

Abstract
Mixed lineage kinase domain‐like (MLKL) is the executioner in the caspase‐independent form of programmed cell death called necroptosis. Receptor‐interacting serine/threonine protein kinase 3 (RIPK3) phosphorylates MLKL, triggering MLKL oligomerization, membrane translocation and membrane disruption. MLKL also undergoes ubiquitylation during necroptosis, yet neither the mechanism nor the significance of this event has been demonstrated. Here, we show that necroptosis‐specific multi‐mono‐ubiquitylation of MLKL occurs following its activation and oligomerization. Ubiquitylated MLKL accumulates in a digitonin‐insoluble cell fraction comprising organellar and plasma membranes and protein aggregates. Appearance of this ubiquitylated MLKL form can be reduced by expression of a plasma membrane‐located deubiquitylating enzyme. Oligomerization‐induced MLKL ubiquitylation occurs on at least four separate lysine residues and correlates with its proteasome‐ and lysosome‐dependent turnover. Using a MLKL‐DUB fusion strategy, we show that constitutive removal of ubiquitin from MLKL licences MLKL auto‐activation independent of necroptosis signalling in mouse and human cells. Therefore, in addition to the role of ubiquitylation in the kinetic regulation of MLKL‐induced death following an exogenous necroptotic stimulus, it also contributes to restraining basal levels of activated MLKL to avoid unwanted cell death.
Keywords: DUB‐fusion, membranes, MLKL, necroptosis, ubiquitylation
Subject Categories: Autophagy & Cell Death, Post-translational Modifications & Proteolysis
Mono‐ubiquitylation of the necroptotic effector molecule MLKL promotes its proteasome‐ and lysosome‐mediated turnover to restrains its necroptosis‐inducing activity.

Introduction
Necroptosis is a type of programmed cell death that shares some molecular components with the better‐known apoptotic cell death pathway, but has distinct morphological features and different physiological consequences. Unlike apoptosis, which is immunologically silent and can be rapidly cleared by neighbouring phagocytic cells (Segawa & Nagata, 2015), necroptosis induces an inflammatory response by releasing cellular contents including DNA and cytosolic proteins (Kaczmarek et al, 2013). Necroptosis can be induced by a number of stimuli, but is mainly studied downstream of tumour necrosis factor (TNF) ligation to its receptor tumour necrosis factor receptor 1 (TNFR1). If inhibitor of apoptosis (IAP) proteins and caspase activities are suppressed, this results in higher order assemblies of receptor‐interacting serine/threonine protein kinase 1 (RIPK1) and RIPK3, and subsequent RIPK3 activation and auto‐phosphorylation (Sun et al, 2002; Wu et al, 2014). MLKL is phosphorylated by RIPK3 and oligomerizes, translocates to biological membranes and induces organelle and cell swelling and membrane rupture (Vanden Berghe et al, 2015; Petrie et al, 2019a).
Ubiquitylation plays a pivotal role in regulating TNF signalling. In addition to RIPK1 ubiquitylation by cIAPs, which forms a platform for MAPK and NF‐κB activation (Bertrand et al, 2008), the linear ubiquitin chain assembly complex (LUBAC), composed of HOIL‐1, HOIP and SHARPIN, generates M1‐linked ubiquitin chains on RIPK1, TRADD, TNFR1 and NEMO. This ubiquitylation leads to full activation of IKK and MAPK, and limits TNF‐induced cell death (Haas et al, 2009; Tokunaga et al, 2009; Ikeda et al, 2011). LUBAC also recruits CYLD, a deubiquitylating enzyme (DUB), that removes M1‐ and K63‐linked ubiquitin chains on RIPK1 and other complex components, while A20 sterically protects M1‐linked ubiquitin chains (Draber et al, 2015; Dondelinger et al, 2016).
Ubiquitylation has been implicated in the regulation of signalling checkpoints during necroptosis. RIPK3 undergoes K63‐linked ubiquitylation on Lys‐5, which is believed to support the formation of necrosome. Removal of this ubiquitin chain by the ubiquitin‐editing enzyme A20 was proposed to negate necroptosis, because A20 loss led to RIPK3‐dependent necroptosis in T cells and fibroblasts (Onizawa et al, 2015). Furthermore, ubiquitylated TRAF2 was reported to be associated with inactive MLKL, and CYLD deubiquitylates TRAF2 following necroptotic stimulation, allowing TRAF2 to dissociate from MLKL and MLKL to engage with, and be activated by, RIPK3 (Petersen et al, 2015).
MLKL has also been shown to undergo ubiquitylation upon necroptotic stimulation (Lawlor et al, 2015; Hildebrand et al, 2020), yet the significance of this post‐translational modification and its signalling function is unknown. Here, we show that necroptotic signalling stimulates MLKL ubiquitylation and that this antagonizes necroptosis via restraining the protein level of activated MLKL. MLKL oligomerization is the crucial stage for MLKL ubiquitylation, but RIPK3 phosphorylation is not necessary because auto‐active oligomerizable MLKL mutants are ubiquitylated. MLKL ubiquitylation was removed in vitro by USP21, but not by other chain‐specific deubiquitylating enzymes, suggesting that MLKL is mono‐ubiquitylated at multiples sites. Upon necroptotic stimulation, MLKL variants that are unable to induce cell death can still be ubiquitylated and this correlates with proteasome/lysosome mediated turnover. Conversely, mutation of four lysine residues identified by mass spectrometry analysis to be ubiquitylated following a necroptotic stimulus did not affect MLKL ubiquitylation or MLKL's cytotoxic activity. We therefore devised a novel approach to completely remove all ubiquitins from MLKL by fusing it to USP21. We showed that this MLKL‐USP21 fusion was resistant to ubiquitylation in both human and mouse cell lines and was more cytotoxic when compared with MLKL fused to a catalytically inactive USP21. Strikingly, human MLKL, which is very resistant to auto‐activation (Tanzer et al, 2016; Petrie et al, 2018), was auto‐activated by the USP21 fusion, suggesting that ubiquitylation is a major brake on the activation of human MLKL.
Results
MLKL becomes ubiquitylated during necroptosis
MLKL has previously been reported to undergo ubiquitylation in mouse bone marrow‐derived macrophages (BMDMs) stimulated with the necroptotic stimulus: LPS/Smac‐mimetic (Compound A)/pan‐caspase inhibitor (Q‐VD‐OPh) (Lawlor et al, 2015). To test whether MLKL ubiquitylation is specific to necroptosis, we stimulated wild‐type mouse dermal fibroblasts (MDFs) with TNF, Smac‐mimetic and the pan‐caspase inhibitor IDN‐6556 (TSI) individually or in combination for 3 h. GST‐UBA immobilized on glutathione sepharose beads (Hospenthal et al, 2015; Stafford et al, 2018) was used to purify ubiquitylated proteins from cell lysates. These purified samples generated a distinct ladder of between 50 and 75 kDa when probed with anti‐MLKL, which corresponds to non‐ubiquitylated MLKL, mono‐ubiquitylated MLKL and multi‐ubiquitylated MLKL (Fig 1A). Though it is unclear why non‐ubiquitylated MLKL is recovered in the UBA pulldown system, it does not affect the observation of ubiquitin ladders. Ubiquitylated MLKL species were only clearly detected for stimuli that promote MLKL phosphorylation (TI and TSI), but not for stimuli that induce apoptotic cell death (TS; Fig 1A). Ubiquitylation probably occurs before or possibly at the same time as membrane permeabilization, because by the time of UBA PD for MDFs (3 h) and HT29 cells (16 h), cells were at least 60% propidium iodide (PI)‐positive (Fig EV1A).
Figure 1. MLKL undergoes ubiquitylation during necroptosis.
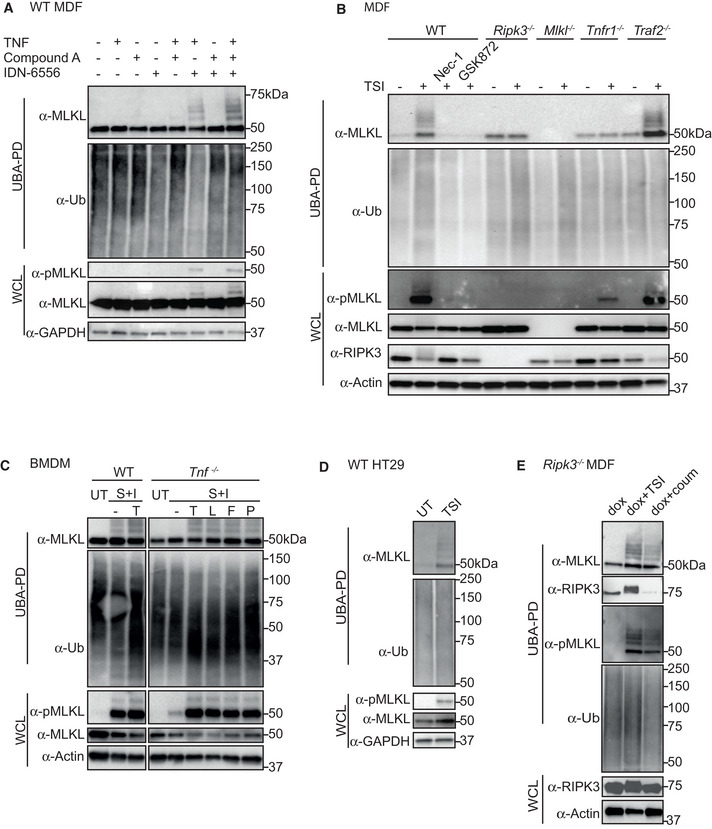
- WT MDFs were treated ± TSI individually or in combination for 3 h. Whole cell lysate (WCL) and UBA pulldown (UBA PD) fractions were analysed by Western blot and probed with antibodies as indicated. Representative of three independent experiments. Samples of UBA pulldown in following figures were analysed in the same way unless otherwise indicated.
- WT, Ripk3 −/−, Mlkl −/−, Tnfr1 −/− and Traf2 −/− MDFs were untreated (UT) or treated with TSI for 3 h. Nec‐1 and GSK872 were added to inhibit RIPK1 and RIPK3 kinase activities, respectively.
- WT and Tnf −/− BMDMs were treated ± death ligands including TNF (T), LPS (L), Fas ligand (F) and Poly I:C (P) in addition to S and I for 3 h.
- WT HT29 cells were untreated (UT) or treated with TSI for 16 h.
