

Abstract
Bacterial phospholipases and sphingomyelinases are lipolytic esterases that are structurally and evolutionarily heterogeneous. These enzymes play crucial roles as virulence factors in several human and animal infectious diseases. Some bacterial phospholipases C (PLCs) have both phosphatidylcholinesterase and sphingomyelinase C activities. Among them, Listeria monocytogenes PlcB, Clostridium perfringens PLC, and Pseudomonas aeruginosa PlcH are the most deeply understood. In silico predictions of substrates docking with these three bacterial enzymes provide evidence that they interact with different substrates at the same active site. This review discusses structural aspects, substrate specificity, and the mechanism of action of those bacterial enzymes on target cells and animal infection models to shed light on their roles in pathogenesis.
Keywords: bacterial pathogenesis, bacterial phospholipases, bacterial sphingomyelinases, bacterial toxins, virulence factors
The aim of this review is to discuss similarities and differences among the most explored bacterial enzymes that have both phospholipase C and sphingomyelinase C activities. The structural aspects of three of those enzymes, their substrate specificity, and in silico substrate docking analysis are discussed, as well as their mechanisms of toxicity in cultured cells and animal models.

Abbreviations
- CHO
Chinese hamster ovary
- CpPLC
Clostridium perfringens PLC
- EDTA
ethylenediaminetetraacetic acid
- EGTA
egtazic acid
- HUVEC
human umbilical vein endothelial
- LLO
listeriolysin O
- LmPlcB
Listeria monocytogenes PLC
- Mpl
Listerial metalloprotease
- PaPlcH
Pseudomonas aeruginosa PlcH
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PI
phosphatidylinositol
- PLCs
phospholipases C
- PS
phosphatidylserine
- ROS
reactive oxygen species
- SM
sphingomyelin
- TrkA
Tyr kinase A receptor
Bacterial phospholipases and sphingomyelinases are a structurally and evolutionarily heterogeneous group of lipolytic esterases [1, 2, 3]. These bacterial enzymes generate metabolites identical to the second messengers produced by eukaryotic enzymes, which play essential roles in physiological processes [1]. They are critical in the pathogenesis of various infectious diseases as they favor bacterial invasion and survival [1, 2, 3]. In some cases, they cause lysis of the host cells, thereby helping pathogens acquire some nutrients from the host, such as iron, phosphate and alternative carbon sources [1, 2, 3]. Some of those bacterial enzymes alter membrane lipid homeostasis by changing the balance of signaling molecules and thus diverting cellular processes driven by lipids to the benefit of the bacteria [1, 2, 3]. They may hydrolyze vacuolar lipids, causing phagosomal escape or phagocytosis hindrance, favoring intracellular survival, immune response evasion, and infection establishment [1, 2, 3].
Although most bacterial lipolytic esterases have either phospholipase or sphingomyelinase activity, some of them cleave glycerophospholipids and sphingomyelin (SM) [1]. Among them, Listeria monocytogenes PLC (LmPlcB), Clostridium perfringens PLC (CpPLC), and Pseudomonas aeruginosa PlcH (PaPlcH) are the most deeply explored. CpPLC and LmPlc are zinc metalloenzymes, whereas PaPlcH belongs to the PLC/phosphatase superfamily. This review aims to discuss the similarities and differences among those bacterial enzymes. Although the regulation of their expression is beyond the scope of this review, the structural aspects of LmPlcB, CpPLC, and PaPlcH, their substrate specificity and mechanisms of toxicity both in cultured cells and animal models are discussed below.
The pathogenic bacteria that produce these phospholipases
L. monocytogenes is a Gram‐positive facultative intracellular bacterium that causes severe foodborne infections [4]. The bacterium begins its intracellular life cycle within a membrane‐bound vacuole from which it must escape to survive [3, 4]. Once into the host cytosol, L. monocytogenes multiplies and spreads from cell to cell using an actin‐based mechanism of motility [3, 4]. This spreading event forms a double‐membrane vacuole from which the bacterium escapes to perpetuate the intracellular life cycle [3, 4]. L. monocytogenes secretes several toxins that favor phagosomal escape, including LmPlcB and a phosphatidylinositol‐PLC (PI‐PLC) [3, 5]. Once bacteria have entered the bloodstream through the intestinal epithelium and lymph nodes, they can invade the liver, the spleen, and the brain [3, 4].
C. perfringens is a Gram‐positive anaerobic bacterium that produces more than 17 toxins [6]. This bacterium causes several diseases in humans and animals [7, 8, 9]. CpPLC, also called alpha‐toxin, is produced by all C. perfringens strains and is the main virulence factor in gas gangrene in humans [10]. This disease is an acute and life‐threatening soft tissue infection characterized by significant thrombosis, severe myonecrosis, and gas accumulation at the infection site [11]. Gas gangrene occurs after the introduction of the bacteria in a deep lesion or in a surgical wound [11]. CpPLC helps C. perfringens escape the phagosome and persist in the cytoplasm of macrophages during the earliest infection stages [11]. Then, CpPLC contributes by creating an anaerobic environment, optimal for bacterial growth, and spread [11].
P. aeruginosa is a Gram‐negative opportunistic pathogen associated with acute and chronic infections in predisposed human subjects [12]. P. aeruginosa lung infections are common and severe in chronic obstructive pulmonary disease and cystic fibrosis [12]. P. aeruginosa PlcH releases phosphocholine from SM and phosphatidylcholine (PC), the major components of the pulmonary surfactant, thus aiding in bacterial dispersion [12].
Structural features comparison
LmPlcB is a 33 kDa single‐domain zinc metalloenzyme secreted as a precursor. Its activation requires the cleavage of an amino‐terminal pro‐peptide by either a listerial metalloprotease (Mpl) or host proteases, resulting in the active 29 kDa enzyme [13, 14, 15]. A His residue in Mpl plays a role in the interaction with LmPlcB for cleavage [16]. LmPlcB shares 38.7% sequence identity as with B. cereus PLC [15], which binds three Zn+2 ions at the active site, although LmPlcB has a considerably weaker affinity for Zn+2 than the B. cereus enzyme [14].
The LmPlcB amino acid sequence (Accession number Q6EAJ1) was retrieved from the UniProt database [17] (https://www.uniprot.org/) to build a structural model with SWISS‐MODEL [18] (http://swissmodel.expasy.org/) using B. cereus PLC (PDB accession No. 1ah7) as a template. The predicted structure shows a α‐helical single‐domain protein with three zinc atoms, as visualized using Discovery Studio Visualizer (Fig. 1A).
Fig. 1.
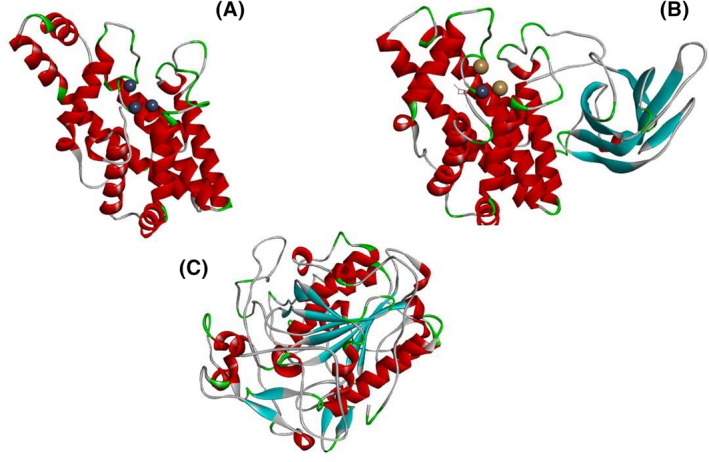
Cartoon representation of the tertiary structure of three bacterial PLCs, which play a role as virulence factors: (A) L. monocytogenes PlcB model built using as template B. cereus PLC (PDB accession No. 1ah7). (B) C. perfringens alpha‐toxin three‐dimensional structure (PDB accession No. 1ca1). (C) P. aeruginosa PaPlcH model built using as template the Francisella tularensis Acid Phosphatase A (PDB accession No 2d1g). The α‐helices, β‐sheets, and loops are labeled in red, cyan, and gray, respectively.
