

Abstract
Metastasis is considered the leading cause of cancer death due to the limited possibilities to therapeutically target this process. How the ubiquitination machinery contributes to metastasis remains underexplored. Angio-associated migratory cell protein (AAMP), a ubiquitously expressed protein involved in cell migration, has been reported to play oncogenic roles in breast and non-small cell lung cancer (NSCLC). However, the role of AAMP in colorectal cancer (CRC) has not been demonstrated. Here, we report that AAMP is aberrantly upregulated in metastatic CRC and that AAMP upregulation is correlated with the poor survival of CRC patients. AAMP knockdown significantly attenuated the migration and invasion of CRC cells, while AAMP overexpression led to the opposite effects. Mechanistically, we identified Ras homolog family member A (RhoA) as a target of AAMP. Smad ubiquitin regulatory factor (SMURF) 2 was previously found to be a CRC suppressor. Notably, we discovered here that SMURF2 acted as an E3 ubiquitin ligase to mediate the ubiquitination and degradation of RhoA. AAMP stabilized RhoA by binding to it and suppressing its SMURF2-mediated ubiquitination and degradation. Subsequently, the level of active RhoA was increased, thereby accelerating CRC cell migration and invasion. These findings indicate a new potential antitumor target for CRC.
Keywords: AAMP, colorectal cancer, metastasis, RhoA, SMURF2, ubiquitination
Graphical abstract
AAMP prevents RhoA from binding to SMURF2, protects RhoA from SMURF2-mediated ubiquitination and degradation, and subsequently induces robust RhoA activity to promote CRC metastasis. Hence, therapeutic interventions that disrupt the functional interplay between AAMP and RhoA or activate SMURF2 may provide promising strategies to treat CRC.
Introduction
Colorectal cancer (CRC) has the second highest mortality rate among cancers, with an estimated 147,950 diagnoses and 53,200 deaths in the United States in 2020.1 Despite advances in surgical resection and systemic chemotherapies, most patients with CRC die as a result of invasion and metastasis.2 However, there is still a lack of effective targeted therapies for metastatic CRC due to its elusive mechanisms. Therefore, defining the mechanisms underlying CRC metastasis is critical for developing effective therapies.
Metastasis is a hallmark of cancer.3 The first step in metastasis is invasion, during which cancer cells migrate away from the primary site and into the surrounding tissues. Changes in cell morphology, protrusive activity, and cell polarity mediated by actions on cytoskeletal dynamics are involved in the acquisition of invasive behavior. Tumor cells can adopt an amoeboid-like migration mode with a rounded cell body phenotype or display a mesenchymal migration mode with an elongated cell body phenotype.4 The Rho guanosine triphosphatase (GTPase) family is a family of ubiquitously expressed GTP-binding proteins involved in regulating cytoskeletal dynamics. Rho GTPases were first identified to participate in cell migration 26 years ago.5 Recently, accumulating data have indicated that Rho GTPases play a vital role in cancer cell migration.6,7 Among the 20 Rho GTPase members in humans, the Ras homolog family member A (RhoA) subfamily is one of the best characterized subfamilies.8 RhoA induces the assembly of the contractile actin cortex and ameboid (bleb) migration by promoting the phosphorylation of myosin light chain 2 (MLC2) and the actin-membrane linkage proteins ezrin/radixin/moesin (ERM).9 Indeed, RhoA was found to promote colon cancer metastasis and correlate with poor clinical outcomes.10, 11, 12 However, the molecular determinants that govern RhoA protein degradation and stability in CRC have yet to be identified.
The ubiquitin-proteasome system is involved in the degradation of more than 80% of proteins in cells, and defects in this system have been shown to cause pathological conditions, including malignant transformation.13 Ubiquitination is a cascade process by which ubiquitin, a ubiquitously expressed protein consisting of 76 amino acids, is ligated to a substrate protein.14 Ubiquitin-activating enzymes (E1s) initially bind to ubiquitin for its activation and then transfer activated ubiquitin to ubiquitin-conjugating enzymes (E2s). Ubiquitin ligases (E3s) finally transfer ubiquitin from E2s to substrate proteins, which ultimately leads to their degradation by the proteasome.14 E3 ligases play a key role throughout the process of ubiquitination because of their substrate specificity.
There are three types of E3s: really interesting new gene (RING) family members, homologous to the E6AP C terminus (HECT) domain E3s and RING-in-between-RING (RBR) E3s.15 Smad ubiquitin regulatory factor 2 (SMURF2) belongs to the homologous HECT domain family of E3s.16 It plays a dual role in cancer, exhibiting both oncogenic and tumor-suppressive functions. Induction of SMURF2 enhances tumor metastasis in a nude mouse model and increases invasion and migration of breast cancer cells17 and inhibits apoptosis by promoting p53 degradation through stabilizing the E3 ligase MDM2.18 In addition, high expression of SMURF2 is related to poor prognosis in esophageal carcinomas.19 Conversely, it has been reported that SMURF2−/− mice can develop various tumors in different tissues and organs, including the liver, blood, lung, pituitary gland, and Harderian gland.20 Moreover, heterozygous SMURF2 mice (SMURF2+/−) are also prone to spontaneous tumor development.21 These findings show that SMURF2 is a potent tumor suppressor that prevents malignant transformation. Indeed, SMURF2 reduces aerobic glycolysis and CRC cell proliferation by promoting ubiquitination and degradation of the glucose-responsive transcription factor carbohydrate response element-binding protein (ChREBP).22 In addition, SMURF2 acts as an E3 ubiquitin ligase that promotes special AT-rich sequence-binding protein-1 (SATB1) degradation by upregulating its ubiquitination. SMURF2 deficiency promotes SATB1 target gene transcription and colon cancer cell proliferation, migration, and tumorigenesis.23 Consistent with these observations, we found that SMURF2 functions as an E3 ligase of RhoA to mediate RhoA ubiquitination and degradation, thereby inhibiting CRC cell migration.
Angio-associated migratory cell protein (AAMP) was initially isolated from a human melanoma cell line during a search for motility-associated cell surface proteins in 1995.24 AAMP was found to be ubiquitously expressed in different human cell lines.25 It contains two important domains: the WD40 repeat domain, which mediates protein-protein interactions, and a heparin-binding consensus sequence, which mediates heparin-sensitive cell adhesion.24 Therefore, AAMP may play a role in cell migration and adhesion. AAMP performs its functions in numerous (patho)physiologic responses. For example, AAMP can interact with TPα and TPβ to promote the migration of primary human coronary artery smooth muscle cells.26 AAMP stimulates endothelial cell migration and angiogenesis by activating RhoA/Rho kinase signaling.27 It was reported that AAMP can directly bind to nucleotide binding oligomerization domain containing-2 (NOD2) via WD40 repeats to regulate innate immune responses.25 Notably, recent studies have revealed that AAMP also plays an oncogenic role. AAMP was found to be overexpressed in gastrointestinal stromal tumors and breast cancer.28,29 Yin et al. reported that high levels of AAMP transcripts were associated with disease progression, metastasis, and poor prognosis in breast cancer patients.29 Another report suggested that AAMP interacted with EGFR and enhanced its phosphorylation, subsequently activating ERK1/2, which endowed non-small cell lung cancer (NSCLC) cells with enhanced proliferation ability and resistance to chemotherapies.30 This research team also found that AAMP interacted with cell division cycle 42 (CDC42) and enhanced CDC42 activation by impairing the interaction of Rho GTPase-activating protein 1 (ARHGAP1) and CDC42, thereby promoting the migration and invasion of NSCLC cells.31 However, the role of AAMP in CRC remains unexplored.
