

Summary
Upregulation and stabilization of Foxp3 expression in Tregs are essential for regulating Treg function and immune homeostasis. In this study, gp96 immunization showed obvious therapeutic effects in a Lyn–/– mouse model of systemic lupus erythematosus. Moreover, gp96 alleviated the initiation and progression of MOG-induced experimental autoimmune encephalomyelitis. Immunization of gp96 increased Treg frequency, expansion, and suppressive function. Gene expression profiling identified the NF-κB family member p65 and c-Rel as the key transcription factors for enhanced Foxp3 expression in Treg by gp96. Mutant gp96 within its Toll-like receptor (TLR) binding domain, TLR2 knockout mice, and mice with cell-specific deletion of MyD88, were used to demonstrate that gp96 activated Tregs and induced Foxp3 expression via a TLR2-MyD88-mediated NF-κB signaling pathway. Taken together, these results show that gp96 immunization restricted antibody-induced and Th-induced autoimmune diseases by integrating Treg expansion and activation, indicating its potential clinical usefulness against autoimmune diseases.
Subject areas: Immune response, Genomics, Molecular biology
Graphical abstract
Highlights
-
•
SLE symptoms in Lyn–/– mice are ameliorated by gp96 immunization
-
•
Tregs expanded by gp96 provide potential in suppressing Th-mediated EAE
-
•
Gp96 promotes Treg proliferation, stability, and suppressive function
-
•
Gp96 binds to and activates Treg in a TLR2-MyD88-NF-кB-Foxp3 pathway
Immune response, Genomics, Molecular biology
Introduction
Regulatory T cells (Tregs) are critical for maintaining immune tolerance and deficiency or impaired suppressive function causes severe autoimmune diseases and chronic inflammation. The emergence and characterization of Tregs have provided novel strategies for developing immunotherapies for autoimmune diseases and chronic inflammation (Sakaguchi et al., 2010, 2012). Based on their differentiation site and precursor cells, Tregs are generally classified into two cell subsets, natural Tregs (nTregs) and induced Tregs (iTregs) (Josefowicz and Rudensky, 2009). iTregs may either be differentiated from conventional CD4+ T cells in the periphery (pTregs) or be generated from naïve T cells in vitro by antigen stimulation in the presence of transforming growth factor-β (TGF-β) and interleukin-2 (Josefowicz and Rudensky, 2009; Zhang et al., 2020).
Many studies showed that soluble factors and/or cellular factors derived from innate immune responses could induce expansion and proliferation of CD4+Foxp3+ Treg cells. Even though tumor necrosis factor (TNF) is a major proinflammatory cytokine, interaction of TNF with TNF receptor type 2 promoted expansion and function of mouse CD4+CD25+ T regulatory cell, indicating TNF played a vital role in modulating autoimmunity (Chen et al., 2007). Especially, natural but not inducible regulatory T cells required TNF-α signaling in mediating suppression of colitis in vivo (Housley et al., 2011). Low-dose IL-2 selectively modulated CD4+ T cell subsets by expanding Treg cells in patients with chronic kidney diseases and systemic lupus erythematosus (SLE) (He et al., 2016; Li et al., 2018). IL-33 also enhanced expansion of Treg cell population by activating type 2 innate lymphoid cells and mast cells, thereby contributing to sepsis-induced long-term immunosuppression and limiting papain-induced allergic inflammation (Morita et al., 2015; Nascimento et al., 2017). CD4+CD25+ Treg subset in TLR2−/− mice was significantly reduced in number compared with WT littermate control mice, indicating a link between Tregs and TLR2. Treatment with the TLR2 ligand Pam3Cys, but not LPS (the TLR4 ligand) or CpG (the TLR9 ligand), could promote Treg cell proliferation in vitro (Netea et al., 2004; Sutmuller et al., 2006).
Forkhead box P3 (Foxp3) is regarded as a master transcription factor for the development, stability, and especially, the suppressive function of Tregs. nTreg and pTreg cells are considered T cell lineages and are characterized by their ability to stably express Foxp3 and other Treg signature genes. In contrast, iTregs exhibit relatively unstable phenotypes because they may lose Foxp3 expression in the absence of TGF-β (Ohkura and Sakaguchi, 2020). In addition, under inflammatory and autoimmune conditions some Treg sub-populations (e.g., CD25low or Foxp3low cell subtype) may undergo transdifferentiation into pathogenic Th17 cells by losing Foxp3 expression (Komatsu et al., 2014; Zhang et al., 2020). Foxp3 expression during nTreg cell development has been shown to be regulated by cytokines, metabolic mediators, and inflammatory factors. Relatively strong T cell receptor (TCR) stimulation and multiple co-stimulatory signals and cytokines are required for the induction of Foxp3 expression (Ono, 2020). Recent studies have also shown that Foxp3 transcription is regulated by three conserved non-coding enhancers known as conserved noncoding sequence 1 (CNS1), CNS2, and CNS3, which each have a distinct role in the development, stability, and function of Tregs (Feng et al., 2014; Zheng et al., 2010).
Tregs maintain immunological self-tolerance and prevent a variety of autoimmune diseases, including SLE and rheumatic diseases, which are characterized by chronic immune activation, elevated autoantibodies by dysregulated B cell responses, and the development of pathogenic T cell populations infiltrating the target organ (Dominguez-Villar and Hafler, 2018). Tregs prevent autoimmunity by inhibiting aberrant activation and proliferation of immune effector cells, including CD8+ T cells, B cells, Th17 cells, and Th1 cells (Nadafi et al., 2020; Zhan et al., 2020). In animal models, autoimmune responses can be controlled by reestablishing T-cell balance to favor Tregs. Patients with rheumatic diseases such as SLE usually show abnormal Treg numbers and function (Zhao et al., 2019). The use of in vitro expansion and in vivo induction of heterogeneous or antigen-specific Treg cells has been regarded as a potentially attractive therapeutic approach for autoimmune diseases (Miyara et al., 2014; Zhao et al., 2019). Therefore, immunotherapies targeting Tregs and their balance with T effector cells are an important strategy for the treatment of autoimmune diseases.
The generation of Tregs in the inflammatory, dysregulated immune system of patients with established autoimmune diseases is a major concern for researchers. Various studies have shown that heat shock protein (HSP)-reactive T cells possess immunoregulatory capacity in models of experimental autoimmune diseases. Particularly, mycobacterial HSP60 and HSP70 induce disease inhibitory IL-10-producing regulatory T cells, which can suppress immune responses that occur in autoimmune diseases, such as rheumatoid arthritis, type 1 diabetes, and possibly atherosclerosis and allergy. First clinical trials with HSPs in rheumatoid arthritis and type 1 diabetes also indicate their potential to restore tolerance in autoimmune diseases (van Eden et al., 2005, 2017). Besides HSP60 and HSP70, heat shock protein gp96 also plays important roles in modulating innate and adaptive immunity, and its use in several immunotherapy studies against cancers has shown no severe adverse effects and moderate improvement in the clinical outcome (Randazzo et al., 2012). We and others have demonstrated that immunization of mice with high-dose gp96 significantly increases Treg frequency and suppressive function and largely abrogates Th1 antitumor immunity and T cell-mediated hepatic injury via interaction with its receptors, Toll-like receptors (TLRs) such as TLR2/4 or CD91 on plasmacytoid dendritic cells (pDCs) (Kinner-Bibeau et al., 2017; Li et al., 2013; Liu et al., 2009; Wu et al., 2012). Our goal in the current study was to rebalance the dysregulated immune system in the context of autoimmunity. We have elucidated a gp96-mediated mechanism of controlling Treg activation in vivo that has therapeutic effects in mice. These results could facilitate the development of Treg-targeting immunotherapies for treating autoimmune diseases.
Results
High-dose gp96 immunization attenuates autoimmunity and lupus nephritis in Lyn–/– mice
We first assessed the effectiveness of gp96 immunization as a potential strategy for treating SLE. Lyn is a Src family kinase that functions as a modulator in multiple aspects of immune signaling (Xu et al., 2005). In addition, recent research has also shown a genetic association between Lyn and SLE (Mohan and Putterman, 2015). The Lyn–/– mouse spontaneously develops a highly penetrant autoimmune and inflammatory disease characterized by anti-double-stranded DNA (anti-dsDNA) IgG antibodies and glomerulonephritis that is similar to human SLE patients (Brodie et al., 2018). Lyn–/– mice were immunized as described in Figure S1A. Figure 1A shows that compared to control mice, anti-dsDNA antibody development was significantly decreased in Lyn–/– mice immunized with gp96 at 16 weeks old (P = 0.002). Similar results were observed in the MRL/lpr mouse model (Figure S1B). Each dose (200 μg/mouse) of recombinant gp96 for immunization only contained around 1.6 EU of LPS, excluding the possibility that contaminated endotoxin may disturb the results.
Figure 1.

Amelioration of systemic lupus erythematosus (SLE)-like symptoms in Lyn–/– mice by heat shock protein gp96 immunization
(A) Female Lyn–/– mice at 16 weeks old were immunized with 200 μg gp96 or saline as control (n = 5/group). Serum anti-dsDNA antibodies were determined using ELISA in Lyn–/– mice 1 week after the last immunization.
(B–E) FACS analysis of Foxp3+CD4+ Treg cells (B), CXCR5+PD-1+Foxp3+ Tfr cells, (C), CXCR5+PD1+Foxp3– Tfh cells (D) and activated conventional CD4+ T cells (CD44hi CD62Llow) (E) in the spleen of mice immunized at 16 weeks of age.
(F–H) FACS analysis of germinal center B cells (F), plasma cells (G) from the spleen, and memory B cells (H) from bone marrow.
(I) Absolute numbers of anti-dsDNA-specific antibody-secreting cells (ASCs) were determined using ELISPOT.
(J) Hematoxylin and eosin (H&E) staining of kidney sections from 32-week-old Lyn–/– mice of indicated groups. Bar, 50 μm.
