

Abstract
Background:
Exposure to plastic-associated endocrine disrupting chemicals (EDCs) has been associated with an increased risk of cardiovascular disease (CVD) in humans. However, the underlying mechanisms for this association are unclear. Many EDCs have been shown to function as ligands of the nuclear receptor pregnane X receptor (PXR), which functions as xenobiotic sensor but also has pro-atherogenic effects in vivo.
Objective:
We sought to investigate the contribution of PXR to the adverse effects dicyclohexyl phthalate (DCHP), a widely used phthalate plasticizer, on lipid homeostasis and CVD risk factors.
Methods:
Cell-based assays, primary organoid cultures, and PXR conditional knockout and PXR-humanized mouse models were used to investigate the impact of DCHP exposure on PXR activation and lipid homeostasis in vitro and in vivo. Targeted lipidomics were performed to measure circulating ceramides, novel predictors for CVD.
Results:
DCHP was identified as a potent PXR-selective agonist that led to higher plasma cholesterol levels in wild-type mice. DCHP was then demonstrated to activate intestinal PXR to elicit hyperlipidemia by using tissue-specific PXR-deficient mice. Interestingly, DCHP exposure also led to higher circulating ceramides in a PXR-dependent manner. DCHP-mediated PXR activation stimulated the expression of intestinal genes mediating lipogenesis and ceramide synthesis. Given that PXR exhibits considerable species-specific differences in receptor pharmacology, PXR-humanized mice were also used to replicate these findings.
Discussion:
Although the adverse health effects of several well-known phthalates have attracted considerable attention, little is known about the potential impact of DCHP on human health. Our studies demonstrate that DCHP activated PXR to induce hypercholesterolemia and ceramide production in mice. These results indicate a potentially important role of PXR in contributing to the deleterious effects of plastic-associated EDCs on cardiovascular health in humans. Testing PXR activation should be considered for risk assessment of phthalates and other EDCs. https://doi.org/10.1289/EHP9262
Introduction
Atherosclerotic cardiovascular disease (CVD) is still the leading cause of mortality and morbidity worldwide despite advanced diagnosis and treatments (Helsley and Zhou 2017; Roth et al. 2015). In the last few decades, our chemical environment has changed substantially, and recent large-scale human studies have implicated a novel link between exposure to endocrine disrupting chemicals (EDCs) and CVD (Helsley and Zhou 2017; Lang et al. 2008; Lind and Lind 2011, 2012; Melzer et al. 2010, 2012b; vom Saal and Myers 2008). Owing to their low cost and variety, plastics have become an essential part in modern life and plastic pollution is a significant environmental and health concern (Eriksen et al. 2014; Seltenrich 2015). Plastic-associated EDCs, including the base chemical bisphenol A (BPA) and numerous plasticizers, are produced in high volume and have been demonstrated to be associated with increased CVD risk in humans (Helsley and Zhou 2017; Lang et al. 2008; Lind and Lind 2011, 2012; Melzer et al. 2010, 2012b; Olsén et al. 2012a; vom Saal and Myers 2008). For example, higher BPA exposure has been associated with the increased CVD risk in humans in a few large-scale cross-sectional (Lang et al. 2008; Melzer et al. 2010, 2012a) and longitudinal (Melzer et al. 2012b) studies. In addition, circulating levels of phthalates and their metabolites have also been associated with increased atherosclerosis or CVD risk factors such as low-density lipoprotein (LDL)-cholesterol levels (Lind and Lind 2011, 2012; Olsén et al. 2012b). However, the underlying mechanisms for EDC-associated CVD are still poorly understood, making it difficult to conduct rational exposure assessment.
We and others have previously identified many plastic-associated EDCs—such as BPA and its analogs [e.g., bisphenol B (BPB)], phthalates [e.g., di(2-ethylhexyl) phthalate (DEHP)], and phthalate substitutes [e.g., tributyl citrate (TBC)]—as potent agonists of pregnane X receptor (PXR) (DeKeyser et al. 2011; Helsley and Zhou 2017; Sui et al. 2012, 2014, 2015, 2018b; Zhou 2016). PXR is a member of the nuclear receptor superfamily and can be activated by many dietary steroids, endogenous hormones, and numerous xenobiotic chemicals, including pharmaceutical drugs and environmental chemicals (Blumberg et al. 1998; Kliewer et al. 1998; Zhou et al. 2009b; Zhou 2016). Activation of PXR by its ligands can lead to the increased expression of enzymes and transporters that regulate xenobiotic metabolism in the liver and intestine (Kliewer et al. 2002; Zhou et al. 2009b). Thus, PXR has been established as a xenobiotic sensor that has been well studied by many groups (Kliewer 2015; Zhou et al. 2009b).
Recent studies by us and others have revealed novel and unexpected functions for PXR in lipid homeostasis and atherogenesis (Cheng et al. 2012; de Haan et al. 2009; Gwag et al. 2019; Helsley et al. 2013; Helsley and Zhou 2017; Meng et al. 2019; Sui et al. 2011, 2014, 2015, 2018b; Zhou et al. 2009a; Zhou 2016). For example, ligand-mediated PXR activation can lead to higher plasma total cholesterol levels and atherogenic LDL levels in mice (Gwag et al. 2019; Helsley et al. 2013; Meng et al. 2019; Sui et al. 2015; Zhou et al. 2009a). PXR has been shown to contribute to increased atherosclerosis in atherosclerosis-prone LDL-receptor–deficient () (Sui et al. 2020) and apolipoprotein E–deficient () mouse models (Sui et al. 2011; Zhou et al. 2009a). Despite these findings, the functions of PXR in mediating EDCs’ pathophysiological effects in humans and animals are still poorly understood. In this study, we used in vitro and in vivo approaches to investigate the impact of a widely used phthalate, dicyclohexyl phthalate (DCHP) on PXR activation and lipid homeostasis.
Methods
Animals
Wild-type (WT) C57BL/6 mice were purchased from The Jackson Laboratory. PXR flox mice () on a C57BL/6 background were generated by using mouse embryonic stem cell clones (C57BL/6 background) containing the conditional PXR flox allele from International Knockout Mouse Consortium (EUMMCR, , EPD0141_1_G04) (Gwag et al. 2019). The targeting vector consisted of a flippase recombination enzyme (Flp)-recognition target (FRT), reporter, and a Cre recombinase recognition target (loxP). After the first loxP site, a neomycin selection cassette containing the human promoter (hBactP) driving the neomycin resistance gene is inserted followed by pA, a second FRT site, and a second loxP site. The last loxP site, a third one, is inserted downstream of the targeted PXR Exon 5 (Gwag et al. 2019). The chimeric mice were crossed with flippase transgenic mice (005703; The Jackson Laboratory) to remove the Frt-flanked cassette. Both the mouse embryonic stem cell clones and the mice used for generating mice were on the C57BL/6 background. Nevertheless, mice carrying PXR flox alleles were further crossed with WT C57BL/6 mice for an additional five generations. Those mice on C57BL/6 background were then bred with mice expressing Cre recombinase to produce tissue-specific PXR-knockout mice. To generate intestinal epithelial cell-deficient ( mice) or hepatocyte-specific PXR-deficient mice ( mice), mice were further crossed with Villin-Cre (004586; The Jackson Laboratory) or Albumin-Cre transgenic mice (003574; The Jackson Laboratory) (Gwag et al. 2019; Meng et al. 2019). In the present study, mice and PXR conditional knockout (i.e., mice and ) mice had the same background (PXR flox alleles) except for one allele of and mice carrying Villin-Cre and Albumin-Cre, respectively. The experiments using and mice also included their corresponding littermates as controls.
The PXR-humanized (huPXR or ) and PXR-null () mice on the C57BL/6 background have been described previously (Ma et al. 2007; Staudinger et al. 2001; Sui et al. 2014). Briefly, PXR-humanized (huPXR, mouse PXR-knockout/human PXR transgenic) mice were generated by transgenesis on PXR-null mice (Staudinger et al. 2001) that were provided by S.A. Kliewer (University of Texas Southwestern Medical Center, Dallas, TX) using a bacterial artificial chromosome clone containing the complete human PXR gene and including 5′- and 3′-flanking sequences at the National Cancer Institute (NCI) (Ma et al. 2007). Those mice were backcrossed on a C57BL/6 genetic background for at least four generations at the NCI and were kindly provided by F. Gonzalez (NCI, Bethesda, Maryland). The mice were backcrossed for another four additional generations onto the C57BL/6 background in our laboratory. For all experiments involving huPXR and mice, the littermates had the same background (mPXR null alleles). The exception to this was the single allele containing the human PXR gene carried by mice.
For DCHP treatment, eight-wk-old male mice were fed a semisynthetic low-fat AIN76 diet containing 4.2% fat and 0.02% cholesterol (D00110804C; Research Diet) and treated by oral gavage with body weight (BW) of DCHP or vehicle (corn oil) daily for 7 d (WT: for control and for DCHP; , for control and for DCHP; ( littermates): for control and for DCHP; : for control and for DCHP; ( littermates): for control and for DCHP; huPXR mice: for control and for DCHP; : for control and for DCHP). For the follow-up outcome studies (e.g., gene and protein analyses), a sufficient number of mice were used based on established protocols or published results for those assays [e.g., for western blotting or real-time quantitative polymerase chain reaction (RT-qPCR) analyses] (Gwag et al. 2019; Lee et al. 2014; Meng et al. 2019).
DCHP has been previously used to treat rodents at relatively high doses, from 10 to BW per day (Ahbab et al. 2017; Lake et al. 1982; Li et al. 2016; Lv et al. 2019). In the present study, we treated the mice with BW per day DCHP for 7 d. Our selected doses were significantly lower than concentrations experimentally used in previous studies. We also took into account the interspecies scaling factor of 12.3 between mice and humans for selecting the drug dosage in the initial clinical trials (Nair and Jacob 2016; Sharma and McNeill 2009). This factor reflects the 12.3-fold difference in surface area-to-BW ratio between mice and humans and thus 12.3 times more drugs or chemicals are required to treat mice to be comparable to the dose used in humans (Nair and Jacob 2016; Sharma and McNeill 2009). We also previously identified another plasticizer, tributyl citrate (TBC), as a selective PXR agonist (Sui et al. 2015). Three different doses of TBC (2.5, 5, or BW per day) were used to treat mice for 1 wk, and the results suggested that the dose of BW per day, but not 2.5 or BW per day, led to intestinal PXR activation (Sui et al. 2015). Therefore, the BW per day dose used in this study was appropriate for studying the impact of DCHP exposure on PXR activation in vivo.
On the day of euthanasia, mice were fasted for the 6 h following the dark cycle (feeding cycle). After the bodyweight was measured, the mice were then anesthetized with ketamine/xylazine ( BW) by intraperitoneal injection and dissected to open the peritoneum and chest cavity. Blood was removed by left ventricle puncture using a syringe. The circulatory system was perfused by injecting of saline into the left ventricle after nicking the right atrium. This also killed the mouse by exsanguination. The major organs/tissues—including liver, kidney, and subcutaneous and epidydimal white adipose tissue—were collected and weighted. The mice were housed in microisolator cages and were provided ad libitum access to a standard chow diet before the treatment with deionized water in a temperature-controlled room () with a 12-h light/dark cycle and humidity ranging from 30% to 70%. All animal studies were performed in compliance with protocols approved by the institutional animal care and use committees of the University of California, Riverside and the University of Kentucky.
