

Abstract
Biomolecular condensates concentrate macromolecules into discrete cellular foci without an encapsulating membrane. Condensates are often presumed to increase enzymatic reaction rates through increased concentrations of enzymes and substrates (mass action), although this idea has not been widely tested and other mechanisms of modulation are possible. Here we describe a synthetic system where the SUMOylation enzyme cascade is recruited into engineered condensates generated by liquid-liquid phase separation of multidomain scaffolding proteins. SUMOylation rates can be increased up to 36-fold in these droplets compared to the surrounding bulk, depending on substrate KM. This dependency produces substantial specificity among different substrates. Analyses of reactions above and below the phase separation threshold lead to a quantitative model in which reactions in condensates are accelerated by mass action and by changes in substrate KM, likely due to scaffold-induced molecular organization. Thus, condensates can modulate reaction rates both by concentrating molecules and by physically organizing them.
Introduction
Biomolecular condensates concentrate proteins and RNA molecules without a surrounding membrane1,2. Condensates appear in a wide range of biological contexts, including mRNA storage/degradation3,4, T-cell activation5,6, and ribosome biogenesis7. Many condensates appear to form through liquid-liquid phase-separation (LLPS), in which oligomerization mediated by multivalent interactions lowers the solubility of proteins and/or nucleic acids sufficiently to form a second phase1,2,8. Where measured, the degree of concentration in this phase ranges from 2- to 150-fold for different constituents9–12. Scaffold-like molecules, those that contribute more strongly to formation of the condensate, are typically the most highly concentrated components9,13. Client-like molecules, which are recruited into the condensate through interactions with scaffolds but do not contribute strongly to formation of the structure, tend to be less concentrated (with exceptions in cases of high affinity for scaffold9,13).
Condensates provide potential mechanisms by which cells can regulate biochemical processes temporally and spatially1,2,14. Because condensates concentrate enzymes and their potential substrates, the compartments are often invoked as accelerating biochemical reactions. Indeed, a number of studies have shown that enzymatic rates can be increased by LLPS, including nucleation and assembly of actin filaments and microtubules5,8,15–18, RNA autosplicing19, production of 3’,5’-cyclic GMP-AMP20, carboxylation of ribulose-1,5-bisphosphate21, activation of Ras22, RNA polymerization23, formate dehydrogenation24, and glucose oxidation/peroxidation25. Since condensates may selectively concentrate certain enzymes and/or substrates, and many substrates often compete for the same enzyme, phase separation has also been invoked as a mechanism to provide specificity in signaling or metabolic networks through accelerating some reactions over others1,14. Such bias has been engineered by clustering a metabolic branchpoint enzyme with only one of its two downstream target enzymes, shifting activity away from the non-clustered pathway26.
Beyond mass action (i.e. the effects of simply concentrating components), phase separation could in principle alter reactions by other mechanisms, for example changing molecular conformations27,28 and/or inducing specific molecular organizations29, and by the effects of crowding on these properties and molecular diffusion. In recent studies of membrane-associated condensates that control actin filament nucleation and Ras activation, phase separation was found to increase the membrane dwell time of key components, consequently increasing their specific activities16,22. Thus, in these systems, phase separation accelerates reactions not only by concentrating molecules, but also by changing the intrinsic activities of those molecules. These complexities, however, have not been generally addressed in three-dimensional droplet systems.
Here we developed a simplified in vitro model of natural condensates, to understand how phase separation can alter enzymatic activity. We used the FKBP-rapamycin-FRB system30,31 to recruit components of the SUMOylation enzymatic cascade32 to polySH3 domain and polyProline-Rich-Motif (polyPRM) scaffold proteins8. We measured SUMOylation activity toward several different substrates at scaffold concentrations above and below the LLPS threshold. Recruiting enzyme and substrate into phase separated droplets can substantially increase reaction rates. This effect is dependent on substrate properties: SUMOylation of high KM substrates is accelerated by LLPS, while SUMOylation of others can be inhibited by LLPS due to a combination of low KM and substrate inhibition, thus affording specificity. We found that condensates can accelerate reactions by two mechanisms: increased concentration, and molecular organization affording a scaffold-dependent decrease in KM. Computationally modeling the combined effects of molecular organization (leading to a change in KM) and concentration enables description of the reaction rates of various substrates under diverse conditions. These data indicate that three-dimensional condensates, like two-dimensional condensates, can accelerate reactions by both concentration-dependent and concentration-independent mechanisms, affording switchable control over reaction rates and specificity.
Results
Design of an inducible condensate-targeted enzyme cascade
We generated a model condensate based on inducible targeting of the SUMOylation enzymatic cascade (Figure 1A)33 into phase separated droplets composed polySH33, containing three tandem SH3 domains, and its polyPRM5 ligand, containing five tandem proline-rich-motifs (Figure 1B)8. The cascade attaches the 12 kDa Small Ubiquitin-like Modifier protein, SUMO, to diverse substrates. Many of the SUMOylating and deSUMOylating enzymes are concentrated in PML Nuclear Bodies, and it has been suggested that these condensates may regulate cellular SUMOylation34. Here we used a minimal version of the cascade, comprising the E1 (SAE1/2 heterodimer) and E2 (Ubc9) enzymes, substrate(s), and SUMO1 (Figure 1A).
Figure 1. Design of an inducible, condensate-targeted enzyme cascade.
