

Abstract
The inflammatory response after traumatic brain injury (TBI) can lead to significant secondary brain injury and chronic inflammation within the central nervous system (CNS). Cell therapies, including mesenchymal stromal cells (MSC), have led to improvements in animal models of TBI and are under investigation in human trials. One potential mechanism for the therapeutic potential of MSC is their ability to augment the endogenous response of immune suppressive regulatory T cells (Treg). We have recently shown that infusion of human cord blood Treg decreased chronic microgliosis after TBI and altered the systemic immune response in a rodent model. These cells likely use both overlapping and distinct mechanisms to modulate the immune system; therefore, combining Treg and MSC as a combination therapy may confer therapeutic benefit over either monotherapy. However, investigation of Treg+MSC combination therapy in TBI is lacking. In this study, we compared the ability MSC+Treg combination therapy, as well as MSC and Treg monotherapies, to inhibit the neuroinflammatory response to TBI in vivo and in vitro. Treg+MSC combination therapy demonstrated increased potency to reduce the neuro- and peripheral inflammatory response compared to monotherapy; furthermore, the timing of infusion proved to be a significant variable in the efficacy of both MSC monotherapy and Treg+MSC combination therapy in vivo and in vitro.
Keywords: Mesenchymal stromal cell, mesenchymal stem cell, Regulatory T cell, cell therapy, traumatic brain injury, neuroinflammation, microglia
Introduction
Traumatic brain injury (TBI) is a leading cause of morbidity and mortality in the United States and worldwide.1,2 After the primary mechanical injury, the subsequent inflammatory response within the brain and systemic immune system contributes to secondary brain injury and further pathology.3 This neuroinflammatory response can persist for years, and TBI survivors often suffer substantial, long-term disabilities.4,5 However, to date, no therapy has been shown to attenuate secondary brain injury and improve long-term outcomes in TBI.6,7
Microglia are the resident myeloid cells within the central nervous system (CNS) parenchyma and are key mediators of the neuroinflammatory response after TBI.3,8,9 Our lab has investigated the use of cell therapies to attenuate the microglia-mediated inflammatory response and have found that human mesenchymal stromal cells (MSC) and, recently, human cord blood-derived regulatory T cell (Treg) therapy both decrease the neuroinflammatory response after TBI.6,10,11 We believe that a primary mechanism by which cell therapies mitigate the inflammatory response is via interaction with the host immune system, specifically endogenous Treg.10,12 Furthermore, several others have shown that the interaction between MSC and Treg likely plays an important role in efficacy of therapy and that Treg+MSC combination therapy can have synergistic effects in vitro and in vivo.13–19 However, the effects of combination therapy on the microglia response after TBI have not been investigated. Therefore, in this study, we examined the ability of human MSC+Treg combination therapy, as well as Treg and MSC monotherapies, to attenuate the neuroinflammatory response after TBI in vivo and in vitro, as well as innate and adaptive peripheral immune responses in vitro. We hypothesized that combination therapy would confer increased potency and efficacy compared to both Treg and MSC monotherapies. We found that Treg+MSC therapy significantly decreases the systemic inflammatory response compared to monotherapy. In addition, we demonstrate that timing of therapy may have a significant impact on efficacy, and that staggered Treg+MSC therapy was more effective in vivo and in vitro.
Materials and methods
All procedures involving the use of research animals were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were reviewed approved by the University of Texas Health Science Center Institutional Animal Care and Use Committee (AWC-18–0121). Peripheral blood was from healthy human adult donors was acquired after informed consent using a protocol approved by the institutional review board (IRB) (HSC-MS-10–0190). Human umbilical cord blood was provided by the MD Anderson Cord Blood Bank (Houston, TX) via a Material Transfer Agreement (MTA).
Animals
Male Sprague Dawley Rats (225–250 g, Envigo Labs) were the primary source of tissue, including CNS and spleen. Animal usage was reviewed and approved by the Animal Welfare Committee at University of Texas Health Science Center at Houston, Texas, protocol: AWC-18–0121. All animal procedures were performed in accordance with the standards of the American Association for the Accreditation of Laboratory Animal Care (AAALAC). For these experiments, five-week old rats were housed in pairs under 12 hour light/dark cycles in temperature-controlled conditions with water and food accessible ad libitum.
1. Treg production and immunophenotpying
Mononuclear cell isolation
Umbilical cord blood (UCB) was acquired from 3 individual donors. Each donor was maintained separate throughout processing and subsequent experiments. UCB mononuclear cells (MNC) were isolated as previously described using SepMate PBMC isolation tubes (SepMate-50, STEMCELL Technologies, Inc., Vancouver, Canada).10 Briefly, 15 mL of Ficoll-Paque was added to the bottom chamber the SepMate tube (GE Healthcare, Chicago, IL). UCB and phosphate buffered saline (PBS) were mixed in a 1:1 ratio (Lonza, Basel, Switzerland), and, within each tube, 30 mL of the mixture was layered on to the Ficoll-Paque. With the brake on, we centrifuged the tubes at 1200g for 20 minutes. The top layer containing the MNC was transferred into a new 50mL centrifuge tube, washed with PBS, and spun down again at 400g for 8 minutes. Cell counts and viability were obtained with a NucleoCounter NC-200 and Via1-Cassettes (ChemoMetec, Allerod, Denmark). The cells were washed a final time with PBS and then resuspended in cryopreservation media [CryoStor CS10 (STEMCELL Technologies, Inc, Vancouver, Canada)] at a cell density of 10×106 cells/mL. The cryovials were placed in a Mr. Frosty Freezing Container (Nalgene Nunc, Rochester, NY) and stored at −80°C overnight. The next day, the cryovials were placed in liquid nitrogen vapor phase and stored until further use.
MNC thaw
Prior to use, MNC were thawed and with washed with HyClone RPMI 1640 (GE Healthcare, Chicago, IL) + 5% human AB serum (Thermo Fisher Scientific, Waltham, MA). We have found that viability and yield are increased when MNC are thawed with media versus PBS. Cell counts and viability were obtained using a NucleoCounter. For Treg expansion, the cells were centrifuged at 300g for 5 minutes, resuspended in Treg expansion media (below).
Treg expansion
Thawed UCB MNC were resuspended in our previously described Treg expansion media [HyClone RPMI 1640 (GE Healthcare, Chicago, IL), 5% human AB serum (Thermo Fisher Scientific, Waltham, MA), 1% GlutaMAX (Gibco, Thermo Fisher Scientific, Waltham, MA), 10 μg/mL of gentamicin (Gibco), 100nM Rapamycin (Thermo Fisher Scientific, Waltham, MA), and 500 IU/mL IL-2 (CellGenix, Freiburg, Germany)] at an initial seeding density of 1×106 cells/mL without any isolation step (Supp Figure 1).10 The MACS GMP ExpAct Treg kit was used for expansion at a ratio of 4:1 bead:cell. On day 0, we plated 2×106 MNC at 1×106 cell/mL in a 12- well culture plate (Costar, Corning Inc., Corning, NY). On day 2, the cells and media were collected. The cells were centrifuged at 300g for 5 minutes, and the supernatant was removed. The cells were counted and resuspended in Treg expansion media at a density of 5×105 cells/mL. Cells were then split every 48–72 hours to keep cell density between 5–7.5×105 cells/mL and transferred to appropriate sized culture flasks.
On day 10, positive selection of CD4+ cells was performed using Miltenyi CD4+ microbeads (Miltenyi Biotec, Auburn, CA 130-045-101). First, the expansion beads were removed. The collected cells were centrifuged at 300g for 5 minutes, resuspended in separation buffer [PBS + 2 mM ethylenediaminetetraacetic acid (EDTA) + 0.5% human serum albumin (HSA; Baxter, Deerfield, Illinois)], and transferred to a 15mL conical tube. The tube was placed in a MACSiMAG Separator to remove the expansion beads (Miltenyi Biotec). The beads were allowed to adhere to the wall of the tube, and the supernatant, containing the cells, was removed and placed into a new tube.
The collected cells were centrifuged at 300g for 5 minutes, resuspended in buffer, and incubated with the CD4 MicroBeads. The cell suspension was added to an MS column; the CD4+ cells were captured in the column and then subsequently flushed out with buffer. This process was repeated to improve purity. Cell count and viability were obtained. The cells were then washed and centrifuged at 300g for 5 minutes. The supernatant was taken off, and the cells were resuspended in Treg expansion media at 5×105 cells/mL. The Treg expansion beads then added to the cell culture at a ratio of 4:1 bead:cell.
