

Abstract
Fragile X syndrome (FXS) is the leading monogenic form of intellectual disability and autism, with patients exhibiting numerous auditory-related phenotypes during their developmental period, including communication, language development, and auditory processing deficits. Despite FXS studies describing excitatory–inhibitory (E–I) imbalance in the auditory circuit and an impaired auditory critical period, evaluation of E–I circuitry maturation in the auditory cortex of FXS models remains limited. Here, we examined GABAA receptor (GABAAR)-mediated inhibitory synaptic transmission within the auditory cortex, characterizing normal intracortical circuit development patterns in wild-type (WT) mice and examining their dysregulation in developing Fmr1 knock-out (KO) mice. Electrophysiological recordings revealed gradual developmental shifts in WT L4-L2/3 connectivity, where circuit excitability significantly increased after critical period onset. KO mice exhibited accelerated developmental shifts related to aberrant GABAergic signaling. Specifically, Fmr1 KO L2/3 pyramidal neurons have enhanced developmental sensitivity to pharmacological GABAAR modulators, altered maturation of GABAAR voltage-dependent conductance, with additional presynaptic GABA release alterations. These differences are further accompanied by alterations in developmental long-term potentiation. Together, our results suggest that altered GABAergic signaling within developing Fmr1 KOs impairs the normal patterning of E–I circuit and synaptic plasticity maturation to contribute to the impaired auditory cortex critical period and functional auditory deficits in FXS.
Keywords: auditory cortex, critical period, Fragile X, GABA inhibition, plasticity
Introduction
Fragile X syndrome (FXS) is the most common monogenic form of intellectual disability and autism (Hersh et al. 2011). The identified mutation in FXS, a hypermethylation of the CGG trinucleotide repeat expansion in the Fmr1 promoter, leads to its silencing and loss of expression of Fragile X mental retardation protein (FMRP), a translational regulator and modulator of excitatory–inhibitory (E–I) balance (Darnell et al. 2011; Tang et al. 2015). FXS patients have deficits in auditory processing (Van der Molen et al. 2012; Rotschafer and Razak 2014), communication, and language development (Finestack et al. 2009; Hersh et al. 2011), as well as hypersensitivity to sensory stimuli (Miller et al. 1999), making the auditory cortex a region of particular interest. Fmr1 knock-out (KO) mice also exhibit auditory phenotypes suggestive of E–I imbalance, including susceptibility to audiogenic seizures (Chen and Toth 2001), altered auditory processing with enhanced auditory cortex excitability (Yun et al. 2006; Rotschafer and Razak 2013), impaired parvalbumin cell development in early auditory cortex maturation (Wen et al. 2018), failed stabilization of auditory cortex long-term potentiation (LTP; Yang et al. 2014), and impaired plasticity in tonotopic frequency mapping during the auditory cortex critical period (Kim et al. 2013).
Critical periods are postnatal developmental windows characterized by heightened experience-dependent synaptogenesis and synaptic plasticity that establish stable and mature neural circuits. In addition to various molecular and neuromodulatory factors, the dynamic regulation of E–I levels in the developing brain by the ionotropic neurotransmitter receptor system mediates the initiation, duration, and capacity of critical periods (Hensch 2005; Rakhade and Jensen 2009). Specifically, inhibitory GABA action induces and ultimately restricts experience-dependent plasticity in the visual and auditory cortex (Fagiolini and Hensch 2000; Jang et al. 2009; Kalish et al. 2020), with signaling by α1-containing GABAA receptor (GABAAR) circuits necessary for regulating visual cortical plasticity (Fagiolini et al. 2004). The maturation of GABAAR inhibitory properties within the auditory cortex further coincides with its critical period and is further modulated by environmental exposure (de Villers-Sidani et al. 2007; Dorrn et al. 2010; Barkat et al. 2011; Sanes and Kotak 2011; Takesian et al. 2012; Mowery et al. 2016; Cisneros-Franco et al. 2018; Kalish et al. 2020). Excitatory circuits also modulate critical periods (Hensch 2005; Oswald and Reyes 2008), and we have reported that hyperexcitability from early-life seizures can disrupt auditory tonotopic critical period plasticity in wild-type (WT) mice by prematurely unsilencing glutamatergic synapses (Sun et al. 2018). In addition to regulating critical periods, achieving proper E–I balance is necessary for auditory processing, with the postnatal maturation of inhibitory synaptic transmission specifically required for refining and increasing the specificity of auditory processing (Wehr and Zador 2003; Froemke and Jones 2011; Blackwell and Geffen 2017).
Despite auditory phenotypes suggestive of E–I imbalance in Fmr1 KO mice, studies directly examining the maturation of synaptic transmission in the auditory cortex are limited. Thus, given the crucial role of inhibitory synaptic signaling in regulating E–I levels for critical periods and auditory processing, we sought to evaluate whether the maturation of GABAergic signaling was dysregulated in the auditory cortex of Fmr1 KO mice. Using electrophysiology, we investigated auditory intracortical L4-L2/3 synaptic transmission across the critical period of auditory forebrain development in WT and Fmr1 KO mice, given the critical role of the L4-L2/3 circuit in tonotopic frequency processing (Barkat et al. 2011). Our studies highlight an altered maturational excitability sequence for L4-L2/3 basal synaptic transmission in Fmr1 KOs that is out of sync with the WT critical period window and related to aberrant GABAergic regulation. Synaptic evaluation of L2/3 pyramidal neurons further revealed enhanced developmental sensitivity to pharmacological GABAAR modulators, altered maturation of GABAAR reversal potentials and voltage-dependent conductance, and impairments in L4 presynaptic GABA release mechanisms. These synaptic GABAergic differences are accompanied by alterations in the stabilization of LTP and the normal patterning of synaptic plasticity across development. Our results suggest that developmental alterations to pre- and postsynaptic GABAergic signaling in the Fmr1 KO auditory cortex contribute to the impaired E–I circuit and synaptic plasticity maturation in the auditory cortex to likely underlie the various auditory-related deficits in the Fmr1 KO mice.
Materials and Methods
Animals
Fmr1 WT and Fmr1 KO mice of the congenic FVB background were obtained from The Jackson Laboratory (stock # 004828; 004624). Heterozygous females were generated from the original breeders, whereby heterozygous female mice were later crossed with WT or hemizygous male mice to obtain Fmr1 WT and KO male littermates. Age-matched WT and KO male littermate mice were used for all experiments to avoid the confounding effects of X-inactivation that would be present in heterozygous females. All procedures were performed in accordance with the guidelines of the NIH and with approval of the Institution of Animal Care and Use Committee with the University of Pennsylvania. All efforts were made to minimize animal suffering and numbers.
Western Blots
To compare the developmental expression patterns of GABAAR subunits, Fmr1 WT and KO littermates were sacrificed at P9, P12, P15, P24, and P56. Brains were removed and the auditory cortex was microdissected. Using a coronal brain matrix, a 1.5–2 mm thick coronal brain slice containing the auditory cortex was extracted as identified by stereotaxic coordinates and anatomical landmarks from Paxinos and Franklin (2004) and the Allen Brain Atlas. The brain slice was cut horizontally above the rhinal fissure, whereby 15° cuts were made bilaterally to isolate the auditory cortex. Tissue was flash-frozen in chilled ethanol and stored at −80°C until homogenization.
For the measurement of membrane-bound protein expression, the auditory cortices from 2 mice were pooled together for a single sample (for western blots, n = total number of pooled samples), as was necessary to generate sufficient protein concentration during membrane preparations. The membrane extraction was performed as described previously (Talos et al. 2006; Rakhade et al. 2012; Yennawar et al. 2019). Total protein amounts were measured with a Bradford protein assay (Bio-Rad) and samples were diluted to obtain equal amounts of proteins in each sample (15 μg total) in lysis buffer and 4× Laemmli Sample Buffer (Bio-Rad). Samples were separated by gel electrophoresis on Criterion TGX 4–20% Tris-Glycine precast gels (Bio-Rad), and transferred to PVDF membranes (Millipore Immobilon-FL). Primary antibodies used are as follows: GABAAα3 (1:1000, Sigma G4291, RRID:AB_477015), GABAAα1 (1:750, NeuroMab 75-136, RRID:AB_2108811), NKCC1 (1:1000, Millipore AB3560P, RRID:AB_91514), KCC2 (1:1000, Millipore 07–432, RRID:AB_310611), and β-actin (1:5000; Sigma A5441; RRID:AB_476744). Secondary antibodies used were goat anti-rabbit IRDye 800CW (1:15,000; LI-COR 926-32212; RRID:AB_621848) and goat anti-mouse (IRDye 680LT; LI-COR 926-68024; RRID:AB_10706168). The Odyssey Infrared Imaging System was used for the quantification of protein expression. β-actin was used for normalization of individual samples, and normalized values are expressed as a percentage of mean age-matched WT samples for direct within-age evaluation, or mean P56 WT samples run on the same blot for developmental comparisons.
Immunohistochemistry
P12 mice were anesthetized and perfused with 4% PFA/PBS. Brains were postfixed for 2 h, cryoprotected in 30% sucrose/PBS, embedded, and frozen in OCT. Free-floating sections were collected at 30 μm in 0.02% sodium azide/PBS. After washing sections in PBS, antigen retrieval was performed, incubating sections in citrate buffer, pH 6.0 for 30 min at 75°C. Sections were maintained in citrate buffer for an additional 15 min at room temperature (RT), washed, and blocked with 10% normal goat serum/PBS. Primary antibody for extracellular targeting GABAAα3 (1:500, Alomone Labs AGA-003, RRID:AB_2039866) was incubated overnight at 4°C in 1% goat serum/PBS. Sections were washed, incubated in goat anti-rabbit 594 (1:1000, Invitrogen A-11012, RRID:AB_141359) for 1 h at RT, and washed. Sections were then permeabilized with 0.2% Triton/PBS for 5 min, washed, and reblocked in 10% goat serum/0.1% Triton/PBS for 1 h at RT. Sections were incubated with anti-vesicular GABA transporter (VGAT) (1:500, Synaptic Systems 131 004, RRID:AB_887873) and anti-microtubule-associated protein 2 (MAP2) (1:500, Abcam ab5392, RRID:AB_2138153) overnight at 4°C in 1% goat serum/0.1% Triton/PBS, and incubated in goat anti-chicken 488 (1:1000, Invitrogen A-11039, RRID:AB_2534096) and goat anti-guinea pig 647 (1:1000, Invitrogen A-21450, RRID:AB_2735091) for 1 h at RT. Sections were PBS washed, mounted on slides, and cover slipped with Fluoromount.
Images were acquired on a Lecia SP5 confocal microscope with a researcher blinded to genotype. The auditory cortex was localized using the hippocampus and rhinal fissure as anatomical landmarks. Images were taken using 63× objective (numerical aperture = 1.4) with a digital zoom of 2. Gain, offset, and laser intensity settings were kept identical during imaging sessions. MAP2 was used to approximate cortical layers, with imaging started in L6 and progressing to L2/3. Images were obtained at 1024 × 1024 pixels bilaterally from both hemispheres from a given brain section, using a line average of 2 and z-step of 0.25 μm.
Images were processed and analyzed blind using FIJI. Five z-planes with the most robust staining were selected from the z-stack for image processing. The despeckling and smooth functions were used to reduce noise from each plane and channel, and then collapsed into a single image using z-project Sum Slices. Background was subtracted using a rolling ball algorithm, with channels auto-thresholded (over/under). To avoid artifact, each channel was thresholded at multiple intervals of the maximum threshold (95, 90, 85, 80, and 75%). Using the Coloc2 plugin (Gao and Heldt 2016; Yennawar et al. 2019), we compared the integrated intensity values for total fluorescent intensities and Mander’s coefficient to quantify colocalization of VGAT and GABAAα3 (Manders et al. 1993). Colocalization was repeated at each threshold to avoid thresholding artifacts (Zhou et al. 2011; Yennawar et al. 2019).
Electrophysiology
Brain Slice Preparation
Acute coronal brain slices for extracellular, whole-cell, and perforated-patch electrophysiology were prepared from Fmr1 WT and KO littermate mice at the following ages: P9–10, P12–13, P15–16, P21–25, and P40–45. Animals were decapitated and brains were removed and placed into a chilled cutting solution containing the following (in mM): 220 sucrose, 3 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 1 MgCl2, 26 NaHCO3, and 10 D-glucose, ~ 300 mOsm, pH 7.4, and bubbled with 95% O2/5% CO2 at 4°C. Coronal slices (350 μm) containing the auditory cortex, identified by anatomical landmarks, were cut using a vibratome (Leica VT1000S) in cutting solution. Slices were incubated in oxygenated artificial cerebrospinal fluid (ACSF) containing the following (in mM): 124 NaCl, 5 KCl, 2 CaCl2, 1.2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 D-glucose, ~ 295–300 mOsm, and transferred to 32°C for 30 min. Slices were maintained at RT for at least 1 h before recordings.