(K and L) Foxp3-GFP knock-in (KI) mice were immunized as in (K), and then Foxp3+ Treg cells were sorted and adoptively transferred into Lyn–/– mice. Serum was collected at the indicated times. Serum anti-dsDNA antibodies were determined using ELISA (L). The data are representative of two independent experiments with similar results. n = 5 mice/group. Mean ± SD is shown. The Student's t-test was used for statistical analysis. P < 0.05 was considered statistically significant.
No significant change of conventional CD4+ T cells and only a slight decrease of CD8+ T cells were observed in the spleen of Lyn–/– mice under gp96 treatment (Figure S1C). The percentage of Tregs and follicular regulatory T cells (Tfr) in spleen was significantly increased by 22.4% and 33.8%, respectively, following gp96 immunization (Figures 1B and 1C). Besides, a similar trend of increased Tfr by gp96 immunization was observed in mesenteric lymph nodes (MLNs) and Peyer's Patches (Figure S1D). As Lyn–/– mice exhibit increased numbers of activated or effector memory CD4+ and CD8+ T cells as autoimmune disease progresses (Hua et al., 2014), we investigated the effects of gp96 in the hyperreactivity of T cells. The percentages of follicular helper T cells (Figure 1D), activated conventional CD4+ T cells (Figure 1E), germinal center B cells (GCB) (Figure 1F), plasma cells (Figure 1G) in the spleen, and memory B cells (Figure 1H) in the bone marrow (BM) were significantly lower in gp96-immunized mice than they were in control mice. No obvious effect on activation of CD8+ T cells was observed under gp96 immunization (Figure S1E). Anti-dsDNA specific antibody-secreting cells (ASCs) in the BM were also fewer in gp96-immunized mice than they were in the control mice, as determined using enzyme-linked immune absorbent spot (ELISpot, Figure 1I). Meanwhile, according to previous studies on eTreg and cTreg category (Sidwell et al., 2020; Toomer et al., 2016), we found that gp96 immunization could induce conversion from cTreg to eTreg in vivo by analyzing Treg markers such as ICOS, CD69, KLRG1, CD62L, CCR7, Ki-67 (Figure S1F).
Moreover, a significantly higher number of thrombocytes and hematocrit level were observed in gp96-immunized mice than in the control mice (Figure S1G). The Lyn–/– mice developed diffuse mesangioproliferative glomerulonephritis, which was characterized by partial enlargement of the epithelium of Bowman's capsule, and was suppressed in the kidney of gp96-immunized mice (Figure 1J). Furthermore, we adoptively transferred gp96-treated Tregs into Lyn–/– mice (Figure 1K), which showed higher inhibitory effects with a longer duration on anti-dsDNA levels than the transfer of control Tregs did (Figure 1L). Moreover, a significant decrease of plasma cells, GCB, and Tfh cells was observed after adoptive transfer of Tregs isolated from gp96-immunized mice (Figure S1H).
High-dose gp96 immunization suppresses Th-induced inflammation in experimental autoimmune encephalomyelitis (EAE) mouse model
Experimental autoimmune encephalomyelitis (EAE) is a mouse Th1/Th17 cell-mediated autoimmune disease of the central nervous system (CNS) used as a model of human multiple sclerosis (MS). We investigated whether gp96-expanded Treg could inhibit Th-induced inflammation. The results illustrated in Figure 2A show that EAE disease initiation and progression were more significantly inhibited in mice immunized with gp96 than they were in the control mice (P = 0.002). Less body weight loss was also observed in gp96-treated mice (Figure 2B). Compared to control mice, gp96-treated mice showed significantly decreased immune cell infiltration to spinal cords (Figure 2C). The number of CD4+ T, Th1, and CD8+ T cells in the spinal cord were significantly decreased by 61.5%, 50%, and 55%, respectively, in gp96-immuized mice relative to control mice. The number of Th17 was slightly lower in gp96-immunized mice than it was in the untreated control mice. No significant changes of the number of CD4+ T cells, Th1, Th17, and CD8+ T cells were observed in the spleen and LNs between the two groups of mice (Figures 2D–2G). Gp96-immunized mice displayed a significantly higher Treg frequency in the spleen and lymph nodes (LNs) than the control mice. However, lower Treg percentage in spinal cord-infiltrated cells was observed in gp96-treated mice (Figure 2H). This may be because of pronouncedly decreased infiltration of CD4+ T cells, Th1, Th17, and CD8+ T cells in the spinal cord in homeostasis. Taken together, these results indicate that gp96 immunization inhibited EAE initiation and progression via systemic expansion of Treg and suppression of Th1, CD8+ T and Th17 cells infiltration in spinal cord.
Figure 2.

Treatment of EAE with heat shock protein gp96
(A and B) Female C57BL/6 mice were immunized with 200 μg gp96 or saline (control) before induction of EAE. Disease score (A) and body weight changes (B) were monitored daily after EAE induction. (C) Spinal cord collected at day 18 from indicated mice were stained with hematoxylin and eosin to assess inflammation. Scale bars, 500 μm.
(D–G) Absolute numbers of CD8+ T cells (D), CD4+ T cells (E), IL-17A-producing CD4+ T cells (F) and IFN-γ-producing CD4+ T cells (G) from indicated organs of mice were evaluated by flow cytometry.
(H) Percentages of Foxp3+ Tregs in the indicated organs of mice. The data are representative of two independent experiments with similar results. n = 5 mice/group. Mean ± SD is shown. The Student's t-test or two-way ANOVA was used for statistical analysis. P < 0.05 was considered statistically significant. ns = not significant.
Gp96 induces Foxp3 expression and increases CD4+Foxp3+ Treg population, stability, and suppressive function
To elucidate the immunoregulatory mechanism of gp96, we first measured T cell subtype, B cell, natural killer (NK) cell, CD11c+, and CD11b+ cell populations in the spleen of gp96-immunized mice using multicolor flow cytometry. Figure S2A shows that only the percentages of the CD4+Foxp3+ and CD8+Foxp3+ populations were significantly increased following gp96 immunization. CD4+Foxp3+ Tregs are well recognized to be involved in maintaining immunological tolerance; therefore, we studied the effect of gp96 on Foxp3+ Tregs.
Indeed, the expansion of Tregs was observed in high-dose (200 or 400 μg) gp96 immunization (Figure 3A). Gp96 immunization led to a pronounced increase of mRNA and protein expression levels of Foxp3 in Tregs (Figures 3B and 3C). Other subsets of regulatory cells, such as the M2-macrophage, and regulatory B cell populations were not significantly changed in gp96-immunized mice compared to the control mice, whereas myeloid-derived supressor cells (MDSCs) were moderately reduced by gp96 immunization (Figures S2B–S2D). Moreover, no significant induction of effector CD8+ T and Th1 cells was observed in gp96-immunized mice, whereas Th2 and Th17 cells were decreased upon gp96 immunization (Figures S2E–S2G). A possible explanation for the increased Treg population by gp96 immunization could be that they were either generated from peripheral expansion or the conversion of conventional T cells. To test this hypothesis, we sorted the peripheral CD4+Foxp3+ Tregs and CD4+Foxp3– conventional T cells (Tconv) cells from Foxp3-GFP knock-in (KI) mice using GFP expression as the criterion. Then, we adoptively transferred each group of cells into Rag2−/− recipient mice. We found that the CD4+Foxp3+ T cells were more stably maintained in the Rag2−/− mice immunized with gp96 than they were in the control mice, whereas very slightly increased percentage of Foxp3+ Treg cells from conventional CD4+ T cells was observed by gp96 treatment, which may be because of homeostasis (Figure 3D). In addition, excessive proliferation was observed in CD4+Foxp3+ T cells treated with gp96 in vivo and in vitro (Figures 3E and S3A). Meanwhile, gp96 treatment also alleviated apoptosis of Treg and promoted its survival (Figure S3B). No obvious increase of Tregs from differentiation from naïve T cells was observed following gp96 treatment both in vitro and in vivo (Figures S3C and S3D). Compared to the control, Foxp3+ Tregs from gp96-immunized mice or those treated with gp96 showed higher levels of H3K27 acetylation (H3K27ac) in the Foxp3 promoter region, higher recruitment of the histone acetyltransferase p300, and lower H3K4 methylation levels of the CNS3 region (Figures 3F and S3E).
Figure 3.

Heat shock protein gp96 induces Foxp3 expression and promotes Treg proliferation
(A) Frequency of CD4+Foxp3+ Treg in spleen of C57BL/6 mice immunized with the indicated dose of recombinant gp96.
(B and C) Foxp3 expression in Tregs from mice immunized with 200 μg gp96 or saline as control was measured using flow cytometry analysis (B) and real-time PCR (C).
(D) FACS-sorted CD4+Foxp3-GFP+ Tregs and CD4+Foxp3-GFP– Tconv cells from Foxp3-GFP KI mice were transferred to Rag2−/− mice. Mice were then immunized with gp96 or saline three times. Foxp3-GFP levels in CD4+ T cells from the spleen were analyzed 1 week after the last immunization.
(E) Proliferation of Tregs was measured using in vivo BrdU labeling in the indicated organs of gp96-immunized and saline-immunized C57BL/6 mice.
(F) Chromatin immunoprecipitation (ChIP) analysis of H3K27ac, p300 and H3K4 monomethylation (H3K4me1) at Foxp3 locus and control loci (Hsp90ab, Hspa2, Rpl30, and Gm5069). The data are representative of two independent experiments with similar results. n = 5 mice/group. Mean ± SD is shown. The Student's t-test was used for statistical analysis. P < 0.05 was considered statistically significant. ns = not significant.
