

Abstract
Post‐translational modifications frequently modulate protein functions. Lysine acetylation in particular plays a key role in interactions between respiratory cytochrome c and its metabolic partners. To date, in vivo acetylation of lysines at positions 8 and 53 has specifically been identified in mammalian cytochrome c, but little is known about the structural basis of acetylation‐induced functional changes. Here, we independently replaced these two residues in recombinant human cytochrome c with glutamine to mimic lysine acetylation and then characterized the structure and function of the resulting K8Q and K53Q mutants. We found that the physicochemical features were mostly unchanged in the two acetyl‐mimetic mutants, but their thermal stability was significantly altered. NMR chemical shift perturbations of the backbone amide resonances revealed local structural changes, and the thermodynamics and kinetics of electron transfer in mutants immobilized on gold electrodes showed an increase in both protein dynamics and solvent involvement in the redox process. We also observed that the K8Q (but not the K53Q) mutation slightly increased the binding affinity of cytochrome c to its physiological electron donor, cytochrome c 1—which is a component of mitochondrial complex III, or cytochrome bc 1—thus suggesting that Lys8 (but not Lys53) is located in the interaction area. Finally, the K8Q and K53Q mutants exhibited reduced efficiency as electron donors to complex IV, or cytochrome c oxidase.
Keywords: acetylation, bioenergetics, cytochrome c , electron transport chain, post‐translational modifications
Acetylation is the main post‐translational modification of proteins in eukaryotic cells, affecting more than 60% of mitochondrial proteins. Mammalian cytochrome c, in particular, is found acetylated at lysines 8 and 53. Here, we characterize two acetyl‐mimetic mutants (Lys‐to‐Gln mutation) of human cytochrome c to gain insight into the structural and functional changes that could further explain the acetylation role in cancer and other diseases.
Abbreviations
- A
pre-exponential factor
- AcK
acetyl-lysine
- ANOVA
one-way analysis of variance
- BLI
Bio-layer interferometry
- Cc
cytochrome c
- Cc 1
cytochrome c 1
- CcO
cytochrome c oxidase
- CIII
complex III
- CIV
complex IV
- CSP
chemical shift perturbation
- DMEM
Dulbecco’s modified Eagle’s medium
- E. coli
Escherichia coli
- E 0
midpoint reduction potential
- E 1/2
midpoint redox potential value
- ETC
electron transport chain
- HSQC
heteronuclear single quantum correlation
- ITC
isothermal titration calorimetry
- K D
dissociation equilibrium constant
- k s
electron transfer rate constant
- LB
luria-Bertani
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- MD
molecular dynamics
- MEF
mouse embryonic fibroblast
- MOA
8-Mercaptooctanoic acid
- MOOL
8-Mercapto-1-octanol
- NHE
normal hydrogen electrode
- NOESY
nuclear Overhauser effect spectroscopy
- P1
first principal component
- PCA
principal component analysis
- PDB
protein data bank
- PMSF
phenylmethanesulfonyl fluoride
- PTMs
post-translational modifications
- RG
radius of gyration
- RMSF
root mean square fluctuation
- RSA
rabbit serum albumin
- SAMs
self-assembled monolayers
- SDS/PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- T
temperature
- T m
midpoint melting temperature
- TOCSY
total correlation spectroscopy
- UV
ultraviolet
- Vis
visible
- WT
wild-type
- ε
extinction coefficient
Post‐translational modifications (PTMs) have traditionally been associated with mechanisms that cells use to regulate and expand the functionality of their biomolecules [1, 2, 3]. These modifications can be reversible or irreversible and can include covalent addition of complex molecules, chemical groups, and proteins/peptides, as well as protein cleavage and amino acid modification. Recent studies however have shown that certain PTMs (including acetylation, glycosylation, methylation, nitration, S‐palmitoylation, phosphorylation, S‐nitrosylation, sumoylation, and ubiquitination) or the loss of modification sites in proteins are responsible for the development of certain diseases [4, 5, 6, 7]. Based on the PTM database (PTMD), phosphorylation is the most frequent PTM associated with human disease (http://ptmd.biocuckoo.org/) [6]. These data are striking because acetylation is the major covalent modification to occur in eukaryotic cells, where over 60% of mitochondrial proteins (soluble and membrane‐embedded) have one or more acetylation sites [8, 9, 10, 11]. Acetylation actually regulates diverse protein properties, including DNA interaction, subcellular localization, transcriptional activity, and stability [12]. Acetylation is a reversible enzymatic process carried out by lysine acetyltransferases, in which the ε‐amino group of lysine residues is reversibly modified by the acetyl group. Interestingly, the acetylation process can take place by means of a non‐enzymatic mechanism in the mitochondrial matrix, where the addition of the acetyl group is promoted by high concentrations of acetyl‐CoA and the alkaline pH [13].
A principal mitochondrial protein affected by PTMs is respiratory cytochrome c (Cc), which has also been described as a target for lysine acetylation [14]. Under homeostasis, Cc is known to be located in the mitochondrial intermembrane space and acts as an electron carrier between complex III (CIII) and complex IV (CIV) in the electron transport chain (ETC). It was recently reported that CIII and CIV host two Cc‐binding sites each: the so‐called proximal, or catalytic site, which is the relevant functional position for electron transfer, and the distal, or non‐productive site, which facilitates the turnover and sliding of Cc molecules [15, 16]. In the case of the respiratory supercomplexes, the close arrangement of CIII and CIV suggests a ‘restrained diffusion pathway’ for Cc molecules between the two complexes [17]. Nevertheless, several internal and external cell stimuli can induce the translocation of Cc to other cell compartments (nucleus, vacuoles, zymogen granules, cytosol, and even the rough endoplasmic reticulum), where Cc behaves as a moonlighting protein able to interact with an ample set of non‐redox proteins [18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28].
As mentioned above, Cc can be acetylated on its lysine residues [29]. Several studies with chemically acetylated hemeprotein showed that lysine acetylation modulates respiratory chain functioning by inducing a substantial drop in the Cc reduction/oxidation rate [29] and, consequently, a decrease in Cc‐mediated electron transfer [30]. Notably, these in vitro acetylation assays simultaneously affect multiple lysines, being difficult to assign any acetylation effect to any particular lysine. Downregulation of mitochondrial electron flow promotes inhibition of cellular respiration, also known as the Warburg effect [31]. So far, in vivo acetylation of both Lys8 and Lys53 has only been described in mammalian Cc. Acetylation of Lys8 was observed in a proteomic analysis of mouse liver mitochondria, whereas acetylation of Lys53 was identified in prostatic cancer cells [8, 32]. This association with prostate cancer led to acetylation of Lys53 being the first to become the object of study [32].
The interaction between Cc and its partners depends strongly on the ionic strength of the media, revealing the importance of charged residues such as lysines in its associations [33]. Lys8 and Lys53, with their side chains exposed to the solvent, are involved in the binding of Cc to most of its metabolic partners as they contribute to a positively charged Lys‐rich patch at the hemeprotein surface [16, 22, 34, 35, 36, 37]. Since acetylation neutralizes the positive charge of lysines, this PTM might induce drastic changes in the physicochemical and structural features of Cc and could therefore alter interactions with its metabolic partners.
The difficulty of preserving the acetylation state of Cc and the low yield obtained from cell extracts make it challenging to analyze. Furthermore, the specific acetyltransferase acting on the hemeprotein remains unknown. Hence, recourse was made to amino acid point mutations to mimic the PTM. The Lys‐to‐Gln mutation can mimic the acetylation‐dependent neutralization of the positive lysine charge, so that the resulting Cc species could be used as acetyl‐mimetic mutants. As part of a strategy to gain insight into the structural and functional analysis of lysine‐acetylated Cc, we here characterize two acetyl‐mimetic mutants of Cc, namely K8Q and K53Q Cc, using K8A and K53A mutants as controls.
Methods
Site‐directed mutagenesis, protein expression, and purification of recombinant proteins
Site‐directed mutagenesis was performed on a pBTR1 plasmid comprising the CYCS gene coding for human Cc, along with the CYC3 gene of the yeast Cc heme lyase [38]. The CYC3 gene is required for the proper cytoplasmic maturation of Cc, as the heme lyase covalently attaches the heme group to the apoprotein of Cc. Primers used for mutagenic PCR are detailed in Table S1.
The expression of wild‐type (WT) and acetyl‐mimetic Cc species was performed as previously described [39]. Briefly, Escherichia coli (E. coli) BL21 (DE3) cells were transformed with either pBTR1‐Cc WT or pBTR1‐Cc acetyl‐mimetic plasmids. The cells were cultured at 30 °C with agitation (150 rpm) in Luria‐Bertani (LB) medium supplemented with 100 μg·mL−1 ampicillin for 20 h. Cells were harvested by centrifugation (9000 g for 10 min), suspended in lysis buffer (10 mm Tricine‐NaOH, pH 8.5, supplemented with 1 mm phenylmethylsulfonyl fluoride (PMSF), 0.02 mg·mL−1 DNase and complete protease inhibitor) and physically ruptured by sonication. Cellular debris was separated by centrifugation at 14 000 g for 30 min at 4 °C. For the NMR experiments, 15N‐labeled protein was expressed in minimal M9 medium supplemented with 16.7 μg·mL−1 δ‐aminolevulinic acid hydrochloride and 15NH4Cl as sole nitrogen source. Protein purification was carried out by ion chromatography in a Nuvia‐S (Bio‐Rad, Hercules, CA, USA) column using a FPLC system (Bio‐Rad). The purity of protein samples was routinely tested by electrophoretic (Fig. S1A) and spectrophotometric analyses (see below). In all cases, the A280/A550 ratio of the resulting Cc preparations in the reduced state was about 1.1—as previously reported to be in pure Cc samples [39]—with a maximum 10% deviation. Immunoblot was performed with rabbit anti‐human Cc serum (obtained by immunizing male rabbits with full‐length recombinant Cc suspended in a 0.85% NaCl solution) as primary antibody. HRP‐conjugated secondary antibody was used for detection (Sigma‐Aldrich Co., St. Louis, MO, USA, catalog number A0545). The immunoreactive bands were detected using Amersham ECL Plus Western Blotting Detection Reagents (GE Healthcare Life Sciences, Chalfont St. Giles, UK). The molecular masses of Cc variants were checked by running a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS/PAGE) and matrix‐assisted laser desorption/ionization‐time of flight (MALDI‐TOF). Tryptic digestion analysis confirmed the point mutations of the Cc species. Protein concentration was determined by visible (Vis) spectrophotometry, using an extinction coefficient (ε550nm) of 28.92 mm −1·cm−1 for reduced Cc [39]. Protein samples were dialyzed against 10 mm sodium phosphate buffer pH 5.5 or pH 7.4 for NMR, or CD and bio‐layer interferometry (BLI) experiments.
