

Abstract
Mitochondrial cristae are extraordinarily crowded with proteins, which puts stress on the bilayer organization of lipids. We tested the hypothesis that the high concentration of proteins drives the tafazzin‐catalyzed remodeling of fatty acids in cardiolipin, thereby reducing bilayer stress in the membrane. Specifically, we tested whether protein crowding induces cardiolipin remodeling and whether the lack of cardiolipin remodeling prevents the membrane from accumulating proteins. In vitro, the incorporation of large amounts of proteins into liposomes altered the outcome of the remodeling reaction. In yeast, the concentration of proteins involved in oxidative phosphorylation (OXPHOS) correlated with the cardiolipin composition. Genetic ablation of either remodeling or biosynthesis of cardiolipin caused a substantial drop in the surface density of OXPHOS proteins in the inner membrane of the mouse heart and Drosophila flight muscle mitochondria. Our data suggest that OXPHOS protein crowding induces cardiolipin remodelling and that remodeled cardiolipin supports the high concentration of these proteins in the inner mitochondrial membrane.
Keywords: Barth syndrome, lipid‐protein interaction, macromolecular crowding, mitochondria, oxidative phosphorylation
Subject Categories: Membranes & Trafficking, Organelles
Mutual crosstalk between membrane lipids and proteins acts as a rheostat to regulate mitochondrial cristae ultrastructure.
Introduction
Mitochondria contain the structurally unique and evolutionarily conserved phospholipid cardiolipin (CL) that is located in the inner membrane (IM). CL has been implicated in several mitochondrial activities, including the formation of respiratory supercomplexes (Zhang et al, 2002; Pfeiffer et al, 2003), maintenance of the membrane potential (Jiang et al, 2000; Koshkin & Greenberg, 2002), cristae biogenesis (Kojima et al, 2019), the fission–fusion cycle (Ban et al, 2017), and oxidative stress (Tyurina et al, 2014), but a unifying mechanism to explain its various functions has remained elusive. However, it has been recognized that the mechanism by which CL affects mitochondria, can be attributed to the unique physicochemical properties of a lipid that has intrinsic negative curvature and that associates tightly with membrane proteins (Lewis & McElhany, 2009; Mileykovskaya & Dowhan, 2009; Ren et al, 2014; Mårtensson et al, 2017; Pennington et al, 2019; Acoba et al, 2020).
Important clues about the function of CL have emerged from the study of Barth syndrome, a disease caused by abnormal fatty acids in CL (Hauff & Hatch, 2006; Ren et al, 2014). Normally, CL receives fatty acids from other phospholipids by tafazzin‐catalyzed transacylation, a process referred to as CL remodeling (Xu et al, 2006a). Deletion of tafazzin alters the CL species composition (Vreken et al, 2000), makes CL prone to degradation (Xu et al, 2016), and causes mitochondrial dysfunction (Ma et al, 2004), which in humans presents with the clinical features of Barth syndrome (Barth et al, 1983; Clarke et al, 2013). These data strongly suggest that CL remodeling is functionally significant but raise questions about its mechanism and its role.
We have recently proposed that CL remodeling is caused by the high density of proteins in the IM, which imposes physical stress on the packing order of lipids. This hypothesis has emerged from our quest to identify the process that induces CL remodeling in mitochondria. We had shown that stress on the physical state of lipids can trigger remodeling reactions in vitro (Schlame et al, 2012a) and were trying to determine the source of lipid stress in vivo. We made the unexpected discovery that reduced expression of proteins of oxidative phosphorylation (OXPHOS) inhibits CL remodeling, having the same effect on CL as the deletion of tafazzin (Xu et al, 2019). Importantly, it was the global expression of IM proteins, rather than the expression or the redox activity of any specific OXPHOS complex, that was critical for CL remodeling.
The idea that the protein content of the IM requires CL remodeling is based on four premises. (i) The IM of mitochondria is extremely crowded, far exceeding the protein concentration of most other biological membranes (Lotan & Nicolson, 1981; Capaldi, 1982; Hatefi, 1985). (ii) Crowding disturbs the organization of lipids and deforms the bilayer, which puts stress on the elastic properties of lipids and therefore incurs a substantial increase in free energy (Andersen & Koeppe, 2007; Phillips et al, 2009; Guigas & Weiss, 2016; Brown, 2017). (iii) Stress in the packing order of lipids is a necessary condition for the tafazzin reaction. For instance, stress resulting from the transition of bilayers into strongly curved hexagonal structures induces the formation of remodeled CL in vitro (Schlame et al, 2012a). (iv) CL remodeling produces unsaturated species (Oemer et al, 2020) that have a larger intrinsic negative curvature and a greater propensity to support non‐bilayer structures than saturated species (Sankaram et al, 1989). As a result, remodeling may reduce curvature stress and the energy penalty of crowding. It is therefore plausible to postulate that CL remodeling stabilizes protein‐crowded membranes and that protein crowding, in turn, provides the driving force for CL remodeling.
In this paper, we tested two predictions made by the crowding hypothesis. First, we tested whether membrane protein crowding in and of itself affects the outcome of the tafazzin reaction and whether the concentration of membrane proteins correlates with the molecular composition of CL. Second, we tested whether genetic deletion of tafazzin prevents protein crowding in the mitochondrial IM and, if so, whether this effect is dependent on CL. Specifically we determined whether deletion of either tafazzin or CL synthase reduces the surface concentration of the most abundant proteins of the IM, i.e. the OXPHOS complexes and the solute carriers.
Results
Protein crowding induces tafazzin‐catalyzed CL remodeling in liposomes
Membrane proteins influence the physical state of lipids, and this effect is dependent on the protein‐to‐lipid ratio (Andersen & Koeppe, 2007; Brown, 2017). The state of lipids, in turn, is critical for the velocity, the specificity, and the equilibrium of the tafazzin reaction (Schlame et al, 2012a, 2017). Thus, the question arises as to whether membrane proteins, by affecting the physical state of lipids, are able to alter quality and quantity of tafazzin‐catalyzed transacylations. This question is not specific to mitochondrial proteins but applies to all membrane proteins that may interfere with the packing order of lipids. Here we determined the effect of myelin basic protein (MBP) and bacteriorhodopsin (BR) because they have a strong influence on their lipid environment (Epand & Moscarello, 1982; Botelho et al, 2006; Verchère et al, 2017) and because they are among the few membrane proteins available in a detergent‐free state. The latter is critical given that detergents alone alter the outcome of the tafazzin reaction (Schlame et al, 2012a, 2017).
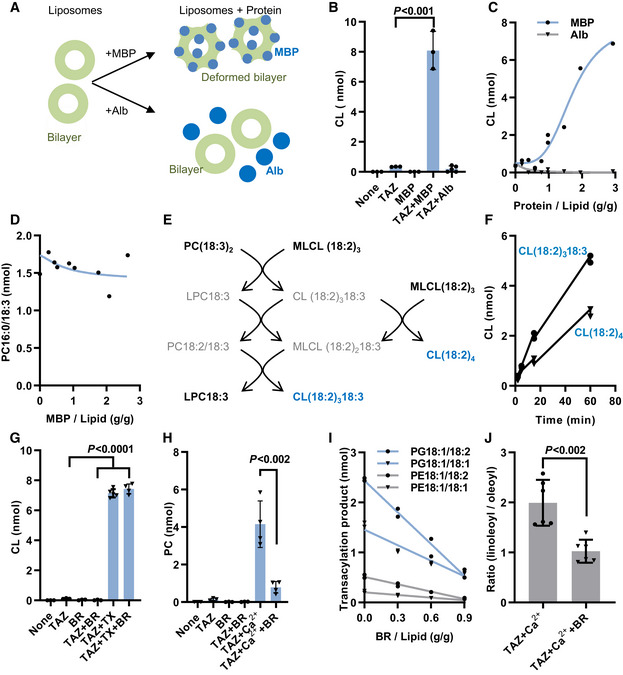
MBP is a monotopic membrane protein that has a strong impact on the organization of lipids. The structural effects of MBP depend on the lipid composition and have not been defined in every case, but studies have consistently shown that large amounts of MBP (protein/lipid mass ratio near unity) alter the physical properties of lipids, including packing conditions, phase transition temperatures, phase segregation behavior, and the propensity for non‐bilayer assemblies (Epand & Moscarello, 1982; Smith & Cornell, 1985; Rosetti et al, 2010). We prepared liposomes from dilinoleoyl‐phosphatidylcholine (PC18:2/18:2) and trilinoleoyl‐monolyso‐cardiolipin (MLCL18:2/18:2/18:2) and let them react with purified recombinant tafazzin in the presence or absence of MBP (Fig 1A). Tafazzin alone did not catalyze any acyl transfer from PC to MLCL, confirming that the enzyme does not react with lipids in the bilayer state (Schlame et al, 2012a, 2017). However, the reaction was strongly activated upon addition of MBP. This was caused by an effect of MBP on the physical environment, presumably on the physical properties of lipids, rather than by inherent acyltransferase activity because MBP alone did not elicit any reaction. The effect of MBP was not duplicated by albumin, a protein that does not interact with membranes (Fig 1B and C) and required high protein‐to‐lipid ratios, suggesting a crowding effect (Fig 1C). MBP did not stimulate the tafazzin reaction with micellar substrates consisting of 85% palmitoyl‐lysophosphatidylcholine (LPC16:0) and 15% PC18:3/18:3 (Fig 1D), which confirms that the physical state of lipids was critical for the MBP mechanism. These data strongly suggest that MBP acted on lipids rather than on tafazzin. When tafazzin reacted with liposomes consisting of PC18:3/18:3 and MLCL18:2/18:2/18:2, MBP triggered the formation of two different CL species, an outcome that requires sequential forward and reverse transacylations among multiple phospholipid species (Fig 1E and F). Thus, MBP activated the tafazzin reaction when added to liposomes at high concentration, prompting a series of acyl exchanges between CL and PC.