- RIPK3‐gyrase was inducibly expressed in Ripk3 −/− MDFs by doxycycline (dox) for 5 h, and cells were then treated ± combination of TSI, or coumermycin (coum) for 3 h.
Data information: Smac‐mimetic Compound A (S), IDN‐6556 (I), wild‐type (WT). TS, SI and TSI are used in combination, as apoptotic or necroptotic stimuli.
Source data are available online for this figure.
Figure EV1. Cell death time course of MDFs and HT29 cells following necroptotic stimulation.
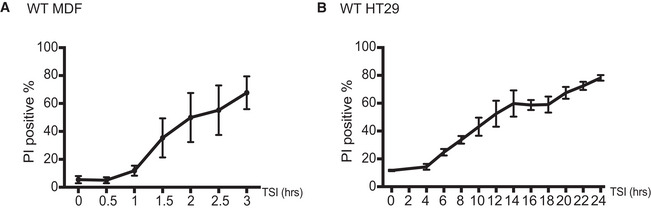
-
A, BMDFs (A) and HT29 (B) cells were treated with TSI. Cell death was measured by PI staining and flow cytometry. Data are plotted as mean ± SEM of at least three independent experiments.
Data information: Propidium iodide (PI). TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
As expected, TSI failed to induce MLKL ubiquitylation in Tnfr1 −/− MDFs, but, interestingly, did not completely prevent all TSI‐induced MLKL phosphorylation (Fig 1B). Furthermore, the kinase activities of RIPK1 and RIPK3 were required for MLKL ubiquitylation because genetic deletion of Ripk3, or treatment with the RIPK1 and RIPK3 inhibitors, Nec‐1 and GSK872, respectively, prevented MLKL ubiquitylation (Fig 1B). MLKL phosphorylation and ubiquitylation were not, however, reduced by genetic deletion of Traf2 (Fig 1B). To test whether MLKL ubiquitylation occurred in other cell types, we examined mouse BMDMs. Necroptosis can be induced in BMDMs by a range of different ligands including LPS, Poly I:C and the TNF receptor superfamily ligand FasL, when they are combined with a Smac‐mimetic and a caspase inhibitor, IDN‐6556 (SI) (Holler et al, 2000; Kaiser et al, 2013). To exclude a role for autocrine TNF, whose synthesis can be induced by a Smac‐mimetic, LPS, poly I:C or FasL, we compared BMDMs isolated from Tnf −/− and WT mice, and saw the same MLKL ubiquitin laddering pattern in both. This ubiquitylation correlated with MLKL phosphorylation on S345, a well‐known hallmark of MLKL activation and necroptosis (Fig 1C) (Murphy et al, 2013). We and others have shown that human and mouse MLKL are regulated distinctly (Li et al, 2015; Tanzer et al, 2016; Davies et al, 2018, 2020; Petrie et al, 2018), therefore to see whether human MLKL was also ubiquitylated during necroptosis, we treated the human colonic adenocarcinoma HT29 cell line with TSI. Although induction of necroptosis takes longer in HT29 than in MDFs (Fig EV1B), we observed similar MLKL phosphorylation and ubiquitylation in HT29 cells as in MDFs with similar levels of cell death (Figs 1D and EV1B).
To determine to what extent upstream necroptotic signalling was required for MLKL ubiquitylation, we used a well‐established RIPK3 dimerization strategy to activate RIPK3 in the absence of necroptotic signals (Moujalled et al, 2014; Orozco et al, 2014). We therefore stably expressed a doxycycline‐inducible RIPK3‐gyrase fusion protein, which can be dimerized and activated by coumermycin (Moujalled et al, 2014), in Ripk3 −/− MDFs. As expected, forced dimerization of RIPK3 induced MLKL phosphorylation but was also able to induce MLKL ubiquitylation independent of TSI stimulation and to similar levels as TSI, indicating that RIPK3 dimerization and activation is sufficient to induce MLKL ubiquitylation (Fig 1E).
MLKL is ubiquitylated on multiple sites during necroptosis
Ubiquitin can be coupled to, predominantly, lysine residues in target proteins. Ubiquitin itself contains eight ubiquitylation sites (M1, K6, K11, K27, K29, K33, K48 and K63), which leads to the formation of ubiquitin chains of varying architectures. Differently linked Ub chains serve distinct functions in cells (Swatek & Komander, 2016). To investigate the ubiquitin architecture on MLKL, we purified ubiquitylated proteins from necroptotic cells using sepharose beads coupled with GST‐UBA and digested them on the beads with a set of linkage selective deubiquitylating enzymes (UbiCRest) (Hospenthal et al, 2015). Each DUB specifically recognizes and removes a known subset of poly‐ubiquitin chain type with validated activity (Fig 2A) (Stafford et al, 2018). The non‐specific DUB and USP21 (Ye et al, 2011) converted MLKL into a non‐ubiquitylated form, but the other chain‐specific DUBs made no discernible difference to the MLKL ubiquitin laddering pattern (Fig 2B). To control that there was sufficient DUB activity, we decreased the ratio of substrate to DUBs to 1/2 and 1/4 (Fig 2C). vOTU, a DUB that can cleave every type of poly‐ubiquitin chain except M1‐linked chains, deubiquitylated most ubiquitylated proteins (see total ubiquitin blot, Fig 2C), yet it only deubiquitylated the highest molecular weight species of MLKL˜Ub adducts, and did not affect bands representing the lower molecular weight MLKL˜Ub species. No changes were observed upon treatment with OTULIN, a DUB specific for M1‐linked ubiquitin chains (Keusekotten et al, 2013), indicating that the residual Ub left by vOTU are not M1‐linked ubiquitin chains. Given that USP21 is the only DUB in the tested panel that can cleave the covalent bond between a protein substrate and the first ubiquitin unit added, these data suggest that ubiquitylated MLKL is most likely mono‐ubiquitylated at multiple sites.
Figure 2. MLKL is mono‐ubiquitylated at multiple sites.
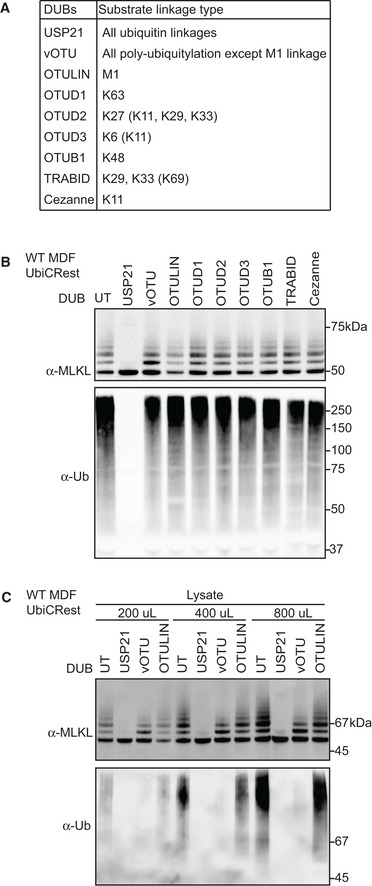
- Deubiquitylating enzymes (DUBs) and their ubiquitin substrates. Less efficiently cleaved substrates are indicated in brackets.
- UBA pulldown from WT MDFs treated with TSI for 3 h were subjected to the DUBs shown in (A). Beads eluates were analysed by Western blot and probed with antibodies as indicated. Representative of three independent experiments.
- 1.4‐ml cleared cell lysate from 4 × 106 TSI‐treated WT MDFs was split into three parts of the indicated volume, followed by UBA pulldown and DUB incubation. Bead eluates were analysed by Western blot and probed with the indicated antibodies. Representative of three independent experiments.
Data information: TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
MLKL ubiquitylation accumulates in the membrane fraction and can be deubiquitylated by USP21 localized to the plasma membrane
During necroptosis, activated MLKL oligomerizes and translocates to biological membranes, ultimately causing membrane rupture and cell death (Cai et al, 2014; Chen et al, 2014; Hildebrand et al, 2014). To understand more about MLKL ubiquitylation, we examined when and where this occurs. WT MDFs were stimulated with TSI over a time course from 30 to 180 min. Cytosolic and crude membrane fractions were generated as previously described using digitonin (Liu et al, 2018) and subjected to UBA pulldown. We refer to the 0.025% digitonin‐insoluble fraction as “crude membrane”, but do not exclude the potential for non‐membrane‐associated large macromolecular/amyloid‐like structures to sediment along with crude membranes (Liu et al, 2017). Ubiquitylated MLKL emerged and accumulated within the whole‐cell lysate and the crude membrane fraction from 90 min post‐stimulus onwards, coinciding precisely with the appearance of phosphorylated MLKL and onset of cell death (Figs 3A and EV1A). On the other hand, the predominant species of MLKL in the cytosol was non‐phosphorylated and non‐ubiquitylated (Fig 3A). These data suggest that MLKL ubiquitylation occurs in the crude membrane fraction but not in the cytosol.
Figure 3. MLKL ubiquitylation accumulates in the crude membrane fraction and can be digested by USP21 located on biological membranes.
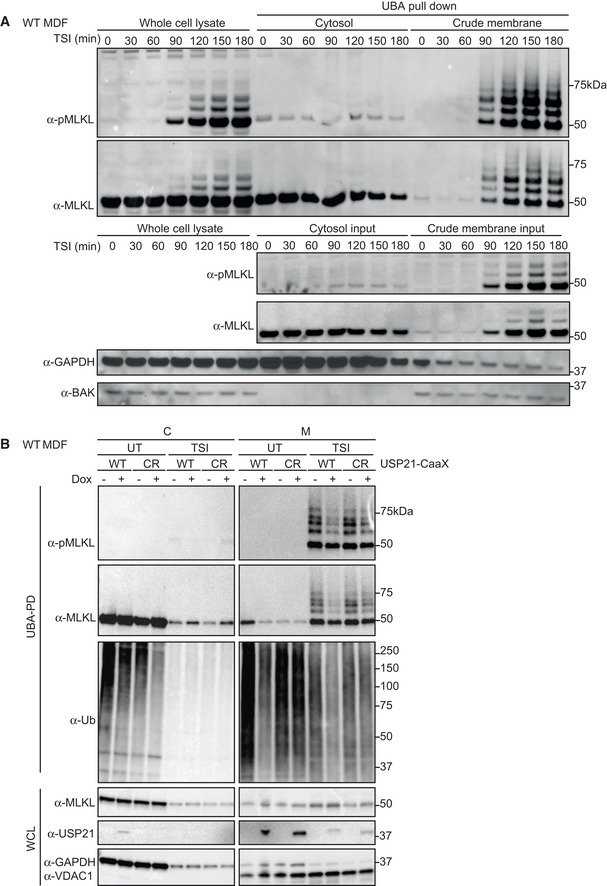
- WT MDFs were treated with TSI for indicated time. Cells were fractionated into cytosol (C) and crude membrane (M), followed by UBA pulldown. All fractions were analysed by Western blot and probed with antibodies as indicated. Representative of three independent experiments.