Sorted Tregs from Foxp3-GFP KI mice after incubation with gp96 showed higher expression of proliferation and activation markers and stronger suppressive function (Figures S3F and S3G). Compared to the control, Tregs from gp96-treated mice exhibited higher inhibitory effects on proliferation of T effector cells ex vivo (Figure 4A). Finally, we investigated whether gp96-induced Tregs competently exerted a suppressive function in vivo. The same number of Tregs (CD45.1+CD45.2+) sorted from the spleen of gp96- or saline-immunized mice was adoptively transferred into Rag2−/− mice that also received naïve CD4+ T cells (CD45.2+, Figure 4B). Mice administered naïve CD4+ T cells developed substantial weight loss associated with colitis (Figure 4C). The adoptive transfer of gp96-induced Treg cells protected mice from developing symptoms associated with colitis more significantly than the transfer of control Tregs did (Figure 4D). Compared to the control, mice that received gp96-induced Treg cells had a significantly lower percentage of CD45.1–CD45.2+ T effector cells and higher percentage of CD45.1+CD45.2+ Tregs in the spleen and MLN (Figure 4E). Taken together, these results led us to conclude that gp96 positively affected Treg through the induction of Foxp3 expression, maintenance of Treg lineage stability in vivo, promotion of Treg expansion and suppressive function. Furthermore, gp96 participated in Treg development by modulating epigenetic modifications.
Figure 4.

Gp96 enhances Treg suppressive function
(A) A total of 5×104 cell trace violet-labeled CD4+CD25– T (Teff) cells were cultured with Tregs at a ratio of 2:1 for 3 days. Teff cell division cycle was measured using FACS (left). Suppression rate of proliferation was calculated (right).
(B–E) Rag2−/− recipient mice were intravenously administered 4×105 naïve CD4+ T cells (CD45.2+) with or without co-transfer of 4×105 Treg cells (CD45.1+CD45.2+) sorted from gp96-immunized or control CD45.1+CD45.2+ mice. Experimental scheme (B). Mouse weight changes were monitored for 6 weeks (C). Hematoxylin and eosin (H&E) staining of colon at end of the experiment. Dashed lines indicate lymphocytes infiltrating areas, scale bar, 2 mm (D). Flow cytometric analysis and quantification of frequency of CD45.1 and CD45.2 cells from spleen and mesenteric lymph nodes (MLN) 6 weeks after transfer (E). Dots represent data from individual mice. The data are representative of two independent experiments with similar results. n = 5 mice/group. Mean ± SD is shown. The Student's t test or two-way ANOVA was used for statistical analysis. P < 0.05 was considered statistically significant.
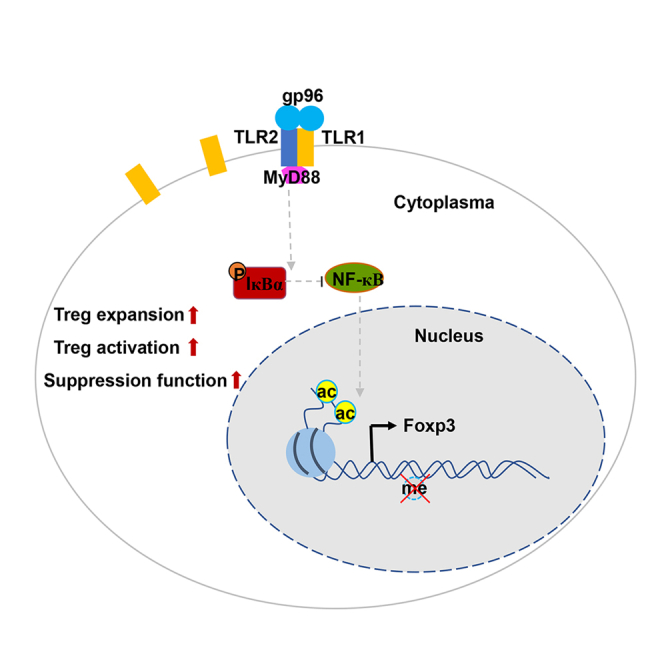
Activated NF-κB elements induce Foxp3 expression in gp96-stimulated Tregs
To explore the mechanism by which gp96 immunization acts on Foxp3 expression, we analyzed gene expression profiles of Foxp3+ Tregs from Foxp3-GFP KI mice immunized with gp96. Compared to the control mice, Tregs in gp96-immunized mice showed an upregulation of 587 genes and downregulation of 1277 genes (log2[fold change] > 1, Figure 5A). In particular, the upregulated genes in Treg cells were significantly enriched in signaling by T cell receptor, JAK-STAT, TNF and NF-κB (Figure 5B). Then, we selected T cell receptor, JAK-STAT, and NF-κB signaling pathways, which are of great importance to the Treg lineage, for gene set enrichment analysis (GSEA, Figures 5C, 5D, S4A, and S4B). Because NF-κB signaling is the major downstream effector pathway of activated CD91 or TLRs in response to gp96 (Li et al., 2013; Pawaria and Binder, 2011), we investigated the effect of gp96-induced NF-κB signaling on Foxp3 expression. The results showed that gp96 significantly promoted IκB-α and p65 phosphorylation in Tregs (Figures 5E and 5F).
Figure 5.

Gp96 induces Foxp3 expression via NF-кB pathway
(A) Volcano map depicting genes upregulated (red) or downregulated (blue) 2-fold or more in Treg cells.
(B) Gene Ontology (GO) terms of the differentially expressed genes in gp96-treated Tregs compared with control Tregs.
(C) Gene set enrichment analysis (GSEA) of NF-кB signaling gene set in gp96-treated Tregs relative to expression in control Treg cells.
(D) Heatmap of NF-кB signaling pathways (Log2 fold change) that are differentially expressed.
(E) FACS analysis of IкB phosphorylation in Foxp3+ Tregs treated with 100 μg/mL of gp96 or saline as control in vitro for 0.5 h.
(F) Western blot analysis of IкB-α and p65 phosphorylation in sorted Tregs treated as in (E).
(G) p65 and c-Rel were detected using immunofluorescent staining. Scale bar, 25 μm.
(H) Chromatin immunoprecipitation (ChIP) analysis of p65 and c-Rel binding at Foxp3 promoter region. Chromatin obtained from spleen Tregs (1×106) of gp96-immunized or control mice were immunoprecipitated with anti-p65 and anti-c-Rel antibodies, followed by real-time PCR analysis.
(I) mRNA levels of Foxp3 in Tregs treated with 100 μg/mL gp96 in vitro and curcumenol or PDTC were analyzed using real-time PCR after 24 h. The data are representative of two independent experiments with similar results. n = 5 mice/group. Mean ± SD is shown. The Student's t-test was used for statistical analysis. P < 0.05 was considered statistically significant. ns = not significant.
The NF-κB family member p65 and c-Rel have been suggested to bind to the promoter region of Foxp3 to promote its transcription (Long et al., 2009). Activation of NF-κB signaling by gp96 was confirmed in gp96-stimulated Treg cells with the nuclear translocation of c-Rel and p65 (Figure 5G). Furthermore, the chromatin immunoprecipitation assay also indicated that binding of c-Rel and p65 to the Foxp3 promoter region was significantly increased by gp96 (Figure 5H). Pre-treatment of Tregs with curcumenol (Cur), a specific inhibitor of p65 translocation (Lo et al., 2015), slightly inhibited gp96-induced Foxp3 expression. Whereas ammonium pyrrolidine dithiocarbamate (PDTC), an inhibitor of IκB phosphorylation and nuclear translocation of NF-κB (Qin et al., 2014), almost completely ablated the upregulation of Foxp3 by gp96 (Figure 5I). In addition, differentially expressed genes of gp96-treated Tregs were correlated to those of the wild-type (WT) and Rela–/–Rel–/– (DKO) Tregs (Oh et al., 2017) (Figure S4C). These data indicate that gp96 induced Foxp3 expression via p65 and c-Rel.
TLRs binding domain is essential for gp96-induced Treg activation
NF-κB is the hallmark downstream pathway for almost all TLRs that have been shown to interact with gp96 (Liu et al., 2016). Moreover, in addition to DCs, B and T cells, an abrupt increased binding of gp96 to Tregs (∼7-fold) was observed at a high dose but not the low dose used in gp96 immunization (Figure 6A). This observation indicates that Tregs can capture gp96 when it is abundant.
Figure 6.

TLR binding domain is essential for gp96-induced Treg activation
(A) Low- (20 μg) or high- (200 μg) dose Cy5-labelled gp96 was subcutaneously injected into the backs of Foxp3-GFP KI mice. Flow cytometry analysis of Cy5-positive Tregs (CD4+Foxp3-GFP+), DCs (CD11c+), B cells (CD3–CD19+), conventional CD4+ T cells (CD4+Foxp3-GFP–) and CD8+ T cells 6 h post treatment.
(B–D) Ovalbumin (OVA)-specific IgG in serum on day 10 and day 21 post OVA treatment was measured using ELISA (B). FACS analysis of Treg cells (C) and Tfr cells (D) in spleen.
(E) Flow cytometry analysis of Foxp3, Helios, CD62L, CD69, ICOS, and CD137 expression on CD4+Foxp3+ Tregs in the spleen of saline, mutant or WT gp96-immunized mice. n = 5 mice/group.
(F) Serum anti-dsDNA antibodies were determined by ELISA in Lyn–/– mice at the indicated weeks of age one week after mutant gp96 treatment.
(G) FACS analysis of surface TLR1/2 expression in CD4+Foxp3+ Tregs from C57BL/6 mice.
(H) Tregs were cultured with 100 μg/mL His-gp96 for 4 h before immunoprecipitation with anti-His antibody and western blotting.
(I) Luciferase assay using reporter plasmids containing the Foxp3 promoter in 293T cells. Expression plasmids of TLR1 and TLR2 were transfected as indicated. Cells were either unstimulated or stimulated with 100 μg/mL gp96 for 12 h before analysis.
(J) Frequency of Tregs in the spleen from Tlr2−/− mice immunized with 200 μg gp96 or saline (control, n = 5/group).
(K) Mean Fluorescence intensity (MFI) of indicated markers in the spleen from Tlr2−/− mice immunized with 200 μg gp96 or saline (control) (n = 5/group).