In this study, we identified AAMP as a CRC-promoting protein and found that its expression was strongly associated with poor clinical outcomes in CRC patients. AAMP promoted CRC cell migration and invasion both in vitro and in vivo. We further revealed RhoA as a direct binding partner of AAMP. AAMP stabilized RhoA by preventing its SMURF2-mediated ubiquitination and degradation, thereby augmenting the activation of RhoA (GTP-RhoA) in CRC. Our study establishes AAMP and RhoA as potential targets for the development of new anti-CRC therapeutics.
Results
AAMP is upregulated in human CRC and correlates with poor clinical outcomes
To determine the clinical significance of AAMP in CRC, we analyzed microarray datasets in the Oncomine database. As shown in Figure 1A, AAMP messenger RNA (mRNA) levels were increased significantly in human CRC tissues compared with adjacent normal colon tissues (p < 0.0001). To verify the microarray analysis results, we performed quantitative reverse-transcription polymerase chain reaction (qRT-PCR) experiments on human CRC specimens and their matched normal tissues. AAMP mRNA was upregulated 1.1- to 5.7-fold in 10 tumor tissues compared with the matched normal tissues (Figure 1B). Importantly, analysis on the Gene Expression Profiling Interactive Analysis (GEPIA)32 website showed that AAMP upregulation was associated with shortened patient overall survival (OS) (p = 0.02) and disease-free survival (DFS) (p = 0.17) times (Figures 1C and 1D).
Figure 1.

Upregulation of AAMP in CRC and its correlation with poor clinical outcomes
(A) Analysis of Oncomine data showing the mRNA expression levels of AAMP in CRC tissues compared with normal tissues. Data were pooled from two datasets (Alon colon and TCGA colorectal). Forty-four normal tissues and 141 cancer tissues were analyzed. Student's t test; ∗∗∗∗p < 0.0001. (B) Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis of AAMP mRNA levels in 10 paired CRC samples and normal tissues (n = 3, mean ± SEM), ∗∗∗p < 0.001. (C and D) Analysis of associations between the tumor AAMP mRNA level and patient overall survival (OS) and disease-free survival (DFS) times via the GEPIA website (http://gepia.cancer-pku.cn/; OS, p = 0.02, n = 189, 188; DFS, p = 0.17, n = 189, 188). (E) Immunoblot showing different protein levels of AAMP in a normal colon mucosal epithelial cell line (NCM460) and colon cancer cell lines. (F) Immunohistochemical (IHC) staining of normal and CRC tissues with an anti-AAMP antibody. Representative patient samples are shown. A total of 101 patient samples were stained and analyzed. (G) Semiquantitative analysis of AAMP IHC staining in 79 paired tumor/normal samples. IOD, integral optical density. Student's t test; ∗∗∗p < 0.001; mean ± SEM. (H) Rates of negative and positive lymph node metastasis status among CRC patients with low and high AAMP expression.
We further assessed the AAMP protein level in a panel of human CRC cell lines using the normal colon epithelial cell line NCM460 as a nonmalignant control. AAMP expression was relatively high in the CRC cell lines compared with NCM460 cells, with the highest expression in HCT116 cells (Figure 1E). To further explore the protein level of AAMP in CRC patients, we performed immunohistochemical (IHC) staining for AAMP in primary human tumors obtained from a large cohort of CRC patients. A detailed description of the clinical features of the CRC samples used in this study is provided in Table S1. Among the specimens obtained from 101 patients, 79 biopsy specimens contained both tumor and matched normal adjacent tissues, whereas the other 22 had only tumor tissues. In the 79 matched samples, semiquantitative analysis showed markedly higher intensities of AAMP staining in CRC than in adjacent normal tissues (Figures 1F and 1G). Notably, as shown in Figure 1H, lymph node metastasis was detected more frequently in patients whose tumor samples had high AAMP expression levels (compared with low AAMP expression; the percentages of positive lymph node metastases were 48.8% and 30.9%, respectively). The relationships between AAMP expression and clinicopathologic features are shown in Table S2. The expression of AAMP was positively correlated with American Joint Committee on Cancer stage and lymph node metastasis, although the correlations were not statistically significant, with p values of 0.092 and 0.071, respectively. Together, our results establish a positive correlation of upregulated AAMP expression with CRC progression and poor survival.
AAMP promotes CRC metastasis
To examine the biological function of AAMP in CRC, we generated three small interfering RNAs (siRNAs), namely, human AAMP (h-AAMP) siRNA1, 2, and 3. siRNA2 exhibited the highest knockdown (KD) efficiency, followed by siRNA3 and 1 (Figures S1A and S1B). We also developed a lentiviral short hairpin RNA (shRNA) targeting the same RNA sequence as siRNA2, and its KD efficiency was verified (Figures S1A and S1B). Mouse AAMP siRNAs and shRNA were generated via the same strategy (Figure S1C). We then employed h-AAMP siRNA2 and 3 in the AAMP-high colon cancer cell line HCT116. As shown in Figures S2A and S2B, AAMP KD in HCT116 cells had little effect on apoptosis, cell-cycle progression, or the proliferation ability (Figure S2C).
We next assessed the role of AAMP in cell migration using Transwell chamber assays. As shown in Figures 2A and 2B, AAMP shRNA transfection reduced the migration of HCT116 (human colon cancer cells; by 84.7%, p < 0.0001) and MC38 (mouse colon cancer cells; by 67.1%, p < 0.001) (compared with control shRNA [sh-control]-transfected cells as controls). The impairment of migration induced by AAMP KD was further validated in LoVo cells. Wound healing assays showed that healing of the open wound area was markedly attenuated upon AAMP KD in LoVo cells (Figure S3A). Transwell chamber assays demonstrated that the number of migrated cells was reduced by 76.1% in AAMP KD LoVo cells compared with control LoVo cells (p < 0.0001) (Figure S3B). We then examined the impact of AAMP inactivation on CRC cell invasion using an independent assay with Matrigel-coated Transwell chambers. Again, downregulation of AAMP impeded the invasion of HCT116, MC38, and LoVo cells by 74.9% (p < 0.001), 57.9% (p < 0.0001), and 74.9% (p < 0.0001) (Figures 2C, 2D, and S3C). Conversely, AAMP overexpression (OE) promoted HCT116 cell migration and invasion, with increases of 4.6- and 2.5-fold after AAMP OE, respectively (Figures 2E and 2F). These data were consistent with our finding that increased AAMP expression was associated with more severe lymph node metastasis in patients with human CRC (Figure 1H).
Figure 2.

AAMP promotes colon cancer cell migration and invasion in vitro
(A) Transwell migration assay of HCT116 cells transfected with sh-control or sh-AAMP. (B) Transwell migration assay of MC38 cells transfected with sh-control or sh-AAMP. (C) Transwell invasion assay of HCT116 cells transfected with sh-control or sh-AAMP. (D) Transwell invasion assay of MC38 cells transfected with sh-control or sh-AAMP. (E) Transwell migration assay of HCT116 cells transfected with Vector or OE-AAMP plasmids. (F) Transwell invasion assay of HCT116 cells transfected with Vector or OE-AAMP plasmids. Student's t test; ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01; n = 5, mean ± SEM. Scale bars, 50 μm.