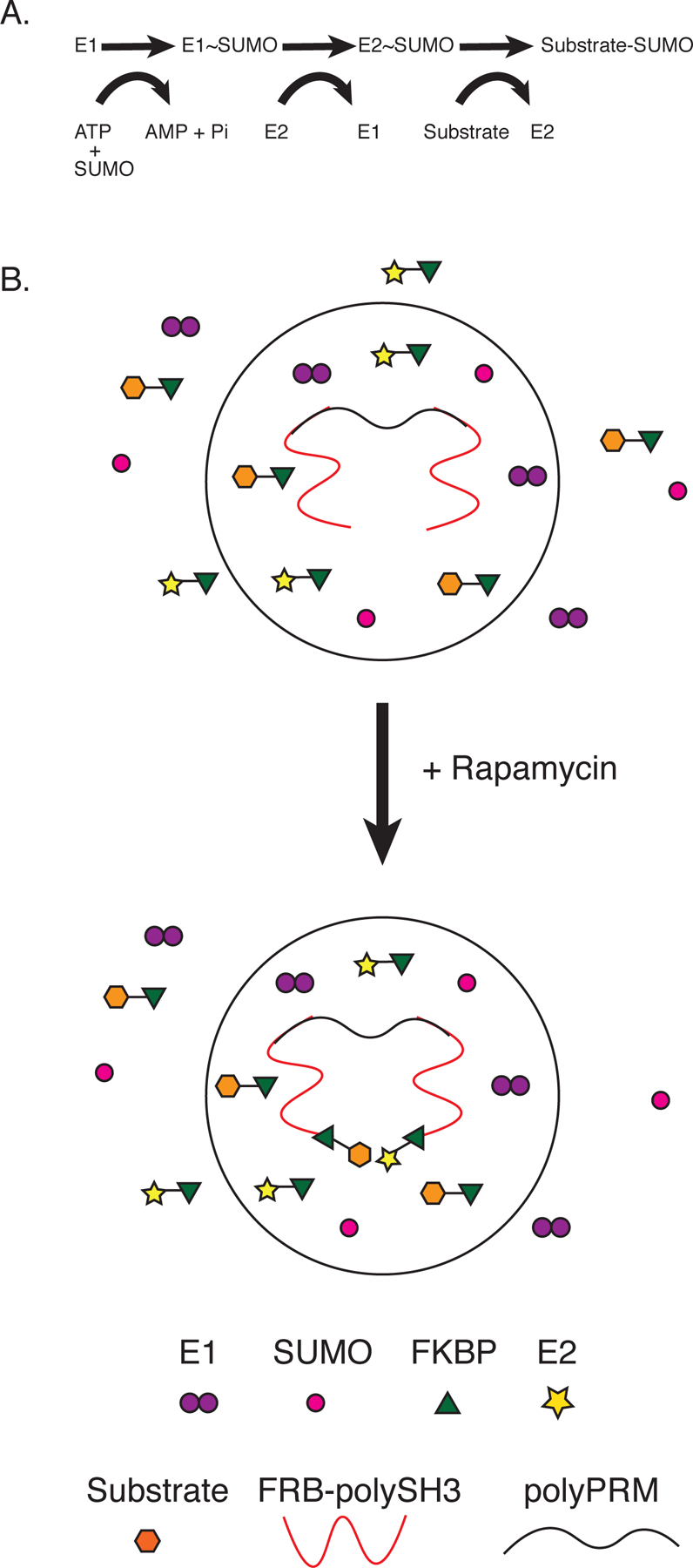
(A) Schematic of the SUMOylation cascade. SUMO is initially conjugated to E1 through a thioester bond, then transferred to E2 through a second thioester, and finally to a lysine on the target protein.
(B) Inducible recruitment of the SUMOylation cascade to polySH3-polyPRM condensates using the FRB-FKBP-rapamycin system. FRB is fused to polySH3, which is concentrated in the condensates. Upon addition of rapamycin, FKBP-E2 and FKBP-substrate enrich in the condensates, while untagged SUMO remains evenly distributed. Although not illustrated in the diagram, untagged E1 weakly enriches due to binding E2 (see text).
PolySH33 and polyPRM5 undergo reversible LLPS when mixed at concentrations above ~5µM8. We fused the FRB domain from mTOR onto the N-terminus of polySH33, and the FKBP12 protein to the N-termini of Ubc9 and substrates. Addition of rapamycin induces heterodimerization of FRB and FKBP12, recruiting Ubc9 and substrate to the polySH33 scaffold. When polySH33 and polyPRM5 are at concentrations below the LLPS threshold they form oligomeric complexes8, and rapamycin will cause binding of Ubc9/substrate to these structures. When the scaffolds are above the LLPS threshold, this rapamycin-induced interaction should recruit Ubc9 and substrates into the droplet phase (Figure 1C). Note that in all experiments E1 and SUMO1 are untethered.
Condensates increase the total SUMOylation rate
Condensates formed by mixing FRB-polySH33 and polyPRM5 strongly recruit E2 (mCherry-FKBP-Ubc9) and substrate (FKBP-EGFP-peptide) in the presence of rapamycin but not in a DMSO control (Figure 2A). The substrate used here is a peptide derived from the PML protein (residues 480–495, SQTQSPRKVIKMESEE), which contains the canonical SUMOylation consensus sequence ΨKXE35, and is recognized directly by Ubc9.
Figure 2. Condensates increase the total SUMOylation rate.