Figure 1. Membrane proteins alter CL remodeling in vitro .
-
AMyelin basic protein (MBP) or albumin (Alb) were added to liposomes that contained purified tafazzin (TAZ). Acyl transfer reactions were measured by mass spectrometry.
-
BCL formation was measured in the presence of 60 nmol MLCL(18:2)3, 40 nmol PC(18:2)2, 4 µg TAZ, and 0.2 mg MBP or 0.2 mg Alb.
-
CCL formation was measured as in B with different amounts of MBP or Alb.
-
DPC16:0/18:3 formation was measured in the presence of 85 nmol LPC16:0, 15 nmol PC18:3/18:3, 4 µg TAZ, and MBP.
-
E, FCL formation was measured in the presence of 60 nmol MLCL(18:2)3, 40 nmol PC(18:3)2, 4 µg TAZ, and 0.2 mg MBP (2 independent measurements per time point).
-
GCL formation was measured in the presence of 40 nmol PC(18:2)2, 10 nmol MLCL(18:2)3, 8 µg TAZ, 1250 nmol Triton X‐100 (TX), and 5.6 nmol bacteriorhodopsin (BR).
-
HPC formation was measured in the presence of 180 nmol CL(18:2)4, 20 nmol LPC16:0, 8 µg TAZ, 20 mM Ca2+, and 5.6 nmol BR.
-
ITransacylations were measured in mixtures containing 90 nmol CL(18:1)4, 90 nmol CL(18:2)4, 10nmol LPG18:1, 10 nmol LPE18:1, 8 µg TAZ, 20 mM Ca2+, and different amounts of BR (2 independent measurements per BR/Lipid ratio).
-
JTransacylations were measured as in I in the presence of 0.9 g BR/ g lipid. The ratio of total linoleoyl transfer over total oleoyl transfer was calculated.
Data information: The graphs show replicas of independent experiments, their mean values, and standard deviations. Means were compared by Student’s t‐test.
In contrast to MBP, BR did not activate the tafazzin reaction when added to PC/MLCL liposomes. As expected, the reaction was activated by Triton X‐100, which converts bilayers into micelles, but again BR had no effect on the reaction in the presence of Triton X‐100 (Fig 1G). To test the effect of BR on the reverse reaction, we prepared liposomes from lysophosphatidylcholine (LPC) and CL. BR also failed to activate the reverse reaction. However, as shown previously (Schlame et al, 2012a), the reverse reaction was activated by Ca2+, which converts CL‐containing liposomes into inverted micelles. Surprisingly, BR inhibited the reverse transacylation in the presence of Ca2+ (Fig 1H). These data demonstrate that the effect of BR on the tafazzin reaction depends on the lipid phase state and is different from the effect of MBP. Integral membrane proteins, like BR, are generally known to stabilize the bilayer phase state and have specifically been shown to prevent the formation of Ca2+‐induced inverted micelles (Taraschi et al, 1983). Thus, the action of BR can be explained by its effect on the organization of lipids. BR acts on different CL/lysophospholipid pairs but only at high BR/lipid ratios, which is consistent with a crowding mechanism (Fig 1I). Importantly, BR altered the acyl specificity of the reaction, suggesting that protein crowding can affect the qualitative outcome of the reaction (Fig 1J).
In summary, the data show that some membrane proteins have the potential to affect the tafazzin reaction when present at high concentration. They can induce or inhibit acyl remodeling, alter the acyl specificity of the reaction, and initiate bidirectional acyl exchanges. These activities mimic CL remodeling in vivo, implying that the density of IM proteins could in theory control the molecular species composition of CL.
IM protein concentration correlates with CL composition in yeast
Since protein crowding affected CL remodeling in liposomes, we were asking whether the same can be demonstrated for mitochondrial membranes. However, it is not straightforward to increase the protein concentration of mitochondria because tight import regulation prevents the over‐accumulation of proteins (Friedman & Nunnari, 2014; Harbauer et al, 2014). One exception where large changes in mitochondrial protein expression are possible is Saccharomyces cerevisiae. This microorganism can alternate between two metabolic states, in which energy is harvested either by glycolysis or by respiration. It has been shown that not only the abundance of mitochondria and the cellular amount of OXPHOS proteins but also the concentration of OXPHOS proteins in mitochondria increases during the transition from the fermentative to the respiratory state (Di Bartolomeo et al, 2020). However, since this transition also leads to a proliferation of cristae, it is not clear whether the transition actually increases the abundance of OXPHOS proteins per surface area of IM.
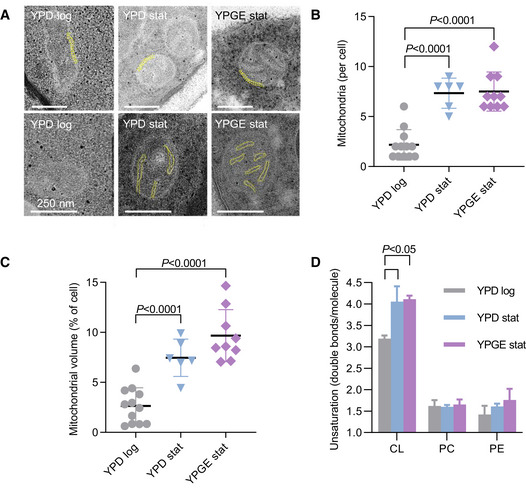
To determine the protein concentration of the IM, we measured the surface area of the IM by electron microscopy and the abundance of IM proteins (OXPHOS complexes and solute carriers) by mass spectrometry using label‐free quantitation. These data permitted the calculation of relative values that were proportional to the average surface density of proteins in the IM. As internal reference we chose the outer membrane (OM) because it forms a uniform envelope around mitochondria regardless of their internal composition and the OM‐associated TOM complex because it shows little variation between respiring and fermentative yeast (Di Bartolomeo et al, 2020) and between yeast with and without CL (Sauerwald et al, 2015). We analyzed S. cerevisiae under three conditions, including logarithmic growth on yeast extract/peptone/dextrose (YPD log), stationary growth on YPD (YPD stat), and stationary growth on yeast extract/peptone/glycerol/ethanol (YPGE stat). These conditions were chosen to produce a progressive shift from the fermentative to the respiratory state because respiration is higher in the stationary than in the logarithmic phase and is higher in glycerol/ethanol than in dextrose. As expected, cristae were present in YPD stat and YPGE stat but nearly absent in YPD log (Fig EV1A). The number of mitochondria per cellular cross section and the total mitochondrial volume increased from YPD log to YPD stat but showed little further increase from YPD stat to YPGE stat (Fig EV1B and C). By tracing mitochondrial membranes in electron micrographs, we determined that the IM/OM area ratio also increased from YPD log to YPD stat but not any further from YPD stat to YPGE stat (Fig 2A). Finally, we found that the relative abundance of solute carriers and OXOHOS proteins increased from YPD log to YPD stat and then increased further from YPD stat to YPGE stat (Fig 2B). Importantly, the abundance of carriers and OXPHOS proteins rose by a much larger factor than the IM surface area, suggesting that their concentration was higher in YPD stat than in YPD log and even higher in YPGE stat.
Figure EV1. Stationary growth increases the density of cristae, the abundance of mitochondria, and the unsaturation of cardiolipin (CL), but not the unsaturation of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) in S. cerevisiae .
-
AThe mitochondrial ultrastructure was analyzed by transmission electron microscopy. The yellow labels show the double membrane of mitochondria (OM and boundary IM) in the upper panels and mitochondrial cristae (IM) in the lower panels. Scale bars have a length of 250 nm.
-
B, CThe number of mitochondrial cross‐sections and the mitochondrial volume were quantified in randomly collected electron micrographs. Multiple micrographs were analyzed per biological replica.
-
DLipids were analyzed by mass spectrometry and the number of double bonds per molecule was calculated from the compositions of CL, PC, and PE.
Data information: The graphs show biological replicas (N = 3), mean values, and standard deviations. Means were compared by Student’s t‐test.
Figure 2. Membrane protein concentration correlates with CL composition in yeast mitochondria.
