

Abstract
Bunyamwera orthobunyavirus and its serogroup can cause several diseases in humans, cattle, ruminants, and birds. The viral M-polyprotein helps the virus to enter the host body. Therefore, this protein might serve as a potential vaccine target against Bunyamwera orthobunyavirus. The present study applied the immunoinformatics technique to design an epitopic vaccine component that could protect against Bunyamwera infection. Phylogenetic analysis revealed the presence of conserved patterns of M-polyprotein within the viral serogroup. Three epitopes common for both B-cell and T-cell were identified, i.e., YQPTELTRS, YKAHDKEET, and ILGTGTPKF merged with a specific linker peptide to construct an active vaccine component. The low atomic contact energy value of docking complex between human TLR4 (TLR4/MD2 complex) and vaccine construct confirms the elevated protein–protein binding interaction. Molecular dynamic simulation and normal mode analysis illustrate the docking complex’s stability, especially by the higher Eigenvalue. In silico cloning of the vaccine construct was applied to amplify the desired vaccine component. Structural allocation of both the vaccine and epitopes also show the efficacy of the developed vaccine. Hence, the computational research design outcomes support that the peptide-based vaccine construction is a crucial drive target to limit the infection of Bunyamwera orthobunyavirus to an extent.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10989-021-10322-9.
Keywords: M-polyprotein, Immunoinformatics, Peptide-vaccine, Epitope, TLR4
Introduction
Bunyamwera orthobunyavirus belongs to the family Bunyaviridae, of genus Bunyavirus. Generally, more than 25 orthobunyaviruses are the leading causative agents of several diseases in the human host, as well as in livestock and bird species (Dutuze et al. 2018). The consequences of the disease is known as Bunyamwera fever, an arthropod-borne viral infection, primarily manifest in human beings (Kohl et al. 2003). Mosquito, ticks, mites etc., acts as a potent carriers of the virus from ruminants to humans. Bunyamwera virus and its serogroup cause many associate diseases like an acute fever in humans, encephalitis, joint pain, arthralgia, myalgia, genetic defects, follicular tonsillitis, bronchopneumonia, abortions, and premature births (Ashford 2001; Lambert and Lanciotti 2009; Medlock et al. 2007; Yanase et al. 2006). This viral serogroup is prevalent primarily in African countries, such as Nigeria, Uganda, Kenya, and Zambia (Fig. 1A) (Cavazzana et al. 2016; Wertheim et al. 2012). The genome of this virus contains an ambisense tripartite RNA genome consisting of three fragments of ambisense RNA, i.e., small (S), medium (M), and large (L) (Mazel-Sanchez and Elliott 2012; Odhiambo et al. 2016; Shi et al. 2016). The S, M, and L fragments lengths are approximately 1 kb −2 kb, 4 kb −5 kb, and 6.9 Kb, respectively. The S fragment encodes NSs protein, while the M fragment encodes a polyprotein, split into three post-transcriptional mRNA encoding three different functionally autonomous polypeptides, namely Gn, Nsm, and Gc. The M-polyprotein, translated from M fragment RNA of Bunyamwra orthobunyavirus can be used as an ideal component for vaccine construction as these encodes for virion surface glycoproteins that mediate the entry of the virus into the host body (Shi et al. 2016; Soldan and González-Scarano 2005). As noted, M-polyprotein is evolutionarily conserved within the serogroup of the Bunyamwera virus (Fig. 1B).
Fig. 1.

A Infection reported and death case by Bunyamwera orthobunyavirus and serogroup in African countries (source WHO report, 1998). B Phylogenetic tree of Bunyamwera orthobunyavirus and serogroup based on M-polyprotein by using the ML method to infer phylogenetic tree in MEGA
There are mainly two promising existing techniques for potent vaccine development, i.e., conventional and reverse vaccinology techniques (He et al. 2010). Reverse vaccinology is a specific, advanced, most effortless, and powerful approach compared to the conventional one (Kazi et al. 2018). Reverse vaccinology utilizes immunoinformatics knowledge to construct an efficient vaccine against pathogenic viruses (Vivona et al. 2008). Using reverse vaccinology technique, multiple computational methods based on immunoinformatics can be employed to construct a vaccine design (Bhattacharya et al. 2020b). This technique exploits several authentic prediction servers and software to find the promising epitopes present within the targeted protein.
The 3D conformational architecture of identified epitopes and vaccine construct significantly impact peptide-based immunoinformatics research. Molecular docking is performed to examine the interaction of epitopes with the immune system. The molecular dynamic simulation study and normal mode analysis are required to validate the rigidity of the vaccine protein docking complex and calculate protein flexibility. More significantly, in-silico cloning was adopted for amplifying the desired vaccine protein, which ultimately leads to establishing a robust immune system to protect the targeted microorganism. Therefore, the methodological efforts will emerge as a significant drive towards controlling Bunyamwera infection and allied diseases by the support of highly advanced immunoinformatics proficiency practices.
The present research aims to limit Bunyamwera infection and abundance to achieve a suitable epitopic vaccine component against Bunyamwera orthobunyavirus.
Material and Methods
The standard methodologies applied to execute existing work are listed in a flow chart (Fig. 2). Every step of this adopted methodology is crucial and promising for successfully designing the desired epitopic vaccine against Bunyamwera orthobunyavirus.
Fig. 2.

Flow chart of the adopted methodology for designing the desired epitopic vaccine
Retrieval of Protein Sequences
The amino acid sequences of M-polyprotein and glycoprotein of Bunyamwera orthobunyavirus serogroup (e.g., Bunyamwera virus, Cache Valley virus, Fort Sherman virus, Ilesha virus, Shokwe virus, Wyeomyia virus, Ngari virus, Germiston virus, Maguari virus, Potosi virus, Northway virus, Biroa virus, Batai virus, Kairi virus, Guaroa virus, Bozo virus, Main drain virus, Playas virus) were retrieved from the NCBI protein database in FASTA format (Geer et al. 2010). These sequences were further processed via multiple immunoinformatic servers and software for developing the epitopic vaccine against B. orthobunyavirus.
