

Abstract
Objective:
Viral infections influence intracellular peptide repertoires available for presentation by HLA-I. Alterations in HLA-I/peptide complexes can modulate binding of killer immunoglobuline-like receptors (KIRs) and thereby the function of natural killer (NK) cells. Although multiple studies have provided evidence that HLA-I/KIR interactions play a role in HIV-1 disease progression, the consequence of HIV-1 infection for HLA-I/KIR interactions remain largely unknown.
Design:
We determined changes in HLA-I presented peptides resulting from HIV-1-infection of primary human CD4+ T cells and assessed the impact of changes in peptide repertoires on HLA-I/KIR interactions.
Methods:
Liquid chromatography-coupled tandem mass spectrometry to identify HLA-I presented peptides, cell-based in-vitro assays to evaluate functional consequences of alterations in immunopeptidome and atomistic molecular dynamics simulations to confirm experimental data.
Results:
A total of 583 peptides exclusively presented on HIV-1-infected cells were identified, of which only 0.2% represented HIV-1 derived peptides. Focusing on HLA-C∗03 : 04/KIR2DL3 interactions, we observed that HLA-C∗03 : 04-presented peptides derived from noninfected CD4+ T cells mediated stronger binding of inhibitory KIR2DL3 than peptides derived from HIV-1-infected cells. Furthermore, the most abundant peptide presented by HLA-C∗03 : 04 on noninfected CD4+ T cells (VIYPARISL) mediated the strongest KIR2DL3-binding, while the most abundant peptide presented on HIV-1-infected cells (YAIQATETL) did not mediate KIR2DL3-binding. Molecular dynamics simulations of HLA-C∗03 : 04/KIR2DL3 interactions in the context of these two peptides revealed that VIYPARISL significantly enhanced the HLA-C∗03 : 04/peptide contact area to KIR2DL3 compared with YAIQATETL.
Conclusion:
These data demonstrate that HIV-1 infection-induced changes in HLA-I-presented peptides can reduce engagement of inhibitory KIRs, providing a mechanism for enhanced activation of NK cells by virus-infected cells.
Keywords: HIV-1 infection, HLA-C∗03 : 04, HLA-I, HLA-I presented peptide repertoire, killer immunoglobuline-like receptor, KIR2DL3, natural killer cells
Introduction
Human leukocyte antigen (HLA) class I (HLA-I) molecules present endogenously produced peptides that reflect the functional state of a cell, and can serve as indicators of cellular stress [1]. Peptide-binding to HLA-I molecules in the endoplasmic reticulum (ER) is fundamental for proper folding and trafficking to the cell surface, and this process is regulated by protein abundance as well as ER stress [2,3]. Peptide repertoires presented on HLA-I molecules are dependent on the respective HLA-I peptide-binding grooves, with conserved hydrogen-bonding networks leading to preferentially binding of peptides with a length of 8–12 amino acids (aa) [4]. Viral infections, such as HIV-1 infection, induce significant intracellular stress responses, including expression of protein chaperones, inhibition of protein synthesis and activation of the ubiquitination machinery [5–8], altering the repertoire of endogenously processed peptides. Resulting changes in HLA-I-presented peptides, including presentation of virus-derived peptides, can not only be detected by adaptive but also by innate immune responses [1].
Natural killer (NK) cells are innate effector cells, and their function is tightly regulated through activating and inhibitory receptors, including receptors of the killer immunoglobuline-like receptor (KIR) family. HLA-I molecules serve as ligands for most KIRs [9], and multiple studies have revealed associations between specific HLA/KIR compound genotypes and progression to AIDS [10]. Previous investigations have shown that peptides presented by HLA-I can modulate interactions with KIRs, with consequences for KIR+ NK cell function [11–16]. However, these studies have mostly focused on modulation of KIR+ NK cell function by individual viral peptides. The impact of overall changes in peptide repertoires presented on virus-infected cells on KIR-binding remains unknown. Here, we investigated the effect of HIV-1 infection on changes in HLA-I presented peptides by mass spectrometry, and show that infection-induced changes in HLA-C∗03 : 04-presented peptides reduced binding of the inhibitory KIR2DL3 receptor. These data suggest that changes in HLA-I peptide repertoires during infections can serve as an ‘innate’ mechanism enabling enhanced recognition of infected cells by NK cells.
Materials and methods
Isolation and infection of primary human CD4+ T cells
Blood was twice drawn from one donor 6 months apart. The study participant provided written informed consent for participation under protocols approved by the Ärztekammer Hamburg (PV4780). CD4+ T cells were isolated using CD4 Microbeads (Miltenyi Biotech, Bergisch Gladbach, Germany), and 50 × 106 cells were cryopreserved and stored at −80°C. Remaining cells were stimulated as described with CD3/CD28 dynabeads (Thermo Fisher Scientific, Waltham, Massachusetts, USA) [17]. At least 100 × 106 stimulated cells were infected via spinoculation with HIV-1 NL4-3[18] using a MOi = 0.01, cultured in T75 flasks for 72 h at 39.5°C, which yielded high infection rates, and subsequently cryopreserved. Infection rates were determined using an antibody directed against HIV-1 p24 Core Antigen-FITC (Beckman Coulter GmbH, Brea, California; USA) and analysed by flow cytometry. Infection rate was 65 and 40% for the two experiments. respectively. An additional 80 × 106 stimulated CD4+ T cells were treated identically but not exposed to HIV-1 and cryopreserved.
Isolation of HLA-I molecules from primary CD4+ T cells and analysis of HLA-I presented peptides using liquid chromatography-coupled tandem mass spectrometry
HLA-I molecules were isolated using standard immunoaffinity purification [19], using the pan-HLA-I-specific mAb W6/32. HLA-I extracts were analysed in five technical replicates as described [20,21], and HLA-I annotation of peptides was performed using NetMHCpan 3.0 [21–23].
Assessment of in-vitro binding of peptides to HLA-C∗03 : 04
Selected peptides were loaded on HLA-C∗03 : 04-transfected 721.221 cells containing a CRISPR/Cas9 generated TAP knockout (221-TAPko-HLA-C∗03 : 04) [13]. Fixed cells were stained using anti pan-HLA-ABC-APC (eBioscience, Frankfurt am Main, Germany) and analysed by flow cytometry. HLA-I stabilization was assessed as median MFI of APC and normalized to background values.