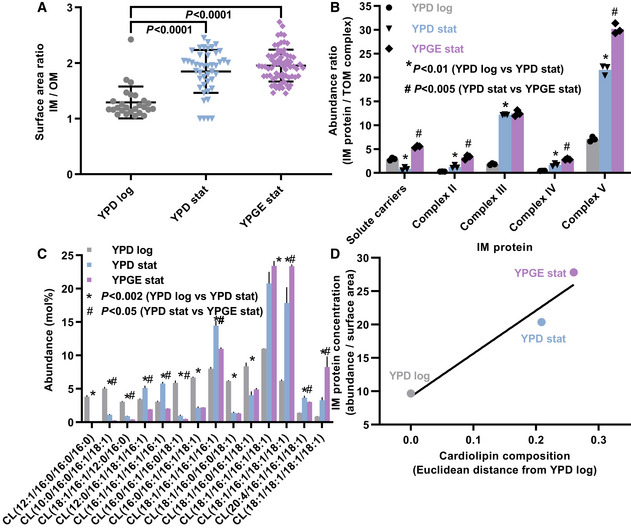
- Inner (IM) and outer (OM) mitochondrial membranes were traced in randomly collected electron micrographs in order to calculate the IM/OM surface area ratio. Multiple micrographs were analyzed per biological replica.
- Abundances of protein complexes were measured by label‐free quantitative proteomics.
- Molecular species of CL were quantified by lipidomics.
- The IM protein concentration was calculated by dividing the sum of the abundance ratios (IM complexes/TOM complex) by the surface area ratio (IM/OM). The CL composition was expressed as Euclidean distance from the CL composition in YPD log cells. A linear regression line is drawn.
Data information: Graphs show mean values and standard deviations of three biological replicas. Means were compared by Student’s t‐test.
To determine whether the change in protein concentration was associated with altered CL remodeling, we analyzed the CL composition under the aforementioned culture conditions. The unsaturation of CL increased from YPD log to YPD stat and from YPD stat to YPGE stat. Specifically, we found an increase in tetraunsaturated species at the expense of diunsaturated species (Fig 2C) consistent with our previous results (Xu et al, 2019). These changes were specific to CL and were not reproduced by PC and phosphatidylethanolamine (PE) (Fig EV1D). Thus, the progressive rise in protein concentration in the IM from YPD log to YPD stat and from YPD stat to YPGE stat was associated with an equally progressive change in the CL composition (Fig 2D).
To further test the concept that a rising IM protein concentration affects CL remodeling, we forced expression of the alternative oxidase (AOX1), an integral IM protein, in Pichia pastoris (Young et al, 2013). In P. pastoris, inducible AOX1 expression can increase the AOX1 abundance substantially while preserving mitochondrial targeting (Vogl et al, 2014). Consistent with the idea that an increase in IM protein concentration can trigger CL remodeling, we found that forced expression of AOX1 caused substantial changes in the CL composition, specifically the rise of all‐C18 unsaturated species at the expense of mixed acyl species (Fig EV2).
Figure EV2. AOX expression alters the molecular species composition of CL in P. pastoris .
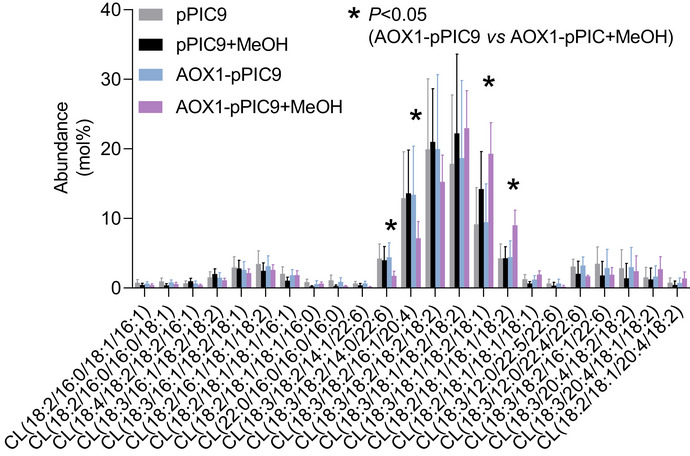
Cells were transformed with alternative oxidase (AOX1) inserted into pPIC9 vector DNA, which confers methanol‐inducible overexpression of AOX1. Cells were grown in MGY medium either in the presence or absence of 1% methanol (MeOH). The molecular species composition of CL was analyzed. Only species > 0.5 mol% are shown. The graph shows biological replicas (N = 4), mean values, and standard deviations. Means were compared by Student’s t‐test.
Taken together, the data support the idea that the dense packing of proteins in the IM is driving CL remodeling and that the CL composition varies with the actual concentration of membrane proteins. They further suggest that this effect can be induced by different membrane proteins.
CL remodeling is necessary to concentrate proteins in the IM of heart mitochondria
If protein crowding induces CL remodeling, remodeled CL may confer an advantage to protein‐crowded membranes. If that is true, genetic ablation of the remodeling enzyme tafazzin (TAZKO) may prevent the membrane from reaching a high protein concentration. To test this conjecture, we determined the effect of TAZKO on the protein concentration of the IM. We measured the IM surface area by electron microscopy and the IM protein abundance by Western blotting and mass spectrometry.
These measurements were carried out in murine hearts because we anticipated heart mitochondria to be particularly susceptible to TAZKO given their high expression of OXPHOS proteins. We confirmed that TAZKO caused substantial changes in the molecular species pattern of CL both in embryonic and adult hearts but also in other organs, such as the liver. The shift from polyunsaturated to partially saturated species was accompanied by the appearance of MLCL, an indicator of excessive CL degradation (Table EV1). TAZKO mice were smaller than controls (22 ± 3 g versus 33 ± 3 g, N = 8, P < 0.0001, t‐test) and experienced increased embryonic and perinatal mortality (15.6% observed versus 50.0% expected homozygous KO males at weaning, N = 77, P < 10−6, Fisher’s exact test), which is consistent with a recently published report of the mouse model (Wang et al, 2020). TAZKO mice also had cardiomyopathy. They accumulated lipid droplets within cardiomyocytes (Fig EV3A) and showed a blunted response to pharmacologic stress of the heart rate and shortening fraction (Fig EV3B). However, the cardiomyopathy was milder than the one reported before (Wang et al, 2020) likely owing to differences in genetic background or breeding conditions. In particular, the ultrastructure of cardiac mitochondria was not affected (Fig 3A). Cardiac mitochondria had normal size and normal IM surface area both in embryonic and in adult TAZKO hearts (Fig 3B).
Figure EV3. TAZKO causes cardiomyopathy in mice.
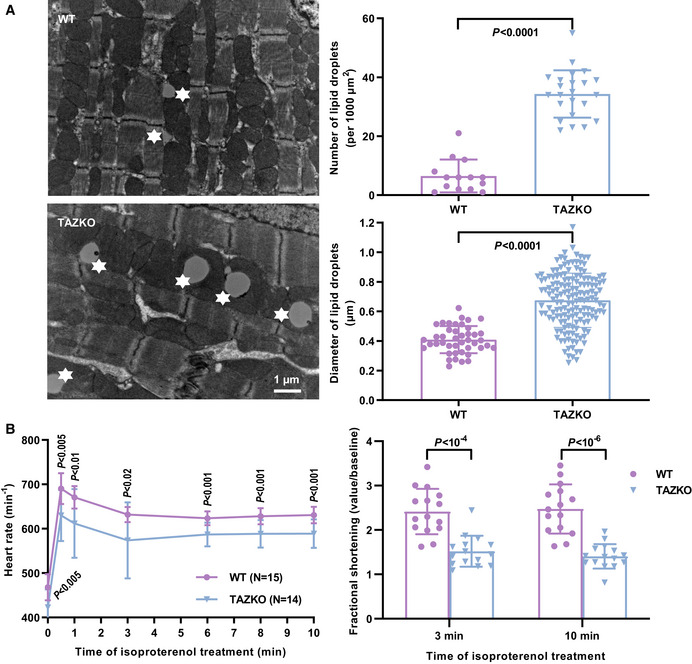
- Heart tissue was analyzed by quantitative electron microscopy. Lipid droplets are marked by asterisks. Graphs show mean values and standard deviations. Multiple micrographs were analyzed in three biological replicas.
- Anesthetized mice were subjected to a pharmacologic stress test by isoproterenol infusion. Cardiac performance was analyzed by echocardiography.
Data information: The graphs show mean values and standard deviations of the indicated number of biological replicas. Means were compared by Student’s t‐test.
Figure 3. TAZKO reduces the concentration of proteins in the IM of mouse heart mitochondria.
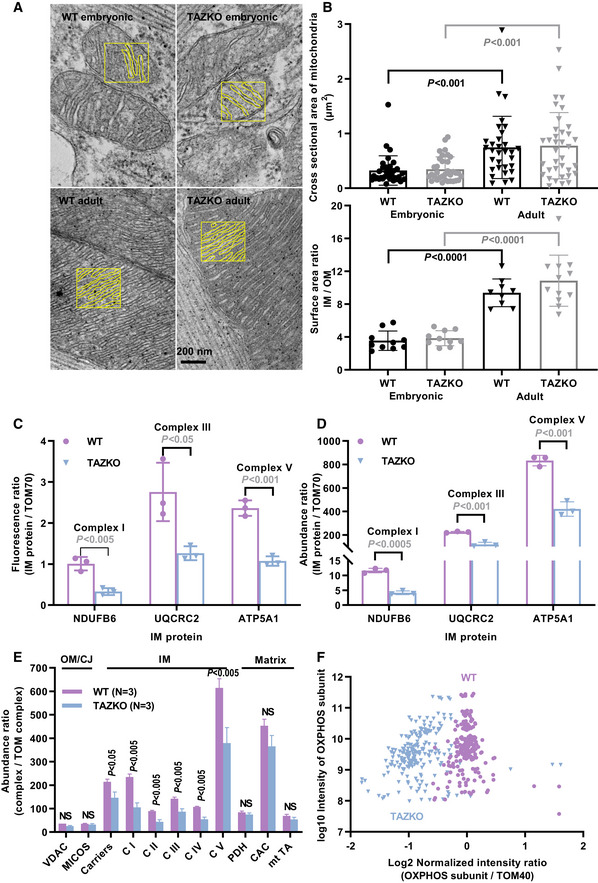
-
AMitochondrial membranes were traced in electron micrographs to determine the IM/OM surface area ratio. Tracings of the IM are shown in yellow boxes.