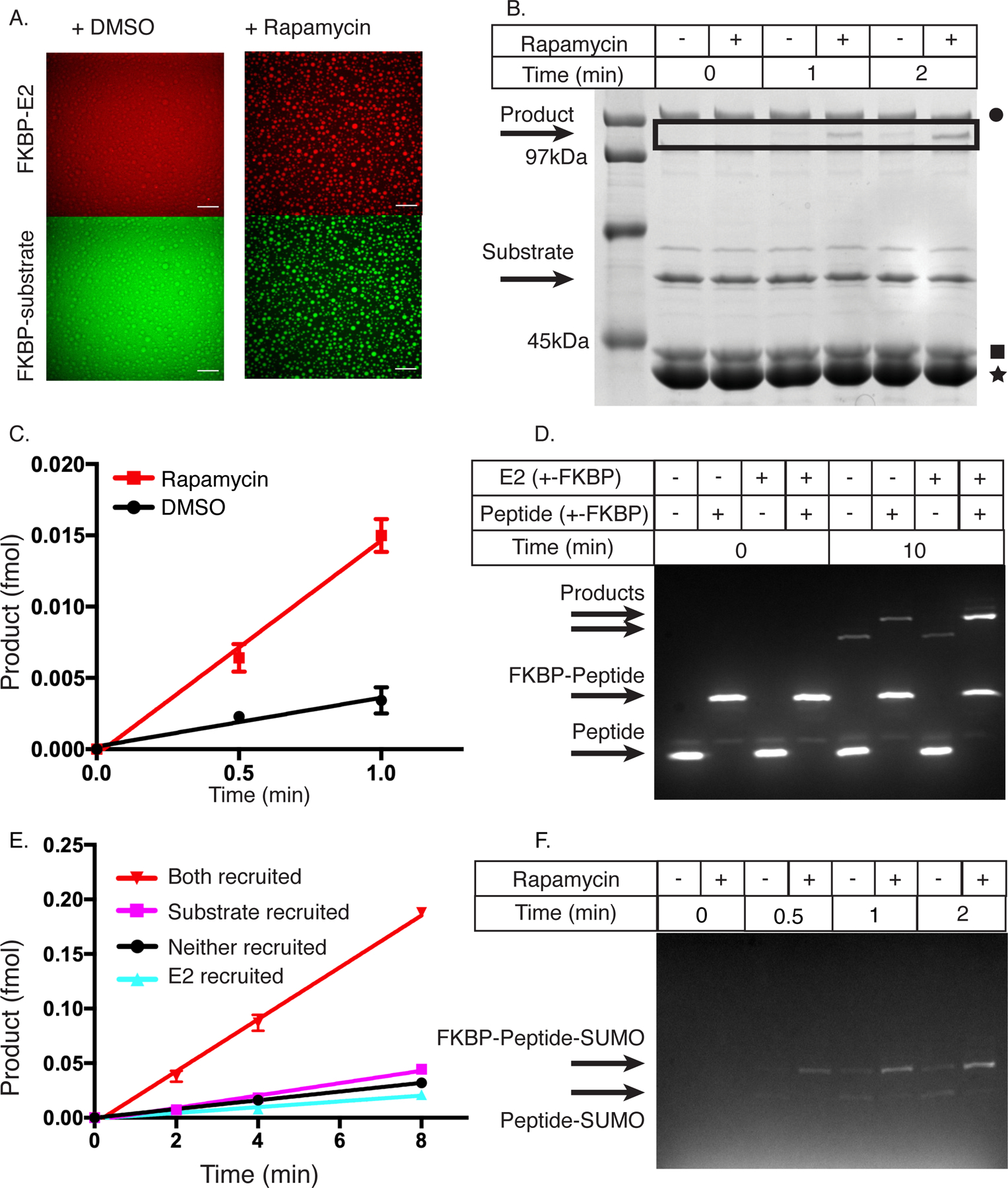
(A) Confocal fluorescence microscopy images of mCherry-FKBP-E2 (top row) and FKBP-EGFP-substrate (bottom row) in the presence of FRB-polySH33:polyPRM5 condensates upon addition of DMSO control (left column) or rapamycin (right column). These images are representative of 3 independent experiments. Scale bar is 50um.
(B) SDS-PAGE gel stained with Coomassie blue illustrating production of SUMOylated substrate as a function of time with either DMSO (−) or rapamycin (+). Black square denotes E2, black star denotes FRB-polySH33, and black circle denotes E1. This gel is representative of 3 independent experiments.
(C) Quantification of data in panel B, showing intensity of the SUMOylated substrate band as a function of time. DMSO = black circles, rapamycin = red squares. Data are plotted as the mean and SD from n=3 independent experiments.
(D) Fluorescence-detected SDS-PAGE gel depicting production of SUMOylated substrate as a function of time when E2, substrate, both or neither are fused to FKBP (and thus are recruited into FRB-polySH33-polyPRM5 droplets). + indicates FKBP-fusion, - indicates non-fused. This gel is representative of 4 independent experiments.
(E) Quantification of data in panel D, showing intensity of the SUMOylated substrate band as a function of time. E2+substrate recruited, inverted red triangles; E2 recruited, cyan triangles; substrate recruited, magenta squares; neither recruited black circles. Absolute amounts quantified by an internal standard. Data are plotted as the mean and SD from n=4 independent experiments.
(F) Fluorescent SDS page gel of SUMOylation of PML peptide or FKBP-PML peptide when co-incubated with FRB-polySH33:polyPRM5 condensates and either DMSO or rapamycin. SUMOylated FKBP-peptide is the upper band, and SUMOylated peptide is the lower band. This gel is representative of 2 independent experiments.
All figure panels have associated raw data.