To verify the positive role of AAMP in CRC metastasis in vivo, we performed in vivo imaging analysis to monitor the effect of AAMP on tumor metastasis in nude mice injected with HCT116-luc cells (luciferase-expressing stable cells). AAMP deficiency significantly suppressed tumor cell metastasis to the lung (Figures 3A and 3B), accompanied by a reduced number and size of metastatic nodules and increased body weight (Figures 3C–3G).
Figure 3.

AAMP silencing inhibits colon cancer cell metastasis in vivo
(A) Representative luminescence images of metastases acquired with an IVIS imaging system. Luminescence imaging in nude mice inoculated with colon cancer cells via tail vein injection at the fifth week (n = 6, 6). (B) Quantitative analysis of fluorescence intensity showing metastases in the control and AAMP KD groups. Student's t test; ∗∗∗p < 0.001; n = 6, mean ± SEM. (C) Representative images of lung metastases. The red arrows indicate the metastatic nodules. (D) Representative H&E staining images of lung metastases. The red arrows indicate the metastatic nodules. Scale bars, 200 μm. (E) The number of metastatic nodules in the lung. Student's t test; ∗∗∗∗p < 0.0001; n = 6, mean ± SEM. (F) The size of metastatic nodules in the lung. (G) Body weight change. Student's t test; ∗p < 0.05; n = 6, mean ± SEM. (H) Representative AAMP IHC staining images in paired primary CRC and metastatic tissues from CRC patients (n = 27). (I) Statistical analysis of paired patient samples (primary versus metastatic tissues in the same patient). Student's t test; ∗p < 0.05; n = 27, mean ± SEM. IVIS, in vivo imaging system.
We further analyzed the expression of AAMP in human CRC patient metastases versus primary tumors via IHC staining of patient tissue array samples (paired primary versus metastatic tissues from the same patient). A detailed description of the clinical features of the patients from whom the metastasis samples used in this study were obtained is provided in Table S3. Among the specimens obtained from the 31 patients, 27 biopsy specimens contained both primary and matched metastatic CRC tissues, whereas the other 4 had only primary CRC tissues. Among the 27 matched samples, we found significantly higher expression of AAMP in metastatic tissues than in primary CRC tissues (p = 0.0431) (Figures 3H and 3I). Thus, our data demonstrate a positive correlation between AAMP expression and CRC metastasis.
AAMP regulates CRC cell morphology and EMT
To explore the mechanisms underlying the promotive effect of AAMP on CRC metastasis, transcriptome sequencing (AAMP KD versus AAMP control HCT116 cells) was conducted. GO analysis showed that pathways associated with regulation of the cytoskeleton and cell migration were significantly enriched (Figure 4A). Upon AAMP KD or OE, we noted prominent morphological changes in colon cancer cells. AAMP KD in HCT116 cells caused a pronounced reduction in the cell area, with reduced F-actin elongation (p < 0.0001), whereas AAMP OE resulted in a significant increase in the cell area, with outward F-actin elongation (p < 0.0001) (Figures 4B and 4C). Cell shape is mainly controlled by the organization of the cytoskeleton, which can be monitored by F-actin staining, and by actomyosin-mediated cell contractility, which can be monitored by assessing the phosphorylation of ERM (p-ERM).9 Cytoskeletal F-actin staining with fluorescent phalloidin confirmed the morphological changes in HCT116 cells upon AAMP KD and OE. In addition, we examined the effect of altered AAMP expression on cell contractility by assessing the phosphorylation status of ERM via immunoblot analysis. The results revealed a decrease in the amount of p-ERM upon AAMP KD. In contrast, AAMP OE in HCT116 cells enhanced ERM phosphorylation (Figure 4D).
Figure 4.

AAMP regulates colon cancer cell morphology and EMT
(A) Transcriptome sequencing of control and AAMP KD HCT116 cells was performed, and a GO Circos plot was generated through analysis in R. (B) Representative images of changes in the morphology of HCT116 cells upon AAMP KD (left). Red represents phalloidin (F-actin) and blue represents DAPI (nucleus). Quantification of the of HCT116 cell area upon AAMP KD (right). Student's t test; ∗∗∗∗p < 0.0001. (C) Representative images of changes in the morphology of HCT116 cells upon AAMP OE (left). Red represents phalloidin (F-actin) and blue represents DAPI (nucleus). Quantification of the HCT116 cell area upon AAMP OE (right). Student's t test; ∗∗∗∗p < 0.0001. (D) Immunoblot analysis of AAMP and p-ERM in HCT116 cells transfected with si-control, si-AAMP, Vector, or OE-AAMP OE plasmids. (E) Immunoblot analysis of AAMP, N-cadherin, E-cadherin, and Snail in HCT116 cells transfected with si-control, si-AAMP, Vector, or OE-AAMP OE plasmids. KD, knockdown; OE, overexpression.
Epithelial-mesenchymal transition (EMT) is a process by which epithelial cells lose their polarity and cell-cell adhesion and undergo cytoskeletal changes to gain migratory and invasive properties to acquire a mesenchymal cell phenotype.33 Therefore, we further explored the effect of AAMP on EMT in CRC cells. We found that AAMP KD significantly increased the level of the epithelial marker E-cadherin but decreased the levels of the mesenchymal marker N-cadherin and the EMT-inducing transcription factor Snail. Conversely, AAMP OE increased the levels of N-cadherin and Snail but decreased that of E-cadherin (Figure 4E).
Together, our data indicate that AAMP facilitates CRC metastasis by remodeling the cytoskeleton and promoting cell contractility and EMT.
AAMP increases the activity of RhoA by inhibiting its degradation
RhoA can participate in the process of EMT and increase the phosphorylation of ERM to induce the assembly of the contractile actin cortex and initiation of amoeboid-like migration.9,34 Notably, RhoA was confirmed to promote colon cancer metastasis.10, 11, 12 Our results also showed that inhibiting RhoA activity with CCG-1423 attenuated the migration of HCT116 cells and RKO cells (Figures S4A and S4B). It was previously reported that AAMP can accelerate endothelial cell migration through increasing the activity of RhoA without a change in the total RhoA level.27 Given these findings, we sought to determine whether AAMP affects RhoA in CRC cells. As shown in Figure 5A, the RhoA G-LISA activation assay indicated reduced RhoA activity after AAMP KD (p = 0.0021). However, the total RhoA protein level was also reduced upon AAMP KD but increased upon AAMP OE (Figures 5B and S4C), which indicated that AAMP may promote CRC metastasis in a way different from the mechanisms reported by others. Under the same conditions, the mRNA levels of RhoA remained unchanged (Figure 5C), thus ruling out a transcriptional regulatory mechanism. To further exclude the possibility that AAMP might regulate the synthesis of the RhoA protein, we examined the changes in the RhoA protein level after AAMP KD in the presence of the protein synthesis inhibitor cycloheximide (CHX) or proteasome inhibitor MG132. We found that inhibition of protein degradation with MG132 but not treatment with the protein synthesis inhibitor CHX restored the RhoA protein level after AAMP KD (Figures 5D and 5E). To further determine whether AAMP affects the stability of RhoA, we added CHX to HCT116 and MC38 cells. RhoA protein degradation was faster in the AAMP KD groups than in the control groups (Figure 5F). Next, we reasoned that the reduction in RhoA activity mediated by AAMP KD in CRC cells resulted from a reduction in the total RhoA protein expression level. RhoA activity was assayed again after AAMP KD in the presence of MG132. As expected, RhoA activity was restored by MG132 (Figure 5G).