The cells were then split every 48–72 hours to keep cell density between 5–7.5×105 cells/mL and transferred to appropriate sized culture flasks. On day 20 of expansion, cells were collected for flow cytometry and cryopreservation. The cells were transferred to 50mL conical tubes, spun down for 5 minutes at 300g and then resuspended in buffer. Expansion beads were removed with the MACSiMAG magnet Separator (Miltenyi Biotec) as previously described.10 A sample was taken for flow cytometric analyses. The remaining cells were stored in cryopreservation media [CryoStor CS10 (STEMCELL Technologies, Inc, Vancouver, Canada)] at a cell density of 10×106 cells/mL. The cryovials were stored in a Mr. Frosty at −80°C overnight and then transferred to liquid nitrogen vapor phase storage until further use.
Treg thaw
Treg were quickly thawed and washed in RPMI + 5% AB serum. Cell counts and viability were obtained with the NucleoCounter NC-200 (Chemometec). For in vitro experiments, cells were resuspended in assay-specific media. For in vivo experiments, cells were resuspended in sterile PBS.
Treg flow cytometry immunophenotyping
To evaluate cell populations and identify Treg, we performed flow cytometry on the thawed Treg using a multi-parameter panel as previously described.10 This panel contained CD3-FITC, CD4-PE, CD8-APC, CD25-PE Cy7, CD127-BV421, CD19-BV510, and 7-AAD cell viability solution. Data were acquired with a Gallios Flow Cytometer (Beckman Coulter, Brea, CA) and analyzed with FlowJo vr10.6.1 (FlowJo, LLC). Single cells were isolated using forward and side scatter parameters, and live versus dead cells were gated on 7-AAD. Treg were identified as live CD3+CD4+CD25+CD127dim cells (Supp Figure 2).10
2. MSC isolation and expansion
MSC were isolated and expanded from human adipose tissue donors (n=3), as previously described (Supp Figure 1).20 Briefly, MSC were isolated, as via enzymatic digestion with collagenase (Worthington Biochemical, Lakewood, NJ) and expanded in αMEM (Gibco) supplemented with 5% human platelet lysate (Cook), 1X Glutamax (Gibco) and 10 μg/mL gentamicin (Gibco). P0 cells were plated at an initial density of 100×103 cell/cm2 in an appropriately sized flask. At 70–90% confluency, cells were washed with PBS (without Ca++ and Mg++) and detached with TrypLE Express (Gibco). Cells were transferred to increasing flask sizes until reaching 70–90% confluence in a T3180 CellStack (Corning) at P3, at which time they were lifted, washed, and cryopreserved in a 1:1 dilution of CryoStor CS10 in expansion medium at a density of 4–5 × 106 cells/cryovial. The cryovials were stored in a Mr. Frosty at −80°C overnight. The next day, the cryovials were transferred to liquid nitrogen vapor phase and stored until further use. Prior to use, MSC were thawed and washed with PBS and then centrifuged for 5 minutes at 300g. Cell counts and viability were acquired using the NucleoCounter. MSC were resuspended in the assay-specific media for in vitro experiments and in sterile PBS for in vivo experiments.
3. In vitro experiments
Rat splenocyte inflammation assay
Splenocytes (Sp) were isolated as previously described.10,21 Briefly, a naïve rat was placed under anesthesia, and then spleen was removed and placed in PBS. The spleen was processed with a gentleMACS Dissociator (Miltenyi Biotech), passed through a 70 μm filter and spun down at 400g for 5 minutes. After a repeat wash with PBS, the cells were resuspended in RPMI + 5% human AB serum. In 96-well plates, 2×105 Sp were added to each well and, to induce an inflammatory response, activated with lipopolysaccharide (LPS) or concanavalin A (ConA). To evaluate the suppressive ability of Treg, MSC, and Treg+MSC, three different MSC donors and three different Treg donors were utilized and added as treatment to the wells.
In the LPS activated plate, Treg (1:2 Treg:Sp), MSC (1:10 MSC:Sp) or combination of Treg (1:2) + MSC (1:10) were added to the wells, and the cell culture supernatant was collected after 24 hours. In the ConA activated plate, Treg (1:2 Treg:Sp), MSC (1:10 MSC:Sp) or combination of Treg (1:2) + MSC (1:10) were added to the wells, and the culture supernatant was collected after 72 hours. The samples were analyzed utilizing rat specific TNFα and IFNγ ELISA kit (BD Biosciences, San Jose, CA) per manufacturer’s protocol. Based on the ELISA results, the most potent Treg+MSC donor combination, in addition to their corresponding monotherapy samples, were analyzed using a 13-plex rat-specific cytokine/chemokine LEGENDplex bead array according to the manufacturer’s protocol (Biolegend, San Diego, CA #740401). The samples were analyzed using a BD LSRII, and analysis was performed using LEGENDplex analysis software (Biolegend).
Rat microglia isolation and activation assay
Microglia were isolated from the brain from a naïve male rat (n=1) as previously described using the Neural Tissue Dissociation Kit and GentleMACS Dissociator (Miltenyi Biotec).10 Using Percoll centrifugation, myelin was removed from the cells. The brain cells were then cultured as previously described.10 First, we coated a 48-well plated with cell matrix basement membrane gel (ATCC, Manassas, VA). We then plated 2.5 ×105 cells/well in microglia-specific media (DMEM + 10% FBS), and media was changed every 48 hours. After 6 days, the cells were activated with LPS (1 μg/mL). 2 hours after activated, Treg (1:2 Treg:microglia), MSC (1:10 MSC:microglia) or combination of Treg (1:2) +MSC (1:10) were added to the wells; MSC were added either 1) concurrently with Treg or 2) 18 hours after activation.
The culture supernatant was collected at 72 hours. All samples were analyzed using a 13-plex rat-specific cytokine/chemokine LEGENDplex bead array according to the manufacturer’s protocol (Biolegend, San Diego, CA #740401). The samples were analyzed using a BD LSRII, and analysis was performed using LEGENDplex analysis software (Biolegend).
Microglia immunophenotyping
In addition, at 72 hours, the rat microglia cells were collected and analyzed using flow cytometry. After cell culture supernatant was removed, the cells were washed with PBS and then detached from the culture plate with TrypLE Express (Thermo Fisher). The cells were washed, centrifuged at 400g, and resuspended in cell staining buffer. The isolated brain cells were immunophenotyped using the multiparametric flow cytometry panel (Figure 2) as we have previously described.10 The cells were then stained with the antibody mixture in Table 1 and incubated in the dark for 20 minutes, at which time 7-AAD was added. After 10 minutes, data were acquired with LSR-2 flow cytometer with Diva acquisition software (BD Biosciences, San Jose, CA). VersaComp Antibody Capture Beads (Beckman Coulter, Inc.) were used to create fluorescence spillover compensation values.
Figure 2. Microglia gating strategy and multicolor flow cytometry panel.