Reagents and Plasmids
DCHP, DEHP, diisononyl phthalate (DiNP), diisodecyl phthalate (DiDP), di--octyl phthalate (DnOP), diisobutyl phthalate (DiBP), di--butyl phthalate (DnBP), diethyl phthalate (DEP), dihexyl phthalate (DHP), diallyl phthalate (DAP), and dimethyl phthalate (DMP) were purchased from Sigma-Aldrich. The chemicals used in this study were dissolved in dimethyl sulfoxide (DMSO). The plasmids used in this study have been described previously, including human (h) and mouse (m) PXR expression vectors (Blumberg et al. 1998; Kliewer et al. 1998), beta galactosidase () expression vector (Blumberg et al. 1998; Umesono et al. 1991), GAL4 DNA-binding domain (DBD)-linked nuclear receptor ligand-binding domain (LBD) vectors (Tabb et al. 2003, 2004; Zhou et al. 2004), GAL4 reporter (VP16-hPXR) (Tabb et al. 2003; Zhou et al. 2004), GAL4 DBD-linked nuclear receptor co-regulators [steroid receptor co-activator-1 (SRC-1), PPAR binding protein (PBP), nuclear receptor co-repressor (NCoR), silencing mediator of retinoid and thyroid hormone receptors (SMRT)] (Grün et al. 2002; Tabb et al. 2003; Zhou et al. 2004), PXR-dependent CYP3A4 promoter reporter (CYP3A4XREM-luciferase) (Drocourt et al. 2002; Zhou et al. 2004), CYP3A2 promoter reporter [] (Blumberg et al. 1998; Kliewer et al. 1998), and GAL4 reporter (MH100-luciferase) (Blumberg et al. 1998; Zhou et al. 2004).
Cell Culture and Transfection Assay
The human intestine epithelial cell line LS180 (ATCC CL-187) was obtained from American Type Culture Collection, and the cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C in 5% carbon dioxide (). Transfection assays were performed as previously described with modifications (Grün et al. 2002; Tabb et al. 2003; Zhou et al. 2004). The cells were seeded into 24-well plates overnight and reached confluence. The cells were then transiently transfected with expression plasmids, corresponding luciferase reporter plasmids, and with control plasmids using FuGENE 6 (E2691; Promega Corporation) in serum-free DMEM. Briefly, the hPXR full-length expression vector was co-transfected with the hPXR-dependent CYP3A4 promoter luciferase reporter plasmid (CYP3A4XREM-luciferase). The mPXR full-length expression vector was co-transfected with the mPXR-dependent CYP3A2 promoter luciferase reporter plasmid []. DBD-linked nuclear receptor LBD vectors were co-transfected with the MH100-luciferase GAL4 reporter. For mammalian two-hybrid assays, LS180 cells were transfected with the MH100-luciferase GAL4 reporter, VP16-hPXR, and GAL4 DBD-linked nuclear receptor co-regulators (SRC-1, PBP, NCoR, and SMRT) plasmids (Grün et al. 2002; Tabb et al. 2003; Zhou et al. 2004). Twenty-four hours posttransfection, the cells were treated with chemicals or DMSO vehicle for 24 h in serum-free DMEM ( replicates per group). Then the cells were lysed with passive lysis buffer (E1941; Promega Corporation), and extracts were prepared for and luciferase assays. Luciferase assays were performed according to the manufacturer’s manual (E1531; Promega Corporation) using a Synergy H1 Hybrid Reader (11120535; BioTek Instruments, Inc.). After adding of solution to the lysate, the lysate was incubated at 37°C for 5–10 min and then the reaction was stopped with of sodium carbonate. The lysate was then read at the optical density of 595 () using a Synergy H1 Hybrid Reader. Reporter gene activity was normalized to the transfection controls and the results expressed as normalized as relative light units per per minute to facilitate comparisons between plates. Fold activation was calculated relative to DMSO controls. Half-maximal effective concentration () values were calculated by curve fitting of data, using GraphPad Prism 8 software.
Competitive Ligand-Binding Assay
For ligand-binding assays, LanthaScreen Time Resolved Fluorescence Resonance Energy Transfer (TR-FRET) competitive binding assays were performed in 384-well solid black plates (Meng et al. 2019). The tested compounds were incubated with of glutathione -transferase (GST)-hPXR LBD (PV4840; Thermo Fisher Scientific), of fluorescent-labeled hPXR agonist (Fluomore PXR Green; PV4843; Thermo Fisher Scientific), and of terbium-labeled anti-GST antibody (LanthaScreen Tb-anti-GST antibody; PV3550; Thermo Fisher Scientific) at 25°C for 1 h. A Synergy H1 Hybrid Reader (11120535; BioTek Instruments, Inc.) was used to measure the terbium emission peak at and fluorescein emission at . The TR-FRET ratio was calculated and expressed as the signal from the fluorescein emission divided by the terbium signal, with per compound per concentration.
Plasma Analysis
Plasma total cholesterol and triglyceride concentrations were measured using the Wako Cholesterol E enzymatic colorimetric assay (999-02601) and the Wako L-Type TG M assay (994-02891; Wako) according to the manufacturer’s protocol (Fujifilm Healthcare Solutions). The blood was collected in tubes containing ethylenediaminetetraacetic acid (EDTA) as an anticoagulant ( of 0.5M EDTA for blood). After gently mixing with EDTA, the blood samples were centrifuged at at 4°C for 15 min. The top clear phase was then transferred into a clean vial by pipetting and stored at . The lipoprotein fractions were isolated by spinning of plasma following a modified protocol (Havel et al. 1955; Zhou et al. 2009a). Briefly, lipoprotein fractions were isolated by centrifuging at for 3 h at 4°C in a Beckman Optima TL-100 tabletop ultracentrifuge at its own density (). The infranatant was then adjusted to a density of with solid potassium bromide to harvest the very-low-density lipoprotein (VLDL) (), LDL (), and high-density lipoprotein (HDL) () fractions by spinning at for 18 h at 4°C. The cholesterol content of the lipoprotein fractions was then measured enzymatically (cholesterol 999-02601; Wako), with (total cholesterol and triglycerides; Tables S9 and S11) and (VLDL-cholesterol, LDL-cholesterol, HDL-cholesterol; Tables S9 and S11).
Lipid Analysis of Plasma Ceramides
The ceramide levels were measured by liquid–liquid extraction and liquid chromatography–tandem mass spectrometry (LC-MS/MS) using a modified method described previously (Deng et al. 2020; Matyash et al. 2008), with mice per genotype per treatment. Twenty microliters of plasma was mixed with of methanol and of internal standard (IS; of C24:0 ; 22789; Cayman Chemical) by vortex for 5 min. The mixture was then incubated with of methyl-tert-butyl ether (MTBE) for 20 min at room temperature (RT) with shaking. This was followed by adding of MS-grade water for phase separation. After 10-min incubation at RT, the sample was centrifuged at for 10 min. The upper (organic/lipids) phase was transferred into a glass vial and the lower phase was reextracted with of MTBE. The combined organic phases were evaporated to dryness under a stream of filtered nitrogen. The extracted lipids were dissolved in of acetonitrile/water (4:1) and stored at until analysis. For the LC-MS/MS analysis, an AB Sciex 4000 Q Trap coupled with an Exion LC system was used. Chromatographic separation was carried out with a reverse-phase column (Waters ACQUITY UPLC , , ) maintained at 40°C, and the flow rate was set to . The mobile phases consisted of water/acetonitrile (A; 60/40, vol/vol), and isopropanol/acetonitrile (B; 90/10, vol/vol), both containing ammonium formate and 0.1% formic acid. A gradient program was used as follows: (T min/%A): 0/48, 1.5/48, 4.0/42, 7.0/42, 11.0/34, 13.0/30, 13.1/3.0, 15.5/3.0, 15.6/48, and 18.5/48. The injection volume was . The mass spectrometer was equipped with an electrospray ionization (ESI) source and operated in positive mode under the following operating parameters except for collision energy (CE): ion spray voltage, 5.0 kV; desolvation temperature, 400°C; ion source gas 1, 40 psi; ion source gas 2, 50 psi; curtain gas, 30 psi; collision gas, medium; declustering potential, 60 V; and entrance potential, 10.0 V. Quantitative analysis was conducted by monitoring the precursor ion to production ion transitions of m/z 482.5/264.3 (C12:0 ceramide, CE 31V), m/z 510.6/264.3 (C14:0 ceramide, CE 33V), m/z 538.5/264.3 (C16:0 ceramide, CE 41V), m/z 566.6/264.3 (C18:0 ceramide, CE 45V), m/z 564.5/264.3 (C18:1 ceramide, CE 35V), m/z 594.5/264.3 (C20:0 ceramide, CE 42V), m/z 622.6/264.3 (C22:0 ceramide, CE 42V), m/z 650.6/264.3 (C24:0 ceramide, CE 49V), m/z 648.6/264.3 (C24:1 ceramide, CE 41V), and m/z 655.9/271.5 (C24:0 ceramide-d7, IS) with a dwell time of 0.1 s.
In Vivo Lipid Uptake and Absorption Assays
The intestinal cholesterol uptake assays were performed as previously described (Meng et al. 2019). Briefly, 8-wk-old male and mice were fasted for 4 h and administered of corn oil containing of by oral gavage (NET139001MC; PerkinElmer) (: for control and for DCHP; , for control and for DCHP). Two hours later, the small intestine was excised, flushed with sodium taurocholate, and cut into segments. The segments were incubated with of 1N sodium hydroxide (NaOH) overnight at 65°C, and mixed with ScintiSafe (6196-95-8; Fisher Scientific) for scintillation counting by a liquid scintillation analyzer (Packard TRI-CARB 2300TR). For in vivo intestinal cholesterol absorption rate measurement (Meng et al. 2019), 8-wk-old male and mice were fasted for 4 h and treated with corn oil containing of (NET139001MC; PerkinElmer) by oral gavage ( mice: for control and for DCHP; mice: for control and for DCHP). The mice were then immediately treated with the lipoprotein lipase inhibitor poloxamer-407 (16758; Sigma-Aldrich) ( BW) by intraperitoneal injection. Blood was collected by retro-orbital bleeding under isoflurane anesthesia at different time points within 6 h, and plasma was measured by liquid scintillation counting.
Enteroid Culture and Cholesterol Uptake Assay
Enteroid isolation and cholesterol uptake assays were performed as previously described in detail (Meng et al. 2019). Briefly, untreated 8-wk-old male , , huPXR, and mice were euthanized, and 8 cm of intestine was collected for crypts isolation ( for each genotype). A total of crypts were placed into of depolymerized Matrigel (354248; Corning) containing of R-Spondin 1, of epidermal growth factor (EGF), and of Noggin. The crypts suspensions and Matrigel were incubated with of minigut culture media (Advanced DMEM/F12; 12634010; Thermo Fisher; containing 1% penicillin–streptomycin, 1% l-glutamine, 1% of nitrogen supplement, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid, and 2% B27 supplement) in a 24-well plate at 37°C in a 5% incubator for 1 d. The media was then replaced with enteroid growth media (minigut media containing of R-Spondin 1, of Noggin, and of EGF) every 3 d until the enteroids were mature to form a villus-like epithelium. The mature enteroids were treated with minigut media containing of DCHP or DMSO vehicle for 24 h. After depolymerizing with ice-cold Dulbecco’s phosphate-buffered saline (PBS), the enteroids were washed with ice-cold PBS to remove the Matrigel. The total RNAs were extracted from enteroids for RT-qPCR analysis. Cholesterol uptake assays were performed as previously described (Meng et al. 2019). Briefly, micelles were prepared by mixing taurocholate, egg yolk , cholesterol, together with of (NET139001MC; PerkinElmer)/ of cholesterol and evaporated under a mild stream of argon. The lipid film was then hydrated in serum-free Modified Eagle Medium (MEM) containing 0.5% fatty acid-free BSA (9048-46-8; Sigma-Aldrich) and incubated at 37°C in a rotating incubator. The solutions were filtered through a surfactant-free cellulose acetate filter (431220; Corning). The enteroids were then incubated with micelles containing (NET139001MC; PerkinElmer) for 1 h at 37°C in a 5% incubator, washed twice with cold PBS, and lysed with NaOH ( of 0.1N). Radioactivity was measured using a liquid scintillation counter and normalized to total protein mass.