Figure 5.

AAMP inhibits degradation and increases the levels of total and active RhoA in colon cancer cells
(A) RhoA activity in control and AAMP KD HCT116 cells. Student's t test; ∗∗p < 0.01; n = 3, mean ± SEM. (B) Immunoblot analysis of RhoA in HCT116 cells transfected with si-control or si-AAMP. (C) qRT-PCR analysis of RhoA mRNA expression in HCT116 and MC38 cells transfected with si-control or si-AAMP. Student's t test; NS, no significance; n = 3, mean ± SEM. (D) Immunoblot analysis of AAMP and RhoA in HCT116 cells after treatment with cycloheximide (CHX) (10 μg/mL, 3 h) or MG132 (10 μM, 3 h). (E) Immunoblot analysis of AAMP and RhoA in MC38 cells after treatment with CHX (10 μg/mL, 3 h) or MG132 (10 μM, 3 h). (F) Immunoblot analysis of AAMP and RhoA in HCT116 or MC38 cells treated with CHX (10 μg/mL) at the indicated time points. Densitometric analysis results are shown on the right. (G) RhoA activity in control and AAMP KD HCT116 cells treated with MG132 (10 μM, 3 h). Student's t test; NS, no significance; n = 3, mean ± SEM. (H) Transwell migration assays (upper) and invasion assay of HCT116 cells transfected with Vector + si-control, OE-AAMP + si-control, or OE-AAMP + si-RhoA. Student's t test; ∗∗p < 0.01, ∗∗∗p < 0.001, NS, no significance; mean ± SEM.
To explore the biological significance of RhoA in the metastasis-promoting function of AAMP, we examined the influence of RhoA KD on the metastasis of AAMP-overexpressing HCT116 cells. As shown in Figure 5H, RhoA KD significantly attenuated the migration and invasion capacities of AAMP OE HCT116 cells. These data suggest that AAMP promotes CRC metastasis at least partially through RhoA.
Collectively, our results show that AAMP promotes CRC metastasis by increasing the level of active RhoA through regulating the degradation of the RhoA protein.
AAMP exerts metastasis-promoting activity by binding to RhoA and inhibiting its SMURF2-mediated ubiquitination and degradation
To gain insights into how AAMP regulates RhoA in the acceleration of CRC metastasis, we searched UbiBrowser (http://ubibrowser.ncpsb.org.cn/ubibrowser/) to predict the E3 ubiquitin ligase that mediates the ubiquitination and degradation of RhoA. As shown in Figures 6A, 20 proteins were predicted to be E3 ligases for RhoA. We then selected the top 3 proteins (NEDD4, NEDD4L, and SMURF2) and further biochemically validated the endogenous interactions between RhoA and the predicted E3 ligases. Coimmunoprecipitation (coIP) assays with an anti-RhoA antibody in HCT116 cells were performed. We found that RhoA could not bind to NEDD4 or NEDD4L (Figures S5A and S5B) but could bind to SMURF2 (Figure 6B). Consistent with this finding, RhoA coenrichment was observed after pull-down with an anti-SMURF2 antibody (Figure 6C). In addition, SMURF2 KD reduced RhoA ubiquitination and increased the amounts of both total and active RhoA protein (Figures 6D and 6E). These results indicate that SMURF2 can mediate RhoA ubiquitination and degradation as its E3 ligase.
Figure 6.

AAMP binds to RhoA and inhibits its SMURF2-mediated ubiquitination and degradation
(A) E3 ubiquitin ligases of RhoA were predicted with UbiBrowser (http://ubibrowser.ncpsb.org.cn/ubibrowser/). (B and C) Co-IP and immunoblot analysis of HCT116 cell extracts with the indicated antibodies. IP was performed with IgG as a negative control. (D) Ubiquitination of RhoA, total RhoA levels, and SMURF2 levels were assayed in HCT116 cells transfected with si-control or si-SMURF2. Cells were treated with MG132 (10 μM) for 10 h before harvesting. (E) Activity of RhoA in HCT116 cells transfected with si-control or si-SMURF2. Student's t test; ∗∗∗p < 0.001; n = 3, mean ± SEM. (F) Ubiquitination of RhoA, total RhoA levels, and SMURF2 levels were assayed in HCT116 cells transfected with si-control, si-AAMP, or si-AAMP + si-SMURF2. Cells were treated with MG132 (10 μM) for 10 h before harvesting. (G) Co-IP and immunoblot analysis of HEK293T cell extracts with the indicated antibodies. (H) Co-IP and immunoblot analysis of HEK293T, HCT116, or LoVo cell extracts with the indicated antibodies. (I) Ubiquitination of RhoA and the interaction between RhoA and SMURF2 were assayed in control and AAMP KD HCT116 cells.
To explore whether SMURF2 is involved in the regulation of RhoA by AAMP, we simultaneously silenced AAMP and SMURF2 in HCT116 cells. MG132 (10 μM, 10 h) was used to accumulate ubiquitinated proteins. We found that the increased RhoA ubiquitination and decreased total RhoA expression levels resulting from AAMP KD were restored to the control levels (Figure 6F). We further investigated the impact of SMURF2 on CRC metastasis. In line with our hypothesis, SMURF2 KD promoted HCT116 and MC38 cell migration and invasion, as determined through Transwell chamber assays and wound healing assays (Figures S5C–S5F). Given that the WD40 repeats of AAMP can mediate its protein-protein interactions, we explored whether AAMP interacts with RhoA or SMURF2. CoIP results showed that AAMP bound to RhoA in HEK293T cells, as well as in HCT116 and LoVo cells (Figures 6G and 6H). Notably, AAMP could not interact with SMURF2 (Figure S5G). We next examined the SMURF2-RhoA interaction and RhoA ubiquitination in HCT116 cells transfected with AAMP siRNA. As shown in Figure 6I, the binding of SMURF2 to RhoA and the ubiquitination of RhoA decreased after AAMP KD, indicating that AAMP KD protected RhoA from SMURF2-mediated ubiquitination and degradation.
Together, our findings suggest that AAMP and SMURF2 competitively bind to RhoA, thereby inhibiting SMURF2-mediated ubiquitination and degradation of RhoA and ultimately promoting CRC metastasis.
The expression of RhoA was positively correlated with that of AAMP but negatively correlated with that of SMURF2
Given that AAMP can stabilize the RhoA protein by inhibiting its SMURF2-mediated degradation, we proposed that the protein expression levels of AAMP and RhoA would be positively correlated but that those of SMURF2 and RhoA would be negatively correlated in CRC. To test this hypothesis, we performed IHC staining in human CRC samples, and representative images are shown in Figure 7A. In line with our hypothesis, linear regression analysis of the IHC scores in human CRC samples revealed a strong positive correlation between AAMP and RhoA protein expression levels (R2 = 0.530, p = 0.016) (Figure 7B), and a strong negative correlation between SMURF2 and RhoA protein expression levels (R2 = −0.578, p = 0.008) (Figure 7C).