- WT USP21‐CaaX and USP21C221R‐CaaX (CR) were inducibly expressed in WT MDFs by doxycycline for 5 h. Ubiquitylated proteins were enriched followed by TSI stimulation and cellular fractionation. All fractions were analysed by Western blot and probed with antibodies as indicated. Representative of three independent experiments.
Data information: Wild‐type (WT), whole cell lysate (WCL). TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
To explore further where ubiquitylated MLKL is located, we took advantage of the fact that ubiquitylated MLKL was digested by USP21 (Fig 2B). We therefore fused a CaaX motif tag (C = cysteine, a = aliphatic amino acid, X = terminal residue) to the C‐terminus of the catalytic domain USP21 to localize it to the plasma membrane (Hancock et al, 1991). We chose “CVLQ” to mimic the CaaX motif found at the C‐terminal end of another DUB, USP32 (Sapmaz et al, 2019), and also generated a catalytically dead USP21 mutant version (USP21C221R) as a control, also fused with CaaX motif. This mutant, like the previously published mutant USP21C221A, lacks the ability to remove ubiquitin (Ye et al, 2011, 2012; Morrow et al, 2018). Using these stably and inducibly expressed constructs, we evaluated TSI‐mediated MLKL ubiquitylation with or without USP21‐CaaX function. Taking into account the amount of MLKL in the crude membrane fraction it was clear that while it did not completely denude MLKL of ubiquitin, the WT USP21‐CaaX fusion did substantially reduce the amount of ubiquitylated MLKL, as well as generally affecting the levels of ubiquitylated proteins (Fig 3B), although it did not reduce the kinetics of either apoptosis or necroptosis (Fig EV2). Taken together, these data suggest that ubiquitylated MLKL is localized to the plasma membrane.
Figure EV2. USP21‐CaaX expression does not alter the kinetics of TNF‐induced apoptosis or necroptosis in MDFs.
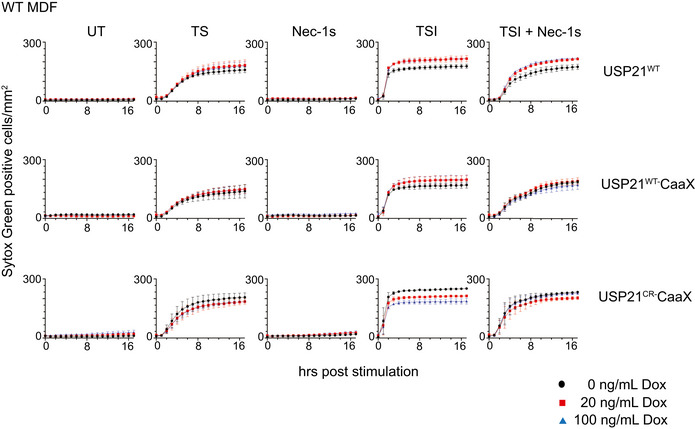
WT USP21, USP21‐CaaX and USP21C221R‐CaaX were inducibly expressed in WT MDFs by doxycycline. TS and Nec‐1s were used to control for apoptotic signalling, and TSI was used to trigger necroptotic signalling. Sytox Green‐positive cells were quantified in real time by live cell imaging. Data are plotted as mean ± SEM of two independent experiments.
Data information: Wild‐type (WT), untreated (UT). TS and TSI are used as apoptotic or necroptotic stimuli respectively.
Source data are available online for this figure.
Necroptosis‐induced MLKL ubiquitylation is driven by its oligomerization
In order to kill, MLKL oligomerizes, translocates to and permeabilizes membranes. To dissect further the drivers of MLKL ubiquitylation, we generated a series of MLKL mutants that were defective in one or more of these essential steps. Previously, we found that alanine replacement of the surface exposed residues R105 and D106 in the four‐helix bundle (4HB) domain of mouse MLKL prevents it from oligomerizing, translocating and inducing cell death (Hildebrand et al, 2014; Tanzer et al, 2016). In the context of full‐length MLKL, we found that these mutations prevent formation of a high molecular weight complex (complex II) in the “crude membrane” fraction following necroptotic stimulation by TSQ (Fig 4A). Although R105A/D106A MLKL was phosphorylated following this necroptotic stimulus, it did not exhibit the distinct TSI‐induced ubiquitylation observed for WT MLKL (Fig 4B). This supports the idea that MLKL only undergoes ubiquitylation post‐activation by RIPK3 and suggests that MLKL does not undergo ubiquitylation without oligomerization.
Figure 4. MLKL oligomerization drives its necroptosis‐specific ubiquitylation.
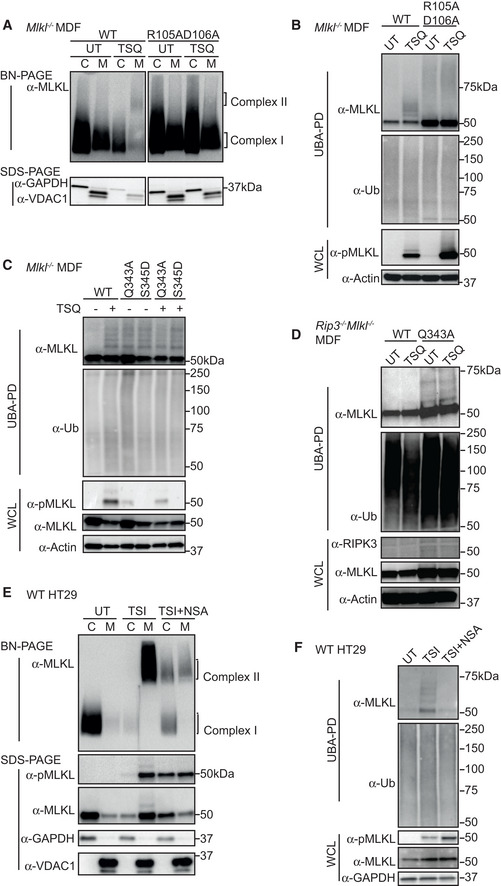
- WT and R105AD106A mutant MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline, at the same time cells were untreated (UT) or treated with TSQ for 6 h. Cells were fractionated into cytosol (C) and crude membrane (M). Fractions were analysed by BN‐ or SDS–PAGE, Western blot and probed with the indicated antibodies. Representative of three independent experiments.
- Cell lysates from (A) were subjected to UBA pulldown and analysed as described above.
- WT, Q343A and S345D mutant MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline; at the same time, cells were treated ± TSQ for 16 h, followed by UBA pulldown. Representative of three independent experiments.
- WT and Q343A mutant MLKL were inducibly expressed in Ripk3 −/− Mlkl −/− MDFs by doxycycline; at the same time, cells were treated ± TSQ for 16 h, followed by UBA pulldown. Representative of three independent experiments.
- HT29 cells were stimulated with TSI, ± NSA (500 nM) or left untreated (UT) for 16 h, followed by cellular fractionation. Fractions were analysed by BN‐ or SDS–PAGE, Western blot and probed with the indicated antibodies. Representative of three independent experiments.
- Cell lysates from (E) were subjected to UBA pulldown and analysed as described above.
Data information: Wild‐type (WT). TSI and TSQ are used as necroptotic stimuli.
Source data are available online for this figure.
The MLKL mutants, Q343A (which perturbs a hydrogen bond with K219 in the ATP binding motif VTIK) and S345D (phospho‐mimetic), are auto‐activated forms of murine MLKL (Murphy et al, 2013). We induced expression of Q343A or S345D mutant MLKL in Mlkl −/− and Ripk3 −/− Mlkl −/− MDFs for 16 h where, as predicted, they killed cells independently of an extrinsic necroptotic stimulus (Fig EV3B). As expected, induction of necroptosis in Mlkl −/− cells reconstituted with wild type MLKL resulted in MLKL ubiquitylation (Fig 4C). However, activated MLKL mutants expressed in the same Mlkl −/− MDFs were ubiquitylated independently of a necroptotic stimulus and to an extent comparable to wild‐type MLKL induced to undergo necroptosis (Fig 4C). Furthermore, Q343A MLKL became ubiquitylated in Ripk3 −/− Mlkl −/− MDFs and therefore independently of any upstream activation (Fig 4D). We conclude that while MLKL ubiquitylation correlates with its oligomerization and phosphorylation, direct RIPK3 association is not required for MLKL ubiquitylation in murine cells.
Figure EV3. MLKL oligomerization drives its necroptosis‐specific ubiquitylation.
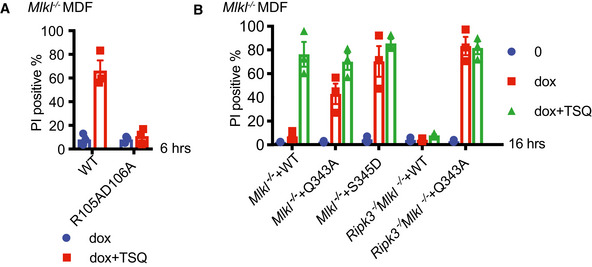
Data information: Propidium iodide (PI). TSQ is used as a necroptotic stimulus.
Source data are available online for this figure.
Necrosulfonamide (NSA) is an inhibitor of human MLKL by forming a covalent bound with Cys86 that only exists in human MLKL but not mouse MLKL (Liu et al, 2017). Consistent with a recent report (Murai et al, 2018; Samson et al, 2020), we found that while it did not inhibit the phosphorylation of MLKL or the formation of high molecular weight MLKL‐containing complex, large proportions of this MLKL species remained in the cytosolic (0.025% digitonin soluble) fraction (C) of cells (Fig 4E). Consistent with the reduction in MLKL oligomer in the crude membrane (0.025% digitonin insoluble) fraction (M), NSA also reduced MLKL ubiquitylation (Fig 4F).