(L and M) Chimeric mouse model was produced and immunized with gp96 (L). Flow cytometry analysis of ICOS, KLRG1, CD69, and CD137 expression on CD4+Foxp3+ Tregs from CD45.1+ cells and CD45.2+ cells (M). Dots represent data from individual mice. The data are representative of two independent experiments with similar results. n = 5 mice/group. Mean ± SD is shown. The Student's t test was used for statistical analysis. P < 0.05 was considered statistically significant. ns = not significant.
Previous studies demonstrated that the YGWSGNMER motif (aa 652-660) within the client-binding domain (CBD, aa 652–678) of gp96 is essential for its interaction with TLRs (Liu et al., 2016; Wu et al., 2012). We therefore generated mutant gp96 by introducing alanine substitution mutations at all of the conserved residues in the first half of the CBD as “YAASAAAAA” (aa 652-660), which has been shown to lose the ability to bind to TLR2. Then, we examined the effect of immunization with WT or mutant gp96 within its TLRs binding site on antibody production as a direct in vivo proof of the immunosuppression activity induced by gp96 (Figure S5A). Figure 6B shows that compared to WT gp96, mice co-immunized with mutant gp96 and 4-hydroxy-3-nitrophenylacetyl ovalbumin (NP-OVA) exhibited a dramatic ∼4-fold increased antibody production. Significantly higher percentages of Treg and Tfr were observed in the spleens of WT gp96-immunized but not mutant gp96-immunized mice or control mice (Figures 6C, 6D, and S5B).
As seen in Figures 6E, S5C, and S5D, Tregs in mice immunized with WT gp96 but not mutant gp96 maintained higher levels of ICOS, CD69, Helios, Foxp3, CD137, and PD-1 and lower levels of CD62L than those of the control mice, indicating that mutant gp96 cannot promote Treg activation. However, expression of GITR, CTLA4, and CD44 on Tregs showed no significant changes under gp96 immunization. We further wondered if immunization with mutant gp96 could attenuate autoimmunity in Lyn–/– mice. Unlike WT gp96, treatment with mutant gp96 failed to decrease serum anti-dsDNA levels (Figure 6F). No significant difference of Treg, Tfr and plasma cells was observed between mutant gp96-treated and control mice (Figure S5E).
TLR2 signaling is responsible for Treg activation in response to gp96
To determine the specific TLRs responsible for Treg activation in response to gp96 immunization, we first examined the Treg TLR expression profiles. We found that TLR1 and TLR2 were more highly expressed on Treg cells than the other subtypes were (Figures 6G, S6A, and S6B). As expected, exogenous gp96 was co-precipitated with TLR1 and TLR2 on Tregs (Figure 6H) and 293T cells (Figure S6C). Co-transfection of TLR1 and TLR2 under gp96 treatment activated the Foxp3 promoter reporter (Figure 6I), indicating that the TLR1/TLR2 heterodimer mediated Foxp3 transcription in response to gp96 stimuli. TLR1 or TLR6 signaling have previously been shown to require heterodimerization with TLR2 (Schumann and Tapping, 2007). Treatment of Treg cells with gp96 activated the TLR2 downstream pathway (Figure S6D), making TLR2 the focus of this investigation.
Furthermore, the expansion of Treg by gp96 immunization was largely abolished in Tlr2−/− mice (Figure 6J). In addition, Tregs in Tlr2−/− mice failed to show changes in activation markers in response to gp96 in contrast to the changes shown by WT mice, as determined using flow cytometry (Figures 6K and S6E). Next, we generated mixed BM chimeric mice by transferring equal numbers of BM cells from CD45.1+ WT and CD45.2+ Tlr2−/− mice. Mice were immunized with gp96 or saline and the result is shown in Figure 6L. Analyses of CD69, ICOS, KLRG1, Foxp3, Helios, and CD137 showed that in the spleen, WT (CD45.1+) but not TLR2-deficient (CD45.2+) Tregs were significantly activated following gp96 immunization (P < 0.05, Figures 6M and S6F). Together, the results demonstrated that Treg activation by gp96 occurred via a TLR1/2-mediated signaling pathway.
To further verify the role of TLR1/2 signal in gp96-induced Treg proliferation and activation in vivo, Myd88fl/fl mice were crossed with Foxp3-GFP-hCre to generate mice that were selectively deficient in MyD88 expression in Foxp3+ Tregs (Foxp3CreMyd88fl/fl). Foxp3CreMyd88fl/fl and Foxp3CreMyd88+/+ (WT) mice were used in a gp96-mediated immunosuppression assay. WT mice showed an abrupt decrease in antibody production, whereas Foxp3CreMyd88fl/fl mice were unable to limit antibody production following gp96 administration (Figure 7A). Immunization with gp96 increased Tfr cells and Tregs in WT mice, whereas this phenomenon was almost completely abrogated by deletion of the MyD88 gene in Foxp3+ Tregs (Figures 7B and 7C). Tregs from Foxp3CreMyd88fl/fl mice failed to exhibit changes in activation markers in response to gp96 immunization in contrast to WT mice that did (Figure 7D). To determine if the effects of gp96 on Treg cell activation are cell-intrinsic or microenvironment-dependent, we generated mixed BM chimeric mice by transferring equal numbers of BM cells from CD45.1+ WT and CD45.2+ Foxp3CreMyd88fl/fl mice. Analyses of CD69, ICOS, KLRG1, and CD137 expression showed that in the spleen, WT (CD45.1+) but not MyD88-deficient (CD45.2+) Tregs were significantly activated following gp96 immunization (P < 0.05, Figure 7E). In addition, unlike Myd88+/+ mice, EAE was not alleviated by immunization with gp96 in Foxp3CreMyd88fl/fl mice (Figure 7F).
Figure 7.

Gp96 activates CD4+Foxp3+ Tregs through MyD88 signal
(A–C) Foxp3CreMyd88fl/fl and Foxp3CreMyd88+/+ mice were treated as in Figure 6B. OVA-specific IgG levels were determined in serum on day 20 post OVA challenge using ELISA (A). FACS analysis of CXCR5+PD-1+ cells (Tfr) (B) in spleen and CD4+Foxp3+ Tregs (C) in the indicated organs.
(D) FACS analysis of Foxp3, Helios, CD62L, CD69, ICOS and CD137 expression on CD4+Foxp3+ Tregs in spleens of Foxp3CreMyd88fl/fl and Foxp3CreMyd88+/+ mice immunized with 200 μg gp96 or saline (control).
(E) Flow cytometry analysis of ICOS, KLRG1, CD69 and CD137 expression in CD4+Foxp3+ Tregs from CD45.1+ cells and CD45.2+ cells.
(F) Foxp3CreMyd88fl/fl and Foxp3CreMyd88+/+ mice were immunized with gp96 or saline (control) before induction of EAE. Disease score was measured daily after EAE induction. Dots represent data from individual mice. The data are representative of two independent experiments with similar results. Mean ± SD is shown. The Student's t test or two-way ANOVA was used for statistical analysis. P < 0.05 were considered statistically significant. ns = not significant.
Next, we performed RNA-sequencing (RNA-seq) in Foxp3CreMyd88fl/fl mice to gain detailed mechanistic insight into MyD88-deficient Tregs following gp96 immunization. There was a sharp difference in gene profile changes in response to gp96 between Foxp3CreMyd88fl/fl and WT mice. Compared with those in WT mice, we identified 1035 upregulated genes and 1315 downregulated genes in gp96 treated-MyD88-deficient Treg cells (Figure S7A). GSEA showed that none of the Treg-signature pathways were significant in MyD88-deficient Tregs (Figures S7B and S7C). Genes correlated with Treg activation, including Cd69, Klrg1, Icos, Ctla4, Ccr5, Ccr4, and Ikzf2, were transcriptionally increased in WT but not Foxp3CreMyd88fl/fl mice following gp96 immunization (Figure S7D). Taken together, these data indicate that activation of Treg by gp96 is dependent on TLR2-MyD88 signaling pathway in a cell-intrinsic manner.
Discussion
Previous studies have shown that immunization with optimal doses of tumor-derived gp96 elicits tumor immunity, whereas immunization with a 5-10-fold higher dose actually downregulates antitumor immune responses (Chandawarkar et al., 1999; Liu et al., 2009). However, the exact underlying mechanisms remain largely unknown. In this study, we found that immunization with high-dose gp96 was effective against antibody-mediated SLE and Th-induced EAE in mice. Both in vivo and in vitro studies showed that gp96 promoted Treg expansion and activation but had no effect on Tconv cells. Our findings further revealed that activation of NF-κB and TLRs by gp96 was responsible for the increased Treg proliferation. This observation was confirmed by the finding that expansion of Treg by gp96 was largely abrogated by a mutation within its TLR binding site and in mice with Treg-specific deletion of MyD88. Because of the high expression of TLR1/2 in Tregs and involvement of the TLR2/MyD88 pathway in functional Treg proliferation (George et al., 2020) and Treg-cell-specific deletion of MyD88 resulted in a deficiency of intestinal Tregs and increase of inflammatory Th17 cells in experimental colitis (Wang et al., 2015), we deduced that the TLR1/2-MyD88 pathway may play a major role in gp96-mediated Treg proliferation.
Indeed, gp96-induced Treg activation was largely abolished in TLR2 KO mice (Figure 6K). Moreover, these gp96-expanded Tregs highly expressed the tTreg marker Helios (Thornton et al., 2019), and exhibited H3K27ac histone modification in the promoter region and H3K4me1 demethylation in the CNS3 enhancer region as seen in tTreg cells (Kitagawa et al., 2017; Ohkura and Sakaguchi, 2020). Therefore, we consider that activation of Treg cells by gp96 may be at least partly because of the expansion of nTregs that appear to be highly stable in Foxp3 expression and maintenance of the suppressive function (Ohkura et al., 2012).