Extracellular Recordings
Multielectrode array (MEA) recording systems (MED64 System, Alpha Med Scientific) were used to perform extracellular field potential recordings. The MEA probes had 64 planar electrodes, arranged in an 8 × 8 pattern with 150 μm interelectrode spacing (MED-P5155). Slices were transferred to the MEA recording chamber and perfused with ACSF at 30–32°C with 3–8 mL/min flow rates. Higher perfusion rates were necessary for older animals to improve O2 supply to brain slices and maintain viability (P9–16: ~ 3–4 mL/min; P21–25: ~ 6 mL/min; P40–45: ~ 8 mL/min) (Hajos et al. 2009; Reinhard et al. 2014; Panuccio et al. 2018). Slices were anchored with a harp and aligned so that the electrodes were parallel with the auditory cortical layers, as identified by anatomical landmarks using the 4× objective of a Nikon Eclipse TE2000-5 microscope. Slices were recovered in the recording chamber for 30 min with minimal stimulation (7–10 μA) every 1–1.5 min to monitor normalization of responses before beginning experiments. Stimulations (0.2 ms pulse) were made in L4 with electrodes positioned approximately 400–450 μm from the pia, with field excitatory postsynaptic potential (fEPSP) response analysis made from the adjacent electrode positioned in L2/3. For each experiment, the main L4 stimulation electrode was selected by identifying the stimulation and recording electrode pair that elicited the smallest synaptic response when stimulated at 10 μA or 20 μA to maintain consistent recording practices across all experiments.
Input–output (I–O) curves were evaluated, examining the relationship between fEPSP in L2/3 to increasing stimulation intensities in L4 (+5 μA every 30 s) until maximal synaptic response was reached, measured by the maximum fEPSP amplitude. Half-maximal stimulus intensities were estimated by identifying the minimum intensity that elicited half-maximal fEPSP amplitude determined from I–O curves. Paired-pulse ratios (PPRs) were calculated by dividing the amplitude of the second fEPSP by the first fEPSP when 2 successive stimuli of 500, 200, 100, 50, 25, 10, and 6 ms intervals were applied every 30 s. For experiments analyzing sensitivity to picrotoxin (PTX, Sigma P1675) or bicuculline (BIC, Tocris 2503), slices were incubated in ACSF with 5 μM PTX or 5 μM BIC for 30 min after initial baseline I–O curve and paired-pulse recordings (Jang et al. 2009). In PTX and BIC experiments the first peak was measured, as the synaptic response often showed multiple peaks. All data collection and analysis were performed in Mobius (MED64).
Whole-Cell Electrophysiology
Whole-cell patch-clamp recordings were made from L2/3 pyramidal neurons in the auditory cortex, visualized with an upright Zeiss Axioscop 2 FsPlus IR-DIC microscope, with the researchers blinded to genotype. Recordings were made at RT with ACSF perfused at a rate of ~ 2 mL/min. Glass electrodes were pulled (P-87, Sutter Instruments) with a resistance of 2–4 MΩ and filled with internal solution containing (in mM): 135 CsCl, 10 EGTA, 10 HEPES, 5 ATP-Mg, and 5 QX-314 (pH 7.3, 290–295 mOsm) (Zhou et al. 2013). Liquid junction potentials were corrected using the Axopatch 200B amplifier. To isolate spontaneous inhibitory postsynaptic currents (sIPSCs), cells were voltage-clamped at −60 mV and recorded for 10–15 min in the presence of 20 μM NBQX (Tocris) and 50 μM D-AP5 (Abcam). In experiments examining sensitivity to a selective α1-subunit GABAAR agonist, we recorded 10 min of traces following 6 min of zolpidem (ZOLP; 100 nM, Sigma). The effects of ZOLP on evoked IPSCs (eIPSCs) were also evaluated by comparing the average trace of 10 evoked responses from L4 stimulation before and after ZOLP. Miniature IPSCs (mIPSCs) were additionally recorded in the presence of 1 μM tetrodotoxin (TTX, Tocris). PPRs of eIPSCs were calculated from an average of 5 sweeps following L4 stimulation using interstimulus intervals of 400, 200, 90, 70, 50, and 20 ms. Access resistance was monitored throughout each experiment, with applied series resistance compensation of 65%, and recordings discarded if access resistance was > 25 MΩ or changed > 20%. Data were collected using Axopatch 200B amplifier and pCLAMP 10 software (Molecular Devices), filtered at 2 kHz, and digitized at 20 kHz using a Digidata 1440A analog to digital converter.
Analysis was performed offline using Clampfit 10.2 (Molecular Devices). Each trace was first low-pass filtered at 1 kHz (Gaussian). The sIPSC events were detected automatically with a threshold of 5–6 pA (2 times the root mean square of the noise) (Rakhade et al. 2008; Zhou et al. 2011; Sun et al. 2013) and were visually confirmed. The weighted decay time constant (τdw) was calculated by first fitting a double-exponential function on the averaged sIPSC trace for each neuron that excluded multiple overlapping sIPSC events: f(t) = Afaste-t/τfast + Aslowe-t/τslow and then using the fit values in the following equation: τdw = [(Afast × τfast) + (Aslow × τslow)]/(Afast + Aslow) (Vislay et al. 2013; Zhou et al. 2013).
Perforated Patch Electrophysiology
EGABA was measured using perforated patch recordings. Gramicidin (Sigma-Aldrich Inc.) was dissolved in methanol to 10 mg/mL and diluted in the internal pipette solution to a final concentration of 150 μg/mL (Ostroumov et al. 2016). The membrane integrity within pipette tips was confirmed by the presence of the sodium currents of recorded neurons (not being blocked by the QX-314 inside of the electrodes). eIPSCs in L2/3 pyramidal neurons were recorded at holding potentials between −100 mV and 0 mV, with stimulation at the border of L3 and L4, increasing in 10 mV steps with access resistance continuously monitored. Stimulation intensities were normalized by first identifying the minimal stimulus intensity necessary to elicit a threshold evoked response, and then using a stimulus of approximately twice the intensity that evoked a monosynaptic response. All recordings were performed in the presence of 20 μM NBQX and 50 μM D-AP5. Given that during early postnatal ages L2/3 pyramidal neurons have a high input resistance that exhibit an age-dependent decrease (P9–10: 523.9 ± 20.56 MΩ; P12–13: 503.5 ± 36.01 MΩ; P15–16: 350.5 ± 27.51 MΩ; P21–25: 216.6 ± 14.65 MΩ), it was necessary to correct the holding potential values for I-V plots (Rheims et al. 2008). The following formula was used:
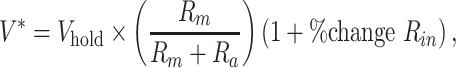 |
where Vhold is the command membrane holding potential; Rm is the average membrane resistance during P9–10 perforated-patch recordings; Ra is the average access resistance during P9–10 recordings; and % change in Rin is the percent change in input resistance for a given age from P9–10 Rin for proper age-dependent corrections. Reversal potentials were estimated from the intersection of current–voltage plots, where the amplitudes of the eIPSCs were plotted against the modified voltage holding potentials (V*).
LTP: Evoked Postsynaptic Currents
For single-cell LTP studies, electrodes were used with a resistance of 2–4 MΩ and filled with internal solution containing (in mM): 110 Cs methanesulfonate, 10 TEA-Cl, 4 NaCl, 2 MgCl2, 0.5 EGTA, 10 HEPES, 4 ATP-Mg, 0.3 GTP, 7 phosphocreatine, creatine phosphokinase (17 units/mL), 1 QX-314, pH 7.3, and 270–280 mOsm. Evoked postsynaptic current (ePSC) recordings were made from whole-cell patch-clamped L2/3 pyramidal neurons with stimulations evoked from L4 in the absence of any pharmacological blockers. We intentionally permitted GABAergic modulation during LTP experiments to evaluate the cell’s net ability for LTP across development as it mediates all E–I synaptic input. Thus, the ePSCs measured were from evoked glutamatergic responses with inhibitory synaptic modulation. Stimulation intensities that evoked 40–60% of maximal ePSC amplitude were used. Cells were held at −60 mV for 5 min baseline recording (10 s intervals), followed by pairing where cells were held at +30 mV with 5 tetani (0.1 ms pulse, 100 Hz for 500 ms, separated by 830 ms), and monitored for 30 min postpairing at −60 mV. Access resistance was monitored throughout each experiment, with applied series resistance compensation of up to 75% and recordings discarded if access resistance was > 25 MΩ or changed > 20%. Analysis was performed offline. Evoked postsynaptic current peak amplitudes were measured, with normalization of ePSCs to the average peak amplitude of the baseline recording.
Experimental Design and Statistical Analyses
For all experiments, the sample size and power were determined based on previously published work in our laboratory and using StatMate software. Mice were excluded if their weight was > 2 standard deviations from the mean weight of age-matched mice.
Statistical analysis was completed using Prism 8 (GraphPad). Data were tested for normality using the Shapiro–Wilk normality test. For within-age comparisons, statistical significance was assessed using a Student’s t-test or Mann–Whitney test depending on normality of the data. For developmental expression analysis, 2-way analysis of variance (ANOVA) and Sidak’s multiple comparisons tests with adjusted P values were used to assess significance. For the analysis of input–output curves in MEA experiments, a comparison of sigmoidal curve fits was used. PPRs were analyzed using a 2-way ANOVA with Sidak’s multiple comparisons test. Changes in amplitude and τdw following ZOLP were analyzed using paired t-test or Wilcoxon matched-pairs signed rank depending on the normality of the data. For within-age genotype comparisons of LTP studies, statistical significance was assessed using 2-way repeated-measures ANOVA. For comparing changes in decay of LTP, a comparison of slopes from linear regressions was performed from 6 to 30 min postpairing values. Data are reported as mean ± standard error of mean with P values reported in Results. All ns are reported in the figure legends or tables, or within figures themselves.
Results
Accelerated Developmental Progression of Basal Synaptic Transmission in the Auditory Intracortical Circuit of Fmr1 KO Mice
Given the auditory phenotypes described in Fmr1 KO mice, we sought to identify whether there exists functional deficits in synaptic excitability between L4 to L2/3 in the developing auditory intracortical circuit as these feedforward projections from L4 are the first stage of higher-level auditory processing within the intracortical circuit following tonotopic thalamic input (Merzenich et al. 1975; Kanold et al. 2014; Li et al. 2014; Javitt and Sweet 2015). Using MEA extracellular field recordings in ex vivo slice preparations, we evaluated L4 to L2/3 basal synaptic transmission by comparing input–output (I–O) curves. To capture the sequence of auditory intracortical maturation, mice were evaluated at P9, prior to ear canal opening, P12 and P15, capturing the start and end, respectively, of the mouse auditory critical period (Barkat et al. 2011), P24, a juvenile development age, and at P56, as adults (Fig. 1A).
Figure 1 .
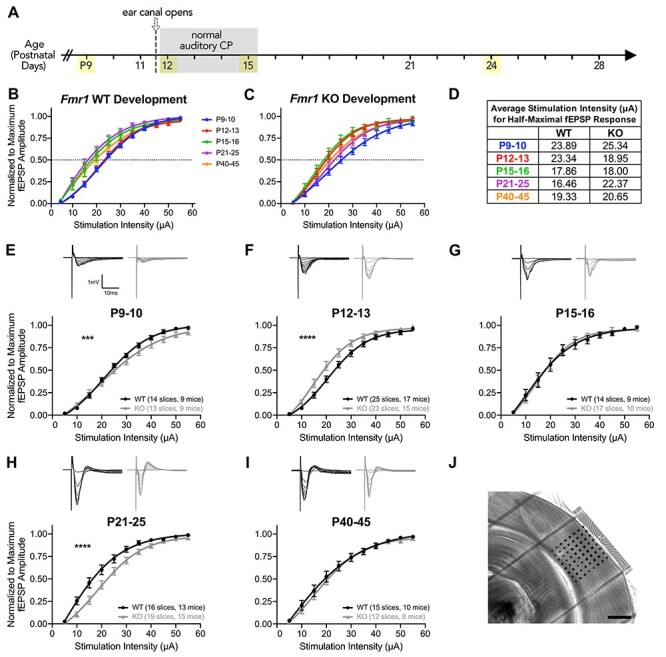
Fmr1 KO mice exhibit dynamic changes in excitability in L4 to L2/3 basal synaptic transmission during auditory cortex development. I–O curves of stimulation intensity to fEPSP amplitude, normalized to maximum amplitude, and fit with sigmoidal curves. (A) The timeline of approximate auditory postnatal maturation in mice with ages analyzed for experiments highlighted in yellow. (B–C) The overlay of I–O curves for WT and KO mice to evaluate developmental shifts in excitability. (D) Average stimulation intensities for half-maximal fEPSP response estimated from sigmoidal curve fits in (B) and (C). (E–I) Top: representative fEPSP traces in L2/3 extracellular recordings from L4 stimulation. Bottom: the comparison of sigmoidal curve fits for I–O curves at each age between WT and Fmr1 KO mice. (F) Fmr1 KO mice are significantly more excitable at P12–13 and (H) significantly less excitable at P21–24. (J) Representative DIC image of coronal slice with MEA overlaid on the auditory cortex. Scale bar: 600 μm. ***P < 0.001, ****P < 0.0001.