Ubiquitylated MLKL undergoes proteasome and lysosome‐dependent turnover
Unlike the R105A/D106A MLKL mutant, E109A/E110A MLKL mutant and N‐terminal FLAG‐tagged MLKL (N‐FLAG MLKL) are able to form higher order oligomers in crude membrane fractions following necroptotic stimulation, yet are, nevertheless, unable to induce necroptosis (Hildebrand et al, 2014; Tanzer et al, 2016) (Figs 5A and B, and EV4A and B). These mutants therefore allowed us to examine whether ubiquitylation was a consequence of necroptosis. Upon induction of necroptosis, E109A/E110A MLKL was more heavily phosphorylated and ubiquitylated than wild‐type MLKL (Fig 5C). Similarly, N‐FLAG MLKL was also phosphorylated and ubiquitylated within 2 h of TSI treatment, although these modified MLKL species diminished over time (Fig 5D).
Figure 5. MLKL ubiquitylation does not lead to cell death, but correlates with the turnover of activated MLKL.
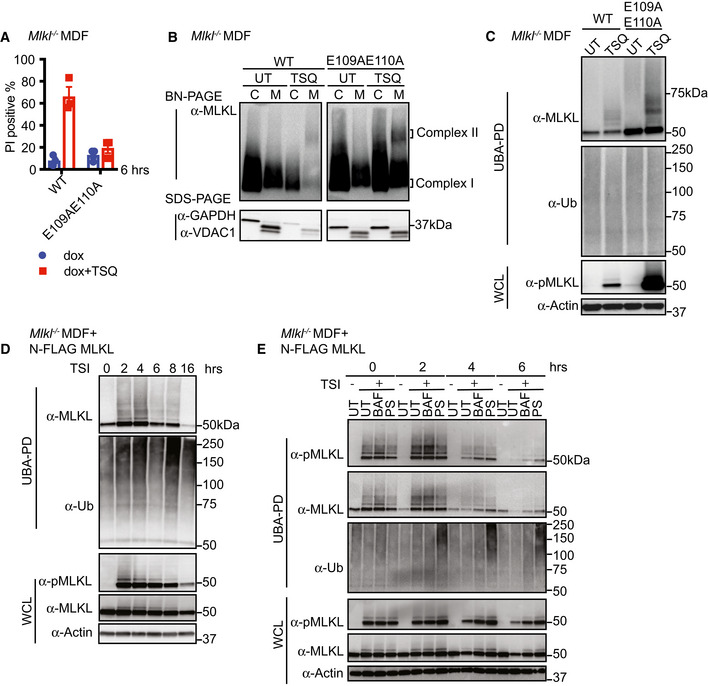
- WT and E109AE110A mutant MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline, and cells were untreated (UT) or treated with TSQ for 6 h (please note that the same WT control was used in Fig EV3A). Cell death was measured by PI staining based on flow cytometry. Data are plotted as mean ± SEM of three independent experiments.
- Cytosol (C) and crude membrane (M) cellular fractions from (A) were analysed by Western blot from BN‐PAGE or SDS–PAGE using antibodies as indicated. The same WT control was used in Fig 4A. Representative of three independent experiments.
- Cell lysates from (A) were subjected to UBA pulldown and analysed as described above.
- N‐FLAG MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline overnight, and cells were treated with TSI for indicated time, followed by UBA pulldown. Representative of three independent experiments.
- N‐FLAG MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline for 16 h. Cells were stimulated ± TSI for 3 h after withdrawal of doxycycline. Then, TSI medium was removed and replaced to medium containing inhibitors Bafilomycin A1 (BAF), PS341 (PS) or left untreated (UT). IDN‐6556 was added to all conditions to block apoptosis. Cells were collected 0, 2, 4 and 6 h after medium replacement, followed by UBA pulldown. Representative of three independent experiments.
Data information: Wild‐type (WT). TSI and TSQ are used as necroptotic stimuli.
Source data are available online for this figure.
Figure EV4. N‐FLAG MLKL behaves like WT MLKL but does not induce cell death following necroptotic stimulation.

- WT MLKL and N‐FLAG MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline for 12 h, and cells were treated with TSI or TSQ. TS treatment controlled that the response to TS was normal. Cell death was measured by PI staining based on flow cytometry. Data are plotted as mean ± SEM of three independent experiments.
- WT MLKL and N‐FLAG MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline for 6 h, and cells were untreated (UT) or treated with TSQ. Crude membrane (M) and cytosolic (C) cellular fractions were analysed by Western blot from BN‐PAGE or SDS–PAGE using antibodies as indicated. Representative of three independent experiments.
Data information: Wild‐type (WT). TS, TSI and TSQ are used as apoptotic or necroptotic stimuli.
Source data are available online for this figure.
The cellular turnover of active forms of MLKL has been observed previously by our group and by others (Gong et al, 2017; Yoon et al, 2017; Zargarian et al, 2017; Hildebrand et al, 2020). Therefore, we examined ubiquitylation of N‐FLAG MLKL in MDFs over a time course following a TSI pulse. N‐FLAG MLKL is phosphorylated, oligomerizes and accumulates in the 0.025% insoluble cell fraction without killing Mlkl −/− MDFs following necroptotic stimulation. This later feature facilitates the study of these ubiquitylated MLKL species and their cellular turnover in situ using proteasome and lysosome inhibitors as previously described for unmodified MLKL (Hildebrand et al, 2020). We found that after 4‐h TSI stimulation, levels of ubiquitylated and phosphorylated N‐FLAG MLKL dramatically decreased. This decrease can be delayed by either the lysosome inhibitor bafilomycin or the proteasome inhibitor PS341 (Fig 5E). This suggests that MLKL ubiquitylation correlates with its proteasome and lysosome‐dependent turnover.
MLKL ubiquitylation antagonizes necroptosis
We sought to identify the precise amino acids on MLKL that were modified by ubiquitin. We enriched for activated and ubiquitylated N‐FLAG MLKL from the “crude membrane” fraction of MDFs by performing a FLAG tag affinity purification. Eluted fractions from FLAG affinity beads were analysed by mass spectrometry, which identified Gly‐Gly conjugates on lysine residues K9, K51, K69 and K77 (Fig EV5A). All four are located in the 4HB domain of MLKL (Fig EV5B), which suggests a role for ubiquitylation in regulating MLKL‐mediated cell death. We did not identify any additional signs of ubiquitin modification on the brace or pseudokinase region of MLKL using this experimental system.
Figure EV5. Simultaneous arginine replacement of four ubiquitylation sites on the mouse MLKL 4HB domain does not prevent necroptosis‐induced ubiquitylation.
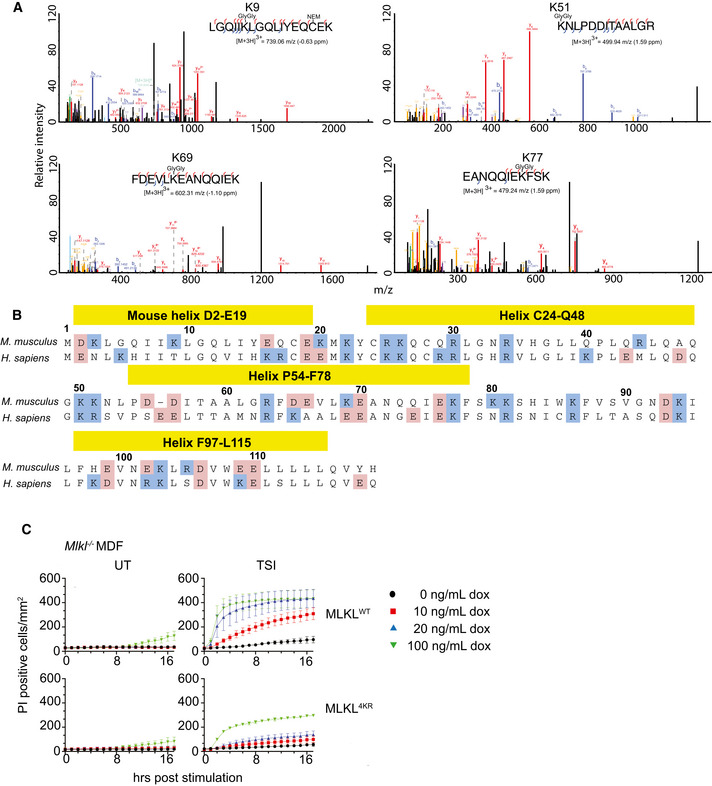
- MS spectra were manually validated to confirm the identification of four Gly‐Gly sites on activated MLKL.
- Alignment of mouse and human MLKL N‐terminal domain. Positively charged residues are labelled in blue, and negatively charged residues are labelled in pink.
- WT and 4KR mutant MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline (at the indicated concentrations), and cells were treated ± TSI (added simultaneously) for 4 h. Sytox Green‐positive cells were quantified in real time by IncuCyte S3 live cell imaging. Data are plotted as mean ± SEM of three independent experiments.
Data information: Wild‐type (WT), untreated (UT). TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
We mutated the four lysines to arginine and inducibly expressed the “4KR” mutant in Mlkl −/− MDFs to explore their role. Surprisingly, the 4KR MLKL was phosphorylated and ubiquitylated in a similar manner to wild‐type MLKL following TSI treatment (Fig 6B). There was a reduction in the ability of this mutant to kill cells in response to TSI treatment (Fig EV5C), but this is seemingly unrelated to its ubiquitylation and more likely due to modification of conserved residues in the cytotoxic domain of MLKL. These results highlight the difficulties in mutational analyses when studying ubiquitylation and rather than mutate any of the other 38 lysines in MLKL we tried a different approach.
Figure 6. Simultaneous arginine replacement of four ubiquitylation sites on the mMLKL 4HB domain does not prevent necroptosis‐induced ubiquitylation.
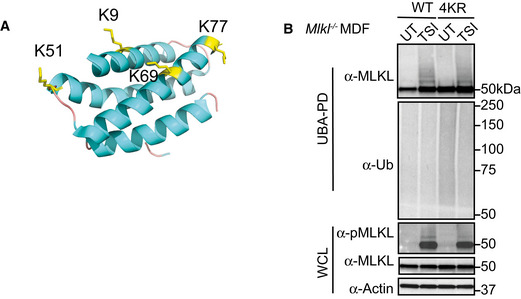
- Cartoon of the N‐terminal region (residues 1–180) of mouse MLKL (PDB accession 4BTF [Murphy et al, 2013]) showing the four lysine residues identified from MS analysis as yellow sticks.