Even though E1 is not tagged with FKBP, it was moderately enriched in the presence of rapamycin (Supplementary Figure 1A), likely due to interaction with E2, since enrichment correlates with E2, but not substrate, recruitment (Supplementary Figures 1A, B). To simplify the microscopy and kinetics of our system, all subsequent experiments used E1 at a saturating concentration, such that the E2~SUMO1 transfer to substrate is rate-limiting, minimizing effects of E1 enrichment (Supplementary Figure 1C). SUMO1 was not enriched in the droplets in any conditions (Supplementary Figure 1D).
We utilized a gel shift assay to determine whether rapamycin-induced enrichment into droplets modulates the reaction rate of the SUMOylation cascade. We mixed all components (E1, mCherry-FKBP-E2, FKBP-EGFP-peptide, SUMO1, FRB-polySH33 and polyPRM5) with either DMSO or rapamycin, incubated for 1 hour, and initiated the reaction with ATP. The production of SUMO-conjugated product was much faster with rapamycin than with the DMSO control (Figures 2B and C). Acceleration required both FRB-polySH33 and polyPRM5 scaffolds and also rapamycin (Supplementary Figure 2A). Thus, recruitment to FRB-polySH33 alone or the presence of droplets without recruitment does not change the reaction rate. Moreover, acceleration required that both E2 and substrate be recruited together (Figures 2D and E). Together these data indicate that the SUMOylation cascade is enhanced by rapamycin-dependent recruitment of E2 and substrate into droplets composed of the FRB-polySH33:polyPRM5 complex.
One proposed activity of condensates is to enhance reaction specificity through selective substrate enrichment. To examine this possibility we co-incubated EGFP-peptide and FKBP-EGFP-peptide in a single reaction with all other components of the SUMOylation cascade (and scaffolds) and either DMSO or rapamycin. When incubated with DMSO (no droplet recruitment), both substrates were SUMOylated equally. In the presence of rapamycin, however, FKBP-EGFP-peptide is SUMOylated much more efficiently due to its selective recruitment into the droplets (Figure 2F and Supplementary Figure 2B). Thus, specificity among otherwise equally reactive substrates can be achieved by selective recruitment into a condensate.
Reaction enhancement is substrate-dependent
We next examined a second substrate, the C-terminal domain of human RanGAP (residues 398–587, hereafter referred to as RanGAP). Unlike the PML peptide, in addition to its SUMOylation motif (ΨKXE), RanGAP also binds with high affinity to a surface of Ubc9 adjacent to the catalytic site36. This interaction affords RanGAP a relatively low KM among substrates (2.5 µM, Supplementary Figure 3B, Supplementary Table 1)37.
In contrast to the peptide substrate, SUMOylation of FKBP-RanGAP is inhibited when the protein is recruited into condensates (Figure 3A and B). This effect is likely significantly due to substrate inhibition at the concentrations achieved in the condensates (Supplementary Figures 3A, B). An additional factor that could also contribute to decreased total reaction rate upon recruitment is high substrate concentration relative to KM, resulting in reaction saturation such that condensate recruitment provides no further enhancement. Consistent with these mechanisms, inhibition was reduced when the assay was performed with a 10-fold lower total concentration of RanGAP (Supplementary Figure 3A).
Figure 3. Rate enhancement is substrate-dependent.
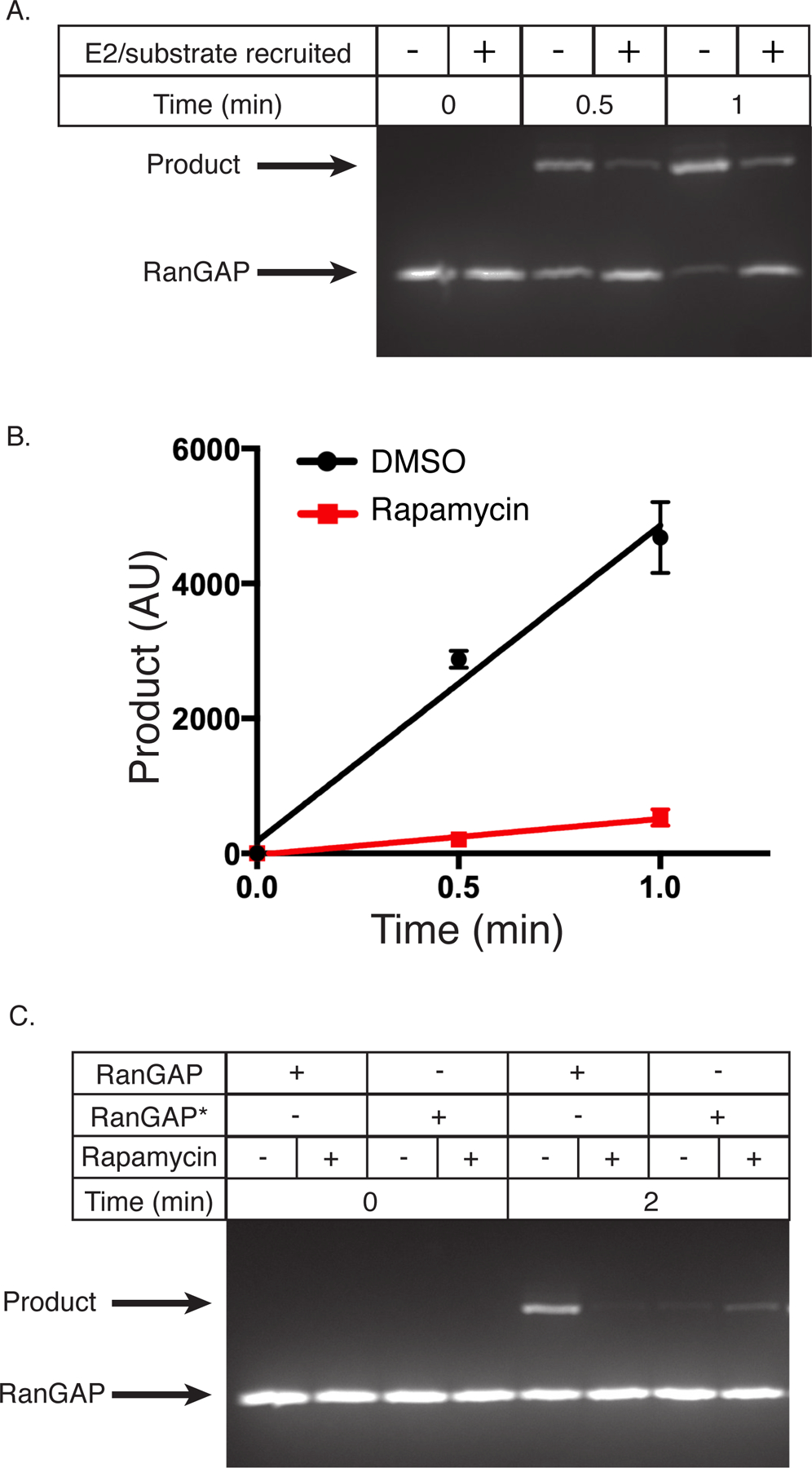
(A) Fluorescent SDS-PAGE gel depicting the production of SUMOylated substrate as a function of time with FKBP-RanGAP, FKBP-E2, and FRB-polySH33:polyPRM5 condensates with DMSO (−) or rapamycin (+). This gel is representative of 3 independent experiments.