The C10A mutant of Arabipdosis thaliana cytochrome c 1 (Cc 1), used to avoid protein dimerization, was expressed as previously described [15]. E. coli BL21 (DE3) cells were co‐transformed with pET‐Cc 1‐C10A and pEC86—coding for the CYC3 gene of the yeast Cc heme lyase—to produce the soluble domain of Cc 1. Cells were grown for 24 h at 150 rpm and 30 °C in LB‐rich medium supplemented with 16.7 μg·mL−1 δ‐aminolevulinic acid hydrochloride, 50 μg·mL−1 kanamycin, and 12 μg·mL−1 chloramphenicol. Cells were collected by centrifugation (9000 g for 10 min) and then suspended in lysis buffer (20 mm Tris/HCl, pH 8.0, supplemented with 1 mm PMSF, 0.02 mg·mL−1 DNase and complete protease inhibitor). The periplasmic fraction was obtained by the freeze‐thaw lysis method. The resulting suspension was centrifuged at 20 000 g for 30 min and loaded onto a Nuvia‐Q column (Bio‐Rad). Cc 1 was eluted with a NaCl linear gradient in 20 mm Tris/HCl, pH 8.0. The fractions containing Cc 1 were further purified by FPLC using a gel filtration ENrich SEC 70 column (Bio‐Rad). Protein concentration was determined by Vis spectrophotometry, using an extinction coefficient (ε552nm) of 28 mm −1·cm−1 for reduced Cc 1 [15]. The recombinant protein was dialyzed against 10 mm sodium phosphate buffer pH 7.4 for BLI experiments.
CD spectroscopy
CD spectra were recorded using a Jasco® J‐815 spectropolarimeter equipped with a Peltier temperature control system. The secondary structure analyses were carried out by recording far‐ultraviolet (UV) CD spectra (185–250 nm) at 25 °C in a 1‐mm quartz cuvette. Samples contained 3 μm protein in 10 mm sodium phosphate (pH 7.4), supplemented with 10 μm potassium ferricyanide. 20 scans were averaged for each sample.
The coordination of the heme iron atom to the S δ atom of Met80 (the sixth axial ligand of heme group) was analyzed by visible (B‐band) recording near‐UV/Vis CD spectra (300–600 nm) in a 10 mm quartz cuvette at 25 °C. Samples contained 30 μm protein in 10 mm sodium phosphate (pH 7.4), supplemented with 100 μm potassium ferricyanide.
Thermal unfolding was monitored between 10 °C and 105 °C (with a heating rate of 1 °C·min−1) by recording the CD signal at far‐UV in a 10 mm quartz cuvette. For these assays, the oxidized Cc species were at 3 μm final concentration in 10 mm sodium phosphate (pH 7.4), supplemented with 10 μm potassium ferricyanide. The heme coordination of the Cc species was monitored from 10 °C and 105 °C (with a heating rate of 1 °C·min−1) by recording near‐UV/Vis CD spectra from solutions in a 10 mm quartz cuvette. Samples contained 30 μm Cc in 10 mm sodium phosphate buffer (pH 7.4), supplemented with 100 μm potassium ferricyanide. The midpoint melting temperature (T m) values were obtained from the fits of principal components obtained from CD and fluorescence spectra measured on temperature ramps.
Electronic absorption spectroscopy
The redox state reversibility of heme group was checked by recording absorption spectra from 350 to 600 nm after the addition of 10 equivalents of potassium ferricyanide as an oxidizing agent and 50 equivalents of sodium ascorbate as a reducing agent. Spectra of 5 µm of Cc species were recorded at 25 °C in 10 mm sodium phosphate buffer (pH 7.4).
The coordination of the heme group of Cc was analyzed by monitoring the absorption changes at 699 nm, indicative of the heme Fe‐Met80(Sδ) bond as reported previously [40]. Electronic absorption spectra were recorded in the 600–750 nm range at 25 °C, using a Jasco® V‐650 spectrophotometer in a 1‐mL quartz cuvette with a path length of 10 mm. Samples contained 0.2 mm oxidized Cc in 10 mm sodium phosphate (pH 5.8), supplemented with 0.2 mm potassium ferricyanide. For pH titration studies, the pH of each sample was adjusted to the reported values by adding aliquots of 0.1–0.5 M NaOH or 0.1–0.5 M HCl. To calculate the pK a value of each protein, the absorbance changes at 699 nm were fitted to the Henderson‐Hasselbalch equation expressed in the following terms [41]:
| (1) |
where Y is the experimental value for absorbance at 699 nm, Absmax and Absmin are the maximum and minimum values for absorbance at 699 nm, X is the pH of the protein sample, pK a is the apparent pK a value for alkaline transition, and NS is the slope of the sigmoid.
Molecular dynamics simulations
The initial structures were based on the NMR structure of reduced human Cc (Protein Data Bank, PDB, ID: 2N9I; [42]). All reduced mutants (K8A, K8Q, K8AcK, K53A, K53Q, and K53AcK) structural models were built using the WT protein as a template with UCSF Chimera software [43]. The force field parameters for acetyl‐lysine [44] and heme group [45] were used to generate the topology and coordinate files, required for the simulation, with TLEAP module of AMBER [46].
Molecular dynamic (MD) simulations were performed using the OpenMM [47] software in CUDA platform with AMBER 14SB force field [48] and particle mesh Ewald electrostatics with an Ewald summation cut‐off of 10 Å. For each simulation, the system was neutralized with chlorine counter‐ions according to the total charge of the protein and solvated with optimal‐3‐charge, 4‐point rigid water model water molecule [49]. The whole system was subjected to 2500 steps of energy minimization at 298 K. Langevin thermostat was used to control the temperature with a friction coefficient of 1 ps−1 and a step size of 0.002 ps. Each system was subjected to 900 ns MD simulation. For the trajectory analysis, the CPPTRAJ module of AMBER was used [50] to analyze the last 50 ns of simulation.
NMR spectroscopy
NMR experiments were performed on a 700 MHz Bruker Avance‐III spectrometer equipped with a cryoprobe. Backbone amide group resonances of Cc variants were monitored by recording 2D 15N‐1H heteronuclear single quantum correlation (HSQC) experiments. Samples contained 150 μm of reduced 15N‐labeled Cc in 20 mm citrate buffer (pH 5.5), 5% D2O and sodium ascorbate to ensure the redox state of the sample. NMR assignments of the 15N and 1H nuclei of reduced WT Cc (BMRB accession number 25907) were taken from previous work [42]. Additional 3D 15N‐1H total correlation spectroscopy (TOCSY)‐HSQC and 3D 15N‐1H nuclear Overhauser effect spectroscopy (NOESY)‐HSQC experiments were recorded to identify the resonances of the Lys‐Gln or Lys‐Ala Cc mutants. The experimental data were processed using Bruker TopSpin NMR 4.1.1 software (Bruker), and chemical shift perturbations (CSPs) were analyzed with NMRFAM‐Sparky NMR assignment tool [51].
Bio‐layer interferometry measurements
BLI measurements were performed on the Octet RED 96e platform (FortéBio) using Ni‐NTA (NTA) Biosensors (FortéBio). The protocol used for the kinetic analysis is detailed in Table S2.
The biosensors were hydrated in a 96‐well black plate for 10 min with BLI buffer containing 10 mm sodium phosphate (pH 7.4) supplemented with 0.01 μm rabbit serum albumin (RSA) and 0.01% Triton X‐100 as blocking agents to reduce non‐specific binding to the biosensor. The biosensors were incubated with 200 μL of His6‐tagged Cc 1 sample (50 μg·mL−1 diluted in BLI buffer) at the loading step for immobilization of the analyte into the NTA matrix. Binding of WT and acetyl‐mimetic Cc variants was analyzed at ligand concentrations between 0.3215 and 10 μm diluted in BLI buffer and performed in duplicate. The surface of the biosensor was regenerated by 10 mm glycine (pH 1.7) using the standard protocol described by the manufacturer. The experimental data were processed with Data Analysis HT 12.0 software (fortébio) and analyzed with Origin Lab Origin(Pro), Version 2019 (OriginLab Corporation, Northampton, MA, USA), using the non‐linear equation described by Müller‐Esparza et al. [52]:
| (2) |
where Y is the wavelength shift at equilibrium, Bmax is the maximum wavelength shift, X is the effector complex concentration, K D is the equilibrium dissociation constant, and NS is the slope of the linear components.
Isolation of mitochondria from mouse embryonic fibroblast cell lines
Mouse embryonic fibroblast (MEF) cell lines were cultured at 37 °C in a humidified atmosphere of 5% CO2. MEF cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma‐Aldrich Co.) containing 4500 mg·L−1 glucose (Sigma‐Aldrich Co.), supplemented with 10% heat‐inactivated fetal bovine serum (Sigma‐Aldrich Co.), 2 mm L‐glutamine, 100 U·mL−1 streptomycin, 100 μg·mL−1 penicillin and 0.11 mg·mL−1 sodium pyruvate (Sigma‐Aldrich Co.). Cells were grown to 80% confluence in 24‐well plates with 500 μL of DMEM. Mitochondria were obtained by differential centrifugation as reported previously [53].
Cytochrome c oxidase activity
To assess the functionality of acetyl mimetics as electron donors, we tested their ability to reduce cytochrome c oxidase (CcO) in a mitochondrial context using a CcO activity kit (Biochain, Newark, CA, USA), as reported previously [54]. Activity was measured on mitochondria isolated from MEF cell lines in a Jasco® V‐650 spectrophotometer in a 0.1‐mL quartz cuvette with 10 mm path length, according to the manufacturer’s instructions. The Abs550 slopes of at least three independent experiments were calculated and correlated with CcO activity values.
Statistical analysis
Data were analyzed using Origin Lab Origin(Pro), Version 2019 (OriginLab Corporation, Northampton, MA, USA). Data are expressed as means ± SD. One‐way analysis of variance (ANOVA) was employed for comparisons of multiple groups.
Electrochemical measurements
Linear scan voltammetric measurements were performed with an AUTOLAB PGSTAT 30, from Eco Chemie B.V, in a three‐electrode undivided glass cell, equipped with a gas inlet and thermostatted with a water jacket. Working electrodes were polycrystalline gold with a 0.0314 cm2 geometric area. Prior to performing measurements, the gold surface was cleaned by successively polishing it with 0.3 and 0.05 µm alumina, rinsed with Millipore water, then sonicated in absolute ethanol to remove residual alumina. Then, the electrode surface was dried and chemically cleaned using ‘piranha’ solution (7 : 3 mixture of concentrated H2SO4 (95%) and 30% v/v H2O2). Gold electrodes were modified with thiol self‐assembled monolayers (SAMs) by immersing the electrode in ethanol solutions containing 1 mm 8‐mercaptooctanoic acid (MOA) and 2.5 mm 8‐mercapto‐1‐octanol (MOOL), from Sigma‐Aldrich Co., for 30 min at room temperature. Protein immobilization was carried out by depositing on the modified electrode a 15 µL drop of a solution containing 20 µm protein, 10 mm sodium phosphate buffer solution (pH 7) for 45 min at 4 °C. After protein incubation, the electrodes were rinsed with water and washed with the working buffer solution. The counter and reference electrodes were a Pt bar and an Ag/AgCl/NaCl‐saturated electrode, respectively. Reported potential values were corrected to the normal hydrogen electrode (NHE) potential scale by adding +192 mV to the experimental potential values. Working solutions contained 20 mm sodium phosphate buffer as supporting electrolyte, at pH 7.0 ± 0.1. All measurements were performed under argon atmosphere.
Results and Discussion
Physicochemical features of cytochrome c are mainly unchanged in the two acetyl‐mimetic mutants
Cc contains 18 lysine residues but only Lys8 and Lys53 are acetylated in vivo [8, 32]. Each residue is located in opposite regions of the hemeprotein (Fig. 1A): Lys8 in helix I and Lys53 in helix II, with their respective side chains well exposed to the solvent. To explore how acetylation of Lys8 and Lys53 influences the physicochemical features of Cc, we designed two acetyl‐mimetic mutants of Cc, replacing the corresponding lysines with glutamines (K8Q and K53Q Cc mutants) to resemble the change in charge that occurs when lysine becomes acetylated (Fig. 1B). To check the specific effect of these point mutations, Lys‐to‐Ala mutants were also studied (K8A and K53A, Fig. 1B).