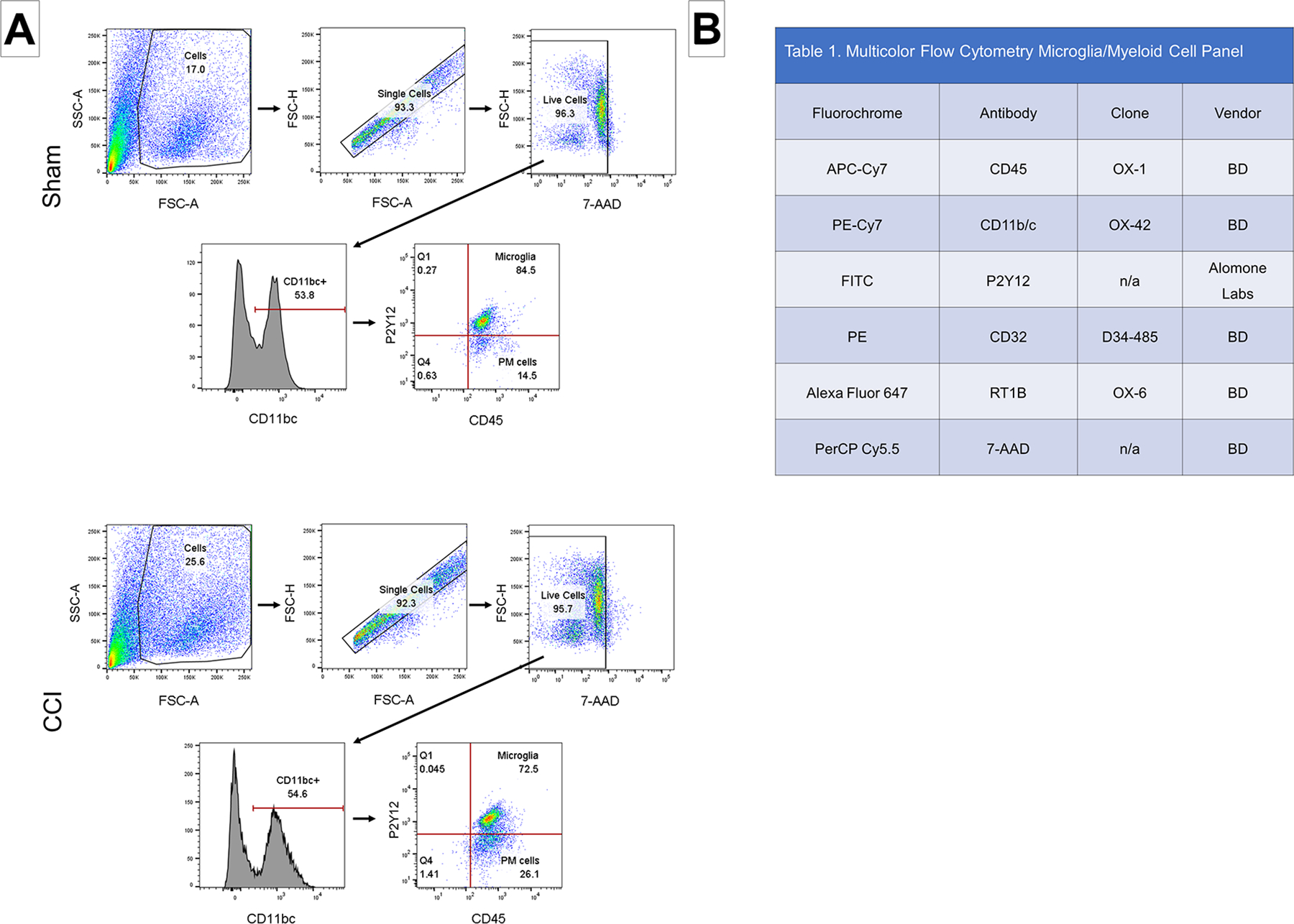
(A): Representative samples from both sham and CCI groups are shown. The single cell population was identified based on SSC and FSC. Live cells were identified as negative for the 7-AAD. Microglia were identified using a two-step method. First, CD11b/c+ cells (PE-Cy7) were selected to identify all myeloid cells. The CD11b/c+ cells were then gated on P2Y12 (FITC) and CD45 (APC-Cy7). Microglia were identified as triple positive cells. SSC, side scatter; FSC, forward scatter. (B): The multiple color flow cytometry panel used to identify and immunophenotype microglia and peripheral myeloid cells.
Table 1.
Multicolor Flow Cytometry Microglia/Myeloid Cell Panel
| Fluorochrome | Antibody | Clone | Vendor |
|---|---|---|---|
| APC-Cy7 | CD45 | OX-1 | BD |
| PE-Cy7 | CD11b/c | OX-42 | BD |
| FITC | P2Y12 | n/a | Alomone Labs |
| PE | CD32 | D34–485 | BD |
| Alexa Fluor 647 | RT1B | OX-6 | BD |
| PerCP Cy5.5 | 7-AAD | n/a | BD |
Microglia gating strategy
Conventional flow cytometry analyses were performed with FlowJo vr10.6.1. Fluorescence-Minus-One (FMO) controls were used to set gate boundaries. Live cells were gated by CD11bc expression and then gated on P2Y12 and CD45 expression in order to identify P2Y12+ microglia (Figure 2). Triple positive cells were identified as microglia.
4. In vivo experiments
Experimental groups
Experiments were conducted in two cohorts, in which two distinct treatment strategies were employed (Figure 1). In each cohort, the were n=6 animals per group.
Figure 1.
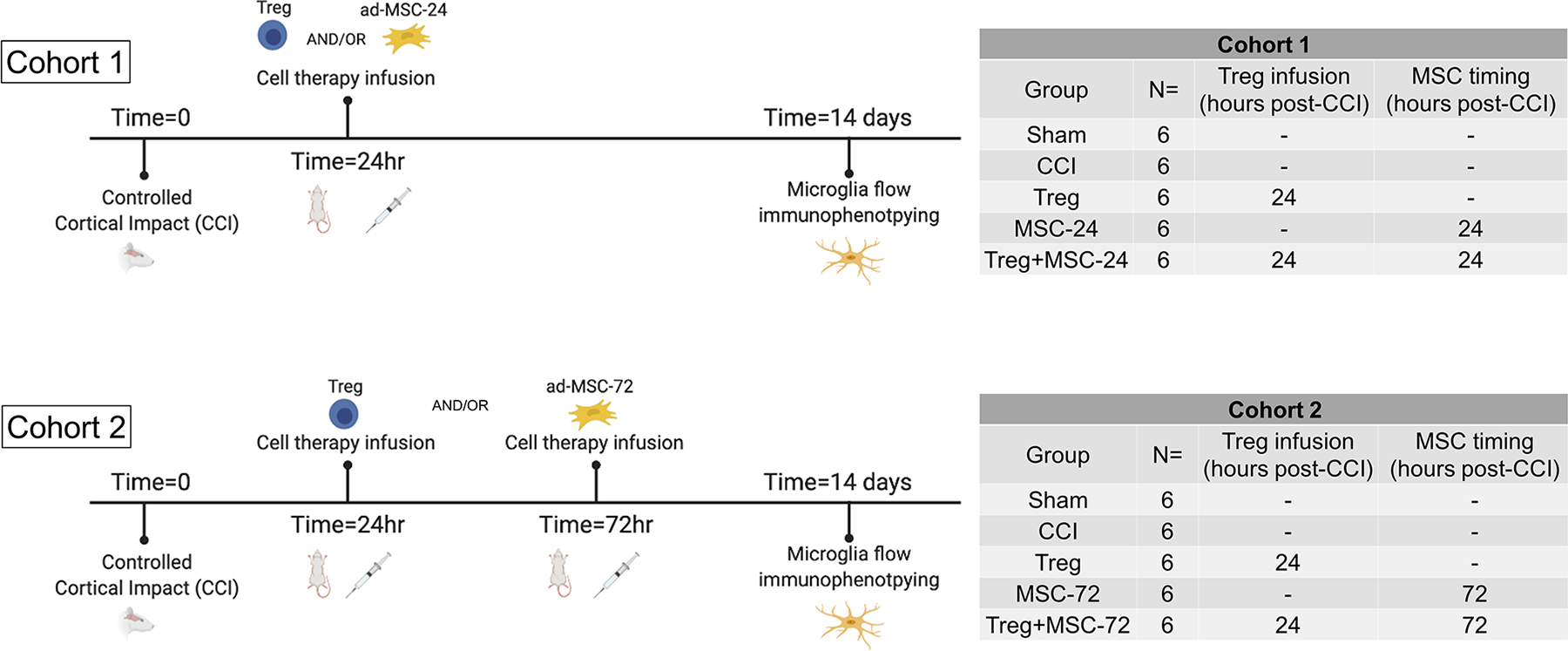
Experimental design and timing of the performed in vivo experiment.
Controlled cortical impact model
Controlled cortical impact (CCI) procedures were conducted as previously described10,22–27. In brief, animals were anesthetized for surgery with 4% isoflurane and oxygen in a vented chamber, then maintained with 2–3% isoflurane during the procedure. A stereotactic frame is used to secure the animal while the surgical site was cleaned and disinfected with alcohol and iodine solution. Local analgesia is provided by subcutaneous 0.25% bupivacaine administered prior to incision. A midline cranial incision exposed the right sided musculature and soft tissue, which were bluntly dissected away for exposure of the calvarium. A 7-mm diameter craniectomy was performed between the right coronal and lambdoid sutures to expose the dura. A CCI device (Impact One Stereotaxic Impactor, Leica Microsystems, Buffalo Grove, IL) was positioned to administer a standardized, unilateral severe brain injury. The 6mm impactor tip traveled to a depth of 3.1 mm with an impact velocity of 5.6 m/s, and a dwell time of 150ms in the parietal association cortex. The incision was closed immediately after impact using sterile wound clips and animals recovered in clean cages. Sham injured animals were anesthetized and then received the midline incision, followed by separating the skin, connective tissue and aponeurosis from the cranium. As before, the incision was closed using sterile wound clips.
Treg and MSC infusion
After injury, animals were randomized to receive either 1) Treg alone, 2) MSC alone or 3) Treg+MSC. Treg and MSC were thawed and washed as described above. Treg and MSC were resuspended in 1mL of sterile PBS at a dose of 10×106 cells/kg for each cell type. All treatment animals underwent CCI and then received Treg, MSC or combination infusion, via tail vein (Figure 1). In Cohort 1, treatment animals received either Treg at 24 hours, MSC at 24 hours, or combination of Treg+MSC at 24 hours. In Cohort 2, treatment animals received either Treg at 24 hours, MSC at 72 hours, or combination of Treg (at 24 hours) + MSC (at 72 hours).