Assessment of KIR2DL3-binding to HLA-C∗03 : 04/peptide complex
KIR-Fc staining was performed as described [12,13,24]. Measurement of KIR-Fc binding was assessed as percentage of PE-positive cells (SUP1). KIR Jurkat reporter cell assays were performed as described [16]. Briefly, peptide pulsed 221-TAPko-HLA-C∗03 : 04 cells were coincubated with Jurkat Reporter cells expressing KIR2DL3, with the intracellular tail exchanged through CD3ξ chain resulting in activation of the cell after ligand binding in a ratio of 1 : 10. After 5 h of incubation at 37°C/5%, CO2 cells were stained with anti-CD69-BV421 (Biolegend, Inc. San Diego, California, USA), anti-CD3-PerCP-Cy5.5 (Biolegend) and anti-KIR2DL3-APC (Miltenyi Biotec). Washed and fixed cells were analysed using flow cytometry (BD LSRFortessa: BD Biosciences Franklin Lakes, New Jersey, USA). Gates were set to only include CD3+/KIR2DL3+ cells. Activation of reporter cells was quantified as median MFI of CD69 expression normalized to background expression.
Molecular dynamics simulations of HLA-C∗03 : 04/peptide/KIR2DL3 complexes
Initial structures were generated via alignment of PDBs 1EFX [25] and 1B6U [26] for HLA-C∗03 : 04/peptide/KIR2DL2 or KIR2DL3. The resulting HLA-C∗03 : 04/peptide/KIR2DL3 ternary complex was solvated in TIP3P [27] water and 150 mmol/l Na+ and Cl- to mimic the physiological condition, annealed over three iterations of 10 000 minimization steps and 5 ns equilibration at 310 K at 1 atm, and finally equilibrated for 50 ns. The 1EFX source peptide was next mutated to either VIYPARISL or YAIQATETL within an equilibrated snapshot. These new complex structures were prepared as described above, equilibrated another 100 ns at 310 K/1 atm, and run at production for 200 ns. Solvent-accessible surface areas (SASAs) and single point energies were computed for each production frame. All MD simulations were conducted with NAMD2.12 [28] using the CHARMM27 [29,30] force field (rigidBonds; 2-fs timestep); temperature and pressure were controlled with a Langevin thermostat (damped at 1/ps) and Langevin Piston barostat, respectively [31]. Energies were computed using the NAMD Energy plugin in VMD, and SASAs via VMD's measure tool; mutants were created with the MUTATOR plugin. Molecular images were generated with VMD-1.9.2 [32].
Data analysis
Data analysis was performed using BD FACS Diva Software, FlowJo V10, GraphPad Prism 7, MATLAB 2017b, GIMP 2.8.16, NetMHCpan 3.0 and Adobe Illustrator CS 5.1.
Results
Liquid chromatography-coupled tandem mass spectrometry reveals changes in HLA-I-presented peptide repertoires of HIV-1-infected primary CD4+ T cells
To identify HLA-I presented peptides on noninfected and HIV-1-infected CD4+ T cells, we infected primary human CD4+ T cells with HIV-1 in vitro. To ensure HIV-1 infection, CD4+ T cells were prestimulated prior to infection (stimulated and HIV-1-infected CD4+ T cells, sHIVCD4+ T cells), as described [33]. HLA-I presented peptides were analysed using LC-MS/MS. As control populations, untreated CD4+ T cells (noninfected/nonstimulated, nCD4+ T cells) and CD3/CD28-bead-stimulated CD4+ T cells (sCD4+ T cells) were used. Experiments were performed twice 6 months apart using PBMCs derived from the same study individual (HLA-A∗02 : 01/∗02 : 01, -B∗27 : 05/∗40 : 01, -C∗02 : 02/∗03 : 04). In total, more than 12 000 HLA-I presented peptides were identified, with 1369 presented on nCD4+ T cells. 4716 on sCD4+ T cells and 3962 on sHIVCD4+ T cells. HIV-1 derived peptides were only detected on HIV-1-infected cells (Fig. 1a).
Fig. 1.
Analysis of the immunopeptidome of primary HIV-1-infected CD4+ T cells by mass spectrometry.
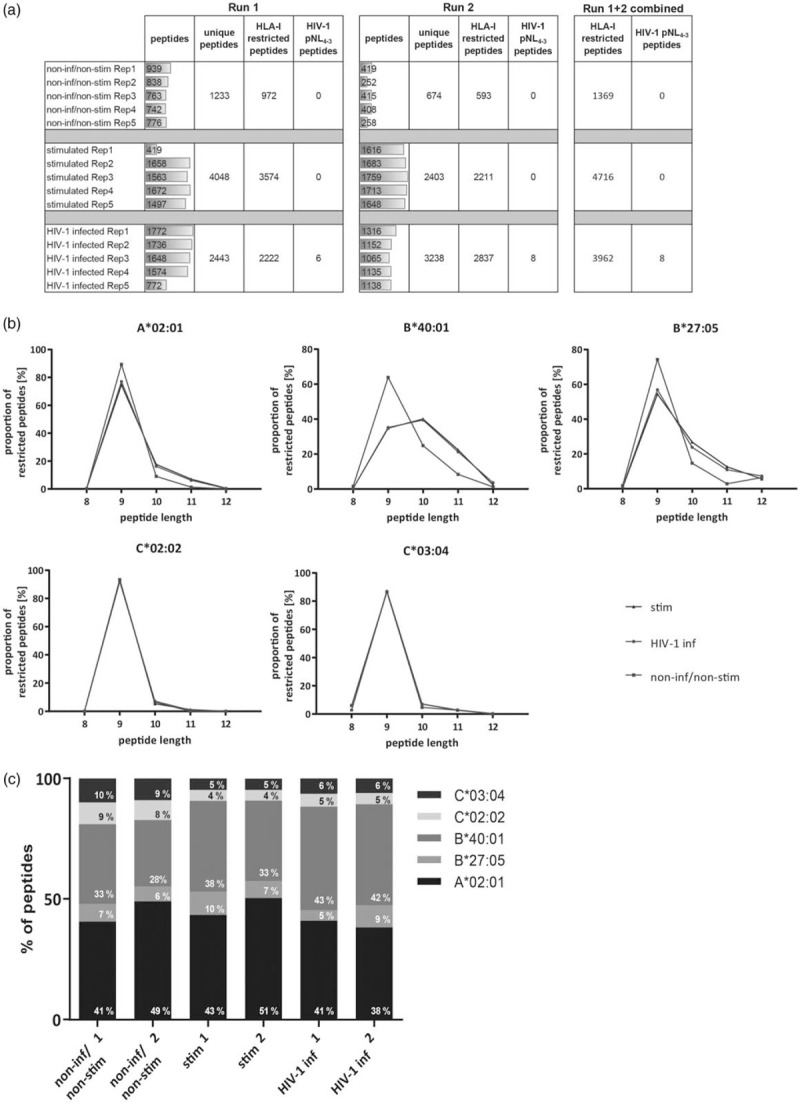
(a) Results of LC MS/MS experiment. nCD4+ T cells (non-stimulated), sCD4+ T cells (stimulated) and sHIVCD4+ T cells (HIV-1 infected). (b) Proportional distribution of peptide length for nCD4+ T cells (green), sCD4+ T cells (blue) and sHIVCD4+ T cells (red) for the different HLA-I alleles. (c) Distribution of HLA-I-restricted peptides according to HLA-I alleles analysing nCD4+ T cells (non inf/non stim 1+2), sCD4+ T cells (stim 1+2) and sHIVCD4+ T cells (HIV-1 inf 1+2). The proportion of HLA-C∗03:04-restricted peptides is highlighted in purple.