To date, the impact and exact role of TLR activation in enhancing or reversing Treg generation, activation, and function are somewhat controversial (Cao et al., 2018; Nguyen et al., 2015; Nyirenda et al., 2015). This may be because of different expression levels of TLRs in Tregs and their related downstream pathways, the hetreogeneity of Treg subtypes, balance between Treg and Th17, modulation by macrophage and DCs, and the different immune processes studied. TLR2 has also been shown to directly regulate Tregs. TLR2 agonist, together with TCR activation, was sufficient to induce proliferation of Treg cells in the absence of antigen-presenting cells (Liu et al., 2006).Viral infection, such as dengue infection, induced the proliferation of functional Treg cells. Viral infection subverted CD8+ T-cell responses through Tregs expanded by TLR2/MyD88 signal (George et al., 2020). DAMPs have also been implicated in initiating essential anti-inflammatory and reparative functions of Treg cells by promoting the expansion of IL-33 receptor-positive Treg and inducing TCR-independent Treg secretion of IL-13 (Dwyer and Turnquist, 2021). In the present study, we identified a gp96-TLR1/2-MyD88-NF-κB-Foxp3 pathway, which validated the notion that gp96 may directly control Treg proliferation and suppressive function by enhancing TLR1/2-mediated Foxp3 transcription. This was further supported by our observations that activated downstream NF-κB family member p65 and c-Rel directly bound to the Foxp3 promoter and promoted its transcription, and mutant gp96 within the TLR binding domain or MyD88 KO in mice failed to induce Foxp3 expression in Tregs. Therefore, our results provide evidence of the mechanisms by which the induction of Foxp3 expression by TLR signaling is vital in enhancing Treg proliferation and function by gp96. Our results therefore demonstrate a previously unrecognized pathway by which the gp96-activated TLR network directly promotes Treg proliferation and functional activation.
Previous studies show that despite their ability to induce Treg proliferation, the regulatory function of Tregs expanded by TLR2 agonist was impaired. The impaired regulatory function of Treg cells can be recovered after TLR2 stimulation eradication (Chen et al., 2009; Liu et al., 2006; Sutmuller et al., 2006). In the current study, we found that gp96 promoted Treg cell proliferation, increased levels of its activation markers (e.g. CD69, Helios, ICOS, Foxp3, etc.), and enhanced Treg function in vitro. The different impact of gp96 and TLR2 agonist on Treg function may be because of their different binding capacity to TLR2 heterodimers and downstream pathway, as well as interaction between Tregs and other immune cells. This needs further investigation.
Presently, we cannot totally exclude the possibility that gp96 indirectly controls Treg expansion and function by interaction with other immune cell types, such as pDCs (Kinner-Bibeau et al., 2017). It is possible that NF-κB activation in Tregs by gp96 may upregulate Sema4a that facilitates its interaction with pDC (Zhang et al., 2019). In addition, more studies are needed to elucidate the role of different TLRs in gp96-induced Foxp3 expression, although our primary data showed that TLR2 may play an essential role in this process.
Various reagents have been reported to upregulate Foxp3 expression, expand nTregs, promote conversion of naïve T cells into Tregs, or activate Tregs, such as retinoic acid (Lee et al., 2018), vitamin D3 (Hayes and Ntambi, 2020), rapamycin (Stallone et al., 2016). This observation suggests they may have potential protective effects against autoimmune diseases. It has been suggested that treatment with low-dose recombinant human IL-2 reduces disease activity in patients with SLE by changing the proportions of Treg and T effector cells in clinical practice (He et al., 2016). In this study, we demonstrated that immunization with high-dose gp96 had a robust therapeutic effect on the spontaneous SLE mouse model by expanding Tregs and suppressing Tfh and plasma cells. In addition, gp96 treatment led to increased Tfr cells that are functional specialized T regulatory cells safeguarding against both self-reactive T and B cells, and Foxp3 induction by gp96 may stabilize Tfr to prevent their converting to pathogenic “ex-TFRs” (Wei et al., 2021). This needs more evidence to support. Moreover, high-dose gp96 immunization was shown to suppress Th1 and Th17 responses and reduce EAE disease severity through Treg expansion. These results provide key information about the immune events controlled by gp96 in its regulation of Treg activation and expansion, suggesting its potential effectiveness as a therapy for autoimmune diseases.
Limitations of the study
Our study examines the role and underlying mechanism of shock protein gp96 in utility of Treg cells for treatment of autoimmune diseases. Our results show that gp96 immunization elicited a therapeutic effect in Lyn–/– mice, used as a mouse model of SLE, and delayed the onset and progression of EAE. Mechanism analyses suggest that gp96 activated Tregs by enhancing Foxp3 expression via TLR2-MyD88-mediated NF-κB signaling. However, the exact mechanism of gp96-orchestrated activation of Treg subsets, and induction of antigen-specific Tregs needs further comprehensive investigation. In addition, Treg-specific TLR2 KO mice should be used instead of systematic knock out mice. Furthermore, the importance of this mechanism in human T cells, especially lupus patients' derived T cells needs further clarification.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD4-PerCP-Cy5.5 (RM4-5) | eBioscience | Cat#45-0042-82;RRID:AB_1107001 |
| CD4-FITC (GK1.5) | Biolegend | Cat#100406; RRID:AB_312691 |
| CD4-PE (GK1.5) | Biolegend | Cat#100408; RRID:AB_312693 |
| CD45R/B220-APC (RA3-6B2) | Biolegend | Cat#103212; RRID:AB_312997 |
| CD45R/B220-PE (RA3-6B2) | eBioscience | Cat#12-0452-82; RRID:AB_465671 |
| CD44-APC (IM7) | eBioscience | Cat#17-0441-81; RRID:AB_469389 |
| CD62L-PE (MEL-14) | Biolegend | Cat#104408; RRID:AB_313095 |
| ICOS-APC (7E.17G9) | eBioscience | Cat#17-9942-82;RRID:AB_2716948 |
| KLRG1-APC (2F1) | eBioscience | Cat#17-5893-81;RRID:AB_469468 |
| CD69-APC (H1.2F3) | Biolegend | Cat#104513; RRID:AB_492844 |
| Helios-PE/Cy7 (22F6) | eBioscience | Cat#25-9883-41;RRID:AB_2637135 |
| CD137-PE/Cy7 (17B5) | eBioscience | Cat#25-1371-80;RRID:AB_2573397 |
| TLR1-PE (eBioTR23(TR23)) | eBioscience | Cat#12-9011-80; RRID:AB_657857 |
| TLR2-PE (6C2) | eBioscience | Cat#12-9021-82;RRID:AB_466230 |
| TLR4-PE (UT41) | eBioscience | Cat#12-9041-80;RRID:AB_466236 |
| TLR5-AF647 (ACT5) | Biolegend | Cat#148103; RRID:AB_2563694 |
| TLR6-PE | R&D Systems | Cat#FAB1533P;RRID:AB_1964728 |
| CD5-PE/Cy7 (53-7.3) | Biolegend | Cat# 100622;RRID:AB_2562773 |
| CD1d-PE (1B1) | eBioscience | Cat#12-0011-82;RRID:AB_465483 |
| CD19-FITC (6D5) | Biolegend | Cat#115506; RRID:AB_313641 |
| Foxp3-APC (FJK-16 s) | eBioscience | Cat#17-5773-82; RRID:AB_469457 |
| Foxp3-PE (FJK-16 s) | eBioscience | Cat#12-5773-82; RRID:AB_465936 |
| Foxp3-eFluor 450 (FJK-16 s) | eBioscience | Cat#48-5773-80; RRID:AB_1518813 |
| IFN-γ-APC (XMG1.2) | eBioscience | Cat#17-7311-81; RRID:AB_469503 |
| IL-17A-FITC (TC11-18H10.1) | Biolegend | Cat#506908; RRID:AB_536010 |
| IL-4-PE (11B11) | Biolegend | Cat#504103; RRID:AB_315317 |
| IL-10-APC (JES5-16E3) | eBioscience | Cat#17-7101-82; RRID:AB_469502 |
| CD45.1-FITC (A20) | Biolegend | Cat#110705; RRID:AB_313494 |
| CD45.2-PerCP/Cy5.5 (104) | Biolegend | Cat#109827; RRID:AB_893352 |
| BrdU-FITC (3D4) | Biolegend | Cat#364103; RRID:AB_2564480 |
| Phospho-IkB alpha (Ser32, Ser36) | eBioscience | Cat#50-9035-42; RRID:AB_2574311 |
| PD-1-FITC (J43) | eBioscience | Cat#11-9985-81; RRID:AB_465471 |
| CXCR5-PE (SPRCL5) | eBioscience | Cat#12-7185-80; RRID:AB_11218887 |
| CD95-AF488 (APO-1/FAS) | eBioscience | Cat#53-0951-82; RRID:AB_10671269 |
| GL7-APC (GL7) | Biolegend | Cat#144617; RRID:AB_2800674 |
| CD138-PerCP/Cy5.5 (281-2) | Biolegend | Cat#142509; RRID:AB_2561600 |
| CD80-APC (16-10A1) | Biolegend | Cat#104714; RRID:AB_313135 |
| CD73-PE/Cy7 (eBioTY/11.8(TY/11.8)) | eBioscience | Cat#25-0731-80; RRID:AB_10870789 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human TGF-β1 | PeproTech | 100-21 |
| Recombinant Murine IL-2 | PeproTech | 212-12 |
| SYBR Green | Takara | RR420DS |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | P8139 |
| Ionomycin | Sigma | I0634 |
| Brefeldin A | eBioscience | 00-4506-51 |
| Pertussis Toxin | Merck Millipore | 516562-50UG |
| NP-OVA (Ovalbumin) | Santa Cruz | Sc-396355 |
| Ultra-LEAF-anti-mouse CD28 (37.51) | Biolegend | 102115 |
| Ultra-LEAF anti-mouse CD3ε (145-2C11) | Biolegend | 100339 |
| DynabeadsTM mouse T-activator CD3/CD28 | Thermo | 11452D |
| Critical commercial assays | ||
| Naïve CD4+ T Cell Isolation Kit | Miltenyi Biotec | 130-104-453 |
| CD4+CD25+ Regulatory T Cell Isolation Kit | Miltenyi Biotec | 130-091-041 |
| Foxp3 Staining Buffer Set | eBioscience | 00-5523-00 |
| FITC BrdU Flow Kit | BD Pharmingen | 559619 |
| Cell Trace Violet Cell Proliferation Kit | Invitrogen | C34571 |
| Deposited data | ||
| RNA-seq data | This study | GSE154174 |
| Experimental models: Organisms/strains | ||
| Mouse: Foxp3-GFP-hCre | Zhou et al., 2009 | N/A |
| Mouse: Foxp3-GFP KI | Zhou et al., 2009 | N/A |
| Mouse: Lyntm1Sor/J (C57BL/6J) | Jackson Laboratory | 003515 |
| Mouse: Tlr2–/– (C57BL/6) | Shanghai Model Organisms | NM-KO-18026 |
| Mouse: Myd88fl/fl | Jackson Laboratory | 00-8888 |
| Mouse: CD45.1 (C57BL/6) | Shanghai Model Organisms | NM-KI-210226 |
| Mouse: Rag2–/– (C57BL/6) | Shanghai Model Organisms | NM-KO-190429 |
| Mouse: MRL/lpr | Shanghai Laboratory Animal Center (SLAC) Co., Ltd. | N/A |
| Oligonucleotides | ||
| MOG35-55 (MEVGWYRSPFSRVVHLYRNGK) | GenScript | N/A |
| All primers used in this study were listed in Table S1. | This study | N/A |
| Recombinant DNA | ||
| Plasmid: pCMV-FLAG-TLR1 | GeneCopoeia | N/A |
| Plasmid: pCMV-HA-TLR2 | GeneCopoeia | N/A |
| Software and algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ |
| FlowJo | BD | https://www.flowjo.com/solutions/flowjo |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact Songdong Meng, E-mail: mengsd@im.ac.cn.