RNA Isolation and RT-qPCR Analysis
Total RNA was isolated from mouse tissues () or cells () using TRIzol Reagent following the manufacture’s protocol (15596026; Thermo Fisher Scientific) and RNA concentrations and quality were evaluated using NanoDrop spectrophotometers (ND-2000; Thermo Fisher Scientific). Total RNA () was reverse transcribed using SuperScript III reverse transcriptase according to the manufacturer’s instructions (18080093; Invitrogen). RT-qPCR was performed using gene-specific primers (Table S1) and SYBR Green Supermix (170-8886; Bio-Rad) using a CFX RT-PCR Instrument (184-5096; Bio-Rad) according to the manufacturer-supplied protocol. Briefly, the complementary DNA template was analyzed following the PCR program per the following steps: 1) denaturation at 95°C for 30 s, 2) 40 cycles of denaturation (at 95°C for 15 s) plus annealing/extension (at 60°C for 60 s) followed by plate reading, and 3) melting curve analysis at 65–95°C (0.5°C increment, 5 s/step). For each biological sample, two technical replicate cycle threshold (Ct) values were collected and averaged. The mean Ct values were normalized to glyceraldehyde-3-phosphate dehydrogenase, and the relative mRNA expression levels were calculated using the comparative method (Livak and Schmittgen 2001). The relative gene expression was presented as mean fold change over control samples. All oligonucleotides were purchased from Sigma-Aldrich, and the sequences of primer sets used in this study are listed in Table S1.
Western Blotting
Protein extraction and western blotting were performed as previously described (Sui et al. 2018a). Animal tissues () were homogenized in of ice-cold lysis buffer (9803; Cell Signaling Technology) containing phosphatase inhibitor cocktails (Sigma-Aldrich; P5726 and P0044) and protease inhibitor cocktails (11836153001; Roche) using a Bullet Blender (BBX24; Next Advance). Lysates were centrifuged at for 15 min at 4°C after homogenization. The top lipid layer was removed, and the remaining supernatant was centrifuged again to remove the lipid. A bicinchoninic acid protein assay kit (23225; Thermo Fisher Scientific) was used to measure protein concentrations. The isolated proteins were then denatured by boiling at 100°C for 5 min in buffer (161-0737; Bio-Rad). The lysates were resolved on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and then electrophoretically transferred to a nitrocellulose membrane. The membrane was incubated with blocking buffer ( containing 0.1% Tween-20 and 5% nonfat dry milk) for 1 h at RT before further incubating with blocking buffer containing anti-β-glucocerebrosidase (anti-GBA1) (1:1000 dilution; G4171; Sigma), anti-microsomal triglyceride transfer protein (anti-MTP; 1:1000 dilution; sc-33316; Santa Cruz Biotechnology), or anti-actin (1:5,000 dilution; A2066; Sigma) antibodies overnight at 4°C. The membranes were then incubated with blocking buffer containing horseradish peroxidase-conjugated anti-rabbit secondary antibodies (1:5,000 dilution; 12-348; Sigma) at RT for 1 h, and then developed with an enhanced chemiluminescence system (32209; Thermo Fisher scientific). The densitometry analyses of the protein bands were performed using the ImageJ software to quantify the relative protein expression levels according to the guidelines (http://rsb.info.nih.gov/ij/). The GBA1 and MTP protein measurements were normalized by actin, the internal control protein, and the ratios between the target protein and the control protein were used for statistical analyses.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) analysis was performed by using a SimpleChIP Enzymatic Chromatin IP Kit (9003; Cell Signaling Technology) according to the manufacturer’s protocol. The intestine tissues () were isolated and cross-linked with formaldehyde in tissue culture dishes at RT with shaking. The cross-linked chromatin was sonicated three times for 15 s on ice. After centrifugation at for 10 min at 4°C, the supernatant of the lysate was incubated with of anti-PXR antibody (sc-25381; Santa Cruz Biotechnology) overnight at 4°C with rotation. The chromatin–antibody complex was eluted and de-cross-linked at 65°C for 3 h. The precipitated genomic DNA was purified by using a spin column purification kit (14209; Cell Signaling Technology). qPCR analysis was then performed with the primers targeting the PXR response element in the Niemann-Pick C1-Like 1 (NPC1L1) and MTP promoters. qPCR was performed using Taq polymerase (R001C; TaKaRa) with denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 45 s (total 25 cycles). The primer sequences used for ChIP analysis are listed in Table S1.
Statistical Analysis
All data are presented as the and the individual data points are also shown in the figures. Individual pairwise comparisons were analyzed by two-sample, two-tailed Student’s -test. One-way analysis of variance (ANOVA) was used when multiple comparisons were made, followed by Dunnett’s -test. Two-way ANOVA was used when multiple comparisons were made followed by a Bonferroni multiple comparisons test. Two-way ANOVAs were performed using SigmaPlot (version 13.0; SYSTAT), and the other analyses were performed using GraphPad Prism 8. was considered statistically significant.
Results
Evaluating the Potential of Commonly Used Phthalates as PXR Agonists
A series of commonly used phthalate plasticizers were tested for PXR activation, including DCHP, DiNP, DEHP, DiDP, DnOP, DiBP, DnBP, DHP, DAP, DME, and DEP) (Figure 1A; Figure S1). One of the widely studied phthalates, DEHP, has been demonstrated to be a PXR agonist (DeKeyser et al. 2011; Sui et al. 2015) and was used a positive control for our assays. Indeed, DEHP activated hPXR and induced hPXR-mediated CYP3A4-luciferase reporter activities. Interestingly, DCHP was found to be a more potent hPXR agonist than any other phthalates tested, including DEHP (Figure 1A). Next, DCHP was tested to determine whether it can dose-dependently affect hPXR and mPXR activity. Dose–response analysis demonstrated that DCHP activated both hPXR and mPXR. The () of DCHP were for hPXR and for mPXR (Figure 1B,C), values within the range of other known PXR ligands (Zhou et al. 2007, 2009b). The ability of DCHP to activate other nuclear receptors was also investigated. DCHP activated three forms of PXR including hPXR, mPXR, and rat PXR (rPXR) but did not activate other nuclear receptors (Figure 1D).
Figure 1.

The effects of DCHP on PXR activity in cell-based transfection assays. (A) Human LS180 intestinal cells were transfected with a full-length hPXR plasmid, hPXR reporter (CYP3A4-luciferase), and () control plasmid. Cells were treated with various phthalates at the indicated concentrations for 24 h (). (B,C) LS180 cells were transfected with (B) hPXR and CYP3A4-luciferase reporter together with plasmid or (C) mPXR and reporter together with plasmid. After transfection, LS180 cells were then treated with DCHP at the indicated concentrations for 24 h (). (D) LS180 cells were transfected with GAL4 plasmids in which the GAL4 DNA-binding domain is linked to the indicated nuclear receptor ligand-binding domain and a GAL4 reporter. After transfection, LS180 cells were treated with DMSO control or DCHP for 24 h (, two-sample, two-tailed Student’s -test, ***, compared with the control group). Reporter gene activity was normalized to the transfection controls, and the results were normalized to relative light units per per minute to facilitate comparisons between plates. Fold activation was calculated relative to vehicle DMSO controls. Mean fold activation is shown. Error bars . The numerical data corresponding to this figure are shown in Tables S2–S5. Note: , beta galactosidase; CARα, constitutive androstane receptor; DAP, diallyl phthalate; DCHP, dicyclohexyl phthalate; DEHP, di(2-ethylhexyl) phthalate; DEP, diethyl phthalate; DHP, dihexyl phthalate; DiBP, diisobutyl phthalate; DiDP, diisodecyl phthalate; DiNP, diisononyl phthalate; DMP, dimethyl phthalate; DMSO, dimethyl sulfoxide; DnBP, di--butyl phthalate; DnOP, di--octyl phthalate; ERα, estrogen receptor alpha; ERβ, estrogen receptor beta; FXR, farnesyl X Receptor; hPXR, human pregnane X receptor; LXR, liver X receptor; mPXR, mouse pregnane X receptor; OD, optical density; PPARα, peroxisome proliferator-activated receptor alpha; PXR, pregnane X receptor; RARα, retinoic acid receptor alpha; rPXR, rat pregnane X receptor; RXR, retinoid X receptor; SEM, standard error of the mean; VDR, vitamin D receptor.
Investigating the Impact of DCHP on PXR and Co-Regulator Interactions
TR-FRET PXR competitive binding assays were next performed to investigate whether DCHP can directly bind to PXR LBD. Like the known PXR agonist, DEHP, cells treated with DCHP exhibited lower PXR LBD fluorescence in a dose-dependent manner (Figure 2A). The half maximal inhibitory concentration () for DCHP binding to PXR was , a value similar to the that we calculated for DCHP to activate hPXR in transfection assays. Because nuclear receptor co-regulators are essential for regulating nuclear receptor activation (Sui et al. 2012), we investigated the effects of DCHP on PXR and co-regulator interactions. DCHP treatment led to more interactions between PXR and the co-activators SRC-1 and PBP (Figure 2B). In the absence of ligands, PXR interacted with the co-repressors, NCoR and SMRT (Figure 2C), and treatment with DCHP disrupted the interactions between PXR and these co-repressors (Figure 2C).
Figure 2.

Binding of DCHP to PXR ligand-binding domain (LBD) and the impact of DCHP on PXR and co-regulator interactions. (A) Competitive ligand binding assays were performed to evaluate the inhibition of FRET between fluorescein-labeled PXR ligand and recombinant GST-PXR LBD by DCHP and DEHP. DEHP was included as a positive control. Results are presented as mean TR-FRET emission ratios based on the signal from the fluorescein emission divided by the terbium signal (). Error bars . (B,C) Human LS180 cells were transfected with a GAL4 reporter, VP16-hPXR vector, and expression vector of GAL4 DNA-binding domain (DBD) or GAL4 DBD linked to the receptor interaction domains of (B) nuclear receptor co-activators (GAL4-SRC1 or GAL4-PBP) or (C) nuclear receptor co-repressors (GAL4-SMRT or GAL4-NCoR). After transfections, LS180 cells were treated with DMSO vehicle control or DCHP at the indicated concentrations for 24 h. Reporter gene activity was normalized to the transfection controls, and the results were normalized to relative light units per per minute to facilitate comparisons between plates (). Mean fold activation was calculated relative to vehicle DMSO controls. Error bars . The numerical data corresponding to this figure are shown in Tables S6–S8. Note: DCHP, dicyclohexyl phthalate; DEHP, di(2-ethylhexyl) phthalate; DMSO, dimethyl sulfoxide; FRET, fluorescence resonance energy transfer; GAL4, galactose-responsive transcription factor; GST, glutathione S-transferase; NCoR, nuclear receptor co-repressor; OD, optical density; PBP, peroxisome proliferator-activated receptor binding protein; PXR, pregnane X receptor; SEM, standard error of the mean; SMRT, silencing mediator of retinoid and thyroid hormone receptors; SRC-1, Steroid receptor co-activator-1; TR-FRET, time-resolved measurement of fluorescence resonance energy transfer.