Figure 7.

Correlations between RhoA and AAMP expression and between RhoA and SMURF2 expression
(A) IHC staining of human CRC tissues with anti-RhoA, anti-AAMP or anti-SMURF2 antibodies. Representative patient samples are shown. A total of 14 patient samples were stained and analyzed. “P” is for patient and the number is for the order in which they appear in the figures. (B) Linear regression analysis of RhoA and AAMP IHC scores. (C) Linear regression analysis of RhoA and SMURF2 IHC scores. IHC score: staining area × staining intensity.
Discussion
Despite developments in the prognosis and treatment of CRC, no significant progress has been made in improving the survival of patients with metastatic CRC. Thus, there is an urgent need for a better understanding of the biology of metastatic CRC. AAMP is a motility-associated protein.24 Previous studies have demonstrated that AAMP can stimulate the migration of endothelial cells and smooth muscle cells as well as tumor cells such as breast cancer cells and NSCLC cells.26,27,29,31 However, the role of AAMP in CRC metastasis has not been studied. Interestingly, the mechanisms underlying the metastasis-promoting function of AAMP in different cell types are quite diverse. AAMP promotes the migration of endothelial cells by activating RhoA without a change in the total RhoA protein level27; smooth muscle cells by binding to TPα and TPβ26; and NSCLC cells by interacting with CDC42.31 Therefore, the mechanism by which AAMP functions in CRC cells could be different from those previously reported. Indeed, we found that AAMP promoted CRC metastasis by increasing the level of total RhoA protein, thus increasing the level of active RhoA protein. More interestingly, we found that RhoA activation was dependent on its reduced ubiquitination and degradation. Through our bioinformatic analysis and experimental verification approaches, we report that AAMP promotes CRC metastasis by competitively binding to RhoA and inhibiting its degradation mediated by SMURF2 (an E3 ligase for RhoA), thereby increasing the level of active RhoA.
Our study provides experimental evidence that AAMP is obviously overexpressed in CRC primary tumors and particularly in CRC metastases. Reduced OS and DFS times were observed in patients with high AAMP expression compared with those with low AAMP expression. Our results also suggested that AAMP is related to metastasis in CRC patients and can be considered a clinical prognostic biomarker in CRC. Consistent with these findings, we confirmed that AAMP enhanced the migration and invasion of colon cancer cells in vitro and in vivo and found that AAMP regulated colon cancer cell morphology (cytoskeletal organization and cell contractility) and EMT.
Cells are remarkably flexible in their migration behavior, adapting rapidly to changing cues in their environment to change their shape. Tumor cells can adopt a mesenchymal migration mode with an elongated cell shape or display ameboid/blebbing motility with a rounded cell morphology during metastasis.35 GTPase and ROCK signaling have been established as central players in regulating the transition between mesenchymal and ameboid motility.36 To investigate the mechanism by which AAMP is involved in the regulation of colon cancer cell shape, we first focused on RhoA, as the RhoA subfamily is one of the most-studied subfamilies of GTPases, and its members have been reported to exert oncogenic effects in many cancers, including CRC.10,11,37,38 Moreover, it was demonstrated that AAMP can increase the level of activated RhoA protein without altering the total RhoA protein level to promote endothelial cell migration and angiogenesis,27 but the underlying mechanism is not understood. In contrast, we reported that AAMP can protect RhoA from ubiquitination and degradation to increase the levels of total and active RhoA protein, thereby facilitating CRC metastasis.
We further explored how AAMP regulates RhoA ubiquitination and degradation. Since no E3 ligase has been reported to mediate RhoA ubiquitination and degradation in CRC, we first predicted the E3 ligase for RhoA with UbiBrowser, which is a resource for known and predicted human ubiquitin ligase (E3)-substrate interaction networks. After verification by coIP, we found that among the top 3 (NEDD4, NEDD4L, and SMURF2) predicted E3 ligases, only SMURF2 can bind to RhoA and mediate RhoA ubiquitination and degradation. Indeed, SMURF2 was reported to hinder CRC promotion by promoting the ubiquitination and degradation of ChREBP and SATB1.22,23 Consistent with these observations, we found that SMURF2 inhibits CRC cell migration and invasion. As AAMP often functions through protein-protein interactions, we sought to determine whether AAMP can bind to RhoA or SMURF2. Interestingly, we found that AAMP bound to RhoA and suppressed the SMURF2-RhoA interaction and RhoA ubiquitination and degradation, which accelerated CRC metastasis. Therefore, our results revealed a vital role of the AAMP-SMURF2-RhoA signaling pathway in CRC metastasis.
Since Rho GTPase pathways regulate the dissemination of malignant cells, key components in these pathways are attractive targets for therapeutic intervention. However, all members of the small GTPase superfamily are generally considered to be undruggable, as they lack stable cavities in addition to nucleotide binding pockets.8 Beneficial effects of ROCK (an Rho downstream kinase effector) inhibitors, such as Y27632 and fasudil, have been observed in many types of cancers.39,40 However, targeting ROCK is accompanied by contraction of blood vessels and a high likelihood of inducing hypotension.39 As the poor pharmacokinetics and specificity of inhibitors targeting Rho GTPases or their downstream effector ROCK severely limit the opportunity for clinical development of these agents, we could focus on the upstream regulator of Rho GTPases. Here, we found that AAMP bound to RhoA and disrupted SMURF2-mediated RhoA ubiquitination and degradation. Thus, blocking the AAMP-RhoA interaction or activating SMURF2 could be a promising strategy to inhibit CRC metastasis.
In addition to the observation that AAMP is involved in the regulation of CRC cell migration and invasion, our transcriptome sequencing data revealed that AAMP might also regulate drug transport and participate in drug resistance (Figure 4A). Indeed, we found that AAMP KD significantly increased the sensitivity of CRC cells to SN38, the active metabolite of irinotecan that is often used to treat CRC (Figures S6A and S6B). In contrast, AAMP OE reduced SN38 sensitivity (Figures S6C and S6D). These results were in line with a previous report that demonstrated that AAMP promoted drug (icotinib and doxorubicin) resistance in NSCLC cells.30 In addition, the metabolic states of cancer cells determine their fates in nutrient-poor environments.41 AAMP KD further disrupted metabolic pathways involved in cancer progression (data not shown). Therefore, our data suggest a multifaceted function of AAMP in regulating drug resistance and metabolism in CRC, and the mechanisms underlying this function will be further investigated in follow-up studies in the near future.