Microglia immunophenotyping
At 14 days after injury, animals were euthanized, and brains were harvested. The ipsilateral hemispheres were processed as previously described above using the Neural Tissue Dissociation Kit and GentleMACS dissociator (Miltenyi Biotec).28
The isolated brain cells were immunophenotyped using the multiparametric flow cytometry panel (Figure 2) as we have previously described.10 The cells were then stained with the antibody mixture in Table 1 and incubated in the dark for 20 minutes, at which time 7-AAD was added. After staining, the red blood cells were lysed in a TQ-Prep (Beckman Coulter, Inc.). Finally, 25μL of counting control beads (Cyto-Cal™) was added to each sample while vortexing. Data were acquired with LSR-2 flow cytometer with Diva acquisition software (BD Biosciences, San Jose, CA). VersaComp Antibody Capture Beads (Beckman Coulter, Inc.) were used to create fluorescence spillover compensation values.
Microglia gating strategy
Conventional flow cytometry analyses were performed with FlowJo vr10.6.1. Fluorescence-Minus-One (FMO) controls were used to set gate boundaries. Live cells were gated by CD11bc expression and then gated on P2Y12 and CD45 expression in order to identify P2Y12+ microglia (Figure 2). Triple positive cells were identified as microglia. Microglia cells were then gated on phenotypic markers CD32 and RT1B. Using the provided reference formula, we calculated absolute microglia cell counts per hemisphere, adjusted for brain hemisphere weight:
t-SNE interpretation of microglia and splenocyte flow cytometry data
t-SNE analyses were performed using FlowJo vr10.6 using the same Flow Cytometry Standard (FCS) files used in the analyses above (FlowJo, LLC). Total live cell events were randomly down-sampled to 3000 events within each sample. Then, the down-sampled events were concatenated to link each individual sample file into a single standard file. The t-SNE analysis and plot were created with the FlowJo plugin. All data points are derived from the same concatenated file in each t-SNE figure. We first analyzed the density plots of the individual groups and then generated antibody heat maps to visualize the fluorescent intensity of each marker in each group.
5. Statistical analysis
Date were analyzed with GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). With respect to microglia flow cytometry data, ROUT analyses identified outliers in the data, based on brain hemisphere weight, which were excluded in the final analysis. 1 sample was removed from the Treg+MSC-72 group based on its brain weight. In order to normalize data from both experimental cohorts, absolute cell count means were scaled to CCI.
Comparisons between means of sham/naïve and injured/activated controls within each group were analyzed using an unpaired T-test to demonstrate the effectiveness of our injury model or effect of in vitro activation. Then, comparisons between means of the injury/activated control and treatment groups were analyzed using ordinary one-way ANOVA with Dunnett’s multiple comparisons test. Values of p ≤ 0.05 were considered significant and indicated with (#) for p ≤ 0.05, (##) for p ≤ 0.01, (###) for p ≤ 0.001. Further post-hoc analysis to compare the means of monotherapy and combination therapy were analyzed using one-way ANOVA with Sidak’s multiple comparisons test. All group data are presented as mean ± standard error. Values of p ≤ 0.05 were considered significant and indicated with (*) for p ≤ 0.05, (**) for p ≤ 0.01, (***) for p ≤ 0.001.
Results
Immunomodulatory effects on rat splenocytes in vitro
The systemic immune response to TBI has significant effects on secondary injury and progression of subacute and chronic neuroinflammation, and we have previously demonstrated the both Treg and MSC can modulate the innate and adaptive immune responses of activated rat splenocytes in vitro.10,24 However, the effect of combination therapy on rat immune responses was unknown; therefore, we tested three different Treg and three different MSC donors, both alone (Treg:Sp 1:2; MSC:Sp 1:10) and in combination therapy (Treg:Sp 1:2 + MSC:Sp 1:10).
After LPS stimulation, Treg and MSC monotherapies and Treg+MSC significantly reduced TNFα production (Figure 3a). Furthermore, Treg+MSC significantly reduced TNFα production compared to both Treg and MSC monotherapies. Out of 9 possible Treg+MSC combinations, only two combinations failed to reach a significant difference in comparison its respective MSC monotherapy, and only one Treg+MSC combination failed to reach a significant difference one in comparison its respective Treg monotherapy. The effects of combination therapy on the adaptive immune response were more striking (Figure 3b). Treg+MSC significantly reduced IFNγ production in comparison to the activated control and both Treg and MSC monotherapies. Furthermore, each combination of Treg+MSC significantly reduced IFNγ production compared to their respective Treg or MSC monotherapy, with the exception of one Treg+MSC compared to its respective Treg monotherapy. Furthermore, two of the individual Treg donors did not significantly reduce IFNγ production; however, when used in combination with MSC, all three Treg+MSC combinations from each donor significantly reduced IFNγ production.
Figure 3. Characterization of Treg, MSC and Treg+MSC combination therapy potency using activated rat splenocytes in vitro.
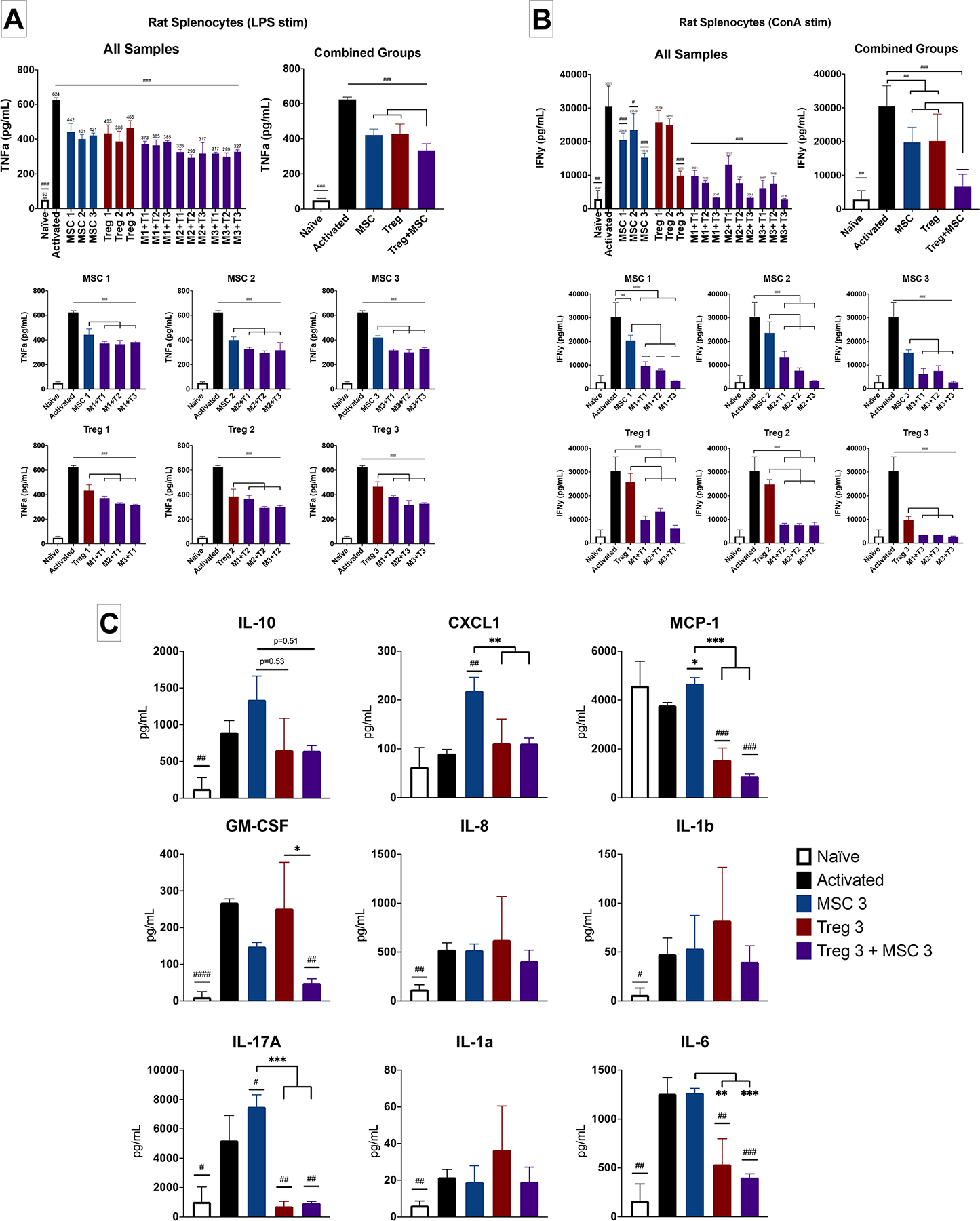
(A): TNFα production (ELISA) by rat splenocytes after LPS stimulation (Treg Donors 1–3, MSC Donors 1–3). (B): IFNγ production (ELISA) by rat splenocytes after ConA stimulation (Treg Donors 1–3, MSC Donors 1–3). (C): Assessment of multiple chemokines/cytokine production (LEGENDplex) by activated rat splenocytes (Treg Donor 3, MSC Donor 3). All samples run in triplicate. Statistical significance between naive/treatment and activated control is indicated with (#) for p ≤ 0.05, (##) for p ≤ 0.01, (###) for p ≤ 0.001. Statistical significance between treatment groups is indicated with (*) for p ≤ 0.05, (**) for p ≤ 0.01, (***) for p ≤ 0.001. Treg, regulatory T cell; MSC, mesenchymal stromal cell; TNFα, tumor necrosis factor alpha; IFNγ, interferon gamma; ELISA, enzyme-linked immunosorbent assay.