(B) Quantification of data in panel A, showing intensity of the SUMOylated substrate band as a function of time. DMSO, black circles; rapamycin, red squares. Data are plotted as the mean and SD from n=3 independent experiments.
(C) Fluorescent SDS-PAGE gel depicting the production of SUMOylated substrate as a function of time with FKBP-RanGAP or FKBP-RanGAP*, FKBP-E2, and FRB-polySH33:polyPRM5 condensates with DMSO (−) or rapamycin (+). This gel is representative of 2 independent experiments.
All figure panels have associated raw data.
To further test this idea, we produced a RanGAP substrate with a higher KM, similar to that of the peptide substrate (~83 ± 26 µM, Supplementary Figure 3C, Supplementary Table 1), by mutating phenylalanine 562 to alanine in the Ubc9-docking surface of RanGAP36 (RanGAP*). Mutation increased KM ~60-fold (to 150 ± 70 µM) and eliminated substrate inhibition (Supplementary Figure 3D, Supplementary Table 1). The mutant also displays apparent positive cooperativity in SUMOylation (Hill coefficient of 1.4 ± 0.3 in fits to the cooperativity-modified Michaelis-Menten equation), although the mechanism underlying this effect remains unclear. When recruited to condensates along with E2, the reaction rate of this mutant was enhanced (Figure 3C). Together, these data suggest that rate enhancement is not a universal property of our condensates but is substrate-dependent. Substrates whose total concentration is low relative to KM are enhanced by recruitment with enzymes into condensates, while substrates whose concentration is high relative to KM and/or display substrate inhibition will not be affected and can even be inhibited by such recruitment.
SUMOylation is greatly accelerated in the droplet phase
We next quantified the reaction rates in the droplet and bulk phases individually. This required measurement of the total reaction rates in droplet and bulk as well as the volume of each phase.
The droplet volume is sufficiently small in our assays (see below) that it was not feasible to measure rates in droplets directly. Thus, we determined droplet rates by the difference between rate in the total phase-separated solution and that in the clarified bulk solution (Figure 4A). For each condition, we incubated paired solutions containing FRB-polySH33 and polyPRM5 (above the phase separation threshold, generating condensates) plus all components of the SUMOylation cascade: E1, mCherry-FKBP-E2, FKBP-EGFP-RanGAP*, SUMO1, and rapamycin. We removed condensates from one solution of each pair by centrifugation, transferred the supernatant to a separate tube for assay, and initiated reactions by addition of ATP (Figure 4A). SUMOylation measured in the clarified solution yields the bulk reaction rate, while the difference between the reaction rates in each pair represents the droplet rate. The clarified supernatants do not contain visible condensates as assessed by confocal fluorescence microscopy (Supplementary Figure 4). Sub-diffraction condensates in the bulk solution would artifactually decrease the droplet rate and increase the bulk rate.
Figure 4. SUMOylation is greatly accelerated in the droplet phase.