Fig. 1.
3D structure of cytochrome c and formula of selected amino acids. (A) Ribbon representation of human Cc structure (PDB, ID: 2N9I [42]), with the heme group (green color) and Lys8, Lys53, and Trp59 represented by sticks. The different foldon units are colored as follows: foldon I in blue, foldon II in green, foldon III in yellow, foldon IV in red, and foldon V in purple. (B) Chemical structure of acetyl‐lysine, lysine, glutamine, and alanine.
Since mutation of lysine to glutamine or alanine neutralizes the positive charge, we first calculated the surface electrostatic potential of the WT, point mutants (K8A, K8Q, K53A, and K53Q), and acetylated forms (K8AcK and K53AcK) of Cc at physiological ionic strength. In all cases, the lysine substitution resulted in a loss of positive potential at the surface area surrounding the mutated residue (Fig. 2), consistent with the charge difference between the ionic amino‐terminal group of lysine in WT Cc and the neutral character of acetyl‐lysine, glutamine, or alanine in the mutants. The similar surface electrostatic potentials of the acetyl‐lysine variants (K8AcK and K53AcK) and the glutamine mutants (K8Q and K53Q) suggest that the Lys‐to‐Gln substitution could lead to good mimetics of acetylated Cc.
Fig. 2.
Surface electrostatic potentials of WT and mutant species of cytochrome c. Electrostatic potentials were calculated at 150 mm ionic strength. Green circles denote mutated residues at position 8 (upper) and 53 (lower).
Glutamine and alanine site‐directed mutants were thus designed, expressed, and purified (see Methods section). The molecular mass of all purified mutants was corroborated by SDS/PAGE (Fig. S1A) and MALDI‐TOF mass spectrometry (Fig. 3A). The peak closest to 12 500 m/z in all mass spectra corresponds to the formation of a Schiff base adduct, as described previously [55]. Since the molecular masses of the glutamine mutants (K8Q and K53Q) were the same as that of the WT Cc, protein spots from SDS/PAGE bands were subjected to trypsin digestion to confirm the Lys‐to‐Gln substitution from the masses of the resulting fragments (Fig. S1B). UV‐Vis absorption spectra clearly confirmed the typical hallmarks of c‐type cytochromes in all mutants (Fig. 3B). The spectra for WT and mutant Cc in the reduced state show a sharp maximum at ca. 415 nm (Soret band), as well as two lower maximums at 520 and 550 nm (α and β bands, respectively) that merge in the oxidized state. The redox state reversibility of the heme group in all samples was checked by recovering original absorption spectra upon addition of potassium ferricyanide or sodium ascorbate as oxidizing or reducing agent, respectively.
Fig. 3.
Mass and visible absorption spectra of WT and mutant species of cytochrome c. (A) MALDI‐TOF spectra of WT and Cc mutants. The molecular mass of WT, K8Q, and K53Q Cc is 12 235 Da, whereas that of K8A and K53A Cc is 12 179 Da. (B) Visible absorption spectra of oxidized (dashed line) and reduced (solid line) Cc in the presence of potassium ferricyanide and sodium ascorbate, respectively. The color code is the same as in panel (A). a.u., arbitrary units.
The effect of mutations on the secondary structure of Cc was analyzed by far‐UV CD spectroscopy of WT and mutant Cc species. The most prominent change was observed in the 195‐nm band with the K8Q mutant, suggesting a substantial increase in α‐helix content (Fig. 4). A detailed analysis using the cdpro software package (Fig. S2) not only confirmed the increase in α‐helix content in K8Q but also revealed a significant increase in K53Q.
Fig. 4.
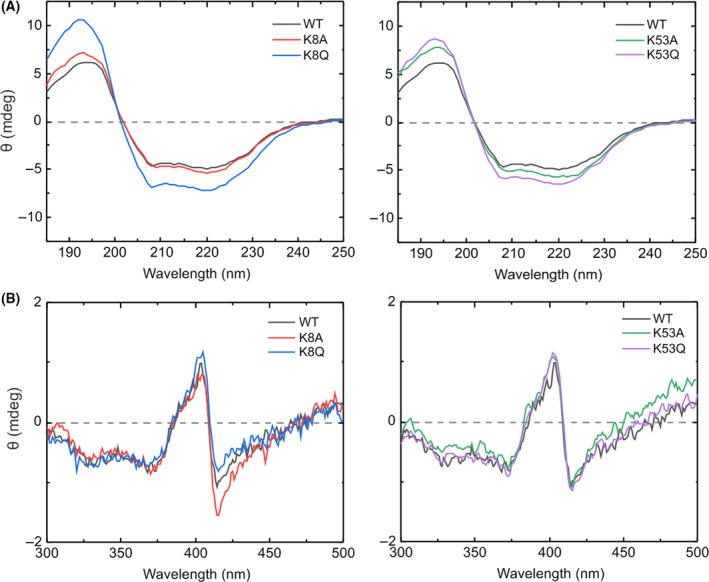
Analysis of the secondary structure and heme environment of the WT and mutant species of cytochrome c. (A) Far‐UV and (B) near‐UV/Vis CD spectra of oxidized WT, K8A, K8Q, K53A, and K53Q Cc at 3 and 30 µm, respectively.
Since subtle changes in the heme group can alter the redox properties and physiological functions of Cc, the CD spectra in the visible region were also recorded to provide further information on the heme environment [56]. In its native form, the heme iron atom of Cc is hexacoordinated, with the fifth and sixth axial ligands being His18 and Met80, respectively [57]. The CD spectra of WT Cc and all mutants clearly exhibited the distinctive B‐band splitting corresponding to the Cotton effect in the Soret absorption region [58]. Taken together, these findings confirm that Lys8 and Lys53 mutations have a negligible effect on the heme environment.
Since the stability of the heme crevice is preserved under physiological conditions, the effect of pH on the heme environment was checked by Vis absorption spectroscopy. In the pH range of 1–12, at least five different conformations of Cc have been described as a result of changes in protein folding and/or heme axial coordination [59]. The transition from state III to state IV, called alkaline transition, is induced by an increase in pH [60]. This state transformation involves replacing the axial ligand Met80 with a lysine residue (Lys72, Lys73, or Lys79) [61, 62]. The interaction between heme iron and Met80 in the oxidized form of Cc causes a maximum charge‐transfer peak in the absorption spectra at 699 nm [40, 63], which disappears as the alkaline transition occurs (Fig. S3, left panels). The presence of the 699 nm maximum at the initial pH value (pH ca. 6.3) indicates the integrity of protein folding [64]. The intensity of this charge‐transfer peak was recorded in the pH 6.0–12.5 range, and the fitting of titration curves (Fig. S3, right panels) provided the pK a values for the alkaline transition of all mutants included in this study. Considering that Tyr48‐phosphomimetic Cc mutants have lower but biologically relevant pK a transition values [40, 65], one would expect that the alkaline transition of mutants at Lys53, which is in the same loop as Tyr48, would also be altered. However, the pK a values of all mutants were very close to that of WT Cc (Table 1). These results suggest that the native heme environment is preserved in acetyl‐mimetic Cc species, consistent with exposure of the side chains of lysine, alanine and glutamine residues at positions 8 and 53 to solvent without altering the hydrogen bond pattern around the heme crevice [40]. Conservation of Met80‐Fe bond distance, as predicted by theoretical MD calculations, also confirms the preservation of heme moiety (Fig. S4).
Table 1.
pK a values for alkaline transition in oxidized WT and mutant cytochrome c.
| pK a | |
|---|---|
| WT | 9.48 ± 0.05 |
| K8A | 9.54 ± 0.03 |
| K8Q | 9.56 ± 0.06 |
| K53A | 9.42 ± 0.04 |
| K53Q | 9.31 ± 0.03 |
Thermal stability of cytochrome c is differentially modified depending on the mutated residue
The conformational stability of Cc depends greatly on the protein environment, as well as the residues surrounding the heme group [66]. Changes in H‐bond networks, hydrophobic interactions, steric restrictions, and even the protein sequence can alter the T m of the protein. To explore the effect of temperature on the secondary structure and heme moiety of Cc species, thermal stability assays were monitored by recording the CD and fluorescence spectra in the far‐UV and near‐UV/Vis ranges, respectively, in the temperature interval 10–105 °C. To ensure the preservation of Fe‐Met80 coordination, the experiments were carried out at pH 7.4. The complex behavior of CD and fluorescence spectra required principal component analysis (PCA) in which the first principal component (P1) of spectra was fitted to a three‐state or four‐state model, depending on the assay (Fig. S5). The fitting of P1 provided T m values for the different thermal transitions that the mutants underwent along the temperature ramp (Table 2).
Table 2.
Midpoint melting temperature (T m) values for oxidized WT and mutant cytochrome c as calculated within the 185–250 nm (UV) and 300–600 nm (Vis) ranges by CD and fluorescence spectroscopy.
| T m (°C) | |||
|---|---|---|---|
| CD spectra | Fluorescence | ||
| 185–250 nm | 300–600 nm | 185–250 nm | |
| WT | 55.5 ± 0.8/87.1 ± 0.2 | 62.4 ± 1.1/87.4 ± 0.3 | 48.6 ± 2.4/58.2 ± 0.9/93.4 ± 1.6 |
| K8A | 52.5 ± 0.2/86.5 ± 0.2 | 28.5 ± 0.8/56.1 ± 0.6/84.1 ± 1.2 | 53.2 ± 0.4/88.4 ± 0.4 |
| K8Q | 53.2 ± 0.3/88.2 ± 0.4 | 29.1 ± 0.3/69.1 ± 3.7/87.8 ± 0.7 | 51.2 ± 0.3/90.1 ± 0.3 |
| K53A | 57.7 ± 4.6/82.1 ± 1.7 | 49.0 ± 0.8/83.5 ± 0.2 | 52.4 ± 0.6/88.8 ± 2.5 |
| K53Q | 55.7 ± 0.4/87.8 ± 0.3 | 53.6 ± 0.5/87.0 ± 0.6 | 57.6 ± 0.5/92.5 ± 0.3 |
The thermal unfolding profile from the representation of P1 and temperature in the far‐UV CD spectra of all mutants showed similar thermal behavior to the WT protein and consisted of two thermal transitions (Fig. S5). These thermal unfolding profiles have been explained using a previous folding model, based on the division of Cc into five folding regions (Fig. 1) with different thermal stabilities [67]. According to the model, the most stable region included helix I and helix IV (foldon I), while the Ω‐loop between Thr19 and Phe36 residues and helix III comprised the second folding unit (foldon II). Denaturation of these two folding units may correspond to the second thermal transition observed in the far‐UV CD spectra, since foldons I and II are helix‐rich regions. The following region (foldon III) corresponds to two short stretches of residues from Phe36 to Lys39 and Gly56 to Gly60, while foldon IV includes the loop extending between helix III and helix IV and contains Met80 residue. The most unstable region (foldon V) extends from Thr40 to Lys55, between the two stretches of foldon III. The configuration of these three last regions is less structured and may thus be responsible for the first thermal transition. Since the Lys8 and Lys53 residues are located in helix I in foldon I and helix II in foldon V, respectively, preservation of the Cc thermal profile in the far‐UV range indicates that the mutation of these residues does not alter foldon stability. The T m values for WT and mutant Cc species were quite similar (Table 2), with small differences consistent with the minor changes in secondary structure previously described in Figs 4 and S2.