We then tested the one of the most potent Treg+MSC combinations (Treg donor 3 and MSC donor 3) using a rat-specific Legendplex bead assay to further investigate the effects of combination therapy on 13 different cytokines and chemokines (Figure 3c). We found that ConA stimulation increased the production of IL-10, GM-CSF, IL-8, IL-1b, IL-17a, IL-1a, and IL-6. MSC monotherapy did not significantly decrease any of these 5 cytokines/chemokines; in fact, there was an increase in CXCL1, MCP-1, and IL-17A compared to activated control. Treg monotherapy significantly decreased MCP-1, IL-17A and IL-6. Treg+MSC combination therapy significantly decreased MCP-1, GM-CSF, IL-17A, and IL-6 production. Furthermore, there were several significant decreases in cytokine production by both Treg and Treg+MSC compared to MSC monotherapy, including CXCL1, MCP-1, IL-17A, and IL-6. Finally, there was a significant decrease in GM-CSF in the Treg+MSC combination therapy compared to Treg monotherapy.
Altogether, these data indicate that Treg+MSC combination therapy may be a more potent modulator of systemic inflammatory responses and inhibitor of pro-inflammatory cytokines than either Treg or MSC monotherapies.
Immunomodulatory effects of Treg+MSC combination therapy on rat microglia in vitro
We also investigated the effects of Treg+MSC combination on primary culture rat microglia in vitro. Given the results of our in vivo data, we studied the effects of Treg and MSC monotherapies and two different combination treatment regimens: Treg were added at 2 hours after activation, and MSC were added either 1) concurrently with Treg (MSC) or 2) 18 hours after activation (MSC-18).
At 72 hours, there were significant differences between naïve and activated microglia in the levels of IL-10, CXCL1, TNFα, GM-CSF, IL-8, IL-1a, and IL-6 (Figure 4a). In comparison to activated microglia, all treatments significantly decreased IL-10 production. Only Treg+MSC-18 decreased CXCL1. MCP-1 was reduced by both MSC monotherapies and both Treg+MSC combination therapies. With respect to GM-CSF, IL-8, and IL-1a, only MSC-18 and Treg+MSC-18 reduced production of these cytokines, while Treg monotherapy increased production of GM-CSF and IL-1a. There were no significant differences between activated control and any treatment with respect to IL-6, although these values were all at upper limit of the assay.
Figure 4. Characterization of Treg, MSC and Treg+MSC combination therapy potency using activated rat microglia in vitro.
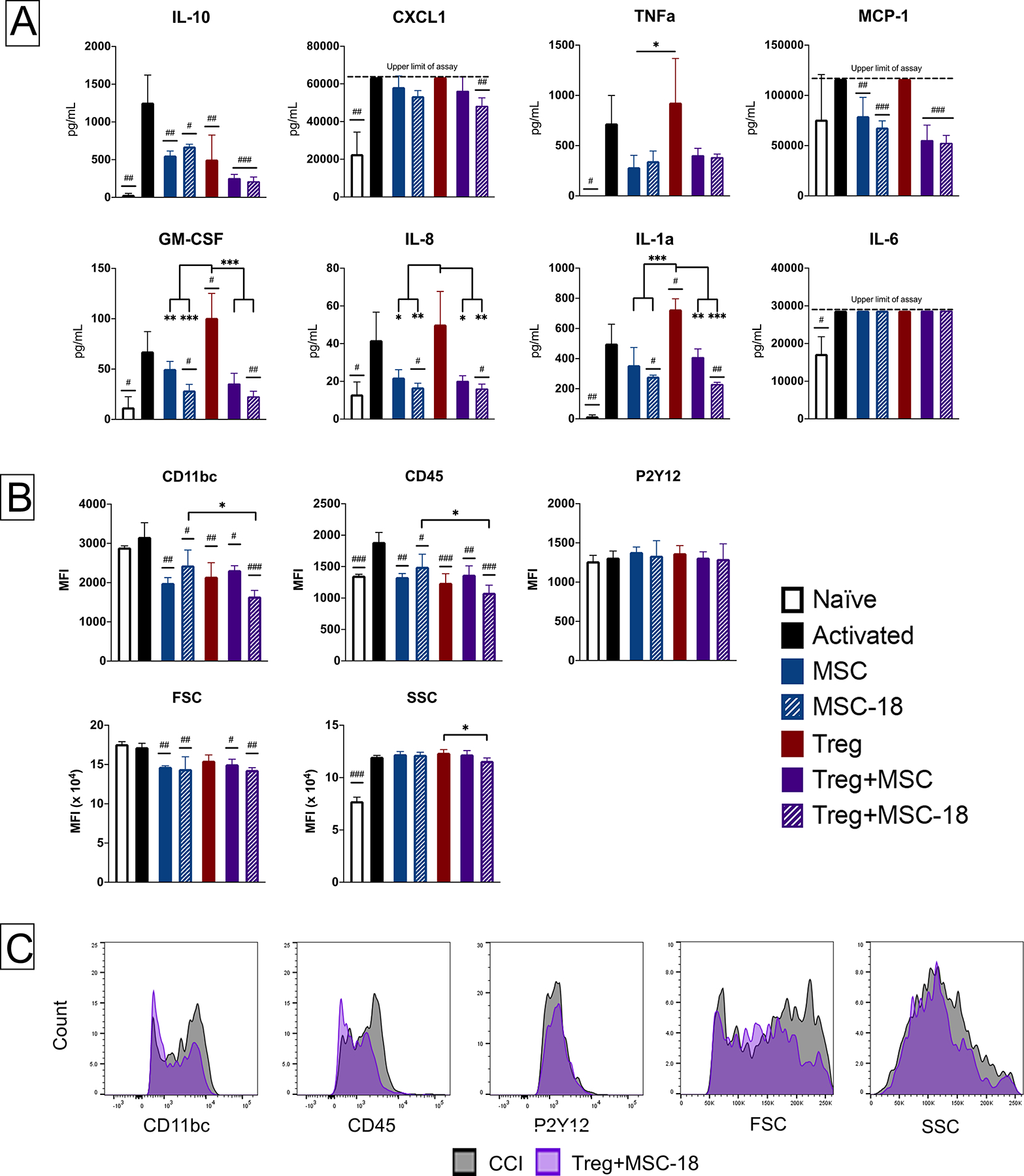
(A): Assessment of multiple chemokines/cytokine production (LEGENDplex) by activated rat microglia. (B-C): Flow cytometry characterization of changes in microglia activation. (B): Changes in microglia cell surface marker expression after injury and treatment. (C): Representation histogram overlays of antibody expression in the injured control and Treg+MSC-18 combination therapy groups. All samples run in triplicate. Statistical significance between naive/treatment and activated control is indicated with (#) for p ≤ 0.05, (##) for p ≤ 0.01, (###) for p ≤ 0.001. Statistical significance between treatment groups is indicated with (*) for p ≤ 0.05, (**) for p ≤ 0.01, (***) for p ≤ 0.001. Treg, regulatory T cell; MSC, mesenchymal stromal cell; ELISA, enzyme-linked immunosorbent assay.
We also performed flow cytometry on the cultured rat microglia in order to evaluate the effects of therapy on the microglial phenotype (Figure 4b). In comparison to naïve microglia, the activated microglia had elevated CD45 expression and higher SSC. All treatments reduced CD11b/c and CD45 MFI; however, there was also a significant decrease in both CD11b/c and CD45 MFI in the Treg+MSC-18 compared to MSC-18 monotherapy. Both MSC monotherapies and combination therapies reduced FSC, a marker of cell size and microglia activation, compared to the activated control. Finally, while treatment did not significant affect SSC compare to the activated control, Treg+MSC-18 did reduce SSC in comparison to Treg monotherapy.