- WT and 4KR mutant MLKL were inducibly expressed in Mlkl −/− MDFs by doxycycline for 6 h and cells were untreated (UT) or treated with TSI, followed by UBA pulldown. Representative of three independent experiments.
Data information: Wild‐type (WT), whole cell lysate (WCL). TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
We had observed that the deubiquitylating enzyme USP21 removed all ubiquitin from MLKL in vitro (Fig 2B). We therefore hypothesized that fusing the catalytic domain of USP21 to MLKL would remove TSI‐induced ubiquitylation from MLKL. We fused it to the C‐terminal end of MLKL, because, as with the FLAG tag, N‐terminal fusions affect the ability of MLKL to kill (Fig EV4A). We fused the catalytic domain of USP21 to the C‐terminus of both human and mouse MLKL (Fig 7A) and expressed the fusion proteins in MLKL −/− HT29 and Mlkl −/− MDF cells, respectively, to determine, firstly, whether it still retained cytotoxic activity and, secondly, whether it would be resistant to necroptosis‐induced ubiquitylation. To specifically control for the loss of ubiquitylation, we also generated MLKL fused to the previously described catalytically dead USP21 mutant (USP21C221R; Figs 3B and 7A). Both the MLKL‐USP21 and MLKL‐USP21C221R fusion proteins were exogenously expressed and the cells were stimulated with TSI for varying amounts of time. Ubiquitylated proteins were enriched via UBA pulldown. Like murine MLKLWT, the catalytically inactive control, MLKL‐USP21C221R, when expressed in Mlkl −/− MDFs, showed high MW laddering indicative of ubiquitylation and the ubiquitylation was enhanced following TSI stimulation (Fig 7B). These high MW MLKL‐USP21C221R species were reduced and collapsed into non‐ubiquitylated form by digestion of recombinant USP21 (Fig EV6C). In contrast, the ubiquitin laddering was not evident for the MLKL‐USP21WT fusion (Fig 7B). MLKL‐USP21 and MLKL‐USP21C221R reconstituted sensitivity to TSI stimulation in Mlkl −/− MDFs with only modestly delayed kinetics when compared to wild‐type MLKL (Fig 7C). However, unlike wild‐type MLKL or MLKL‐USP21C221R, MLKL‐USP21 could induce cell death independent of TSI stimulation (Fig 7C), even when expressed in Ripk3 −/− Mlkl −/− MDFs (Fig 7C).
Figure 7. MLKL ubiquitylation antagonizes necroptosis.
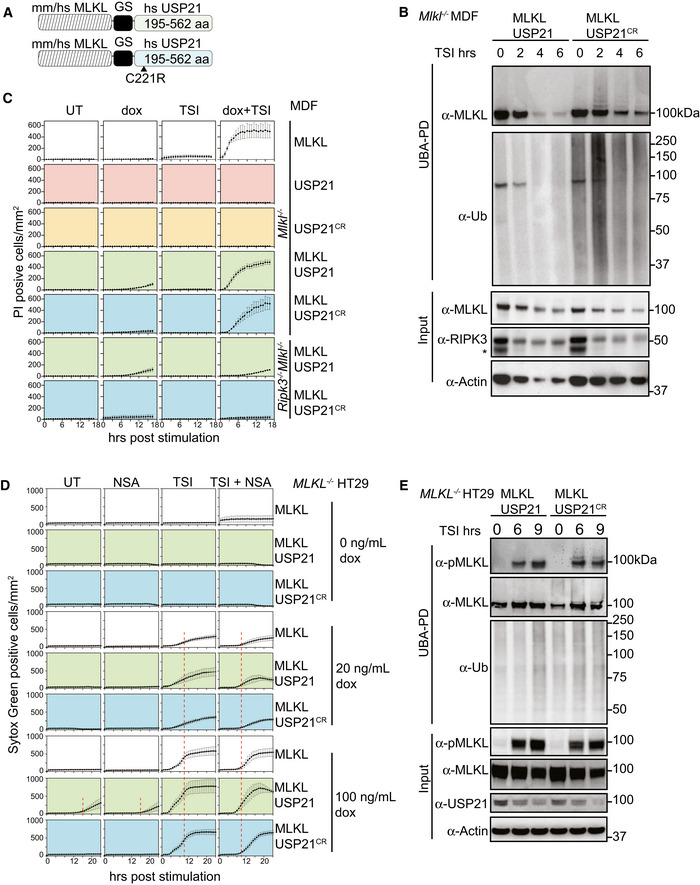
- Schematic of the MLKL‐USP21 and USP21 catalytic dead mutant proteins.
- Mouse MLKL‐USP21 and MLKL‐USP21C221R were inducibly expressed in Mlkl −/− MDFs by doxycycline (10 ng/ml) for 6 h with addition of a necroptotic stimulus (TSI) for the indicated time, followed by UBA pulldown. Non‐specific band is indicated with an asterisk in the blot of RIPK3. Representative of three independent experiments.
- Mlkl −/− MDFs and Ripk3 −/− Mlkl −/− MDFs stably transfected with constructs encoding MLKL, USP21, USP21C221R, MLKL‐USP21 and MLKL‐USP21C221R were treated with doxycycline, TSI or in combination. Propidium iodide‐positive cells were quantified in real time by IncuCyte live cell imaging. Data are plotted as mean ± SEM of three independent experiments.
- MLKL −/− HT29 cells stably transfected with constructs encoding human MLKL‐USP21 and MLKL‐USP21C221R, were treated with doxycycline, NSA (1 μM), TSI or combinations thereof (added simultaneously). Sytox Green‐positive cells were quantified in real time by live cell imaging. Data are plotted as mean ± SEM of six independent experiments (A red dashed line is shown to highlight the delay in death kinetics upon treatment with NSA).
- Human MLKL‐USP21 and MLKL‐USP21C221R were inducibly expressed in MLKL −/− HT29 cells by doxycycline (10 ng/ml) for 16 h, and TSI was added for the indicated times but all samples were collected 25 h post‐induction, followed by UBA pulldown. Representative of three independent experiments.
Data information: Wild‐type (WT). TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
Figure EV6. MLKL ubiquitylation antagonizes necroptosis.
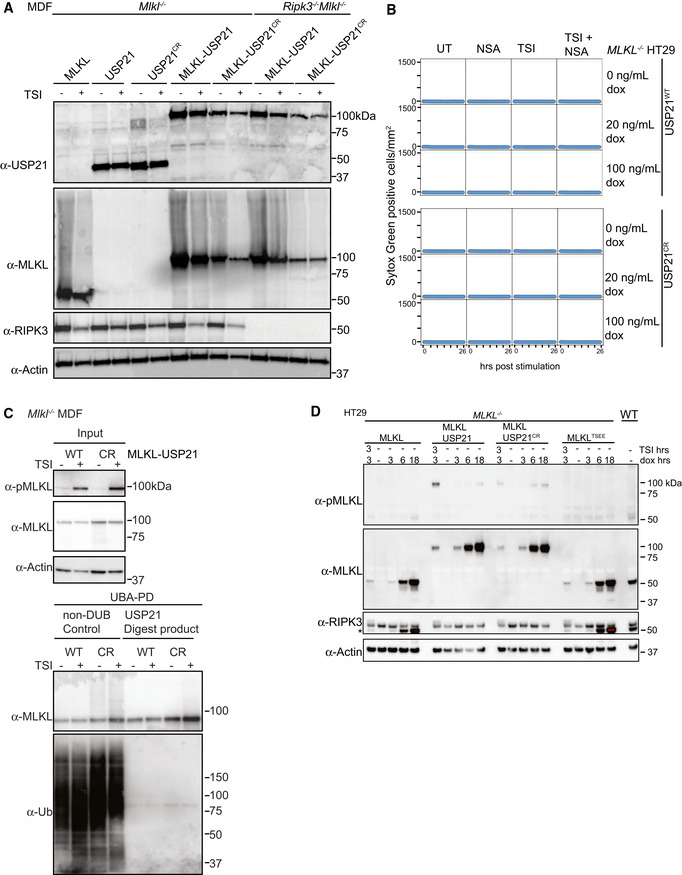
- Mouse MLKL, USP21, USP21C221R, MLKL‐USP21 and MLKL‐USP21C221R fusions were inducibly expressed in Mlkl −/− MDFs and Ripk3 −/− Mlkl −/− MDFs as indicated, following doxycycline addition (20 ng/ml) for 6 h ± TSI. Representative of two independent experiments.
- MLKL −/− HT29 cells stably transfected with doxycycline‐inducible constructs encoding human USP21 and human USP21C221R were treated with doxycycline, NSA (1 μM), TSI or combinations thereof (added simultaneously). Sytox Green‐positive cells were quantified in real time by live cell imaging. Representative of two independent experiments.
- Mouse MLKL‐USP21 and MLKL‐USP21C221R fusions were inducibly expressed in Mlkl −/− MDFs by doxycycline (10 ng/ml) for 8 h with addition of a necroptotic stimulus (TSI) for 3 h, followed by UBA pulldown and USP21 digestion. Antibody (D6E3G, Cell Signaling Technology) was used here to detect MLKL phosphorylation. Representative of two independent experiments.
- MLKL −/− HT29 cells were stably transfected with indicated doxycycline‐inducible MLKL alleles (phospho‐mimetic human MLKL mutant T357E/S358E indicated as MLKLTSEE) and treated with doxycycline (100 ng/ml) ± TSI (added simultaneously). A residue band from MLKL blot is indicated by an asterisk in RIPK3 blot due to reprobing. Representative of three independent experiments.
Data information: TSI is used as a necroptotic stimulus.
Source data are available online for this figure.
We made similar observations for human MLKL‐USP21 fusions expressed in MLKL −/− HT29 cells. Human MLKL‐USP21C221R, but not MLKL‐USP21 fusions, became ubiquitylated following TSI stimulation (Fig 7E). Furthermore, like human MLKL, human MLKL‐USP21 and MLKL‐USP21C221R fusions were able to reconstitute the capacity of MLKL −/− HT29 cells to undergo TSI‐induced necroptosis when their expression was induced by doxycycline (Fig 7D), while USP21 controls did not (Fig EV6B). NSA was able to delay TSI‐induced cell death for all MLKL species, but became less effective over time, presumably overwhelmed by ongoing expression of MLKL. Induced expression of MLKL‐USP21 also caused TSI‐independent cell death from 16 h, without detectable MLKL phosphorylation (t 0 = 25‐h induction, Fig 7E), and this could also be delayed by NSA (Fig 7D). These data indicate that ubiquitylation of acts as an important kinetic regulator of the necroptosis pathway following oligomerization and membrane association.