Evaluating the Impact of Exposure to DCHP on Plasma Lipid Levels in WT Mice
To investigate whether DCHP exposure can also affect plasma lipid levels in vivo, C57BL/6 WT mice were treated with BW per day of DCHP or vehicle control by oral gavage for 7 d. Short-term exposure to DCHP did not affect body weight or major organ weight (Figure S2). WT mice treated with DCHP had significantly higher plasma total cholesterol levels but similar triglyceride levels, as compared with control mice (Figure 3A,B). Lipoprotein fraction analysis was also performed, and mice treated with DCHP had significantly higher atherogenic LDL-cholesterol levels but similar HDL-cholesterol levels, as compared with control mice (Figure 3C).
Figure 3.

The effects of DCHP exposure on plasma lipid levels in wild-type (WT) mice. Eight-wk-old male WT mice were treated with vehicle control or BW per day of DCHP daily by oral gavage for 7 d. Fasting plasma (A) total cholesterol and (B) triglyceride levels were measured by enzymatically colorimetric methods. (C) Lipoprotein fractions (VLDL-C, LDL-C, and HDL-C) were isolated from the plasma and the cholesterol levels of each fraction were then measured. (, two-sample, two-tailed Student’s -test, **, , and ***, ). Results represent mean values. Error bars . The numerical data corresponding to this figure are shown in Table S9. Note: BW, body weight; DCHP, dicyclohexyl phthalate; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; SEM, standard error of the mean; VLDL-C, very-low-density lipoprotein-cholesterol.
Determining the Contribution of Intestinal or Hepatic PXR Signaling toward DCHP-Elicited Hyperlipidemia
(Gwag et al. 2019) and mice (Meng et al. 2019) were previously generated to study the tissue-specific role of PXR in xenobiotic or lipid metabolism. To determine the contribution of intestinal PXR signaling to DCHP-induced hyperlipidemia, male and control littermates were treated with BW per day of DCHP or vehicle control by oral gavage for 7 d. qPCR analysis showed that the expression levels of known PXR target genes cytochrome P450, family 3, subfamily A, polypeptide 11 (CYP3A11); GST alpha 1 (GSTA1); and multidrug resistance protein 1a (MDR1a) were higher in the intestine of DCHP-treated mice but not in littermates (Figure 4A), suggesting the activation of intestinal PXR signaling by DCHP treatment in vivo. Consistent with WT mouse results, exposure to DCHP led to significantly higher plasma total cholesterol but similar triglyceride levels in mice (Figure 4B,C). The deficiency of intestinal PXR abolished the impact of DCHP exposure on plasma cholesterol levels (Figure 4B). DCHP treatment was also able to lead to higher atherogenic VLDL- and LDL-cholesterol levels in control mice but had no effects on these lipoprotein levels in mice (Figure 4D).
Figure 4.

The impact of DCHP exposure on intestinal PXR target gene expression and plasma lipid levels in and mice. Eight-wk-old male and littermates were administered vehicle control or BW per day of DCHP daily by oral gavage for 7 d. (A) Intestinal expression of PXR target genes was analyzed by qPCR (, two-sample, two-tailed Student’s -test, *, ). Fasting plasma (B) total cholesterol and (C) triglyceride levels were analyzed enzymatically by colorimetric methods. (D) Lipoprotein fractions (VLDL-C, LDL-C, and HDL-C) were isolated from the plasma, and the cholesterol levels of each fraction were then measured enzymatically. (, two-way ANOVA, Bonferroni multiple comparisons test for multiple comparisons, *, ). The numerical data corresponding to this figure are shown in Tables S10–S11. Results represent mean values. Error bars . Note: ANOVA, analysis of variance; BW, body weight; CYP3A11, cytochrome P450, family 3, subfamily A, polypeptide 11; DCHP, dicyclohexyl phthalate; , glutathione -transferase; MDR1a, multidrug resistance protein 1a; PXR, pregnane X receptor; , intestinal epithelial cell-specific pregnane X receptor deficient; , pregnane X receptor flox; qPCR, quantitative polymerase chain reaction; SEM, standard error of the mean.
In addition to mice, and littermates were also treated with vehicle control and the same dose of DCHP for 7 d. Unlike mice, the deficiency of hepatic PXR did not affect DCHP-elicited hypercholesterolemia given that DCHP exposure had similar effects on plasma cholesterol levels in mice as that in mice (Figure S3).
Evaluating the Effects of DCHP-Mediated PXR Activation on Cholesterol Absorption in Vivo and ex Vivo
We evaluated the impact of DCHP exposure on the expression of PXR-regulated intestinal genes that mediate lipid homeostasis. The intestinal expression of key lipogenic genes, the essential cholesterol transporter NPC1L1 and MTP (Meng et al. 2019; Sui et al. 2015), was higher with DCHP treatment in but not in mice (Figure 5A). We then performed ChIP assays, and the results demonstrated that DCHP exposure led to the recruitment of PXR onto the NPC1L1 and MTP promotor regions containing PXR-responsive elements (Meng et al. 2019; Sui et al. 2015) in the intestine of but not mice (Figure 5B).
Figure 5.
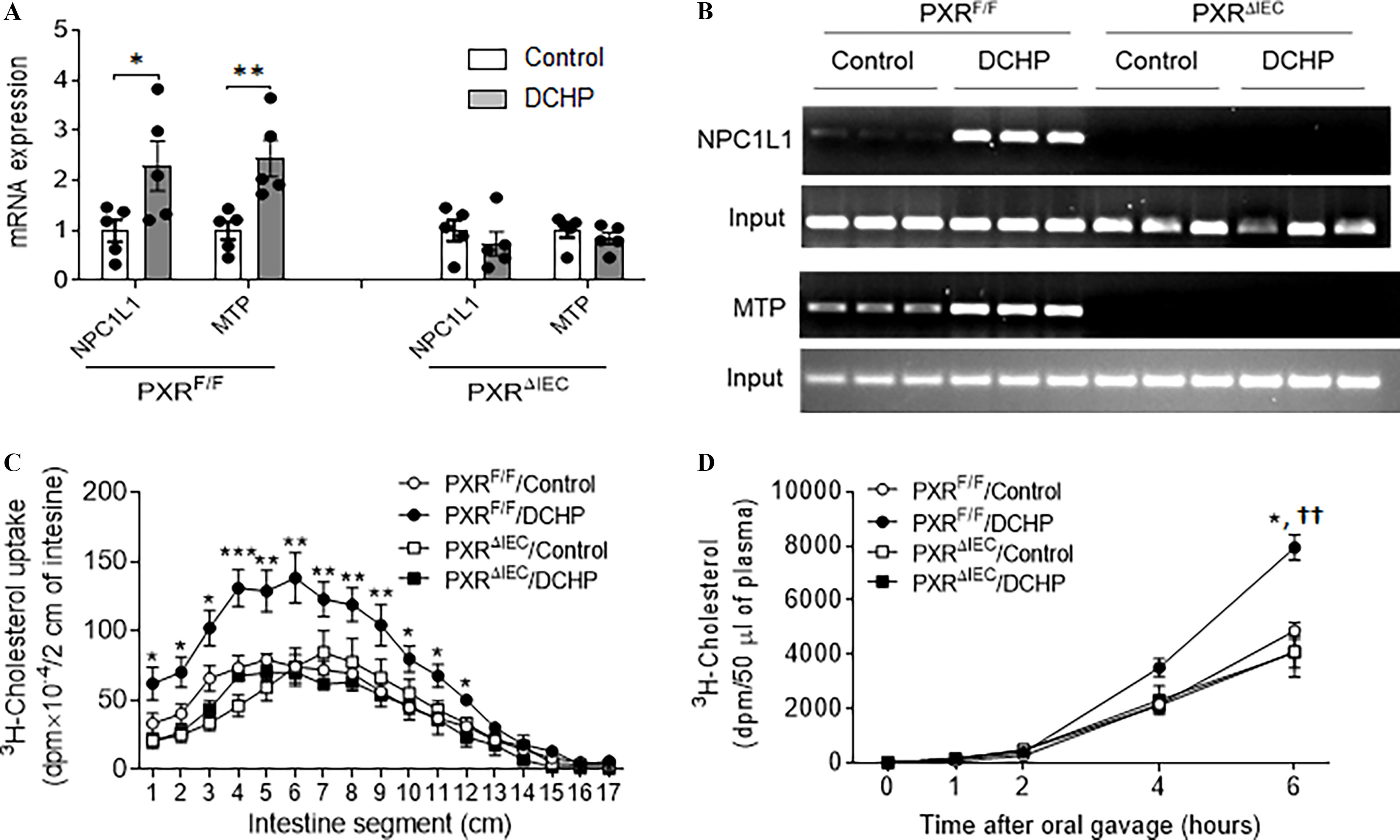
The effects of DCHP exposure on intestinal NPC1L1 and MTP expression and lipid absorption in and mice. Eight-wk-old male and littermates were administered vehicle control or BW per day of DCHP daily by oral gavage for 7 d. (A) Intestinal expression of NPC1L1 and MTP was measured by qPCR (, two-sample, two-tailed Student’s -test, *, and **, ). (B) The recruitment of PXR onto the NPC1L1 and MTP promoter in the intestine was evaluated by chromatin immunoprecipitation (ChIP) assays. The PCR products of the ChIP assays were visualized by loading onto 2% agarose gel (). (C) Control or DCHP-treated and mice were given an oral challenge of oil containing , and the distribution of radioactivity in intestinal segments of the mice was evaluated after 2 h (, two-way ANOVA, Bonferroni multiple comparisons test for multiple comparisons, *, , **, and ***, ). (D) Control or DCHP-treated and mice were injected with lipase inhibitor poloxamer-407 and then given an oral challenge of oil containing . Plasma samples were collected from those mice at several time points within 6 h, and the presence of was measured (, two-way ANOVA, Bonferroni multiple comparisons test for multiple comparisons. *, , compared with mice treated with control; ††, , compared with mice treated with DCHP). The numerical data corresponding to this figure are shown in Tables S12–S14. Results represent mean values. Error bars . Note: ANOVA, analysis of variance; BW, body weight; DCHP, dicyclohexyl phthalate; MTP, microsomal triglyceride transfer protein; NPCIL1, Niemann-Pick C1-Like 1; , intestinal epithelial cell-specific pregnane X receptor deficient; , pregnane X receptor flox; qPCR, quantitative polymerase chain reaction; SEM, standard error of the mean.
Given that NPC1L1 and MTP are required for intestinal lipid absorption and lipoprotein assembly and secretion (Abumrad and Davidson 2012; Hussain et al. 2012), we then performed in vivo intestinal cholesterol absorption assays using . Indeed, DCHP exposure led to significantly higher cholesterol uptake in the proximal intestine of mice, but the deficiency of intestinal PXR abolished the impact of DCHP exposure on cholesterol uptake (Figure 5C). Cholesterol absorption rates following DCHP exposure were also measured. Six hours after the gavage, the concentration of in the plasma was significantly higher in DCHP-exposed mice, as compared with control mice (Figure 5D). Consistent with cholesterol update results, the cholesterol absorption rates in mice were not affected by DCHP exposure (Figure 5D).