CpPLC is a 45 kDa zinc metalloenzyme composed of two domains joined by a short flexible linker region [19, 20]. The N‐terminal domain (residues 1–246) contains the active site and consists of nine tightly packed alpha‐helices [19]. CpPLC contains three essential Zn+2 atoms in its N‐terminal domain, as also observed for B. cereus [21] and L. monocytogenes PLC [13], and these could be removed by ethylenediaminetetraacetic acid (EDTA) or o‐phenanthroline. Asp56 is a critical residue in the Zn+2 binding site of CpPLC [22], and the D56G substitution changes the secondary structure and abolishes toxicity [23]. The C‐terminal domain (residues 256–370) consists of an eight‐stranded antiparallel beta‐sandwich required for the Ca+2‐dependent interaction with substrates [19, 24]. A ‘closed’ form of CpPLC has been described, with one Zn+2 lost and a hidden catalytically active site, and with an additional alpha helix that binds to the N‐ and C‐terminal domains [20]. An in silico study of CpPLC evidences at least six pairs of amino acid contacts between N‐ and C‐terminal domains [25]. The structure of C. perfringens alpha‐toxin (PDB accession No. 1ca1) is shown in Fig. 1B.
Structurally, CpPLC lacks two helices in the N‐terminal domain present in B. cereus PLC [19], resulting in a planar surface that interacts directly with the target membrane. CpPLC could can anchor the membrane by its Ca+2 binding sites in the C‐terminal domain and orienting its active catalytic site to interact directly with membrane phospholipids [19]. The absent B. cereus helix hairpin is replaced with a tryptophan residue, which, together with several hydrophobic residues in the C‐terminal domain (Phe334, Tyr 331), is well placed to interact with the hydrophobic membrane interior. In addition, the C‐terminal domain, analogous to C2 domains, binds Ca+2 ions but does not complete their coordination spheres, and has many positively charged residues (such as Lys300) placed to interact with the phosphate at the membrane surface [1, 19]. These adaptations for membrane interaction likely explain the higher cytotoxicity of CpPLC compared to its B. cereus homologue [7, 19].
PaPlcH does not share any structural similarity with LmPlcB or CpPLC, but has 23% sequence identity, as the acid phosphatase AcpA from F. tularensis [26, 27]. Thus, PaPlcH is the paradigm member of the PLC/phosphatase superfamily, with members in various prokaryotic species, including M. tuberculosis, Bordetella spp., F. tularensis, and Burkholderia spp, and homologs in fungi (Aspergillus fumigatus) and plants (Arabidopsis) [27]. PaPlcH is secreted via the twin‐arginine translocation and type II Xcp‐dependent systems, and it forms a multimeric complex with the chaperones PlcR1 or PlcR2 [28, 29, 30, 31].
To build a structural model of PaPlcH, its amino acid sequence (Accession number P06200) was retrieved from the UniProt database (https://www.uniprot.org/) [17]. The PaPlcH three‐dimensional structure was modeled with SWISS‐MODEL using the F. tularensis Acid Phosphatase A (PDB accession No 2d1g) as a template [18] (http://swissmodel.expasy.org/). The predicted structure (Fig. 1C) was visualized using Discovery Studio Visualizer.
Substrate specificity comparison
LmPlcB hydrolyzes PC, SM, phosphatidylethanolamine (PE), phosphatidylserine (PS), PI, cardiolipin, phosphatidylglycerol (PG), plasmalogens, and plasmenylethanolamine [13, 32]. The hydrolysis mechanism of PI by LmPlcB differs from that of L. monocytogenes PI‐PLC because it generates a different product [32]. Accordingly, recombinant LmPlcB hydrolyses a range of phospholipids with different head groups [14]. On large unilamellar vesicles, LmPlcB has the highest activity on PE, PS‐rich mixtures, which are expected to be the main phospholipids on the inner membrane of the double‐membrane vacuole that is in contact with L. monocytogenes during cell‐to‐cell spread [33]. Its enzymatic activity has been widely related to disrupting the primary vacuole in certain cell types [34, 35, 36, 37, 38] and spreading vacuoles in others [39, 40]. The dual enzymatic activity of LmPlcB allows membrane fusion, which could be important for L. monocytogenes cell‐to‐cell spreading [33]. Interestingly, phosphocholine, a product of LmPLC enzymatic activity on PC, is a potent inhibitor of Listeriolysin O (LLO), a pore‐forming toxin secreted by L. monocytogenes that has cytotoxic effects on host cells during infection, thus promoting bacterial intracellular survival [41].
Docking studies with PC and SM were used for visualizing their interaction with the LmPlcB catalytically active site (Fig. 2).
Fig. 2.
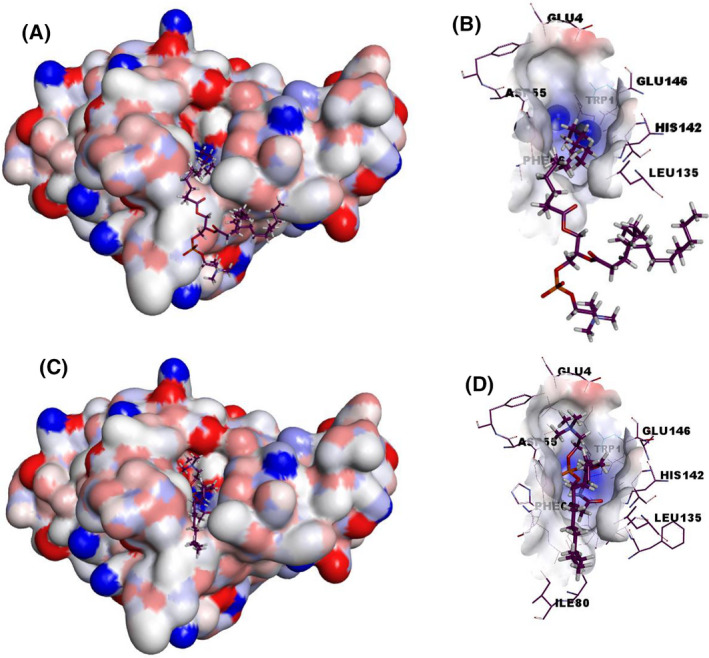
The electrostatic potential surface of LmPlcB with docked PC and SM. The generated model of LmPlcB (Fig. 1A) was used as a target for docking studies. SwissDock web service (http://www.swissdock.ch/docking) was used to predict the molecular interactions that may occur with different phospholipids: PC (PubChem CID:5497103) (A and B) and SM (ZINC56870813) (C and D). The docked structures were visualized using Discovery Studio Visualizer.
LmPlcB is catalytically active at a wide pH range (5.0–8.0) [13, 32], although the recombinant protein exhibits optimal activity at acidic pH [14]. During cell‐to‐cell spread of Listeria, a decrease in pH triggers the metalloprotease Mpl, known to activate pro‐LmPlcB [42]. This pH change increases the active mature form of LmPlcB, which helps bacteria escape from the double‐membrane vacuole [42]. One mechanism by which pH regulates Mpl protease activity is by influencing the charge of a His residue that acts as a pH sensor [16]. Activation of pro‐LmPlcB is also mediated by an Mpl‐independent pathway, utilizing host cysteine proteases [43]. Finally, LmPlcB catalytic activity is regulated by vacuolar pH, and its compartmentalization to the spreading vacuole is critical for intracellular survival in neutrophils [44].
CpPLC hydrolyzes PC and SM [45, 46, 47, 48], with phosphatidylcholinesterase activity being five‐fold higher than sphingomyelinase C activity [49]. This enzyme also hydrolyzes PE, PI, PS, and PG [50]. This broader substrate specificity might allow C. perfringens to infect different tissues and help the bacterium escape the early phagosome toward the macrophage cytoplasm [50, 51]. Using Langmuir monolayer, vesicles, and red blood cells, it has been shown that CpPLC acts in a two‐stage process with an initial, rapid hydrolysis stage followed by membrane insertion when the hydrolysis products change the membrane dipole potential [52].
Docking studies with PC and SM were used for visualizing their interaction with the CpPLC catalytically active site (Fig. 3).
Fig. 3.
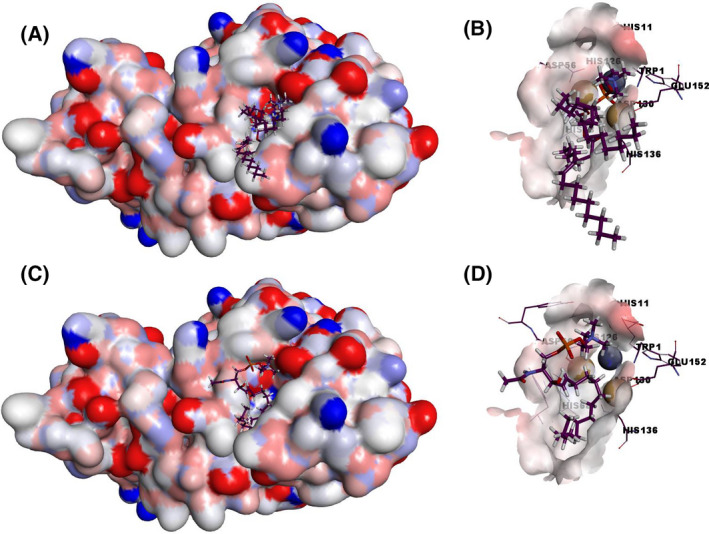
The electrostatic potential surface of CpPLC with docked PC and SM. The structure of CpPLC (PDB accession no. 1ca1) was used as a target for docking studies. SwissDock web service (http://www.swissdock.ch/docking) was used to predict the molecular interactions that may occur with PC (PubChem CID:5497103) (A and B) and SM (ZINC56870813) (C and D). The docked structures were visualized using the Discovery Studio Visualizer.
