used for quantitative analysis. To determine the production of ROS, the fluorescence intensity in each group was analyzed with the Image-Pro Plus 6.0 analysis system.
Measurement of NO and MDA
The concentration of NO in the culture supernatants was determined by measuring the amount of nitrite generated by the Griess reagent (1% sulfanilamide/0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5% H3PO4), as previously de-scribed (Ha et al., 2013; Lee et al., 2013). NO concentrations were measured in crude extracts containing equal amounts of protein. Optical density at 540 nm was measured using a microplate reader (Molecular Devices Corp., Sunnyvale, CA) and NO concentrations were calculated relative to a standard curve of sodium nitrite with known concentrations of NO (31).
MDA concentrations were measured in BV-2 cell homogenates as described (32). Briefly, cells were treated with a reaction mixture containing trichloroacetic acid (15%, 400 μl) and 2-thiobarbituric acid (TBA) 0.67%/butylated hydroxytoluene 0.01% (800 μl). MDA reacts with TBA to form a fluorescent adduct. Samples were then boiled for 1 h at 95°C and centrifuged at 4000 g for 10 min. Fluorescence intensity was measured at excitation and emission wavelengths of 430 and 550 nm, respectively.
Statistical analysis
Statistical comparisons were performed using single-factor ANOVA followed by Tukey’s post hoc tests. Data from three independent experiments were analyzed and are represented as means ± standard deviations. Statistical significance was defined as a P-value < 0.01.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2018R1A2B6001743 and 2021R1F1A1051920), and by a Student Research Grant from the University of Ulsan College of Medicine, Seoul, Korea.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Lee CM, Lee DS, Jung WK, et al. Benzyl isothiocyanate inhibits inflammasome activation in E. coli LPS-stimulated BV2 cells. Int Mol Med. 2016;38:912–918. doi: 10.3892/ijmm.2016.2667. [DOI] [PubMed] [Google Scholar]
- 2.Budai MM, Varga A, Milesz S, et al. Aloe vera downregulates LPS-induced inflammatory cytokine production and expression of NLRP3 inflammasome in human macrophages. Mol Immunol. 2013;56:471–479. doi: 10.1016/j.molimm.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 3.inarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 4.Jeng KCG, Hou RCW, Wang JC, et al. Sesamin inhibits lipopolysaccharide-induced cytokine production by suppression of p38 mitogen-activated protein kinase and nuclear factor-kappaB. Immunol Lett. 2005;97:101–106. doi: 10.1016/j.imlet.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Fu YY, Zhang F, Zhang L, et al. Mangiferin regulates interleukin-6 and cystathionine-β-synthase in lipopolysaccharide-induced brain injury. Cell Mol Neurobiol. 2014;34:651–657. doi: 10.1007/s10571-014-0039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–379. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang F, Wang Z, Wei X, et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab. 2014;34:660–667. doi: 10.1038/jcbfm.2013.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C, Wei YZ, He XM, et al. Naringenin produces neuroprotection against LPS-induced dopamine neurotoxicity via the inhibition of microglial NLRP3 inflammasome activation. Front Immunol. 2019;10:936. doi: 10.3389/fimmu.2019.00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Kim SM, Na JM, et al. Protective effect of 3-(naphthalen-2-yl(propoxy)methyl)azetidine hydrochloride on hypoxia-induced toxicity by suppressing microglial activation in BV-2 cells. BMB Rep. 2016;49:687–692. doi: 10.5483/BMBRep.2016.49.12.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim EA, Na JM, Kim J, et al. Neuroprotective effect of 3-(naphthalen-2-yl(propoxy)methyl)azetidine hydrochloride on brain ischaemia/reperfusion injury. J Neuroimmune Pharmacol. 2017;12:447–461. doi: 10.1007/s11481-017-9733-x. [DOI] [PubMed] [Google Scholar]
- 11.Kim EA, Kim H, Ahn JY, et al. Suppression of lipopolysaccharide-induced microglial activation by a benzothiazole derivative. Mol Cells. 2010;30:51–57. doi: 10.1007/s10059-010-0087-y. [DOI] [PubMed] [Google Scholar]
- 12.Sorbara MT, Girardin SE. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011;21:558–560. doi: 10.1038/cr.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang JW, Yang SJ, Na JM, et al. 3-(Naphthalen-2-yl (propoxy)methyl)azetidine hydrochloride attenuates NLRP3 inflammasome-mediated signaling pathway in lipopolysaccharide-stimulated BV2 microglial cells. Biochem Biophys Res Commun. 2018;495:151–156. doi: 10.1016/j.bbrc.2017.10.131. [DOI] [PubMed] [Google Scholar]