However, the P1 plot of the near‐UV/Vis CD spectra showed that the Lys8 mutations, especially in the K8A mutant, considerably modified the thermal behavior of Cc. While structural features of the heme moiety, such as heme plane bending, aromatic residue packing, and heme internal electric field [68, 69], determine the visible B‐band spectra, the thermal profile variations in the near‐UV/Vis CD region could correspond to conformational changes in the heme crevice. The appearance of a new thermal transition at ca. 28 °C indicates the influence of the Lys8 mutation on the heme moiety (Fig. S5, Table 2). This residue is located in helix I in foldon I, which is where the Cys14 and Cys17 residues are located. Cys14 and Cys17 are covalently linked to the heme group, and so perturbations of the foldon I residues could affect the redox cofactor. This possible alteration of the redox cofactor was not predicted by theoretical MD calculations (Fig. S4), which yielded a value at 25 °C lower than that of the new thermal transition. On the other hand, there is no substantial evidence of changes in the thermal profile after Lys53 mutation. The transitions for WT and Lys53 mutants in the near‐UV/Vis CD region are consistent with the equivalent ones in the far‐UV CD region. The T m values of Lys53 variants differed slightly from those of the WT form (Table 2), a fact that can be attributed to small heme moiety variations resulting from the changes in Cc conformation, mentioned below.
Trp59 fluorescence of Cc, which is quenched by the heme porphyrin ring in the native state, was recorded [70]. Although the fluorescence profiles of Lys8 mutants are close to that of the WT form, those of Lys53 mutants showed differences at temperatures between 60 and 80 °C (Fig. S5). Since Lys53 and Trp59 are located in contiguous regions (foldons V and III, respectively), mutation at Lys53 could destabilize foldon V and, in turn, foldon III, causing the change in Trp59 fluorescence. The final decrease in fluorescence intensity of all Cc species probably corresponds to the exposure of the Trp59 indole ring to the solvent as the protein unfolds [71]. The fitting of P1 fluorescence gave three T m values for WT and only two for Cc mutants (Table 2). A detailed analysis of the fluorescence P1 curves of mutants revealed the coupling of two close low‐temperature transitions that cannot be distinguished with the model used.
Taken together, these findings suggest that the thermal stability of Cc is affected differently depending on the mutated position, since Lys8 and Lys53 are located on opposite faces of the Cc surface.
Mimicking acetylation induces local structural changes in cytochrome c
To understand the effect of the point mutations on the spatial structure of Cc, the reduced forms of K8A, K8Q, K53A, and K53Q Cc mutants were characterized by recording the 1D 1H, 2D 1H–15N HSQC, 3D 15N‐1H TOCSY‐HSQC, and 3D 15N‐1H NOESY‐HSQC NMR spectra to monitor the backbone amide signals and compare them with those from the WT protein. The 1D 1H NMR spectra of all mutants showed a resonance at ca. −3.5 ppm, which corresponds to the 1H signal of Met80‐ε‐CH3 group and corroborates the preservation of native heme axial coordination, in accordance with previous data. The backbone amide resonances of the Cc mutants identified in 2D 1H‐15N HSQC spectra were confirmed by recording 3D 15N‐1H TOCSY‐HSQC and 3D 15N‐1H NOESY‐HSQC spectra for each of the mutants. The difference in CSPs calculated from the 2D 1H‐15N HSQC of each reduced mutant with respect to that of the reduced WT is illustrated in Fig. 5. Generally speaking, Lys‐to‐Ala mutation results in substantial CSPs compared with Lys‐to‐Gln one; not only in terms of the magnitude of the CSP but also the number of affected residues. These differences can be followed in Fig. S6–S9. A plausible explanation is that the Lys‐to‐Ala substitution induces significant changes in the chemical environment of those residues close to the new alanine residue, which differs from lysine in a loss of charge and side‐chain length [72, 73]. On the other hand, the chemical environment is less affected by the Lys‐to‐Gln substitution, albeit with new resonances corresponding to the appearance of side‐chain amide groups of Gln in the NMR spectra (Figs S7 and S9.
Fig. 5.
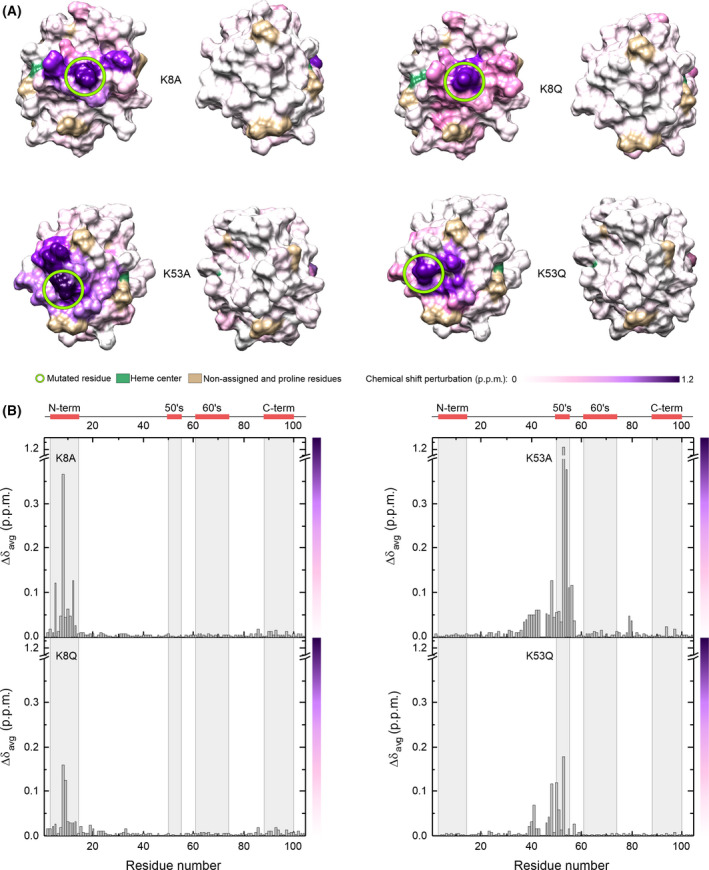
NMR chemical shift perturbations of reduced cytochrome c mutants. (A) Each surface map shows residues colored according to CSPs calculated previously from 2D 1H‐15N HSQC spectra of reduced K8A, K8Q, K53A, and K53Q Cc with respect to the 2D 1H‐15N HSQC spectra of reduced WT Cc. (B) Plot of NMR CSPs of reduced Cc mutants. CSP values as a function of residue number calculated from 2D 1H‐15N HSQC spectra of reduced K8A, K8Q, K53A, and K53Q Cc with respect to the 2D 1H‐15N HSQC spectrum of reduced WT Cc. The color bar represents the color scale used in panel A.
A surface map comparison of NMR CSPs of acetyl‐mimetic Cc mutants revealed that the K8Q spectrum showed a smaller number of affected residues with respect to the WT protein (Fig. 5). In K8Q, only Lys5, Ile9, Phe10, Ile11, and Met12 (located in the same helix I as Gln8) experienced significant CSPs (Fig. S7). However, the superposition of the K53Q and WT HSQC spectra (Fig. S9) revealed that the loss of charge at position 53 caused by the mutation drastically alters the chemical environment of nearby residues in the sequence and close amino acidic neighbors in the space. Some of them (Thr40, Gly41, Gln42, Ser47, and Tyr48) are located in the nested Ω‐loop, with the side chain of Gln53 pointing toward it. Like K8Q, other residues located in the same helix II as Gln53 (Ala50, Ala51, and Asn54, including its amide side chain) undergo large CSPs (Fig. S9). Consistent with the thermal stability assays, the spatial location of the mutated residues in the protein structure could explain the functional changes shown by the acetyl‐mimetic mutants.
Binding between the acetyl‐mimetic cytochrome c species and its redox partner cytochrome c1 is subtly dependent on the spatial location of the mutated residues
The interaction of Cc with its metabolic redox and non‐redox partners takes place mainly through the Gln16, Ala50, Ala51, Lys72, Gly77, Ile81, and Lys86 residues, which surround the heme crevice and constitute the binding surface. It was previously reported that lysines 8, 13, 27, 72, 86, and 87 are all in contact with Cc 1, whereas lysines 5, 7, 8, 22, 25, 53, 72, 73, 86, and 88 contact the SET/TAF‐Iβ histone chaperone. [16, 22, 34, 37]. Considering that Lys8 and Lys53 are involved in Cc binding to its partners, one would expect this interaction to be modulated by mutation of those residues.
Our previous NMR data on the plant Cc 1‐human Cc complex indicated that lysine at position 8 is substantially perturbed by binding to Cc 1, suggesting that it is involved in interface formation [16]. The question arises whether acetyl‐mimetic Cc could alter Cc binding to Cc 1. In this context, BLI was used to estimate the binding affinity values for the plant Cc 1‐human Cc complex. The N‐terminal soluble domains of human and plant Cc 1 share a 62% of sequence identity and similar charge distributions. Previous NMR and Isothermal titration calorimetry (ITC) analyses indicated that Arabidopsis thaliana Cc 1 shows almost identical affinity toward human and Arabidopsis thaliana Cc [15, 16]. The maximum wavelength shifts at saturation were plotted against protein concentration and fitted to a non‐linear equation to calculate the K D (Fig. S10). The K D values obtained for the WT protein and the Lys53 acetyl‐mimetic variant were similar (Table 3), although the binding profile for K53Q Cc suggests the existence of two binding events. However, the K D value for the Cc 1‐K8Q Cc complex was slightly lower than for the WT complex. This could be explained by the fact that Lys8 is directly involved in the interaction with Cc 1 [16, 74].
Table 3.
Dissociation constant (K D) values for the interaction of reduced cytochrome c 1 with reduced WT, K8Q, and K53Q cytochrome c.
| K D (µm) | |
|---|---|
| WT | 0.137 ± 0.032 |
| K8Q | 0.099 ± 0.004 |
| K53Q | 0.116 ± 0.014 |
Thermodynamics and kinetics of interfacial electron transfer in cytochrome c mutants
It has been shown that PTMs of Cc are able to regulate its electron donor function [14, 32, 39, 54, 75, 76, 77, 78, 79, 80]. Since the patch of positively charged lysine residues of Cc contributes to electrostatic interactions with redox partners in mammals, plants, and yeasts [15, 16, 34, 35, 37, 74, 81, 82, 83, 84], the redox functionality of Cc may be influenced by lysine mutation.
Consequently, the redox functionality of acetyl mimetics of Cc in the adsorbed state was also assessed. Cc actually performs its many biological functions upon partner binding, so protein film voltammetry was used as a suitable technique to get further information on the redox properties of bound Cc. A common strategy consists in the immobilization of the redox protein onto a metal electrode modified with SAM of organic molecules to reproduce the essential features of protein/protein interfaces. For this, WT, K8Q, and K53Q Cc species were adsorbed onto polycrystalline gold electrodes modified with mixed SAMs made up of 1 : 2.5 mixtures of MOA and MOOL to mimic the biological interface of Cc in its primary redox function. The thermodynamics and kinetics of interfacial electron transfer were determined by performing variable temperature cyclic voltammetry experiments. The estimated characteristic interfacial electron transfer parameters are shown in Table 4.
Table 4.