Both the cytokine and flow cytometry data reveal that Treg and MSC monotherapy have differing effects on activated microglia with respect to cytokine production and surface phenotypes. Treg+MSC combination therapy more consistently inhibited the microglia inflammatory response compared to either monotherapy.
Effects of treatment on the microglia response to TBI at 14 days after injury
Given the significant improvement in potency of Treg+MSC combination therapy compared to monotherapy, we then examined the effects of treatment on microgliosis and microglia immunophenotypes, using conventional flow cytometry and t-SNE visualization, at 14 days after injury. In Cohort 1, treatment animals received either Treg at 24 hours, MSC at 24 hours, or combination of Treg+MSC at 24 hours (Figure 2a). In Cohort 2, treatment animals received either Treg at 24 hours, MSC at 72 hours, or combination of Treg (at 24 hours) + MSC (at 72 hours) (Figure 2b).
We have previously demonstrated the Treg monotherapy decreased microgliosis at 30 days, but not at 7 days, after CCI.10 Furthermore, we and others have shown that MSC monotherapy can also decrease microglia activation after CCI.11,25 However, Treg+MSC combination therapy has yet to be tested in a CCI model. Using conventional flow cytometry logic gating analysis, the Treg+MSC-72 group, but no other treatment group, significantly reduced microglia cell counts compared to CCI in the ipsilateral hemisphere (Figure 5a; Supp Figure 3). Interestingly, while there were several significant differences between sham and CCI with respect to phenotypic markers (Figure 5b and Supp Figure 3), the only significant differences after any treatment were a reduction in CD45 and CD11bc expression in the MSC-72 monotherapy group (Figure 5b).
Figure 5: Flow cytometry characterization of changes in microglia and peripheral myeloid cells populations after injury and treatment.
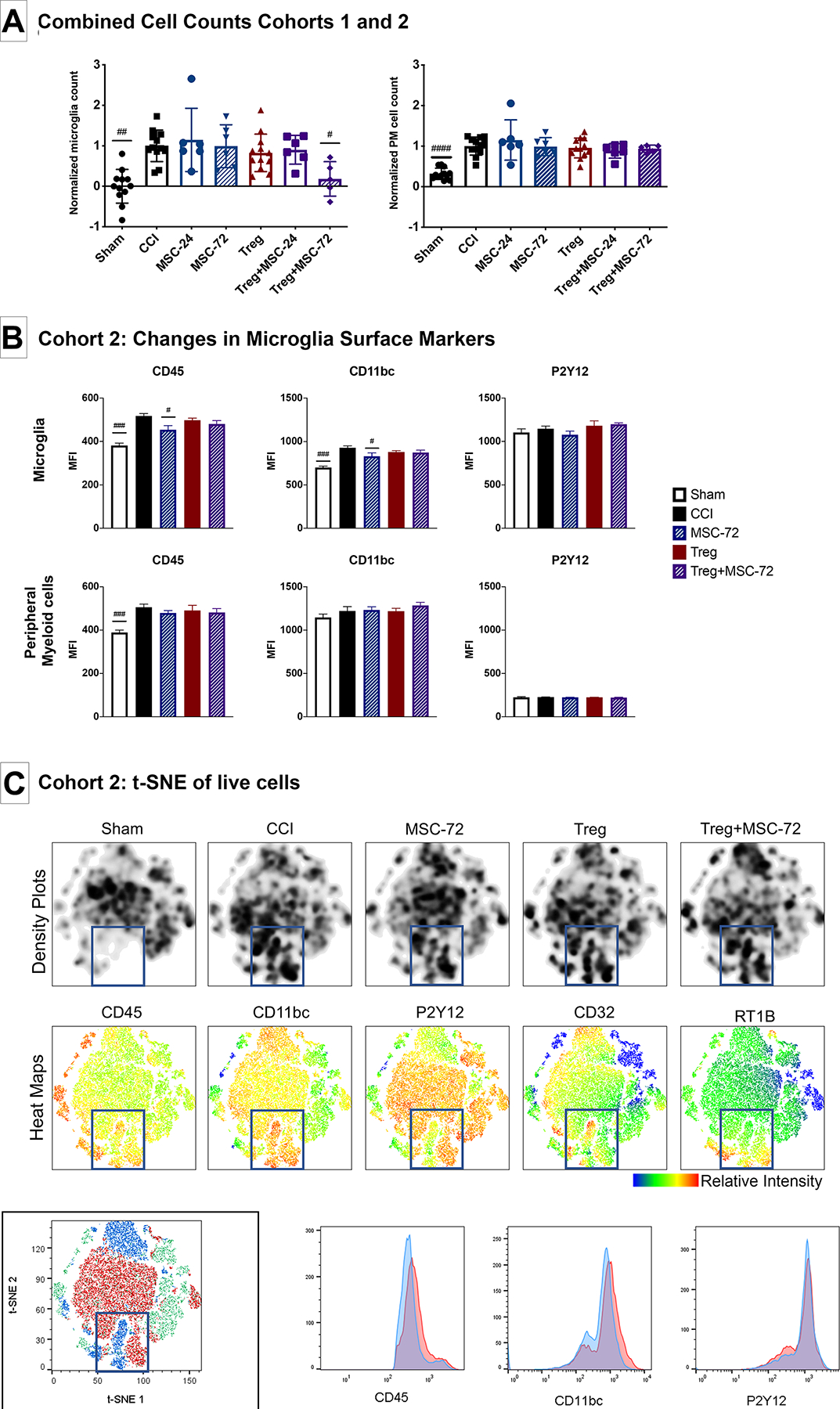
(A-B): Assessment of microglia activation using conventional flow cytometry logic gating analysis. (A): Combined absolute microglia (left) and PM (right) cell counts from Cohorts 1 and 2, normalized to CCI. (B): Changes in microglia and PM flow cytometry surface marker expression in Cohort 2. (C): t-SNE visualization of changes in microglia and PM cell populations after injury and treatment. Density plots (top row) demonstrate changes in cell populations between sham and CCI. Analysis of antibody heat maps (middle row), cell clusters and antibody histograms (bottom row) demonstrate that there are increases in both microglia and PM cell populations and markers of activation in the injured brain at 14 days after injury. No substantial differences between the injured control and treatments groups are appreciated. Statistical significance between sham/treatment and CCI is indicated with (#) for p ≤ 0.05, (##) for p ≤ 0.01, (###) for p ≤ 0.001. Statistical significance between treatment groups is indicated with (*) for p ≤ 0.05, (**) for p ≤ 0.01, (***) for p ≤ 0.001. PM, peripheral myeloid cell; CCI, controlled cortical impact; t-SNE, t-distributed stochastic neighbor embedding.
In addition, we observed significant increases in the number of peripheral myeloid cells (PM; CD11bc+CD45+P2Y12-) in the ipsilateral hemisphere at 14 days after injury (Figure 5a); however, treatment did not significantly effect PM cells counts (Figure 5a). Furthermore, after injury, there was a significant increase in the percentage and MFI of RT1B+ PM and a decrease in CD32+ PM, indicating an increase in antigen presenting cells (RT1B) and decrease in phagocytic cells (CD32) after injury (Supp Figure 3). The Treg+MSC72 group was the only treatment group that demonstrated a trend towards normalization of these phenotypes (i.e. decreased RT1B+ and increased CD32+ PM) (Supp Figure 4).
Using the same FCS files used in the analyses above, we also performed t-SNE analyses to visualize changes in microglia marker expression after CCI and treatment (Figure 5c). At 14 days after injury, there are clear visual differences between the sham and CCI (blue boxes), with activated microglia (P2Y12+CD11highCD45high) and peripheral myeloid cell (P2Y12-CD11highCD45high) populations in the injured group that are not present in the sham controls. This confirms our conventional flow cytometry findings that microgliosis, microglia activation, and invasion of peripheral myeloid cells is present in the injured hemisphere at 14 days. However, we did not observe any significant visual changes between the injured controls and any of the treatment groups. Of note, when performing t-SNE analysis, equal numbers of cells are used and randomly selected from each sample, so any changes in absolute cell counts between groups may not be appreciable.