To elucidate auditory intracortical circuit maturation, we first examined the natural shifts in excitability during postnatal development by overlaying all of the I–O curves across the ages for each genotype (Fig. 1B,C). In WT mice, we observed a clear progression of shifts in the I–O curves that correspond with pre- and postcritical period (Fig. 1B). Specifically, WT I–O curves at P9–10 and P12–13 did not differ from one another (Tukey’s multiple comparisons: P9–10 vs. P12–13, P = 0.9998), but after P12–13, I–O curves subsequently exhibited a significant leftward shift indicating increased excitability within the circuit (Tukey’s: P12–13 vs. P15–16, P < 0.0001; P15–16 vs. P21–25, P = 0.5837) (Fig. 1B,D). At the subsequent ages of P40–45, the I–O curve had a slight rightward shift relative to the P21–25 I–O curve (Tukey’s: P21–25 vs. P40–45, P = 0.0316), likely attributable to the slower GABAergic maturation that occurs in the postnatal brain (Rakhade and Jensen 2009). In contrast, the aforementioned developmental shifts occurred at earlier ages in Fmr1 KO mice (Fig. 1C). Fmr1 KO P12–13 mice I–O curves revealed earlier enhanced excitability with a significant leftward shift compared with P9–10 and were similar to P15–16 (Tukey’s: P9–10 vs. P12–13, P < 0.0001; P12–13 vs. P15–16, P = 0.9675). I–O curves in the Fmr1 KO mice also exhibited an earlier shift that yielded decreased excitability at P21–25 comparable with that of WT at P40–45 (Tukey’s: P15–16 vs. P21–25, P = 0.001; P21–25 vs. P40–45, P = 0.7375). These results suggest that Fmr1 KO mice have accelerated developmental shifts in excitability in their intracortical L4-L2/3 synaptic transmission relative to the normal maturation of the WT auditory critical period.
We further compared I–O curves of Fmr1 KO mice directly to their age-matched WT littermates to more closely examine differences in excitability at each age. Fmr1 KO mice exhibited dynamic differences in L4-L2/3 excitability compared with WT mice across postnatal auditory cortex development. At P9–10, differences in the gain of I–O curves, rather than threshold excitability, resulted in significantly different I–O curves between the genotypes (comparison of fits: F(4,274) = 4.913, P = 0.0008), where KO mice were slower to reach maximal amplitudes compared with WT mice (Fig. 1E). The major differences in excitability between WT and KO mice occurred at P12–13 and P21–25, where Fmr1 KO were significantly more excitable at P12–13, the start of the auditory critical period, in their L4-L2/3 connectivity relative to WT mice (F(4,442) = 11.44, P < 0.0001) (Fig. 1F). In contrast, by P21–25, Fmr1 KO were significantly less excitable in their basal synaptic transmission compared with WT (F(4,329) = 15.15, P < 0.0001) (Fig. 1H). No differences in excitability were observed between genotypes at either P15–16 or P40–45.
We also compared fEPSP PPRs in the L4-L2/3 auditory synaptic circuit to evaluate possible presynaptic alterations. Consistent with previous cortical reports, we broadly observed short-term depression (PPR < 1) across most ages. Differences in PPR are observed between genotypes at P12–13 (Supplementary Fig. 1B) and P40–45 (Supplementary Fig. 1F). PPR values tend to be more depressed in KO mice, particularly at the interstimulus intervals of 500, 50, and 25 ms. These results suggest possible alterations in presynaptic-related transmitter release mechanisms within the L4-L2/3 auditory cortex circuit in Fmr1 KO mice.
Fmr1 KO Mice Have an Enhanced Age-Dependent Sensitivity to PTX
Given that GABAARs affect circuit excitability and are crucial regulators of critical periods, we assessed whether altered GABAergic signaling contributes to the excitability differences in L4-L2/3 basal synaptic transmission. To determine the GABAAR signaling component regulating the I–O curve fEPSPs, we analyzed the shifts of the I–O curves to a subthreshold concentration (5 μM) of the GABAAR antagonist PTX that did not cause epileptiform discharges (Jang et al. 2009; Stebbings et al. 2016). We thus examined the evolution and strength of inhibition over auditory cortex development and identified potential differences in pharmacological sensitivity within the L4-L2/3 auditory cortex circuit in WT and Fmr1 KO mice.
Depending on whether GABAAR-mediated chloride anion currents are outward (depolarizing) or inward (hyperpolarizing) during development, the application of GABAAR antagonists can respectively decrease or increase neuronal activity and synaptic responses (Scheyer et al. 2020). In the auditory cortex of WT mice, subthreshold PTX concentrations did not alter the I–O curves at either P9–10 or P12–13 (Fig. 2A,B). However, at P15–16 in WT mice, PTX exposure resulted in a significant decrease in fEPSP amplitudes, manifested by suppressed I–O curves compared with the ACSF baseline (F(4,163) = 3.803, P = 0.0055) (Fig. 2C). Our results in WT mice are suggestive of the antagonism of depolarizing GABA that is prevalent early in brain development (Ben-Ari 2002) and embedded in the fEPSPs. At P21–25, PTX application did not alter I–O curves for WT mice (F(4,161) = 0.7609, P = 0.5522) (Fig. 2D), but at P40–45, PTX increased the fEPSP amplitudes of the I–O curves, consistent with the blocking of hyperpolarizing GABA present in the fEPSPs (F(4,231) = 5.088, P = 0.0006) (Fig. 2E).
Figure 2 .
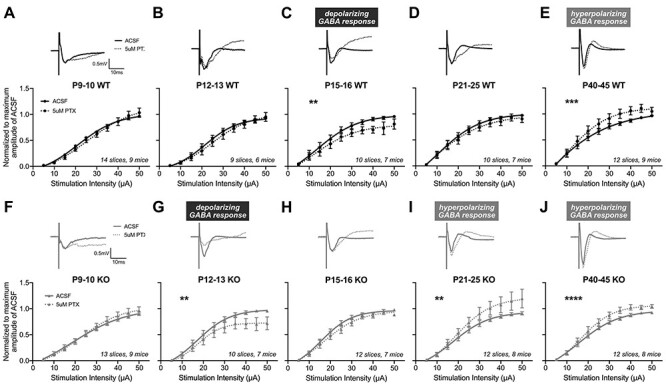
The auditory cortex of Fmr1 KO mice exhibits an accelerated age-dependent shift in their sensitivity to PTX. I–O curves from MEA field recordings were analyzed following 5 μM PTX. (A–E) Top: representative traces from Fmr1 WT mice at 40 μA stimulation. Bottom: I–O curves before and after PTX, normalized to the maximum amplitude in ACSF and fit with sigmoidal curves. (C) P15–16 Fmr1 WT mice exhibit decreased fEPSP amplitudes following PTX, suggestive of the presence and subsequent blockade of depolarizing GABA by PTX. (E) Fmr1 WT exhibit significantly increased fEPSP amplitudes to PTX at P40–45, indicating PTX’s blockade of inhibitory hyperpolarizing GABA. (F–J) Representative traces and I–O curves from Fmr1 KO mice. (G) Fmr1 KO mice exhibit significantly decreased responses to PTX at P12–13 but not at (H) P15–16. (I–J) At both P21–25 and P40–45, Fmr1 KO mice exhibit significantly increased amplitudes to PTX. The number of slices and mice analyzed for each age and genotype are indicated in each graph. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Similar effects of GABAAR antagonism by PTX were observed across development in Fmr1 KO mice, where I–O curve responses correlated with the blockade of depolarizing GABA to progressively more hyperpolarizing GABA with postnatal age, but with notable age-specific differences in sensitivity compared with WT mice. Unlike the lack of change in WT mice at P12–13, Fmr1 KO mice exhibited diminished excitability in response to PTX with significantly suppressed I–O curves compared with ACSF baseline (F(4,164) = 4.162, P = 0.0031) (Fig. 2G). This is coincident with the increased L4-L2/3 excitability in KO mice (Fig. 1F) and suggests that depolarizing GABA may contribute to this enhanced excitability. We further confirmed Fmr1 KO’s enhanced sensitivity to GABAAR antagonists at this age by also evaluating responses to bicuculline (5 μM) (Supplementary Fig. 2B). We found consistent effects to PTX where bicuculline significantly suppressed I–O curves in KO mice, suggestive of a more sensitive blockade of depolarizing GABA within fEPSPs compared with WT. By P15–16, PTX did not alter the I–O curve in either direction (F(4,198) = 1.603, P = 0.175) (Fig. 2H), at which time WT mice showed PTX-induced suppression of I–O curves. Furthermore, Fmr1 KO mice displayed PTX-induced hyperexcitability starting at P21–25, where the I–O curves shifted to significantly increased fEPSP responses to PTX (P21–25: F(4,220) = 4.771, P = 0.001; P40–45: F(4,240) = 7.039, P < 0.0001) (Fig. 2I–J), suggesting the presence of hyperpolarizing GABA in the circuit at an earlier age than WT mice.
Notably, the ages at which Fmr1 KO mice exhibited significantly different PTX-induced I–O curves compared with WT are consistent with the ages we observed the overall excitability differences between genotypes (Fig. 1), suggesting that altered GABAergic signaling may mediate Fmr1 KO P12–13 hyperexcitability and P21–25 decreased excitability relative to WT. Additionally, when examining the pattern of PTX-induced I–O curve shifts across development, we found that the blockade of apparent depolarizing and hyperpolarizing GABAAR signaling within the fEPSPs occurred at earlier stages in Fmr1 KO mice compared with WT (i.e., P12 KO looks like P15 WT; P21 KO looks like P40 WT). Consistent with the faster developmental progression of L4-L2/3 basal synaptic transmission, Fmr1 KO mice also undergo an overall pattern of accelerated age-dependent pharmacological sensitivity to a GABAAR antagonist at the L4-L2/3 circuit within the auditory cortex.
Fmr1 KO Mice Exhibit Altered Regulation of GABAAR α-Subunit Expression during Auditory Cortex Maturation
We next examined the developmental regulation of GABAAR subunits in the auditory cortex of Fmr1 WT and KO mice, as they influence GABAergic maturation and network excitability (Marowsky et al. 2012). We performed western blot analysis to compare the protein expression of membrane-bound GABAAR α1 and α3 subunits given that their developmental regulation determines GABA sensitivity and the kinetics of chloride ion conductance (Laurie et al. 1992; Okada et al. 2000; Bosman et al. 2005). Additionally, the α1 and α3 subunits influence critical period plasticity (Fagiolini et al. 2004), and often exhibit altered expression in neurodevelopmental disorders and epilepsy (Rakhade and Jensen 2009; Brooks-Kayal 2011). We normalized each of the developmental ages to P56 WT levels to evaluate the dynamic developmental expression pattern of these subunits during the postnatal maturation of the auditory cortex and found a significant effect of age across all subunits (2-way ANOVA, age, P < 0.0001) (Fig. 3A–C). Consistent with Laurie et al. (1992), the most prominent developmental changes in α-subunits were observed around ear canal opening. Notably, during WT auditory cortex development, GABAAα3 peaked in expression at P12, at the start of the WT auditory critical period soon after ear canal opening (Barkat et al. 2011; Hackett et al. 2015), and subsequently rapidly declined (Fig. 3A). In contrast, GABAAα1 expression was relatively low early in postnatal development in WT mice and increased most rapidly between P9 and P12, peaking at P15, the end of the auditory critical period, before reaching adult-like levels (Fig. 3B). The ratio of GABAAR α1:α3, indicative of GABAAR maturity, developmentally increases past the fourth postnatal week in WT mice to reach adult levels (Fig. 3C).