Multiple Sequence Alignment and Phylogenetic Analysis of Bunyamwera Serotypes
Multiple sequence alignment (MSA) of the stored protein sequences was performed via the M-Coffee server, which is originally an extension of Tree-based Consistency Objective Function for alignment Evaluation (T-COFFEE), Version_11.00.d625267 (Notredame et al. 2000; Wallace et al. 2006). This specific server is a widely accepted sequence alignment server that carries out MSA by generating a local and global pairwise alignment library of amino acid sequences. The query sequences of the FASTA file are deposited to the specified server for accurate alignment of sequences. This server attributes a specific color code for each aligned amino acid as the output result. The fundamentality of MSA is to find conserved regions within proteins, map out evolutionary linkage, and apply for the protein homology modeling (Thompson and Plewniak 1999). The phylogenetic relationship among these 18 M-polyprotein of Bunyamwera serotypes was constructed on the maximum likelihood (ML) method with the help of the Molecular Evolutionary Genetics Analysis (MEGA) version X software in global alignment (Kumar et al. 2018). MEGA v.X implements many analytical methods and reliable tools for constructing a phylogenetic tree that reflects the relationship between the species groups based on protein sequence (in FASTA format) as input data. This software can also construct phylogenetic trees considering neighbor-joining, maximum parsimony, UPGMA, and maximum evolution tree methods.
Prediction of B-Cell Epitopic Region
B-cell epitopes are molecules recognized by the antibodies of the immune system and can elicit a proper humoral immune response. Identification of B-cell epitopes is one of the crucial steps in the generation of a functional epitopic vaccine. To identify the B-cell epitopes present within M-polyprotein, the IEDB immune database is used (Nielsen et al. 2003). The IEDB linear B-cell prediction can employ several methods to recognize promising epitopes. However, the present study adopted the Bepipred Linear Epitope Prediction method to identify potential epitopic elements that are further used for vaccine design construction (Larsen et al. 2006).
T-Cell Epitope Prediction Within the B-Cell Epitope
The presence of a T-cell epitope within a protein chain can initiate cell-mediated cytotoxicity. Instead of separating B and T-cell epitope, the common epitopes are much more effective in an immune system (Bhattacharya et al. 2019). To get the common B and T-cell epitopes, identified B-cell epitopes are processed through the Proped-I and Proped servers for MHC-I and MHC-II molecule, respectively (Bhasin and Raghava 2007; Dikhit et al. 2017, 2018; Singh and Raghava 2003). The servers identify T-cell epitopes that can bind with 51 MHC-II and 47 MHC-I allele components (Mustafa and Shaban 2006). The servers use a matrices prediction algorithm to acquire the efficient T-cell epitope (Lin et al. 2008).
3D Structure Prediction of Epitopes
3D structure of an epitope has a pivotal role in the construction of purposeful vaccine design. For constructing the epitope model, the DISTILL 2.0 server was used (Baú et al. 2006). The server possesses two types of recurrent neural networks for structure prediction that are bidirectional (Pollastri et al. 2002). The result is filtered through two succeeding stages before output. The input was magnified via prophecies in the first stage and averaged in multiple adjoining windows.
If σj = (αj, βj, γj) is an output of j, as helix, strand and coil correspondingly as input then in the second stage, Ij will be the input rather than j-
where kf = j + f(2ω + 1), 2ω + 1 shown the window size (ω = 7) and 2p + 1 signify considered window numbers (p = 7) (Pollastri and Mclysaght 2005).
Vaccine Construction and Modeling
Further, to construct a multi-epitopic vaccine design against Bunyamwera orthobunyavirus virus, adjuvant and selected epitopes are joined together with the help of linker peptides. The 3D structure of a vaccine protein is critical for gaining access to the immune system. Therefore, the 3D structure of the vaccine protein has been modeled via RaptorX web server, a self-activating system for 3D structure modeling of amino acid sequences (Källberg et al. 2012). It only needs a FASTA input to form a 3D model of the query sequence. Protein modeling is currently served as the leading technique to generate dependable 3D protein structure models and is customarily employed in several practical applications (Rout et al. 2020; Sushila et al. 2020).
Structure Validation
Validation of the protein model is remarkable for the establishment of its 3D structure. The superior structure of the protein model was validated through the ProSA and PROCHECK web-based server. The PROCHECK was used to evaluate the stereochemical parameters of polypeptide sequences and overall structure geometry (Laskowski et al. 1993). All residues within the Ramachandran plot output are critical for the validation of protein structure. Accordingly, the ProSA server was appointed to compute the ‘Z’ score of the protein model within the known ‘Z’ score range (Wiederstein and Sippl 2007). ProSA also estimates the energy of amino acid residues and furnishes an energy plot that is critical for model validation. Both servers need a standard PDB file of the desired model as an input component to assess model quality.
Antigenicity Analysis
The antigenic appearance of an epitope is a decisive feature to gain access within the immune system. The VaxiJen (v2.0) server was used to characterize the antigenic quality of the selected epitopes (Doytchinova and Flower 2007a). The server predicts immunogens applying independent alignment methods (Droppa-Almeida et al. 2018). Thus, the prediction of antigenic epitope was solely based on physiochemical parameters of the protein rather than the existing amino acid sequence (Mehla and Ramana 2016). The prediction technique has five targeted fields based on the bio-component (viz. virus, bacteria, fungi, parasite and tumor). The real-time accuracy of the server fluctuates from 70% to 89% with the change of targeted substances (Doytchinova and Flower 2007b). An auto cross-covariance (ACC) process was also incorporated to maintain the uniform length of the deposited sequence (Doytchinova and Flower 2007a). The following formulas anticipate the ACC
| 1 |
| 2 |
where ‘j’ is the presented by z-scale (j = 1,2,3), ‘l’ is the lag (l = 1, 2….L), ‘n’ is the amino acid represented (n = 1, 2…n). Two different scales are evaluated by Eq. (2).
Molecular Docking Analysis
Molecular docking is one of the critical steps for successful vaccine development. In this research work, PatchDock web server has been allotted to perform the protein–protein docking of the selected molecule (Duhovny et al. 2002). A geometry-based algorithm was utilized for the completion of molecular docking (Schneidman‐Duhovny et al. 2003). For docking analysis, the PDB files of both proteins were used. Best docking complexes are ranked against geometric shape complimentary with Atomic Contact Energy (ACE) value. The ACE is computed by the following formula.