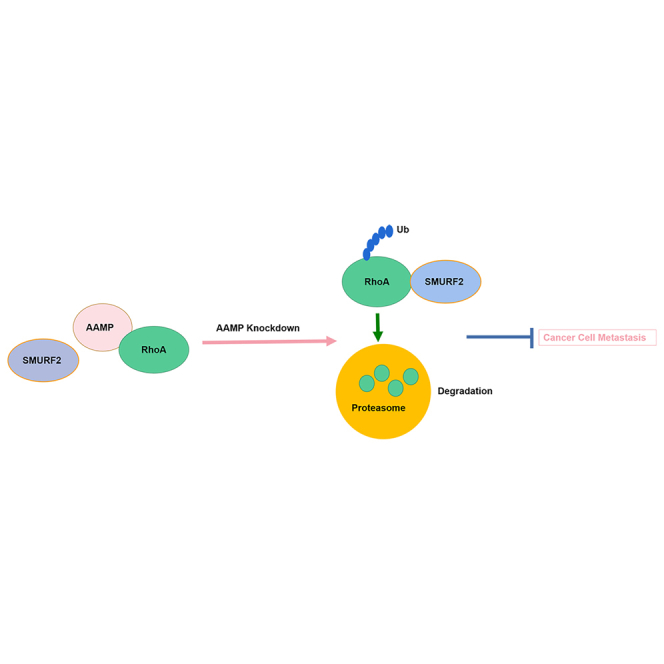
In summary, we discover the role of AAMP in CRC and identify SMURF2 as an E3 ligase for RhoA ubiquitination and degradation. Our study revealed that AAMP facilitates CRC metastasis by modulating RhoA-associated signaling pathways. AAMP prevents RhoA from binding to SMURF2, protects RhoA from SMURF2-mediated ubiquitination and degradation and subsequently induces robust RhoA activity to promote ameboid (bleb) migration (Figure 8). Hence, therapeutic interventions that disrupt the functional interplay between AAMP and RhoA or activate SMURF2 might provide promising strategies to treat CRC. However, the binding sites by which AAMP or SMURF2 interact with RhoA are still unclear and need to be studied. In addition, whether other mechanisms in addition to the AAMP-SMURF2-RhoA pathway also participate in the CRC metastasis-promoting effect of AAMP remains to be further explored.
Figure 8.

Proposed model showing the mechanism by which AAMP promotes CRC cell metastasis
AAMP stabilizes RhoA by inhibiting the interaction between RhoA and SMURF2 and the subsequent SMURF2-mediated ubiquitination and degradation of RhoA, thereby promoting phosphorylation of ERM, ameboid (bleb) migration, and CRC metastasis. AAMP deficiency results in dissociation of RhoA from SMURF2 and results in ubiquitination and degradation of RhoA, which inhibits ameboid (bleb) migration and CRC metastasis.
Materials and methods
Patient samples and IHC assays
This study was approved by the Sir Run Run Shaw Hospital and Zhejiang University Ethics Committee. For analysis of AAMP mRNA levels in human CRC and adjacent normal colon tissues, 10 pairs of samples were obtained during surgery at Sir Run Run Shaw Hospital. To assess the correlations between the AAMP and RhoA and the RhoA and SMURF2 IHC scores, 14 fresh samples of human CRC tissue were obtained during surgery at Sir Run Run Shaw Hospital. To assess the correlations between AAMP IHC scores and clinical characteristics of patients with CRC, 180-spot, paraffin-embedded tissue array chips (HColA180Su15), including 79 paired CRC and normal tissues and 22 tumor tissues obtained from patients with 8–9 years of follow-up information, were purchased from Shanghai Outdo Biotech (Shanghai, China). Detailed clinical features of the CRC samples are summarized in Tables S1 and S2. To compare AAMP protein levels between primary and metastatic tissues of CRC, 75-spot, paraffin-embedded tissue array chips (HLin-Ade075Met-01)—including 15 paired primary, metastatic tumor and normal tissues, 12 paired primary and metastatic tumor tissues, and 4 metastatic tumor tissues (2 tissues were lost) —were also purchased from Shanghai Outdo Biotech.
For the samples from Sir Run Run Shaw Hospital, IHC scores were calculated as the IHC staining intensity scores multiplied by the staining area scores. The staining intensity was divided into four levels: 0 indicated negative staining, 1 indicated weakly positive staining, 2 indicated moderately positive staining, and 3 indicated strongly positive staining. The staining area was divided into five levels: 0 indicated no staining, 1 indicated a positive staining area of 1%–25%, 2 indicated a positive staining area of 26%–50%, 3 indicated a positive staining area of 51%–75%, and 4 indicated a positive staining area of 76%–100%. For samples from Shanghai Outdo Biotech, IHC scores were calculated as the IHC staining intensity multiplied by the positive staining rate. The staining intensity was divided into four levels: 0 indicated negative staining, 0.5 indicated weakly positive staining, 1 indicated moderately positive staining, and 2 indicated strongly positive staining. The positive staining rate was the ratio of the number of positive cells to the total number of cells.
For large-scale data analysis of the AAMP mRNA level in CRC and normal colon tissues, data were obtained from the Oncomine database. For survival analysis of patients with CRC, data were obtained from the GEPIA website.
Cell lines
HEK293T cell line, NCM460 cell, human colon cancer cell lines HCT116, HT29, LoVo, SW480, SW620, SW48, and RKO, and mouse colon cancer cell line MC38, were purchased from the Chinese Academy of Sciences (Shanghai, China). NCM460, LoVo, SW480, SW620, and RKO cells were cultured in RPMI 1640 medium (Gibco, USA); HEK293T, MC38, HT29, and SW48 cells were cultured in DMEM with high glucose (Gibco); HCT116 cells were cultured in McCoy's 5A medium (Gibco). All above media contained 10% fetal bovine serum (FBS, Gibco). All cell lines were cultured in a 37°C humidified incubator with a mixture of 95% air and 5% CO2. All cell lines were tested for mycoplasma contamination.
Mouse model of lung metastasis
Animal experiments were approved by the Animal Care and Use Committee of Zhejiang University. Male nude mice, 4–6 weeks old, were purchased from Shanghai SLAC Laboratory Animal (Shanghai, China). Luciferase-expressing HCT116 cells transduced with lentivirus-expressing sh-control or sh-AAMP (1 × 106 cells/mouse) were injected via the tail vein. D-Luciferin (15 mg/mL, 150 mg/kg) obtained from Meilunbio (Dalian, China) was injected intraperitoneally into mice 15 min before imaging. Metastasis was monitored by bioluminescence imaging using the IVIS imaging system (PerkinElmer, USA). Endpoint assays were conducted 8 weeks after inoculation.
Reagents and antibodies
Cycloheximide was purchased from Beyotime Biotechnology (Hangzhou, China). MG132 was purchased from Sigma-Aldrich (USA). Rhodamine phalloidin was purchased from Cytoskeleton (USA). A G-LISA RhoA Activation Assay Biochem Kit was purchased from Cytoskeleton. Antibodies against AAMP (used for western blot [WB]) and RhoA (used for WB and IHC staining) were purchased from Acbam. Antibodies against β-actin, p-ERM, N-cadherin, E-cadherin, Snail, Ubiquitin, NEDD4, NEDD4L, and SMURF2 were obtained from Cell Signal Technology. Antibody against AAMP (used for IHC staining) was obtained from GeneTex. Antibody against RhoA (used for IP) was purchased from Santa Cruz Biotechnology.