Midpoint redox potential, thermodynamic and kinetic parameters as calculated from electron transfer of WT, K8Q, and K53Q cytochrome c immobilized on a chemically modified polycrystalline gold electrode. , Midpoint redox potential value, , Reduction entropy, , Reduction enthalpy, k s, Electron transfer rate constant, A, Pre‐exponential factors, , Activation enthalpy.
|
(mV vs. NHE) |
(J K−1·mol−1) |
(kJ·mol−1) |
k s (s−1) |
10‐5·A (s−1) |
(kJ·mol−1) |
||||
|---|---|---|---|---|---|---|---|---|---|
| WT | 184 ± 5 | –142 ± 8 | –60 ± 6 | 1300 ± 50 | 3.1 ± 0.9 | 13.6 ± 0.5 | |||
| K8Q | 178 ± 5 | –130 ± 7 | –56 ± 4 | 1100 ± 50 | 12.1 ± 1.8 | 17.2 ± 0.8 | |||
| K53Q | 172 ± 5 | –108 ± 8 | –49 ± 5 | 1600 ± 50 | 9.7 ± 1.3 | 15.8 ± 0.7 |
Measured at 25 °C.
The WT, K8Q, and K53Q Cc species showed the typical voltammetric response of an isolated population of surface‐immobilized species (Fig. S11A). The standard potential of the WT Cc species at 25 °C was determined from the average of the cathodic and anodic peak potentials of the experimental voltammograms at low potential scan rate (0.1 V·s−1). The standard potential associated with the Fe3+ / Fe2+ redox conversion of the heme group was ˜ 185 mV vs. NHE, which was significantly more negative than the one in solution (˜ 265 mV) [85], revealing a further relative stabilization of the ferric form of the native heme conformation upon electrostatic immobilization. The midpoint redox potential values (E 1/2) at 25 °C for the three Cc species were not significantly different (Table 4), thus suggesting that the acetyl‐mimetic mutations do not substantially modify the heme moiety.
The variation of the standard redox potential with temperature (Fig. S11B) provided the reduction entropy () and enthalpy () values for the redox conversion of immobilized proteins (Table 4). More specifically, the and values were estimated from the slopes of E 1/2 vs. temperature (T) and E 1/2/T vs. 1/T plots, respectively. The redox thermodynamics of the adsorbed Cc species turned out to be characterized by highly negative and values, revealing large differences in protein solvation and/or structure after its change in oxidation state. It can also be observed that the and values were relatively insensitive to the mutation. For electron transfer proteins with positively charged redox centers, less negative reduction entropies are generally associated with the greater role of solvent reorganization accompanying the electron transfer step. The less negative values of the mutants point either to an increase in water accessibility to the heme, which was not predicted for the proteins in solution (Fig. S4), or to a perturbation of the Fe‐ligand bond favoring the ferric over the ferrous heme. Notably, the perfect enthalpy‐entropy compensation observed in the three proteins also points to the substantial contribution of solvent exchange between protein and solution to the energetics of protein reduction.
The interfacial electron transfer kinetics of adsorbed Cc species was also explored by determining their standard heterogeneous electron transfer rate constants (). To obtain the activation parameter of the electron transfer process, we investigated the temperature dependence of for immobilized Cc species (Fig. S11C). Pre‐exponential factors (A) and activation enthalpies () obtained from the intercept and slopes of the linear Arrhenius plots are shown in Table 4. The electron exchange rates between electrode and the different Cc species were essentially the same. Interestingly, the glutamine mutation drives a markedly higher activation enthalpy, whose effect is partially offset by an increase in the pre‐exponential factor. The higher values obtained for the mutants are consistent with greater solvent participation in the activation process [86], whereas their higher A values could come from the increased dynamics of the immobilized K8Q and K53Q mutants.
Analyses of trajectories from MD simulations revealed that both the overall folding and heme environment are very well preserved in the two Cc mutants. RMSD and radius of gyration (RG) values also displayed a similar behavior (Table 5), which indicates the absence of mutation‐induced protein unfolding or significant structural changes. The close similarity of the heme group solvent‐accessible surface and the Met80‐Fe distance in WT and mutant Cc species indeed confirmed that the heme environment is practically unaltered (Fig. S4), in agreement with visible CD spectra and E 1/2 values. Interestingly, the different values for atomic root mean square fluctuation (∆RSMF) obtained with the mutants as compared with WT Cc revealed an increase in protein dynamics of residues at the interaction surface of Cc with its metabolic partners (Fig. 6). In particular, the RMSF values of the 25–35, 40–60, and 72–85 stretches in the two acetyl mimetics are higher than those of WT Cc, explaining the increase in the pre‐exponential factor of immobilized acetyl‐mimetic mutants.
Table 5.
Statistics of molecular dynamics (MD) simulation trajectories with WT and mutant Cc species. Average values were calculated for the plateau of the MD simulation (last 50 ns). For all simulations, RMSD drift was smaller than 0.003 Å/ns.
| Average RMSD (Å) | Average RG (Å) | Average SAS (Å2) | Met80‐Fe distance (Å) | |
|---|---|---|---|---|
| Cc WT | 0.79 ± 0.14 | 12.93 ± 0.05 | 49.68 ± 16.26 | 2.46 ± 0.03 |
| Cc K8A | 1.00 ± 0.21 | 12.94 ± 0.05 | 57.19 ± 14.89 | 2.46 ± 0.03 |
| Cc K8Q | 0.87 ± 0.16 | 12.98 ± 0.05 | 51.78 ± 18.50 | 2.46 ± 0.03 |
| Cc K8AcK | 0.92 ± 0.17 | 13.00 ± 0.05 | 52.66 ± 18.24 | 2.45 ± 0.03 |
| Cc K53A | 0.90 ± 0.17 | 13.00 ± 0.05 | 60.15 ± 12.71 | 2.45 ± 0.03 |
| Cc K53Q | 0.97 ± 0.19 | 12.99 ± 0.04 | 48.20 ± 17.64 | 2.45 ± 0.03 |
| Cc K53AcK | 0.91 ± 0.16 | 12.99 ± 0.05 | 40.27 ± 13.45 | 2.45 ± 0.03 |
Fig. 6.
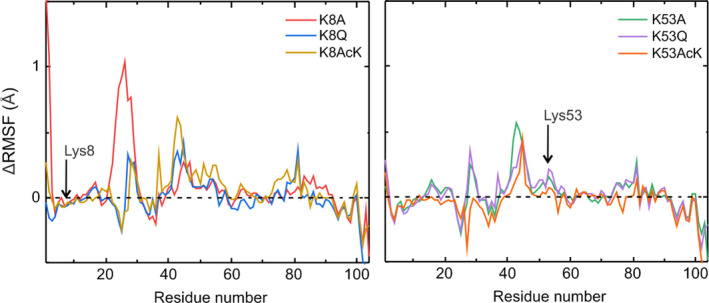
Simulation of molecular fluctuations in cytochrome c species. Comparison of the ∆RMSF of reduced K8A, K8Q, K8AcK, K53A, K53Q, and K53AcK mutants with respect to reduced WT Cc over the last 50 ns of their respective MD trajectories.
The impact of Lys8 and Ly53 mutations on the electron donor capacity of Cc in the mitochondrial context (where the hemeprotein acts as mediator between CIII and CIV) was also explored. Mitochondrial CcO activity assays revealed that electron flow was significantly lower with the two acetyl mimetics than with the WT Cc species (Fig. 7). These data are consistent with the in vitro electron transfer rate of K53Q to CIV recorded by Bazylianska and colleagues [32] and the involvement of Lys8 at the interface of the Cc‐CIV complex reported previously [36, 74, 84, 87]. It has actually been published that there are two binding sites for Cc—the so‐called proximal and distal sites—on the surface of CIV, where the surface residues exposed to the binding interface differ [15]. A detailed analysis of this binding interface showed the involvement of Lys8 and Lys53 residues at the proximal and distal Cc‐binding sites, respectively, in the Cc‐CIV complex (Fig. S12). This finding suggests that mutation of Lys8 could modulate electron transfer in the Cc‐CIV complex, whereas mutation of Lys53 could slow down the turnover of Cc molecules. Although electron carriers display a certain degree of ‘promiscuity’ due to the fact that their surfaces are often recognized by multiple partners, their redox centers are usually close to a hydrophobic patch that optimizes electron transfer between redox partners, as is the case in a high number of metabolic processes and reactions [88, 89, 90, 91, 92, 93, 94, 95]. Hence, the observed alteration of electron transfer to CIV when Lys8 or Lys53 are modified can be explained by a direct or indirect modification of the recognition mechanism between the two partners [36].
Fig. 7.
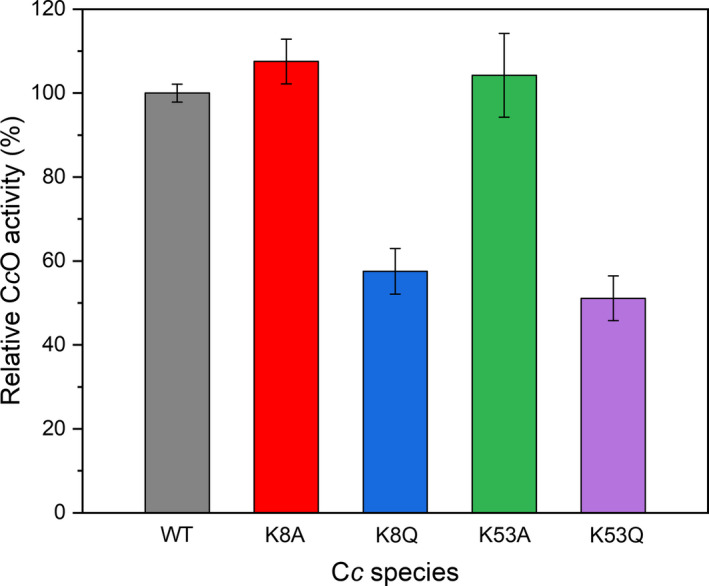
Cytochrome c oxidase activity with WT and mutant cytochrome c as electron donors. CcO activity was determined in mitochondria isolated from MEF cells upon addition of exogenous WT, K8Q, K8A, K53Q, and K53A Cc. Experimental data were normalized with respect to that obtained with WT Cc. Each value represents the average of three individual experiments ± SD. See the Methods section for details.
Conclusion
Structural and functional analysis of site‐directed acetyl‐mimetic mutants is a helpful approach to acetylated Cc characterization. Our CD and Vis spectroscopy results indicated that acetyl‐mimetic mutants at residues Lys8 and Lys53 keep the overall physicochemical features of Cc. However, the thermal stability of Cc species is slightly altered, with changes dependent on the spatial location of mutated residue. The observed differences may be related to the backbone resonance changes detected by NMR, since the magnitude and number of affected residues also depend on the position of the mutated amino acid.
Interestingly, mimetic acetylation enhances Cc dynamics and solvent involvement in the redox process in the immobilized state, as observed by cyclic voltammetry. With respect to redox functionality, both mimetic Cc species exhibit a significant decrease in CcO activity in a mitochondrial context, suggesting that these residues are involved in the interaction with CIV and, consequently, that alteration of protein functionality by lysine acetylation is likely. On the other hand, when binding to the Cc 1 partner, only the Lys8 mutants show an increased binding affinity for the Cc 1‐Cc complex; this can be attributed to the fact that Lys8 is in direct contact with Cc 1.
These results lay the groundwork for more detailed studies on the effect of acetylation on the multiple extra‐mitochondrial functions of Cc and its implication for the development of certain diseases.
Conflict of interest
All authors declare no conflict of interest.
Author contributions
All authors contributed to conception and experimental design, discussed findings and wrote the manuscript. IM, GPM, AGC and JLOS performed experimental research work.