To determine whether HIV-1 infection resulted in changes in peptide length distributions, we compared peptide length between the different conditions and HLA-I allotypes (Fig. 1b). 9aa long peptides represented the most abundant peptides for all HLA-I molecules. For HLA-A∗02 : 01, -B∗40 : 01 and -B∗27 : 05, peptides presented on stimulated (sHIVCD4+ and sCD4+ T cells) showed a shift towards presentation of longer peptides, ranging from 9 to 11aa length. There was no difference in peptide lengths between the different conditions for HLA-C∗02 : 02- and -C∗03 : 04-presented peptides (Fig. 1b). The highest proportion of peptides was assigned to HLA-A∗02 : 01 (average of 44%), followed by HLA-B∗40 : 01 (average of 36%) (Fig. 1c). HLA-B∗27 : 05-presented peptides contributed very little (average of 7%) to the total number of detected peptides, HLA-C∗02 : 02 (average of 6%) and HLA-C∗03 : 04 (average of 7%) showed similar proportions. The contributions of individual HLA-I molecules to the detected HLA-I presented peptides were consistent between the two experiments, with a small reduction of HLA-C-presented and an increase in HLA-B∗40 : 01-presented peptide on stimulated (sCD4+ and sHIV) CD4+ T cells (Fig. 1c). Specifically, there was an increase of HLA-B∗40 : 01-presented peptides in sHIVCD4+ (42.5%) compared with sCD4+ (35.5%) and nCD4+ T cells (30.5%) (Fig. 1c). Taken together, these data show that HIV-1 infection alone has no significant effect on the length of peptides presented by HLA-C molecules, but resulted in an increase in the proportion of peptides assigned to HLA-B∗40 : 01.
HIV-1 infection of CD4+ T cells leads to presentation of distinct peptides by HLA-I including a small subset of HIV-1-derived peptides
Recognition of HIV-1-infected cells by CD8+ T cells relies on the presentation of virus-derived peptides by HLA-I. In line with previous studies [34–37]; only 0.2% of all identified peptides on HIV-1-infected cells were of viral origin (Fig. 1a). Identified HIV-1-derived peptides were 8–12 aa in length; three were restricted to HLA-A∗02 : 01 and four to HLA-B∗40 : 01, two of which represented overlapping peptides (SESARINTIL and SESARINTI) (Fig. 2a). Only one identified peptide did not exhibit any known HLA-I-binding motif (RSLFGSDPSSQ). The six HIV-1 derived peptides identified in the first experiment were also present in the second experiment 6 months later, during which two additional peptides were identified, indicating a very consistent pattern of peptide presentation (Fig. 2a). Relative quantity of HIV-1 derived peptides compared with host-derived peptides revealed that the HLA-B∗40 : 01-restricted peptide SESARINTIL was presented at very high quantities in both experiments, while the other HIV-1 peptides were presented at moderate levels (Fig. 2b). Overall, these data demonstrate that a small minority of HLA-I presented peptides on HIV-1-infected CD4+ T cells are derived from HIV-1, suggesting that changes in HLA-I presented host-derived peptides induced by infection may play a more prominent role in recognition of infected cells by NK cells.
Fig. 2.
HLA-I-presented peptides derived from HIV-1.
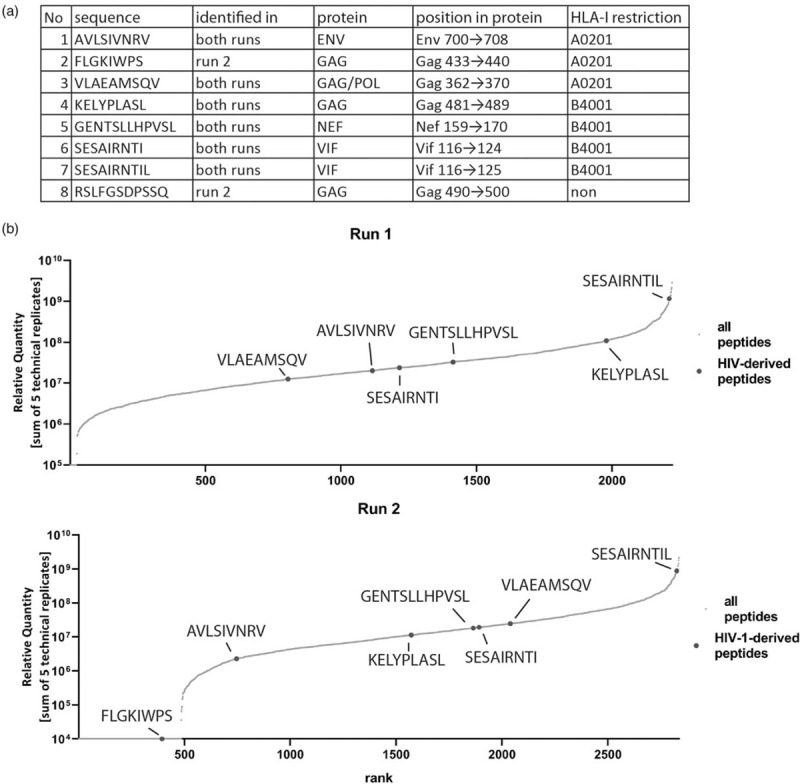
(a) Sequence, occurrence, source protein and HLA-I-restriction of the identified peptides. (b) Relative quantity of HIV-1 peptides compared with host-derived peptides.