-
BCross sectional areas of mitochondria and IM/OM surface area ratios were determined in randomly collected electron micrographs. Multiple micrographs were analyzed per biological replica.
-
CProtein abundances were measured in adult hearts by quantitative Western blotting with fluorescence‐labeled secondary antibodies. The abundances of IM proteins (NDUFB6, UQCR2, ATP5A1) are expressed relative to the abundance of the OM protein TOM70.
-
D–FRelative abundances of mitochondrial proteins were measured in adult hearts by label‐free quantitative proteomics. C I‐V, complex I‐V; CAC, citric acid cycle enzymes; MICOS, contact site complex; mt TA, mitochondrial transaminases; NS, not significant; PDH pyruvate dehydrogenase; VDAC, voltage‐dependent anion channel.
Data information: The graphs show mean values and standard deviations of three biological replicas. Means were compared by Student’s t‐test. Data in F represent 77 OXPHOS subunits measured in three biological replicas.
In order to determine the effect of TAZKO on the concentration of IM proteins, we measured the abundance of three OXPHOS subunits (NDUFB6, UQCR2, ATP5A1) and the abundance of a subunit of the TOM complex (TOM70) as reference. First, we performed quantitative Western blotting with fluorescence‐labeled antibodies (Fig EV4), which showed that the three OXPHOS/TOM70 abundance ratios were substantially lower in TAZKO hearts than in controls (Fig 3C). Then we repeated the measurement by label‐free relative quantitative proteomics, which confirmed the effect of TAZKO on the OXPHOS/TOM70 abundance ratios (Fig 3D). Further analysis of the proteomics data revealed that TAZKO reduced the relative abundance of several IM proteins, including solute carriers and all OXPHOS complexes. No such effect was observed on proteins residing in the OM, cristae junctions (CJ), or the matrix (Fig 3E). A scatter plot of 77 OXPHOS subunits confirmed that TAZKO decreased the mitochondrial concentration of OXPHOS proteins by about 50 percent (Fig 3F). This decrease applied equally to subunits encoded by the nuclear or the mitochondrial DNA (Fig EV5A). Since only nuclear but not mitochondrial subunits have to be imported from the cytosol, our data suggest that TAZKO inhibited protein assembly into the membrane but not protein import.
Figure EV4. TAZKO reduces the abundance of OXPHOS complexes I, III, and V in heart mitochondria.
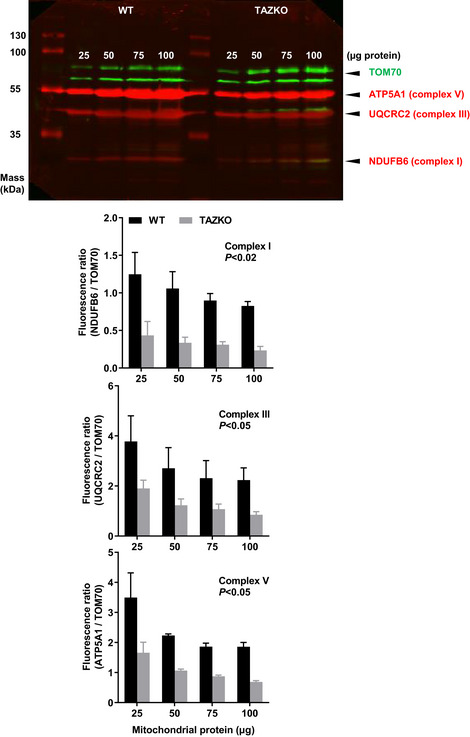
Heart mitochondria were isolated from 5 months old mice (WT and TAZKO). Different amounts of mitochondria (25–100 µg protein) were analyzed by quantitative Western blotting with fluorescence‐labeled secondary antibodies. The graphs show mean values and standard deviations of three biological replicas. Means were compared by Student’s t‐test.
Figure EV5. CL deficiency reduces the abundance of OXPHOS subunits encoded by nuclear and mitochondrial DNA.
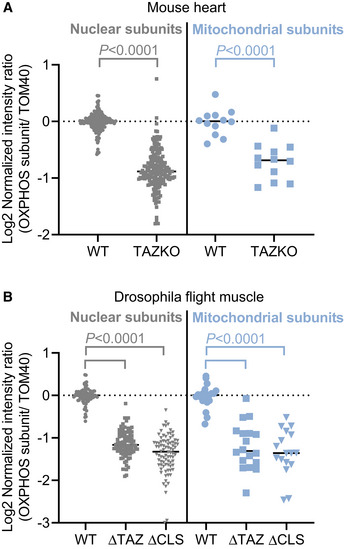
- Hearts were harvested from WT or TAZKO mice at the age of 5 months and analyzed by label‐free quantitative proteomics.
- Indirect flight muscles were harvested from adult flies with mutations in the tafazzin gene (∆TAZ) or the CL synthase gene (∆CLS) or from wild type (WT) and analyzed by label‐free quantitative proteomics.
Data information: The graphs show individual and mean values. Data represent 73 nuclear and four mitochondrial OXPHOS subunits (A) or 28 nuclear and seven mitochondrial OXPHOS subunits (B) of three biological replicas each. Means were compared by Student’s t‐test.
In summary, TAZKO reduced the mitochondrial concentration of OXPHOS proteins and carriers but not the IM surface area. Consequently, TAZKO caused a substantial decrease in the surface density of IM proteins, suggesting that the IM fails to incorporate large amounts of membrane proteins when CL is not remodeled.
CL deficiency reduces the protein concentration in the IM of flight muscle mitochondria
To test the effect of tafazzin on IM proteins in a different model, we created a tafazzin mutant (∆TAZ) in Drosophila melanogaster. We studied mitochondria of indirect flight muscles because, like mouse heart, it is a tissue with very high OXPHOS capacity. We compared ∆TAZ with the wild‐type and with another mutant, in which CL was deleted (inactivation of CL synthase, ∆CLS) in order to confirm that CL was indeed involved in the mechanism. Again, we measured IM surface area and IM protein abundance, and we analyzed the mitochondrial morphology in the mutants and the wild type.
∆TAZ and ∆CLS did not affect the characteristic lamellar morphology of IMs in flight muscle mitochondria. However, the mutant mitochondria contained localized defects with dilated cristae and aggregated IMs, often coalescing into empty cavities (Fig 4A). These defects were found in 20–30% of the mutant mitochondria (Fig 4B) but comprised only 2–5% of the total mitochondrial volume (Fig 4C). The proportion of tubular cristae was somewhat higher in ∆CLS (Fig 4D) but lamellar cristae occupied > 90% of the intramitochondrial space in all strains. As a result, the IM/OM surface area ratio did not change between the mutants and the control (Fig 4E). In contrast, ∆TAZ and ∆CLS caused a large (50–70%) decrease in the mitochondrial concentration of IM proteins, including solute carriers and OXPHOS complexes I‐V. Again, no such effect was observed on proteins residing in the OM, CJ, or matrix, respectively (Fig 4F). A scatter plot of 35 OXPHOS subunits confirmed that their relative abundance decreased by more than 50% in both mutants (Fig 4G). Again, subunits encoded by the nuclear DNA and subunits encoded by the mitochondrial DNA were equally affected (Fig EV5B), suggesting that ∆TAZ and ∆CLS inhibited the assembly of proteins into the IM rather than protein import.
Figure 4. CL deficiency causes reduced protein concentration and localized defects in the IM of Drosophila flight muscle mitochondria.
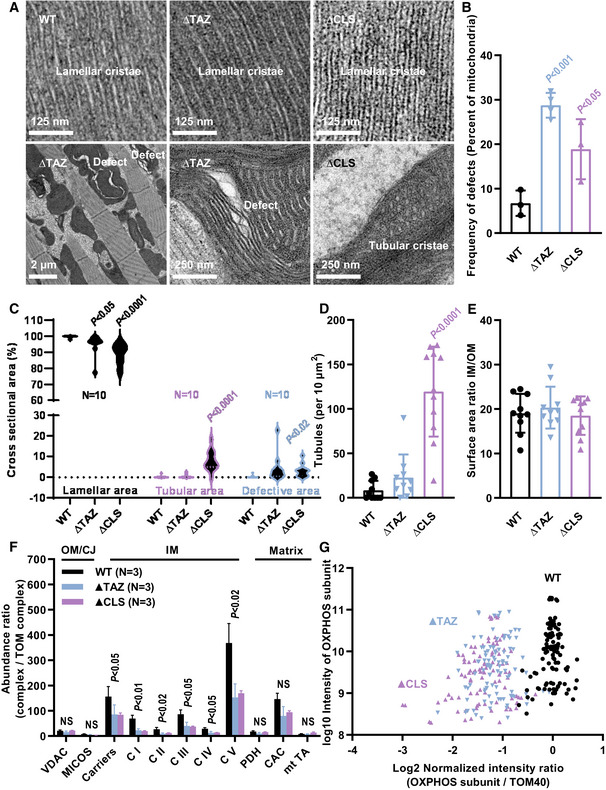
-
A–ETissues were analyzed by quantitative electron microscopy. Multiple micrographs were analyzed per biological replica.