Figure 3 .
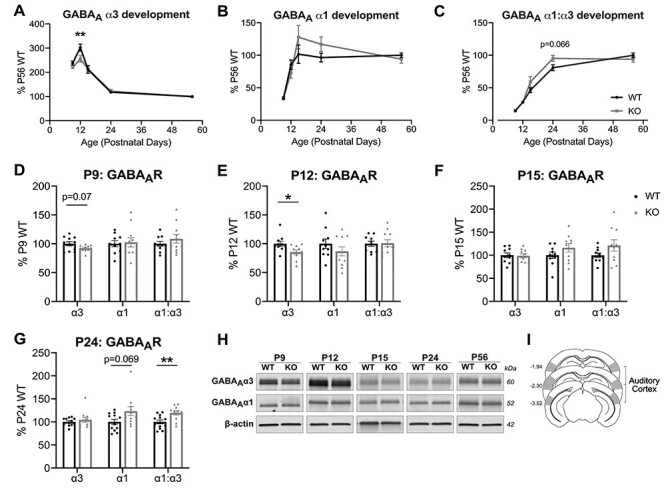
Fmr1 KO mice exhibit altered developmental regulation in the expression of GABAAR α1 and α3 subunits in the auditory cortex. (A–C) Western blot analysis of membrane-bound expression of GABAAR α3, GABAAR α1, and GABAAR α1:α3, with all timepoints normalized to P56 WT to analyze the developmental expression of the GABAAR α-subunits in the auditory cortex. (A) Developmental expression of GABAAR α3 exhibits an overall interaction between age and genotype, with significantly decreased expression for KO compared with WT at P12. (C) Developmental GABAAR α1:α3 expression exhibits an overall interaction between age and genotype. (D–G) GABAAR α-subunits expressed relative to age-matched WT littermates. (D) Fmr1 KO exhibit a trending decrease in GABAAR α3 expression at P9 (n = 11 WT, 11 KO). (E) GABAAR α3 is significantly decreased at P12 (n = 10, 11). (F) No differences in expression observed at P15 (n = 10, 10). (G) P24 Fmr1 KO exhibit a trending increase in GABAAR α1, with significantly increased GABAAR α1:α3 expression (n = 12, 11). No differences were observed at P56 (n = 18, 15), data not shown. (H) Representative western blots of GABAAR α3 and GABAAR α1 with β-actin used as loading controls. (I) Schematic of approximate auditory cortex dissections as described in Materials and Methods. *P < 0.05, **P < 0.01.
Fmr1 KO mice exhibit altered α-subunit developmental patterns in the auditory cortex. GABAAα3 expression was significantly different from WT development (2-way ANOVA, interaction: F(4,109) = 2.897, P = 0.025; genotype: F(1,109) = 4.801, P = 0.031), where α3 does not peak as high in the KO auditory cortex (Sidak’s multiple comparisons: P12 WT 302.11 ± 14.18% vs. KO 255.48 ± 13.25%, P = 0.002) (Fig. 3A). Although α1 development did not have statistically significant differences between genotypes (Fig. 3B), the GABAAR α1:α3 ratio during development was significantly different (2-way ANOVA, interaction: F(4,108) = 2.601, P = 0.04; genotype: F(1,108) = 2.899, P = 0.092), where Fmr1 KO mice had faster increases to adult WT levels at earlier stages in development (Sidak’s multiple comparisons: P24 WT 80.75 ± 4.53% vs. KO 95.25 ± 4.68%, P = 0.066) (Fig. 3C).
Direct comparisons of Fmr1 KO mice to their age-matched WT littermates showed significantly reduced GABAAα3 in Fmr1 KO mice at P12 (WT: 100 ± 4.48%, KO: 85.66 ± 3.62%, P = 0.021) (Fig. 3E). Changes in α3-subunit expression may manifest even earlier, where trending decreases were observed at P9, days before the ear canal is open (WT: 100 ± 3.58%, KO: 92.47 ± 1.63%, P = 0.069) (Fig. 3D). No differences in GABAAα3 were observed at later ages. However, we observed significantly increased GABAAR α1:α3 ratios in Fmr1 KO mice at P24 (WT: 100 ± 4.33%, KO: 119.7 ± 4.7%, P = 0.006), along with a trending increase in membrane-bound α1 (WT: 100 ± 5.44%, KO: 123.3 ± 9.85%, P = 0.069) (Fig. 3G). Neither the GABAAα1 level nor the ratio of GABAAR α1:α3 subunits were different at the early ages. These collective differences in the α3 and α1 expression suggest a dynamic alteration in GABAAR developmental expression in the auditory cortex of Fmr1 KO mice during the critical period of development.
Reduced Fraction of GABAARα3 Staining Puncta in the Auditory Cortex of P12 Fmr1 KO Mice
Given the importance of P12 as the start of the normal WT auditory critical period, we wanted to determine whether the decreased expression of membrane-bound GABAARα3 levels we observed by western blot was reflective of synaptic expression across the auditory cortex, or was specific to a particular cortical layer. We performed immunohistochemistry using an extracellular-targeting GABAARα3 antibody to examine colocalization with the inhibitory presynaptic marker VGAT (Fig. 4A). We quantified the fluorescence intensities of GABAARα3 and VGAT immunoreactivity, and calculated the fraction of VGAT puncta that colocalized with surface GABAARα3 using Mander’s coefficient (Manders et al. 1993; Gao and Heldt 2016; Yennawar et al. 2019). Compared with WT mice, P12 Fmr1 KO mice had significantly reduced fluorescence intensity of GABAARα3 across all cortical layers of the auditory cortex (2-way ANOVA, genotype, F(1,56) = 7.516, P = 0.0082) (Fig. 4B), indicating reduced expression consistent with the differences observed by western blot. Meanwhile, WT and KO mice had similar levels of VGAT immunoreactivity (2-way ANOVA, genotype, F(1,56) = 0.0066, P = 0.9357) (Fig. 4C). Thus, when examining the fraction of VGAT+ puncta colocalizing with GABAARα3, Fmr1 KO mice had significantly reduced levels in the auditory cortex (2-way ANOVA, genotype, F(1,56) = 8.005, P = 0.0065) (Fig. 4D). Together, with the western blot data, our expression data show that Fmr1 KO mice have decreased synaptic α3-subunit-containing GABAARs across all layers of the auditory cortex at P12.
Figure 4 .
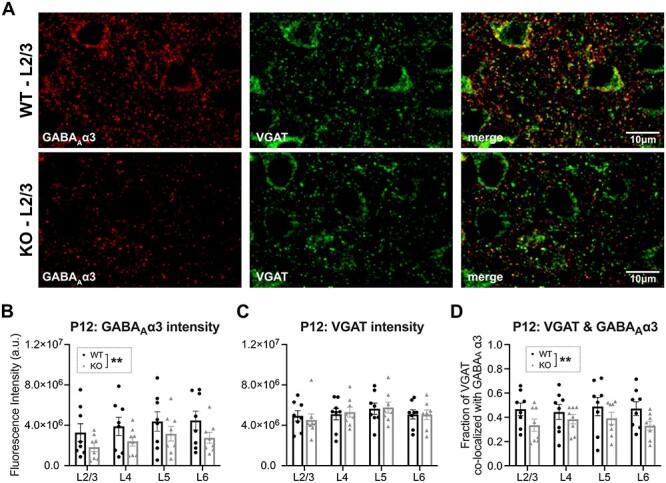
Decreased colocalization of GABAAR α3 and VGAT in auditory cortex of P12 Fmr1 KO mice. (A) Representative confocal micrographs of immunohistochemistry of extracellular targeting GABAAR α3 and presynaptic VGAT in L2/3. (B–D) Quantification of results using Coloc2 plugin from FIJI (n = 8 WT, 8 KO). Total fluorescence intensity of (B) GABAAR α3 and (C) VGAT immunoreactivity. (D) Mander’s coefficient to calculate the fraction of VGAT immunoreactivity that colocalizes with GABAAR α3. Two-way ANOVA indicates a significant genotype effect in (B) and (D); however, there were no significant differences in any particular cortical layer as analyzed by Sidak’s multiple comparisons test. No differences were found in VGAT intensity. **P < 0.01.
P12 Fmr1 KO Mice Exhibit Impaired Inhibitory Presynaptic Regulation of L2/3 Pyramidal Neurons
Given the altered expression of GABAAR α-subunits and regulation of L4-L2/3 basal synaptic transmission in the Fmr1 KO auditory cortex, we next asked whether the development of inhibitory synaptic function in the auditory cortex was dysregulated specifically in L2/3 pyramidal neurons of Fmr1 KO mice. The L2/3 pyramidal neurons are crucial in both processing tonotopic information from L4 and integrating input from other brain regions before sending their own projections to deeper layers and other cortical regions (Guo et al. 2012; Meng et al. 2020). We performed whole-cell patch-clamp recordings from ex vivo slices across development, measuring sIPSC properties to detect any overall abnormalities to inhibitory synaptic function and neuronal excitability (Fig. 5; Supplementary Table 1). Consistent with the overall progression of sIPSC properties during development in the rodent cortex (Bosman et al. 2005; Takesian et al. 2012), WT mice exhibited a clear age-dependent increase in the frequency of sIPSCs and a decrease in the weighted decay τ, indicative of faster decay kinetics (Fig. 5C,D). The sIPSC amplitudes were unchanged across WT development (Fig. 5B). Fmr1 KO mice exhibited similar developmental patterns of sIPSC amplitudes and decay τ measures. However, Fmr1 KO mice did exhibit a trending increase of sIPSC frequency at P12–13 (P = 0.063), soon after the ear canal opens (Fig. 5C; Supplementary Table 1), suggestive of developmental alterations of sIPSC maturation in Fmr1 KO mice.
Figure 5 .
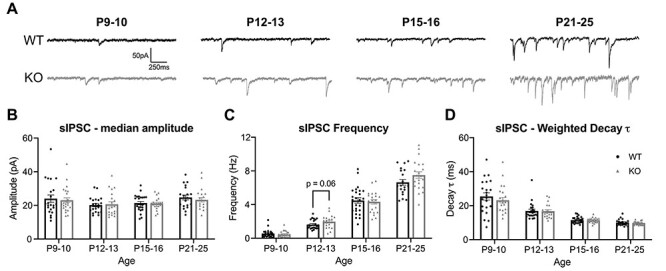
Increasing trend for sIPSC frequency in P12–13 Fmr1 KO mice. (A) Representative traces of sIPSCs recorded across development from L2/3 pyramidal neurons in the auditory cortex. (B) Amplitudes of sIPSCs were unaltered in Fmr1 KO mice at any age. (C) P12 KO mice exhibit a trend toward increased sIPSC frequency. (D) No differences in the weighted decay τ were observed between WT and KO mice at any age. Developmental maturation of inhibitory transmission is observed with decreasing weighted decay τ times across ages. All values are summarized in Supplementary Table 1.
Given that spontaneous action potential firing is not blocked during sIPSC recording, we further characterized the possible GABA-mediated dysregulation in P12 Fmr1 KO mice by examining L2/3 pyramidal neuron mIPSCs and PPR of eIPSCs to identify contributors to the elevated trend in sIPSC frequency (Supplementary Fig. 3). The mIPSC amplitudes and weighted decay τ were similar between Fmr1 WT and KO (Supplementary Fig. 3B–C). Unlike the trending increase of sIPSC frequency in Fmr1 KO mice, there was no difference in mIPSC frequency (Supplementary Fig. 3D), which suggests that presynaptic firing alterations or network excitability driven action potentials likely contribute to the trending sIPSC frequency difference.
We additionally sought to evaluate whether there exist L4 inhibitory presynaptic deficits. We therefore compared the PPRs of eIPSCs in L2/3 pyramidal neurons from stimulation evoked in L4 with a range of interstimulus intervals (Supplementary Fig. 3E,F). Both P12–13 WT and KO mice generally exhibit paired-pulse depression (PPR < 1), but Fmr1 KO mice had significantly more depressed responses compared with WT littermates. Notably, the intervals at which more depressed responses were observed in Fmr1 KO mice were at 400, 50, and 20 ms, similar to the intervals where more enhanced paired-pulse depression were observed in fEPSP paired-pulse measures (Supplementary Fig. 1). Our findings suggest that the L4-L2/3 circuit maturation in Fmr1 KO mice is further impacted by impairments in L4 inhibitory presynaptic mechanisms.