To further evaluate changes in host-derived peptides that resulted from HIV-1 infection, we compared overall peptide repertoires between nCD4+ T cells and sHIVCD4+ T cells. To control for changes in peptide repertoires resulting from stimulation of CD4+ T cells with CD3/CD28 beads, we excluded any peptides also presented on sCD4+ T cells. Overall, 833 host-derived peptides were uniquely presented on sHIVCD4+ T cells and were classified as HIV-1 infection-induced host-derived stress-peptides, while 282 peptides were unique to nCD4+ T cells and classified as naturally presented self-peptides (Fig. 3a). As previous studies have shown that peptides presented by HLA-C molecules can significantly impact binding of inhibitory KIR2DL receptors [13,14,24,38], we focused subsequent analyses on the 43 and 30 HLA-C∗03 : 04-restricted peptides uniquely presented by sHIVCD4+ and nCD4+ T cells, respectively (Fig. 3b).
Fig. 3.
Overlap analysis illustrating the numbers of HLA-I/HLA-C∗03 : 04-restricted peptides identified under the different conditions.
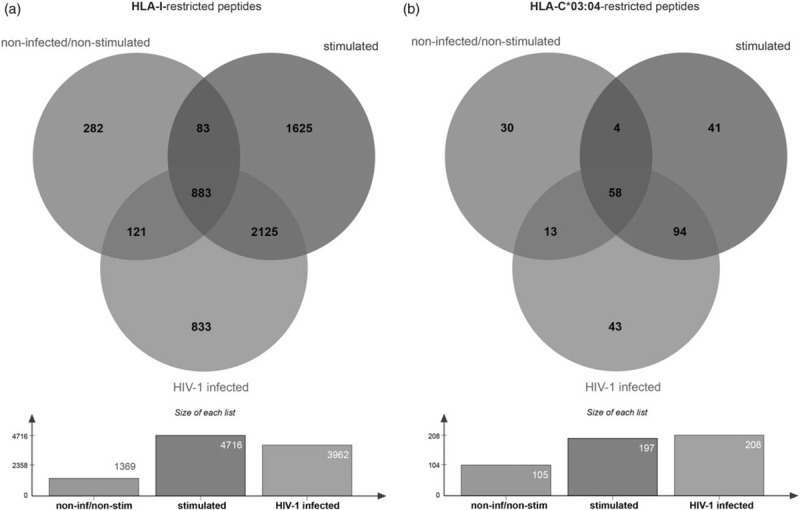
Peptides identified on noninfected/nonstimulated cells in green, peptides identified on stimulated cells in blue and peptides identified on HIV-1-infected cells in red. (a) All HLA-I-restricted peptides. (b) HLA-C∗03 : 04 restricted peptides.
The most abundant HLA-C∗03 : 04-restricted peptides presented on noninfected cells mediated strong binding to inhibitory KIR2DL3
To assess whether changes in host-derived peptides presented by HLA-C∗03 : 04 on HIV-1-infected cells can modulate KIR2DL3-binding, we first determined the KIR2DL3-binding capacity of identified peptides when presented by HLA-C∗03 : 04. Of the 73 HLA-C∗03 : 04-restricted peptides that were unique to either nCD4+ or sHIVCD4+ T cells, we selected 32 peptides (16 nCD4+ T cells, n1-n16; 16 sHIVCD4+ T cells, h1-h16) for functional studies, including peptides consistently detected in the two independent experiments and additional peptides that exhibited high binding scores for HLA-C∗03 : 04 using NetMHC rank [39] (SUP2). TAP-deficient 721.221 cells transfected with HLA-C∗03 : 04 (221-TAPko-HLA-C∗03 : 04) were used to validate that the selected peptides were indeed binding to HLA-C∗03 : 04, and all 32 peptides stabilized HLA-C∗03 : 04-expression on 221-TAPko-HLA-C∗03 : 04 in vitro with an average increase of 3.3-fold compared with no peptide control (Fig. 4a). We subsequently assessed whether HLA-C∗03 : 04 presented peptides modulated KIR-binding by staining peptide-pulsed 221-TAPko-HLA-C∗03 : 04 cells with recombinant KIR2DL3 (CD158b2) Fc Chimera Protein (KIR2DL3-Fc). Despite similar stabilization of HLA-C∗03 : 04 expression, the tested peptides exhibited strong differences in KIR2DL3-Fc binding (Fig. 4b). One peptide identified exclusively on nCD4+ T cells (peptide n3, VIYPARISL) showed very high binding of KIR2DL3 (36.9%), and also a number of additional peptides from nCD4+ T cells (n4, n8, n9 and n10) enabled high binding of KIR2DL3-Fc, ranging from 9.3 to 14.6%. In contrast, HLA-C∗03 : 04-restricted peptides identified uniquely on sHIVCD4+ T cells mediated lower binding of KIR2DL3-Fc (average of 3.5%). Overall, HLA-C∗03 : 04-restricted peptides identified uniquely on sHIVCD4+ T cells resulted in a 50% decrease in overall KIR2DL3-Fc binding, indicating a binding loss to the inhibitory KIR2DL3 during HIV-1 infection (Fig. 4b, P = 0.042).
Fig. 4.
Interaction of peptides, HLA-C∗03 : 04 and KIR2DL3 in vitro.