Next, enteroids isolated from intestinal crypts of and mice were also used in an ex vivo approach to evaluate the in vivo findings (Meng et al. 2019). As expected, exposure to DCHP also led to higher expression of NPC1L1 and MTP in enteroids isolated from mice (Figure S4A). By contrast, DCHP did not affect the expression of these genes in PXR-deficient enteroids isolated from mice (Figure S4A). We also performed cholesterol uptake assays using micelles containing . After DCHP treatment, the enteroids isolated from mice exhibited a greater uptake of (Figure S4B). However, DCHP treatment did not affect the uptake of by PXR-deficient enteroids isolated from mice (Figure S4B).
Investigating the Impact of Exposure of DCHP on Plasma Ceramide Levels
In addition to plasma lipid profiles, recent studies have identified novel predictors for CVD such as circulating ceramides (Kurz et al. 2019; Laaksonen et al. 2016; Meeusen et al. 2018; Wang et al. 2017). We employed targeted lipidomics to analyze various ceramide species, including C12:0, C14:0, C16:0, C18:0, C18:1, C20:0, C22:0, C24:0, and C24:1 (Deng et al. 2020; Dobierzewska et al. 2017; Federico et al. 2018). Interestingly, exposure to DCHP led to higher plasma levels of two major ceramide species—C16:0 and C24:0 ceramides—in but not in mice (Figure 6A,B). Interestingly, DCHP-mediated PXR activation led to a higher intestinal expression of the genes, GBA1 and neuraminidase 3 (NEU3), which mediate the salvage pathway of ceramide synthesis in but not mice (Figure 6C). Next, we exposed enteroids isolated from and mice to DCHP or vehicle control. Consistently, DCHP treatment led to a higher expression of the known PXR target genes (e.g., CYP3A11, GSTA1, and MDR1a), as well as GBA1 and NEU3, in control enteroids but not in PXR-deficient enteroids (Figure S4C).
Figure 6.

The impact of DCHP exposure on circulating ceramide levels and ceramide-synthesis–related gene expression in and mice. Eight-wk-old male and littermates were treated with vehicle control or BW per day of DCHP daily by oral gavage for 7 d. Plasma ceramide levels of (A) and (B) mice were measured by LC-MS/MS (, two-sample, two-tailed Student’s -test, *, ). (C) Intestinal expression of ceramide-synthesis–related genes were measured by qPCR (, two-sample, two-tailed Student’s -test, *, ). The numerical data corresponding to this figure are shown in Tables S15–S16. Results represent mean values. Error bars . Note: BW, body weight; CERS4, ceramide synthase 4; DCHP, dicyclohexyl phthalate; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1; GBA1, glucosylceramidase beta 1; LC-MS/MS, liquid chromatography–tandem mass spectrometry; NEU3, neuraminidase 3; , intestinal epithelial cell-specific pregnane X receptor deficient; , pregnane X receptor flox; SMPD1, sphingomyelin Phosphodiesterase 1; SMPD3, sphingomyelin Phosphodiesterase 3; qPCR, quantitative polymerase chain reaction; SEM, standard error of the mean.
Evaluating the Effects of DCHP Exposure on Plasma Lipid and Ceramide Levels in PXR-Humanized Mice
The effects of DCHP on plasma lipid and ceramide levels were further evaluated in PXR-humanized (huPXR; or ) mice (Ma et al. 2007; Sui et al. 2014) owing to PXR’s pharmacological differences across species (Blumberg et al. 1998; Zhou et al. 2009b). Male huPXR and littermates were exposed to vehicle control or DCHP at a dose of BW per day for 7 d. Similar to results, exposure to DCHP led to higher expression of PXR target genes, CYP3A11 and MDR1a, and the key lipogenic genes, NPC1L1 and MTP, in the intestine of huPXR but not in littermates (Figure 7A). Exposure to DCHP also caused huPXR mice to have higher plasma total cholesterol levels but did not affect the plasma cholesterol levels in mice (Figure 7B). DCHP treatment did not affect plasma triglyceride levels in huPXR or mice (Figure 7C). Analysis of lipoprotein fractions (VLDL, LDL, and HDL) revealed that DCHP treatment led to significantly higher atherogenic VLDL- and LDL-cholesterol levels but not HDL cholesterol levels in huPXR mice (Figure 7D). By contrast, DCHP exposure did not affect any of the lipoprotein levels in mice (Figure 7D).
Figure 7.

The effects of DCHP exposure on PXR target gene expression and plasma lipid levels in PXR-humanized (huPXR) mice. Eight-wk-old male huPXR and littermates were treated with BW per day of DCHP or DMSO control daily by oral gavage for 7 d. (A) The intestinal mRNA expression of PXR target genes and lipogenic genes were measured by qPCR analysis (, two-sample, two-tailed Student’s -test, *, and ***, ). Fasting plasma (B) total cholesterol and (C) triglyceride levels were analyzed by enzymatically colorimetric methods. (D) Lipoprotein fractions VLDL, LDL, and HDL were isolated from the plasma, and the cholesterol levels of VLDL, LDL, and HDL were examined by colorimetric methods. (, two-way ANOVA, Bonferroni multiple comparisons test for multiple comparisons, *, ). Error bars . The numerical data corresponding to this figure are shown in Tables S17–S18. Note: ANOVA, analysis of variance; CYP3A11, cytochrome P450, family 3, subfamily A, polypeptide 11; DCHP, dicyclohexyl phthalate; DMSO, dimethyl sulfoxide; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; MDR1a, multidrug resistance protein 1a; MTP, microsomal triglyceride transfer protein; NPC1L1, Niemann-Pick C 1-Like 1; PXR, pregnane X receptor; , pregnane X receptor deficient; qPCR, quantitative polymerase chain reaction; SEM, standard error of the mean; VLDL, very-low-density lipoprotein-cholesterol.
Next, lipidomics analysis was performed to measure ceramide species in these mice. Consistent with mouse treatment results (Figure 6A,B), exposure to DCHP also led to higher C16:0 and C24:0 ceramide levels in huPXR mice but not in mice (Figure 8A,B). The ceramide-synthesis–related genes in the intestine of huPXR and mice were then evaluated. DCHP treatment stimulated the expression of GBA1, which regulates the salvage pathway of ceramide synthesis (Figure 8C). The protein levels of GBA1 and key lipogenic protein MTP were also analyzed by western blotting, and the quantification results showed that exposure to DCHP led to higher protein levels of GBA1 and MTP in the intestine of huPXR mice (Figure 8D).
Figure 8.

The impact of DCHP exposure circulating ceramide levels and ceramide-synthesis–related gene expression in PXR-humanized (huPXR) mice. Eight-wk-old male huPXR and littermates were treated with BW per day of DCHP or vehicle control daily by oral gavage for 7 d. Plasma ceramide levels of (A) huPXR and (B) mice were measured by LC-MS/MS. (, two-sample, two-tailed Student’s -test, *, ). (C) Intestinal expression of ceramide-synthesis–related genes were measured by qPCR (, two-sample, two-tailed Student’s -test, *, ). (D) Immunoblotting of GBA1, MTP, and Actin proteins in the intestine of huPXR and mice. The densitometry analyses of the protein bands were shown below the immunoblotting panel (, two-way ANOVA, Bonferroni multiple comparisons test for multiple comparisons, *, , **, , and ***, ). The numerical data corresponding to this figure are shown in Tables S19–S21. Results represent mean values. Error bars . Note: ANOVA, analysis of variance; DCHP, dicyclohexyl phthalate; GBA1, glucosylceramidase beta 1; LC-MS/MS, liquid chromatography–tandem mass spectrometry; MTP, microsomal triglyceride transfer protein; , pregnane X receptor deficient; qPCR, quantitative polymerase chain reaction; SEM, standard error of the mean.
Last, enteroids were isolated from huPXR and mice for ex vivo DCHP exposure. Gene expression analysis demonstrated that DCHP treatment led to higher expression levels of known PXR target genes (CYP3A11, GSTA1, and MDR1a), lipogenic genes (NPC1L1 and MTP), and ceramide-synthetic–related gene (GBA1) in enteroids of huPXR but not in that of mice (Figure S5A). In addition, cholesterol update assays showed that exposure to DCHP led to more uptake of by enteroids of huPXR mice but did not affect the cholesterol update by enteroids of mice (Figure S5B).
Discussion
Since first introduced in the 1920s, phthalates have been widely used as plasticizers to increase the flexibility and durability of diverse materials in making polyvinylchloride (PVC) polymers (Autian 1973). Phthalates are also used in numerous products, including food packaging, personal cosmetics, nutritional supplements, cleaning materials, pharmaceuticals, insecticides, and children’s toys (Schettler 2006). In the present study, a widely used phthalate, DCHP, was identified as a potent agonist for the nuclear receptor PXR. Although the adverse health effects of several well-known phthalates, such as DEHP, have attracted considerable attention and research, little is known about DCHP’s potential impact on human health. Therefore, DCHP has recently been proposed by the U.S. Environmental Protection Agency as a high-priority substance for risk evaluation (U.S. EPA 2019). Interestingly, we found that both WT and PXR-humanized mice treated with DCHP exhibited hyperlipidemia. Whole-body PXR-knockout and intestine-specific PXR-deficient mice did not develop hyperlipidemia, suggesting that the effects of DCHP on lipid levels were mediated by intestinal PXR signaling. In addition to hyperlipidemia, mice treated with DCHP also exhibited higher circulating ceramides, a novel CVD predictor (Kurz et al. 2019; Laaksonen et al. 2016; Meeusen et al. 2018; Wang et al. 2017), a finding that appeared to be both dose and PXR dependent. DCHP-mediated PXR activation stimulated the expression of several key lipogenic genes and ceramide-synthetic–related genes in the intestine. The present study, to the best of our knowledge, is the first to demonstrate the impact of DCHP exposure on plasma lipid and ceramide levels in vivo. These findings indicate that PXR may play an important role in mediating the adverse effects of certain phthalates on cardiovascular health in humans (Figure 9).
Figure 9.