Materials availability
This study did not generate new unique materials or reagents.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Mice
Foxp3-GFP-hCre, and Foxp3-GFP KI mice have been described previously (Lin et al., 2007; Zhang et al., 2015; Zhou et al., 2009). Lyntm1Sor/J and Myd88fl/fl transgenic mice were purchased from the Jackson Laboratory. WT CD45.1, Tlr2–/– and Rag2–/– mice were purchased from the Shanghai Model Organisms. 12-week-old female MRL/lpr mice were purchased from Shanghai Laboratory Animal Center (SLAC) Co., Ltd. Female mice were used in this study. The age of Lyn–/– mice has been indicated in Figure Legends. Other mice used were 6 to 8 weeks old. The experiments in mice were conducted in strict accordance with the regulation of the Institute of Microbiology, Chinese Academy of Sciences of Research Ethics Committee. The protocol was approved by the Research Ethics Committee (permit number PZIMCAS2011001).
Method details
Construction and purification of recombinant gp96 and gp96 mutant protein
Constructs of recombinant human heat shock protein gp96 and TLR2 binding site mutant gp96 were subcloned into the pFastBac1 vector. The Bac-to-Bac Baculovirus expression system used to express the recombinant gp96 proteins was used as described previously (Liu et al., 2014). Cell culture and protein purification were carried out under a GMP environment (SINOVAC, China). Soluble recombinant gp96 and mutant gp96 proteins were isolated as follows. After filtration and concentration, the supernatant was collected and loaded onto a HiTrap Q column (GE Healthcare, USA). The eluent was changed into PB buffer by ultrafiltration and subsequently purified using a ceramic hydroxyapatite column (GE Healthcare, USA). The purified proteins were desalted and concentrated using an Amicon Ultra [15ml 50KD] (Millipore, USA) and stored at -80°C. The purity of WT and mutant gp96 protein is more than 95% as determined by SDS-PAGE. The lipopolysaccharide (LPS) content is 8.237±1.162 EU/mg protein determined by kinetic turbidimetric limulus amebocyte lysate (LAL) assay (Zhanjiang A&C Biological LTD., China). The immunization dose of recombinant gp96 was 200 μg/mouse, and each dose contain around 1.6 EU of LPS.
Serum Ig and autoantibody measurement
Levels of serum anti-dsDNA IgG were measured using ELISA-based quantification kits (Alpha Diagnostic) according to the manufacturer's instructions. Diluted serum samples were added to the plates and incubated for 2 h at room temperature. After washing with PBS containing 0.05% Tween 20, auto-antibodies were detected using horseradish peroxidase (HRP)-conjugated anti-mouse total IgG (Fcγ fragment specific, Bethyl Laboratories). The assays were developed by adding HRP substrate 3,3′,5,5′-tetramethylbenzidine (TMB) to initiate the reaction and stopped by adding 2 M sulfuric acid. The absorbance at 450 nm was measured using a microplate reader (Biotek).
ELISPOT assay
ELISpot analysis to determine the absolute numbers of anti-dsDNA-specific antibody-secreting cells (ASCs) was performed according to a standard procedure. Briefly, ELISPOT plates were pre-coated with methyl-bovine serum albumin (BSA, 10 μg/ml,) for 2 h at 37°C and subsequently coated with calf thymus DNA (10 μg/ml) overnight. BM single-cell suspensions were filtered twice through a Falcon cell strainer (70 μm), washed, and then resuspended in RPMI 1640 medium supplemented with 10% fetal calf serum. After blocking the plates, 1×106 cells were pipetted onto the plates and incubated for 5 h at 37°C with 5% CO2. The plates were washed and incubated with biotin-labeled goat anti-mouse immunoglobulin G (IgG) for 2 h, followed by alkaline phosphatase for 1 h. The spots were developed using AEC (Merck Millipore) and analyzed using an automated ELISpot reader.
Histology analysis
For histological analysis, tissues were fixed in 10% formalin, embedded in paraffin and stained with hematoxylin and eosin (H&E). Glomerulonephritis and interstitial nephritis were scored using a 0–3 scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe), which for the glomeruli was based on glomerular size and hypercellularity and the presence of glomerular sclerosis, whereas for intestinal disease it was based on the degree of inflammatory infiltrate and alteration in tissue architecture.
T cell transfer colitis model
Naïve CD4+ T cells were isolated using a Naïve CD4+ T cell isolation kit. A total of 4×105 naïve Tregs were then adoptively transferred intravenously into Rag2–/– mice. For cotransfer experiments, 4×105 naïve Tregs were co-injected with 4×105 sorted CD4+CD25+ Treg cells into Rag2–/– mice. Recipient mice were subsequently weighed every 7 days to evaluate inflammatory bowel disease (IBD) development. Mice were euthanized 8-10 weeks after transfer when substantial weight loss occurred in the control groups. Colonic specimens from the distal colon were analyzed by histopathology. Lymphocytes were isolated from the spleen MLN and analyzed using flow cytometry.
In vitro induction of Treg differentiation
Naïve CD4+ T cells were prepared from the spleen and LNs of mice using the Naïve CD4+ T cell isolation kit. Naïve CD4+ T cells (1×105 cells) were cultured in a 96-well plate pre-coated with anti-CD3 (2 μg/ml) and anti-CD28 (2 μg/ml) in T cell medium (RPMI, 10% fetal bovine serum, 25 mM glutamine, 55 μM 2-mercaptoethanol, 100 U/ml penicillin, 100 mg/ml streptomycin). For Treg culture, T cells were cultured with the addition of 100 U/ml of IL-2 (PeproTech) and various concentrations of TGF-β (PeproTech) with or without gp96 (100 μg/ml).
In vivo differentiation of naïve CD4+ T cells
Naïve CD4+ T cells (CD4+CD25–CD62LhiCD44lo) were intravenously injected into 8-week-old Rag2–/– mice treated with PBS or gp96 (100 μg/ml). On day 21 after adoptive transfer of naïve CD4+ T cells, spleens and LNs were removed and lymphocytes were prepared for determination of CD4+Foxp3+ Tregs using FACS analysis.
In vitro suppression assay
A total of 5×104 naive Teff (CD4+CD25–) were labeled with cell trace violet (Life Technology) and stimulated with 2 μg/ml soluble anti-CD3ε antibody in the presence of 5×104 irradiated Rag2–/– splenocytes. Tregs from PBS- or gp96-treated mice were isolated using Treg isolation kit and added to the culture to achieve Treg/Teff cell ratios of 1:2. Teff cells only, without Tregs, were used as a positive control for T cell proliferation. Cells were cultured in a 96-well u-bottom plate at 37°C, 5% CO2 for 3 days. Violet dilutions of T cells were analyzed and quantified using flow cytometry.
In vivo Treg stability assay
CD4+Foxp3-GFP+ and CD4+Foxp3-GFP– T cells were sorted from splenocytes of Foxp3-GFP KI mice using FACS Aria III. A total of 2×105 cells were then adoptively transferred intravenously into Rag2–/– mice and 4 weeks later, Tregs in the recipient mice were analyzed using flow cytometry based on the Foxp3-GFP intensity or IC staining of Foxp3.
In vitro Treg cell culture
Sorted Treg cells were labeled with cell trace violet (Life Technology). Tregs were activated with DynabeadsTM mouse T-activator CD3/CD28 for T cell expansion (Life Technologies) using a beads-to-cell ratio of 2:1 in the presence of 2000 U/mL rhIL-2. 4 days later, violet dilutions of Treg cells were analyzed and quantified using flow cytometry.
BrdU incorporation assay
BrdU incorporation assay was conducted using a BrdU flow kit according to the manufacture’s instruction. Briefly, 2 mg BrdU was injected into mice intraperitoneally and 4 h later, lymphocytes from various organs were prepared and surface stained with the indicated markers on ice for 30 minutes. After permeabilization and treatment with DNase I, the cells were stained with FITC-conjugated anti-BrdU antibody and analyzed using flow cytometry.
Reporter assay
The Foxp3 reporter constructs were subcloned into a PGL4 basic reporter construct. Cells were transfected in 12-well plates with 0.75 μg pGL4 reporter and 0.25 μg pRL-TK as the control. The transfected cells were cultured for 24 h and then stimulated with gp96 (100 μg/ml) for 0.5 h before analysis. Firefly luciferase and Renilla luciferase activities were measured consecutively using the dual luciferase reporter system (Promega), and the firefly luciferase activity was normalized to that of Renilla luciferase.