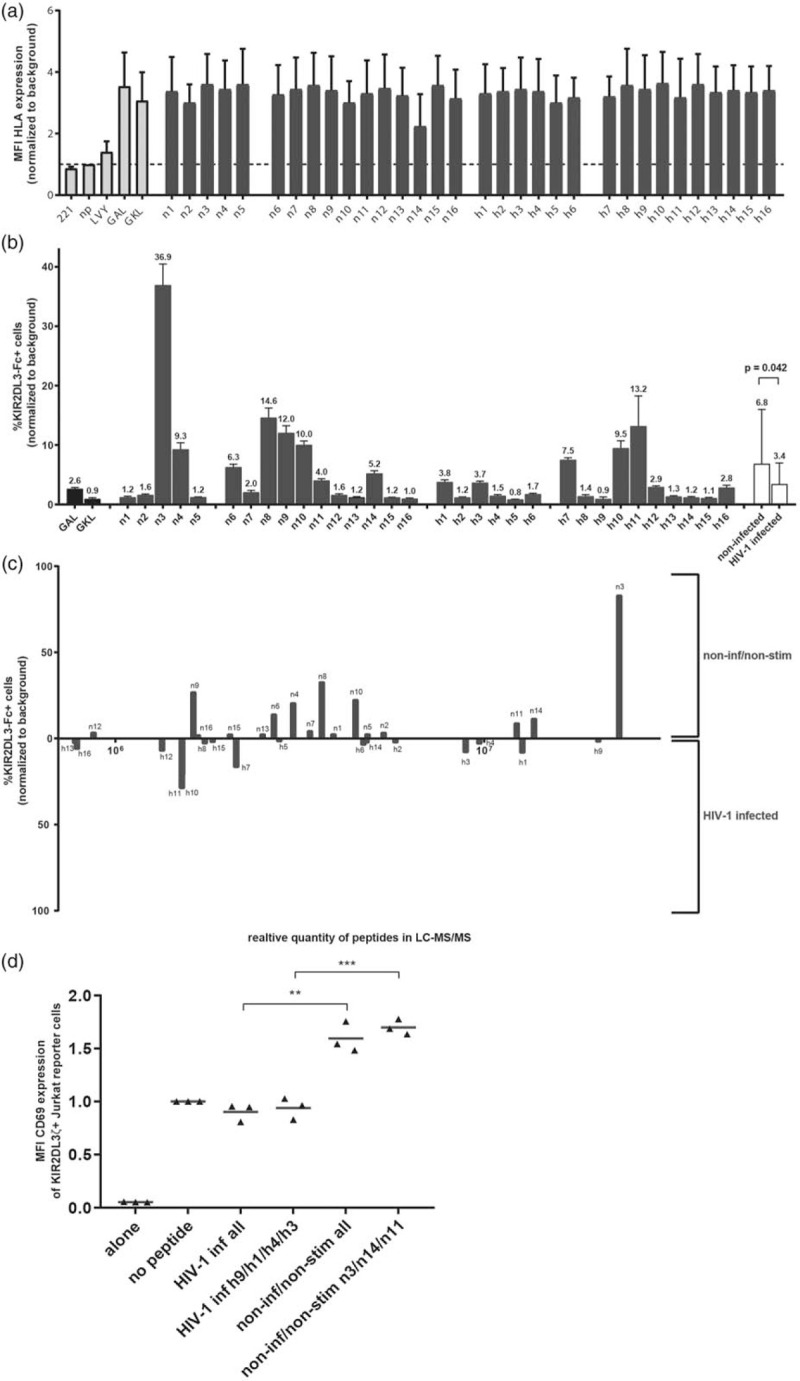
Peptides identified on nCD4+ T cells in green, peptides identified on sHIVCD4+ T cells in red. (a) Stabilization of HLA-C∗03:04. (b) Binding of KIR2DL3-Fc to HLA-C∗03:04/peptide complex. Average of KIR2DL3 positive cells of non-infected/non-stimulated peptides and HIV-1-infected peptides in white. P-value calculated using Mann-Whitney test. (c) KIR2DL3-binding capacity of peptides relative to their relative quantity. (d) CD69 expression of KIR2DL3ζ+ Jurkat reporter cells. Red bar demonstrates median of each group. P-values were calculated using unpaired, two-tailed t-test.
Given the differential ability of HLA-C∗03 : 04-restricted peptides to mediate KIR2DL3-binding, we next analysed KIR2DL3-binding in relation to the relative abundancy by which these peptides were presented by HLA-I (Fig. 4c). Remarkably, the strongest KIR2DL3-Fc binder (peptide n3) also represented the most abundant peptide detected on nCD4+ T cells, whereas the most abundant HLA-C∗03 : 04-restricted peptides identified on sHIVCD4+ T cells only mediated no or very minimal KIR2DL3-Fc-binding (Fig. 4b,c). Finally, we measured the ability of the HLA-C∗03 : 04-restricted peptides to activate KIR2DL3ζ+ Jurkat reporter cells. Combining all 16 peptides derived either from nCD4+ or sHIVCD4+ T cells, or only the three to four most abundant peptides identified under each condition (n3/n14/n11 for nCD4+ T cells or h9/h1/h4/h3 for sHIVCD4+ T cells), we observed higher activation of KIR2DL3ζ+ Jurkat reporter cells by peptides derived from nCD4+ T cells (Fig. 4d), in line with the results using KIR2DL3-Fc. Taken together, these data demonstrate that the most abundant peptides presented by HLA-C∗03 : 04 on nonactivated CD4+ T cells more strongly engaged the inhibitory KIR2DL3 receptor than the most abundantly presented peptides on HIV-1-infected CD4+ T cells.
Molecular dynamics simulations of HLA-C∗03 : 04/n3/KIR2DL3 and HLA-C:03 : 04/h9/KIR2DL3 show structural evidence for peptide-mediated functional differences
To establish a structural basis for the observed differences between HLA-C∗03 : 04-restricted peptides derived from nCD4+ T cells and sHIVCD4+ T cells in mediating KIR2DL3-binding, we carried out atomistic molecular dynamics simulations of the HLA-C∗03 : 04/peptide/KIR2DL3 binding complexes associated with either the n3 (VIYPARISL) or the h9 (YAIQATETL) peptide. Initial complex structures were generated in the canonical HLA-I-peptide-KIR configuration (Fig. 5a), with each peptide anchored in the HLA-C∗03 : 04 peptide-binding groove and presented to the KIR interface. Simulation trajectories revealed clear differences between the structural epitopes of the n3 and h9 peptides (Fig. 5b–d). Most significantly, binding between the HLA-C∗03 : 04/n3 complex and KIR2DL3 was dominated by both electrostatic and van der Waals (vdW) interactions involving the arginine in position 6 of the n3 peptide (VIYPARISL), which facilitated additional interactions at positions 7, 8 and 9. By contrast, the HLA-presented h9 peptide was defined by vdW interactions most prominent at the threonine in position 8 (YAIQATETL) (Fig. 5c). In the case of peptide n3, the side chain of the arginine in position 6 (R6) formed a persistent salt bridge with E21 of KIR2DL3, a glutamate near the KIR-peptide interaction interface. The R6 side chain extension induced by this salt bridge had the effect of ‘flattening’ the peptide-KIR interface, facilitating a multitude of peptide backbone and side chain interactions between peptide positions 6–9 and KIR2DL3 (Fig. 5b). The resulting HLA-C∗03 : 04/peptide complex contained a number of hydrophobic/vdW interactions, as well as an additional salt bridge between the peptide's C-terminal carboxylate and K44 of KIR2DL3, which competes with preexisting salt bridges between K44 and D183/E187 of KIR2DL3. The two salt bridges at P6 and P9 served to staple the n3 C-terminus in place, and, despite some degree of solvent exposure, likely accounted for a considerable amount of favourable binding free energy. On the contrary, no net stabilizing electrostatic interactions with KIR2DL3 were observed for peptide h9, despite the presence of polar/charged groups at positions 6–8 (Fig. 5d). Rather, the h9 peptide predominantly involved the burial of the T8 side chain methyl group in a generic hydrophobic pocket in KIR2DL3 with some additional contacts between T6/KIR2DL3-M70 and E7/KIR2DL3-Q71. This combination of interactions places the h9 C-terminal carboxylate in close proximity to KIR2DL3-D183, partially accounting for the lack of favorable electrostatics interactions (Fig. 5b). Although peptide n3 only exhibited a small surplus of vdW interaction energy over peptide h9, on average, peptide n3 did engage the KIR2DL3 with nearly 60% more contact area and with a large stabilizing contribution from net electrostatic interactions (Fig. 5d). These observations derived from atomistic molecular dynamics simulations provide a structural correlate for the observed stronger binding of KIR2DL3 to the HLA-C∗03 : 04/n3 complex as compared to the HLA-C∗03 : 04/h9 complex.