-
F, GTissues were analyzed by label‐free quantitative proteomics. C I‐V, complex I‐V; CAC, citric acid cycle enzymes; MICOS, contact site complex; mt TA, mitochondrial transaminases; NS, not significant; PDH pyruvate dehydrogenase; VDAC, voltage‐dependent anion channel.
Data information: The graphs show mean values and standard deviations of three biological replicas. Mutants were compared to the wild type by Student’s t‐test. In B, each data point is the mean value of 40–70 mitochondrial cross sections. In C, data were collected from 10 electron micrographs per strain. In G, data represent 35 OXPHOS subunits measured in three biological replicas.
Taken together, our data demonstrate a drop in the surface density of IM proteins in mutant mitochondria, with little difference between ∆TAZ and ∆CLS. This conclusion follows from the reduced abundance of OXPHOS proteins and carriers in the face of an unchanged IM surface area. The decrease in protein concentration was specific to the IM because it was not observed in other mitochondrial compartments. Thus, remodeled CL is necessary to concentrate membrane proteins in the IM and either the lack of CL remodeling or the lack of CL biosynthesis prevents the assembly of protein‐rich mitochondrial IMs.
Discussion
The presence of CL in mitochondria has long inspired speculations as to why this structurally unique phospholipid is necessary in the powerhouses of eukaryotic cells. Our data support the hypothesis that CL allows mitochondria to form tightly packed IMs with a high surface density of proteins and that CL, in order to fulfill this function, has to have a particular fatty acid composition.
The conclusion is supported by three key findings. First protein crowding triggered the fatty acid remodeling of CL in vitro, second there was a link between the protein concentration and the CL composition in the IM of mitochondria, and third either the lack of CL or the lack of CL remodeling prevented the IM from reaching a high surface density of proteins. We believe that the crowding hypothesis explains most of the hitherto reported effects of CL deficiency on mitochondrial bioenergetics, such as reduced respiratory activity, lower membrane potential, and poor energy coupling (Jiang et al, 2000; Baile et al, 2014), and therefore establishes a unifying mechanism for the functions of CL in mitochondria.
The underlying biophysical principles have not been addressed in this paper, but they can be inferred from what is known about transmembrane proteins and their surrounding lipids. Mismatch between the natural thickness of the lipid bilayer and the hydrophobic length of the imbedded proteins (Fig 5A) force bilayer deformations, which are best described in terms of the material properties of lipids (Phillips et al, 2009; Brown, 2017). In order to cover all hydrophobic regions of large transmembrane proteins, such as OXPHOS complexes, the bilayer has to stretch locally, which forces the surface of each monolayer to bend (Fig 5B). Since these effects deform the bilayer out of its relaxed state, they incur a certain energy penalty (Andersen & Koeppe, 2007). The more proteins the membrane contains, the more curved the monolayer surfaces will become (Fig 5C). As a result of the increased bilayer deformation, a sharp increase in energy penalty can be expected (Andersen & Koeppe, 2007; Brown, 2017). This is why protein crowding of biological membranes strains the cohesive force that holds the lipid bilayer together (Guigas & Weiss, 2016). In case of the mitochondrial IM, where protein crowding is extreme, it is easy to see that adaptive mechanisms are required to preserve membrane stability.
Figure 5. Proposed model of how CL supports protein crowding.
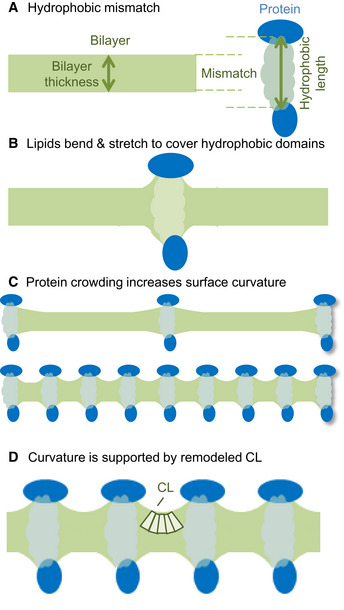
- In biological membranes, there is mismatch between the natural thickness of the lipid bilayer and the hydrophobic length of membrane proteins.
- In order to cover hydrophobic protein regions, the bilayer has to deform (to bend and to stretch).
- If multiple proteins are present, the mismatch imposes curvature stress on each monolayer. The higher the protein concentration the higher is the curvature.
- We hypothesize that remodeled CL stabilizes the deformation of the bilayer because it has both intrinsic negative curvature (wedge‐shaped molecule) and high protein affinity. By stabilizing bilayer deformations, remodeled CL increases the cohesion of protein‐crowded membranes.
We are proposing that CL provides such an adaptive mechanism. Experimental and computational studies have shown that CL is attracted to proteins more (Beyer & Klingenberg, 1985; Duncan et al, 2016) and supports bilayer deformations better (de Kruijff & Cullis, 1980; Boyd et al, 2017) than other phospholipids. It is therefore ideally suited to mitigate the stress of crowding in mitochondrial IMs. Remodeled (unsaturated) CL is better equipped for this function than non‐remodeled (saturated) CL because it has higher intrinsic curvature (Sankaram et al, 1989). The high intrinsic curvature of remodeled CL is likely to reduce the energy required for lipid bending (Fig 5D). Furthermore, the acyl chains of remodeled CL may be more flexible than the acyl chains of non‐remodeled CL and therefore might theoretically adapt better to the shape of proteins.
The proposed mechanism is also consistent with the thermodynamics of CL remodeling. Tafazzin catalyzes a reversible chemical reaction that lacks an internal driving force. However, the necessary force can be provided by the energy of crowding. Such driving force naturally selects unsaturated CL species because they spontaneously form curved monolayers that can easily cover the hydrophobic regions on the protein, reducing the energy penalty of crowding.
This study was limited by technical difficulties to adjust the surface density of membrane proteins, both in vitro and in vivo. This made it challenging to rigorously test the relationship between CL composition and protein concentration over a continuous range of values. In vitro, most integral membrane proteins require detergents to be reconstituted into liposomes and they cannot be concentrated ad libitum. In vivo, mitochondria resist manipulations of their protein concentration and composition due in part to a tight control of protein import (Harbauer et al, 2014; Eisenberg‐Bord & Schuldiner, 2017). This limits the spectrum of feasible approaches. While in their entirety our data support the conclusion that tafazzin is sensing membrane stress, different proteins (MBP and BR) had opposite effects, and our study was not designed to elucidate the underlying mechanisms.
Our findings touch on a fundamental aspect of mitochondrial physiology, namely the ability to boost the quantity of OXPHOS enzymes within a fixed organelle volume. The data suggest that mitochondria augment their OXPHOS capacity first by increasing the IM surface area and then by increasing the concentration of OXPHOS proteins per surface area. The essential role of CL in the latter may explain why the activation of CL biosynthesis is critical for the upregulation of thermogenesis in brown adipose tissue (Sustarsic et al, 2018). It also explains why CL synthesis and remodeling are crucial for T cell plasticity, where adaptations of energy metabolism are required for the T cell cycle (Corrado et al, 2020). Finally, the crowding hypothesis is largely consistent with the clinical presentation of Barth syndrome, which specifically afflicts organs with high peak energy expenditure, such as skeletal muscle and heart (Clarke et al, 2013). In summary, our data suggest that the biosynthesis and the remodeling of CL enable OXPHOS proteins to accumulate at high surface concentration in the mitochondrial IM. This lends further support to the notion that CL is the ‘green phospholipid’, a critical ingredient to achieve energy efficiency in mitochondria (Claypool et al, 2008).
Materials and Methods
Tafazzin reactions in vitro
The S. cerevisiae gene encoding tafazzin (TAZ1) was inserted into the pGEX‐EF vector and expressed in E. coli (BL21) as a glutathione‐S‐transferase fusion protein. Recombinant tafazzin was solubilized in Triton X‐100 and affinity‐purified as described (Schlame et al, 2017). Purified tafazzin was stored at −80°C at a protein concentration of about 1 g/l and a Triton concentration of 1 mM. Purified tafazzin was added to 10 mM Tris buffer (pH 7.4) containing lipids in 0.1–0.2 ml. The final lipid concentration was 1 mM, the final tafazzin concentration was 0.04 g/l (0.05 µM), and the final Triton concentration was 0.04 mM. Lipids were mixed in an organic solvent, the solvent was evaporated under a stream of nitrogen, and the buffer was added. Liposomes were created by sonication in a sonicator bath (Branson model 8210). Liposomes were converted into micelles or inverted micelles by adding Triton X‐100 or Ca2+ respectively. Membrane proteins, including MBP and BR were added as specified in the legends. MBP was purchased from Sigma (www.sigmaaldrich.com), and BR was purchased from Halotek Applied Biotechnologies (www.halotek.de). Samples were incubated at 37°C for 90 min, unless specified otherwise. Reactions were terminated by addition of 2 ml methanol and 1 ml chloroform. Lipids were analyzed by matrix‐assisted laser‐desorption time‐of‐flight (MALDI‐TOF) mass spectrometry (MS) as described below.