qRT-PCR
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA) reagent. cDNA was generated from 1,000 ng of total RNA using PrimeScript RT reagent (Takara, Japan). Real-time qPCR was performed using a SYBR Premix Ex Taq II (Takara, Japan). The 2−ΔΔCt method was used to calculate the relative amounts of the target RNAs. The primer sequences used in this study are as follows: human β-actin: forward primer 5′-ACTCTTCCAGCCTTCCTTCC-3′, reverse primer 5′-CGTCATACTCCTGCTTGCTG-3′; mouse β-actin: forward primer 5′-AGAGGGAAATCGTGCGTGAC-3′, reverse primer 5′-CAATAGTGATGACCTGGCCGT-3′; human AAMP: forward primer 5′-CTTTGCATTGCACTCAGCAT-3′, reverse primer 5′-GCTGAAACCAGCACAAGTCA-3′; mouse AAMP: forward primer 5′-AGTGGATCGGTAGCTTTCGC-3′, reverse primer 5′-AGATCATCTGCCGGGTCC-3′; human RhoA: forward primer 5′-GAGCACACAAGGCGGGAG-3′, reverse primer 5′-TGCCATATCTCTGCCTTCTTCA-3′; mouse RhoA: forward primer 5′-AGCTTGTGGTAAGACATGCTTG-3′, reverse primer 5′-GTGTCCCATAAAGCCAACTCTAC-3′; human SMURF2: forward primer 5′-GGCAATGCCATTCTACAGATACT-3′, reverse primer 5′-CAACCGAGAAATCCAGCACCT-3′; mouse SMURF2: forward primer 5′-AAACAGTTGCTTGGGAAGTCA-3′, reverse primer 5′-TGCTCAACACAGAAGGTATGGT-3′.
F-Actin staining
Cells were cultured in glass-bottom dishes in complete medium. Cells were then fixed with 4% paraformaldehyde solution at room temperature for 10 min and permeabilized at room temperature with 0.5% Triton X-100 for 5 min. Subsequently, cells were incubated in rhodamine phalloidin (200 nM) for 30 min at 4°C. Cells were then washed three times with PBS and incubated with DAPI. Samples were then visualized using a Nikon A1 microscope, and cell areas were analyzed via a high content cell imaging analysis system.
Transwell assay
For the migration assay, cell culture inserts with an 8-μm pore size were used directly. For the invasion assay, 25 μL of Matrigel (Corning, USA) was mixed with 1 mL of serum-free culture medium; 100 μL of the mixture was added to the bottom of inserts and incubated at 37°C for 1 h.
Cells were harvested and resuspended at 1 × 106 cells/mL in FBS-free culture medium. A 200 μL volume of the cell suspension was added to the upper chambers containing the cell culture inserts. Then, 600 μL of culture medium containing 10% FBS was added to the lower chambers. After 24 h, cell migration or invasion was assessed. Cells on the top surface of the inserts were removed with a cotton swab. Cells on the bottom surface of the inserts were fixed with 4% paraformaldehyde for 5 min at room temperature and then washed with PBS. Migrated or invaded cells were stained with 0.05% crystal violet in distilled water for 30 min or with DAPI for 10 min. After two washes with PBS, migrated or invaded cells were visualized and photographed.
Wound healing assay
Cells were plated and cultured in six-well plates to 100% confluence. Wounds were made in the confluent cell layer by making a vertical scratch in each wells. Marker lines were made in the horizontal direction. Cell migration was assessed every 24 h.
Immunoprecipitation assay
Cells were lysed in lysis buffer containing 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.5% NP40, 0.09% NaN3, 20 nM NaF, 1 mM Na3VO4, 10 mM PMSF, and a protease and phosphatase inhibitor cocktail. Lysates were incubated with anti-flag M2 magnetic beads (Sigma, USA) overnight at 4°C. Alternatively, lysates were immunoprecipitated with the indicated antibody for 4 h at 4°C. Then, samples were incubated with protein A/G Sepharose beads (GE Healthcare) overnight at 4°C. After washing three times with wash buffer containing 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.5% NP40, and 0.018% NaN3, 2× SDS loading buffer was added. Then, the samples were boiled, and immunoblot analyses were performed.
RNA interference, plasmid construction, and transfections
Cells were transfected with siRNA using RNAiMAX (Thermo Fisher Scientific) according to the manufacturer's protocol. siRNAs were purchased from RiboBio (Guangzhou, China), and the target sequences are as follows: h-AAMP-siRNA no. 1: 5′-GGAGGTCTGGTCCTTTGAA-3′; h-AAMP-siRNA no. 2: 5′-GTGCTGGTTTCAGCCATGA-3′; h-AAMP-siRNA no. 3: 5′-CGCAGACTCTTAGGCATCA-3′; m-AAMP-siRNA no. 1: 5′-GCTGTGGTGTACACCTGTA-3′; m-AAMP-siRNA no. 2: 5′-GCAGGTAGACACCAAGGAA-3′; m-AAMP-siRNA no. 3: 5′-GGGAGGAGAGTGAGTCTAA-3′; h-SMURF2-siRNA no. 1: 5′-CACTCCAATTAGTGGAACA-3′; h-SMURF2-siRNA no. 2: 5′-ACAGCAAGTGGTATCGTTA-3′; h-SMURF2-siRNA no. 3: 5′-GTCAGGTCACAACGACATA-3′; m-SMURF2-siRNA: 5′-GTGCCATTCTACAGATACTTT-3′; h-RhoA-siRNA no. 1: 5′-GAAGGATCTTCGGAATGAT-3′; h-RhoA-siRNA no. 2: 5′-GACCAAAGATGGAGTGAGA-3′; h-RhoA-siRNA no. 3: 5′-AGAACTATGTGGCAGATAT-3′.
Cells were transfected with plasmids with jetPRIME purchased from Polyplus (USA) according to the manufacturer's protocol. The plasmids used in this study were purchased from WZ Biosciences (Shandong, China).
Statistical analysis
Unless otherwise noted, quantitative data are presented as the mean ± standard error values. Data were analyzed with Student's t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). p < 0.05 was considered to indicate a significant difference.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81972745), the Ten Thousand Plan Youth Talent Support Program of Zhejiang Province (ZJWR0108009), and Zhejiang Medical Innovative Discipline Construction Project-2016.
Author contributions
J.X. and W.H. conceived and designed the study. Y.W., B.L., W.L., and R.Z. performed the experiments. Y.W. and B.L. analyzed the data and prepared the figures. Y.W. wrote the paper. J.X. and W.H. modified the paper. All authors reviewed the manuscript, and the manuscript was approved by all authors for publication.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.11.007.
Contributor Information
Weidong Han, Email: hanwd@zju.edu.cn.