Discussion
MLKL undergoes ubiquitylation during necroptosis. In this study, we identified a distinctive ubiquitylation of both mouse and human MLKL that could be induced by a range of necroptotic stimuli. This signature ubiquitin ladder was essentially resistant to cleavage by a host of DUBs. Only USP21, the DUB that can remove all ubiquitin modifications, including mono‐ubiquitylation, was able to deubiquitylate MLKL. Therefore, we propose that MLKL is mono‐ubiquitylated at multiple sites. Mono‐ubiquitylation is typically associated with endosome‐lysosome trafficking (Haglund et al, 2003; Mosesson & Yarden, 2006) and is less likely to play a scaffolding role or result in proteasomal degradation (Wilkinson et al, 1995; Oh et al, 2018).
Full activation of MLKL's killing activity is a multi‐step and protracted process, the choreography of which has not yet been fully deduced. Nevertheless, there is a broad consensus, based on analysis of MLKL mutants and a range of different techniques, that phosphorylation by RIPK3 leads to MLKL oligomerization, translocation to biological membranes and subsequent membrane permeabilization (Tanzer et al, 2016; Petrie et al, 2018, 2020; Samson et al, 2020). Recent single‐cell imaging approaches examining human MLKL show that phosphorylated MLKL clusters in cytoplasmic vesicles together with RIPK1. These vesicles are actively transported to the plasma membrane, where they begin to coalesce in hotspots, some hours prior to membrane lysis (Samson et al, 2020). Consistent with data showing that NSA does not prevent MLKL phosphorylation or initial oligomerization, but does block higher order oligomerization (Liu et al, 2017), NSA appears to prevent MLKL clustering (Samson et al, 2020). To define where and when ubiquitylation of MLKL occurred, we fractionated cells into cytosolic and crude membrane fractions, and only detected ubiquitylated MLKL in the 0.025% digitonin‐insoluble (crude membrane) fraction. We also examined a number of different MLKL mutants that we had previously characterized for their ability to form oligomers, translocate to the crude membrane fraction and promote necroptosis. The MLKL mutants, Q343A and S345D, which are able to form 0.025% digitonin‐insoluble high molecular weight oligomers and induce cell death without necroptotic stimulation (Murphy et al, 2013; Tanzer et al, 2015), were ubiquitylated independently of RIPK3 or a necroptotic TSQ stimulus. On the other hand, the R105A/D106A mutant that is unable to form high molecular weight oligomers on BN‐PAGE did not become ubiquitylated following a necroptotic stimulus. This implies that MLKL ubiquitylation occurs after MLKL oligomerization and does not specifically require RIPK3 or upstream signalling.
MLKL has been variously reported to traffic to the nucleus (Yoon et al, 2016; Weber et al, 2018), lysosomes (Wang et al, 2014; Yoon et al, 2017; Fan et al, 2019), mitochondria (Wang et al, 2012) and the plasma membrane (Cai et al, 2014; Chen et al, 2014; Hildebrand et al, 2014; Samson et al, 2020) following necroptotic activation. Crude membrane fractionation enriches membrane and insoluble protein aggregates, therefore in an attempt to determine more precisely where MLKL ubiquitylation occurred, we generated a plasma membrane‐targeted, CaaX‐fused USP21 (Wright & Philips, 2006). Because ubiquitin modification plays a role in the initial, plasma membrane‐located, TNF/TNFR1‐induced necroptotic signalling, we anticipated that expressing USP21‐CaaX might affect induction of necroptosis. However, necroptosis was induced in cells expressing a non‐targeted USP21 or a catalytically inactive USP21C221R‐CaaX to equal levels as in USP21‐CaaX cells, despite the fact that USP21‐CaaX was clearly active and reduced total ubiquitin levels. Consistent with MLKL being targeted to the plasma membrane, MLKL was less ubiquitylated in USP21‐CaaX cells than in cells expressing USP21C221R‐CaaX following a necroptotic stimulus. MLKL ubiquitylation was, however, not completely prevented by USP21‐CaaX expression leaving open several interpretations of this data, viz. not all MLKL is ubiquitylated at the plasma membrane; or USP21‐CaaX was unable to access all MLKL in the membrane; or MLKL is ubiquitylated prior to reaching the plasma membrane and can only be deubiquitylated once it arrives there. Therefore, while we cannot exclude that ubiquitylated MLKL sediments as part of large amyloid‐like polymers or large cytoskeletal structures in the crude membrane fraction without being directly associated with a biological membrane (Liu et al, 2017), our data suggest that at least some ubiquitylated MLKL is accessible to a plasma membrane localized DUB.
As previously observed (Murai et al, 2018), we found that in the presence of NSA, the high molecular weight human MLKL complex that is normally found exclusively in the crude membrane fraction was now also observed in the cytosolic fraction. Combined with other observations (Samson et al, 2020), this indicates that NSA interferes with MLKL membrane association. Since NSA treatment also inhibited MLKL ubiquitylation, this suggests that membrane association is required for MLKL ubiquitylation and that the relevant E3 ligase is also membrane associated.
Neither N‐terminally FLAG‐tagged MLKL nor the E109A/E110A mutant MLKL are capable of killing cells, although both are phosphorylated and form 0.025% digitonin‐insoluble high molecular weight oligomers in a similar manner to wild‐type MLKL following induction of necroptosis. Since both are also ubiquitylated, we can conclude that ubiquitylation is not sufficient to cause necroptosis. To more precisely define the role of MLKL ubiquitylation during necroptosis, we used mass spectrometry to identify ubiquitylated lysines in mouse MLKL. We found four such lysines in the 4HB domain, but none in the pseudokinase domain. Only one of these four, K77, is conserved in human MLKL; however, this is not particularly surprising because homology of charged residues in MLKL is not well conserved between mouse and human (Fig EV5B). While this manuscript was in preparation, another group reported activation dependent MLKL ubiquitylation at residues K51, K77, K172 and K219, showing more possible MLKL ubiquitylation sites in other contexts (Garcia et al, 2021).
Mutation of these lysines did not prevent either MLKL‐induced killing or MLKL ubiquitylation. The persistence of ubiquitin modification is a relatively common scenario when trying to define the role of ubiquitylation by mutating lysine. This is because E3 ligase mediated ubiquitylation is rarely tightly restricted to a motif and can therefore be promiscuous in target modification (Petroski & Deshaies, 2003; Wu et al, 2003). This means that if a favoured lysine in the target is mutated, the E3 ligase may nevertheless ubiquitylate another lysine. We therefore tried a more innovative approach. Since we had shown that the DUB USP21 was able to remove necroptosis‐induced ubiquitylation of MLKL in vitro, we generated an MLKL C‐terminal USP21 fusion that we predicted would be constitutively deubiquitylated. A similar approach fusing the K63 specific DUB AMSH to EGFR has been used to investigate EGFR degradation (Huang et al, 2013); however, it was not clear whether a DUB that removes all ubiquitin would be as well tolerated by cells. Stable inducible expression of USP21 alone was not toxic to cells over 24 h and fusion of the wild‐type or catalytically dead USP21 did not markedly affect MLKL's cytotoxic activity following a TSI death stimulus. Furthermore, catalytically active USP21 fused to MLKL prevented TSI‐induced ubiquitylation of MLKL. Interestingly, in both human and mouse cells, loss of MLKL ubiquitylation also allowed MLKL to kill cells without a necroptotic stimulus, albeit less potently when compared with such a stimulus. This further supports the idea that ubiquitylation of MLKL is an important “insurance policy” against low level activation of MLKL by other cellular kinases or low‐level spontaneous transition to the active conformation. Furthermore, our results indicate that the approach of directly fusing a DUB, even a pan DUB‐like USP21, to a protein of interest may be a widely applicable technique to evaluate the role of ubiquitin modification in other systems.
Other mechanisms have been proposed for MLKL turnover post‐activation. The ESCRT‐III machinery was proposed to mediate plasma membrane shedding alongside active MLKL during necroptosis (Gong et al, 2017), and cells were reported to release active MLKL‐containing vesicles via endosomal trafficking (Yoon et al, 2017; Zargarian et al, 2017; Fan et al, 2019). It has been proposed that this turnover helps set a threshold of activated MLKL required for necroptosis and that it delays the onset of cell death to allow production of essential cytokines and damage‐associated molecular patterns (DAMPs) and an inflammatory response (Vandenabeele et al, 2017). Another possibility is that the prolonged multi‐step path from MLKL phosphorylation to membrane permeabilization and cell death allows multiple points of regulation that ensure that a cell only commits to necroptosis under precisely defined conditions (Jacobsen et al, 2016; Dovey et al, 2018; Hildebrand et al, 2020; Samson et al, 2020). For this to work, the cell must have mechanisms to deactivate already activated MLKL, and the ubiquitylation of MLKL may serve as one such mechanism. Our results, showing that MLKL‐USP21, but not a wild‐type MLKL expressed to similar levels, kills cells in the absence of a necroptotic stimulus, suggest that MLKL may be continually translocating to membranes and turned over by MLKL ubiquitylation. Like the rapid degradation of inflammatory cytokine mRNAs (Lacey et al, 2015; Menon & Gaestel, 2018) although energetically costly, this might allow a more rapid response to a pathogen than an on/off mechanism focused solely on initiation and may also provide another mechanism to detect and act on attempts by pathogens to interfere with this anti‐pathogen response.
Materials and Methods
Compound and cytokines
Human TNF‐Fc made in‐house, Smac‐mimetics Compound A (Tetralogic), IDN‐6556 (Tetralogic), Q‐VD‐OPh (R & D Systems), Lipopolysaccharide (LPS, Sigma), Poly I:C (Sigma), Fas Ligand (a gift from Lorraine O'Reilly, WEHI), doxycycline (Sigma), Necrostatin‐1 (Nec‐1, Sigma), GSK872 (a gift from Anaxis Pty, Ltd.), coumermycin (Sigma), Necrosulfonamide (NSA, Merck Millipore), propidium iodide (Sigma), Sytox Green (Thermo Fisher), puromycin (Thermo Fisher), N‐ethylmaleimide (NEM, Sigma), deubiquitylase (DUBs) made in house (Hospenthal et al, 2015), complete protease inhibitor cocktail (Roche), Bafilomycin A1 (BAF, Enzo), PS341 (Sigma).