Fig. 5.
Molecular dynamics simulations of HLA-I/peptide-KIR complexes.
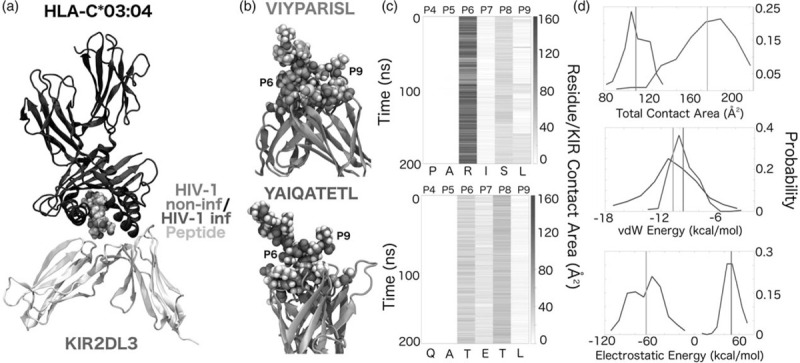
(a) Illustration of canonical HLA-C∗03:04-n3-KIR2DL3 arrangement. (b) Equilibrated peptide-KIR2DL3 for peptides n3 (green) and h9 (purple). (c) KIR-peptide contact area, as a function of time and peptide residue position. (d) Total peptide-KIR contact area, peptide-KIR vdW interaction energy, and peptide-KIR electrostatic energy. Mean values are indicated by vertical lines that are colour-coded according to the convention established in (b).
Discussion
NK cells have been suggested to be involved in the control of HIV-1 infection [40], but the precise mechanisms that lead to NK cell mediated activation by infected cells are not fully understood. Increasing data indicate that not only reduction of HLA-I expression on infected cells triggers NK cell activation, but also that presentation of peptides by HLA-I can abrogate binding of inhibitory KIRs such as KIR2DL3 [11–13,15]. We investigated changes in HLA-I presented peptide repertoires caused by HIV-1 by mass spectrometry and their impact on binding to the inhibitory receptor KIR2DL3, and observed reduced KIR2DL3-binding to HLA-C∗03 : 04 molecules presenting peptides derived from infected versus uninfected cells.
Intracellular infections lead to massive changes of cell metabolism, resulting in differences of endogenously produced peptides presented by HLA-I molecules on infected cells. Comparing naturally HLA-I presented peptides from HIV-1-infected, activated but noninfected and resting CD4+ T cells using mass spectrometry confirmed HIV-1 induced changes in HLA-I-presented peptides, in line with previous studies [35,37]. CD4+ T cells activated using anti-CD3/CD28 produce IFN-γ [41], which facilitates the presentation of longer peptides by HLA-I molecules [42,43]. In our experimental setup, sCD4+ T cells indeed presented longer peptides by HLA-A and -HLA-B molecules. In contrast, activation did not affect the length of peptides presented by HLA-C, in line with previous studies reporting that HLA-C molecules preferentially sample 9meres [44]. Apart from changes in peptide length, activation with and without HIV-1 infection also resulted in moderate shifts in the allocation of peptides presented by respective HLA-I molecules. Remarkably, HLA-B∗27 : 05-restricted peptides only represented 5–10% of the identified peptides. As the proportion of HLA-B∗27 : 05-restricted peptides did not change after infection, this phenomenon appears to be independent of HIV-1 infection. Although the factors responsible for this limited contribution of identified HLA-B∗27 : 05-restricted peptides to the overall number of identified peptides are not understood, one potential explanation is that HLA-B∗27 : 05 has been described to form homodimers presenting peptides in the absence of β2m [45], which might lead to a decreased number of presented peptides resulting from two free HLA-B∗27 : 05 heavy chains forming one homodimer presenting one peptide. One consistent observation emerging from studies of HLA-I presented peptides on virus-infected cells is that only a very small fraction of peptides are actually derived from virus proteins [34–37]. In the current study, indeed only 0.2% of the peptides presented on HIV-1-infected cells were derived from HIV-1. The in-depth analysis of HLA-I-presented peptides is cell and work-intensive, and our analysis was limited to two time points in a single individual with suitable HLA-I type. However, despite this limitation, six out of the eight identified HIV-1-derived peptides were consistently detected in the two independent experiments within the over 5600 peptides identified, suggesting a high level of reproducibility.