Inside two atoms set S1 and S2 with distance d. ACE is
where, |s–t| significance Euclidean distance amid s and t, T [s,t] signifies the prearranged score of s and t atom pair.
T[s,t] is estimated using the subsequent formula
Here, ‘0’ indicates the solvent. (Ns,t) represent a connection between s, t and another connection of s-0 show (Nt,0) appropriated connection number of expected complexes. In addition, Cs,t, and Cs,0 indicate connections of s,t, and s-0 (Guo et al. 2012).
Molecular Dynamics Simulation
Molecular dynamics simulation was applied to appraise vaccine stability using NAMD2.14b2_Win64-multicore software package to select the suitable CHARMM force-field parameters (Kalé et al. 1999; MacKerell et al. 2000). In this prospect, the designed vaccine was a solvated water molecule in a cubic box of 5 × 7 × 9 nm3 performed on VMD1.9.3 software (Humphrey et al. 1996). Here, we set the NVT thermodynamic parameter for running NAMD simulation and ensembled it at 310 K and 1.013 atm pressure. RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation) assessed the whole dynamic simulation of the vaccine protein and showed its actual stability for its binding to the receptor molecule. The molecular dynamics study technique and parameters were adapted from earlier research (Rout et al. 2019; Rout el at. 2021).
Normal Mode Analysis (NMA) Analysis
The iMODS server was used for Normal Mode Analysis (López-Blanco et al. 2014). The iMODS server assists two homologous structures and large macromolecules for molecular structure exploration. The internal coordinate of NMA naturally arranges the atomic motion of biological macromolecules with respect to functional activities. The characteristic of internal coordinate formulation progresses effectively for NMA and extends its entire stereochemistry maintenance (Kovacs et al. 2004). The server is provided with B-factor, the deformability of structural complex, elastic network, variance, co-variance map, and Eigenvalue required energy for structural distortion. The sated equations give the potential energy of atomic link (harmonic springs).
| 3 |
where Fij represents the spring stiffness matrix, rij represents the atomic distance of I and j; 0, t stands for equilibrium and current formation. The modes are generated from Lagrangian equations using eigenvalue problem (Noguti and Gō 1983).
| 4 |
where H indicates Hessian derivative of potency, T indicates kinetic energy, matrix X = (x1, x2, … xn) holds eigenvectors and Λ = diag (λ1, λ2, …, λn) indicates diagonal matrix having eigenvalues. The common elucidation in internal coordinates, q, is given by
| 5 |
where ak and δk vary with initial conditions, xk is kth eigenvector, and its allied frequency is vk. The docking complex is deposited to iMODS with default parameters.
In Silico Cloning of Vaccine
In order to conduct the cloning of vaccine construct and its expression within an expression vector, the reverse translation and optimization of the codon is a vital footstep. For reverse translation and codon optimization, the Java Codon Adaptation Tool (Jcat) was utilized (Grote et al. 2005). The server provides optimized nucleotide sequence, codon adaptive index (CAI), and GC content in percentile value (Pandey and Prajapati 2019). The optimum CAI value for gene expression within target bacteria is 1.0, but a value between 0.8 and 1.0 is considerable (Narula et al. 2018). Henceforth, the GC content of the optimized codon should have resided within 30–70% range for successful expression. The expression vector pET28a ( +) was selected for cloning, and its sequences were retrieved from ‘addgene’ vector database (Kamens 2015). Finally, precise cloning was performed using SnapGene v5.1.7 software (Biotech, 2015).
Results
Multiple Sequence Alignment and Phylogenetic Analysis of Bunyamwera Serotypes
The MSA analysis results show a high level of sequence similarities between the M-polyprotein of Bunyamwera serogroup. The MSA is shown in the supplementary file (F-1), demonstrating a consensus score of 97, which is significant. The amino acids with violet and green color represent bad alignment, whereas yellow and red signify average and good alignments, respectively. Most of the residues within the result are depicted in red color, indicating a good alignment. From the result of MSA, it can be supposed that the amino acid compositions of these proteins follow a more or less similar pattern. As the properties and functions of the proteins solely rely upon their amino acid composition, these proteins resemble their characteristics and functionality (Edgar and Batzoglou 2006). Therefore, it can be concluded that any therapeutics element developed against M-polyprotein will be effective for all the serogroup of Bunyamwera. Phylogenetic tree of the 18 nos. of M-polyprotein sequences was constructed using MEGA-X software through the maximum likelihood method (Jones et al. 1992). The main aim for employing the maximum likelihood method is to establish the pattern of branching i.e., tree topology, length of the branches, and parameters of the evolutionary model that maximize the chances of proper sequence study (Strimmer and Haeseler 1996). The phylogenetic tree of all the Bunyamwera serogroup reflects their evolutionary history. The tree’s branching shares 99% of sequence identity (Fig. 1B), reflecting that they are present in proximity to each other. The tree also elaborates that the M-polyprotein is evolutionarily conserved within the serogroup of the Bunyamwera virus.
B-Cell Epitopes Prediction
The FASTA sequence of M-polyprotein of Bunyamwera orthobunyavirus was deposited to the IEDB online server with default parameters for obtaining potential B-cell epitopes. Residues with a higher value than the given threshold of 0.35 are designated as B-cell epitope and are graphically presented in Fig. 3. At this given threshold, the sensitivity and specificity of the server are 49% and 75%, respectively (Larsen et al. 2006). The IEDB server predicted 55 nos. of B-cell epitopes, which are presented in supplementary Table S1 along with their amino acid sequence, length, and position.
Fig. 3.

Graphical interpretation of B-cell epitopes (red line indicates the given threshold while yellow and green represents epitopic and non-epitopic portions, respectively) (Color figure online)
T-Cell Epitope Prediction Within the B-Cell Epitope
The T-cell epitopes are predicted from earlier identified B-cell epitopes using Proped and Proped-I servers. Three 9meric epitopes YQPTELTRS, YKAHDKEET, and ILGTGTPKF, emerged as potential epitopes common to both the B-cell and T-cell. The B-cell epitopes are reduced to nine amino acids sequence in common epitopes, as the Proped predicted only 9meric epitopes (Patra et al. 2020). The 9meric epitopes that are shown in Table 1 are highly conserved T-cell epitopes and common to MHC- I, MHC-II allele, and B-cell. These epitopes are the most potent candidate for vaccine construction as they can trigger both B-cell and T-cell mediated immunogenicity.