- 14.Ha JS, Choi HR, Kim IS, et al. Hypoxia-induced S100A8 expression activates microglial inflammation and promotes neuronal apoptosis. Int J Mol Sci. 2021;22:1205. doi: 10.3390/ijms22031205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang SJ, Han AR, Choi HR, et al. N-Adamantyl-4-methylthiazol-2-amine suppresses glutamate-induced autophagic cell death via PI3K/Akt/mTOR signaling pathways in cortical neurons. BMB Rep. 2020;53:527–532. doi: 10.5483/BMBRep.2020.53.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Kwak HJ, Cha JY, et al. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J Biol Chem. 2014;289:23246–23255. doi: 10.1074/jbc.M114.577908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, Choi J, Ryu J, et al. Activation of autophagy during glutamate-induced HT22 cell death. Biochem Biophys Res Commun. 2009;388:339–344. doi: 10.1016/j.bbrc.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Yin WY, Ye Q, Huang HJ. Salidroside protects cortical neurons against glutamate-induced cytotoxicity by inhibiting autophagy. Mol Cell Biochem. 2016;419:53–64. doi: 10.1007/s11010-016-2749-3. [DOI] [PubMed] [Google Scholar]
- 20.Kim EA, Han AR, Choi J, et al. Anti-inflammatory mechanisms of N-adamantyl-4-methylthiazol-2-amine in lipopolysaccharide-stimulated BV-2 microglial cells. Int Immunopharmacol. 2014;22:73–83. doi: 10.1016/j.intimp.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 21.Murad F. The nitric oxide-cyclic GMP signal transduction system for intracellular and intercellular communication. Recent Prog Horm Res. 1994;49:239–248. doi: 10.1016/B978-0-12-571149-4.50016-7. [DOI] [PubMed] [Google Scholar]
- 22.Possel H, Noack H, Putzke J, et al. Selective upregulation of inducible nitric oxide synthase (iNOS) by lipopolysaccharide (LPS) and cytokines in microglia: in vitro and in vivo studies. Glia. 2000;32:51–59. doi: 10.1002/1098-1136(200010)32:1<51::AID-GLIA50>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev. 2006;12:105–112. doi: 10.1002/mrdd.20102. [DOI] [PubMed] [Google Scholar]
- 24.Cho CH, Kim EA, Kim J, et al. N-Adamantyl-4-methylthiazol-2-amine suppresses amyloid β-induced neuronal oxidative damage in cortical neurons. Free Radic Res. 2016;50:678–690. doi: 10.3109/10715762.2016.1167277. [DOI] [PubMed] [Google Scholar]
- 25.Urabe T, Yamasaki Y, Hattori N, et al. Accumulation of 4-hydroxynonenal-modified proteins in hippocampal CA1 pyramidal neurons precedes delayed neuronal damage in the gerbil brain. Neuroscience. 2000;100:241–250. doi: 10.1016/S0306-4522(00)00264-5. [DOI] [PubMed] [Google Scholar]
- 26.Ha SC, Han AR, Kim DW, et al. Neuroprotective effects of the antioxidant action of 2-cyclopropylimino-3-methyl-1,3-thiazoline hydrochloride against ischemic neuronal damage in the brain. BMB Rep. 2013;46:370–375. doi: 10.5483/BMBRep.2013.46.7.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SJ, Cha JY, Kang HS, et al. Corosolic acid ameliorates acute inflammation through inhibition of IRAK-1 phosphorylation in macrophages. BMB Rep. 2016;49:276–281. doi: 10.5483/BMBRep.2016.49.5.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KJ, Kim YK, Krupa M, et al. Crotamine stimulates phagocytic activity by inducing nitric oxide and TNF-α via p38 and NFκ-B signaling in RAW 264.7 macrophages. BMB Rep. 2016;49:185–190. doi: 10.5483/BMBRep.2016.49.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H, Youn GS, An SY, et al. 2,3-Dimethoxy-2'-hydroxychalcone ameliorates TNF-α-induced ICAM-1 expression and subsequent monocyte adhesiveness via NF-kappaB inhibition and HO-1 induction in HaCaT cells. BMB Rep. 2016;49:57–62. doi: 10.5483/BMBRep.2016.49.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim EA, Choi J, Han AR, et al. Anti-oxidative and anti-inflammatory effects of 2-cyclopropylimino-3-methyl-1,3-thiazoline hydrochloride on glutamate-induced neurotoxicity in rat brain. Neurotoxicology. 2013;38:106–114. doi: 10.1016/j.neuro.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Yang SJ, Lee WJ, Kim EA, et al. Effects of N-ada-mantyl-4-methylthiazol-2-amine on hyperglycemia, hyperlipidemia and oxidative stress in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2014;736:26–34. doi: 10.1016/j.ejphar.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 32.Han AR, Yang JW, Na JM, et al. Protective effects of N,4,5-trimethylthiazol-2-amine hydrochloride on hypoxia-induced β-amyloid production in SH-SY5Y cells. BMB Rep. 2019;52:439–444. doi: 10.5483/BMBRep.2019.52.7.231. [DOI] [PMC free article] [PubMed] [Google Scholar]