PaPlcH hydrolyzes mainly PC and SM, but it also acts on cardiolipin, PE, and PG when assayed on large unilamellar vesicles [53, 54]. However, the catalytic mechanism of PaPlcH is different from that of the zinc‐metalloPLCs. PaPlcH enzymatic activity is not affected by cation chelators [EDTA and egtazic acid (EGTA)], and instead of requiring zinc for activity, it is inhibited by Zn+2 and several divalent cations [27]. Furthermore, PaPlcH is not affected by the zinc‐metalloPLCs inhibitor D609 [27].
Figure 4 shows docking studies of PaPlcH with PC and SM. The results show that both ligands bind to a similar location of the phosphate‐interacting and active sites residues of the ApcA structure, including the putative PaPlcH nucleophile Thr178.
Fig. 4.
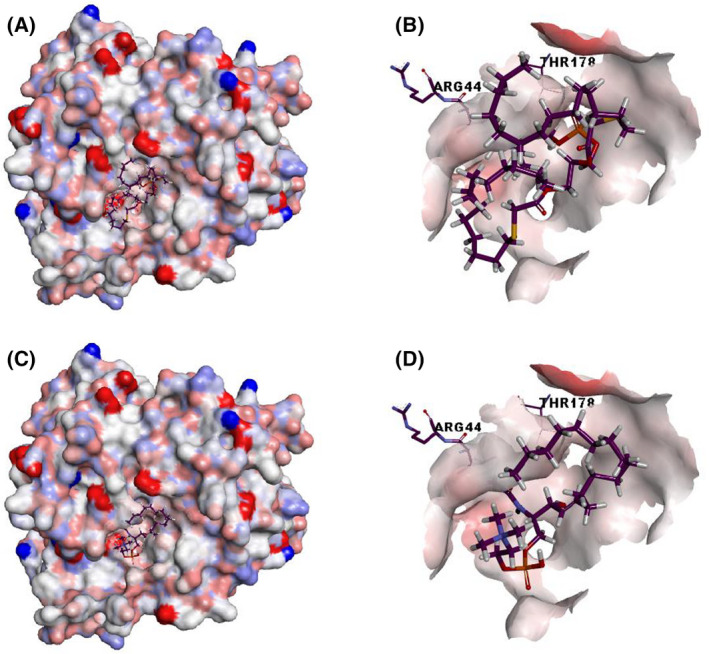
The electrostatic potential surface of PaPlcH with docked PC and SM. The generated model of PaPlcH (Fig. 1C) was used as a target for docking studies. SwissDock web service (http://www.swissdock.ch/docking) was used to predict the molecular interactions that may occur with PC (PubChem CID:5497103) (A and B) and SM (ZINC56870813) (C and D). The docked structures were visualized using Discovery Studio Visualizer.
PaPlcH partially purified from a clinical isolate showed enzymatic activity from pH 5.5–8, optimal at 7–8 [54]. However, studies on the effect of pH on the enzymatic activity with recombinant PlcH remain to be done.
The substrates for the bacterial enzymes discussed in this review and their corresponding optimal pH are summarized in Table 1.
Table 1.
Broad substrate specificities and optimal pH from LmPlcB, CpPLC, and PaPlcH.
| Enzyme | Substrates | pH activity | References |
|---|---|---|---|
| L. monocytogenes PlcB | PC, SM, PE, PG, PI, Cardiolipin, Plasmalogens, Plasmenylethanolamine (Glycerol acetal), PS |
5.0–8.0 optimal: 5.0–6.0 |
[13, 14, 32, 33] |
| C. perfringens PLC | PC, SM, PE, PG, PI, PS |
4.5–7.5 PC: optimal 5.0–6.0 SM: optimal 7.0 |
[45, 47, 49, 50] |
| P. aeruginosa PlcH | PC, SM, PE, PG, Cardiolipin, Plasmalogens, Lyso‐PC |
5.5–8.0 Optimal 7.0–8.0 |
[27, 53, 54] |
Mechanisms of cytotoxicity
Effects on erythrocytes
Iron is an essential resource for bacteria, and in vertebrate hosts, high amounts of iron are incorporated into hemoglobin [2]. Thus, accessing the iron stored in erythrocytes may constitute a significant advantage for bacterial growth during infections [2]. The three described Phospholipases C (PLCs) are hemolytic to a certain degree, and other effects on red blood cells are just starting to be elucidated, as described below.
LmPlcB is hemolytic, and this activity requires the presence of PC in the plasma membrane of the target erythrocytes [55]. Erythrocytes containing PC in their membranes, such as those of guinea pig, horse, and human, are hemolyzed by the enzyme. In contrast, sheep erythrocytes, which almost lack PC in their membrane, are resistant to hemolysis [13, 14].
Initial studies with C. perfringens supernatants evidenced their hemolytic activity when injected into pigeons, sheep, and rabbits [56]. This activity was optimal at 37 °C on the rabbit and human erythrocytes, whereas the hemolysis increased after cooling down the preparations to 4 °C in sheep erythrocytes [56, 57]. Further studies with highly purified CpPLC confirmed its hot‐cold hemolytic activity toward sheep erythrocytes [58, 59, 60]. CpPLC hemolyses horse erythrocytes by hydrolyzing mainly PC and sheep erythrocytes by hydrolyzing SM [57, 61]. Later studies using recombinant CpPLC showed that it causes hot‐cold hemolysis in red blood cells from different species [62, 63, 64, 65, 66]. Site‐directed mutants of CpPLC affecting either the Ca+2 or the Zn+2 binding domain, which affect catalytic activity against aggregated substrates, lack hemolytic activity [22, 62, 63, 64]. CpPLC lacking the linker between the C‐ and the N‐Terminal domains does not induce hemolysis of murine red blood cells, demonstrating that the interaction of both domains is necessary for its hemolytic activity [64, 65, 66].
A two‐stage interaction of CpPLC and human erythrocytes was shown using thermal shape fluctuation spectroscopy [52]. This finding supports a model in which the toxin is first incorporated into the membrane and second exerts its hydrolytic action [52]. At low concentrations, CpPLC induces morphological changes and a decrease of the membrane dipole potential that affects the erythrocyte membrane function [52]. At higher CpPLC concentrations, the morphological changes are accompanied by hemoglobin loss [52]. Additionally, CpPLC impairs erythropoiesis by inhibiting erythroid differentiation in mouse bone marrow cells [67].
PaPlcH caused hot‐cold hemolysis of human and sheep erythrocytes [27]. The hot‐cold hemolysis induced by this enzyme also occurs on goat erythrocytes lacking PC, but not on horse erythrocytes with PC but almost no SM. It suggests that SMase activity is critical for inducing hot‐cold hemolysis [68]. Accordingly, a neutral ceramidase encoded by a gene adjacent to PaPlcH enhances its hemolytic activity [69].
Effects on platelets
CpPLC causes platelet aggregation in vitro and in vivo [24, 70, 71, 72]. This aggregation is induced by the translocation of the fibrinogen receptor gpIIbIIIa from internal stores to the membrane [73]. The effect of CpPLC on gpIIbIIIa involves intracellular calcium increments mediated by plasma membrane calcium channels [74]. Similarly, PaPlcH causes human platelet aggregation, which is dependent on its enzymatic activity [75].
Effects on nucleated cells
The cellular effects of PLCs with phosphatidylcholinesterase and SMase activities have been evaluated on different cell lines, as discussed below.
A double mutant of both the LmPlcB and the PI‐PLC genes shows severely diminished cell‐to‐cell spread effect in a plaque assay with L2 fibroblasts [5]. Accordingly, an LmPlcB mutant induces fewer zones of dead cells when compared to the wild‐type bacteria [39]. J744 macrophage monolayers treated with an LmPlcB mutant of L. monocytogenes observed under electron microscopy showed significantly less lysis of the two‐membrane vacuoles that surround the bacteria after cell‐to‐cell spread than the wild‐type [39]. Infection studies on J744 macrophages have also helped to elucidate the proteolytic pathways that lead to activation of LmPlcB [43].