Table 1.
Common epitopes for B-cell and T-cell along with the encountered MHC alleles and amino acids position of epitopes
| Sequence | MHC-I allele | MHC-II allele | Position |
|---|---|---|---|
| YQPTELTRS |
HLA-B*2702 HLA-B*5401 HLA-B*51 |
DRB1_0401 DRB1_0426 |
716–724 |
| YKAHDKEET |
HLA-A20 Cattle HLA-B*5401 |
DRB1_1114 DRB1_1302 |
782–790 |
| ILGTGTPKF |
HLA-A3 HLA-A2.1 HLA-B*2702 HLA-B*5301 HLA-B*51 HLA-B62 MHC-Ld |
DRB1_0421 | 1172–1180 |
Modeling of the Epitopes
The DISTILL (V.2) webserver was specially appointed for giving precise three-dimensional architecture of amino acid having small sequences (≥ 400 aa) (Baú et al. 2006). The chosen epitopic sequences (YQPTELTRS, YKAHDKEET, and ILGTGTPKF) were submitted to DISTILL for structural configurations. The server offers five structures of the query sequence in a PDB file format, but only the top-ranked structure of each epitope is used for further structural analysis. The 3D images of epitopes are graphically generated with the help of UCSF Chimera software from the PDB data and are presented in supplementary Fig. S1(A, B, and C) (Pettersen et al. 2004).
Vaccine Construction and Modeling
In this work, the three common epitopes were merged with the help of A(EAAAK)4ALEA(EAAAK)4A peptide linker. The linker peptide has the ability to improve the biological activity and expression of the targeted proteins (Chen et al. 2013). These epitopes are also linked with an adjuvant using the same linker component for constructing the vaccine element. The proposed vaccine model is graphically represented in Fig. 4. In that context, the bacterial flagellin (Vibrio cholerae V51) was taken as a vaccine adjuvant to enhance the immune response. The tertiary structure of the vaccine construct has been modeled in the RaptorX web server and represented in Fig. 5.
Fig. 4.
Graphical arrangement of final vaccine construct from epitopes using different linkers
Fig. 5.

3D structure of the final vaccine candidate construct (helices are in yellow color while beta-sheets are colored green) (Color figure online)
Justification of Vaccine Model
Proper justification is critically helpful for establishing the 3D configuration of predicted vaccine construct. The Ramachandran plot by PROCHECK validation server reveals that 91.2%, 8.2%, 0.6%, and 0% non-glycin and non-proline residues exist in most favored, additionally allowed, generously allowed, and disallowed regions, respectively (Table 2). The Ramachandran plot of all residues has been represented in Fig. 6. The presence of more than 90% residues in the most favored region justified the model as a valid one (Laskowski et al. 2006). Within the ‘Z’ score plot of another validation server ProSA, the model lays within ‘Z’ score range of experimentally established proteins (Fig. 7) and ‘Z’ score for this model is −3.36. The energy for this model seems to be significantly low (Fig. 8) that is also a characteristic of good quality protein model (Ramatenki et al. 2015).
Table 2.
Plot statistics of all amino acids of vaccine construct
| Amino acid position | Number | Percentage (%) | Total |
|---|---|---|---|
| Favored region | 145 | 91.2 | 159 |
| Addition allowed region | 13 | 8.2 | |
| Generously allowed region | 1 | 0.6 | |
| Disallowed region | 0 | 0 | |
| End-residues | 2 | ||
| Glycine residues | 2 | ||
| Proline residues | 2 | ||
| Total residues | 165 |
Fig. 6.

Ramachandran plot for all residues of vaccine construct
Fig. 7.
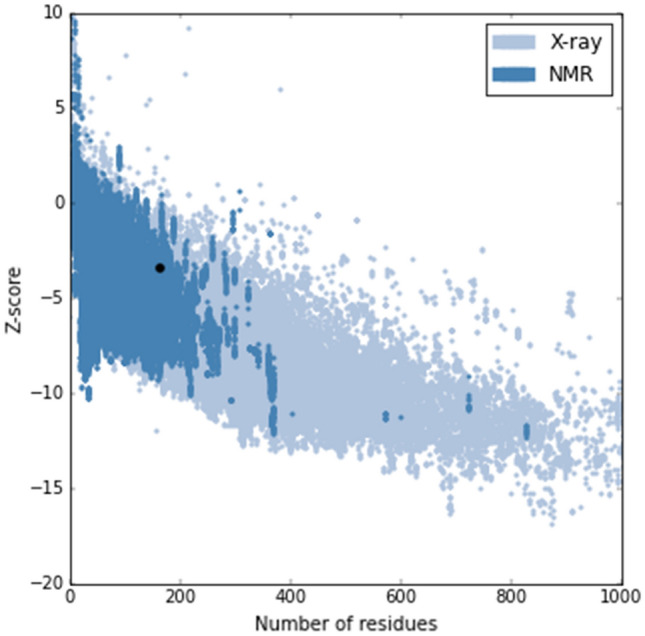
The ‘Z’ score of vaccine construct (black dot) within the ‘Z’ score range of experimentally proved structure (Color figure online)
Fig. 8.

Energy plot of amino acid of vaccine construct
Antigenicity Prediction
Antigenicity is a fundamental parameter to express the pathogenicity and construction of vaccines over the epitopic region (Chen et al. 2007). The constructed vaccine sequence was deposited to VaxiJen v.2.0 server selecting the virus as a targeted organism. The computed antigenic score of the vaccine construct is 0.4657, and the server designated it as a probable antigen being laid over the threshold score of 0.4.
Molecular Docking Analysis
The PDB file of both TLR4 (TLR4/MD2 complex) and vaccine construct are submitted to PatchDock server for efficient molecular docking. The significantly low Atomic Contact Energy (ACE) value-containing complex was chosen for analysis among the provided docking complexes. The selected docking complex having an ACE value of −43.49 kcal/mol, concluded high reactivity between the vaccine construct and TLR4 (TLR4/MD2 complex). Furthermore, the PyMOL molecular visualization program had been applied to analyze and visualize the docking complex (Schrodinger 2010). The vaccine constructs and TLR4 (TLR4/MD2 complex) docking complex are shown in Fig. 9. Protein–protein docking has a significant role in regulating multiple cellular functions (Chen et al. 2016). In the present work, the docking complex with a notably low ACE value implies that the vaccine construct will indeed combine with the TLR4 (TLR4/MD2 complex) and initiate the required inflammatory immune response (Fig. 10).