Discussion
In this study, we have demonstrated that Treg+MSC combination therapy mitigates the neuroinflammatory response to TBI in vivo and in vitro and that combination therapy may confer increased potency and ability to modulate neuro- and systemic inflammatory responses compared to both Treg and MSC monotherapies. Furthermore, we have shown that timing of infusion(s) may have a significant effect on the efficacy of both combination therapy and MSC monotherapy. To our knowledge, this is the first study to examine the effects of human-derived and expanded Treg and MSC combination therapy on microglia activation in vivo or in vitro.
We first examined the effects of Treg+MSC combination therapy, using three different Treg and MSC donors, to attenuate inflammatory responses by activated rat splenocytes. As we have previously demonstrated, this assay provides a reproducible platform to evaluate potency and the interaction between cell therapy and endogenous splenocytes is critical for therapeutic efficacy.12,21 We found that Treg+MSC combination therapy significantly improved potency compared to either monotherapy. As demonstrated in Figure 3, nearly all of the Treg+MSC combinations decreased TNFα or IFNγ production compared to the respective Treg or MSC monotherapy. We then examined the effects one of the most potent combinations of Treg+MSC and the individual monotherapies (Treg donor 3 and MSC donor 3) on a broader range of cytokines and chemokines, using a LEGENDplex multi-analyte kit, after ConA stimulation. In this in vitro model, ConA stimulation induced changes in splenocyte production of several cytokines/chemokines, representing an aggregate increase in the inflammatory response. We found that MSC and Treg monotherapies had differing effects on production of individual cytokines and chemokines, indicating that these cells may act through differing pathways. Furthermore, Treg+MSC combination therapy demonstrated greater ability to attenuate the inflammatory response compared to MSC and Treg monotherapies.
We then examined the ability of cell therapy to modulate the microglia response after TBI in vivo using flow cytometric immunophenotyping. Infusion of combination therapy, with Treg at 24 hours and MSC at 72 hours, was the only treatment, out of the 5 unique treatments examined, to decrease microgliosis in the brain at 14 days. Furthermore, MSC monotherapy given at 72 hours was the only treatment to reduce any cell surface markers of microglia activation. We have previously shown that Treg monotherapy given at 24 hours decreased microgliosis in the ipsilateral hemisphere at 30, but not 7, days after injury and that MSC monotherapy given at 72 hours reduces microgliosis at 7 days after injury.10,24,25 Although we have not previously examined the effects of CCI or cell therapy at 14 days after CCI, our conventional flow cytometry data and t-SNE analyses effectively demonstrate that at 14 days post-CCI there is a significant neuroinflammatory response present in the ipsilateral hemisphere, including persistent microgliosis and presence of peripheral myeloid cells. Furthermore, previous rodent studies have demonstrated that CCI results in a bimodal increase in microgliosis and activation and that 14 days is the potential beginning of the chronic, harmful neuroinflammatory response.29–31 As chronic neuroinflammation can persist for years after a single TBI in humans, treatments that prevent the initiation of this response may lead to improvements in long term outcomes.4
Interestingly, combination therapy of Treg+MSC at 24 hours did not affect microgliosis or cell surface marker expression, indicating that timing of therapy is likely a critical variable for cell therapy efficacy in this model. It is possible that the beneficial effects of the combination therapy were due to the fact that these animals received multiple infusions of cells at different time points. The initial dose may have attenuated the early, pro-inflammatory, toxic milieu, rendering the subsequent the subsequent cell therapy dose more effective.6,32,33 Further investigation is required to distinguish whether the specific combination of Treg at 24 hours and MSC at 72 hours or the multiple doses of anti-inflammatory cell infusions conferred therapeutic efficacy.
Finally, we examined the effects of the cell therapies on cultured rat microglia in vitro using both the LEGENDplex multi-analyte kit and flow cytometry immunophenotyping. Given our in vivo microglia data above, we examined the effects of two different in vitro treatment regimens by adding MSC either at the same timepoint as Treg or at 18 hours after the initial stimulation. After LPS stimulation, there were increases in production of several cytokines/chemokines, indicating an overall increase in the microglia inflammatory response. In addition, we performed flow cytometry analyses on these same cultured microglia to examine the effects of therapy on their immunophenotype. We found that all therapies reduced CD11b/c and CD45 expression, marker of microglia activation.34–36 Interestingly, only Treg+MSC-18 reduced surface markers expression compared to either MSC or Treg monotherapies, further demonstrating that timing of therapy may play in important role in efficacy.
Previous studies have demonstrated that TBI causes a significant alteration in gene expression and upregulation of multiple immune response pathways.3 Recently, in a mouse model, Izzy et al demonstrated that at 2 and 14 days post-TBI there were, respectively, 152 and 127, genes with statistically significant altered expression; however, we do not know the relevance or translatable nature of such findings to the effects of TBI in humans.37 Therefore, while we take caution to make conclusions about the efficacy and translatable potential of these, or any, cell therapies based on in vitro changes in an array of cytokines/chemokines and flow cytometry surface markers, these data do demonstrate a significant increase in overall inflammatory signaling in our model, which these cell therapies were able to attenuate. Furthermore, this in vitro data demonstrates that combination therapy and timing of MSC therapy had significant effects on the microglia inflammatory response.
Our study has several limitations. We utilized two different treatment regimens in our in vivo experiments. This was not intended; however, the timing of our in vivo microglia experiments coincided with the progression of COVID-19 in our region. We intended to examine the effects of Treg+MSC combination therapy given together at 24 hours (i.e. the Treg+MSC-24 group) at 7 and 30 days in order to compare our results to our own previous data. However, we made the decision to change our planned experiment and, instead, utilized the two different treatment regimens and experimental protocol presented here in order to minimize time spent in the lab and best comply with CLAMC and University protocols. Future experiments will examine that effects of Treg+MSC combination therapy on long-term outcomes, including microglia analysis and behavioral testing, to improve translatability of these data. Furthermore, we only utilized male animals in our in vitro and in vivo experiments; future studies should incorporate both sexes, as there are likely important sex differences in TBI and the microglia response.38
Additionally, there are many questions uncovered and left unanswered by this study that should drive future investigations. Does combination therapy change the fate of these infused Treg or MSC? We and others have previously shown that the majority of infused MSC do not actually reach the brain or other target organs, but become trapped in the pulmonary microvasculature.39 However, human studies of adoptive Treg transfer have demonstrated that donor Treg cells can survive and persistent in receipts for extended periods of time.40,41 The effects of combination therapy on the fate of both Treg and MSC deserves further study. Furthermore, does combination therapy affect the xenogeneic immune response by the recipient animal? We believe that the interaction between host immune system and cell therapy products is critical for therapeutic efficacy; furthermore, it has been increasingly recognized that xenogeneic/allogeneic MSC might be a barrier to clinical translation.42 It is possible that the increased efficacy of the staggered Treg+MSC combination therapy was due, in part, to the dampening of the host immune system by the initial Treg infusion, allowing for improved efficacy of the MSC infused 48 hours later. Future studies investigating the potential for repeated dosing, combination therapy, and staggered dosing of Treg and MSC are certainly warranted.
Conclusion
In comparison to both Treg and MSC monotherapies, we have demonstrated that Treg+MSC combination therapy attenuated microgliosis in the injured hemisphere at 14 days after TBI in vivo and the inflammatory responses of activated microglia and splenocytes in vitro. Furthermore, our in vivo and in vitro data demonstrate that the timing of therapy has significant effects on efficacy and, specifically, that combination therapy may be more effective if MSC are infused at a second and later time point after Treg infusion. Taken together, we believe that Treg+MSC combination therapy may confer therapeutic benefits over Treg or MSC monotherapy in the treatment of the inflammatory response after TBI.
Supplementary Material
Treg and MSC isolation and expansion methods. The methods by which both MSC and Treg are isolated and expanded are depicted.
Flow cytometric characterization of human UCB Treg populations. Treg were identified as CD3 positive, CD4 positive, CD8 negative, CD25 positive, and CD127 dim cells.
Additional flow cytometric characterization of microglia at 14 days after injury from Cohort 1 (A) and Cohort 2 (B).
Additional flow cytometric characterization of peripheral myeloid cells at 14 days after injury from Cohort 1 (A) and Cohort 2 (B).
Significance statement:
This study demonstrates the human regulatory T cell (Treg) and mesenchymal stromal cell (MSC) combination therapy improves potency and attenuation of inflammatory responses by activated rat immune cells in comparison to monotherapy. Furthermore, in vivo, only staggered Treg+MSC therapy significantly reduced microgliosis after TBI, indicating that both the combination and timing of cell therapy has significant effects.
Acknowledgements
This study was supported by the NIH 2T32 grant GM0879201-11A1. We would like to thank the MD Anderson Cord Blood Bank for their generosity and support with this study. Figure 1, Supplemental Figure 1, and the graphical abstract were created with BioRender.com.