In murine bone marrow‐derived macrophages, both PI‐PLC and LmPlcB facilitate membrane disruption by acting on the inner membrane of the spreading vacuole, whereas LLO is required to degrade the outer membrane [40]. Studies with L. monocytogenes mutants lacking LLO and LmPlcB or Mpl showed that the last two are required for bacterial growth and lysis of the primary vacuole in epithelial cells, suggesting that the relative importance of LLO, the phospholipases, and Mpl varies in different cell types [34]. Accordingly, LLO is not required for lysis of L.fnd="ER"> monocytogenes‐containing primary vacuoles in some epithelial cell lines, such as Hep‐2 and HeLa [35, 36] or dendritic cells [37]. LmPlcB is specifically required for lysis of the primary vacuole of Hep‐2 and Hela cells in the absence of LLO [38]. An LLO negative‐LmPlcB inducible double mutant was also unable to establish cell‐to‐cell spread on Henle 407 cells without continuous LmPlcB induction during infection [38]. In a plaque assay created to visualize cell‐to‐cell spread from human macrophage U937 toward human epithelial Hep‐2 cells, it was determined that both PI‐PLC and LmPlcB are required to mediate escape from heterologous double‐membrane spreading vacuoles in the absence of LLO expression [40]. Furthermore, LmPlcB and PI‐PLC are critical for increasing ceramide, activating NFkB, and up‐regulating E‐selectin, as required for neutrophil adhesion to endothelial cells [76, 77].
The first report of the action of CpPLC on nucleated cells was to determine the membrane damage effect on human diploid fibroblasts and Hela cells [78]. By labeling these cells with 3H‐uridine and detecting the leakage of radioactive compounds, CpPLC treatment for 30 min was found to result in 25% of radioactivity released at concentrations of 16 µg·mL−1, reaching 100% maximum release after 1 h associated with the hydrolysis of membrane phospholipids [78]. Sarcoma 180 cells showed increased sensitivity toward CpPLC, whereas L1210 leukemia cells were relatively resistant to the cytotoxic effects of this enzyme [79]. CpPLC‐treated sarcoma cells also exhibited PC hydrolysis, alterations in size and shape, microvilli loss, and fistulas in cell membranes [79]. Later, a newly isolated fibroblast mutant, Don Q, had higher sensitivity to CpPLC than the wild‐type [80] due to a single‐point mutation in the UDP‐glucose pyrophosphorylase gene leading to UDP‐glucose deficiency [81, 82]. Since UDP‐glucose is a precursor of ganglioside synthesis, this mutation reduces cellular gangliosides in the outer cell membrane, which explains the increased susceptibility of Don Q to CpPLC. The gangliosides present in the cell membrane probably exert a steric effect or induce an electrostatic charge preventing the toxin from reaching its substrate [83]. Muscle is known to have the lowest concentration of complex gangliosides among all mammalian tissues [84, 85]. This may explain why muscle fibers present higher susceptibility to the cytotoxic effect of CpPLC during gas gangrene [83]. This hypersensitive cell line has been used to evaluate the cytotoxic activity of different CpPLC mutants and as a model to elucidate the mechanism of action of this toxin [62, 64, 83, 86, 87].
Several other cell types, such as cultured intestinal epithelial cells, Chinese hamster ovary (CHO), and Hela cells, have been used to study the effects of DAG generation induced by CpPLC, such as arachidonic acid release, PKC, PLA2, and calcium activation [88, 89, 90, 91, 92]. CpPLC action on endothelial cells showed DAG‐mediated induction of prostacyclin and platelet‐activating factor and increased adhesion molecules, both in endothelial cells and neutrophils, contributing to the process of vascular permeability and neutrophil recruitment [93]. Further studies in neutrophils showed that CpPLC induces respiratory burst through different signaling pathways, involving Tyr kinase A receptor (TrkA) and G proteins, leading to PKC theta and MEK‐ERK activation [94, 95]. Accordingly, CpPLC induces reactive oxygen species (ROS) production in hypersensitive cells through PKC or MEK‐ERK‐NFkB pathways that play an essential role in its cytotoxic/myotoxic effect [87, 96]. Epithelial cells have been used to demonstrate that CpPLC, TrkA, and GM1a ganglioside clustering activates the p38 MAPK pathway and PLC‐gamma1, ERK‐NFkB pathways to generate TNF‐alpha and IL‐8, important mediators of inflammation [97, 98, 99]. CpPLC induces the expression of granulocyte colony‐stimulating factor in human umbilical vein endothelial (HUVEC) cells via JNK activation [100]. Further studies showed that CpPLC causes apoptosis of endothelial cells by increasing ceramide [101]. Interestingly, the myoblast C2C12 cells are more resistant to ceramide‐induced cell death than HUVEC cells, suggesting that endothelial damage could be a primary effect caused by CpPLC [101].
PaPlcH induces the release of prostaglandins and leukotrienes in human granulocytes and mouse peritoneal cells [102]. PaPlcH is cytotoxic to mouse peritoneal macrophages and human mononuclear leukocytes and neutrophils [103]. Purified PaPlcH at low concentrations induces a dose‐dependent increase in IL‐8 expression in human isolated monocytes, which could be important during pathogenesis because IL‐8 is probably the main cause of the excessive neutrophil recruitment associated with pulmonary inflammation and destruction [104]. A PaPlcH mutant was significantly less cytotoxic to a human monocytic cell line (THP‐1) and a human T‐cell line (Jurkat) than the wild‐type strain, having a synergistic effect with the P. aeruginosa extracellular protease LepA [105].
PaPlcH suppresses respiratory burst in neutrophils by interfering with a PKC‐dependent pathway [106]. Thus, a bacterial strain mutant containing a targeted deletion of the plcHR operon induces a more robust oxidative burst than the wild‐type [106]. Furthermore, the addition of purified PaPlcH to the DeltaHR‐stimulated neutrophils suppresses superoxide production induced by phorbol esters to levels stimulated by wild‐type bacteria [106].
PaPlcHR is slightly cytotoxic to lung epithelial cells from a patient with cystic fibrosis and to HeLa cells, and highly toxic (at picomolar concentrations) to CHO and HUVEC [107]. PaPlcHR induces calcium signaling and caspase 3 activation on endothelial cells, and the selectivity toward endothelial cells is mediated by an RGD motif, probably by binding a subset of integrins [107]. The toxin also inhibits migration, invasion, and tubule formation by HUVEC, which points toward an antiangiogenic effect of PaPlcH [107].
Effects on animal models of infection
Studying the effects of bacterial PLCs with phosphatidylcholinesterase and SMase activities or bacterial mutant strains lacking their corresponding genes in animal models has revealed important insights of these enzymes in virulence.
While LmPlcB is not toxic even when up to 25 µg is injected intravenously into mice [13], a L. monocytogenes mutant strain lacking LmPlcB is 20‐fold less lethal to mice [5]. Mice infected with the LmPlcB mutant survived cerebral listeriosis longer and have a reduced intracerebral bacterial load than mice infected with the wild‐type strain [108]. An mpl mutant that fails to produce mature LmPlcB showed impaired virulence in mice compared with a wild‐type strain and, at later stages of infection, exhibits less growth in mouse liver and spleen [109]. It has been shown that LmPlcB activation by Mpl inside vacuoles is important for its virulence because an LmPlcB pro‐mutant, secreted as an already mature and active enzyme, is strongly attenuated in a mouse infection model [110]. Later, it was found that the LmPlcB pro‐mutant interferes less with mitochondrial homeostasis than wild‐type strain, and is more susceptible to intracellular killing by peritoneal mice neutrophils, which explains its impaired virulence [44].
CpPLC is the main virulence factor in gas gangrene [7, 10]. Intramuscular injection of recombinant CpPLC causes myonecrosis and histopathological features of gas gangrene [93]. Immunization with the recombinant C‐terminal domain of this toxin protects mice from the intramuscular challenge of C. perfringens [111]. A C. perfringens plc mutant cannot induce gas gangrene in mice [10, 112]. CpPLC induces myonecrosis, leucocyte accumulation, thrombosis, and neutrophil arrestment in experimental gas gangrene [10, 112]. CpPLC variants, lacking the C‐terminal domain, mutated in residues of Ca+2 binding, or lacking surface‐exposed hydrophobic residues, failed to produce myonecrosis in a gas gangrene murine model [48, 62, 64, 113]. The importance of inflammatory mediators, such as TNF‐alpha, and the role of ROS production in CpPLC toxic effect has been demonstrated in vivo [96, 114]. Studies indicated that granulocyte colony‐stimulating factor does not reduce CpPLC induced myonecrosis in mice [100]. On the other hand, CpPLC decreases endothelial cell counts in mouse muscles infected with C. perfringens, which could contribute to myonecrosis progression [101].