Antibodies
Mouse phospho‐MLKL (Ser345) rabbit monoclonal ERP9515(2) (Abcam)
Mouse phospho‐MLKL (Ser345) rabbit monoclonal (D6E3G; Cell Signalling Technology)
MLKL rat monoclonal 3H1 (available from Millipore MABC604; generated in‐house)
β‐actin mouse monoclonal AC‐15 (Sigma A‐1978)
BAK NT rabbit polyclonal #06‐536 (EMD Millipore)
GAPDH rabbit monoclonal 14C10 (Cell Signalling Technology)
Pan‐ubiquitin mouse monoclonal #3939 (Cell Signalling Technology)
Mouse/human RIPK1 mouse monoclonal #610458 (BD transduction Laboratories)
Mouse RIPK3 rabbit polyclonal PSC‐2283‐c100 (Axxora [Pro Sci])
Human RIPK3 rat monoclonal 1H2 (generated in‐house) (Petrie et al, 2019b)
Human phospho‐MLKL (Ser358) rabbit monoclonal ab 187091 (Abcam)
VDAC1 rabbit polyclonal AB10527 (Millipore)
Human Usp21 rabbit polyclonal 17856‐I‐AP (Proteintech)
HRP‐conjugated secondary antibodies
Cell lines immortalization and transfection
Cells were cultured in DMEM + 8% FCS at 37°C. HT29 cells were a kind gift from Mark Hampton. MDFs were isolated from tails of mice bearing different genotype, and immortalized by SV40 large T antigen via lentivirus transduction. MLKL mutant constructs were generated as described previously (Murphy et al, 2013; Hildebrand et al, 2014; Moujalled et al, 2014). All oligonucleotides for PCR and mutagenesis were synthesized by IDT. Constructs encoding USP21 catalytic domain with GS linker were made by GenScript (Nanjing, CN). Genes encoding WT mouse MLKL and PCR‐derived mutants were cloned into pF TRE3G PGK puro, a puromycin‐selectable, doxycycline‐inducible vector as previously described (and kindly supplied by Toru Okamoto) (Murphy et al, 2013; Hildebrand et al, 2014; Moujalled et al, 2014; Tanzer et al, 2016). Lentiviruses were generated in HEK293T cells before infection of target cells. Puromycin (5 μg/ml) was added for selection and maintenance of lines stably transduced with lentivirus.
For BMDMs, femora and tibiae were collected from WT and Tnf −/− mice at 6 weeks old, cut and flushed by PBS. Bone marrow was incubated in Petri dishes with DMEM supplemented with 10% FCS and 20% conditioned L929 medium for 7 days. Cells were fed with fresh medium after 3 days of plating.
Concentrations of stimuli and inhibitors
TNF (100 ng/ml), Smac‐mimetic Compound A (500 nM), IDN‐6556 (5 μM), Q‐VD‐OPh (5 μM), doxycycline (20 ng/ml), Necrostatin‐1 (50 μM), GSK872 (5 mM), LPS (50 ng/ml), Fas ligand (6.4 ng/ml), Poly I:C (1 mg/ml), coumermycin (700 nM), Bafilomycin (5 μM), PS341 (50 nM), MG132 (200nM), Chloroquine (50 μM), NH4Cl (2 mM), Ca‐074 Me (20 μM), TLCK (100 μM), AEBSF (100 μM) and concentrations otherwise indicated.
Cell death measured by flow cytometry
Cells were plated at 50,000/well in 24‐well plates before treatment, collected by trypsin and spun down, resuspended and stained with PI (propidium iodide, 1 μg/ml) in PBS buffer and quantified by Flow cytometry.
Cell death measured by live cell imaging
MDFs were plates at 10,000/well or 15,000/well (96‐well plates) and HT29s were plated at 40,000/well (48‐well plates) or 15,000/well (96‐well plates) and allowed to settle for 4 h (MDFs) and 24 h (HT29s), respectively. Cells were stimulated as indicated in media containing propidium iodide (PI, 200 ng/ml) or Sytox Green (500 nM, Thermo Fisher Scientific) and imaged at 45‐min or 1‐h intervals using an IncuCyte S3 Live cell imager. Numbers of PI or Sytox‐positive cells were quantified and plotted using IncuCyte software.
Cellular fractionation and Blue Native‐PAGE
Cells were collected by scraping, spun down and washed in pre‐chilled PBS. MELB buffer (20 mM 4‐(2‐Hydroxyethyl) piperazine‐1‐ethanesulfonic acid (HEPES) pH = 7.5, 100 mM sucrose, 2.5 mM MgCl2, 100 mM KCl) containing 0.025% digitonin was used to permeabilize cells to extract the cytosol fraction. Non‐soluble part was further solubilized by 1% digitonin buffer (Schagger & von Jagow, 1991; Liu et al, 2018). Both fractions were separated by Bis‐Tris Native PAGE and transferred to PVDF membrane. After transfer, the membrane was then destained (50% methanol and 25% acetic acid) and denatured (in 6 M guanidine HCl, 5 mM β‐ME) to maximize epitope exposure.
UBA pulldown assay and UbiCRest
Recombinant GST‐UBA fusion protein (Hjerpe et al, 2009) purified from Eschercihia coli in house was prebound to glutathione sepharose beads (10 μl/condition). Cells were lysed in DISC buffer (30 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% glycerol) containing 1% Triton X‐100 with 10 mM NEM and protease inhibitor. Cleared lysate was then coupled with beads overnight. SDS–PAGE sample loading buffer was used to elute the UBA pulldown fraction. For UbiCRest, washed beads were then incubated with DUBs at previously established concentrations (Hospenthal et al, 2015) and incubated at 37°C for the indicated times. Digestion product was eluted in SDS sample buffer for Western blot analysis.
FLAG‐tagged protein immunoaffinity precipitation and Mass Spectrometry sample preparation
N‐FLAG WT mouse MLKL was inducibly expressed in Mlkl −/− MDFs by doxycycline overnight and stimulated with TSI for 3 h. Cells were collected by scraping and permeabilized by 0.025% digitonin in MELB buffer together with NEM (10 mM) and protease inhibitors. The crude membrane fraction was pelleted by centrifugation and dissolved in 1% SDS in DISC buffer. Cleared lysate of crude membrane was then applied to M2‐FLAG beads for affinity precipitation. FLAG‐tagged protein on beads was eluted by heating at 56°C in 0.5% SDS solution. Eluted material was subjected to tryptic digestion using the FASP method (Wisniewski et al, 2009). Peptides were lyophilized using CentriVap (Labconco) prior to reconstituting in 80 μl 0.1% FA/2% acetonitrile (ACN). Peptide mixtures (1 µl) were analysed by nanoflow reversed‐phase liquid chromatography tandem mass spectrometry (LC‐MS/MS) on an M‐Class HPLC (Waters) coupled to a Q‐Exactive Orbitrap mass spectrometer (Thermo Fisher). Peptide mixtures were loaded in buffer A (0.1% formic acid, 2% acetonitrile, Milli‐Q water) and separated by reverse‐phase chromatography using C18 fused silica column (packed emitter, I.D. 75 μm, O.D. 360 μm × 25 cm length, IonOpticks, Australia) using flow rates and data‐dependent methods as previously described (Delconte et al, 2016; Kedzierski et al, 2017). Raw files consisting of high‐resolution MS/MS spectra were processed with MaxQuant (version 1.5.8.3) for feature detection and protein identification using the Andromeda search engine (Cox et al, 2011). Extracted peak lists were searched against the UniProtKB/Swiss‐Prot Mus musculus database (October 2016) and a separate reverse decoy database to empirically assess the false discovery rate (FDR) using strict trypsin specificity allowing up to two missed cleavages. The minimum required peptide length was set to seven amino acids. The mass tolerance for precursor ions and fragment ions was 20 ppm and 0.5 Da, respectively. The search included variable modifications of oxidation (methionine), amino‐terminal acetylation, carbamidomethyl (cysteine), Gly‐Gly or ubiquitylation (lysine), phosphorylation (serine, threonine or tyrosine) and N‐ethylmaleimide (cysteine).
Author contributions
ZL, JMH, LFD and KS‐A designed and performed experiments, and analysed data. SNY, AB, XW, MT, CAS, CF and SEG performed experiments. JH, UN, JMM and AIW provided methods and resources. ZL, JMH and JS analysed the data and wrote the manuscript. DK conceived the DUB fusion experiment and provided reagents. All authors read and commented on the manuscript.
Conflict of interest
SNY, AB, UN, CF, SEG, JMM, JMH and JS contribute to, or have contributed to, a project with Anaxis Pty Ltd to develop necroptosis inhibitors.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We would like to thank Jiami Han, Yueyuan Li and the WEHI mouse facility for technical assistance. This work was funded by NHMRC grants 1025594 (JS), 1046984 (JS) and 1105023 (JS and JMH) and fellowships 1172929 (JMM), 1058190 (JS), 1107149 (JS) and 110574 (JS) and was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS (9000587).
The EMBO Journal (2021) 40: e103718.
Contributor Information
Joanne M Hildebrand, Email: jhildebrand@wehi.edu.au.
John Silke, Email: silke@wehi.edu.au.
Data availability
The raw mass spectrometric data and the MaxQuant result files have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier: PXD015537 (http://www.ebi.ac.uk/pride/archive/projects/PXD015537).