Funding:
NIH 2T32 grant GM0879201-11A1; Clare A Glassell Family Pediatric Surgery Research Fund; Senator Lloyd and B.A. Bentsen Center for Stroke Research; Grace Reynolds Wall Research Endowment
Disclosure of Potential Conflicts of Interest
SDO has received research support from Athersys, CBR Systems, Hope Bio and Biostage. CSC has received research support from Athersys, CBR Systems, Hope Bio, Biostage, and Cellvation, and is on the Scientific Advisory Board of Cellvation, Biostage, CBR, and Hope Bio.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Faul M, Xu L, Wald MM, Coronado V. Traumatic brain injury in the United States. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. 2010. [Google Scholar]
- 2.Prevention CfDCa. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation. United States: Epidemiology and Rehabilitation National Center for Injury Prevention and Control; Division of Unintentional Injury Prevention Atlanta, GA. 2015. [DOI] [PubMed] [Google Scholar]
- 3.Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70(3):374–383. [DOI] [PubMed] [Google Scholar]
- 5.Maas AIR, Menon DK, Adelson PD, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. The Lancet Neurology. 2017;16(12):987–1048. [DOI] [PubMed] [Google Scholar]
- 6.Cox CS Jr., Juranek J, Bedi S. Clinical trials in traumatic brain injury: cellular therapy and outcome measures. Transfusion. 2019;59(S1):858–868. [DOI] [PubMed] [Google Scholar]
- 7.Kochanek PM, Dixon CE, Mondello S, et al. Multi-Center Pre-clinical Consortia to Enhance Translation of Therapies and Biomarkers for Traumatic Brain Injury: Operation Brain Trauma Therapy and Beyond. Front Neurol. 2018;9:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26(8):1191–1201. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225–242. [DOI] [PubMed] [Google Scholar]
- 10.Caplan HW, Prabhakara KS, Kumar A, et al. Human cord blood-derived regulatory T-cell therapy modulates the central and peripheral immune response after traumatic brain injury. Stem Cells Transl Med. 2020:e12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson ML, Srivastava AK, Cox CS Jr. Preclinical progenitor cell therapy in traumatic brain injury: a meta-analysis. J Surg Res. 2017;214:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker PA, Shah SK, Jimenez F, et al. Intravenous multipotent adult progenitor cell therapy for traumatic brain injury: preserving the blood brain barrier via an interaction with splenocytes. Exp Neurol. 2010;225(2):341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Ianni M, Del Papa B, De Ioanni M, et al. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol. 2008;36(3):309–318. [DOI] [PubMed] [Google Scholar]
- 14.Engela AU, Baan CC, Dor FJ, Weimar W, Hoogduijn MJ. On the interactions between mesenchymal stem cells and regulatory T cells for immunomodulation in transplantation. Front Immunol. 2012;3:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engela AU, Baan CC, Peeters AM, Weimar W, Hoogduijn MJ. Interaction between adipose tissue-derived mesenchymal stem cells and regulatory T-cells. Cell Transplant. 2013;22(1):41–54. [DOI] [PubMed] [Google Scholar]
- 16.Lee ES, Lim JY, Im KI, et al. Adoptive Transfer of Treg Cells Combined with Mesenchymal Stem Cells Facilitates Repopulation of Endogenous Treg Cells in a Murine Acute GVHD Model. PLoS One. 2015;10(9):e0138846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Negi N, Griffin MD. Effects of mesenchymal stromal cells on regulatory T cells: Current understanding and clinical relevance. Stem Cells. 2020;38(5):596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi T, Tibell A, Ljung K, et al. Multipotent mesenchymal stromal cells synergize with costimulation blockade in the inhibition of immune responses and the induction of Foxp3+ regulatory T cells. Stem Cells Transl Med. 2014;3(12):1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Singh AK, Hoyt RF, Jr., et al. Regulatory T cells enhance mesenchymal stem cell survival and proliferation following autologous cotransplantation in ischemic myocardium. J Thorac Cardiovasc Surg. 2014;148(3):1131–1137; discussiom 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George MJ, Prabhakara K, Toledano-Furman NE, et al. Clinical Cellular Therapeutics Accelerate Clot Formation. Stem cells translational medicine. 2018;7(10):731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz MF, Evans SM, Olson SD, Cox CS Jr., Wenzel PL. A Co-culture Assay to Determine Efficacy of TNF-alpha Suppression by Biomechanically Induced Human Bone Marrow Mesenchymal Stem Cells. Bio Protoc. 2017;7(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma. 1988;5(1):1–15. [DOI] [PubMed] [Google Scholar]
- 23.Bedi SS, Aertker BM, Liao GP, et al. Therapeutic time window of multipotent adult progenitor therapy after traumatic brain injury. Journal of neuroinflammation. 2018;15(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kota DJ, Prabhakara KS, Toledano-Furman N, et al. Prostaglandin E2 Indicates Therapeutic Efficacy of Mesenchymal Stem Cells in Experimental Traumatic Brain Injury. Stem Cells. 2017;35(5):1416–1430. [DOI] [PubMed] [Google Scholar]
- 25.Kota DJ, Prabhakara KS, van Brummen AJ, et al. Propranolol and Mesenchymal Stromal Cells Combine to Treat Traumatic Brain Injury. Stem Cells Transl Med. 2016;5(1):33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prabhakara KS, Kota DJ, Jones GH, Srivastava AK, Cox CS Jr., Olson SD. , Teriflunomide Modulates Vascular Permeability and Microglial Activation after Experimental Traumatic Brain Injury. Molecular therapy : the journal of the American Society of Gene Therapy. 2018;26(9):2152–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava AK, Prabhakara KS, Kota DJ, et al. Human umbilical cord blood cells restore vascular integrity in injured rat brain and modulate inflammation in vitro. Regen Med. 2019;14(4):295–307. [DOI] [PubMed] [Google Scholar]
- 28.Toledano Furman NE, Prabhakara KS, Bedi S, Cox CS, Olson SD. Rat Microglia Isolation and Characterization Using Multiparametric Panel for Flow Cytometric Analysis. In: Srivastava AK, Cox CS, eds. Pre-Clinical and Clinical Methods in Brain Trauma Research. New York, NY: Springer New York; 2018:191–199. [Google Scholar]
- 29.Bai R, Gao H, Han Z, et al. Long-Term Kinetics of Immunologic Components and Neurological Deficits in Rats Following Repetitive Mild Traumatic Brain Injury. Med Sci Monit. 2017;23:1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai R, Gao H, Han Z, et al. Flow Cytometric Characterization of T Cell Subsets and Microglia After Repetitive Mild Traumatic Brain Injury in Rats. Neurochem Res. 2017;42(10):2892–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7(7):e41892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolli R Repeated Cell Therapy: A Paradigm Shift Whose Time Has Come. Circ Res. 2017;120(7):1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castro LL, Kitoko JZ, Xisto DG, et al. Multiple doses of adipose tissue-derived mesenchymal stromal cells induce immunosuppression in experimental asthma. Stem Cells Transl Med. 2020;9(2):250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopperton KE, Mohammad D, Trepanier MO, Giuliano V, Bazinet RP. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry. 2018;23(2):177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melief J, Koning N, Schuurman KG, et al. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60(10):1506–1517. [DOI] [PubMed] [Google Scholar]
- 36.Schittenhelm L, Hilkens CM, Morrison VL. beta2 Integrins As Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front Immunol. 2017;8:1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izzy S, Liu Q, Fang Z, et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Front Cell Neurosci. 2019;13:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caplan HW, Cox CS, Bedi SS. Do microglia play a role in sex differences in TBI? J Neurosci Res. 2017;95(1–2):509–517. [DOI] [PubMed] [Google Scholar]
- 39.Fischer UM, Harting MT, Jimenez F, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18(5):683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol. 2020;38:541–566. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Fueyo A, Whitehouse G, Grageda N, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant. 2020;20(4):1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caplan H, Olson SD, Kumar A, et al. Mesenchymal Stromal Cell Therapeutic Delivery: Translational Challenges to Clinical Application. Front Immunol. 2019;10:1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Treg and MSC isolation and expansion methods. The methods by which both MSC and Treg are isolated and expanded are depicted.
Flow cytometric characterization of human UCB Treg populations. Treg were identified as CD3 positive, CD4 positive, CD8 negative, CD25 positive, and CD127 dim cells.
Additional flow cytometric characterization of microglia at 14 days after injury from Cohort 1 (A) and Cohort 2 (B).
Additional flow cytometric characterization of peripheral myeloid cells at 14 days after injury from Cohort 1 (A) and Cohort 2 (B).
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.