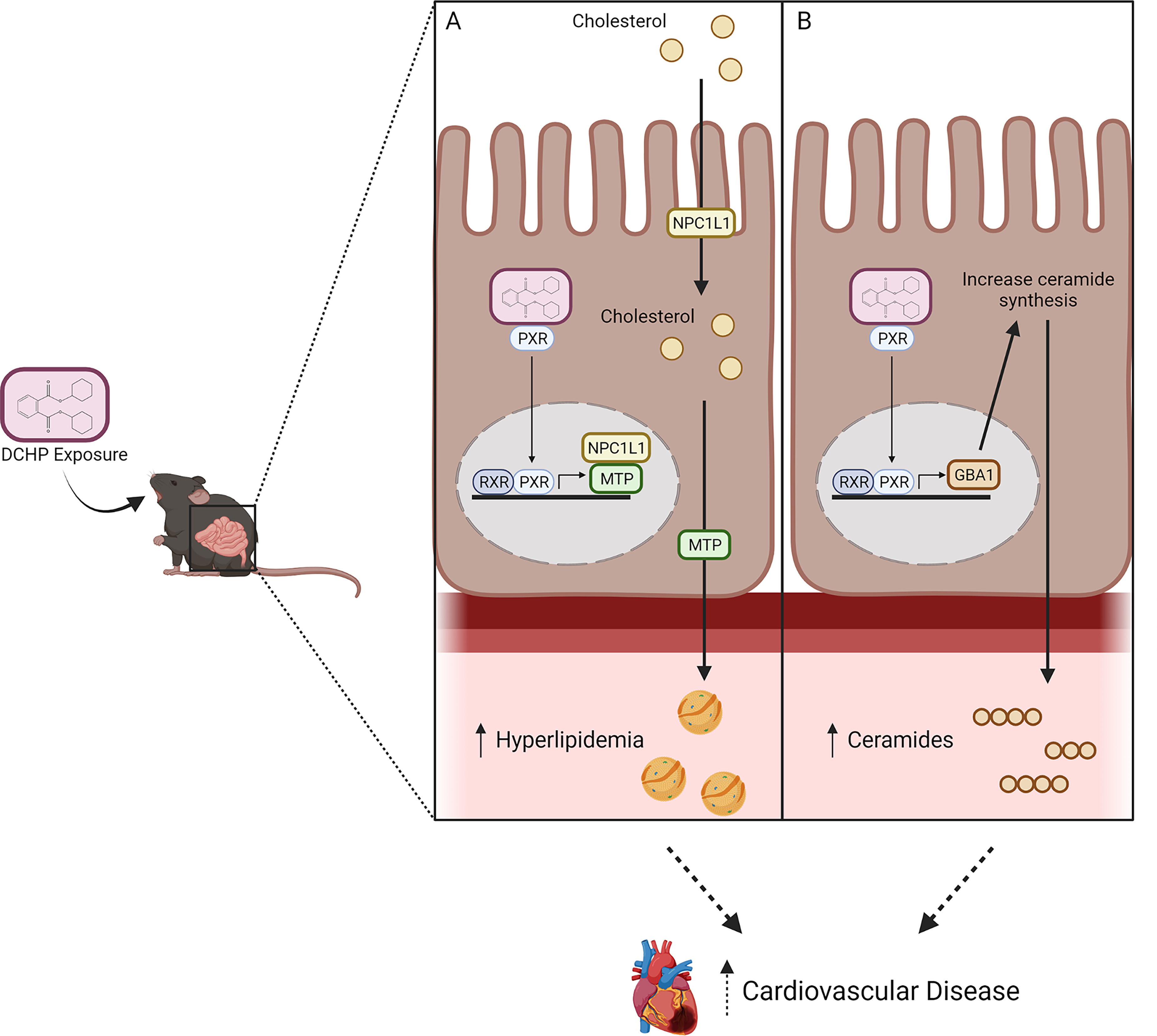
Schematic representation of the potential role of PXR in mediating the adverse effects of DCHP exposure on plasma lipid and ceramide levels. DCHP, a widely used phthalate, is a potent agonist of PXR. Exposure to DCHP leads to higher plasma cholesterol and ceramide levels in mice in an intestinal PXR-dependent manner. DCHP-mediated PXR activation regulates the expression of key lipogenic genes (e.g., NPC1L1 and MTP) (A) and ceramide-synthesis–related genes (e.g., GBA1) (B) in the intestine. Intestinal PXR signaling may contribute to adverse effects of DCHP on cardiovascular health through both hyperlipidemia-dependent and -independent mechanisms. This figure was created using BioRender.com. Note: DCHP, dicyclohexyl phthalate; GBA1, glucosylceramidase beta 1; MTP, microsomal triglyceride transfer protein; NPCIL1, Niemann-Pick C1-Like 1; PXR, pregnane X receptor.
As a key xenobiotic receptor, PXR can be activated by a diverse array of drugs, xenobiotics, and dietary chemicals, and the function of PXR in xenobiotic metabolism has been well studied (Zhou et al. 2009b). In addition to xenobiotic metabolism, PXR signaling was recently implicated in mediating lipid homeostasis, and the activation of PXR by various ligands has been shown to affect plasma lipid levels in various mouse models (Zhou 2016). For example, activation of PXR by the potent ligand PCN led to significantly higher plasma total cholesterol levels and atherogenic VLDL or LDL lipoprotein levels in WT and mice (de Haan et al. 2009; Zhou et al. 2009a). Several human immunodeficiency virus drugs, such as amprenavir (Helsley et al. 2013) and efavirenz (Gwag et al. 2019), have been identified as ligands of PXR that can stimulate hypercholesterolemia in mice in a PXR-dependent manner. Consistent with previous studies, treatment with DCHP also led to higher plasma cholesterol and atherogenic LDL levels in WT mice. Unlike other typical nuclear receptors, PXR exhibits considerable pharmacological differences across mammalian species (e.g., human vs. mouse) owing to its poorly conserved LBD (Zhou et al. 2009b; Zhou 2016). We previously reported that certain EDCs (e.g., BPA) were potent agonists for hPXR but not for mPXR or rPXR (Sui et al. 2012). In the present study, DCHP activated both hPXR and mPXR in vitro by transfection assays, and we then evaluated the impact of DCHP-mediated human PXR activation on lipid homeostasis in vivo using PXR-humanized mice. Consistent with WT mice, exposure to DCHP also led to hypercholesterolemia in huPXR but not in littermates. These results indicate that DCHP-mediated PXR activation may have clinically relevant impact on lipid homeostasis in humans.
PXR is expressed at high levels in both the liver and the intestine (Kliewer 2005; Zhou et al. 2009a), two organs that are essential for whole-body lipid homeostasis (Abumrad and Davidson 2012). Although small intestine lipid absorption is the key step for lipid accumulation in the body (Abumrad and Davidson 2012), most studies on PXR have focused on its function in the liver. For example, we and others have previously demonstrated that PXR can regulate the expression of multiple hepatic lipogenic genes to affect lipid homeostasis in mice (Bachmanov et al. 2002; Gwag et al. 2019; He et al. 2013; Moreau et al. 2009; Nakamura et al. 2007; Roth et al. 2008; Zhou 2016; Zhou et al. 2006). In contrast, much less is known about the role of PXR in the regulation of intestinal lipid homeostasis. Interestingly, our present study demonstrates that DCHP stimulated hyperlipidemia by activating intestinal PXR and that the deficiency of intestinal, but not hepatic, PXR abolished the impact of DCHP on plasma lipid levels. We previously identified cholesterol transporter NPC1L1 and MTP as transcriptional targets of PXR (Meng et al. 2019; Sui et al. 2015). As expected, DCHP-mediated PXR activation also led to the higher intestinal expression of NPC1L1 and MTP.
Both NPC1L1 and MTP have been established as important for the regulation of intestinal lipid homeostasis, including lipid absorption and lipoprotein assembly and secretion (Abumrad and Davidson 2012; Altmann et al. 2004; Davis et al. 2004; Hussain et al. 2012). Gene variants of NPC1L1 and MTP have also been associated with plasma lipid levels in humans (Lundahl et al. 2000; Ledmyr et al. 2002; Myocardial Infarction Genetics Consortium Investigators et al. 2014). Consistent with their function in intestinal lipid homeostasis, we found that DCHP exposure also led to higher intestinal lipid absorption in vivo. To further investigate the impact of DCHP exposure on intestinal lipid homeostasis, an ex vivo approach was also employed by isolating enteroids from intestinal crypts. These enteroids have been considered as “mini-intestines” that exhibit cellular composition and function similar to the gastrointestinal epithelium (Meng et al. 2019; Zachos et al. 2016). Indeed, we found that DCHP stimulated NPC1L1 and MTP expression and resulted in higher cholesterol uptake by enteroids in a PXR-dependent manner ex vivo. These results suggest that intestinal PXR plays an important role in mediating the dyslipidemic effects of DCHP and possibly other relevant EDCs.
In addition to hyperlipidemia, DCHP-mediated PXR activation also led to higher plasma levels of two major ceramide specifics, C16:0 and C24:0 ceramides, in both control and huPXR mice. Ceramides are complex sphingolipids that are components of the cell membrane and also play an important role in regulating cellular inflammatory signaling, stress responses, and apoptosis (Meeusen et al. 2017, 2018; Wang et al. 2017). Circulating ceramide levels have been independently associated with an increased CVD risk in multiple human studies, and ceramides can also be detected in human atherosclerotic plaques (Bismuth et al. 2008; Havulinna et al. 2016; Laaksonen et al. 2016; Meeusen et al. 2018; Uchida et al. 2017; Wang et al. 2017). For example, both C16:0 and C24:0 ceramides have been associated with the risk of major adverse cardiovascular events, including death, independent of other risk factors, such as LDL levels, in humans (Bismuth et al. 2008; Havulinna et al. 2016; Meeusen et al. 2018; Wang et al. 2017; Zhao et al. 2019). Interestingly, and huPXR mice treated with DCHP had higher intestinal expression of the genes (e.g., GBA1, NEU3) that mediate the salvage pathway of ceramide synthesis, and this was observed to occur in a PXR-dependent manner. By contrast, DCHP did not affect other genes regulating the de novo and sphingomyelin phosphodiesterase pathways. Although both and huPXR mice treated with DCHP exhibited higher GBA1 expression, NEU3 expression was higher in DCHP-treated mice but not in huPXR mice, a finding that is likely due to PXR’s species-specific effects given that PXR is remarkably divergent across mammalian species. It would be interesting to study the detailed mechanisms through which intestinal PXR signaling regulates key genes or pathways mediating EDC-induced ceramide production in mouse models vs. humans in the future.
Although hypercholesterolemia is a main risk factor for atherosclerosis (Lusis 2000), the function of ceramide species in regulating atherosclerosis development is still poorly understood. In the present study, we investigated the impact of short-term exposure to DCHP on plasma lipid and ceramide levels in mice, but we did not study whether long-term DCHP exposure can lead to increased atherosclerosis development. Nevertheless, we previously demonstrated that chronic exposure to other PXR ligands, including certain EDCs (e.g., BPA), can induce atherosclerosis development in hyperlipidemic mice (Sui et al. 2014; Zhou et al. 2009a). Interestingly, chronic activation of PXR by these ligands can also affect macrophage function and increase foam cell formation, contributing to atherosclerosis development (Sui et al. 2014; Zhou et al. 2009a). Future studies are required to investigate whether chronic DCHP-mediated ceramide production can affect the functions of macrophage or other cell types (e.g., endothelial cells) to promote atherosclerosis. It is plausible that DCHP and other PXR-agonistic EDCs may increase CVD risk in humans through both hypercholesterolemia-dependent and -independent mechanisms.
Phthalates are a diverse group of compounds that are produced in high volume, and humans can be exposed to certain phthalates at considerable levels (Halden 2010). Although DEHP is a principal phthalate causing human health concerns, DCHP can be found in water, foods, indoor particulate matter, and numerous consumer products (Cao et al. 2015; Cheng et al. 2016; U.S. EPA 2019; Sakhi et al. 2014; Schecter et al. 2013). DCHP and its monoester metabolite monocyclohexyl phthalate (MCHP) have been detected in human samples (Blount et al. 2000; Hartmann et al. 2018; Huang et al. 2014; Saravanabhavan et al. 2013; Wang et al. 2013). For example, several studies demonstrated high levels of DCHP exposure in certain susceptible populations, with urinary MCHP concentrations often (Hartmann et al. 2018; Wang et al. 2013). Another study also detected DCHP in of cord blood samples in a cohort of pregnant women, with a mean concentration of (Huang et al. 2014). Despite these findings in a human population, there were very few studies examining the in vivo effects of DCHP exposure in rodent models and previous studies that also used relatively high doses of DCHP (e.g., ) BW per day (Ahbab et al. 2017; Lake et al. 1982; Li et al. 2016; Lv et al. 2019). Although the dose used in our study still seems to be high, it is important to note that many plastic-associated EDCs (e.g., BPA, BPB, DEHP, TBC) have been identified by us and others as potent PXR agonists (Helsley and Zhou 2017; Sui et al. 2012, 2014, 2015; Zhou 2016). The selected dose serves as a tool to study the mechanisms underlying the links between phthalates and dyslipidemia. Further, we have previously reported that certain EDCs can synergistically activate PXR (Sui et al. 2012). Therefore, future in vitro and in vivo studies using EDC mixtures at human-relevant exposure doses must further elucidate the role of PXR as a mediator of EDC-elicited dyslipidemia and CVD risk in humans. These studies will be important for future environmental chemical risk assessment.