Supporting information
Fig. S1. Expression and purification of WT and mutant cytochrome c species. (A) Left panel: SDS‐PAGE of the 15N‐labelled WT, K8A, K8Q, K53A and K53Q Cc samples used for NMR experiments (5 μg of protein loaded in each lane). The bands within the red rectangle (below 15 kDa) correspond to the different Cc species. M: Molecular mass markers. Right panel: Western blot of purified WT, K8A, K8Q, K53A and K53Q Cc showing the detection of the Cc band in each lane. (B) Tryptic digestion of proteins extracted from the bands of an SDS‐PAGE similar to that in (A) but loaded with 14N‐labelled Cc samples. The calculated masses of AIFIMK, QIFIMK, KTGQAPGYSYTAANANK and KTGQAPGYSYTAANQNK fragments are 722, 779, 1741 and 1799 Da, respectively.
Fig. S2. Secondary structure analysis of WT and mutant cytochrome c species. Percentage of secondary structure for oxidized WT, K8A, K8Q, K53A and K53Q Cc is calculated from far‐UV CD data with the CDPro software package (SP43, SMP50 and CLSTR reference sets) [1]. The results are expressed as the mean ± SD.
Fig. S3. Alkaline transition of WT and mutant cytochrome c species. Electronic absorption spectra were recorded at different pH values (left panels), and titration curves were determined by following the absorbance changes at 695 nm (right panels) of the oxidized species. Process reversibility was checked by recording the last spectrum at the initial pH value. The full data set was fitted to the Henderson–Hasselbalch equation to calculate the pK a values (red lines; see details in Methods section).
Fig. S4. Molecular dynamic simulations of WT and mutant cytochrome c species. Average of the Met80Fe distance (upper) and solvent accessibility of the heme group (lower) within the last 50 ns of their respective MD trajectories are shown for WT, K8A, K8Q, K8AcK, K53A, K53Q and K53AcK Cc species. The results are expressed as the mean ± SD.
Fig. S5. Thermal stability of WT and mutant cytochrome c species. The first principal component (P1) of CD and fluorescence spectra of oxidized WT, K8A, K8Q, K53A and K53Q Cc recorded at varying temperature is plotted.
Fig. S6. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K8A (red) cytochrome c. Insets: Zooms of the signals framed in yellow boxes. The green arrows stand for the CSPs of mutant Cc as compared with the WT species. The yellow circle denotes the mutated residue.
Fig. S7. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K8Q (blue) cytochrome. Insets: Zooms of the signals framed in yellow boxes. The red arrows stand for the CSPs of mutant Cc as compared with the WT species. The red dotted line shows the 15N chemical shift at which the 1H of the Q8 ε‐amine group appears. The yellow circle denotes the mutated residue.
Fig. S8. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K53A (green) cytochrome c. Insets: Zooms of the signals framed in yellow boxes. The red arrows stands for the CSPs of mutant Cc as compared with the WT species. The yellow circle surrounds the mutated residue.
Fig. S9. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K53Q (purple) cytochrome c. Insets: Zooms of the signals framed in yellow boxes. The green arrows stand for the CSPs of mutant Cc as compared with the WT species. The green dotted line shows the 15N chemical shift at which the 1H of the Q53 ε‐amine group appears. The yellow circle surrounds the mutated residue.
Fig. S10. Interaction of WT and mutant cytochrome c with cytochrome c 1 as determined by BLI. Wavelength shift over time (left panels) and calculated dissociation equilibrium constant (K D; right panels) for the interaction between reduced Cc 1 and reduced WT, K8Q or K53Q Cc were determined at increasing Cc concentration. The dotted gray line in the left panels indicates the time at which the wavelength shifts were taken for calculation of the K D values in the right panels. The data set was fitted to a non‐linear equation to obtain the K D values (red lines; see details in Methods section). The results are expressed as the mean ± SD.
Fig. S11. Electrochemical characterization of WT and mutant cytochrome c. (A) Cyclic voltammograms of reduced Cc species immobilized onto a chemically modified polycrystalline gold electrode (scan rate: 0.1 V s‐1). (B) Midpoint redox potential and (C) electron transfer rate constant for immobilized Cc as determined at varying temperature under the same experimental conditions as in panel A. The results are expressed as the mean ± SD.
Fig. S12. Binding sites for cytochrome c on complex IV at the respiratory supercomplex. The representative structure of WT Cc at the distal site of complex IV is in orange, where Lys53 (green) points towards the complex IV surface (light gray), and that at the proximal site is in red, where Lys8 (blue) points towards the complex IV surface.
Table S1. Primers used in the design of cytochrome c mutants.
Table S2. BLI protocol for cytochrome c 1 – cytochrome c interaction.
Acknowledgements
This work was supported by the Spanish Government (PGC2018‐096049‐B‐I00, FEDER/Ministerio de Ciencia e Innovación–Agencia Estatal de Investigación), European Regional Development Fund (FEDER), Andalusian Government (BIO‐198; US‐1254317, US‐1257019, P18‐FR‐3487 and P18‐HO‐4091, US/JUNTA/FEDER, UE), University of Seville (VI PPIT‐US) and the Ramón Areces Foundation. GPM was awarded a PhD fellowship from the Spanish Ministry of Education, Culture and Sport (FPU17/04604).
Gonzalo Pérez‐Mejías and Alejandra Guerra‐Castellano contributed equally to the article
Data accessibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Walsh C (2006) Posttranslational modification of proteins: Expanding nature’s inventory. Roberts and Company Publishers, Greenwood Village CO. [Google Scholar]
- 2. Karve TM and Cheema AK (2011) Small changes huge impact: The role of protein posttranslational modifications in cellular homeostasis and disease. J Amino Acids 2011, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Müller MM (2018) Post‐translational modifications of protein backbones: Unique functions, mechanisms, and challenges. Biochemistry 57, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang YC, Peterson SE and Loring JF (2014) Protein post‐translational modifications and regulation of pluripotency in human stem cells. Cell Res 24, 143–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li S, Iakoucheva LM, Mooney SD and Radivojac P (2010) Loss of post‐translational modification sites in disease. Pac Symp Biocomput 2010, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu H, Wang Y, Lin S, Deng W, Peng D, Cui Q and Xue Y (2018) PTMD: a database of human disease‐associated post‐translational modifications. Genomics Proteomics Bioinformatics 16, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guerra‐Castellano A, Márquez I, Pérez‐Mejías G, Díaz‐Quintana A, De la Rosa MA and Díaz‐Moreno I (2020) Post‐Translational modifications of cytochrome c in cell life and disease. Int J Mol Sci 21, 8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pe J, Cheng T, Kho Y, Xiao H, Xiao L et al. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- 9. Anderson KA and Hirschey MD (2012) Mitochondrial protein acetylation regulates metabolism. Essays Biochem 52, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith‐Hammond CL, Hoyos E and Miernyk JA (2014) The pea seedling mitochondrial Nε‐lysine acetylome. Mitochondrion 19 B, 154–165. [DOI] [PubMed] [Google Scholar]
- 11. Ali I, Conrad RJ, Verdin E and Ott M (2018) Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem Rev 118, 1216–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hosp F, Lassowskat I, Santoro V, De Vleesschauwer D, Fliegner D, Redestig H, Mann M, Christian S, Hannah MA and Finkemeier I (2017) Lysine acetylation in mitochondria: from inventory to function. Mitochondrion 33, 58–71. [DOI] [PubMed] [Google Scholar]
- 13. Wada K and Okunuki K (1968) Studies on chemically modified cytochrome c. I. The acetylated cytochrome c. J Biochem 64, 667–687. [DOI] [PubMed] [Google Scholar]
- 14. Kalpage HA, Bazylianska V, Recanati MA, Fite A, Liu J, Wan J, Mantena N, Malek MH, Podgorski I, Heath EI et al. (2019) Tissue‐specific regulation of cytochrome c by post‐translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J 33, 1540–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moreno‐Beltrán B, Díaz‐Quintana A, González‐Arzola K, Velázquez‐Campoy A, De la Rosa MA and Díaz‐Moreno I (2014) Cytochrome c 1 exhibits two binding sites for cytochrome c in plants. Biochim Biophys Acta 1837, 1717–1729. [DOI] [PubMed] [Google Scholar]
- 16. Moreno‐Beltrán B, Díaz‐Moreno I, González‐Arzola K, Guerra‐Castellano A, Velázquez‐Campoy A, De la Rosa MA and Díaz‐Quintana A (2015) Respiratory complexes III and IV can each bind two molecules of cytochrome c at low ionic strength. FEBS Lett 589, 476–483. [DOI] [PubMed] [Google Scholar]
- 17. Pérez‐Mejías G, Guerra‐Castellano A, Díaz‐Quintana A, De la Rosa MA and Díaz‐Moreno I (2019) Cytochrome c: surfing off of the mitochondrial membrane on the tops of complexes III and IV. Comput Struct Biotechnol J 17, 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soltys BJ, Andrews DW, Jemmerson R and Gupta RS (2001) Cytochrome‐c localizes in secretory granules in pancreas and anterior pituitary. Cell Biol Int 25, 331–338. [DOI] [PubMed] [Google Scholar]
- 19. Martínez‐Fábregas J, Díaz‐Moreno I, González‐Arzola K, Janocha S, Navarro JA, Hervás M, Bernhardt R, Díaz‐Quintana A and De la Rosa MA (2013) New Arabidopsis thaliana cytochrome c partners: a look into the elusive role of cytochrome c in programmed cell death in plants. Mol Cell Proteomics 12, 3666–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martínez‐Fábregas J, Díaz‐Moreno I, González‐Arzola K, Janocha S, Navarro JA, Hervás M, Bernhardt R, Velázquez‐Campoy A, Díaz‐Quintana A and De la Rosa MA (2014) Structural and functional analysis of novel human cytochrome c targets in apoptosis. Mol Cell Proteomics 13, 1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martínez‐Fábregas J, Díaz‐Moreno I, González‐Arzola K, Díaz‐Quintana A and De la Rosa MA (2014) A common signalosome for programmed cell death in humans and plants. Cell Death Dis 5, e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. González‐Arzola K, Díaz‐Moreno I, Cano‐González A, Díaz‐Quintana A, Velázquez‐Campoy A, Moreno‐Beltrán B, López‐Rivas A and De la Rosa MA (2015) Structural basis for inhibition of the histone chaperone activity of SET/TAF‐Iβ by cytochrome c . Proc Natl Acad Sci USA 112, 9908–9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. González‐Arzola K, Díaz‐Quintana A, Rivero‐Rodríguez F, Velázquez‐Campoy A, De la Rosa MA and Díaz‐Moreno I (2017) Histone chaperone activity of Arabidopsis thaliana NRP1 is blocked by cytochrome c . Nucleic Acids Res 45, 2150–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. González‐Arzola K, Velázquez‐Cruz A, Guerra‐Castellano A, Casado‐Combreras MA, Pérez‐Mejías G, Díaz‐Quintana A, Díaz‐Moreno I and De la Rosa MA (2019) New moonlighting functions of mitochondrial cytochrome c in the cytoplasm and nucleus. FEBS Lett 593, 3101–3119. [DOI] [PubMed] [Google Scholar]