Fig. 9.

Molecular docking complex of final vaccine construct and TLR4 (TLR4/MD2 complex) (green color shows the final vaccine structure and red color is the receptor protein) (Color figure online)
Fig. 10.

The TLR4 (TLR4/MD2 complex) protein-mediated inflammatory immune response pathway initiated by the vaccine construct
Molecular Dynamics Simulations of Vaccine Construct
Energy minimizing of vaccine construct uses CHARMM force-field parameter; performed in NAMD 2.14 software. In order to measure the vaccine residue’s flexibility during the time of simulation, the RMSF of all residues was computed and presented in Fig. 11A. Slight fluctuations of most of the residues indicate that vaccine molecules might be recognized perdurable interactions with the receptor protein. RMSD fluctuation plot also defines the structure stability and is represented in Fig. 11B. NAMD plot of Timestep (TS) and protein atomic (Cα) angle are demonstrated in Fig. 11C.
Fig. 11.

Molecular dynamics (MD) simulation of vaccine constructs. A Root mean square fluctuation (RMSF) plot. B Root mean square deviations (RMSD) plot, C) NAMD plot of TS vs. atomic angle (Cα)
Normal Mode Analysis
The docking complex’s stability and its large-scale motility are analyzed through Normal Mode Analysis (NMA). Each residue of the proteins is dynamic to a particular direction, presented by arrows. The resultant motion is shown with a large arrow. From the result of NMA, it was confirmed that the vaccine construct is directed towards the TLR4 (TLR4/MD2 complex) and in vice-versa mode (Fig. 12A). The resultant B-factor shows consistency with the NMA, and in some portions, it is a little bit fluctuating (Fig. 12F). The presence of a few hinges or deformable residues within the deformability plot reflects that the docking complex shows a higher degree of structural stability (Fig. 12E). The connection matrix of paired atomic residue is presented in the covariance matrix map, where the rigidness of the protein complex increases with the color gradient from blue to red (Fig. 12B) (Stetz and Verkhivker 2015). The connection springs are displayed as an elastic network where every dot is representative of springs, while the stiffness of springs increases or decreases with the darkness of every dot (Fig. 12C). The Eigenvalue can be stated as the value proportional to the energy required for deforming the stability of the docking complex (Hasan et al. 2019). The Eigenvalue for the docking complex is 6.067450e−06 which seems to be significantly high for deforming this complex (Fig. 12D).
Fig. 12.

NMA analysis A Mobility of the docking complex vaccine construct and TLR4 (TLR4/MD2 complex) are indicated with arrows, B Covariance matrix map of atomic pair of residues, C Connection spring map of the elastic network model, D Eigenvalue for the docking complex, E Deformability plot of the docking complex, F Calculated B-factor of NMA and PDB B-factor
In Silico Cloning of Vaccine
In silico cloning of the vaccine construct was performed inside the pET28a ( +) expression vector within E. coli K12 strain after codon optimization within the JCat server. The CAI value of the optimized codon is 1.0, which is the most appropriate for the expression of the vaccine construct. The GC content of the optimized sequence is 50.73%, which resides within the optimum range of 30–70%. After confirming the CAI and GC value of the optimized codon, Xho1 and NcoI restriction sites were combined with the optimized codon. Finally, the optimized codon was cloned in pET28a ( +) expression vector (Fig. 13).
Fig. 13.

In silico cloning and structure of the vaccine construct in E. coli (k12 strain) using vector pET28a ( +), restrictions sites
Discussion
Subjective scientific findings and precise construction of potential drug targets are decisive steps to discover the present-day drug. Despite the available antiviral drugs, the vaccine has a central role in eradicating viral infection effectively and precisely (Shepard et al. 2006). Existing research architecture focuses on embarking the probable vaccine targets design against B. orthobunyavirus, which might be mainly responsible for manifesting severe illness in humans, birds, and domestic animals (Dutuze et al. 2018; Vasconcelos and Calisher 2016). Remarkably, vaccine development through the immunoinformatics approach could be a functional and necessary strategy to resist pathogenic infection. Therefore, this biocomputational work focused on the reverse vaccinology approach specifically to develop an epitopic vaccine against the Bunyamwera orthobunyavirus. The MSA outputs of the M-polyprotein protein of Bunyamwera serogroup demonstrate higher levels of amino acid sequence similarity. Based on the constructed phylogeny in MEGA v.X, it is clear that the experimental protein is present in a highly conserved manner among the Bunyamwera serogroup. They show overall 99% similarities in their amino acids’ composition within the M-polyprotein component. Thus, the M-polyprotein was served as a prime target for vaccine designing as it is conserved within the Bunymwera orthobunyavirus. The protein was subjected to B-cell epitope analysis within the IEDB server using the BepiPred method. Subsequently, the 55 emerging B-cell epitopes were again finely processed in Proped and Proped-I server to identify the common epitopes that can interact with B-cell, MHC-II, and MHC-I molecules. The B-cells are able to generate a humoral immune response against viral infections and subsequently develop antibodies with the help of T-helper cells (Dörner and Radbruch 2007). T-cell epitopes generate a cellular immune response with the help of cytotoxic T-cells that is much more effective against viral infections (Rojas et al. 2017). Only three 9mer epitopes (YQPTELTRS, YKAHDKEET, and ILGTGTPKF) were identified as common B-cell and T-cell epitopes, which were further mapped out using the DISTILL v 2.0 server. Together with A(EAAAK)4ALEA(EAAAK)4A peptide linker element, these three epitopes were linked together. Bacterial flagellin (Vibrio cholerae V51) protein was linked with the designed epitopes as an adjuvant applying the same linker to trigger a proper inflammatory response. Subsequently, the constructed vaccine was modeled through the RaptorX web server and validated through the ProSA and PROCHECK servers. The presence of 91.2% residues within the most favored region and the ‘Z’ score value for the model -3.36 within the experimental ‘Z’ score range establishes the model quality. The studies of MD simulation assess the structural stability of the vaccine construct (Figs. 11 and 12). The result of molecular simulation indicates that vaccine construct might be docked with receptor with its proper dimension. The VaxiJen score for vaccine construct is 0.4657, which may also designate as a potential antigen. While the molecular docking between TLR4 (TLR4/MD2 complex) protein and vaccine construct exhibited a highly negative ACE value (−43.49 kcal/mol), it is too critical for spontaneous and high reactivity. Subsequently, such docking also reveals that the vaccine adjuvant will combine with TLR4 (TLR4/MD2 complex) and generate an inflammatory immune response, a promising milestone to control the viral infection (Bhattacharya et al. 2020a). The protein simulation consequence shows the docking complex as stable, as reflected by the high Eigenvalue (6.067450e−06). Besides that, the in-silico cloning techniques clear out as an artificial procedure to amplify the vaccine element for its accurate application. The constructed vaccine has marked effectiveness to limit the robust infectivity of Bunyamwera orthobunyavirus.