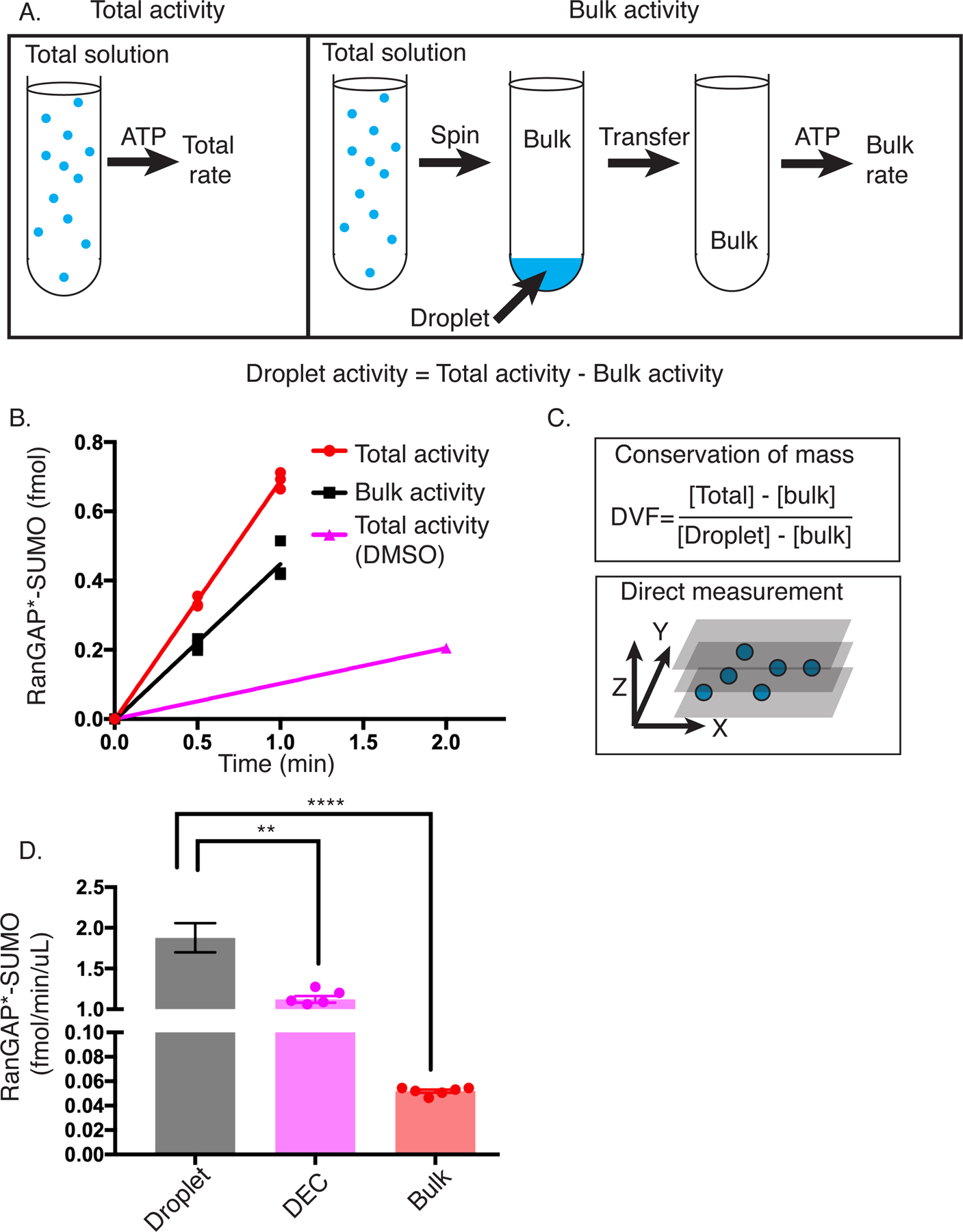
(A) Schematic of workflow to measure SUMOylation rate in the droplet phase. Total SUMOylation rate is measured by simply mixing all components and adding ATP. Bulk rate is measured by centrifuging the total mixture prior to addition of ATP to sediment droplets, transferring the supernatant to a new tube, and then initiating the reaction with ATP. The difference between Total and Bulk rate yields the Droplet rate.
(B) Representative plot showing the production of SUMOylated RanGAP* over time in the total (+rap), bulk (+rap), and total (+DMSO) solutions. Error bars on total (+rap) and bulk (+rap) represent the SEM of 3 experiments.
(C) Schematic depicting the two approaches used to calculate droplet volume fraction. Top panel shows the equation used based on conservation of mass. [ ] represent concentrations in the indicated phase measured by fluorescence imaging. Bottom panel illustrates the direct measurement approach based on confocal imaging of a three-dimensional volume.
(D) Volume-normalized rate toward RanGAP* for the droplet, droplet equivalent concentration (DEC), and bulk phases. Error bars represent the SEM from 6 independent experiments. The statistical significance was assessed by a two-tailed, unpaired Student’s t-test. ** represents a p-value < 0.01 (0.0022), and **** represents a p-value < 0.0001 (1.4 × 10−6). Droplet rate was determined from the difference between the average total reaction rate and average bulk reaction rate, with errors propagated accordingly.
Panels B and D have associated raw data.
Addition of rapamycin results in an approximately 7.6-fold increase in total rate in the solution volume (Figure 4B). Analysis of the total and bulk activities revealed that activity is distributed roughly 2:1 between the bulk and droplet compartments. Thus, the reactions are appreciably faster in the droplet phase, since the droplets account for ~33% of total activity, but only ~1% of the total volume (see below).
We next used two independent approaches to determine the precise droplet volume (Figure 4C). In the first, we used calibrated fluorescence intensities to determine the absolute concentrations of enzyme and substrate in the droplet and bulk phases (Supplementary Table 2) and used conservation of mass to determine their volumes. This approach yielded a droplet fraction of 1.3 ± 0.1 % when based on FRB-polySH33-EGFP, and 1.1 ± 0.2 % based on FKBP-EGFP-RanGAP* fluorescence, showing consistency for two components with different partition coefficients and different mechanisms of droplet enrichment. The concentrations of all components and their partition coefficients are summarized in Supplementary Table 2.
As an alternative method, we measured droplet volumes directly by imaging a large z-stack through a droplet-containing sample using a spinning disk confocal fluorescence microscope. For droplets containing the complete reaction mixture and imaged through FKBP-EGFP-RanGAP*, this procedure yielded a droplet volume fraction of 0.9 ± 0.1 %. The similarity of the values determined by the two orthogonal methods lends credence to the approaches and quantitative results. We used the conservation of mass approach in all analyses below.