PaPlcH causes dermonecrosis, vascular permeability, and death when injected intraperitoneally into mice [115]. This enzyme induces an inflammatory response characterized by the accumulation of inflammatory cells and the release of arachidonic acid metabolites [102]. PaPlcH induces recruitment and activation of platelets at the endothelium, leading to thrombotic lesions similar to those observed in P. aeruginosa sepsis [107]. PaPlcH in the respiratory tract induces inflammation, high levels of tumor necrosis factor‐alpha, interleukin 1‐beta, IL‐6, gamma interferon, MIP‐1 alpha, and MIP‐2 in the lungs [116]. A P. aeruginosa strain PaPlcH mutant exhibits decreased virulence in a murine burn infection model [117]. Similarly, in a mouse model of acute systemic infection by P. aeruginosa, a PaPlcH mutant showed reduced virulence compared to the wild‐type strain [105]. The numbers of mutant bacteria recovered in peritoneal lavage fluid and blood are lower than the numbers of the wild‐type strain [105].
Phosphorylcholine release from the pulmonary surfactant lipids by PlcH during lung infections provides nutrients to P. aeruginosa, and activates biofilm formation and anaerobic metabolism [118, 119, 120]. Furthermore, Cer accumulation can contribute to the pathogenesis of P. aeruginosa lung infections by inhibiting the function of the cystic fibrosis transmembrane conductance regulator Cl channel in epithelial cells, leading to thick mucus production that clogs the airways, which fosters bacterial growth [121].
Concluding remarks
Bacterial PLCs with phosphatidylcholinesterase and sphingomyelinase C activities, such as LmPlcB, CpPLC, and PaPlcH, can exert diverse toxic effects. As observed in in silico docking assays while acting over different substrates, they generate 1,2‐diacylglycerol and ceramide, which change the biophysical properties of cellular target membranes. These products can undergo spontaneous transbilayer movement and recruit signaling proteins, affecting cellular processes such as cell cycle arrest, stress responses, autophagy, apoptosis, and cytokine production. The particular role of the bacterial enzymes in pathogenesis depends on the context of the infection. LmPlcB is activated when L. monocytogenes reaches the acidic environment of late endosomes‐lysosomes, aiding bacterial escape toward the cytosol and cell‐to‐cell propagation. On the other hand, when C. perfringens reaches a certain density in soft tissues and CpPLC is produced, this toxin induces endothelial damage and platelet aggregation, which favors ischemia, thus providing an optimal environment for bacterial growth. Finally, PaPlcH secreted by P. aeruginosa, hydrolyzes target phospholipids where the infection is being established, helps bacteria to obtain substrates from the host, and triggers signaling pathways that lead to inflammation. Several other bacteria such as Burkholderia pseudomallei and Mycobacterium tuberculosis produce enzymes homologous to PaPlcH, but their role in pathogenesis remains to be clarified. The data already available for LmPlcB, CpPLC, and PaPlcH can provide insights to further our understanding of their mechanism of action.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
LM‐G wrote the first draft of the manuscript; FV‐R did the docking studies; MF‐D and AA‐G review and substantively edited the manuscript. All authors read and approved the final version and take responsibility for the overall content and integrity of the work.
Acknowledgements
This work was supported by Vicerrectoría de Investigación, Universidad de Costa Rica UCR (grants 741‐B8‐137, 741‐C0‐067, and 741‐C1‐309) and Fondos del Sistema FEES‐CONARE (grant B8657).
Data accessibility
The data that support the findings of this study are available on request from the corresponding author.
References
- 1. Flores‐Díaz M, Monturiol‐Gross L, Naylor C, Alape‐Girón A, Flieger A. Bacterial sphingomyelinases and phospholipases as virulence factors. Microbiol Mol Biol Rev. 2016;80:597–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hinkel LA, Wargo MJ. (2020) Participation of bacterial lipases, sphingomyelinases and phospholipases in Gram‐positive bacterial pathogenesis. In Health Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids (Goldfine H, ed), pp. 181–202. Springer, Singapore. ISBN 978‐3‐030‐15147‐8. [Google Scholar]
- 3. Goldfine H. (2020) Participation of bacterial lipases, sphingomyelinases and phospholipases in Gram‐positive bacterial pathogenesis. In Health Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids (Goldfine H, ed), pp. 205–221. Springer, Singapore. ISBN 978‐3‐030‐15147‐8. [Google Scholar]
- 4. David DJV, Cossart P. Recent advances in understanding listeria monocytogenes infection: the importance of subcellular and physiological context. F1000Research 2017;6:1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell‐to‐cell spread. Infect Immun. 1995;63:4231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petit L, Gibert M, Popoff MR. Clostridium perfringens: toxinotype and genotype. Trends Microbiol. 1999;7:104–10. [DOI] [PubMed] [Google Scholar]
- 7. Flores‐Díaz M, Alape‐Girón A. Role of Clostridium perfringens phospholipase C in the pathogenesis of gas gangrene. Toxicon. 2003;42:979–86. [DOI] [PubMed] [Google Scholar]
- 8. Sakurai J, Nagahama M, Oda M. Clostridium perfringens alpha‐toxin: characterization and mode of action. J Biochem. 2004;136:569–74. [DOI] [PubMed] [Google Scholar]
- 9. Navarro MA, Mcclane BA, Uzal FA. Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins (Basel). 2018;10:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Awad M, Bryant A, Stevens D, Rood J. Virulence studies on chromosomal alpha‐toxin and theta‐toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha‐toxin in Clostridium perfringens‐mediated gas gangrene. Mol Microbiol. 1995;15:191–202. [DOI] [PubMed] [Google Scholar]
- 11. Stevens DL, Aldape MJ, Bryant AE. Life‐threatening clostridial infections. Anaerobe. 2012;18:254–9. [DOI] [PubMed] [Google Scholar]
- 12. Klockgether J, Tümmler B. Recent advances in understanding Pseudomonas aeruginosa as a pathogen. F1000Research. 2017;6:1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geoffroy C, Raveneau J, Beretti J, Lecroisey A, Alouf JE , Berche P, et al. Purification and characterization of an extracellular 29‐kilodalton phospholipase C from Listeria monocytogenes . Infect Immun. 1991;59:2382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Q, Gershenson A, Roberts MF. Recombinant broad‐range phospholipase C from Listeria monocytogenes exhibits optimal activity at acidic pH. Biochim Biophys Acta. 2016;1864:697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zückert WR, Marquis H, Goldfine H. Modulation of enzymatic activity and biological function of Listeria monocytogenes broad‐range phospholipase C by amino acid substitutions and by replacement with the Bacillus cereus ortholog. Infect Immun. 1998;66:4823–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Forster BM, Bitar AP, Marquis H. A non‐catalytic histidine residue influences the function of the metalloprotease of Listeria monocytogenes . Microbiology. 2014;160:142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Apweiler R, Bairoch A, Wu C, Barker W, Boeckmann B, Ferro S, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:D115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naylor CE, Eaton JT, Howells A, Justin N, Moss DS, Titball RW, et al. Structure of the key toxin in gas gangrene. Nat Struct Biol. 1998;5:738–46. [DOI] [PubMed] [Google Scholar]
- 20. Eaton JT, Naylor CE, Howells AM, Moss DS, Titball RW, Basak AK. Crystal structure of the C. perfringens alpha‐toxin with the active site closed by a flexible loop region. J Mol Biol. 2002;319:275–81. [DOI] [PubMed] [Google Scholar]
- 21. Hough E, Hansen LK, Birknes B, Jynge K, Hansen S, Hordvik A, et al. High‐resolution (1.5 Å) crystal structure of phospholipase C from Bacillus cereus . Nature. 1989;338:357–60. [DOI] [PubMed] [Google Scholar]