Conclusions
The vaccine targeted for the M-polyprotein of Bunyamwera orthobunyavirus might substantially contain the viral pathogenicity. Hence, the current research design will prove a precious drive to lead the modern therapeutics to resist the infection of Bunyamwera orthobunyavirus and other related diseases. Common B-cell and T-cell epitopes (YQPTELTRS, YKAHDKEET, and ILGTGTPKF) will be the key elements to evoke humoral and cell-mediated immunity aligned with the viral infection. The conjugated adjuvant will trigger the TLR4 (TLR4/MD2 complex) protein pathway and generate an inflammatory immune response against the viral infection of Bunyamwera orthobunyavirus. Therefore, this immunoinformatic research might be a powerful and potent technique for constructing epitopic vaccines against several viral diseases.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 (TIFF 1048 kb)
Fig. S1: 3D structure of Epitopes A) YQPTELTRS, B) YKAHDKEET and C) ILGTGTPKF
Supplementary file3 (PDF 159 kb)
File F1: Multiple Sequence Alignment (MSA) result of M polyprotein, Bunyamwera orthobunyavirus and serogroup (T-Coffee Server)
Author Contributions
PG, MB and ARS conceptualized the methodology and data retrieval, GS, BCP, PP did the formal analysis validation. PG, MB. ARS and CC wrote, reviewed, and edited the manuscript. CC and SSL supervised the whole study. All the authors finalized and approved the manuscript.
Funding
None.
Data Availability
All data includes within the manuscript.
Declarations
Conflict of interest
The authors declare no competing interests.
Ethical Approval
Not required.
Consent to Participate
Not required.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Pratik Ghosh and Manojit Bhattacharya have contributed equally.
Contributor Information
Ashish Ranjan Sharma, Email: researchskeletal@gmail.com.
Chiranjib Chakraborty, Email: drchiranjib@yahoo.com.
References
- Ashford RW (2001) Encyclopedia of arthropod-transmitted infections of man and domesticated animals. CABI ISBN:9781845933166; 608
- Baú D, Martin AJ, Mooney C, Vullo A, Walsh I, Pollastri G (2006) Distill: a suite of web servers for the prediction of one-, two-and three-dimensional structural features of proteins BMC Bioinformatics 7:402 [DOI] [PMC free article] [PubMed]
- Bhasin M, Raghava G. A hybrid approach for predicting promiscuous MHC class I restricted T cell epitopes. J Biosci. 2007;32:31–42. doi: 10.1007/s12038-007-0004-5. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Malick RC, Momdal N, Patra P, Pal BB, Patra BC, Das BK. Computational characterization of epitopic region within the outer membrane protein candidate in Flavobacterium Columnare for vaccine development. J Biomol Struct Dyn. 2019;38:450–459. doi: 10.1080/07391102.2019.1580222. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Sharma AR, Mallick B, Sharma G, Lee S-S, Chakraborty C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex Infection. Genet Evol. 2020;85:104587. doi: 10.1016/j.meegid.2020.104587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M, Sharma AR, Sharm G, Patra P, Mondal N, Patr BC, Lee S-S, Chakraborty C. Computer aided novel antigenic epitopes selection from the outer membrane protein sequences of Aeromonas hydrophila and its analyses Infection. Genet Evol. 2020;82:104320. doi: 10.1016/j.meegid.2020.104320. [DOI] [PubMed] [Google Scholar]
- Biotech G (2015) SnapGene® software (from Insightful Science; available at snapgene.com)
- Cavazzana M, Six E, Lagresle-Peyrou C, Andre-Schmutz I, Hacein-Bey-Abina S. Gene therapy for X-linked severe combined immunodeficiency: where do we stand? Hum Gene Ther. 2016;27:108–116. doi: 10.1089/hum.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Liu H, Yang J, Chou K-C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids. 2007;33:423–428. doi: 10.1007/s00726-006-0485-9. [DOI] [PubMed] [Google Scholar]
- Chen X, Zaro JL, Shen W-CJAddr (2013) Fusion protein linkers: property, design and functionality. Adv Drug Del Rev 65:1357–1369 [DOI] [PMC free article] [PubMed]
- Chen F, Liu H, Sun H, Pan P, Li Y, Li D, Hou T (2016) Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking Physical Chemistry Chem Phy 18:22129–22139 [DOI] [PubMed]
- Dikhit MR, Kumar A, Das S, Dehury B, Rout AK, Jamal F, Sahoo GC, Topno RK, Pandey K, Das VN, Bimal S. Identification of potential MHC Class-II-restricted epitopes derived from Leishmania donovani antigens by reverse vaccinology and evaluation of their CD4+ T-cell responsiveness against visceral leishmaniasis. Front Immunol. 2017;8:1763. doi: 10.3389/fimmu.2017.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikhit MR, Das S, Mahantesh V, Kumar A, Singh AK, Dehury B, Rout AK, Ali V, Sahoo GC, Topno RK, Pandey K (2018) The potential HLA Class I-restricted epitopes derived from LeIF and TSA of Leishmania donovani evoke anti-leishmania CD8+ T lymphocyte response. Scientific Rep 8(1):1–3. [DOI] [PMC free article] [PubMed]
- Dörner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity. 2007;27:384–392. doi: 10.1016/j.immuni.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR (2007a) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform 8:4. 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed]
- Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007;25:856–866. doi: 10.1016/j.vaccine.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Droppa-Almeida D, Franceschi E, Padilha FF. Immune-informatic analysis and design of peptide vaccine from multi-epitopes against Corynebacterium Pseudotuberculosis. Bioinform Biol Insights. 2018;12:1177932218755337. doi: 10.1177/1177932218755337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhovny D, Nussinov R, Wolfson HJ (2002) Efficient unbound docking of rigid molecules. In: International workshop on algorithms in bioinformatics. Springer, pp 185–200, Berlin, Heidelberg.