In summary, a widely used phthalate, DCHP, was identified as a potent PXR-selective agonist. Interestingly, short-term exposure to DCHP led to hypercholesterolemia and elevated circulation ceramide levels in mice, and deficiency of intestinal PXR abolished these adverse effects of DCHP. DCHP-mediated PXR activation affected intestinal expression of several key lipogenic and ceramide-synthetic–related genes in the intestine. These findings provide novel mechanistic insights explaining how exposure to certain EDCs increases CVD risk in humans and increases our understanding of gene–EDC interactions in predisposing individuals to CVD and, potentially, other chronic diseases.
Supplementary Material
Acknowledgments
We thank F. Gonzalez at the National Institutes of Health (NIH) National Cancer Institute for providing PXR-humanized mice and C. Yang at Rockefeller University for help with the PXR flox mouse generation. This work was supported in part by grants from the NIH (R01ES023470, R01HL131925, and R01HL123358 to C.Z.) and the American Heart Association grant (19TPA34890065 to C.Z.). R.H. was supported by an NIH National Research Service Award T32 training grant (T32ES018827).
References
- Abumrad NA, Davidson NO. 2012. Role of the gut in lipid homeostasis. Physiol Rev 92(3):1061–1085, PMID: , 10.1152/physrev.00019.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahbab MA, Güven C, Koçkaya EA, Barlas N. 2017. Comparative developmental toxicity evaluation of di-n-hexyl phthalate and dicyclohexyl phthalate in rats. Toxicol Ind Health 33(9):696–716, PMID: , 10.1177/0748233717711868. [DOI] [PubMed] [Google Scholar]
- Altmann SW, Davis HR Jr, Zhu L-J, Yao X, Hoos LM, Tetzloff G, et al. 2004. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 303(5661):1201–1204, PMID: , 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- Autian J. 1973. Toxicity and health threats of phthalate esters: review of the literature. Environ Health Perspect 4:3–26, PMID: , 10.1289/ehp.73043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG. 2002. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav Genet 32(6):435–443, PMID: , 10.1023/a:1020884312053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bismuth J, Lin P, Yao Q, Chen C. 2008. Ceramide: a common pathway for atherosclerosis? Atherosclerosis 196(2):497–504, PMID: , 10.1016/j.atherosclerosis.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount BC, Silva MJ, Caudill SP, Needham LL, Pirkle JL, Sampson EJ, et al. 2000. Levels of seven urinary phthalate metabolites in a human reference population. Environ Health Perspect 108(10):979–982, PMID: , 10.1289/ehp.00108979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg B, Sabbagh W Jr, Juguilon H, Bolado J Jr, van Meter CM, Ong ES, et al. 1998. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev 12(20):3195–3205, PMID: , 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X-L, Zhao W, Dabeka R. 2015. Di-(2-ethylhexyl) adipate and 20 phthalates in composite food samples from the 2013 Canadian Total Diet Study. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 32(11):1893–1901, PMID: , 10.1080/19440049.2015.1079742. [DOI] [PubMed] [Google Scholar]
- Cheng J, Krausz KW, Tanaka N, Gonzalez FJ. 2012. Chronic exposure to rifaximin causes hepatic steatosis in pregnane X receptor-humanized mice. Toxicol Sci 129(2):456–468, PMID: , 10.1093/toxsci/kfs211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Li H-H, Wang H-S, Zhu X-M, Sthiannopkao S, Kim K-W, et al. 2016. Dietary exposure and human risk assessment of phthalate esters based on total diet study in Cambodia. Environ Res 150:423–430, PMID: , 10.1016/j.envres.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Davis HR Jr, Zhu L-J, Hoos LM, Tetzloff G, Maguire M, Liu J, et al. 2004. Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem 279(32):33586–33592, PMID: , 10.1074/jbc.M405817200. [DOI] [PubMed] [Google Scholar]
- de Haan W, de Vries-van der Weij J, Mol IM, Hoekstra M, Romijn JA, Jukema JW, et al. 2009. PXR agonism decreases plasma HDL levels in ApoE3*Leiden.CETP mice. Biochim Biophys Acta 1791(3):191–197, PMID: , 10.1016/j.bbalip.2008.12.008. [DOI] [PubMed] [Google Scholar]
- DeKeyser JG, Laurenzana EM, Peterson EC, Chen T, Omiecinski CJ. 2011. Selective phthalate activation of naturally occurring human constitutive androstane receptor splice variants and the pregnane X receptor. Toxicol Sci 120(2):381–391, PMID: , 10.1093/toxsci/kfq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P, Hoffman JB, Petriello MC, Wang C-Y, Li X-S, Kraemer MP, et al. 2020. Dietary inulin decreases circulating ceramides by suppressing neutral sphingomyelinase expression and activity in mice. J Lipid Res 61(1):45–53, PMID: , 10.1194/jlr.RA119000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobierzewska A, Soman S, Illanes SE, Morris AJ. 2017. Plasma cross-gestational sphingolipidomic analyses reveal potential first trimester biomarkers of preeclampsia. PLoS One 12(4):e0175118, PMID: , 10.1371/journal.pone.0175118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drocourt L, Ourlin J-C, Pascussi J-M, Maurel P, Vilarem MJ. 2002. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J Biol Chem 277(28):25125–25132, PMID: , 10.1074/jbc.M201323200. [DOI] [PubMed] [Google Scholar]
- Eriksen M, Lebreton LC, Carson HS, Thiel M, Moore CJ, Borerro JC, et al. 2014. Plastic pollution in the world’s oceans: more than 5 trillion plastic pieces weighing over 250,000 tons afloat at sea. PLoS One 9(12):e111913, PMID: , 10.1371/journal.pone.0111913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico L, Yang L, Brandon J, Panchatcharam M, Ren H, Mueller P, et al. 2018. Lipid phosphate phosphatase 3 regulates adipocyte sphingolipid synthesis, but not developmental adipogenesis or diet-induced obesity in mice. PLoS One 13(6):e0198063, PMID: , 10.1371/journal.pone.0198063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün F, Venkatesan RN, Tabb MM, Zhou C, Cao J, Hemmati D, et al. 2002. Benzoate X receptors α and β are pharmacologically distinct and do not function as xenobiotic receptors. J Biol Chem 277(46):43691–43697, PMID: , 10.1074/jbc.M206553200. [DOI] [PubMed] [Google Scholar]
- Gwag T, Meng Z, Sui Y, Helsley RN, Park S-H, Wang S, et al. 2019. Non-nucleoside reverse transcriptase inhibitor efavirenz activates PXR to induce hypercholesterolemia and hepatic steatosis. J Hepatol 70(5):930–940, PMID: , 10.1016/j.jhep.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halden RU. 2010. Plastics and health risks. Annu Rev Public Health 31:179–194, PMID: , 10.1146/annurev.publhealth.012809.103714. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Uhl M, Weiss S, Scharf S, König J. 2018. Austrian reference values for phthalate metabolite exposure in children/adolescents and adults. Int J Hyg Environ Health 221(6):985–989, PMID: , 10.1016/j.ijheh.2018.06.001. [DOI] [PubMed] [Google Scholar]
- Havel RJ, Eder HA, Bragdon JH. 1955. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest 34(9):1345–1353, PMID: , 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K, et al. 2016. Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arterioscler Thromb Vasc Biol 36(12):2424–2430, PMID: , 10.1161/ATVBAHA.116.307497. [DOI] [PubMed] [Google Scholar]
- He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O’Doherty RM, et al. 2013. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 62(6):1876–1887, PMID: , 10.2337/db12-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsley RN, Sui Y, Ai N, Park S-H, Welsh WJ, Zhou C. 2013. Pregnane X receptor mediates dyslipidemia induced by the HIV protease inhibitor amprenavir in mice. Mol Pharmacol 83(6):1190–1199, PMID: , 10.1124/mol.113.085753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsley RN, Zhou C. 2017. Epigenetic impact of endocrine disrupting chemicals on lipid homeostasis and atherosclerosis: a pregnane X receptor-centric view. Environ Epigenet 3(4):dvx017, PMID: , 10.1093/eep/dvx017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Li J, Garcia JM, Lin H, Wang Y, Yan P, et al. 2014. Phthalate levels in cord blood are associated with preterm delivery and fetal growth parameters in Chinese women. PLoS One 9(2):e87430, PMID: , 10.1371/journal.pone.0087430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. 2012. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) 9:14, PMID: , 10.1186/1743-7075-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA. 2005. Cholesterol detoxification by the nuclear pregnane X receptor. Proc Natl Acad Sci USA 102(8):2675–2676, PMID: , 10.1073/pnas.0500159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA. 2015. Nuclear receptor PXR: discovery of a pharmaceutical anti-target. J Clin Invest 125(4):1388–1389, PMID: , 10.1172/JCI81244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Goodwin B, Willson TM. 2002. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev 23(5):687–702, PMID: , 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, et al. 1998. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92(1):73–82, PMID: , 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- Kurz J, Parnham MJ, Geisslinger G, Schiffmann S. 2019. Ceramides as novel disease biomarkers. Trends Mol Med 25(1):20–32, PMID: , 10.1016/j.molmed.2018.10.009. [DOI] [PubMed] [Google Scholar]
- Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. 2016. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J 37(25):1967–1976, PMID: , 10.1093/eurheartj/ehw148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BG, Foster JR, Collins MA, Stubberfield CR, Gangolli SD, Srivastava SP. 1982. Studies on the effects of orally administered dicyclohexyl phthalate in the rat. Acta Pharmacol Toxicol (Copenh) 51(3):217–226, PMID: , 10.1111/j.1600-0773.1982.tb01017.x. [DOI] [PubMed] [Google Scholar]
- Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, et al. 2008. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 300(11):1303–1310, PMID: , 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- Ledmyr H, Karpe F, Lundahl B, McKinnon M, Skoglund-Andersson C, Ehrenborg E. 2002. Variants of the microsomal triglyceride transfer protein gene are associated with plasma cholesterol levels and body mass index. J Lipid Res 43(1):51–58, PMID: , 10.1016/S0022-2275(20)30186-3. [DOI] [PubMed] [Google Scholar]
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, et al. 2014. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516(7529):112–115, PMID: , 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen X, Hu G, Li L, Su H, Wang Y, et al. 2016. Effects of in utero exposure to dicyclohexyl phthalate on rat fetal Leydig cells. Int J Environ Res Public Health 13(3):246, PMID: , 10.3390/ijerph13030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind L, Lind PM. 2012. Can persistent organic pollutants and plastic-associated chemicals cause cardiovascular disease? J Intern Med 271(6):537–553, PMID: , 10.1111/j.1365-2796.2012.02536.x. [DOI] [PubMed] [Google Scholar]
- Lind PM, Lind L. 2011. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis 218(1):207–213, PMID: , 10.1016/j.atherosclerosis.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔC(T) method. Methods 25(4):402–408, PMID: , 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lundahl B, Leren TP, Ose L, Hamsten A, Karpe F. 2000. A functional polymorphism in the promoter region of the microsomal triglyceride transfer protein (MTP-493G/T) influences lipoprotein phenotype in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 20(7):1784–1788, PMID: , 10.1161/01.atv.20.7.1784. [DOI] [PubMed] [Google Scholar]