- 22. Nagahama M, Nakayama T, Michiue K, Sakurai J. Site‐specific mutagenesis of clostridium perfringens alpha‐toxin: replacement of Asp‐56, Asp‐130, or Glu‐152 causes loss of enzymatic and hemolytic activities. Infect Immunity. 1997;65:3489–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu C, She Y, Fu F, Lin Y, Xu C. Study of the structure and biological activity of the amino ‐ terminus of the α ‐ toxin from Clostridium welchii type A. Curr Microbiol. 2019;76:1175–85. [DOI] [PubMed] [Google Scholar]
- 24. Sugahara T, Takahashi T, Yamaya S, Ohsaka A. In vitro aggregation of platelets induced by alpha toxin (Phospholipase C) of Clostridium perfringens . Japan J Med Sci Biol. 1976;29:255–63. [PubMed] [Google Scholar]
- 25. Uppalapati SR, Kingston JJ, Qureshi IA, Murali HS, Batra HV. In silico, in vitro and in vivo analysis of binding affinity between n and c‐domains of clostridium perfringens alpha toxin. PLoS One. 2013;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montes L, Ibarguren M, Goñi FM, Stonehouse M, Vasil ML, Alonso A. Leakage‐free membrane fusion induced by the hydrolytic activity of PlcHR 2, a novel phospholipase C/sphingomyelinase from Pseudomonas aeruginosa. Biochim Biophys Acta. 2007;1768:2365–72. [DOI] [PubMed] [Google Scholar]
- 27. Stonehouse MJ, Cota‐Gomez A, Parker SK, Martin WE, Hankin J, Murphy RC, et al. A novel class of microbial phosphocholine‐specific phospholipases C. Mol Microbiol. 2002;46:661–76. [DOI] [PubMed] [Google Scholar]
- 28. Truan D, Vasil A, Stonehouse M, Vasil ML, Pohl E. High‐level over‐expression, purification, and crystallization of a novel phospholipase C/sphingomyelinase from Pseudomonas aeruginosa. Protein Expr Purif. 2013;90:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cota‐Gomez A, Vasil AI, Kadurugamuwa J, Beveridge TJ, Schweizer HP, Vasil ML. plcR1 and plcR2 are putative calcium‐binding proteins required for secretion of the hemolytic phospholipase C of Pseudomonas aeruginosa. Infect Immun. 1997;65:2904–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Voulhoux R, Ball G, Ize B, Vasil ML, Lazdunski A, Wu LF, et al. Involvement of the twin‐arginine translocation system in protein secretion via the type II pathway. EMBO J. 2001;20:6735–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snyder A, Vasil AI, Zajdowicz SL, Wilson ZR, Vasil ML. Role of the Pseudomonas aeruginosa PIcH Tat signal peptide in protein secretion, transcription, and cross‐species Tat secretion system compatibility. J Bacteriol. 2006;188:1762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldfine H, Johnston NC, Knob C. Nonspecific phospholipase‐C of Listeria‐monocytogenes ‐ activity on phospholipids in triton X‐100‐mixed micelles and in biological‐membranes. J Bacteriol. 1993;175:4298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Montes LR, Goñi FM, Johnston NC, Goldfine H, Alonso A. Membrane fusion induced by the catalytic activity of a phospholipase C/sphingomyelinase from Listeria monocytogenes. Biochemistry. 2004;43:3688–95. [DOI] [PubMed] [Google Scholar]
- 34. Marquis H, Doshi V, Portnoy DA. The broad‐range phospholipase C and a metalloprotease mediate listeriolysin O‐independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect Immunity. 1995;63:4531–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hense M, Domann E, Krusch S, Wachholz P, Dittmar K, Rohde M, et al. Eukaryotic expression plasmid transfer from the intracellular bacterium Listeria monocytogenes to host cells. Cell Microbiol. 2001;3:599–609. [DOI] [PubMed] [Google Scholar]
- 36. O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. PNAS. 2002;99:13861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paschen A, Dittmar KEJ, Grenningloh R, Rohde M, Domann E, Chakraborty T, et al. Human dendritic cells infected by Listeria monocytogenes: induction of maturation, requirements for phagolysosomal escape and antigen presentation capacity. Eur J Immunol. 2000;30:3447–56. [DOI] [PubMed] [Google Scholar]
- 38. Gründling A, Gonzalez MD, Higgins DE. Requirement of the Listeria monocytogenes during infection of human epithelial cells. J Bacteriol. 2003;185:6295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vazquez‐Boland J, Kocks C, Dramsi S, Ohayon H, Geoffroy C, Mengaud J, et al. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell‐to‐cell spread. Infect Immun. 1992;60:219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alberti‐Segui C, Goeden KR, Higgins DE. Differential function of Listeria monocytogenes listeriolysin O and phospholipases C in vacuolar dissolution following cell‐to‐cell spread. Cell Microbiol. 2007;9:179–95. [DOI] [PubMed] [Google Scholar]
- 41. La Pietra L, Hudel M, Pillich H, Mraheil MA, Berisha B, Aden S, et al. Phosphocholine antagonizes listeriolysin O‐induced host cell responses of Listeria monocytogenes . J Infect Dis. 2020;222:1505–16. [DOI] [PubMed] [Google Scholar]
- 42. Marquis H, Hager EJ. pH‐regulated activation and release of a bacteria‐associated phospholipase C during intracellular infection by Listeria monocytogenes . Mol Microbiol. 2000;35:289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marquis H, Goldfine H, Portnoy DA. Proteolytic pathways of activation and degradation of a bacterial phospholipase c during intracellular infection by Listeria monocytogenes . J Cell Biol. 1997;137:1381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blank BS, Abi Abdallah DS, Park JJ, Nazarova EV, Pavinski Bitar A, Maurer KJ, et al. Misregulation of the broad‐range phospholipase C activity increases the susceptibility of Listeria monocytogenes to intracellular killing by neutrophils. Microbes Infect. 2014;16:104–13. [DOI] [PubMed] [Google Scholar]
- 45. MacFarlane MG. The biochemistry of bacterial toxins. The lecithinase activity of Clostridium haemolyticum toxin. Biochem J. 1950;47:267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krug EL, Kent C. Phospholipase C from Clostridium perfringens: Preparation and characterization of homogeneous enzyme. Arch Biochem Biophys. 1984;231:400–10. [DOI] [PubMed] [Google Scholar]
- 47. Nagahama M, Michiue K, Sakurai J. Membrane‐damaging action of Clostridium perfringens alpha‐toxin on phospholipid liposomes. Biochim Biophys Acta. 1996;1280:120–6. [DOI] [PubMed] [Google Scholar]
- 48. Jepson M, Howells A, Bullifent HL, Bolgiano B, Crane D, Miller J, et al. Differences in the carboxy‐terminal (putative phospholipid binding) domains of Clostridium perfringens and Clostridium bifermentans phospholipases C influence the hemolytic and lethal properties of these enzymes. Infect Immun. 1999;67:3297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Urbina P, Flores‐Díaz M, Alape‐Girón A, Alonso A, Goni FM. Phospholipase C and sphingomyelinase activities of the Clostridium perfringens alpha‐toxin. Chem Phys Lipids. 2009;159:51–7. [DOI] [PubMed] [Google Scholar]
- 50. Urbina P, Collado M, Alonso A, Goñi FM, Flores‐Díaz M, Alape‐Girón A, et al. Unexpected wide substrate specificity of C. perfringens α‐toxin phospholipase C. Biochim Biophys Acta. 2011;1808:2618–27. [DOI] [PubMed] [Google Scholar]
- 51. O’Brien DK & Melville SB. Effects of clostridium perfringens alpha‐toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect Immun. 2004;72:5204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jewell SA, Titball RW, Huyet J, Naylor CE, Basak AK, Gologan P, et al. Clostridium perfringens alpha toxin interaction with red cells and model membranes. Soft Matter. 2015;11:7748–61. [DOI] [PubMed] [Google Scholar]
- 53. López DJ, Collado MI, Ibarguren M, Vasil AI, Vasil ML, Goñi FM, et al. Multiple phospholipid substrates of phospholipase C/sphingomyelinase HR2 from Pseudomonas aeruginosa . Chem Phys Lipids. 2011;164:78–82. [DOI] [PubMed] [Google Scholar]
- 54. Sherif‐Elleboudy N, Mobrouk‐Aboulwafa M, Haleen‐Hassouna NA. Phospholipase C from Pseudomonas aeruginosa and Bacillus cereus; characterization of catalytic activity. Asian Pac J Trop Med. 2014;7:860–6. [DOI] [PubMed] [Google Scholar]
- 55. Fuzi M, Pillis I. Production of opacity in egg‐yolk medium by Listeria monocytogenes . Nature. 1962;196:195.13993473 [Google Scholar]