- 25. Díaz‐Moreno I, Velázquez‐Cruz A, Curran‐French S, Díaz‐Quintana A and De la Rosa MA (2018) Nuclear cytochrome c ‐ a mitochondrial visitor regulating damaged chromatin dynamics. FEBS Lett 592, 172–178. [DOI] [PubMed] [Google Scholar]
- 26. Elena‐Real CA, Díaz‐Quintana A, González‐Arzola K, Velázquez‐Campoy A, Orzáez M, López‐Rivas A, Gil‐Caballero S, De la Rosa MA and Díaz‐Moreno I (2018) Cytochrome c speeds up caspase cascade activation by blocking 14‐3‐3ε‐dependent Apaf‐1 inhibition. Cell Death Dis 9, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elena‐Real CA, González‐Arzola K, Pérez‐Mejías G, Díaz‐Quintana A, Velázquez‐Campoy A, Desvoyes B, Gutiérrez C, De la Rosa MA and Díaz‐Moreno I (2020) Proposed mechanism for regulation of H2O2 ‐induced programmed cell death in plants by binding of cytochrome c to 14‐3‐3 proteins. Plant J 6, 74–85. [DOI] [PubMed] [Google Scholar]
- 28. Rivero‐Rodríguez F, Díaz‐Quintana A, Velázquez‐Cruz A, González‐Arzola K, Gavilan MP, Velázquez‐Campoy A, Ríos RM, De la Rosa MA and Díaz‐Moreno I (2021) Inhibition of the PP2A activity by the histone chaperone ANP32B is long‐range allosterically regulated by respiratory cytochrome c . Redox Biol 43, 101967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minakami S, Titani K and Ishikura H (1958) The structure of cytochrome c. II. Properties of acetylated cytochrome c . J Biochem 45, 341–348. [Google Scholar]
- 30. Azzi A, Montecucco C and Richter C (1975) The use of acetylated ferricytochrome c for the detection of superoxide radicals produced in biological membranes. Biochem Biophys Res Commun 65, 597–603. [DOI] [PubMed] [Google Scholar]
- 31. Warburg O (1956) On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- 32. Bazylianska V, Kalpage HA, Wan J, Vaishnav A, Mahapatra G, Turner AA, Chowdhury DD, Kim K, Morse PT, Lee I et al. (2021) Lysine 53 acetylation of cytochrome c in prostate cancer: Warburg metabolism and evasion of apoptosis. Cells 10, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Purring‐Koch C and McLendon G (2000) Cytochrome c binding to Apaf‐1: the effects of dATP and ionic strength. Proc Natl Acad Sci USA 97, 11928–11931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. König BW, Osheroff N, Wilms J, Muijsers AO, Dekker HL and Margoliash E (1980) Mapping of the interaction domain for purified cytochrome c 1 on cytochrome c . FEBS Lett 111, 395–398. [DOI] [PubMed] [Google Scholar]
- 35. Kokhan O, Wraight CA and Tajkhorshid E (2010) The binding interface of cytochrome c and cytochrome c 1 in the bc 1 complex: rationalizing the role of key residues. Biophys J 99, 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimada S, Shinzawa‐Itoh K, Baba J, Aoe S, Shimada A, Yamashita E, Kang J, Tateno M, Yoshikawa S and Tsukihara T (2017) Complex structure of cytochrome c‐cytochrome c oxidase reveals a novel protein‐protein interaction mode. EMBO J 36, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pérez‐Mejías G, Olloqui‐Sariego JL, Guerra‐Castellano A, Díaz‐Quintana A, Calvente JJ, Andreu R, De la Rosa MA and Díaz‐Moreno I (2020) Physical contact between cytochrome c 1 and cytochrome c increases the driving force for electron transfer. Biochim Biophys Acta 1861, 148277. [DOI] [PubMed] [Google Scholar]
- 38. Olteanu A, Patel CN, Dedmon MM, Kennedy S, Linhoff MW, Minder CM, Potts PR, Deshmukh M and Pielak GJ (2003) Stability and apoptotic activity of recombinant human cytochrome c . Biochem Biophys Res Commun 312, 733–740. [DOI] [PubMed] [Google Scholar]
- 39. Guerra‐Castellano A, Díaz‐Moreno I, Velázquez‐Campoy A, De la Rosa MA and Díaz‐Quintana A (2016) Structural and functional characterization of phosphomimetic mutants of cytochrome c at threonine 28 and serine 47. Biochim Biophys Acta 1857, 387–395. [DOI] [PubMed] [Google Scholar]
- 40. Guerra‐Castellano A, Díaz‐Quintana A, Moreno‐Beltrán B, López‐Prados J, Nieto PM, Meister W, Staffa J, Teixeira M, Hildebrandt P, De la Rosa MA et al. (2015) Mimicking tyrosine phosphorylation in human cytochrome c by the evolved tRNA synthetase technique. Chem Eur J 21, 15004–15012. [DOI] [PubMed] [Google Scholar]
- 41. Reijenga J, van Hoof A, van Loon A and Teunissen B (2013) Development of methods for the determination of pKa values. Anal Chem Insights 8, 53–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Imai M, Saio T, Kumeta H, Uchida T, Inagaki F and Ishimori K (2016) Investigation of the redox‐dependent modulation of structure and dynamics in human cytochrome c . Biochem Biophys Res Commun 469, 978–984. [DOI] [PubMed] [Google Scholar]
- 43. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC and Ferrin TE (2004) UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 44. Papamokos GV, Tziatzos G, Papageorgiou DG, Georgatos SD, Politou AS and Kaxiras E (2012) Structural role of RKS motifs in chromatin interactions: a molecular dynamics study of HP1 bound to a variably modified histone tail. Biophys J 102, 1926–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Autenrieth F, Tajkhorshid E, Baudry J and Luthey‐Schulten Z (2004) Classical force field parameters for the heme prosthetic group of cytochrome c . J Comput Chem 25, 1613–1622. [DOI] [PubMed] [Google Scholar]
- 46. Case DA, Belfon K, Ben‐Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, Cruzeiro VWD, Darden TA, Duke RE, Giambasu G et al. (2020) AMBER 2020. University of California, San Francisco, CA. [Google Scholar]
- 47. Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD et al. (2017) OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE and Simmerling C (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Izadi S, Anandakrishnan R and Onufriev AV (2014) Building water models: A different approach. J Phys Chem Lett 5, 3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roe DR and Cheatham TE (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 51. Lee W, Tonelli M and Markley JL (2015) NMRFAM‐SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Müller‐Esparza H, Osorio‐Valeriano M, Steube N, Thanbichler M and Randau L (2020) Bio‐Layer Interferometry analysis of the target binding activity of CRISPR‐Cas effector complexes. Front Mol Biosci 7, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Frezza C, Cipolat S and Scorrano L (2007) Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- 54. Guerra‐Castellano A, Díaz‐Quintana A, Pérez‐Mejías G, Elena‐Real CA, González‐Arzola K, García‐Mauriño SM, De la Rosa MA and Díaz‐Moreno I (2018) Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc Natl Acad Sci USA 115, 7955–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Isom AL, Barnes S, Wilson L, Kirk M, Coward L and Darley‐Usmar V (2004) Modification of cytochrome c by 4‐hydroxy‐ 2‐nonenal: Evidence for histidine, lysine, and arginine‐aldehyde adducts. J Am Soc Mass Spectrom 15, 1136–1147. [DOI] [PubMed] [Google Scholar]
- 56. Santucci R and Ascoli F (1997) The Soret circular dichroism spectrum as a probe for the heme Fe(III)‐Met(80) axial bond in horse cytochrome c . J Inorg Biochem 68, 211–214. [DOI] [PubMed] [Google Scholar]
- 57. Banci L, Bertini I, Gray HB, Luchinat C, Reddig T, Rosato A and Turano P (1997) Solution structure of oxidized horse heart cytochrome c . Biochemistry 36, 9867–9877. [DOI] [PubMed] [Google Scholar]
- 58. Blauer G, Sreerama N and Woody RW (1993) Optical activity of hemoproteins in the Soret region. Circular dichroism of the heme undecapeptide of cytochrome c in aqueous solution. Biochemistry 32, 6674–6679. [DOI] [PubMed] [Google Scholar]
- 59. Boffi F, Bonincontro A, Cinelli S, Congiu Castellano A, De Francesco A, Della Longa S, Girasole M and Onori G (2001) pH‐dependent local structure of ferricytochrome c studied by x‐ray absorption spectroscopy. Biophys J 80, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ying T, Zhong F, Xie J, Feng Y, Wang ZH, Huang ZX and Tan X (2009) Evolutionary alkaline transition in human cytochrome c . J Bioenerg Biomembr 41, 251–257. [DOI] [PubMed] [Google Scholar]
- 61. Wilson MT and Greenwood C (1996) The alkaline transition in ferricytochrome c . In Cytochrome c: A Multidisciplinary Approach (Scott RA and Mauk AG, eds), pp. 611–634. University Science Books, Sausalito, CA. [Google Scholar]
- 62. Assfalg M, Bertini I, Dolfi A, Turano P, Mauk AG, Rosell FI and Gray HB (2003) Structural model for an alkaline form of ferricytochrome c . J Am Chem Soc 125, 2913–2922. [DOI] [PubMed] [Google Scholar]
- 63. Schejter A and George P (1964) The 695‐mμ band of ferricytochrome c and its relationship to protein conformation. Biochemistry 3, 1045–1049. [DOI] [PubMed] [Google Scholar]
- 64. Dickerson R and Timkovich R (1975) Cytochrome c . In The Enzymes (Boyer P, ed), pp. 397–472. Academic Press, New York, NY. [Google Scholar]
- 65. García‐Heredia JM, Díaz‐Quintana A, Salzano M, Orzáez M, Pérez‐Payá E, Teixeira M, De la Rosa MA and Díaz‐Moreno I (2011) Tyrosine phosphorylation turns alkaline transition into a biologically relevant process and makes human cytochrome c behave as an anti‐apoptotic switch. J Biol Inorg Chem 16, 1155–1168. [DOI] [PubMed] [Google Scholar]
- 66. Poulos TL (1996) The role of the proximal ligand in heme enzymes. J Biol Inorg Chem 1, 356–359. [Google Scholar]
- 67. Maity H, Maity M and Englander SW (2004) How cytochrome c folds, and why: submolecular foldon units and their stepwise sequential stabilization. J Mol Biol 343, 223–233. [DOI] [PubMed] [Google Scholar]
- 68. Maity H, Rumbley JN and Englander SW (2006) Functional role of a protein foldon –an Omega‐loop foldon controls the alkaline transition in ferricytochrome c . Proteins 63, 349–355. [DOI] [PubMed] [Google Scholar]
- 69. Dragomir I, Hagarman A, Wallace C and Schweitzer‐Stenner R (2007) Optical band splitting and electronic perturbations of the heme chromophore in cytochrome c at room temperature probed by visible electronic circular dichroism spectroscopy. Biophys J 92, 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fisher WR, Taniuchi H and Anfinsen CB (1973) On the role of heme in the formation of the structure of cytochrome c . J Biol Chem 248, 3188–3195. [PubMed] [Google Scholar]
- 71. Kirby EP and Steiner RF (1970) The tryptophan microenvironments in apomyoglobin. J Biol Chem 245, 6300–6306. [PubMed] [Google Scholar]
- 72. Jameson C (1996) Understanding NMR chemical shifts. Annu Rev Phys Chem 47, 135–169. [Google Scholar]
- 73. Wüthrich K (1986) NMR of proteins and nucleic acids. John Wiley and Sons, Inc, New York, NY. [Google Scholar]
- 74. Rieder R and Bosshard HR (1980) Comparison of the binding sites on cytochrome c for cytochrome c oxidase, cytochrome bc 1, and cytochrome c 1. Differential acetylation of lysyl residues in free and complexed cytochrome c . J Biol Chem 255, 4732–4739. [PubMed] [Google Scholar]