Fmr1 KO Mice Exhibit Enhanced Sensitivity to GABAAR α1-Specific Agonist
The decay kinetics of IPSCs are highly dependent on the subunit composition of GABAARs and may determine the tonic inhibition of cortical neurons (Marowsky et al. 2012). The α3-subunit that is highly expressed at birth mediates relatively long-lasting currents, whereas the postnatally upregulated α1-subunit mediates faster, mature IPSCs (Bosman et al. 2005; Pangratz-Fuehrer et al. 2016). In addition to measuring the weighted decay τ of sIPSCs to functionally assess the relative contribution of α1 and α3 subunits (Fig. 5D), we also examined pharmacological sensitivity using subunit-specific agonists.
In a subset of the L2/3 pyramidal neurons from Fig. 5, we analyzed changes in both sIPSC amplitude and weighted decay τ in response to ZOLP, a positive modulator of α1-containing GABAARs (Sanna et al. 2002; Baur and Sigel 2007; Xu et al. 2010) (Fig. 6; Supplementary Table 2). In WT mice, we observed age-dependent increases in ZOLP sensitivity that correspond with pre- and postcritical period. At both P9–10 and P12–13, when α1 expression is relatively low, ZOLP did not elicit any significant changes to sIPSC amplitude in WT mice, but did significantly increase the decay kinetics (Fig. 6C,D), indicating that there was some functional α1 activity even with low expression. Starting at P15, ZOLP was capable of significantly increasing both sIPSC amplitudes and decay τ in WT neurons with greater consistency compared with the younger ages. Fmr1 KO mice also exhibited increasing age-dependent sensitivity to ZOLP. However, at P12–13, Fmr1 KO pyramidal neurons displayed enhanced pharmacological sensitivity to the α1-agonist, where ZOLP significantly increased sIPSC amplitude, unlike WT neurons, and additionally increased decay τ with slightly more enhanced sensitivity (WT: P = 0.0133; KO: P = 0.0074). Although P21–25 KO neurons failed to show a significant amplitude enhancement, neurons were still very responsive to ZOLP with relatively similar increases to decay τ like WT neurons.
Figure 6 .
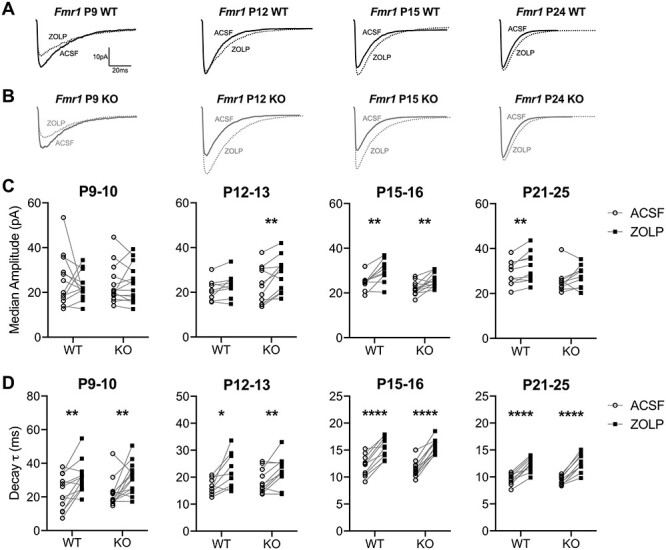
Fmr1 P12 KO mice exhibit enhanced sensitivity to ZOLP. (A,B) Representative traces of averaged sIPSCs from L2/3 pyramidal neurons before and after 100 nM ZOLP. (C) The effects of ZOLP on sIPSC amplitude across ages. No effects observed at P9–10, but significantly increased amplitude following ZOLP observed in Fmr1 KO mice starting at P12–13. (D) The weighted decay τ of averaged sIPSCs significantly increases with ZOLP across all ages for both WT and KO mice. The average values are summarized in Supplementary Table 2. *P < 0.05, **P < 0.01, ****P < 0.0001.
To elucidate whether the enhanced ZOLP sensitivity in P12–13 KO mice is mediated by pre- or postsynaptic mechanisms, we evaluated eIPSCs of L2/3 pyramidal neurons following L4 stimulation. Consistent with the sIPSC studies, ZOLP significantly increased eIPSC amplitudes in Fmr1 KO neurons but not in WT slices (Supplementary Fig. 4B). Overall, our whole-cell patch-clamp data suggest that although the maturation of sIPSC properties in L2/3 pyramidal neurons of the auditory cortex in Fmr1 KO mice was largely similar to that of WT development, Fmr1 KO pyramidal neurons have an enhanced sensitivity to ZOLP and presynaptic alterations at P12, soon after ear canal opening and the start of the WT critical period.
Altered Developmental Changes in Voltage-Dependent eIPSC Conductance in Fmr1 KO Mice
Fmr1 KO mice have a delayed developmental switch in GABAAR polarity in the somatosensory cortex (He et al. 2014), and our MEA studies indicate an enhanced age-dependent fEPSP response to PTX. Therefore, we wanted to further evaluate whether the Fmr1 KO auditory cortex exhibits altered EGABA maturation. We performed gramicidin-perforated patch recordings from L2/3 pyramidal neurons to measure EGABA across development (Fig. 7). Consistent with other rodent developmental GABA maturation studies in cortical neurons (Rheims et al. 2008), the auditory cortex of WT mice also exhibits an age-dependent transition of EGABA from relatively depolarizing to hyperpolarizing values (P9–10: −53.68 ± 1.39 mV; P12–13: −52.58 ± 2.40 mV; P15–16: −66.04 ± 3.09 mV; P21–25: −75.07 ± 3.4914 mV) (Fig. 7C). Notably, the largest EGABA change occurs between P12 and P15, which is between ear canal opening with the start of the auditory critical period and its closure (Tukey’s multiple comparisons: P12–13 vs. P15–16, P = 0.006; P15–16 vs. P21–25, P = 0.1774). Fmr1 KO mice also exhibit similar maturational changes in the auditory cortex, with no significant differences between WT and KO at any age (P9–10: −49.85 ± 2.47 mV; P12–13: −56.29 ± 1.68 mV; P15–16: −71.64 ± 1.46 mV; P21–25: −79.85 ± 2.64 mV) (Fig. 7C). KO mice also had the largest EGABA change between P12 and P15 (Tukey’s: P12–13 vs. P15–16, P = 0.0004; P15–16 vs. P21–25, P = 0.2965). Interestingly, when we excluded the P9–10 group to specifically examine EGABA for ages after ear canal opening, when the auditory circuit more robustly modulates auditory input, there was a significant difference between Fmr1 KO and WT mice, where KO mice tended to have relatively more hyperpolarized EGABA values (2-way ANOVA, genotype effect, F(1,83) = 5.0, P = 0.0276). These data suggest that Fmr1 KO mice exhibit relatively stronger hyperpolarizing GABAAR-mediated inhibition after the critical period onset, which may contribute to altered E–I circuit in the auditory cortex.
Figure 7 .
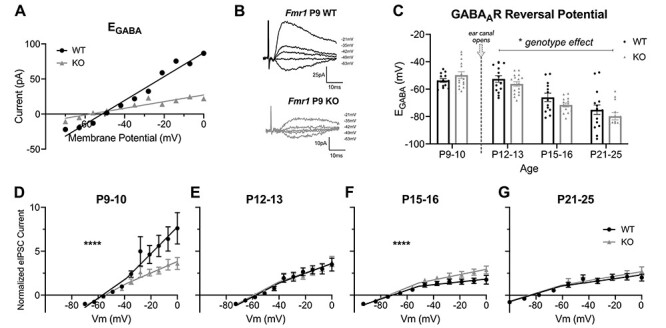
Fmr1 KO mice exhibit altered voltage-dependent eIPSC conductance in L2/3 pyramidal neurons during auditory cortex development. (A,B) Representative example of (A) GABAAR I-V plot and (B) traces of eIPSC gramicidin-perforated patch recordings from P9 WT and KO mice. EGABA is estimated from linear fit from I-V plot. (C) Group data of EGABA from I-V plots across all Fmr1 WT and KO mice and ages. (D–G) Averaged GABAAR I-V plots across ages for WT and KO mice to evaluate developmental regulation of GABAAR responses. Currents for each cell were normalized to the eIPSC peak amplitude of the first holding potential. P9–10: n = 13 cells/5 WT mice and 15 cells/6 KO mice. P12–13: n = 15 cells/5 WT mice and 17 cells/6 KO mice. P15–16: 13 cells/5 WT mice and 14 cells/6 KO mice. P21–25: n = 16 cells/6 WT mice and 14 cells/5 KO mice. *P < 0.05, ****P < 0.0001.
To identify potential contributors to our EGABA genotype differences, the expression of chloride transporters was evaluated in auditory cortex lysates across development. Although we observed a significant developmental expression pattern of membrane-bound NKCC1:KCC2 ratio consistent with general brain maturation (Supplementary Fig. 5A) (Ben-Ari 2002; Dzhala et al. 2005), there were no differences at any age in either NKCC1 or KCC2 expression in Fmr1 KO mice compared with WT littermates (Supplementary Fig. 5C–E). However, we did not perform a more detailed analysis of chloride transporter expression specific to either cortical layers or neuronal subtypes, which could reveal differential expression.
The I-V plots for EGABA evaluations were further normalized and averaged for each of the age groups to assess altered patterns of the relative eIPSC magnitudes in Fmr1 KO compared with WT mice across development (Fig. 7D–G). The I-V plots for WT mice indicated a clear and statistically significant developmental progression in the rectification pattern of the I-V curves. In P9–10 WT mice, there was a strong outward rectification of the GABAAR current beginning around −40 mV (Fig. 7D), consistent with the voltage-dependent activation and modulation of GABAARs (Pavlov et al. 2009; O’Toole and Jenkins 2012). At P12–13, the outward rectification was significantly diminished at the depolarized holding potentials (Fig. 7E), which transitioned to an inward rectification at the later P15–16 and P21–25 ages in WT mice (Fig. 7F,G). In contrast, Fmr1 KO P9–10 mice did not have a strong outward rectification like the age-matched WT mice (segmented linear regression, F(3,297) = 14.70, P < 0.0001). Rather, the I-V curve is already inwardly rectifying and similar to that of P12–13 WT mice. P15–16 KO mice were not as strongly inwardly rectifying compared with WT (F(3,283) = 11.96, P < 0.0001), whereas P21–25 KO mice had similar inward rectification patterns as WT mice (F(3,312) = 0.9949, P = 0.3955). The perforated-patch clamp experiments collectively suggest that auditory cortex L2/3 pyramidal neurons of Fmr1 KO mice undergo dysregulated developmental transitions in their rectification of evoked GABAAR currents prior to ear canal opening and have a tendency toward more hyperpolarized GABAAR reversal potentials during postnatal development compared with WT mice.
Fmr1 KO Mice Have an Altered Progression of Synaptic Plasticity at L4-L2/3 Pyramidal Neuron Synapses in the Auditory Cortex
The experience-dependent plasticity characteristic of critical periods is linked to lasting forms of synaptic plasticity, such as LTP (Crair and Malenka 1995; Kirkwood et al. 1995; Citri and Malenka 2008). Postnatal GABAergic maturation is crucial in regulating the onset and closure of sensory critical periods (Hensch 2005), with age-dependent increases in inhibition known to restrict LTP (Jang et al. 2009; Harauzov et al. 2010). Given the various dysregulations we identified related to synaptic GABAergic maturation of L2/3 pyramidal neurons, we sought to evaluate whether Fmr1 KO mice also exhibited alterations in long-term synaptic plasticity that could contribute to the impaired auditory cortex critical period previously reported in Fmr1 KO mice (Kim et al. 2013). We analyzed LTP of L2/3 pyramidal neurons from L4 synaptic input, evaluating ePSCs in the absence of pharmacological blockers to measure the L2/3 pyramidal neuron’s net ability to potentiate, as it mediates all E–I synaptic input. This net ability is physiologically relevant given that E–I levels dynamically change across development to regulate critical period plasticity, and that cortical inhibition modulates frequency selectivity of excitatory neurons (Froemke and Jones 2011; Blackwell and Geffen 2017).