Yeast
Wild‐type S. cerevisiae cells were cultured in YPD (10 g/l yeast extract, 20 g/l peptone, 20 g/l glucose) or in YPGE (10 g/l yeast extract, 20 g/l peptone, 3% glycerol, 3% ethanol) in an incubator shaker operated at 250 rpm and 30°C. Growth was monitored by measuring optical density (OD) at 600 nm. Cells were harvested either in the logarithmic (OD = 0.6–1.3) or in the stationary (OD > 3) phase of growth. Repeated OD measurements were made to confirm that the cultures no longer expanded during the stationary phase. Before lipid extraction, yeast cell pellets were digested in a medium containing 1.2 M glycerol, 100 mM sodium thioglycolate, 50 mM Tris‐SO4 (pH 7.5), and 1 g/l zymolyase for 15 min at room temperature to break the cell wall. For proteomics analysis, an optimized method was used to lyse and extract proteins under alkaline conditions before solubilization with SDS (Von der Haar, 2007).
Yeast Pichia pastoris cells were genetically engineered to over‐express the alternative oxidase (AOX1), a mitochondrial IM protein, following Invitrogen protocols. The AOX1 cDNA was amplified by RT‐PCR from the yeast cell total RNA with the primers 5′‐ATCGGGATCCAAAACAATGGATTACAAGGATGACGACGATAAGTTAAAACTGTACGCAATAAGG‐3′ and 5′‐ATCGGAATTCTCATAAAACGAGCTCATCTCTTTCCCATCC‐3′, cut with Bam HI and Eco RI, and sub‐cloned into a modified pPIC9 expression vector, from which the α‐factor signal sequence was removed. About 5–20 µg of AOX1‐pPIC9 DNA or control DNA was linearized by Stu I digestion and transformed into freshly made GS115 P. pastoris (GS115 His4‐Mut+) competent cells by electroporation using the BioRad GenePulser. Immediately afterwards, the transformed cells were diluted in 0.6 ml 1 M sorbitol, spread on His‐free RDB plates (1 M sorbitol, 2% glucose, 1.34% yeast nitrogen base with ammonium sulfate, 0.00004% biotin, 0.005% amino acids, 2% Agar), and grown at 28°C for 4–5 days. Transformed colonies were collected, and stored in 15% glycerol at −80°C. For expression experiments, clones were cultured in MGY (1.34% yeast nitrogen base with ammonium sulfate without amino acids, 1% glycerol, 0.00004% biotin) in an incubator shaker operated at 250 rpm and 30°C. Cultures were adjusted to an optical density of 1.0, and AOX1 expression was induced by addition of 1% methanol. Methanol was replenished after 1 day, and cells were harvested after 2 days.
Mice
All protocols were approved by the Institutional Animal Care and Use Committee of the NYU School of Medicine and conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH). Mice were housed under temperature‐controlled conditions under a 12‐h light/dark cycle with free access to drinking water and food. The TAZKO mouse model was developed by D. Strathdee of the Cancer Research UK Beatson Institute (Glasgow, UK). In this model, two loxP sites flank exons 5–10 of the TAZ gene. We obtained floxed sperm in order to generate the KO model by Cre recombination. The model was maintained on a C57BL/6 background. TAZKO (Taz‐/y) male mice were generated from crosses between wild type (Taz+/y) males and heterozygote (Taz+/−) females.
Standard transthoracic resting and pharmacological (β‐adrenergic) stress echocardiography was performed on anesthetized mice as previously described (Phoon et al, 2012; Phoon & Turnbull, 2016). In brief, mice were anesthetized with isoflurane via nose cone (induction: 2.5% isoflurane; maintenance: 1% isoflurane at a flow rate of 1 l/min) with strict thermoregulation (37 ± 0.5°C) to optimize physiological conditions and reduce hemodynamic variability. Two‐dimensional and spectral Doppler echocardiography was performed using the Vevo 770 (Visual Sonics, Toronto, ON, Canada) with the RMV‐704, 40 MHz center frequency transducer. To minimize bias, we performed all echocardiographic measurements offline while blinded to the genotype. While lying on the heated imaging platform and immediately following baseline echocardiography, anesthetized mice received an injection of 2 mg/kg isoproterenol into the peritoneum. We then imaged mice in the parasternal short‐axis view for fractional shortening at serial time points.
To harvest adult hearts, mice were euthanized following CO2 narcosis/asphyxiation. Hearts were excised through a vertical anterior thoracotomy, trimmed from attached non‐cardiac tissue, and rinsed in cold PBS. To harvest embryonic hearts, pregnant dams were euthanized following CO2 narcosis/asphyxiation. The gravid uterus was rapidly dissected and placed in cold PBS. Embryos were localized with respect to the bifurcation of the uterine horns at the vagina. Each embryo was isolated from its uterine sac and separated from its associated membranes and decidua. Using fine scissors, the head was removed and an anterior, vertical incision was made in the chest to expose the thoracic cavity. Under a stereomicroscope, the heart was then carefully extracted from the chest using a pair of very fine curved dissecting tweezers. Heart samples were stored at −80°C.
Flies
Strain w1118; PBac{PB} CG4774c01874/TM6B, Tb1, with a transposon insertion in the coding region of the last exon of the cardiolipin synthase gene (ΔCLS), was obtained from the Bloomington Drosophila Stock Center (No. 10741). The tafazzin mutant (ΔTAZ) and the precise‐excision control (WT) were created in our laboratory (Xu et al, 2006b). To avoid confounding effects, we re‐derived all strains in identical genetic backgrounds. To this end, the ΔCLS and ΔTAZ alleles were backcrossed to the WT control background for 6 generations. The ΔCLS allele was identified by the mini‐w eye color. The ΔTAZ allele was identified by PCR genotyping with the primers: 5′‐GTC CAA ACA TCA GTT GGA TG‐3′ and 5′‐GAA GTA GTT AGG CAA CAG TC‐3′, which yields a 230 bp DNA fragment. The isogenic strains were rebalanced by crossing with w; Sco/CyO; Sb/TM6B. Fly culture and crosses were performed on a standard fly food containing yeast, cornmeal, and molasses in 3 inch culture vials at 24°C. For electron microscopy of flight muscles, the thoraces of CO2‐anesthetized flies were dissected and stored in fixative containing 2.5% glutaraldehyde and 2% paraformaldehyde in PBS. For proteome and lipidome analyses, 15 flies were collected per measurement and stored at −80⁰C. For proteomics, thoraces were dissected, homogenized in 100 µl lysis buffer (0.1 M NaOH, 0.05 M EDTA, 2% SDS, 2% β‐mercaptoethanol), incubated at 90°C for 10 min, and neutralized by adding 2.5 µl of 4 M acetic acid. For lipidomics, thoraces were dissected, homogenized in water, and extracted into chloroform/methanol (Bligh & Dyer, 1959).
Lipid analysis
In‐vitro samples or homogenized tissue samples were extracted into chloroform/methanol as described (Bligh & Dyer, 1959). In brief, samples were suspended in methanol/chloroform (2:1) and incubated at 37°C for 30 min to denature proteins. Chloroform and water were added, the samples were vortexed, and phase separation was achieved by centrifugation. The lower phase was collected, dried under nitrogen, and re‐dissolved in 0.1 ml chloroform/methanol (1:1). Lipids were analyzed by MS using either MALDI‐TOF‐MS or liquid chromatography coupled directly to electrospray‐ionization tandem mass spectrometry (LC‐ESI‐MS/MS).