- Dutuze MF, Nzayirambaho M, Mores CN, Christofferson RC. A review of bunyamwera Batai, and Ngari viruses: understudied orthobunyaviruses with potential one health implications. Front Veterinary Sci. 2018;5:69. doi: 10.3389/fvets.2018.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Batzoglou S. Multiple sequence alignment. Curr Opin Struct Biol. 2006;16:368–373. doi: 10.1016/j.sbi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Geer LY, et al. The NCBI biosystems database. Nucleic Acids Res. 2010;38:D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:W526–W531 [DOI] [PMC free article] [PubMed]
- Guo F, Li SC, Wang L, Zhu D. Protein-Protein binding site identification by enumerating the configurations. BMC Bioinform. 2012;13:158. doi: 10.1186/1471-2105-13-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J, Khan MMH (2019) Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microbial Pathogene 130:19–37 [DOI] [PubMed]
- He Y, Xiang Z, Mobley HLJJoB, Biotechnology (2010) Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol 2010: 297505. 10.1155/2010/297505 [DOI] [PMC free article] [PubMed]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graphics 14:33–38 [DOI] [PubMed]
- Jones DT, Taylor WR, Thornton JMJB. The rapid generation of mutation data matrices from protein sequences. J Mol Graphics. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Kalé L et al. (1999) NAMD2: greater scalability for parallel molecular dynamics. J Comput Phys 151:283–312
- Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamens J. The Addgene repository: an international nonprofit plasmid and data resource. Nucleic Acids Res. 2015;43:D1152–D1157. doi: 10.1093/nar/gku893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi A, Chuah C, Majeed ABA, Leow CH, Lim BH, Leow CY. Current progress of immunoinformatics approach harnessed for cellular-and antibody-dependent vaccine design. Pathogens Global Health. 2018;112:123–131. doi: 10.1080/20477724.2018.1446773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl A, Clayton RF, Weber F, Bridgen A, Randall RE, Elliott RM (2003) Bunyamwera virus nonstructural protein NSs counteracts interferon regulatory factor 3-mediated induction of early cell death. J Virol 77:7999–8008 [DOI] [PMC free article] [PubMed]
- Kovacs JA, Chacón P, Abagyan R. Predictions of protein flexibility: first-order measures. Proteins Struct Funct Bioinformat. 2004;56:661–668. doi: 10.1002/prot.20151. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA x: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Lanciotti RSJJOCM. Consensus amplification and novel multiplex sequencing method for S segment species identification of 47 viruses of the Orthobunyavirus Phlebovirus, and Nairovirus Genera of the Family Bunyaviridae. J Clin Microbiol. 2009;47:2398–2404. doi: 10.1128/JCM.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Research. 2006;2:1–7. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallography 26:283–291
- Laskowski R, MacArthur M, Thornton J (2006) PROCHECK: validation of protein-structure coordinates in International Tables of Crystallography, Volume F. In Crystallography of Biological Macromolecules (eds Rossmann, M. G. & Arnold, E. D.) 722–725 (Kluwer Academic Publishers, The Netherlands)
- Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008;9:1–3. doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P (2014) iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res 42:W271-W276 [DOI] [PMC free article] [PubMed]
- MacKerell AD, Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolym Origin Res Biomol. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Mazel-Sanchez B, Elliott RM. Attenuation of bunyamwera orthobunyavirus replication by targeted mutagenesis of genomic untranslated regions and creation of viable viruses with minimal genome segments. J Virol. 2012;86:13672–13678. doi: 10.1128/JVI.02253-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlock J, Snow K, Leach S. Possible ecology and epidemiology of medically important mosquito-borne arboviruses in Great Britain. Epidemiol Infect. 2007;135:466–482. doi: 10.1017/S0950268806007047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehla K, Ramana J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: a comparative genomics and immunoinformatics approach. Mol BioSyst. 2016;12:890–901. doi: 10.1039/c5mb00745c. [DOI] [PubMed] [Google Scholar]
- Mustafa AS, Shaban FA. ProPred analysis and experimental evaluation of promiscuous T-cell epitopes of three major secreted antigens of Mycobacterium tuberculosis. Tuberculosis. 2006;86:115–124. doi: 10.1016/j.tube.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Narula A, Pandey RK, Khatoon N, Mishra A, Prajapati VK. Excavating chikungunya genome to design B and T cell multi-epitope subunit vaccine using comprehensive immunoinformatics approach to control chikungunya infection Infection. Genet Evol. 2018;61:4–15. doi: 10.1016/j.meegid.2018.03.007. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguti T, Gō N. Dynamics of native globular proteins in terms of dihedral angles. J Phys Soc Jpn. 1983;52:3283–3288. [Google Scholar]
- Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217 [DOI] [PubMed]
- Odhiambo C, Venter M, Lwande O, Swanepoel R, Sang R (2016) Phylogenetic analysis of Bunyamwera and Ngari viruses (family Bunyaviridae, genus Orthobunyavirus) isolated in Kenya Epidemiology & Infection 144:389–395 [DOI] [PubMed]
- Pandey RK, Prajapati VK. Exploring sand fly salivary proteins to design multiepitope subunit vaccine to fight against visceral leishmaniasis. J Cell Biochem. 2019;120:1141–1155. doi: 10.1002/jcb.26719. [DOI] [PubMed] [Google Scholar]