- 56. Bull CG, Pritchett IW. Toxin and antitoxin of and protective inoculation against Bacillus welchii . J Exp Med. 1917;26:119–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Taguchi R, Ikezawa H. Studies on the hemolytic and hydrolytic actions of phospholipases against mammalian erythrocyte membranes. Arch Biochem Biophys. 1976;173:538–45. [DOI] [PubMed] [Google Scholar]
- 58. Sakurai J, Ochi S, Tanaka H. Regulation of Clostridium perfringens alpha toxin activated phospholipase C in rabbit erythrocyte membranes. Infect Immun. 1994;62:717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ochi S, Hashimoto K, Nagahama M, Sakurai J. Phospholipid metabolism induced by Clostridium perfringens alpha‐toxin elicits a hot‐cold type of hemolysis in rabbit erythrocytes. Infect Immun. 1996;64:3930–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ochi S, Oda M, Nagahama M, Sakurai J. Clostridium perfringens alpha‐toxin‐induced hemolysis of horse erythrocytes is dependent on Ca2+ uptake. Biochim Biophys Acta. 2003;1613:79–86. [DOI] [PubMed] [Google Scholar]
- 61. Ochi S, Oda M, Matsuda H, Ikari S, Sakurai J. Clostridium perfringens alpha‐toxin activates the sphingomyelin metabolism system in sheep erythrocytes. J Biol Chem. 2004;279:12181–9. [DOI] [PubMed] [Google Scholar]
- 62. Alape‐Girón A, Flores‐Díaz M, Guillouard I, Naylor CE, Titball RW, Rucavado A, et al. Identification of residues critical for toxicity in Clostridium perfringens phospholipase C, the key toxin in gas gangrene. Eur J Biochem. 2000;267:5191–7. [DOI] [PubMed] [Google Scholar]
- 63. Guillouard I, Garnier T, Cole ST. Use of site‐directed mutagenesis to probe structure‐function relationships of alpha‐toxin from clostridium perfringens. Infect Immunity. 1996;64:2440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walker N, Holley J, Naylor CE, Flores‐Diaz M, Alape‐Giron A, Carter G, et al. Identification of residues in the carboxy‐terminal domain of Clostridium perfringens alpha‐toxin ( Phospholipase C ) which are required for its biological activities. Arch Biochem Biophys. 2000;384:24–30. [DOI] [PubMed] [Google Scholar]
- 65. Titball RW, Fearn AM, Williamson ED. Biochemical and immunological properties of the C‐terminal domain of the alpha‐toxin of Clostridium perfringens . FEMS Microbiol Lett. 1993;110:45–50. [DOI] [PubMed] [Google Scholar]
- 66. Nagahama M, Mukai M, Morimitsu S, Ochi S, Sakurai J. Role of the C‐domain in the biological activities of Clostridium perfringens alpha‐toxin. Microbiol Immunol. 2002;46:647–55. [DOI] [PubMed] [Google Scholar]
- 67. Takagishi T, Takehara M, Seike S, Miyamoto K, Kobayashi K, Nagahama M. Clostridium perfringens α ‐toxin impairs erythropoiesis by inhibition of erythroid differentiation. Sci Rep. 2017;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Montes L‐R, López DJ, Sot J, Bagatolli LA, Stonehouse MJ, Vasil ML, et al. Ceramide‐enriched membrane domains in red blood cells and the mechanism of sphingomyelinase‐induced hot‐cold hemolysis. Biochemistry. 2008;47:11222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Okino N, Ito M. Ceramidase enhances phospholipase C‐induced hemolysis by Pseudomonas aeruginosa . J Biol Chem. 2007;282:6021–30. [DOI] [PubMed] [Google Scholar]
- 70. Ohsaka A, Tsuchiya M, Oshio C, Miyairi M, Suzuki K, Yamakawa Y. Aggregation of platelets in the mesenteric microcirculation of the rat induced by alpha toxin (Phospholipase C) of Clostridium perfringes . Toxicon. 1978;16:333–41. [DOI] [PubMed] [Google Scholar]
- 71. Sakurai J, Fujii Y, Torii K, Kobayashi K. Dissociation of various biological activities of Clostridium perfringens alpha toxin by chemical modification. Toxicon. 1989;27:317–23. [DOI] [PubMed] [Google Scholar]
- 72. Bryant AE, Chen RY, Nagata Y, Wang Y, Lee CH, Finegold S, et al. Clostridial gas gangrene. I. Cellular and molecular mechanisms of microvascular dysfunction induced by exotoxins of Clostridium perfringens . J Infect Dis. 2000;182:799–807. [DOI] [PubMed] [Google Scholar]
- 73. Bryant AE, Chen RY, Nagata Y, Wang Y, Lee CH, Finegold S, et al. Clostridial gas gangrene. II. Phospholipase C‐induced activation of platelet gpIIbIIIa mediates vascular occlusion and myonecrosis in Clostridium perfringens gas gangrene. J Infect Dis. 2000;182:808–15. [DOI] [PubMed] [Google Scholar]
- 74. Bryant AE, Bayer CR, Hayes‐Schroer SM, Stevens DL. Activation of platelet gpIIbIIIa by phospholipase C from Clostridium perfringens involves store‐operated calcium entry. J Infect Dis. 2003;187:408–17. [DOI] [PubMed] [Google Scholar]
- 75. Coutinho I, Berk R, Mammen E. Platelet aggregation by a phospholipase C from Pseudomonas aeruginosa . Thromb Res. 1988;51:495–505. [DOI] [PubMed] [Google Scholar]
- 76. Drevets DA. Listeria monocytogenes virulence factors that stimulate endothelial cells. Infect Immunity. 1998;66:232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schwarzer N, Nöst R, Seybold J, Parida SK, Fuhrmann O, Krüll M, et al. Two distinct phospholipases C of Listeria monocytogenes induce ceramide generation, nuclear factor‐kappa B activation, and E‐selectin expression in human endothelial cells. J Immunol. 1998;161:3010–8. [PubMed] [Google Scholar]
- 78. Mollby R, Thelestam M, Wadstrom T. Effect of Clostridium perfringens phospholipase C (Alpha‐toxin) on the human diploid fibroblast membrane. J Membr Biol. 1974;16:313–30. [DOI] [PubMed] [Google Scholar]
- 79. Lee MH, Sartorelli AC. Combination chemotherapy with Clostridium perfringens phospholipase C and cytosine antimetabolites : complementary inhibition directed at membrane lipids. Cancer Drug Deliv. 1983;1:21–36. [DOI] [PubMed] [Google Scholar]
- 80. Florin I. Isolation of a fibroblast mutant resistant to Clostridium difficile toxins A and B. Microb Pathog. 1991;11:337–46. [DOI] [PubMed] [Google Scholar]
- 81. Flores‐Díaz M, Alape‐Girón A, Persson B, Pollesello P, Moos M, von Eichel‐Streiber C, et al. Cellular UDP‐glucose deficiency caused by a single point mutation in the UDP‐glucose pyrophosphorylase gene. J Biol Chem. 1997;272:23784–91. [DOI] [PubMed] [Google Scholar]
- 82. Flores‐Díaz M, Alape‐Girón A, Titball RW, Moos M, Guillouard I, Cole S, et al. UDP‐glucose deficiency causes hypersensitivity to the cytotoxic effect of Clostridium perfringens phospholipase C. J Biol Chem. 1998;273:24433–8. [DOI] [PubMed] [Google Scholar]
- 83. Flores‐Díaz M, Alape‐Girón A, Clark G, Catimel B, Hirabayashi Y, Nice E, et al. A cellular deficiency of gangliosides causes hypersensitivity to Clostridium perfringens phospholipase C. J Biol Chem. 2005;280:26680–9. [DOI] [PubMed] [Google Scholar]
- 84. Müthing J, Cacic M. Glycosphingolipid expression in human skeletal and heart muscle assessed by immunostaining thin‐layer chromatography. Glycoconj J. 1997;14:19–20. [DOI] [PubMed] [Google Scholar]
- 85. Müthing J, Maurer U, Neumann U, Kniep B, Weber‐Schürholz S. Glycosphingolipids of skeletal muscle : I. Subcellular distribution of neutral glycosphingolipids and gangliosides in rabbit skeletal muscle. Carbohydr Res. 1998;307:135–45. [DOI] [PubMed] [Google Scholar]
- 86. Monturiol‐Gross L, Flores‐Díaz M, Campos‐Rodríguez D, Mora R, Rodríguez‐Vega M, Marks DL, et al. Internalization of Clostridium perfringens α‐toxin leads to ERK activation and is involved on its cytotoxic effect. Cell Microbiol. 2014;16:535–47. [DOI] [PubMed] [Google Scholar]
- 87. Monturiol‐Gross L, Flores‐Díaz M, Pineda‐Padilla MJ, Castro‐Castro AC, Alape‐Giron A. Clostridium perfringens phospholipase C induced ROS production and cytotoxicity require PKC, MEK1 and NFκB activation. PLoS One. 2014;9:e86475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gustafson C, Tagesson C. Phospholipase C from Clostridium perfringens stimulates phospholipase A 2 ‐mediated arachidonic acid release in cultured intestinal epithelial cells. Scand J Gastroenterol. 1990;25:363–71. [DOI] [PubMed] [Google Scholar]
- 89. Kent C, Evers A, Haun SSL. Diacylglycerol metabolism in phospholipase C‐treated mammalian cells. Arch Biochem Biophys. 1986;250:519–25. [DOI] [PubMed] [Google Scholar]