Specifically, lipids formed in vitro were analyzed by MALDI‐TOF‐MS on a Bruker Autoflex instrument (Bruker Daltonics) operated in the reflectron mode (Sun et al, 2008). Extracts were diluted 1:11 in 2‐propanol/acetonitrile (3:2) and then mixed 1:1 with matrix solution containing 9‐aminoacridine (20 g/l). Aliquots of 1 µl or less were spotted on the target plate. Data from 1,000 acceptable laser shots (resolution > 2,500) were acquired in increments of 50 shots per laser position. The laser position was changed randomly. Tissue lipids were analyzed by LC‐ESI‐MS/MS on a QExactive HF‐X instrument coupled directly to a Vanquish UHPLC (Thermo Scientific). An aliquot of 7 µl was injected into a Restek Ultra C18 reversed‐phase column (100 × 2.1 mm; particle size 3 µm) that was kept at a temperature of 50°C. Chromatography was performed with solvents A and B at a flow rate of 0.15 ml/min. Solvent A contained 600 ml acetonitrile, 399 ml water, 1 ml formic acid, and 0.631 g ammonium formate. Solvent B contained 900 ml 2‐propanol, 99 ml acetonitrile, 1 ml formic acid, and 0.631 g ammonium formate. The chromatographic run time was 40 min, changing the proportion of solvent B in a nonlinear gradient from 30 to 35% (0–2 min), from 35 to 67% (2–5 min), from 67 to 83% (5–8 min), from 83 to 91% (8–11 min), from 91 to 95% (11–14 min), from 95 to 97% (14–17 min), from 97 to 98% (17–20 min), from 98 to 100% (20–25 min), and from 100 to 30% (25–26 min). For the remainder of the run time the proportion of solvent B stayed at 30% (26–40 min). The mass spectrometer was operated in negative ion mode. The spray voltage was set to 4 kV and the capillary temperature was set to 350°C. MS1 scans were acquired at a resolution of 120,000, an AGC target of 1e6, a maximal injection time of 65 ms, and a scan range of 300–2,000 m/z. MS2 scans were acquired at a resolution of 30,000, an AGC target of 3e6, a maximal injection time of 75 ms, a loop count of 11, and an isolation window of 1.7 m/z. The normalized collision energy was set to 30 and the dynamic exclusion time to 13 s. For lipid identification and quantitation, data were analyzed by the software LipidSearch 4.1 SP1 (Thermo Scientific). The general database was searched with a precursor tolerance of 2 ppm, a product tolerance of 0.2 Da, an intensity threshold of 1.0%, and an m‐score threshold of 5. In negative ion mode, [M+HCO2] adducts were identified for PC and [M‐H] ions for all other lipids.
Protein analysis
Protein abundances were determined by label‐free relative quantitative proteomics. Proteins were purified and concentrated by a brief SDS‐PAGE run designed to focus all proteins into a single band of about 1 cm. Gels were washed three times in double‐distilled water for 15 min each. Proteins were visualized and fixed by staining with EZ‐Run Protein staining solution (ThermoFisher Scientific, USA). The stained protein gel regions were excised and destained with water. In‐gel digestion was performed overnight with mass spectrometry grade Trypsin (Trypsin gold, Promega, Madison, WI, USA) at 5 ng/µl in 50 mM NH4HCO3 digest buffer. After acidification with 10% formic acid, peptides were extracted with 5% formic acid/50% acetonitrile (v/v) and concentrated to a small droplet using vacuum centrifugation. Desalting of peptides was done using hand packed SPE Empore C18 Extraction Disks (aka Stage Tips, 3 M, St. Paul, MN, USA) as described (Rappsilber et al, 2007). Desalted peptides were again concentrated and reconstituted in 10 µl 0.1% formic acid in water. An aliquot of the peptides was analyzed by nanoliquid chromatography followed by tandem mass spectrometry (nano‐LC‐MS/MS) using an Easy nLC 1,000 equipped with a self‐packed 75 µm × 20 cm reverse phase column (ReproSil‐Pur C18, 3 µm, Dr. Maisch GmbH, Germany) coupled online to a QExactive HF Orbitrap mass spectrometer via a Nanospray Flex source (all insturments from ThermoScientific, Waltham, MA, USA). Analytical column temperature was maintained at 50°C by a column oven (Sonation GmBH, Germany). Peptides were eluted with a 3–40% acetonitrile gradient over 110 min at a flow rate of 250 nl/min. The mass spectrometer was operated in the DDA mode with survey scans acquired at a resolution of 70,000 (at m/z 200) over a scan range of 300–1,750 m/z. Up to 10 of the most abundant precursors from the survey scan were selected with an isolation window of 1.6 Th for fragmentation by higher‐energy collisional dissociation with a normalized collision energy of 27. The maximum injection times for the survey and MS/MS scans were 60 ms, and the ion target value for both scan modes was set to 3e6. Data were analyzed by the software MaxQuant (version 1.5.5.1). We used the Andromeda search engine (Cox et al, 2011; Tyanova et al, 2016) to search the Uniprot Drosophila (downloaded on 01/09/2015; 3,214 entries), mouse (downloaded on 03/06/2018; 16,950 entries), and yeast (downloaded on 12/15/2015; 6,729 entries) protein sequence database, respectively. To determine the relative abundance of a protein across a set of samples, we used its label‐free quantitation (MaxLFQ) intensity calculated by MaxQuant (Cox et al, 2014) which normalizes the protein intensity by normalizing peptide intensities (minimum peptide ratio count = 2) across samples. To determine the relative abundance of a protein complex, we added the raw iBAQ intensities of all observed subunits.
Protein abundances were also determined by quantitative Western blot analysis with fluorescence‐labeled secondary antibodies. To this end, mouse hearts were homogenized, and crude mitochondria were isolated by differential centrifugation. Proteins were resolved on 10% SDS‐PAGE gels and transferred to polyvinylidene difluoride membranes. Membranes were incubated in Odyssey blocking buffer (LI‐COR Biotechnology) with primary antibodies at a concentration of 1 µg/ml. These included monoclonal antibodies to NDUFB6 (complex I), UQCRC2 (complex III), and ATP5A1 (complex V) from ABCAM and a polyclonal antibody to TOM70 (TOM complex) from Invitrogen. Fluorescent GAR‐IRDye800cw and GAM‐IRDye680 secondary antibodies (ABCAM) were used at a dilution of 1:10,000. Protein signals were quantified with the LiCor scanner (LI‐COR Biotechnology).
Transmission electron microscopy
Samples were fixed in glutaraldehyde and osmium tetraoxide. Fixed samples were stained with uranyl acetate. The buffer was gradually exchanged with ethanol, followed by resin exchange and heat polymerization. Sections of 50–100 nm were cut with a Leica Ultracut UCT microtome and collected on EM grids. Sections were stained with uranyl acetate and Sato Lead stains and imaged with a Philips CM12 transmission electron microscope. Random images were collected at different magnifications. Quantitative analyses were typically performed at a magnification of 17,500. To determine the IM/OM surface area ratio, the OM and IM were traced in randomly selected electron micrographs using the software package Image J. The ratio was calculated by dividing the sum of all IM perimeters (cristae plus inner boundary membrane) by the OM perimeter. In the Drosophila experiments, mitochondrial cross sections were classified as lamellar (parallel longitudinal cristae), tubular (circular cross‐sectional cristae), or defective (vacuole‐like areas). The areas of these sections were quantified in Image J.
Statistical analysis
Data were displayed as bar graphs (means ± standard deviation), as violin plots, or as scatter plots. Means were compared by Student’s t‐test. Relations between variables were determined by linear regression analysis. Euclidean distances between CL compositions were calculated as described (Schlame et al, 2012b).
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Acknowledgements
This work was supported in part by the National Institutes of Health (grant R01 GM115593 to MS, shared instrumentation S10 grants RR027990 and OD023659, and core center P30 grant NS050276 to TAN). The authors also acknowledge the NYU Langone Health Microscopy Laboratory for consultation and assistance with transmission electron microscopy. This shared resource was partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. The authors are grateful to Prof. Marcos Alcocer (School of Life and Environmental Sciences, University of Nottingham, Nottingham, NG7 2RD, UK) for advice regarding the transformation of P. pastoris.
The EMBO Journal (2021) 40: e108428.
Data availability
Primary datasets produced in this study are available in the public database MassIVE (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=b191fbb535d94501aeb843496ed27e2a).