In WT mice, L2/3 pyramidal neurons of the auditory cortex exhibited age-dependent LTP that is synchronized with the sequence of postnatal maturation of the auditory circuit, specifically ear canal opening and the start of the auditory tonotopic critical period (Barkat et al. 2011). Notably, LTP was not induced in L2/3 pyramidal neurons at P9–10 when the ear canal is still closed. A potentiated response was initially observed soon after pairing at P9–10, but the ePSC response subsequently decreased, often resulting in a depressed ePSC amplitude (15 min postpairing: 84.40 ± 11.87% relative to baseline) (Fig. 8A). Starting at P12, soon after the ear canal opens and the start of the auditory cortex critical period, LTP was induced in the L4-L2/3 synaptic circuit (128.29 ± 5.67%) (Fig. 8B). LTP was still capable of being induced at the later ages, but there was a gradual decline in magnitude with age (P15–16: 116.66 ± 4.36%; P21–26: 110.38 ± 5.89%). The age-dependent regulation of synaptic plasticity in WT mice was also observed with changes in the LTP decay slopes and spread of amplitude responses (Fig. 8E). P9–10 mice had a sharp decline in decay (linear regression, slope: −1.43 ± 0.19) with high variability in ePSC response. The decay slope relatively attenuated at P12 (−0.39 ± 0.08), and subsequently increased with age (P15–16: −0.47 ± 0.05; P21–25: −0.79 ± 0.07), with notable decreases in variability across maturation. These results indicate that in the auditory cortex, the synaptic plasticity of L4 inputs to L2/3 pyramidal neurons is timed to maximize LTP immediately after ear canal opening, which subsequently decreases in its capacity with age.
Figure 8 .
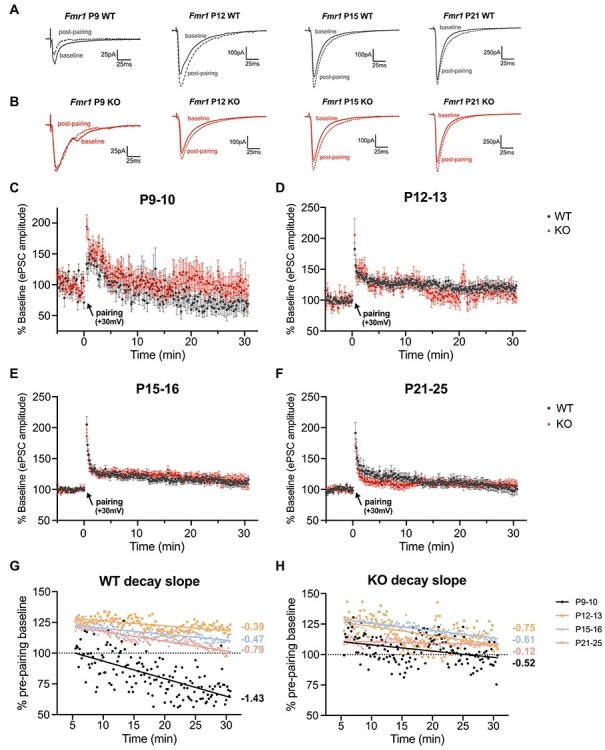
Fmr1 KO mice exhibit an altered progression of L4-L2/3 synaptic plasticity during auditory cortex maturation. (A–D) Top: Representative ePSC traces from L2/3 pyramidal neurons with averaged recordings from baseline (prepairing) and averaged recordings from 15 min postpairing from the auditory cortex of WT and Fmr1 KO mice across development. Bottom: Time course of normalized ePSC amplitude changes in LTP experiments. (A) P9–10 mice fail to exhibit LTP in the L4-L2/3 circuit. (B–D) LTP is capable of being induced from P12–25. (E,F) Overlay of the averaged decay data (6–30 min postpairing) by genotype. In WT mice, LTP does not occur until P12, whereby enhancement is attenuated with age (decay slopes increase). Fmr1 KO mice exhibit an altered pattern of LTP synaptic plasticity with increased variability. P9–10: n = 9 cells/7 WT mice and 10 cells/7 KO mice. P12–13: n = 12 cells/7 WT mice and 11 cells/7 KO mice. P15–16: n = 11 cells/8 WT mice and 10 cells/6 KO mice. P21–25: n = 10 cells/7 WT mice and 9 cells/7 KO mice.
We further compared the developmental regulation of L4-L2/3 synaptic plasticity in Fmr1 KO mice. Similar to P9–10 WT mice, Fmr1 KO mice were unable to readily induce enduring LTP, with no overall genotype difference when comparing the entire 30 min postpairing period (2-way repeated-measures ANOVA). However, L2/3 pyramidal neurons in P9–10 KO mice exhibited significantly increased posttetanic potentiation immediately following pairing (KO: 195 ± 18.69% vs. WT: 133.9 ± 16.52%, unpaired t-test, P = 0.0267) and tended to have attenuated depression of eEPSC responses following pairing (15 min postpairing: 93.44 ± 12.50% relative to baseline) (Fig. 8A). P12 Fmr1 KO mice were able to express LTP but exhibited greater variability in their ePSC responses and their ability to sustain potentiation (15 min postpairing: 109.35 ± 8.59) compared with their age-matched WT littermates (Fig. 8B). The significant variability of ePSCs was also observed prepairing in the average baseline ePSC responses, with P12 KO mice exhibiting significantly increased variance in the raw amplitudes compared with WT mice (Supplementary Fig. 6). Similar to WT mice, LTP was more reliably maintained at older ages in KO mice with reduced variability (P15–16: 123.16 ± 4.23%; P21–25: 109.50 ± 4.32%).
Compared with the developmental progression of decay slopes of WT mice, KO mice exhibited a significantly altered postmaturational sequence, in which, P9–10 KO mice did not have the steep decay slope seen in WT mice (linear regression, −0.52 ± 0.17, P = 0.0004) (Fig. 8F). Fmr1 KO mice had a stronger decay at P12–13 compared with WT littermates (−0.75 ± 0.12, P = 0.0097) and do not exhibit the age-dependent increase in decay slope values (P15–16: −0.61 ± 0.07, P = 0.113; P21–25: −0.12 ± 0.06, P < 0.0001). Overall, we found that the stabilization of L2/3 pyramidal neuron synaptic plasticity is developmentally regulated and that Fmr1 KO mice have an altered regulation during the early postnatal maturation of the auditory cortex.
Discussion
FXS patients exhibit communication and auditory processing deficits suggestive of altered auditory cortex development and function. Although Fmr1 KO mice have impaired auditory critical period plasticity (Kim et al. 2013), mechanisms underlying such phenotypes are not entirely known. Here, we characterized auditory intracortical maturation in WT and Fmr1 KO mice, identifying Fmr1 KO changes in auditory intracortical circuit excitability accompanied by GABAAR subunit alterations, inhibitory synaptic maturation dysregulation, and pharmacological sensitivity differences. We also identified alterations in the developmental patterning of L4-L2/3 LTP, which occurs early in Fmr1 KO development. Our studies highlight altered synaptic GABAergic maturation in the developing Fmr1 KO auditory cortex (Fig. 9), which contribute to impaired E–I balance and synaptic plasticity maturation that could manifest as auditory-related phenotypes in Fmr1 KO mice.
Figure 9 .
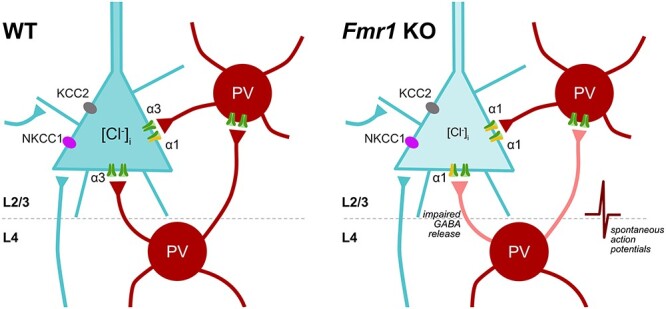
Summary of altered GABAergic inhibitory maturation in the auditory cortex of P12 Fmr1 KO mice. (Left) The depiction of postsynaptic and presynaptic features of GABA-mediated inhibition in a L2/3 pyramidal neuron of WT mice. (Right) Fmr1 KO mice exhibit dynamic alterations in GABAergic maturation of L2/3 pyramidal neurons during auditory cortex development. Postsynaptic GABA-mediated differences include altered GABAAR α-subunit regulation that increases pharmacological sensitivity to ZOLP, and more hyperpolarized EGABA values across development; however, no differences were observed in NKCC1 and KCC2 expression from total auditory cortex lysates. Fmr1 KO mice at P12–13 also exhibit presynaptic differences with increased spontaneous network activity and impaired L4 presynaptic GABA-release mechanisms. Such GABAergic maturational alterations disrupt the normal patterning of L4-L2/3 E–I circuit and synaptic plasticity development in the auditory cortex.
We evaluated the L4-L2/3 auditory cortex circuit, given its role in tonotopic plasticity and cortical auditory processing (Barkat et al. 2011; Li et al. 2014; Javitt and Sweet 2015) and characterized a progression of excitability in WT mice consistent with the general excitability maturation patterns in the developing brain (Rakhade and Jensen 2009; Meng et al. 2020) and E–I frequency tuning in the rat auditory cortex. Specifically, excitatory response to tonal frequency tuning matures to adult levels by P15, whereas inhibitory response tuning develops around P25–30 (Dorrn et al. 2010). In WT mice, the most significant increase in I–O curve excitability occurred at the end of the critical period (P15–16), while excitability relatively decreased at P40–45, consistent with delayed inhibitory maturation. However, relative to WT development, Fmr1 KOs exhibited dynamic L4-L2/3 excitability differences, attributable in part to alterations in GABAergic modulation of excitatory field potentials, namely with the enhanced depolarizing to hyperpolarizing GABA signaling transition identified by our PTX studies.
We analyzed GABA-mediated maturation of L2/3 pyramidal neurons given that their inhibitory regulation is crucial in tonotopic frequency processing from L4 and input integration from other brain regions (Guo et al. 2012; Li et al. 2014; Meng et al. 2020). Notably, we observed Fmr1 KO differences at P12, soon after ear canal opening and the start of the WT critical period. Fmr1 KOs have increased spontaneous network activity, suggested by the trending increase in sIPSC frequency without mIPSC changes. We additionally identified defects consistent with the impaired inhibitory control of barrel cortex L2/3 neurons in Fmr1 KOs (Paluszkiewicz et al. 2011). P12 Fmr1 KO mice have enhanced eIPSC paired-pulse depression, but no differences in VGAT immunoreactivity, suggestive of presynaptic deficits related to GABA vesicle release or calcium influx that could impair inhibitory regulation of pyramidal neurons (Wilcox and Dichter 1994; Cea-Del Rio and Huntsman 2014). Given that fast-spiking inhibitory interneurons distinctly display short-term depression to repetitive stimulation (Beierlein et al. 2003), it is likely that our GABAergic deficits relate to the impaired parvalbumin cell and perineuronal net development in the Fmr1 KO auditory cortex (Wen et al. 2018).
Wen et al. (2018) reported reduced P14 auditory cortex parvalbumin cell numbers in Fmr1 KO mice, and we found reduced P12 GABAAR α3-subunit expression. Surprisingly, there were no differences in baseline s/mIPSC properties between KO and WT L2/3 pyramidal neurons. Fmr1 KOs may undergo compensatory inhibitory synaptic development, like adaptive mechanisms from homeostatic feedback during circuit development, and/or compensatory GABAAR subunit incorporation to maintain GABAergic signaling (Bosman et al. 2005; Kralic et al. 2006; Mullins et al. 2016). Despite similar s/mIPSC properties, P12 Fmr1 KO neurons exhibited enhanced ZOLP sensitivity. This suggests that α1-subunit incorporation may be increased in a small subset of synaptic GABAARs to compensate for decreased α3 expression in KOs, causing enhanced ZOLP sensitivity but no difference in baseline decay τ. Interestingly the basolateral amygdala of Fmr1 KO mice exhibits dynamic alterations in sIPSC properties and diminished ZOLP sensitivity across development (Vislay et al. 2013), indicating region-specific differences in inhibitory development likely influenced by local cell types and circuitry.
A more comprehensive understanding of inhibitory dysregulation in FXS auditory cortex requires the examination of additional cell types and cortical layer (Kalish et al. 2020). Evaluating both local inhibitory input and excitatory projections onto inhibitory interneurons could reveal additional GABA impairments since Fmr1 KO L4 barrel cortex has deficits in excitatory drive onto inhibitory neurons (Gibson et al. 2008). Additionally, Goswami et al. (2019) showed local L2/3 auditory cortex hyperexcitability in P19–23 Fmr1 KO mice, attributed to increased intrinsic excitability but not directly measured. Given our observations of decreased L4-L2/3 excitability in P21–25 KO mice, intrinsic property characterizations could identify whether L2/3 hyperexcitability is compensatory for decreased L4-L2/3 excitability.