- Lusis AJ. 2000. Atherosclerosis. Nature 407(6801):233–241, PMID: , 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Y, Fang Y, Chen P, Duan Y, Huang T, Ma L, et al. 2019. Dicyclohexyl phthalate blocks Leydig cell regeneration in adult rat testis. Toxicology 411:60–70, PMID: , 10.1016/j.tox.2018.10.020. [DOI] [PubMed] [Google Scholar]
- Ma X, Shah Y, Cheung C, Guo GL, Feigenbaum L, Krausz KW, et al. 2007. The pregnane X receptor gene-humanized mouse: a model for investigating drug-drug interactions mediated by cytochromes p450 3A. Drug Metab Dispos 35(2):194–200, PMID: , 10.1124/dmd.106.012831. [DOI] [PubMed] [Google Scholar]
- Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. 2008. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res 49(5):1137–1146, PMID: , 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen JW, Donato LJ, Bryant SC, Baudhuin LM, Berger PB, Jaffe AS. 2018. Plasma ceramides: a novel predictor of major adverse cardiovascular events after coronary angiography. Arterioscler Thromb Vasc Biol 38(8):1933–1939, PMID: , 10.1161/ATVBAHA.118.311199. [DOI] [PubMed] [Google Scholar]
- Meeusen JW, Donato LJ, Jaffe AS. 2017. Lipid biomarkers for risk assessment in acute coronary syndromes. Curr Cardiol Rep 19(6):48, PMID: , 10.1007/s11886-017-0863-9. [DOI] [PubMed] [Google Scholar]
- Melzer D, Gates P, Osborn NJ, Henley WE, Cipelli R, Young A, et al. 2012a. Urinary bisphenol A concentration and angiography-defined coronary artery stenosis. PLoS One 7(8):e43378, PMID: , 10.1371/journal.pone.0043378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer D, Osborne NJ, Henley WE, Cipelli R, Young A, Money C, et al. 2012b. Urinary bisphenol A concentration and risk of future coronary artery disease in apparently healthy men and women. Circulation 125(12):1482–1490, PMID: , 10.1161/CIRCULATIONAHA.111.069153. [DOI] [PubMed] [Google Scholar]
- Melzer D, Rice NE, Lewis C, Henley WE, Galloway TS. 2010. Association of urinary bisphenol A concentration with heart disease: evidence from NHANES 2003/06. PLoS One 5(1):e8673, PMID: , 10.1371/journal.pone.0008673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Gwag T, Sui Y, Park S-H, Zhou X, Zhou C. 2019. The atypical antipsychotic quetiapine induces hyperlipidemia by activating intestinal PXR signaling. JCI Insight 4(3):e125657, PMID: , 10.1172/jci.insight.125657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau A, Téruel C, Beylot M, Albalea V, Tamasi V, Umbdenstock T, et al. 2009. A novel pregnane X receptor and S14-mediated lipogenic pathway in human hepatocyte. Hepatology 49(6):2068–2079, PMID: , 10.1002/hep.22907. [DOI] [PubMed] [Google Scholar]
- Myocardial Infarction Genetics Consortium Investigators, Stitziel NO, Won H-H, Morrison AC, Peloso GM, Do R, et al. 2014. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med 371(22):2072–2082, PMID: , 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AB, Jacob S. 2016. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7(2):27–31, PMID: , 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Moore R, Negishi M, Sueyoshi T. 2007. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem 282(13):9768–9776, PMID: , 10.1074/jbc.M610072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsén L, Lampa E, Birkholz DA, Lind L, Lind PM. 2012a. Circulating levels of bisphenol A (BPA) and phthalates in an elderly population in Sweden, based on the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS). Ecotoxicol Environ Saf 75(1):242–248, PMID: , 10.1016/j.ecoenv.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Olsén L, Lind L, Lind PM. 2012b. Associations between circulating levels of bisphenol A and phthalate metabolites and coronary risk in the elderly. Ecotoxicol Environ Saf 80:179–183, PMID: , 10.1016/j.ecoenv.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Roth A, Looser R, Kaufmann M, Blättler SM, Rencurel F, Huang W, et al. 2008. Regulatory cross-talk between drug metabolism and lipid homeostasis: constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol Pharmacol 73(4):1282–1289, PMID: , 10.1124/mol.107.041012. [DOI] [PubMed] [Google Scholar]
- Roth GA, Forouzanfar MH, Moran AE, Barber R, Nguyen G, Feigin VL, et al. 2015. Demographic and epidemiologic drivers of global cardiovascular mortality. N Engl J Med 372(14):1333–1341, PMID: , 10.1056/NEJMoa1406656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhi AK, Lillegaard ITL, Voorspoels S, Carlsen MH, Løken EB, Brantsæter AL, et al. 2014. Concentrations of phthalates and bisphenol A in Norwegian foods and beverages and estimated dietary exposure in adults. Environ Int 73:259–269, PMID: , 10.1016/j.envint.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Saravanabhavan G, Guay M, Langlois É, Giroux S, Murray J, Haines D. 2013. Biomonitoring of phthalate metabolites in the Canadian population through the Canadian Health Measures Survey (2007–2009). Int J Hyg Environ Health 216(6):652–661, PMID: , 10.1016/j.ijheh.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Schecter A, Lorber M, Guo Y, Qian W, Yun SH, Kannan K, et al. 2013. Phthalate concentrations and dietary exposure from food purchased in New York State. Environ Health Perspect 121(4):473–494, PMID: , 10.1289/ehp.1206367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schettler T. 2006. Human exposure to phthalates via consumer products. Int J Androl 29(1):134–139, PMID: , 10.1111/j.1365-2605.2005.00567.x. [DOI] [PubMed] [Google Scholar]
- Seltenrich N. 2015. New link in the food chain? Marine plastic pollution and seafood safety. Environ Health Perspect 123(2):A34–A41, PMID: , 10.1289/ehp.123-A34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, McNeill JH. 2009. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol 157(6):907–921, PMID: , 10.1111/j.1476-5381.2009.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. 2001. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA 98(6):3369–3374, PMID: , 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Ai N, Park S-H, Rios-Pilier J, Perkins JT, Welsh WJ, et al. 2012. Bisphenol A and its analogues activate human pregnane X receptor. Environ Health Perspect 120(3):399–405, PMID: , 10.1289/ehp.1104426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Helsley RN, Park S-H, Song X, Liu Z, Zhou C. 2015. Intestinal pregnane X receptor links xenobiotic exposure and hypercholesterolemia. Mol Endocrinol 29(5):765–776, PMID: , 10.1210/me.2014-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Liu Z, Park S-H, Thatcher SE, Zhu B, Fernandez JP, et al. 2018a. IKKβ is a β-catenin kinase that regulates mesenchymal stem cell differentiation. JCI Insight 3(2):e96660, PMID: , 10.1172/jci.insight.96660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Meng Z, Park S-H, Lu W, Livelo C, Chen Q, et al. 2020. Myeloid-specific deficiency of pregnane X receptor decreases atherosclerosis in LDL receptor-deficient mice. J Lipid Res 61(5):696–706, PMID: , 10.1194/jlr.RA119000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Park S-H, Helsley RN, Sunkara M, Gonzalez FJ, Morris AJ, et al. 2014. Bisphenol A increases atherosclerosis in pregnane X receptor-humanized ApoE deficient mice. J Am Heart Assoc 3(2):e000492, PMID: , 10.1161/JAHA.113.000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Park S-H, Wang F, Zhou C. 2018b. Perinatal bisphenol A exposure increases atherosclerosis in adult male PXR-humanized mice. Endocrinology 159(4):1595–1608, PMID: , 10.1210/en.2017-03250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Xu J, Rios-Pilier J, Zhou C. 2011. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J Lipid Res 52(9):1652–1659, PMID: , 10.1194/jlr.M017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb MM, Kholodovych V, Grün F, Zhou C, Welsh WJ, Blumberg B. 2004. Highly chlorinated PCBs inhibit the human xenobiotic response mediated by the steroid and xenobiotic receptor (SXR). Environ Health Perspect 112(2):163–169, PMID: , 10.1289/ehp.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb MM, Sun A, Zhou C, Grün F, Errandi J, Romero K, et al. 2003. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J Biol Chem 278(45):43919–43927, PMID: , 10.1074/jbc.M303136200. [DOI] [PubMed] [Google Scholar]
- U.S. EPA (U.S. Environmental Protection Agency). 2019. Proposed Designation of Dicyclohexyl Phthalate (CASRN 84-61-7) as a High-Priority Substance for Risk Evaluation. https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P100XPOZ.txt [accessed 30 October 2021].
- Uchida Y, Uchida Y, Kobayashi T, Shirai S, Hiruta N, Shimoyama E, et al. 2017. Detection of ceramide, a risk factor for coronary artery disease, in human coronary plaques by fluorescent angioscopy. Circ J 81(12):1886–1893, PMID: , 10.1253/circj.CJ-17-0363. [DOI] [PubMed] [Google Scholar]
- Umesono K, Murakami KK, Thompson CC, Evans RM. 1991. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 65(7):1255–1266, PMID: , 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vom Saal FS, Myers JP. 2008. Bisphenol A and risk of metabolic disorders. JAMA 300(11):1353–1355, PMID: , 10.1001/jama.300.11.1353. [DOI] [PubMed] [Google Scholar]
- Wang DD, Toledo E, Hruby A, Rosner BA, Willett WC, Sun Q, et al. 2017. Plasma ceramides, Mediterranean diet, and incident cardiovascular disease in the PREDIMED trial (Prevención con Dieta Mediterránea). Circulation 135(21):2028–2040, PMID: , 10.1161/CIRCULATIONAHA.116.024261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhou Y, Tang C, He Y, Wu J, Chen Y, et al. 2013. Urinary phthalate metabolites are associated with body mass index and waist circumference in Chinese school children. PLoS One 8(2):e56800, PMID: , 10.1371/journal.pone.0056800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, de Jonge HR, et al. 2016. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem 291(8):3759–3766, PMID: , 10.1074/jbc.R114.635995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wang X, Deik AA, Hanna DB, Wang T, Haberlen SA, et al. 2019. Elevated plasma ceramides are associated with antiretroviral therapy use and progression of carotid artery atherosclerosis in HIV infection. Circulation 139(17):2003–2011, PMID: , 10.1161/CIRCULATIONAHA.118.037487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C. 2016. Novel functions of PXR in cardiometabolic disease. Biochim Biophys Acta 1859(9):1112–1120, PMID: , 10.1016/j.bbagrm.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, King N, Chen KY, Breslow JL. 2009a. Activation of PXR induces hypercholesterolemia in wild-type and accelerates atherosclerosis in apoE deficient mice. J Lipid Res 50(10):2004–2013, PMID: , 10.1194/jlr.M800608-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Poulton E-J, Grün F, Bammler TK, Blumberg B, Thummel KE, et al. 2007. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol 71(1):220–229, PMID: , 10.1124/mol.106.029264. [DOI] [PubMed] [Google Scholar]
- Zhou C, Tabb MM, Sadatrafiei A, Grün F, Blumberg B. 2004. Tocotrienols activate the steroid and xenobiotic receptor, SXR, and selectively regulate expression of its target genes. Drug Metab Dispos 32(10):1075–1082, PMID: , 10.1124/dmd.104.000299. [DOI] [PubMed] [Google Scholar]
- Zhou C, Verma S, Blumberg B. 2009b. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl Recept Signal 7:e001, PMID: , 10.1621/nrs.07001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, et al. 2006. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281(21):15013–15020, PMID: , 10.1074/jbc.M511116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.