- 75. Yu H, Lee I, Salomon AR, Yu K and Hüttemann M (2008) Mammalian liver cytochrome c is tyrosine‐48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim Biophys Acta 1777, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pecina P, Borisenko GG, Belikova NA, Tyurina YY, Pecinova A, Lee I, Samhan‐Arias AK, Przyklenk K, Kagan VE and Hüttemann M (2010) Phosphomimetic substitution of cytochrome c tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry 49, 6705–6714. [DOI] [PubMed] [Google Scholar]
- 77. Hildick‐Smith GJ, Downey MC, Gretebeck LM, Gersten RA and Sandwick RK (2011) Ribose 5‐Phosphate glycation reduces cytochrome c respiratory activity and membrane affinity. Biochemistry 50, 11047–11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aluri HS, Simpson DC, Allegood JC, Hu Y, Szczepanek K, Gronert S, Chen Q and Lesnefsky EJ (2014) Electron flow into cytochrome c coupled with reactive oxygen species from the electron transport chain converts cytochrome c to a cardiolipin peroxidase: Role during ischemia‐reperfusion. Biochim Biophys Acta 1840, 3199–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mahapatra G, Varughese A, Ji Q, Lee I, Liu J, Vaishnav A, Sinkler C, Kapralov AA, Moraes CT, Sanderson TH et al. (2017) Phosphorylation of cytochrome c threonine 28 regulates electron transport chain activity in kidney: Implications for amp kinase. J Biol Chem 292, 64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Moreno‐Beltrán B, Guerra‐Castellano A, Díaz‐Quintana A, Del Conte R, García‐Mauriño SM, Díaz‐Moreno S, González‐Arzola K, Santos‐Ocaña C, Velázquez‐Campoy A, De La Rosa MA et al. (2017) Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc Natl Acad Sci USA 114, E3041–E3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Smith HT, Ahmed AJ and Millett F (1981) Electrostatic interaction of cytochrome c with cytochrome c1 and cytochrome oxidase. J Biol Chem 256, 4984–4990. [PubMed] [Google Scholar]
- 82. Lange C and Hunte C (2002) Crystal structure of the yeast cytochrome bc 1 complex with its bound substrate cytochrome c . Proc Natl Acad Sci USA 99, 2800–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hunte C, Solmaz S and Lange C (2002) Electron transfer between yeast cytochrome bc 1 complex and cytochrome c: a structural analysis. Biochim Biophys Acta ‐ Bioenerg 1555, 21–28. [DOI] [PubMed] [Google Scholar]
- 84. Sakamoto K, Kamiya M, Imai M, Shinzawa‐Itoh K, Uchida T, Kawano K, Yoshikawa S and Ishimori K (2011) NMR basis for interprotein electron transfer gating between cytochrome c and cytochrome c oxidase. Proc Natl Acad Sci USA 108, 12271–12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rodríguez‐Roldán V, García‐Heredia JM, Navarro JA, Hervás M, De la Cerda B, Molina‐Heredia FP and De la Rosa MA (2006) A comparative kinetic analysis of the reactivity of plant, horse, and human respiratory cytochrome c towards cytochrome c oxidase. Biochem Biophys Res Commun 346, 1108–1113. [DOI] [PubMed] [Google Scholar]
- 86. Olloqui‐Sariego JL, Zakharova G, Poloznikov AA, Calvente JJ, Hushpulian DM, Gorton L and Andreu R (2019) The Fe (III)/Fe(II) redox couple as a probe of immobilized tobacco peroxidase: effect of the immobilization protocol. Electrochim Acta 299, 55–61. [Google Scholar]
- 87. Sato W, Hitaoka S, Inoue K, Imai M, Saio T, Uchida T, Shinzawa‐Itoh K, Yoshikawa S, Yoshizawa K and Ishimori K (2016) Energetic mechanism of cytochrome c‐cytochrome c oxidase electron transfer complex formation under turnover conditions revealed by mutational effects and docking simulation. J Biol Chem 291, 15320–21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Medina M, Hervás M, Navarro JA, De la Rosa MA, Gómez‐Moreno C and Tollin G (1992) A laser flash absorption spectroscopy study of Anabaena sp. PCC 7119 flavodoxin photoreduction by photosystem I particles from spinach. FEBS Lett 313, 239–242. [DOI] [PubMed] [Google Scholar]
- 89. De la Cerda B, Navarro JA, Hervás M and De la Rosa MA (1997) Changes in the reaction mechanism of electron transfer from plastocyanin to photosystem I in the cyanobacterium Synechocystis sp. PCC 6803 as induced by site‐directed mutagenesis of the copper protein. Biochemistry 36, 10125–10130. [DOI] [PubMed] [Google Scholar]
- 90. Sun J, Xu W, Hervás M, Navarro JA, Rosa MA and Chitnis PR (1999) Oxidizing side of the cyanobacterial photosystem I. Evidence for interaction between the electron donor proteins and a luminal surface helix of the PsaB subunit. J Biol Chem 274, 19048–19054. [DOI] [PubMed] [Google Scholar]
- 91. Frazão C, Enguita FJ, Coelho R, Sheldrick GM, Navarro JA, Hervás M, De la Rosa MA and Carrondo MA (2001) Crystal structure of low‐potential cytochrome c 549 from Synechocystis sp. PCC 6803 at 1.21 Å resolution. J Biol Inorg Chem 6, 324–332. [DOI] [PubMed] [Google Scholar]
- 92. Díaz‐Moreno I, Díaz‐Quintana A, De la Rosa MA, Crowley PB and Ubbink M (2005) Different modes of interaction in cyanobacterial complexes of plastocyanin and cytochrome f . Biochemistry 44, 3176–3183. [DOI] [PubMed] [Google Scholar]
- 93. Díaz‐Moreno I, Díaz‐Quintana A, Molina‐Heredia FP, Nieto PM, Hansson O, De la Rosa MA and Karlsson BG (2005) NMR analysis of the transient complex between membrane photosystem I and soluble cytochrome c 6 . J Biol Chem 280, 7925–7931. [DOI] [PubMed] [Google Scholar]
- 94. Díaz‐Moreno I, Díaz‐Quintana A, Ubbink M and De la Rosa MA (2005) An NMR‐based docking model for the physiological transient complex between cytochrome f and cytochrome c 6 . FEBS Lett 579, 2891–2896. [DOI] [PubMed] [Google Scholar]
- 95. Goñi G, Herguedas B, Hervás M, Peregrina JR, De la Rosa MA, Gómez‐Moreno C, Navarro JA, Hermoso JA, Martínez‐Júlvez M and Medina M (2009) Flavodoxin: a compromise between efficiency and versatility in the electron transfer from Photosystem I to Ferredoxin‐NADP(+) reductase. Biochim Biophys Acta ‐ Bioenerg 1787, 144–154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression and purification of WT and mutant cytochrome c species. (A) Left panel: SDS‐PAGE of the 15N‐labelled WT, K8A, K8Q, K53A and K53Q Cc samples used for NMR experiments (5 μg of protein loaded in each lane). The bands within the red rectangle (below 15 kDa) correspond to the different Cc species. M: Molecular mass markers. Right panel: Western blot of purified WT, K8A, K8Q, K53A and K53Q Cc showing the detection of the Cc band in each lane. (B) Tryptic digestion of proteins extracted from the bands of an SDS‐PAGE similar to that in (A) but loaded with 14N‐labelled Cc samples. The calculated masses of AIFIMK, QIFIMK, KTGQAPGYSYTAANANK and KTGQAPGYSYTAANQNK fragments are 722, 779, 1741 and 1799 Da, respectively.
Fig. S2. Secondary structure analysis of WT and mutant cytochrome c species. Percentage of secondary structure for oxidized WT, K8A, K8Q, K53A and K53Q Cc is calculated from far‐UV CD data with the CDPro software package (SP43, SMP50 and CLSTR reference sets) [1]. The results are expressed as the mean ± SD.
Fig. S3. Alkaline transition of WT and mutant cytochrome c species. Electronic absorption spectra were recorded at different pH values (left panels), and titration curves were determined by following the absorbance changes at 695 nm (right panels) of the oxidized species. Process reversibility was checked by recording the last spectrum at the initial pH value. The full data set was fitted to the Henderson–Hasselbalch equation to calculate the pK a values (red lines; see details in Methods section).
Fig. S4. Molecular dynamic simulations of WT and mutant cytochrome c species. Average of the Met80Fe distance (upper) and solvent accessibility of the heme group (lower) within the last 50 ns of their respective MD trajectories are shown for WT, K8A, K8Q, K8AcK, K53A, K53Q and K53AcK Cc species. The results are expressed as the mean ± SD.
Fig. S5. Thermal stability of WT and mutant cytochrome c species. The first principal component (P1) of CD and fluorescence spectra of oxidized WT, K8A, K8Q, K53A and K53Q Cc recorded at varying temperature is plotted.
Fig. S6. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K8A (red) cytochrome c. Insets: Zooms of the signals framed in yellow boxes. The green arrows stand for the CSPs of mutant Cc as compared with the WT species. The yellow circle denotes the mutated residue.
Fig. S7. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K8Q (blue) cytochrome. Insets: Zooms of the signals framed in yellow boxes. The red arrows stand for the CSPs of mutant Cc as compared with the WT species. The red dotted line shows the 15N chemical shift at which the 1H of the Q8 ε‐amine group appears. The yellow circle denotes the mutated residue.
Fig. S8. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K53A (green) cytochrome c. Insets: Zooms of the signals framed in yellow boxes. The red arrows stands for the CSPs of mutant Cc as compared with the WT species. The yellow circle surrounds the mutated residue.
Fig. S9. Superimposition of 2D 1H–15N HSQC NMR spectra of WT (black) and K53Q (purple) cytochrome c. Insets: Zooms of the signals framed in yellow boxes. The green arrows stand for the CSPs of mutant Cc as compared with the WT species. The green dotted line shows the 15N chemical shift at which the 1H of the Q53 ε‐amine group appears. The yellow circle surrounds the mutated residue.
Fig. S10. Interaction of WT and mutant cytochrome c with cytochrome c 1 as determined by BLI. Wavelength shift over time (left panels) and calculated dissociation equilibrium constant (K D; right panels) for the interaction between reduced Cc 1 and reduced WT, K8Q or K53Q Cc were determined at increasing Cc concentration. The dotted gray line in the left panels indicates the time at which the wavelength shifts were taken for calculation of the K D values in the right panels. The data set was fitted to a non‐linear equation to obtain the K D values (red lines; see details in Methods section). The results are expressed as the mean ± SD.
Fig. S11. Electrochemical characterization of WT and mutant cytochrome c. (A) Cyclic voltammograms of reduced Cc species immobilized onto a chemically modified polycrystalline gold electrode (scan rate: 0.1 V s‐1). (B) Midpoint redox potential and (C) electron transfer rate constant for immobilized Cc as determined at varying temperature under the same experimental conditions as in panel A. The results are expressed as the mean ± SD.
Fig. S12. Binding sites for cytochrome c on complex IV at the respiratory supercomplex. The representative structure of WT Cc at the distal site of complex IV is in orange, where Lys53 (green) points towards the complex IV surface (light gray), and that at the proximal site is in red, where Lys8 (blue) points towards the complex IV surface.
Table S1. Primers used in the design of cytochrome c mutants.
Table S2. BLI protocol for cytochrome c 1 – cytochrome c interaction.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.