Jiansheng Xie, Email: xiejiansheng@zju.edu.cn.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Tauriello D.V.F., Palomo-Ponce S., Stork D., Berenguer-Llergo A., Badia-Ramentol J., Iglesias M., Sevillano M., Ibiza S., Canellas A., Hernando-Momblona X., et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Clark A.G., Vignjevic D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015;36:13–22. doi: 10.1016/j.ceb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Ridley A.J., Comoglio P.M., Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol. Cell Biol. 1995;15:1110–1122. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu D.D., Chen X., Sun K.X., Wang L.L., Chen S., Zhao Y. Role of the lncRNA ABHD11-AS(1) in the tumorigenesis and progression of epithelial ovarian cancer through targeted regulation of RhoC. Mol. Cancer. 2017;16:138. doi: 10.1186/s12943-017-0709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J., Kim D.H., Shah S.R., Kim H.N., Kshitiz K.P., Quiñones-Hinojosa A., Levchenko A. Switch-like enhancement of epithelial-mesenchymal transition by YAP through feedback regulation of WT1 and Rho-family GTPases. Nat. Commun. 2019;10:2797. doi: 10.1038/s41467-019-10729-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen S., Gosens R., Wieland T., Schmidt M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018;183:1–21. doi: 10.1016/j.pharmthera.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Tan P., Ye Y., He L. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 2018;16:e3000051. doi: 10.1371/journal.pbio.3000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X., Wang D., Wang X., Sun S., Zhang Y., Wang S., Miao R., Xu X., Qu X. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J. Exp. Clin. Cancer Res. 2019;38:32. doi: 10.1186/s13046-018-1014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeong D., Park S., Kim H., Kim C.J., Ahn T.S., Bae S.B., Kim H.J., Kim T.H., Im J., Lee M.S., et al. RhoA is associated with invasion and poor prognosis in colorectal cancer. Int. J. Oncol. 2016;48:714–722. doi: 10.3892/ijo.2015.3281. [DOI] [PubMed] [Google Scholar]
- 12.Al-Haidari A.A., Syk I., Thorlacius H. MiR-155-5p positively regulates CCL17-induced colon cancer cell migration by targeting RhoA. Oncotarget. 2017;8:14887–14896. doi: 10.18632/oncotarget.14841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun T., Liu Z., Yang Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer. 2020;19:146. doi: 10.1186/s12943-020-01262-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 15.Morreale F.E., Walden H. Types of ubiquitin ligases. Cell. 2016;165:248–248.e1. doi: 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Kavsak P., Rasmussen R.K., Causing C.G., Bonni S., Zhu H., Thomsen G.H., Wrana J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 17.Jin C., Yang Y.-a., Anver M.R., Morris N., Wang X., Zhang Y.E. Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancing migration and invasiveness. Cancer Res. 2009;69:735–740. doi: 10.1158/0008-5472.CAN-08-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nie J., Xie P., Liu L., Xing G., Chang Z., Yin Y., Tian C., He F., Zhang L. Smad ubiquitylation regulatory factor 1/2 (Smurf1/2) promotes p53 degradation by stabilizing the E3 ligase MDM2. J. Biol. Chem. 2010;285:22818–22830. doi: 10.1074/jbc.M110.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukuchi M., Fukai Y., Masuda N., Miyazaki T., Nakajima M., Sohda M., Manda R., Tsukada K., Kato H., Kuwano H. High-level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res. 2002;62:7162–7165. [PubMed] [Google Scholar]
- 20.Blank M., Tang Y., Yamashita M., Burkett S.S., Cheng S.Y., Zhang Y.E. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat. Med. 2012;18:227–234. doi: 10.1038/nm.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramkumar C., Kong Y., Cui H., Hao S., Jones S.N., Gerstein R.M., Zhang H. Smurf2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. 2012;72:2714–2719. doi: 10.1158/0008-5472.CAN-11-3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y., Yang D., Tian N., Zhang P., Zhu Y., Meng J., Feng M., Lu Y., Liu Q., Tong L., et al. The ubiquitination ligase SMURF2 reduces aerobic glycolysis and colorectal cancer cell proliferation by promoting ChREBP ubiquitination and degradation. J. Biol. Chem. 2019;294:14745–14756. doi: 10.1074/jbc.RA119.007508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu L., Dong L., Wang Y., Liu L., Long H., Li H., Li J., Yang X., Liu Z., Duan G., et al. Reversible regulation of SATB1 ubiquitination by USP47 and SMURF2 mediates colon cancer cell proliferation and tumor progression. Cancer Lett. 2019;448:40–51. doi: 10.1016/j.canlet.2019.01.039. [DOI] [PubMed] [Google Scholar]
- 24.Beckner M.E., Krutzsch H.C., Stracke M.L., Williams S.T., Gallardo J.A., Liotta L.A. Identification of a new immunoglobulin superfamily protein expressed in blood vessels with a heparin-binding consensus sequence. Cancer Res. 1995;55:2140–2149. [PubMed] [Google Scholar]
- 25.Bielig H., Zurek B., Kutsch A., Menning M., Philpott D.J., Sansonetti P.J., Kufer T.A. A function for AAMP in Nod2-mediated NF-kappaB activation. Mol. Immunol. 2009;46:2647–2654. doi: 10.1016/j.molimm.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 26.Reid H.M., Wikstrom K., Kavanagh D.J., Mulvaney E.P., Kinsella B.T. Interaction of angio-associated migratory cell protein with the TPalpha and TPbeta isoforms of the human thromboxane A(2) receptor. Cell Signal. 2011;23:700–717. doi: 10.1016/j.cellsig.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Hu J., Qiu J., Zheng Y., Zhang T., Yin T., Xie X., Wang G. AAMP regulates endothelial cell migration and angiogenesis through RhoA/Rho kinase signaling. Ann. Biomed. Eng. 2016;44:1462–1474. doi: 10.1007/s10439-015-1442-0. [DOI] [PubMed] [Google Scholar]
- 28.Allander S.V., Nupponen N.N., Ringner M., Hostetter G., Maher G.W., Goldberger N., Chen Y., Carpten J., Elkahloun A.G., Meltzer P.S. Gastrointestinal stromal tumors with KIT mutations exhibit a remarkably homogeneous gene expression profile. Cancer Res. 2001;61:8624–8628. [PubMed] [Google Scholar]
- 29.Yin Y., Sanders A.J., Jiang W.G. The impact of angio-associated migratory cell protein (AAMP) on breast cancer cells in vitro and its clinical significance. Anticancer Res. 2013;33:1499–1509. [PubMed] [Google Scholar]
- 30.Yao S., Shi F., Wang Y., Sun X., Sun W., Zhang Y., Liu X., Liu X., Su L. Angio-associated migratory cell protein interacts with epidermal growth factor receptor and enhances proliferation and drug resistance in human non-small cell lung cancer cells. Cell Signal. 2019;61:10–19. doi: 10.1016/j.cellsig.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Yao S., Shi F., Mu N., Li X., Ma G., Wang Y., Sun X., Liu X., Su L. Angio-associated migratory cell protein (AAMP) interacts with cell division cycle 42 (CDC42) and enhances migration and invasion in human non-small cell lung cancer cells. Cancer Lett. 2020;502:1–8. doi: 10.1016/j.canlet.2020.11.050. [DOI] [PubMed] [Google Scholar]
- 32.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 34.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan P., Ye Y., He L., Xie J., Jing J., Ma G., Pan H., Han L., Han W., Zhou Y. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 2018;16:e3000051. doi: 10.1371/journal.pbio.3000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridley A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015;36:103–112. doi: 10.1016/j.ceb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaoui K., Boudhraa Z., Khalifé P., Carmona E., Provencher D., Mes-Masson A.M. Ran promotes membrane targeting and stabilization of RhoA to orchestrate ovarian cancer cell invasion. Nat. Commun. 2019;10:2666. doi: 10.1038/s41467-019-10570-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D.K., Kim E.K., Jung D.W., Kim J. Cytoskeletal alteration modulates cancer cell invasion through RhoA-YAP signaling in stromal fibroblasts. PLoS One. 2019;14:e0214553. doi: 10.1371/journal.pone.0214553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kale V.P., Hengst J.A., Desai D.H., Amin S.G., Yun J.K. The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett. 2015;361:185–196. doi: 10.1016/j.canlet.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 40.Rath N., Olson M.F. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13:900–908. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.