Fmr1 KO mice further exhibited EGABA alterations where L2/3 pyramidal neurons were more hyperpolarized across ages with open ear canals. Although inhibition is important for critical periods, excessive inhibitory GABAergic transmission could limit plasticity (Harauzov et al. 2010). Despite this functional difference, chloride transporter expression, which regulates EGABA by establishing internal chloride concentrations, was not altered in auditory cortex lysates. However, the whole auditory cortical samples used for western blots may not detect layer or cell-specific alterations in transporter expression that might cause functional differences (Wang et al. 2002; Rheims et al. 2008), including KCC2 posttranslational modifications that modulate EGABA (Lee et al. 2007; Medina et al. 2014). Interestingly, intracellular chloride concentrations can influence GABAAR subunit composition by regulating α3 and α1 surface expression, where lower intracellular chloride levels decrease α3 expression, thereby increasing α1-mediated currents (Succol et al. 2012). This is consistent with our EGABA alterations, decreased α3 expression, and increased ZOLP sensitivity in Fmr1 KO mice.
We also identified a previously unreported WT developmental progression from our auditory cortex eIPSC-EGABA I-V plots. eIPSCs transitioned from outwardly to inwardly rectifying with age, similar to the developmental regulation of GABA currents in rat intrinsic cardiac ganglion neurons that likely provide additional chloride current-related mechanisms to control neuronal excitability (Fischer et al. 2005). The rectification of eIPSCs can be regulated by chloride channels and GABAAR subunits (Verdoorn et al. 1990; Smith et al. 1995; Jensen et al. 2002; Pavlov et al. 2009). Developmental RNA-editing of GABAAR-α3 renders more α1-like properties that switches I-V plots from outwardly rectifying to linear (Rula et al. 2008; Nimmich et al. 2009). Meanwhile, Fmr1 KOs lacked the outward rectification and instead resembled older developmental ages, consistent with our α-subunit expression data. Such rectification differences of voltage-dependent conductance could make pyramidal neurons more excitable and generate action potentials (Pavlov et al. 2009) further dysregulating GABAergic inhibition.
The GABAergic regulation of critical periods is related to the appropriate maturation of depolarizing and hyperpolarizing GABA, including their timing, kinetics, and magnitudes (Ben-Ari 2002; He et al. 2014; Deidda et al. 2015). Coincident with the auditory cortex GABA-mediated dysregulation in Fmr1 KOs are alterations in the developmental profile of synaptic plasticity of L2/3 pyramidal neurons. WT mice have LTP timed with ear canal opening, where it cannot be reliably induced at P9–10 but is capable starting P12, followed by age-dependent declines in magnitude. This differs from plasticity studies in the mouse visual cortex, where L2/3 LTP could be induced as early as P8 (Jiang et al. 2007; Li et al. 2017). Meanwhile, P9–10 Fmr1 KOs had larger immediate posttetanic potentiation and attenuated depressed responses postpairing, while P12–13 exhibited high variability in synaptic responses and ability to sustain potentiation. KO mice also failed to exhibit the clear developmental decay slope progression of LTP like in WT mice, similar to the destabilized decay described by Yang et al. (2014). Despite tonotopic critical period plasticity deficits (Kim et al. 2013), we found that LTP was still present in the L4-L2/3 Fmr1 KO circuit, but with the aforementioned moderate abnormalities, suggesting that greater deficits likely arise from other parts of the circuit, like thalamocortical synapses. Alterations characterized in this study may be secondary and consequences of impaired maturation at the earlier stages of intracortical processing. Alternatively, other forms of LTP may be dysregulated to a larger extent, such as mGluR-mediated signaling (Kim et al. 2013; Yang et al. 2014).
We hypothesize that Fmr1 KO dysregulation of synaptic GABAergic maturation influences local auditory cortex circuit excitability and synaptic plasticity. The aberrant GABAergic regulation is likely not directly caused by FMRP loss and is rather secondary to the genetic mutation since GABAAR α-subunits are not direct FMRP targets (Darnell et al. 2011). Instead, the dysregulation may manifest from hyperexcitability in developing FXS brains. Across multiple models early-life seizures increase α1-subunit expression, accelerate GABA polarity switches, and occlude plasticity (Ben-Ari 2002; Galanopoulou 2008; Brooks-Kayal 2011; Sun et al. 2018). Hyperexcitability in lower stages of the auditory circuit could drive enhanced cortical excitability to disrupt synaptic maturation and alter auditory input refinement within the cortex (Li et al. 2014). Fmr1 KOs have enhanced excitatory connectivity in the auditory brainstem even prior to hearing onset (Garcia-Pino et al. 2017; Rotschafer and Cramer 2017), and audiogenic seizures originating from the inferior colliculus and subcortical regions, suggestive of hyperexcitable auditory circuits (Gonzalez et al. 2019). The barrel cortex in KO mice is also hyperexcitable (Contractor et al. 2015), potentially influencing excitability given the integration of somatosensory and auditory cortex circuitry (Fu et al. 2003). Altered critical periods are common with E–I imbalance, with enhanced GAD67 expression causing precocious visual critical periods in Mecp2-null mice (Krishnan et al. 2015), and early-life seizures disrupting auditory critical period plasticity in WT mice (Sun et al. 2018).
In conclusion, our studies extensively characterize WT developmental maturational patterns of the L4-L2/3 auditory cortex circuit, and reveal alterations in synaptic GABAergic maturation in the Fmr1 KO auditory cortex that disrupt the normal development of L4-L2/3 E–I circuit and synaptic plasticity. Such differences occur at early ages, causing inappropriate circuit maturation relative to ear canal opening and sensory input mediation to impair the auditory cortex critical period. Our findings suggest that a broader modulation of E–I imbalance in early development may secondarily ameliorate auditory phenotypes observed in FXS.
Supplementary Material
Contributor Information
Yeri J Song, Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Bo Xing, Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Aaron J Barbour, Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Chengwen Zhou, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Frances E Jensen, Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Notes
We thank Drs Jocelyn Lippman-Bell and Anne E. Takesian for helpful comments on the manuscript. Conflict of Interest: none declared.
Funding
National Institutes of Health (R01NS080565 and R37NS115439 to F.E.J, F31DC016192 to Y.J.S.).
References
- Barkat TR, Polley DB, Hensch TK. 2011. A critical period for auditory thalamocortical connectivity. Nat Neurosci. 14:1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur R, Sigel E. 2007. Replacement of histidine in position 105 in the alpha(5) subunit by cysteine stimulates zolpidem sensitivity of alpha(5)beta(2)gamma(2) GABA(A) receptors. J Neurochem. 103:2556–2564. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, Connors BW. 2003. Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J Neurophysiol. 90:2987–3000. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. 2002. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 3:728–739. [DOI] [PubMed] [Google Scholar]
- Blackwell JM, Geffen MN. 2017. Progress and challenges for understanding the function of cortical microcircuits in auditory processing. Nat Commun. 8:2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman LW, Heinen K, Spijker S, Brussaard AB. 2005. Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol. 94:338–346. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal A. 2011. Molecular mechanisms of cognitive and behavioral comorbidities of epilepsy in children. Epilepsia. 52(Suppl 1):13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cea-Del Rio CA, Huntsman MM. 2014. The contribution of inhibitory interneurons to circuit dysfunction in fragile X syndrome. Front Cell Neurosci. 8:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toth M. 2001. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 103:1043–1050. [DOI] [PubMed] [Google Scholar]
- Cisneros-Franco JM, Ouellet L, Kamal B, de Villers-Sidani E. 2018. A brain without brakes: reduced inhibition is associated with enhanced but dysregulated plasticity in the aged rat auditory cortex. eNeuro. 5:ENEURO.0051–ENEU18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Malenka RC. 2008. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 33:18–41. [DOI] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera-Cailliau C. 2015. Altered neuronal and circuit excitability in fragile X syndrome. Neuron. 87:699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. 1995. A critical period for long-term potentiation at thalamocortical synapses. Nature. 375:325–328. [DOI] [PubMed] [Google Scholar]
- Darnell JC, van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. 2011. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 146:247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villers-Sidani E, Chang EF, Bao S, Merzenich MM. 2007. Critical period window for spectral tuning defined in the primary auditory cortex (A1) in the rat. J Neurosci. 27:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda G, Allegra M, Cerri C, Naskar S, Bony G, Zunino G, Bozzi Y, Caleo M, Cancedda L. 2015. Early depolarizing GABA controls critical-period plasticity in the rat visual cortex. Nat Neurosci. 18:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrn AL, Yuan K, Barker AJ, Schreiner CE, Froemke RC. 2010. Developmental sensory experience balances cortical excitation and inhibition. Nature. 465:932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. 2005. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 11:1205–1213. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Fritschy JM, Low K, Mohler H, Rudolph U, Hensch TK. 2004. Specific GABAA circuits for visual cortical plasticity. Science. 303:1681–1683. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK. 2000. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 404:183–186. [DOI] [PubMed] [Google Scholar]
- Finestack LH, Richmond EK, Abbeduto L. 2009. Language development in individuals with fragile X syndrome. Top Lang Disord. 29:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Harper AA, Anderson CR, Adams DJ. 2005. Developmental changes in expression of GABAA receptor-channels in rat intrinsic cardiac ganglion neurones. J Physiol. 564:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Jones BJ. 2011. Development of auditory cortical synaptic receptive fields. Neurosci Biobehav Rev. 35:2105–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu KM, Johnston TA, Shah AS, Arnold L, Smiley J, Hackett TA, Garraghty PE, Schroeder CE. 2003. Auditory cortical neurons respond to somatosensory stimulation. J Neurosci. 23:7510–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanopoulou AS. 2008. Dissociated gender-specific effects of recurrent seizures on GABA signaling in CA1 pyramidal neurons: role of GABA(A) receptors. J Neurosci. 28:1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Heldt SA. 2016. Enrichment of GABAA receptor alpha-subunits on the axonal initial segment shows regional differences. Front Cell Neurosci. 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pino E, Gessele N, Koch U. 2017. Enhanced excitatory connectivity and disturbed sound processing in the auditory brainstem of fragile X mice. J Neurosci. 37:7403–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM. 2008. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 100:2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez D, Tomasek M, Hays S, Sridhar V, Ammanuel S, Chang CW, Pawlowski K, Huber KM, Gibson JR. 2019. Audiogenic seizures in the Fmr1 knock-out mouse are induced byFmr1Deletion in subcortical, VGlut2-expressing excitatory neurons and require deletion in the inferior colliculus. J Neurosci. 39:9852–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S, Cavalier S, Sridhar V, Huber KM, Gibson JR. 2019. Local cortical circuit correlates of altered EEG in the mouse model of fragile X syndrome. Neurobiol Dis. 124:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Chambers AR, Darrow KN, Hancock KE, Shinn-Cunningham BG, Polley DB. 2012. Robustness of cortical topography across fields, laminae, anesthetic states, and neurophysiological signal types. J Neurosci. 32:9159–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett TA, Guo Y, Clause A, Hackett NJ, Garbett K, Zhang P, Polley DB, Mirnics K. 2015. Transcriptional maturation of the mouse auditory forebrain. BMC Genomics. 16:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Ellender TJ, Zemankovics R, Mann EO, Exley R, Cragg SJ, Freund TF, Paulsen O. 2009. Maintaining network activity in submerged hippocampal slices: importance of oxygen supply. Eur J Neurosci. 29:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauzov A, Spolidoro M, DiCristo G, De Pasquale R, Cancedda L, Pizzorusso T, Viegi A, Berardi N, Maffei L. 2010. Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J Neurosci. 30:361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Nomura T, Xu J, Contractor A. 2014. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci. 34:446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. 2005. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 6:877–888. [DOI] [PubMed] [Google Scholar]
- Hersh JH, Saul RA, Committee on G . 2011. Health supervision for children with fragile X syndrome. Pediatrics. 127:994–1006. [DOI] [PubMed] [Google Scholar]