References
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30: 689–700 [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nat Cell Biol 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P et al (2014) Translocation of mixed lineage kinase domain‐like protein to plasma membrane leads to necrotic cell death. Cell Res 24: 105–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Davies KA, Tanzer MC, Griffin MDW, Mok YF, Young SN, Qin R, Petrie EJ, Czabotar PE, Silke J, Murphy JM (2018) The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ 25: 1567–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KA, Fitzgibbon C, Young SN, Garnish SE, Yeung W, Coursier D, Birkinshaw RW, Sandow JJ, Lehmann WIL, Liang LY et al (2020) Distinct pseudokinase domain conformations underlie divergent activation mechanisms among vertebrate MLKL orthologues. Nat Commun 11: 3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang JG, Teh C, Firth M et al (2016) CIS is a potent checkpoint in NK cell‐mediated tumor immunity. Nat Immunol 17: 816–824 [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Darding M, Bertrand MJ, Walczak H (2016) Poly‐ubiquitination in TNFR1‐mediated necroptosis. Cell Mol Life Sci 73: 2165–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo H, Brown NW Jr, Cao JY, Grace CR, Gough PJ et al (2018) MLKL requires the inositol phosphate code to execute necroptosis. Mol Cell 70: 936–948.e937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, Spilgies L, Surinova S, Taraborrelli L, Hartwig T et al (2015) LUBAC‐recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep 13: 2258–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Guo J, Gao B, Zhang W, Ling L, Xu T, Pan C, Li L, Chen S, Wang H et al (2019) Flotillin‐mediated endocytosis and ALIX‐syntenin‐1‐mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci Signal 12: eaaw3423 [DOI] [PubMed] [Google Scholar]
- Garcia LR, Tenev T, Newman R, Haich RO, Liccardi G, John SW, Annibaldi A, Yu L, Pardo M, Young SN et al (2021) Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis‐induced tissue injury and pathogen clearance. Nat Commun 12: 3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR (2017) ESCRT‐III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169: 286–300.e216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T et al (2009) Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF‐R1 signaling complex and is required for TNF‐mediated gene induction. Mol Cell 36: 831–844 [DOI] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5: 461–466 [DOI] [PubMed] [Google Scholar]
- Hancock JF, Cadwallader K, Paterson H, Marshall CJ (1991) A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J 10: 4033–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI et al (2014) Activation of the pseudokinase MLKL unleashes the four‐helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111: 15072–15077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JM, Kauppi M, Majewski IJ, Liu Z, Cox AJ, Miyake S, Petrie EJ, Silk MA, Li Z, Tanzer MC et al (2020) A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat Commun 11: 3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz‐Otsoa F, Lang V, England P, Rodriguez MS (2009) Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin‐binding entities. EMBO Rep 10: 1250–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative, caspase‐8‐independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495 [DOI] [PubMed] [Google Scholar]
- Hospenthal MK, Mevissen TET, Komander D (2015) Deubiquitinase‐based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat Protoc 10: 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, Gygi SP, Yates NA, Sorkin A (2013) Lysine 63‐linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci USA 110: 15722–15727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz‐Wachtel M, van Wijk SJ, Goswami P, Nagy V, Terzic J et al (2011) SHARPIN forms a linear ubiquitin ligase complex regulating NF‐kappaB activity and apoptosis. Nature 471: 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen AV, Lowes KN, Tanzer MC, Lucet IS, Hildebrand JM, Petrie EJ, van Delft MF, Liu Z, Conos SA, Zhang JG et al (2016) HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis 7: e2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV (2013) Necroptosis: the release of damage‐associated molecular patterns and its physiological relevance. Immunity 38: 209–223 [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES (2013) Toll‐like receptor 3‐mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288: 31268–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski L, Tate MD, Hsu AC, Kolesnik TB, Linossi EM, Dagley L, Dong Z, Freeman S, Infusini G, Starkey MR et al (2017) Suppressor of cytokine signaling (SOCS)5 ameliorates influenza infection via inhibition of EGFR signaling. Elife 6: e20444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, Wauer T, Hospenthal MK, Gyrd‐Hansen M, Krappmann D et al (2013) OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1‐linked polyubiquitin. Cell 153: 1312–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey D, Hickey P, Arhatari BD, O'Reilly LA, Rohrbeck L, Kiriazis H, Du XJ, Bouillet P (2015) Spontaneous retrotransposon insertion into TNF 3'UTR causes heart valve disease and chronic polyarthritis. Proc Natl Acad Sci USA 112: 9698–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL et al (2015) RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6: 6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z (2015) A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA 112: 5017–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu H, Johnston A, Hanna‐Addams S, Reynoso E, Xiang Y, Wang Z (2017) MLKL forms disulfide bond‐dependent amyloid‐like polymers to induce necroptosis. Proc Natl Acad Sci USA 114: E7450–E7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Silke J, Hildebrand JM (2018) Methods for studying TNF‐mediated necroptosis in cultured cells. Methods Mol Biol 1857: 53–61 [DOI] [PubMed] [Google Scholar]
- Menon MB, Gaestel M (2018) MK2‐TNF‐signaling comes full circle. Trends Biochem Sci 43: 170–179 [DOI] [PubMed] [Google Scholar]
- Morrow ME, Morgan MT, Clerici M, Growkova K, Yan M, Komander D, Sixma TK, Simicek M, Wolberger C (2018) Active site alanine mutations convert deubiquitinases into high‐affinity ubiquitin‐binding proteins. EMBO Rep 19: e45680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosesson Y, Yarden Y (2006) Monoubiquitylation: a recurrent theme in membrane protein transport. Isr Med Assoc J 8: 233–237 [PubMed] [Google Scholar]
- Moujalled DM, Cook WD, Murphy JM, Vaux DL (2014) Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis 5: e1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai S, Yamaguchi Y, Shirasaki Y, Yamagishi M, Shindo R, Hildebrand JM, Miura R, Nakabayashi O, Totsuk M, Tomida T et al (2018) A FRET biosensor for necroptosis uncovers two different modes of the release of DAMPs. Nat Commun 9: 4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez‐Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI et al (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453 [DOI] [PubMed] [Google Scholar]
- Oh E, Akopian D, Rape M (2018) Principles of Ubiquitin‐dependent signaling. Annu Rev Cell Dev Biol 34: 137–162 [DOI] [PubMed] [Google Scholar]
- Onizawa M, Oshima S, Schulze‐Topphoff U, Oses‐Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula R et al (2015) The ubiquitin‐modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 16: 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A (2014) RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ 21: 1511–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47(D1): D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SL, Chen TT, Lawrence DA, Marsters SA, Gonzalvez F, Ashkenazi A (2015) TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ 22: 1846–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, Dai W, Young SN, Tanzer MC, Wardak A et al (2018) Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun 9: 2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie EJ, Czabotar PE, Murphy JM (2019a) The structural basis of necroptotic cell death signaling. Trends Biochem Sci 44: 53–63 [DOI] [PubMed] [Google Scholar]
- Petrie EJ, Sandow JJ, Lehmann WIL, Liang LY, Coursier D, Young SN, Kersten WJA, Fitzgibbon C, Samson AL, Jacobsen AV et al (2019b) Viral MLKL homologs subvert necroptotic cell death by sequestering cellular RIPK3. Cell Rep 28: 3309–3319.e3305 [DOI] [PubMed] [Google Scholar]
- Petrie EJ, Birkinshaw RW, Koide A, Denbaum E, Hildebrand JM, Garnish SE, Davies KA, Sandow JJ, Samson AL, Gavin X et al (2020) Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc Natl Acad Sci USA 117: 8468–8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ (2003) Context of multiubiquitin chain attachment influences the rate of Sic1 degradation. Mol Cell 11: 1435–1444 [DOI] [PubMed] [Google Scholar]
- Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, Whitehead LW, Frank D, Garnish SE, Fitzgibbon C et al (2020) MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun 11: 3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapmaz A, Berlin I, Bos E, Wijdeven RH, Janssen H, Konietzny R, Akkermans JJ, Erson‐Bensan AE, Koning RI, Kessler BM et al (2019) USP32 regulates late endosomal transport and recycling through deubiquitylation of Rab7. Nat Commun 10: 1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, von Jagow G (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199: 223–231 [DOI] [PubMed] [Google Scholar]
- Segawa K, Nagata S (2015) An apoptotic 'eat me' signal: phosphatidylserine exposure. Trends Cell Biol 25: 639–650 [DOI] [PubMed] [Google Scholar]
- Stafford CA, Lawlor KE, Heim VJ, Bankovacki A, Bernardini JP, Silke J, Nachbur U (2018) IAPs regulate distinct innate immune pathways to co‐ordinate the response to bacterial peptidoglycans. Cell Rep 22: 1496–1508 [DOI] [PubMed] [Google Scholar]
- Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor‐interacting protein (RIP) by RIP3. J Biol Chem 277: 9505–9511 [DOI] [PubMed] [Google Scholar]
- Swatek KN, Komander D (2016) Ubiquitin modifications. Cell Res 26: 399–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C, Alexander WS, Hildebrand JM, Silke J, Murphy JM (2015) Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem J 471: 255–265 [DOI] [PubMed] [Google Scholar]
- Tanzer MC, Matti I, Hildebrand JM, Young SN, Wardak A, Tripaydonis A, Petrie EJ, Mildenhall AL, Vaux DL, Vince JE et al (2016) Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ 23: 1185–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S et al (2009) Involvement of linear polyubiquitylation of NEMO in NF‐kappaB activation. Nat Cell Biol 11: 123–132 [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P (2015) Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol 2: e975093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Riquet F, Cappe B (2017) Necroptosis: (last) message in a bubble. Immunity 47: 1–3 [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148: 228–243 [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X (2014) Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133–146 [DOI] [PubMed] [Google Scholar]
- Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P (2018) Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol 1: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KD, Tashayev VL, O'Connor LB, Larsen CN, Kasperek E, Pickart CM (1995) Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry 34: 14535–14546 [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- Wright LP, Philips MR (2006) Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res 47: 883–891 [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP (2003) Structure of a beta‐TrCP1‐Skp1‐beta‐catenin complex: destruction motif binding and lysine specificity of the SCF(beta‐TrCP1) ubiquitin ligase. Mol Cell 11: 1445–1456 [DOI] [PubMed] [Google Scholar]
- Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H, Song Y, Chen X, Shao J, Han J (2014) Distinct roles of RIP1‐RIP3 hetero‐ and RIP3‐RIP3 homo‐interaction in mediating necroptosis. Cell Death Differ 21: 1709–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Akutsu M, Reyes‐Turcu F, Enchev RI, Wilkinson KD, Komander D (2011) Polyubiquitin binding and cross‐reactivity in the USP domain deubiquitinase USP21. EMBO Rep 12: 350–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Blaser G, Horrocks MH, Ruedas‐Rama MJ, Ibrahim S, Zhukov AA, Orte A, Klenerman D, Jackson SE, Komander D (2012) Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature 492: 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Bogdanov K, Kovalenko A, Wallach D (2016) Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ 23: 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Kovalenko A, Bogdanov K, Wallach D (2017) MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47: 51–65.e57 [DOI] [PubMed] [Google Scholar]
- Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir‐Birin Y, Croker BA, Regev‐Rudzki N, Edry‐Botzer L, Gerlic M (2017) Phosphatidylserine externalization, necroptotic bodies release, and phagocytosis during necroptosis. PLoS Biol 15: e2002711 [DOI] [PMC free article] [PubMed] [Google Scholar]