References
- Acoba MG, Senoo N, Claypool SM (2020) Phospholipid ebb and flow makes mitochondria go. J Cell Biol 219: e202003131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE (2007) Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 36: 107–130 [DOI] [PubMed] [Google Scholar]
- Baile MG, Sathappa M, Lu YW, Pryce E, Whited K, McCaffery JM, Han X, Alder NN, Claypool SM (2014) Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J Biol Chem 289: 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 19: 856–863 [DOI] [PubMed] [Google Scholar]
- Barth PG, Scholte HR, Berden JA, Van Der Klei‐Van Moorsel JM, Luyt‐Houwen I, Van'T Veer‐Korthof ETh, Van Der Harten JJ, Sobotka‐Plojhar MA (1983) An X‐linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 62: 327–355 [DOI] [PubMed] [Google Scholar]
- Beyer K, Klingenberg M (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24: 3821–3826 [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917 [DOI] [PubMed] [Google Scholar]
- Botelho AV, Huber T, Sakmar TP, Brown MF (2006) Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys J 91: 4464–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd KJ, Alder NN, May ER (2017) Buckling under pressure: curvature‐based lipid segregation and stability modulation in cardiolipin‐containing bilayers. Langmuir 33: 6937–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MF (2017) Soft matter in lipid‐protein interactions. Annu Rev Biophys 46: 379–410 [DOI] [PubMed] [Google Scholar]
- Capaldi RA (1982) Arrangement of proteins in the mitochondrial inner membrane. Biochim Biophys Acta 694: 291–306 [DOI] [PubMed] [Google Scholar]
- Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury‐Ecob R, Clayton N, Martin RP, Tsai‐Goodman B, Garratt V, Ashworth M et al (2013) Barth syndrome. Orphanet J Rare Dis 8: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol 182: 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado M, Edwards‐Hicks J, Villa M, Flachsmann LJ, Sanin DE, Jacobs M, Baixauli F, Stanczak M, Anderson E, Azuma M et al (2020) Dynamic cardiolipin synthesis is required for CD8(+) T cell immunity. Cell Metabol 32: 981–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Di Bartolomeo F, Malina C, Campbell K, Mormino M, Fuchs J, Vorontsov E, Gustafsson CM, Nielsen J (2020) Absolute yeast mitochondrial proteome quantification reveals trade‐off between biosynthesis and energy generation during diauxic shift. Proc Natl Acad Sci USA 117: 7524–7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AL, Robinson AJ, Walker JE (2016) Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc Natl Acad Sci USA 113: 8687–8692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg‐Bord M, Schuldiner M (2017) Ground control to major TOM: mitochondria–nucleus communication. FEBS J 284: 196–210 [DOI] [PubMed] [Google Scholar]
- Epand RM, Moscarello MA (1982) The effect of bovine myelin basic protein on the phase transition properties of sphingomyelin. Biochim Biophys Acta 685: 230–232 [DOI] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505: 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas G, Weiss M (2016) Effects of protein crowding on membrane systems. Biochim Biophys Acta 1858: 2441–2450 [DOI] [PubMed] [Google Scholar]
- Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C (2014) The protein import machinery of mitochondria‐a regulatory hub in metabolism, stress, and disease. Cell Metabol 19: 357–372 [DOI] [PubMed] [Google Scholar]
- Hatefi Y (1985) The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem 54: 1015–1069 [DOI] [PubMed] [Google Scholar]
- Hauff KD, Hatch GM (2006) Cardiolipin metabolism and Barth syndrome. Prog Lipid Res 45: 91–101 [DOI] [PubMed] [Google Scholar]
- Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, Pfanner N, Greenberg ML (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem 275: 22387–22394 [DOI] [PubMed] [Google Scholar]
- Kojima R, Kakimoto Y, Furuta S, Itoh K, Sesaki H, Endo T, Tamura Y (2019) Maintenance of cardiolipin and crista structure requires cooperative functions of mitochondrial dynamics and phospholipid transport. Cell Rep 26: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshkin V, Greenberg ML (2002) Cardiolipin prevents rate‐dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem J 364: 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kruijff B, Cullis PR (1980) Cytochrome c specifically induces non‐bilayer structures in cardiolipin‐containing model membranes. Biochim Biophys Acta 602: 477–490 [DOI] [PubMed] [Google Scholar]
- Lewis RNAH, McElhaney RN (2009) The physicochemical properties of cardiolipin bilayers and cardiolipin‐containing lipid membranes. Biochim Biophys Acta 1788: 2069–2079 [DOI] [PubMed] [Google Scholar]
- Lotan R, Nicolson GL (1981) Plasma membranes of eukaryotes. In Schwartz LM, Azar MM (eds.), Advanced cell biology, pp 129–154. Princeton, NJ: Van Nostrand‐Reinhold; [Google Scholar]
- Ma L, Vaz FM, Gu Z, Wanders RJ, Greenberg ML (2004) The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem 279: 44394–44399 [DOI] [PubMed] [Google Scholar]
- Mårtensson CU, Doan KN, Becker T (2017) Effects of lipids on mitochondrial functions. Biochim Biophys Acta 1862: 102–113 [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W (2009) Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta 1788: 2084–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oemer G, Koch J, Wohlfarter Y, Alam MT, Lackner K, Sailer S, Neumann L, Lindner HH, Watschinger K, Haltmeier M et al (2020) Phospholipid acyl chain diversity controls the tissue‐specific assembly of mitochondrial cardiolipins. Cell Rep 30: 4281–4291 [DOI] [PubMed] [Google Scholar]
- Pennington ER, Funai K, Brown DA, Shaikh SR (2019) The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim Biophys Acta 1864: 1039–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278: 52873–52880 [DOI] [PubMed] [Google Scholar]
- Phillips R, Ursell T, Wiggins P, Sens P (2009) Emerging roles for lipids in shaping membrane‐protein function. Nature 459: 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoon CKL, Acehan D, Schlame M, Stokes DL, Edelman‐Novemsky I, Yu D, Xu Y, Viswanathan N, Ren M (2012) Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc 1: jah3‐e000455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoon CKL, Turnbull DH (2016) Cardiovascular imaging in mice. Curr Protoc Mouse Biol 6: 15–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906 [DOI] [PubMed] [Google Scholar]
- Ren M, Phoon CKL, Schlame M (2014) Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res 55: 1–16 [DOI] [PubMed] [Google Scholar]
- Rosetti CM, Maggio B, Wilke N (2010) Micron‐scale phase segregation in lipid monolayers induced by myelin basic protein in the presence of a cholesterol analog. Biochim Biophys Acta 1798: 498–505 [DOI] [PubMed] [Google Scholar]
- Sankaram MB, Powell GL, Marsh D (1989) Effect of acyl chain composition on salt‐induced lamellar to inverted hexagonal phase transitions in cardiolipin. Biochim Biophys Acta 980: 389–392 [DOI] [PubMed] [Google Scholar]
- Sauerwald J, Jores T, Eisenberg‐Bord M, Chuartzman SG, Schuldiner M, Rapaport D (2015) Genome‐wide screens in Saccharomyces cerevisiae highlight a role for cardiolipin in biogenesis of mitochondrial outer membrane multispan proteins. Mol Cell Biol 35: 3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Acehan D, Berno B, Xu Y, Valvo S, Ren M, Stokes DL, Epand RM (2012a) The physical state of lipid substrates provides transacylation specificity for tafazzin. Nature Chem Biol 8: 862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Blais S, Edelman‐Novemsky I, Xu Y, Montecillo F, Phoon CKL, Ren M, Neubert TA (2012b) Comparison of cardiolipins from Drosophila strains with mutations in putative remodeling enzymes. Chem Phys Lipids 165: 512–519 [DOI] [PubMed] [Google Scholar]
- Schlame M, Xu Y, Ren M (2017) The basis for acyl specificity in the tafazzin reaction. J Biol Chem 292: 5499–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Cornell BA (1985) Myelin basic protein induces hexagonal phase formation in dispersions of diacylphosphatidic acid. Biochim Biophys Acta 818: 275–279 [DOI] [PubMed] [Google Scholar]
- Sun G, Yang K, Zhao Z, Guan S, Han X, Gross RW (2008) Matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometric analysis of cellular glycerophospholipids enabled by multiplexed solvent dependent analyte‐matrix interactions. Anal Chem 80: 7576–7585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sustarsic EG, Ma T, Lynes MD, Larsen M, Karavaeva I, Havelund JF, Nielsen CH, Jedrychowski MP, Moreno‐Torres M, Lundh M et al (2018) Cardiolipin synthesis in brown and beige fat mitochondria is essential for systemic energy homeostasis. Cell Metabol 28: 159–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraschi TF, de Kruijff B, Verkleij AJ (1983) The effect of an integral membrane protein on lipid polymorphism in the cardiolipin‐Ca2+ system. Eur J Biochem 129: 621–625 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW et al (2014) A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem 6: 542–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verchère A, Ou WL, Ploier B, Morizumi T, Goren MA, Bütikofer P, Ernst OP, Khelashvili G, Menon AK (2017) Light‐independent phospholipid scramblase activity of bacteriorhodopsin from Halobacterium salinarum. Sci Rep 7: 9522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T, Thallinger GG, Zellnig G, Drew D, Cregg JM, Glieder A, Freigassner M (2014) Towards improved membrane protein production in Pichia pastoris: general and specific transcriptional response to membrane protein overexpression. New Biotechnol 31: 538–552 [DOI] [PubMed] [Google Scholar]
- Von der Haar T (2007) Optimized protein extraction for quantitative proteomics of yeasts. PLoS One 2(10): e1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 279: 378–382 [DOI] [PubMed] [Google Scholar]
- Wang S, Li Y, Xu Y, Ma Q, Lin Z, Schlame M, Bezzerides VJ, Strathdee D, Pu WT (2020) AAV gene therapy prevents and reverses heart failure in a murine knockout model of Barth syndrome. Circ Res 126: 1024–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Anjaneyulu M, Donelian A, Yu W, Greenberg ML, Ren M, Owusu‐Ansah E, Schlame M (2019) Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin. Proc Natl Acad Sci USA 116: 11235–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Condell M, Plesken H, Edelman‐Novemsky I, Ma J, Ren M, Schlame M (2006b) A Drosophila model of Barth syndrome. Proc Natl Acad Sci USA 103: 11584–11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Malhotra A, Ren M, Schlame M (2006a) The enzymatic function of tafazzin. J Biol Chem 281: 39217–39224 [DOI] [PubMed] [Google Scholar]
- Xu Y, Phoon CK, Berno B, D'Souza K, Hoedt E, Zhang G, Neubert TA, Epand RM, Ren M, Schlame M (2016) Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat Chem Biol 12: 641–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L, Shiba T, Harada S, Kita K, Albury MS, Moore AL (2013) The alternative oxidases: simple oxidoreductase proteins with complex functions. Biochem Soc Trans 41: 1305–1311 [DOI] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277: 43553–43556 [DOI] [PubMed] [Google Scholar]