RNA-seq and analysis
CD4+Foxp3-GFP+ Tregs were sorted to a typical purity of >95%. RNA-seq and bioinformatics analysis were conducted by Shanghai Biotechnology Corporation. The P-values were adjusted using the Benjamini-Hochberg method. Corrected q value=0.05 and log2 (fold change) =1 were set as the threshold for a significantly differential expression.
Real-time PCR
Total RNA was extracted with Trizol reagent and quantified using real-time PCR using the SYBR Green Premix reagent (Takara Bio Inc., Shiga, Japan). The mRNA levels of Foxp3 were analyzed and normalized to Actb.
Flow cytometry
The spleen and LNs were collected and single-cell suspensions of lymphocytes were prepared by mechanical disruption in 1640 medium supplemented with 2% (v/v) FBS. For intracellular cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA, 10 ng/mL), ionomycin (0.5 mM), and Brefeldin A (10 μg/mL) for 4 h at 37°C and 5% CO2 condition. Then, they were first stained for surface markers and then permeabilized and stained with interferon (IFN)-γ, IL-17A, and IL-4 antibodies. For the expression of Foxp3, cells were treated with the Foxp3 Staining Buffer Set (eBioscience) and stained with anti-Foxp3 (identified below). Data were acquired using a FACS Calibur or FACS Aria III system (BD Biosciences) and analyzed using FlowJo software.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assays were performed according to a standard protocol. In brief, cells were fixed for 10 min with 1% formaldehyde, followed by the addition of 0.125 M glycine to quench the formaldehyde. Cells were lysed and the chromatin was collected and fragmented by sonication at a concentration of 106 cells per ChIP sample. Chromatin was immunoprecipitated with 2 μg of ChIP or IgG control antibodies at 4°C overnight and incubated with protein G magnetic beads (Cell Signaling Technology) at 4°C for 2 h, washed and eluted in 150 μl elution buffer. Eluate DNA and input DNA were incubated at 65°C to reverse the crosslinking. After digestion with proteinase K, the DNA was purified and the relative abundance of precipitated DNA fragments was analyzed using qPCR.
Generation of BM chimeras
To generate mixed BM chimeras, BM cells were isolated from the femurs and tibias of CD45.1+ WT, Tlr2–/– or Foxp3CreMyd88fl/fl mice. WT recipient mice (CD45.1+CD45.2+) were sub-lethally irradiated (7 Gy) and intravenously transplanted with a mixture (1:1) of WT (CD45.1) and KO (TLR2 KO or MyD88 KO) BM cells (5×106 BM cells total). Tregs were analyzed 11 weeks after reconstitution.
EAE disease model
On day 0, 8-week-old female mice were immunized subcutaneously with 50 mg MOG35-55 peptide emulsified in complete Freund’s adjuvant containing 2 mg/ml Mycobacterium tuberculosis. On days 0 and 2, 200 ng per mouse pertussis toxin (Merck Millipore) was administered by intraperitoneal injection. The mice were assigned scores daily on a scale of 0–6 with the following criteria: 0, no disease; 1, tail paralysis; 2, wobbly gait; 3, hind limb paralysis; 4, forelimb paralysis; 5, moribund; 6, dead. Gradations of 0.5 were assigned to mice exhibiting signs that fell between two of the scores listed above.
Quantification and statistical analysis
FACS data were collected and processed using FACS software (FlowJo). GraphPad Prism 6 software was used to analyze data using a two-tailed paired Student’s t-test or two-way ANOVA. Data are represented as mean ± SD. P < 0.05 were considered significant.
Additional resources
This study has not generated or contributed to a new website/forum and is not part of a clinical trial.
Acknowledgments
We would like to thank Shuai Bi for technical assistance, Tong Zhao for FACS sorting, and Xiaolan Zhang for performing the confocal laser scanning microscopy. This work was supported by a grant from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29040000) and grants from the National Natural Science Foundation of China (81761128002, 81871297, and 32070163).
Author contributions
S. Meng and X. Li conceived the project. Z. Li and X. Zhou supervised the project. X. Li, Y. Xu, and E. Liu performed the experiments and analyzed the data. X. Xie, J. Wang, H. Zheng, Y. Ju, L. Chen, and C. Li assisted with the performance of some experiments. X. Li and S. Meng wrote the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: December 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103445.
Contributor Information
Xin Li, Email: lix@im.ac.cn.
Songdong Meng, Email: mengsd@im.ac.cn.
Supplemental information
Data and code availability
The raw RNA-seq data reported in this paper were submitted to the Gene Expression Omnibus database available at https://www.ncbi.nlm.nih.gov/geo/ under the accession code GSE154174.
References
- Brodie E.J., Infantino S., Low M.S.Y., Tarlinton D.M. Lyn, lupus, and (B) lymphocytes, a lesson on the critical balance of kinase signaling in immunity. Front Immunol. 2018;9:401. doi: 10.3389/fimmu.2018.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C., Yin C.F., Chai Y.F., Jin H., Wang L.J., Shou S.T. Ulinastatin mediates suppression of regulatory T cells through TLR4/NF-kappa B signaling pathway in murine sepsis. Int. Immunopharmacol. 2018;64:411–423. doi: 10.1016/j.intimp.2018.09.025. [DOI] [PubMed] [Google Scholar]
- Chandawarkar R.Y., Wagh M.S., Srivastava P.K. The dual nature of specific immunological activity of tumor-derived gp96 preparations. J. Exp. Med. 1999;189:1437–1442. doi: 10.1084/jem.189.9.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Davidson T.S., Huter E.N., Shevach E.M. Engagement of TLR2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J. Immunol. 2009;183:4458–4466. doi: 10.4049/jimmunol.0901465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Baumel M., Mannel D.N., Howard O.M., Oppenheim J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007;179:154–161. doi: 10.4049/jimmunol.179.1.154. [DOI] [PubMed] [Google Scholar]
- Dominguez-Villar M., Hafler D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018;19:665–673. doi: 10.1038/s41590-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer G.K., Turnquist H.R. Untangling local pro-inflammatory, reparative, and regulatory damage-associated molecular-patterns (DAMPs) pathways to improve transplant outcomes. Front Immunol. 2021;12:611910. doi: 10.3389/fimmu.2021.611910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Arvey A., Chinen T., van der Veeken J., Gasteiger G., Rudensky A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. 2014;158:749–763. doi: 10.1016/j.cell.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J.A., Park S.O., Choi J.Y., Uyangaa E., Eo S.K. Double-faced implication of CD4(+) Foxp3(+) regulatory T cells expanded by acute dengue infection via TLR2/MyD88 pathway. Eur. J. Immunol. 2020;50:1000–1018. doi: 10.1002/eji.201948420. [DOI] [PubMed] [Google Scholar]
- Hayes C.E., Ntambi J.M. Multiple sclerosis: lipids, lymphocytes, and vitamin D. Immunometabolism. 2020;2:e200019. doi: 10.20900/immunometab20200019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Zhang X., Wei Y., Sun X., Chen Y., Deng J., Jin Y., Gan Y., Hu X., Jia R., et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 2016;22:991–993. doi: 10.1038/nm.4148. [DOI] [PubMed] [Google Scholar]
- Housley W.J., Adams C.O., Nichols F.C., Puddington L., Lingenheld E.G., Zhu L., Rajan T.V., Clark R.B. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J. Immunol. 2011;186:6779–6787. doi: 10.4049/jimmunol.1003868. [DOI] [PubMed] [Google Scholar]
- Hua Z., Gross A.J., Lamagna C., Ramos-Hernandez N., Scapini P., Ji M., Shao H., Lowell C.A., Hou B., DeFranco A.L. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J. Immunol. 2014;192:875–885. doi: 10.4049/jimmunol.1300683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz S.Z., Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinner-Bibeau L.B., Sedlacek A.L., Messmer M.N., Watkins S.C., Binder R.J. HSPs drive dichotomous T-cell immune responses via DNA methylome remodelling in antigen presenting cells. Nat. Commun. 2017;8:15648. doi: 10.1038/ncomms15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y., Ohkura N., Kidani Y., Vandenbon A., Hirota K., Kawakami R., Yasuda K., Motooka D., Nakamura S., Kondo M., et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat. Immunol. 2017;18:173–183. doi: 10.1038/ni.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N., Okamoto K., Sawa S., Nakashima T., Oh-hora M., Kodama T., Tanaka S., Bluestone J.A., Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014;20:62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- Li X., Liu Z., Yan X., Zhang X., Li Y., Zhao B., Wang S., Zhou X., Gao G.F., Meng S. Induction of regulatory T cells by high-dose gp96 suppresses murine liver immune hyperactivation. PLoS ONE. 2013;8:e68997. doi: 10.1371/journal.pone.0068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Park K., Kim J., Min H., Seong R.H. Foxp3 expression in induced regulatory T cells is stabilized by C/EBP in inflammatory environments. EMBO Rep. 2018;19:e45995. doi: 10.15252/embr.201845995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.Y., Liu X.Y., Wang W., Wang S.H., Zhang J.C., Jiang S., Wang Y., Li L.P., Li J.H., Zhang Y.K., et al. Low-dose IL-2 expands CD4(+) regulatory T cells with a suppressive function in vitro via the STAT5-dependent pathway in patients with chronic kidney diseases. Ren. Fail. 2018;40:280–288. doi: 10.1080/0886022X.2018.1456462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Haribhai D., Relland L.M., Truong N., Carlson M.R., Williams C.B., Chatila T.A. Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- Liu F., Wu X., Li L., Liu Z., Wang Z. Formation of peste des petits ruminants spikeless virus-like particles by co-expression of M and N proteins in insect cells. Res. Vet. Sci. 2014;96:213–216. doi: 10.1016/j.rvsc.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Liu W.W., Chen M., Li X.H., Zhao B., Hou J.W., Zheng H.G., Qiu L.P., Li Z.H., Meng S.D. Interaction of toll-like receptors with the molecular chaperone Gp96 is essential for its activation of cytotoxic T lymphocyte response. PLoS ONE. 2016;11:e0155202. doi: 10.1371/journal.pone.0155202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Komai-Koma M., Xu D., Liew F.Y. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc. Natl. Acad. Sci. U S A. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Li X.H., Qiu L.P., Zhang X.J., Chen L.Z., Cao S., Wang F.S., Meng S.D. Treg suppress CTL responses upon immunization with HSP gp96. Eur. J. Immunol. 2009;39:3110–3120. doi: 10.1002/eji.200939593. [DOI] [PubMed] [Google Scholar]