- 90. Jones G, Kent C. The role of protein kinase C in the stimulation of phosphatidylcholine synthesis by phospholipase C. Arch Biochem Biophys. 1991;288:331–6. [DOI] [PubMed] [Google Scholar]
- 91. Schiess K, Kaszkin M, Jordan P, Seider L, Kinzel V. Mobilization of diacylglycerol in intact HeLa cells by exogenous phospholipase C from C. perfringens is accompanied by release of fatty acids including arachidonic acid. Biochim Biophys Acta. 1992;1137:82–94. [DOI] [PubMed] [Google Scholar]
- 92. Igal RA, Caviglia JM, Gomez‐Dumm INT, Coleman RA. Diacylglycerol generated in CHO cell plasma membrane by phospholipase C is used for triacylglycerol synthesis. J Lipid Res. 2001;42:88–95. [PubMed] [Google Scholar]
- 93. Bunting M, Lorant DE, Bryant AE, Zimmerman GA, McIntyre TM, Stevens DL, et al. Alpha‐toxin from Clostridium perfringens induces proinflammatory changes in endothelial cell. J Clin Invest. 1997;100:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ochi S, Miyawaki T, Matsuda H, Oda M, Nagahama M, Sakurai J. Clostridium perfringens alpha‐toxin induces rabbit neutrophil adhesion. Microbiology. 2002;148:237–45. [DOI] [PubMed] [Google Scholar]
- 95. Oda M, Ikari S, Matsuno T, Morimune Y, Nagahama M, Sakurai J. Signal transduction mechanism involved in Clostridium perfringens alpha‐toxin‐induced superoxide anion generation in rabbit neutrophils. Infect Immun. 2006;74:2876–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Monturiol‐Gross L, Flores‐Díaz M, Araya‐Castillo C, Pineda‐Padilla MJ, Clark GC, Titball RW, et al. Reactive oxygen species and the MEK/ERK pathway are involved in the toxicity of Clostridium perfringens α‐toxin, a prototype bacterial phospholipase C. J Infect Dis. 2012;206:1218–26. [DOI] [PubMed] [Google Scholar]
- 97. Oda M, Kabura M, Takagishi T, Suzue A, Tominaga K, Urano S, et al. Clostridium perfringens alpha‐toxin recognizes the GM1a/TrkA complex. J Biol Chem. 2012;287:33070–33079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Oda M, Shiihara R, Ohmae Y, Kabura M, Takagishi T, Kobayashi K, et al. Clostridium perfringens alpha‐toxin induces the release of IL‐8 through a dual pathway via TrkA in A549 cells. Biochim Biophys Acta. 2012;1822:1581–9. [DOI] [PubMed] [Google Scholar]
- 99. Takagishi T, Oda M, Kabura M, Kurosawa M, Tominaga K, Urano S, et al. Clostridium perfringens alpha‐toxin induces Gm1a clustering and trka phosphorylation in the host cell membrane. PLoS One. 2015;10:e0120497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Takehara M, Sonobe Y, Bandou H, Kobayashi K, Nagahama M. Granulocyte colony‐stimulating factor does not influence Clostridium perfringens α‐Toxin‐induced myonecrosis in mice. Toxins (Basel). 2019;11:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Takehara M, Bandou H, Kobayashi K, Nagahama M. Clostridium perfringens α‐toxin specifically induces endothelial cell death by promoting ceramide‐mediated apoptosis. Anaerobe. 2020;65:102262. [DOI] [PubMed] [Google Scholar]
- 102. Meyers DJ, Berk RS. Characterization of phospholipase C from Pseudomonas aeruginosa as a potent inflammatory agent. Infect Immun. 1990;58:659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Meyers DJ, Palmer KC, Bale LA, Kernacki K, Preston M, Brown T, et al. In vivo and in vitro toxicity of phospholipase C from Pseudomonas aeruginosa . Toxicon. 1992;30:161–9. [DOI] [PubMed] [Google Scholar]
- 104. Konig B, Vasil ML, Konlg W. Role of haemolytic and non‐haemolytic phospholipase C from Pseudomonas aeruginosa in interleukin‐8 release from human monocytes. J Med Microbiol. 1997;46:471–8. [DOI] [PubMed] [Google Scholar]
- 105. Kida Y, Shimizu T, Kuwano K. Cooperation between LepA and PlcH contributes to the in vivo virulence and growth of Pseudomonas aeruginosa in mice. Infect Immun. 2011;79:211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Terada LS, Johansen KA, Nowbar S, Vasil AI, Vasil ML. Pseudomonas aeruginosa hemolytic phospholipase C suppresses neutrophil respiratory burst activity. Infect Immun. 1999;67:2371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vasil ML, Stonehouse MJ, Vasil AI, Wadsworth SJ, Goldfine H, Bolcome RE, et al. A complex extracellular sphingomyelinase of Pseudomonas aeruginosa inhibits angiogenesis by selective cytotoxicity to endothelial cells. PLoS Pathog. 2009;5:e1000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schlüter D, Domann E, Buck C, Hain T, Hof H, Chakraborty T, et al. Phosphatidylcholine‐specific phospholipase C from Listeria monocytogenes is an important virulence factor in murine cerebral listeriosis. Infect Immun. 1998;66:5930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Raveneau J, Geoffroy C, Bereiti J, Gaillard J, Alouf JE, Berchel P. Reduced Virulence of a Listeria monocytogenes Phospholipase‐ Deficient Mutant Obtained by Transposon Insertion into the Zinc Metalloprotease Gene. Infect Immunity. 1992;60:916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yeung PSM, Na Y, Kreuder AJ, Marquis H. Compartmentalization of the broad‐range phospholipase C activity to the spreading vacuole is critical for Listeria monocytogenes virulence. Infect Immun. 2007;75:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Williamson E, Titball R. A genetically engineered vaccine against the alpha‐toxin of Clostridium perfringens protects mice against experimental gas gangrene. Vaccine. 1993;11:1253–8. [DOI] [PubMed] [Google Scholar]
- 112. Ellemor DM, Baird RN, Awad MM, Boyd RL, Rood JI, Emmins JJ. Use of genetically manipulated strains of Clostridium perfringens reveals that both alpha‐toxin and theta‐toxin are required for vascular leukostasis to occur in experimental gas gangrene. Infect Immun. 1999;67:4902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jepson M, Bullifent HL, Crane D, Flores‐Diaz M, Alape‐Giron A, Jayasekeera P, et al. Tyrosine 331 and phenylalanine 334 in Clostridium perfringens K ‐toxin are essential for cytotoxic activity. FEBS Lett. 2001;495:172–7. [DOI] [PubMed] [Google Scholar]
- 114. Oda M, Kihara A, Yoshioka H, Saito Y, Watanabe N, Uoo K, et al. Effect of erythromycin on biological activities induced by Clostridium perfringens alpha‐toxin. J Pharmacol Exp Ther. 2008;327:934–40. [DOI] [PubMed] [Google Scholar]
- 115. Berk RS, Brown D, Coutinho I, Meyers D. In vivo studies with two phospholipase C fractions from Pseudomonas aeruginosa . Infect Immun. 1987;55:1728–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wieland CW, Siegmund B, Senaldi G, Vasil ML, Dinarello CA, Fantuzzi G. Pulmonary inflammation induced by Pseudomonas aeruginosa lipopolysaccharide, phospholipase C, and exotoxin A: role of interferon regulatory factor 1. Infect Immun. 2002;70:1352–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ostroff RM, Wretlind B, Vasil ML. Mutations in the hemolytic‐phospholipase C operon result in decreased virulence of Pseudomonas aeruginosa PAO1 grown under phosphate‐limiting conditions. Infect Immun. 1989;57:1369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wargo MJ. Choline catabolism to glycine betaine contributes to Pseudomonas aeruginosa survival during murine lung infection. PLoS One. 2013;8:e56850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sun Z, Kang Y, Norris MH, Troyer RM, Son MS, Schweizer HP, et al. Blocking phosphatidylcholine utilization in Pseudomonas aeruginosa, via mutagenesis of fatty acid, glycerol and choline degradation pathways, confirms the importance of this nutrient source in vivo . PLoS One. 2014;9:e103778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jackson AA, Gross MJ, Daniels EF, Hampton TH, Hammond JH, Vallet‐Gely I, et al. Anr and its activation by PlcH activity in Pseudomonas aeruginosa host colonization and virulence. J Bacteriol. 2013;195:3093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ramu Y, Xu Y, Lu Z. Inhibition of CFTR Cl channel function caused by enzymatic hydrolysis of sphingomyelin. Proc Natl Acad Sci USA. 2007;104:6448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.