- Patra P, Bhattacharya M, Sharma AR, Ghosh P, Sharma G, Patra BC, Mallick B, Lee SS, Chakraborty C. Identification and design of a next-generation multi epitopes bases peptide vaccine candidate against prostate cancer: an in silico approach. Cell Biochem Biophys. 2020;78:495–509. doi: 10.1007/s12013-020-00912-7. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis J Comput Chem 25:1605–1612 [DOI] [PubMed]
- Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins Struct Funct Bioinformatics. 2002;47:228–235. doi: 10.1002/prot.10082. [DOI] [PubMed] [Google Scholar]
- Pollastri G, Mclysaght A (2005) Porter: a new, accurate server for protein secondary structure prediction Bioinformatics 21:1719–1720 [DOI] [PubMed]
- Ramatenki V, Potlapally SR, Dumpati RK, Vadija R, Vuruputuri U (2015) Homology modeling and virtual screening of ubiquitin conjugation enzyme E2A for designing a novel selective antagonist against cancer. J Receptors Signal Transduct 35:536–549 [DOI] [PubMed]
- Rojas J-M, Rodríguez-Calvo T, Sevilla N. Recall T cell responses to bluetongue virus produce a narrowing of the T cell repertoire. Vet Res. 2017;48:38. doi: 10.1186/s13567-017-0444-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout AK, Mishra J, Dehury B, Maharana J, Acharya V, Karna SK, Parida PK, Behera BK, Das BK. Structural bioinformatics insights into ATP binding mechanism in zebrafish (Danio rerio) cyclin-dependent kinase-like 5 (zCDKL5) protein. J Cell Biochem. 2019;6:9437–9447. doi: 10.1002/jcb.28219. [DOI] [PubMed] [Google Scholar]
- Rout AK, Paramanik S, Dehury B, Acharya V, Swain HS, Pradhan SK, Behera B, Pati SK, Behera BK, Das BK (2020). Elucidating the molecular interaction of Zebrafish (Danio rerio) peptidoglycan recognition protein 2 with diaminopimelic acid and lysine type peptidoglycans using in silico approaches. J Biomol Struct Dyn 38(12):3687–99. [DOI] [PubMed]
- Rout AK, Acharya V, Maharana D, Dehury B, Udgata SR, Jena R, Behera B, Parida PK, Behera BK (2021) Insights into structure and dynamics of extracellular domain of Toll-like receptor 5 in Cirrhinus mrigala (mrigala): a molecular dynamics simulation approach. PloS One. 16(1):e0245358. [DOI] [PMC free article] [PubMed]
- Schneidman-Duhovny D, Inbar Y, Polak V, Shatsky M, Halperin I, Benyamini H, Barzilai A, Dror O, Haspel N, Nussinov R, Wolfson HJ. Taking geometry to its edge: fast unbound rigid (and hinge-bent) docking. Proteins Struct Funct Bioinformat. 2003;52:107–112. doi: 10.1002/prot.10397. [DOI] [PubMed] [Google Scholar]
- Schrodinger LJV (2010) The PyMOL molecular graphics system 1:0. Version 1.3r1,” Schrodinger, LLC, New York
- Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP. Hepatitis B virus infection: epidemiology and vaccination. Epidemiologic Rev. 2006;28:112–125. doi: 10.1093/epirev/mxj009. [DOI] [PubMed] [Google Scholar]
- Shi X, et al. Bunyamwera orthobunyavirus glycoprotein precursor is processed by cellular signal peptidase and signal peptide peptidase. Proc Natl Acad Sci. 2016;113:8825–8830. doi: 10.1073/pnas.1603364113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Raghava G (2003) ProPred1: prediction of promiscuous MHC class-I binding sites. Bioinformatics 19:1009–1014 [DOI] [PubMed]
- Soldan SS, González-Scarano F. Emerging infectious diseases: the bunyaviridae. J NeuroVirol. 2005;11:412–423. doi: 10.1080/13550280591002496. [DOI] [PubMed] [Google Scholar]
- Stetz G, Verkhivker GM (2015) Dancing through life: Molecular dynamics simulations and network-centric modeling of allosteric mechanisms in Hsp70 and Hsp110 chaperone proteins PLoS One 10:e0143752 [DOI] [PMC free article] [PubMed]
- Strimmer K, Von Haeseler A. Quartet Puzzling: a Quartet Maximum-Likelihood Method for Reconstructing Tree. Topologies Mol Biol Evol. 1996;13:964–969. [Google Scholar]
- Sushila N, Das BK, Mir IN, Rout AK, Prasad KP, Tripathi G (2020). Cloning, characterization and ontogenetic expression profile of RAG-2 and IgM in angelfish (Pterophyllum scalare). Gene 739:144496. [DOI] [PubMed]
- Thompson JD, Plewniak F, Poch OJNar. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 1999;27:2682–2690. doi: 10.1093/nar/27.13.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos PF, Calisher CH (2016) Emergence of human arboviral diseases in the Americas, 2000–2016. Vector-Borne Zoonotic Dis 16:295–301 [DOI] [PubMed]
- Vivona S, Gardy JL, Ramachandran S, Brinkman FS, Raghava G, Flower DR, Filippini F. Computer-aided biotechnology: from immuno-informatics to reverse Vaccinology. Trends Biotechnol. 2008;26:190–200. doi: 10.1016/j.tibtech.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Wallace IM, O’sullivan O, Higgins DG, Notredame C (2006) M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res 34:1692–1699 [DOI] [PMC free article] [PubMed]
- Wertheim HF, Horby P, Woodall JP. Atlas of human infectious diseases. Wiley; 2012. [Google Scholar]
- Wiederstein M, Sippl M (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407–W410 [DOI] [PMC free article] [PubMed]
- Yanase T, Kato T, Yamakawa M, Takayoshi K, Nakamura K, Kokuba T, Tsuda T. Genetic characterization of Batai virus indicates a genomic reassortment between orthobunyaviruses in nature. Adv Virol. 2006;151:2253–2260. doi: 10.1007/s00705-006-0808-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file2 (TIFF 1048 kb)
Fig. S1: 3D structure of Epitopes A) YQPTELTRS, B) YKAHDKEET and C) ILGTGTPKF
Supplementary file3 (PDF 159 kb)
File F1: Multiple Sequence Alignment (MSA) result of M polyprotein, Bunyamwera orthobunyavirus and serogroup (T-Coffee Server)
Data Availability Statement
All data includes within the manuscript.