- Jang HJ, Cho KH, Kim HS, Hahn SJ, Kim MS, Rhie DJ. 2009. Age-dependent decline in supragranular long-term synaptic plasticity by increased inhibition during the critical period in the rat primary visual cortex. J Neurophysiol. 101:269–275. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Sweet RA. 2015. Auditory dysfunction in schizophrenia: integrating clinical and basic features. Nat Rev Neurosci. 16:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ML, Timmermann DB, Johansen TH, Schousboe A, Varming T, Ahring PK. 2002. The beta subunit determines the ion selectivity of the GABAA receptor. J Biol Chem. 277:41438–41447. [DOI] [PubMed] [Google Scholar]
- Jiang B, Trevino M, Kirkwood A. 2007. Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J Neurosci. 27:9648–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish BT, Barkat TR, Diel EE, Zhang EJ, Greenberg ME, Hensch TK. 2020. Single-nucleus RNA sequencing of mouse auditory cortex reveals critical period triggers and brakes. Proc Natl Acad Sci U S A. 117:11744–11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanold PO, Nelken I, Polley DB. 2014. Local versus global scales of organization in auditory cortex. Trends Neurosci. 37:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Gibboni R, Kirkhart C, Bao S. 2013. Impaired critical period plasticity in primary auditory cortex of fragile X model mice. J Neurosci. 33:15686–15692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Lee HK, Bear MF. 1995. Co-regulation of long-term potentiation and experience-dependent synaptic plasticity in visual cortex by age and experience. Nature. 375:328–331. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Sidler C, Parpan F, Homanics GE, Morrow AL, Fritschy JM. 2006. Compensatory alteration of inhibitory synaptic circuits in cerebellum and thalamus of gamma-aminobutyric acid type a receptor alpha1 subunit knockout mice. J Comp Neurol. 495:408–421. [DOI] [PubMed] [Google Scholar]
- Krishnan K, Wang BS, Lu J, Wang L, Maffei A, Cang J, Huang ZJ. 2015. MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex. Proc Natl Acad Sci U S A. 112:E4782–E4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH. 1992. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 12:4151–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ. 2007. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem. 282:29777–29784. [DOI] [PubMed] [Google Scholar]
- Li DK, Zhang C, Gu Y, Zhang SH, Shi J, Chen XH. 2017. The spatial-temporal interaction in the LTP induction between layer IV to layer II/III and layer II/III to layer II/III connections in rats' visual cortex during the development. Neuroscience. 350:39–53. [DOI] [PubMed] [Google Scholar]
- Li LY, Ji XY, Liang F, Li YT, Xiao Z, Tao HW, Zhang LI. 2014. A feedforward inhibitory circuit mediates lateral refinement of sensory representation in upper layer 2/3 of mouse primary auditory cortex. J Neurosci. 34:13670–13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of objects in dual colour confocal images. J Microsc. 169:375–382. [DOI] [PubMed] [Google Scholar]
- Marowsky A, Rudolph U, Fritschy JM, Arand M. 2012. Tonic inhibition in principal cells of the amygdala: a central role for alpha3 subunit-containing GABAA receptors. J Neurosci. 32:8611–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, Pellegrino C. 2014. Current view on the functional regulation of the neuronal K(+)-Cl(-) cotransporter KCC2. Front Cell Neurosci. 8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Solarana K, Bowen Z, Liu J, Nagode DA, Sheikh A, Winkowski DE, Kao JPY, Kanold PO. 2020. Transient subgranular hyperconnectivity to L2/3 and enhanced pairwise correlations during the critical period in the mouse auditory cortex. Cereb Cortex. 30:1914–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzenich MM, Knight PL, Roth GL. 1975. Representation of cochlea within primary auditory cortex in the cat. J Neurophysiol. 38:231–249. [DOI] [PubMed] [Google Scholar]
- Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, Hagerman RJ. 1999. Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet. 83:268–279. [PubMed] [Google Scholar]
- Mowery TM, Kotak VC, Sanes DH. 2016. The onset of visual experience gates auditory cortex critical periods. Nat Commun. 7:10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins C, Fishell G, Tsien RW. 2016. Unifying views of autism Spectrum disorders: a consideration of autoregulatory feedback loops. Neuron. 89:1131–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmich ML, Heidelberg LS, Fisher JL. 2009. RNA editing of the GABA(A) receptor alpha3 subunit alters the functional properties of recombinant receptors. Neurosci Res. 63:288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole KK, Jenkins A. 2012. The apparent voltage dependence of GABAA receptor activation and modulation is inversely related to channel open probability. Mol Pharmacol. 81:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Onodera K, Van Renterghem C, Sieghart W, Takahashi T. 2000. Functional correlation of GABA(A) receptor alpha subunits expression with the properties of IPSCs in the developing thalamus. J Neurosci. 20:2202–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroumov A, Thomas AM, Kimmey BA, Karsch JS, Doyon WM, Dani JA. 2016. Stress increases ethanol self-administration via a shift toward excitatory GABA Signaling in the ventral tegmental area. Neuron. 92:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald AM, Reyes AD. 2008. Maturation of intrinsic and synaptic properties of layer 2/3 pyramidal neurons in mouse auditory cortex. J Neurophysiol. 99:2998–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluszkiewicz SM, Martin BS, Huntsman MM. 2011. Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev Neurosci. 33:349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangratz-Fuehrer S, Sieghart W, Rudolph U, Parada I, Huguenard JR. 2016. Early postnatal switch in GABAA receptor alpha-subunits in the reticular thalamic nucleus. J Neurophysiol. 115:1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panuccio G, Colombi I, Chiappalone M. 2018. Recording and modulation of epileptiform activity in rodent brain slices coupled to microelectrode arrays. J Vis Exp. 10.3791/57548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I, Savtchenko LP, Kullmann DM, Semyanov A, Walker MC. 2009. Outwardly rectifying tonically active GABAA receptors in pyramidal cells modulate neuronal offset, not gain. J Neurosci. 29:15341–15350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. 2004. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA, USA: Elsevier. [Google Scholar]
- Rakhade SN, Fitzgerald EF, Klein PM, Zhou C, Sun H, Huganir RL, Jensen FE. 2012. Glutamate receptor 1 phosphorylation at serine 831 and 845 modulates seizure susceptibility and hippocampal hyperexcitability after early life seizures. J Neurosci. 32:17800–17812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Jensen FE. 2009. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 5:380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Zhou C, Aujla PK, Fishman R, Sucher NJ, Jensen FE. 2008. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J Neurosci. 28:7979–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard K, Tikidji-Hamburyan A, Seitter H, Idrees S, Mutter M, Benkner B, Munch TA. 2014. Step-by-step instructions for retina recordings with perforated multi electrode arrays. PLoS One. 9:e106148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheims S, Minlebaev M, Ivanov A, Represa A, Khazipov R, Holmes GL, Ben-Ari Y, Zilberter Y. 2008. Excitatory GABA in rodent developing neocortex in vitro. J Neurophysiol. 100:609–619. [DOI] [PubMed] [Google Scholar]
- Rotschafer S, Razak K. 2013. Altered auditory processing in a mouse model of fragile X syndrome. Brain Res. 1506:12–24. [DOI] [PubMed] [Google Scholar]
- Rotschafer SE, Cramer KS. 2017. Developmental emergence of phenotypes in the auditory brainstem nuclei of Fmr1 knockout mice. eNeuro. 4:ENEURO.0264–ENEU17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotschafer SE, Razak KA. 2014. Auditory processing in fragile x syndrome. Front Cell Neurosci. 8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rula EY, Lagrange AH, Jacobs MM, Hu N, Macdonald RL, Emeson RB. 2008. Developmental modulation of GABA(A) receptor function by RNA editing. J Neurosci. 28:6196–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes DH, Kotak VC. 2011. Developmental plasticity of auditory cortical inhibitory synapses. Hear Res. 279:140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna E, Busonero F, Talani G, Carta M, Massa F, Peis M, Maciocco E, Biggio G. 2002. Comparison of the effects of zaleplon, zolpidem, and triazolam at various GABA(A) receptor subtypes. Eur J Pharmacol. 451:103–110. [DOI] [PubMed] [Google Scholar]
- Scheyer AF, Borsoi M, Wager-Miller J, Pelissier-Alicot AL, Murphy MN, Mackie K, Manzoni OJJ. 2020. Cannabinoid exposure via lactation in rats disrupts perinatal programming of the gamma-aminobutyric acid trajectory and select early-life Behaviors. Biol Psychiatry. 87:666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RL, Clayton GH, Wilcox CL, Escudero KW, Staley KJ. 1995. Differential expression of an inwardly rectifying chloride conductance in rat brain neurons: a potential mechanism for cell-specific modulation of postsynaptic inhibition. J Neurosci. 15:4057–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbings KA, Choi HW, Ravindra A, Caspary DM, Turner JG, Llano DA. 2016. Ageing-related changes in GABAergic inhibition in mouse auditory cortex, measured using in vitro flavoprotein autofluorescence imaging. J Physiol. 594:207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Succol F, Fiumelli H, Benfenati F, Cancedda L, Barberis A. 2012. Intracellular chloride concentration influences the GABAA receptor subunit composition. Nat Commun. 3:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Kosaras B, Klein PM, Jensen FE. 2013. Mammalian target of rapamycin complex 1 activation negatively regulates polo-like kinase 2-mediated homeostatic compensation following neonatal seizures. Proc Natl Acad Sci U S A. 110:5199–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Takesian AE, Wang TT, Lippman-Bell JJ, Hensch TK, Jensen FE. 2018. Early seizures prematurely unsilence auditory synapses to disrupt thalamocortical critical period plasticity. Cell Rep. 23:2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takesian AE, Kotak VC, Sanes DH. 2012. Age-dependent effect of hearing loss on cortical inhibitory synapse function. J Neurophysiol. 107:937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. 2006. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J Comp Neurol. 497:42–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Wang T, Wan H, Han L, Qin X, Zhang Y, Wang J, Yu C, Berton F, Francesconi W, et al. 2015. Fmr1 deficiency promotes age-dependent alterations in the cortical synaptic proteome. Proc Natl Acad Sci U S A. 112:E4697–E4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Molen MJ, Van der Molen MW, Ridderinkhof KR, Hamel BC, Curfs LM, Ramakers GJ. 2012. Auditory and visual cortical activity during selective attention in fragile X syndrome: a cascade of processing deficiencies. Clin Neurophysiol. 123:720–729. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Draguhn A, Ymer S, Seeburg PH, Sakmann B. 1990. Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron. 4:919–928. [DOI] [PubMed] [Google Scholar]
- Vislay RL, Martin BS, Olmos-Serrano JL, Kratovac S, Nelson DL, Corbin JG, Huntsman MM. 2013. Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J Neurosci. 33:7548–7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Shimizu-Okabe C, Watanabe K, Okabe A, Matsuzaki H, Ogawa T, Mori N, Fukuda A, Sato K. 2002. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 139:59–66. [DOI] [PubMed] [Google Scholar]
- Wehr M, Zador AM. 2003. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 426:442–446. [DOI] [PubMed] [Google Scholar]
- Wen TH, Afroz S, Reinhard SM, Palacios AR, Tapia K, Binder DK, Razak KA, Ethell IM. 2018. Genetic reduction of matrix metalloproteinase-9 promotes formation of perineuronal nets around parvalbumin-expressing interneurons and normalizes auditory cortex responses in developing Fmr1 knock-out mice. Cereb Cortex. 28:3951–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox KS, Dichter MA. 1994. Paired pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J Neurosci. 14:1775–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yu L, Cai R, Zhang J, Sun X. 2010. Early continuous white noise exposure alters auditory spatial sensitivity and expression of GAD65 and GABAA receptor subunits in rat auditory cortex. Cereb Cortex. 20:804–812. [DOI] [PubMed] [Google Scholar]
- Yang S, Yang S, Park JS, Kirkwood A, Bao S. 2014. Failed stabilization for long-term potentiation in the auditory cortex of FMR1 knockout mice. PLoS One. 9:e104691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yennawar M, White RS, Jensen FE. 2019. AMPA receptor dysregulation and therapeutic interventions in a mouse model of CDKL5 deficiency disorder. J Neurosci. 39:4814–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SW, Platholi J, Flaherty MS, Fu W, Kottmann AH, Toth M. 2006. Fmrp is required for the establishment of the startle response during the critical period of auditory development. Brain Res. 1110:159–165. [DOI] [PubMed] [Google Scholar]
- Zhou C, Huang Z, Ding L, Deel ME, Arain FM, Murray CR, Patel RS, Flanagan CD, Gallagher MJ. 2013. Altered cortical GABAA receptor composition, physiology, and endocytosis in a mouse model of a human genetic absence epilepsy syndrome. J Biol Chem. 288:21458–21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Lippman JJ, Sun H, Jensen FE. 2011. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J Neurosci. 31:18211–18222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.