Based on the total reaction rates in the droplet and bulk solutions and the known volume fractions, we calculated the reaction rate in each phase, yielding 1.8 ± 0.2 fmol/min/µl and 0.05 ± 0.001 fmol/min/µl for the droplet and bulk phases, respectively. All errors for droplet rates were propagated from uncertainties in droplet volume and total and bulk rates. Thus, under the conditions employed here, the SUMOylation reaction is accelerated ~36-fold within the droplet phase relative to the surrounding bulk phase (Figure 4D).
Condensates are more active than predicted by concentration
We next asked whether the acceleration in condensates is solely due to their higher concentrations of enzymes and substrates, or if additional factors might also modulate activity. To address this question, we carried out reactions at the concentrations of E2 and RanGAP* measured within the droplets above (Supplementary Table 2) but lacking the scaffolds. In these Droplet Equivalent Concentration (DEC) reactions, the SUMOylation rate was 1.1 ± 0.1 fmol/min/µl, a value 1.6-fold below that in the droplets (Figure 4D). Thus, while a significant portion of the reaction acceleration produced by condensation derives from the increased concentrations of enzymes and substrates, the higher condensate rate cannot be fully explained by concentration alone. The condensates impart excess activity beyond that dictated by mass action.
Excess activity is due to a scaffold-induced decrease in KM
We initially examined macromolecular crowding as a potential source of excess activity38. The combined concentration of all scaffolds, enzymes, and substrates in the condensates is ~30 mg/mL, corresponding to ~3% w/v. To mimic this condition without scaffolds, we added 3% w/v of either PEG3350 or Ficoll70 to the DEC reaction. Neither agent caused a significant change in reaction rate compared to the DEC conditions alone, suggesting that crowding is not a major contributing factor to excess activity (Figure 5A).
Figure 5. Excess activity is due to a scaffold-induced decrease in KM.
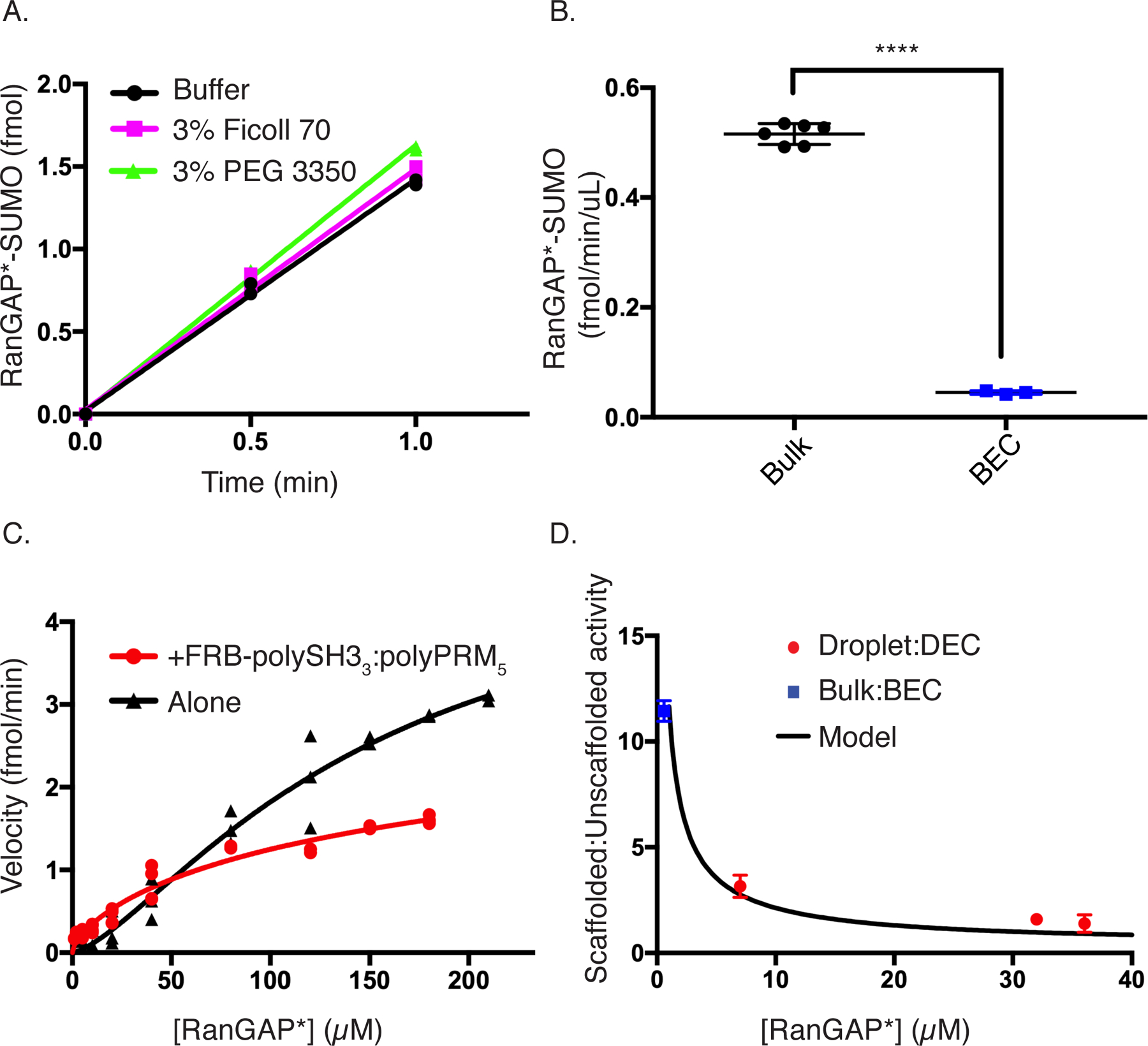
(A) Time course showing the production of SUMOylated substrate with E1, E2, RanGAP*, and SUMO1 at droplet equivalent concentrations (black circles), +3% Ficoll 70 (magenta squares), or + 3% PEG 3350 (green triangles). Each datapoint shown in duplicate.