Although virus-derived peptides constituted only a small minority of infection-induced alterations of the immunopeptidome of HIV-1-infected CD4+ T cells, we observed very clear differences in host-derived peptides between infected and uninfected cells, including over 800 peptides uniquely detected on HIV-1-infected CD4+ T cells. Furthermore, over 2000 peptides were detected on activated (sHIV and sCD4+ T cells) but not on resting CD4+ T cells (nCD4+ T cells). To determine whether the presentation of these host-derived stress peptides can impact sensing of infected cells by KIR+ NK cells, we focused the analysis on HLA-C∗03 : 04-restricted peptides, as changes in HLA-C∗03 : 04/KIR2DL interactions have been associated with differential NK cell mediated control of HIV-1 replication [12,13,24]. Using a subset of 32 peptides selected from infected and uninfected cells based on consistent detection and high in-silico binding scores to HLA-C∗03 : 04, we demonstrated that while all peptides similarly stabilized HLA-C∗03 : 04 expression, peptides uniquely presented on nCD4+ T cells led to significantly stronger binding of KIR2DL3. In particular, the most abundant peptide, VIYPARISL (n3), induced the highest binding of KIR2DL3 and activation of KIR2DL3+ reporter cells, while the most abundant peptide presented on sHIVCD4+ T cells, YAIQATETL (h9) did not. These results support a model in which HLA-I-presented host-peptides on nonstressed cells provide a strong inhibitory signal to NK cells expressing inhibitory KIRs, while changes in these peptide repertoires during infection-induced stress result in reduced engagement of inhibitory KIRs, facilitating NK cell activation. This activation can lead to killing of infected cells by the release of cytolytic granules or, as NK cells are also endowed with immunomodulatory function, also result in enhanced immune activation through the release of cytokines.
The current study was limited by a small sample size, the necessity of stimulating CD4+ T cells prior to infection with HIV-1, and by the use of in-vitro systems limiting the investigated interactions to one inhibitory NK cell receptor and its HLA-C ligand. We therefore used molecular dynamics simulations of the HLA-C∗03 : 04/KIR2DL3 complexes with the dominant peptide presented on nCD4+ T cells (VIYPARISL, n3) and sHIVCD4+ T cells (YAIQATETL, h9) to further characterize the structural components that resulted in reduced KIR2DL3-binding to HLA-C∗03 : 04-restricted peptide presented on HIV-1-infected cells. This modelling demonstrates that VIYPARISL binds with almost twice the equilibrated contact area with KIR2DL3 as compared to YAIQATETL. This increased contact area was particularly due to interactions at P6 and P9 of the 9mer peptide that yielded more favourable single point vdW and electrostatic energies at the peptide n3-KIR interface. The critical relevance of residues P6 and P9 was not expected, given previous studies based on crystal structures of KIR2DL1 [46], KIR2DL2 [25], KIR2DL3 [26] and KIR2DS2 [47], where the peptide residues at P7-P8 were shown to have the most significant impact on KIR/HLA-I interactions [48]. However, we previously showed an impact of peptide residue P9 on KIR/HLA-I interactions, as changes in P9 influenced binding of HLA-C∗06 : 02 to KIR2DL1 and KIR2DS1 without impacting presentation by HLA-C∗06 : 02 [16]. These results suggest that changes in additional residues of HLA-I presented peptides can have an important impact on KIR-binding due to consequences for tertiary peptide structures, which has already been described for TCR engagement [49]. Taken together, the results of this study demonstrate that changes in HLA-I presented peptide repertoires resulting from infection and cellular stress can result in reduced engagement of inhibitory KIRs, facilitating NK cell activation and offering a potential mechanism of innate immune-surveillance of virus-infected cells.
Acknowledgements
These studies were supported by the German Center for Infectious Diseases Research (DZIF). The authors thank all the donors and coordinators of the healthy donor cohort at the Heinrich Pette Institute (HPI) in Hamburg for their participation in our study. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 NL4-3 Infectious Molecular Clone (pNL4-3) from Dr Malcolm Martin (Cat# 114). We thank Dr Christian Körner for his helpful comments to the experiments.
The National Institutes of Health (NIH) supported this work through material supply.
M.Z. and M.A. conceived and designed the experiments. M.Z., S.S., S.L., A.N. and C.S. performed experiments and analysed the data. J.K.W., T.H. and R.Z. performed computational simulations and analysed the data. G.M provided materials and helped with assay design. M.J.B. assisted with statistical analysis. M.A. designed and supervised the study. All authors provided continuous critical review of the data and commented on the manuscript.
All figures have associated raw data, stored at the Heinrich Pette Institute, Leibniz Institute for Experimental Virology (HPI) and Institute for Cell Biology, University of Tübingen. All primary data files and materials are available upon request from the corresponding author.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Supplementary Material
Supplementary Material
Footnotes
Supplemental digital content is available for this article.
References
- 1.Gleimer M, Parham P. Stress management: MHC class I and class I-like molecules as reporters of cellular stress. Immunity 2003; 19:469–477.. [DOI] [PubMed] [Google Scholar]
- 2.Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics 2015; 14:658–673.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Granados DP, Tanguay P-L, Hardy M-P, Caron É, de Verteuil D, Meloche S, et al. ER stress affects processing of MHC class I-associated peptides. BMC Immunol 2009; 10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrows SR, Rossjohn J, McCluskey J. Have we cut ourselves too short in mapping CTL epitopes?. Trends Immunol 2006; 27:11–16.. [DOI] [PubMed] [Google Scholar]
- 5.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 2001; 13:349–355.. [DOI] [PubMed] [Google Scholar]
- 6. Bukau, B., Cell, A. H.-, and 1998, undefined The Hsp70 and Hsp60 chaperone machines. [online] https://www.cell.com/fulltext/S0092-8674(00)80928-9 [Accessed 29 May 2019] [Google Scholar]
- 7.Welihinda A, Tirasophon W. Expression, R. K.-G., and 1999, undefined the cellular response to protein misfolding in the endoplasmic reticulum. [online] https://www.ingentaconnect.com/content/cog/ge/1999/00000007/F0030004/art00009 [Accessed 29 May 2019]. [Google Scholar]
- 8.Wilkinson KD. Ubiquitin-dependent signaling: the role of ubiquitination in the response of cells to their environment. J Nutr 1999; 129:1933–1936.. [DOI] [PubMed] [Google Scholar]