- Lo J.Y., Kamarudin M.N., Hamdi O.A., Awang K., Kadir H.A. Curcumenol isolated from Curcuma zedoaria suppresses Akt-mediated NF-kappaB activation and p38 MAPK signaling pathway in LPS-stimulated BV-2 microglial cells. Food Funct. 2015;6:3550–3559. doi: 10.1039/c5fo00607d. [DOI] [PubMed] [Google Scholar]
- Long M., Park S.G., Strickland I., Hayden M.S., Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Miyara M., Ito Y., Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2014;10:543–551. doi: 10.1038/nrrheum.2014.105. [DOI] [PubMed] [Google Scholar]
- Mohan C., Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat. Rev. Nephrol. 2015;11:329–341. doi: 10.1038/nrneph.2015.33. [DOI] [PubMed] [Google Scholar]
- Morita H., Arae K., Unno H., Miyauchi K., Toyama S., Nambu A., Oboki K., Ohno T., Motomura K., Matsuda A., et al. An interleukin-33-mast cell-interleukin-2 Axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. 2015;43:175–186. doi: 10.1016/j.immuni.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadafi R., Gago de Graca C., Keuning E.D., Koning J.J., de Kivit S., Konijn T., Henri S., Borst J., Reijmers R.M., van Baarsen L.G.M., et al. Lymph node stromal cells generate antigen-specific regulatory T cells and control autoreactive T and B cell responses. Cell. Rep. 2020;30:4110–4123 e4114. doi: 10.1016/j.celrep.2020.03.007. [DOI] [PubMed] [Google Scholar]
- Nascimento D.C., Melo P.H., Pineros A.R., Ferreira R.G., Colon D.F., Donate P.B., Castanheira F.V., Gozzi A., Czaikoski P.G., Niedbala W., et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 2017;8:14919. doi: 10.1038/ncomms14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea M.G., Sutmuller R., Hermann C., Van der Graaf C.A.A., Van der Meer J.W.M., van Krieken J.H., Hartung T., Adema G., Kullberg B.J. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J. Immunol. 2004;172:3712–3718. doi: 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- Nguyen V., Pearson K., Kim J.H., Kamdar K., DePaolo R.W. Retinoic acid can exacerbate T cell intrinsic TLR2 activation to promote tolerance. PLoS ONE. 2015;10:e0118875. doi: 10.1371/journal.pone.0118875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyirenda M.H., Morandi E., Vinkemeier U., Constantin-Teodosiu D., Drinkwater S., Mee M., King L., Podda G., Zhang G.X., Ghaemmaghami A., et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J. Immunol. 2015;194:5761–5774. doi: 10.4049/jimmunol.1400472. [DOI] [PubMed] [Google Scholar]
- Oh H., Grinberg-Bleyer Y., Liao W., Maloney D., Wang P., Wu Z., Wang J., Bhatt D.M., Heise N., Schmid R.M., et al. An NF-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity. 2017;47:450–465.e5. doi: 10.1016/j.immuni.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura N., Hamaguchi M., Morikawa H., Sugimura K., Tanaka A., Ito Y., Osaki M., Tanaka Y., Yamashita R., Nakano N., et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37:785–799. doi: 10.1016/j.immuni.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Ohkura N., Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020;30:465–474. doi: 10.1038/s41422-020-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology. 2020;160:24–37. doi: 10.1111/imm.13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawaria S., Binder R.J. CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat. Commun. 2011;2:521. doi: 10.1038/ncomms1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.D., Cao Z.H., Li X.F., Kang X.L., Xue Y., Li Y.L., Zhang D., Liu X.Y., Xue Y.Z. Effect of ammonium pyrrolidine dithiocarbamate (PDTC) on NF-kappaB activation and CYP2E1 content of rats with immunological liver injury. Pharm. Biol. 2014;52:1460–1466. doi: 10.3109/13880209.2014.898075. [DOI] [PubMed] [Google Scholar]
- Randazzo M., Terness P., Opelz G., Kleist C. Active-specific immunotherapy of human cancers with the heat shock protein Gp96-revisited. Int. J. Cancer. 2012;130:2219–2231. doi: 10.1002/ijc.27332. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Miyara M., Costantino C.M., Hafler D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Powrie F., Ransohoff R.M. Re-establishing immunological self-tolerance in autoimmune disease. Nat. Med. 2012;18:54–58. doi: 10.1038/nm.2622. [DOI] [PubMed] [Google Scholar]
- Schumann R.R., Tapping R.I. Genomic variants of TLR1 - it takes (TLR-)two to tango. Eur. J. Immunol. 2007;37:2059–2062. doi: 10.1002/eji.200737604. [DOI] [PubMed] [Google Scholar]
- Sidwell T., Liao Y., Garnham A.L., Vasanthakumar A., Gloury R., Blume J., Teh P.P., Chisanga D., Thelemann C., de Labastida Rivera F., et al. Attenuation of TCR-induced transcription by Bach2 controls regulatory T cell differentiation and homeostasis. Nat. Commun. 2020;11:252. doi: 10.1038/s41467-019-14112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallone G., Infante B., Di Lorenzo A., Rascio F., Zaza G., Grandaliano G. mTOR inhibitors effects on regulatory T cells and on dendritic cells. J. Transl Med. 2016;14:152. doi: 10.1186/s12967-016-0916-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutmuller R.P.M., den Brok M.H.M.G.M., Kramer M., Bennink E.J., Toonen L.W.J., Kullberg B.J., Joosten L.A., Akira S., Netea M.G., Adema G.J. Toll-like receptor 2 controls expansion and function of regulatory T cells. J. Clin. Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton A.M., Lu J., Korty P.E., Kim Y.C., Martens C., Sun P.D., Shevach E.M. Helios(+) and Helios(-) Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. Eur. J. Immunol. 2019;49:398–412. doi: 10.1002/eji.201847935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomer K.H., Yuan X.M., Yang J., Dee M.J., Yu A.X., Malek T.R. Developmental progression and interrelationship of central and effector regulatory T cell subsets. J. Immunol. 2016;196:3665–3676. doi: 10.4049/jimmunol.1500595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eden W., Jansen M.A.A., Ludwig I., van Kooten P., van der Zee R., Broere F. The enigma of heat shock proteins in immune tolerance. Front Immunol. 2017;8:1599. doi: 10.3389/fimmu.2017.01599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eden W., van der Zee R., Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 2005;5:318–330. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- Wang S., Charbonnier L.M., Rivas M.N., Georgiev P., Li N., Gerber G., Bry L., Chatila T.A. MyD88 adaptor-dependent microbial sensing by regulatory T cells promotes mucosal tolerance and enforces commensalism. Immunity. 2015;43:289–303. doi: 10.1016/j.immuni.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X., Zhang J., Zhou X. Ex-TFRs: a missing piece of the SLE puzzle? Front Immunol. 2021;12:662305. doi: 10.3389/fimmu.2021.662305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Hong F., Gewirth D., Guo B., Liu B., Li Z. The molecular chaperone gp96/GRP94 interacts with Toll-like receptors and integrins via its C-terminal hydrophobic domain. J. Biol. Chem. 2012;287:6735–6742. doi: 10.1074/jbc.M111.309526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Harder K.W., Huntington N.D., Hibbs M.L., Tarlinton D.M. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22:9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Zhan T., Wang X., Ouyang Z., Yao Y., Xu J., Liu S., Liu K., Deng Q., Wang Y., Zhao Y. Rotating magnetic field ameliorates experimental autoimmune encephalomyelitis by promoting T cell peripheral accumulation and regulating the balance of Treg and Th1/Th17. Aging (Albany NY) 2020;12:6225–6239. doi: 10.18632/aging.103018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wei Q., Xiang X., Zhou B., Chen J., Li J., Li Q., Xiong H., Liu F. Semaphorin 4A acts in a feed-forward loop with NF-kappaB pathway to exacerbate catabolic effect of IL-1beta on chondrocytes. Int. Immunopharmacol. 2019;69:88–94. doi: 10.1016/j.intimp.2019.01.006. [DOI] [PubMed] [Google Scholar]
- Zhang X., Olsen N., Zheng S.G. The progress and prospect of regulatory T cells in autoimmune diseases. J. Autoimmun. 2020;111:102461. doi: 10.1016/j.jaut.2020.102461. [DOI] [PubMed] [Google Scholar]
- Zhang Y.L., Wu B.X., Metelli A., Thaxton J.E., Hong F., Rachidi S., Ansa-Addo E., Sun S.L., Vasu C., Yang Y., et al. GP96 is a GARP chaperone and controls regulatory T cell functions. J. Clin. Invest. 2015;125:859–869. doi: 10.1172/JCI79014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Chu Y., Liang Z., Zhang B., Wang X., Jing X., Hao M., Wang Y., An J., Zhang X., et al. Low dose of IL-2 combined with rapamycin restores and maintains the long-term balance of Th17/Treg cells in refractory SLE patients. BMC Immunol. 2019;20:32. doi: 10.1186/s12865-019-0305-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Josefowicz S., Chaudhry A., Peng X.P., Forbush K., Rudensky A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.Y., Bailey-Bucktrout S.L., Jeker L.T., Penaranda C., Martinez-Llordella M., Ashby M., Nakayama M., Rosenthal W., Bluestone J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009;10:1000–U1104. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA-seq data reported in this paper were submitted to the Gene Expression Omnibus database available at https://www.ncbi.nlm.nih.gov/geo/ under the accession code GSE154174.