- 9.Holzemer A, Garcia-Beltran WF, Altfeld M. Natural killer cell interactions with classical and non-classical human leukocyte antigen class I in HIV-1 infection. Front Immunol 2017; 8:1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin MP, Carrington M. Immunogenetics of HIV disease. Immunol Rev 2013; 254:245–264.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopalan S, Long EO. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J Exp Med 1997; 185:1523–1528.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Teijlingen NH, Hölzemer A, Körner C, García-Beltrán WF, Schafer JL, Fadda L, et al. Sequence variations in HIV-1 p24 Gag-derived epitopes can alter binding of KIR2DL2 to HLA-C∗03. AIDS 2014; 28:1399–1408.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hölzemer A, Thobakgale CF, Jimenez Cruz CA, Garcia-Beltran WF, Carlson JM, van Teijlingen NH, et al. Selection of an HLA-C∗03:04-restricted HIV-1 p24 Gag sequence variant is associated with viral escape from KIR2DL3+ natural killer cells: data from an observational cohort in South Africa. PLOS Med 2015; 12:e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fadda L, Körner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw∗0102-restricted HIV-1 p24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS Pathog 2012; 8:e1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lunemann S, Martrus G, Hölzemer A, Chapel A, Ziegler M, Körner C, et al. Sequence variations in HCV core-derived epitopes alter binding of KIR2DL3 to HLA-C∗03:04 and modulate NK cell function. J Hepatol 2016; 65:252–258.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapel A, Garcia-Beltran WF, Hölzemer A, Ziegler M, Lunemann S, Martrus G, et al. Peptide-specific engagement of the activating NK cell receptor KIR2DS1. Sci Rep 2017; 7:2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trickett A, Kwan YL. T cell stimulation and expansion using anti-CD3/CD28 beads. J Immunol Methods 2003; 275:251–255.. [DOI] [PubMed] [Google Scholar]
- 18.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 1986; 59:284–291.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelde A, Kowalewski DJ, Stevanovic S. Purification and identification of naturally presented MHC class I and II ligands. Methods Mol Biol 2019; 1988:123–136.. [DOI] [PubMed] [Google Scholar]
- 20.Nelde A, Kowalewski DJ, Backert L, Schuster H, Werner J-O, Klein R, et al. HLA ligandome analysis of primary chronic lymphocytic leukemia (CLL) cells under lenalidomide treatment confirms the suitability of lenalidomide for combination with T cell-based immunotherapy. Oncoimmunology 2018; 7:e1316438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, et al. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci U S A 2015; 112:E166–E175.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 2016; 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009; 61:1–13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Körner C, Simoneau CR, Schommers P, Granoff M, Ziegler M, Hölzemer A, et al. HIV-1-mediated downmodulation of HLA-C impacts target cell recognition and antiviral activity of NK cells. Cell Host Microbe 2017; 22:111–119.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 2000; 405:537–543.. [DOI] [PubMed] [Google Scholar]
- 26.Maenaka K, Juji T, Stuart DI, Jones EY. Crystal structure of the human p58 killer cell inhibitory receptor (KIR2DL3) specific for HLA-Cw3-related MHC class I. Structure 1999; 7:391–398.. [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983; 79:926–935.. [Google Scholar]
- 28.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005; 26:1781–1802.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 1983; 4:187–217.. [Google Scholar]
- 30.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 2004; 25:1400–1415.. [DOI] [PubMed] [Google Scholar]
- 31.Bussi G. Communications, M. P.-C. P., and 2008, undefined stochastic thermostats: comparison of local and global schemes. Elsevier [online]. https://www.sciencedirect.com/science/article/pii/S0010465508000106 [Accessed 11 November 2019]. [Google Scholar]
- 32.Humphrey W, Dalke A. Graphics, K. S.-J. of molecular, and 1996, undefined VMD: visual molecular dynamics. Elsevier [online] https://www.sciencedirect.com/science/article/pii/0263785596000185 [Accessed 11 November 2019]. [Google Scholar]
- 33.Polacino PS, Liang HA, Firpo EJ, Clark EA. T cell activation influences initial DNA synthesis of simian immunodeficiency virus in resting T lymphocytes from macaques. J Virol 1993; 67:7008–7016.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaciuk JC, Skaley M, Bardet W, Schafer F, Mojsilovic D, Cate S, et al. Direct interrogation of viral peptides presented by the class I HLA of HIV-infected T cells. J Virol 2014; 88:12992–13004.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ternette N, Yang H, Partridge T, Llano A, Cedeño S, Fischer R, et al. Defining the HLA class I-associated viral antigen repertoire from HIV-1-infected human cells. Eur J Immunol 2016; 46:60–69.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rucevic M, Kourjian G, Boucau J, Blatnik R, Garcia Bertran W, Berberich MJ, et al. Analysis of major histocompatibility complex-bound HIV peptides identified from various cell types reveals common nested peptides and novel T cell responses. J Virol 2016; 90:8605–8620.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hickman HD, Luis AD, Bardet W, Buchli R, Battson CL, Shearer MH, et al. Cutting edge: class I presentation of host peptides following HIV infection. J Immunol 2003; 171:22–26.. [DOI] [PubMed] [Google Scholar]
- 38.Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 2011; 476:96–100.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol 2017; 199:3360–3368.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alter G, Altfeld M. NK cells in HIV-1 infection: evidence for their role in the control of HIV-1 infection. J Intern Med 2009; 265:29–42.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verhoef CM, Van Roon JA, Vianen ME, Glaudemans CA, Lafeber FP, Bijlsma JW. Lymphocyte stimulation by CD3-CD28 enables detection of low T cell interferon-gamma and interleukin-4 production in rheumatoid arthritis. Scand J Immunol 1999; 50:427–432.. [DOI] [PubMed] [Google Scholar]
- 42.Chong C, Marino F, Pak H, Racle J, Daniel RT, Müller M, et al. High-throughput and sensitive immunopeptidomics platform reveals profound interferon(-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol Cell Proteomics 2018; 17:533–548.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rist MJ, Theodossis A, Croft NP, Neller MA, Welland A, Chen Z, et al. HLA peptide length preferences control CD8+ T cell responses. J Immunol 2013; 191:561–571.. [DOI] [PubMed] [Google Scholar]
- 44.Gfeller D, Guillaume P, Michaux J, Pak H-S, Daniel RT, Racle J, et al. The length distribution and multiple specificity of naturally presented HLA-I ligands. J Immunol 2018; 201:3705–3716.. [DOI] [PubMed] [Google Scholar]
- 45.Allen RL, O’Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. J Immunol 1999; 162:5045–5048.. [PubMed] [Google Scholar]
- 46.Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol 2001; 2:452–460.. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Xiao Z, Ko HL, Shen M, Ren EC. Activating killer cell immunoglobulin-like receptor 2DS2 binds to HLA-A∗11. Proc Natl Acad Sci U S A 2014; 111:2662–2667.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyington JC, Sun PD. A structural perspective on MHC class I recognition by killer cell immunoglobulin-like receptors. Mol Immunol 2002; 38:1007–1021.. [DOI] [PubMed] [Google Scholar]
- 49.Singh NK, Riley TP, Baker SCB, Borrman T, Weng Z, Baker BM. Emerging concepts in TCR specificity: rationalizing and (maybe) predicting outcomes. J Immunol 2017; 199:2203–2213.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.