(B) Bar chart showing the volume-normalized reaction rate of bulk (black) or bulk equivalent concentration (BEC, blue). Error bars represent the SEM from 6 experiments. Statistical significance was assessed by a two-tailed, unpaired Student’s t-test. **** represents a p-value < 0.0001 (1.3 × 10−9).
(C) Rate of production of SUMOylated RanGAP* as a function of substrate concentration with (red squares) or without (black circles; same data as shown in Supplementary Figure 3D) sub-threshold concentrations of FRB-polySH33-polyPRM5. Each symbol represents the mean and standard deviation from n=3 (<150uM) and n=2 (≥150uM) independent experiments. Points without errors bars have standard deviations too small to show.
(D) Ratio of scaffolded:unscaffolded reaction rate (black curve) calculated by dividing the fit of the red curve from the fit of the black curve in Panel C. Overlaid on this curve are the scaffolded:unscaffolded rate ratios from bulk vs BEC (blue square) and droplet vs DEC (red circles). Error bars represent SEM from a total of 6 independent measurements for the bulk:BEC point and the droplet:DEC datapoints at ~7 and 32uM. The 36uM datapoint is from 3 independent experiments.
All figure panels have associated raw data.
Another possibility is that E2 and substrate may be organized by FRB-polySH33:polyPRM5 oligomers, as in certain signaling cascades39. Since FRB-polySH33:polyPRM5 oligomers form both above and below the LLPS concentration threshold8, we explored this effect by comparing the bulk phase reaction rates with those produced by the same enzyme and substrate concentrations in the absence of scaffolds (Bulk Equivalent Concentration, BEC; Figure 5B). This showed that the scaffolds increase the reaction rate by 11.4 ± 0.5-fold (Figure 5B). As in the experiments above on droplets, this enhancement requires both scaffolds and rapamycin-mediated recruitment of both E2 and substrate to them (Supplementary Figure 5). Thus, the excess activity is not only preserved in the bulk phase, it is even more pronounced there than in the droplets.
To understand the biochemical basis of the scaffold-induced excess activity, we titrated FKBP-EGFP-RanGAP* into the SUMOylation reactions in the presence and absence of sub-LLPS threshold concentrations of FRB-polySH33 and poly-PRM5. As shown in Figure 5C, the scaffolds shift the curve to the left, indicating a decrease in apparent KM. In contrast to the positive cooperativity of the unscaffolded reaction, the scaffolded reaction displayed apparent negative cooperativity (Hill coefficient = 0.69 ± 0.1), yielding a KM value of 50 ± 15 µM (versus 150 µM for the unscaffolded reaction), with no statistically significant change in Vmax (Supplementary Table 1). This negative cooperativity may derive from the necessity of having both enzyme and substrate bound simultaneously to proximal sites in a scaffold oligomer to enhance the reaction. Such dual binding becomes less frequent at high FKBP-EGFP-RanGAP* concentrations, effectively returning the system to the unscaffolded state. We note that the scaffolded KM is likely a weighted average of a distribution of different KM values due to different oligomeric states of the scaffolds and organization of enzyme and substrate attached to them.
One consequence of the shift in KM is that the ratio of scaffolded to unscaffolded reaction rate, ([S]h/(KM,Sh + [S]h)/([S]/(KM,US + [S]) where h is the Hill coefficient, is largest (=[S]h-1*KM,US/ KM,Sh) at low concentrations and decreases to 1 as the substrate approaches saturation (Figure 5D). This behavior is qualitatively consistent with our observation that excess activity is higher in the low-concentration bulk than in the high-concentration droplets. Thus, we asked whether the excess activity in the droplet phase could also be described by a scaffold-induced change in KM from 150 µM to 50 µM. As shown in Figure 5D, for three different total concentrations of substrate (0.5, 1.0, 2.0 µM), producing droplet concentrations spanning from 7 µM to 36 µM, the excess activity (now defined as the ratio of scaffolded (droplet or bulk) to unscaffolded (DEC or BEC) rates) in the droplets and bulk fall on the same curve. Together, these data suggest that excess activity arises from changes in KM due to organization of E2 and substrate by the oligomeric FRB-polySH33:poly-PRM5 scaffold. This effect is analogous in droplet and bulk, but manifests differently due to differences in substrate concentration relative to KM in the two phases.
Activity enhancement is scaffold-specific
To learn whether these effects differ between scaffolds we repeated the substrate titration in the presence of a pentameric SH3 scaffold, FRB-polySH35 (vs FRB-polySH33 previously), and polyPRM5 below the LLPS threshold concentration. In contrast to the trimeric scaffold, the pentameric FRB-polySH35 had a more subtle effect on reaction rates, and KM and Vmax are less affected (Figure 6A, Supplementary Table 1). The two curves are virtually identical in the range of our experiments (< 40 µM substrate). The smaller effect is not due to a tethering defect, since condensates formed by polySH35:polyPRM5 recruit mCherry-FKBP-E2 and FKBP-EGFP-RANGAP* to virtually the same degree as those formed by the trimeric scaffold (1.3 vs 1.4 µM, and 32 vs 31 µM, respectively).
Figure 6. Activity enhancement is scaffold-specific.
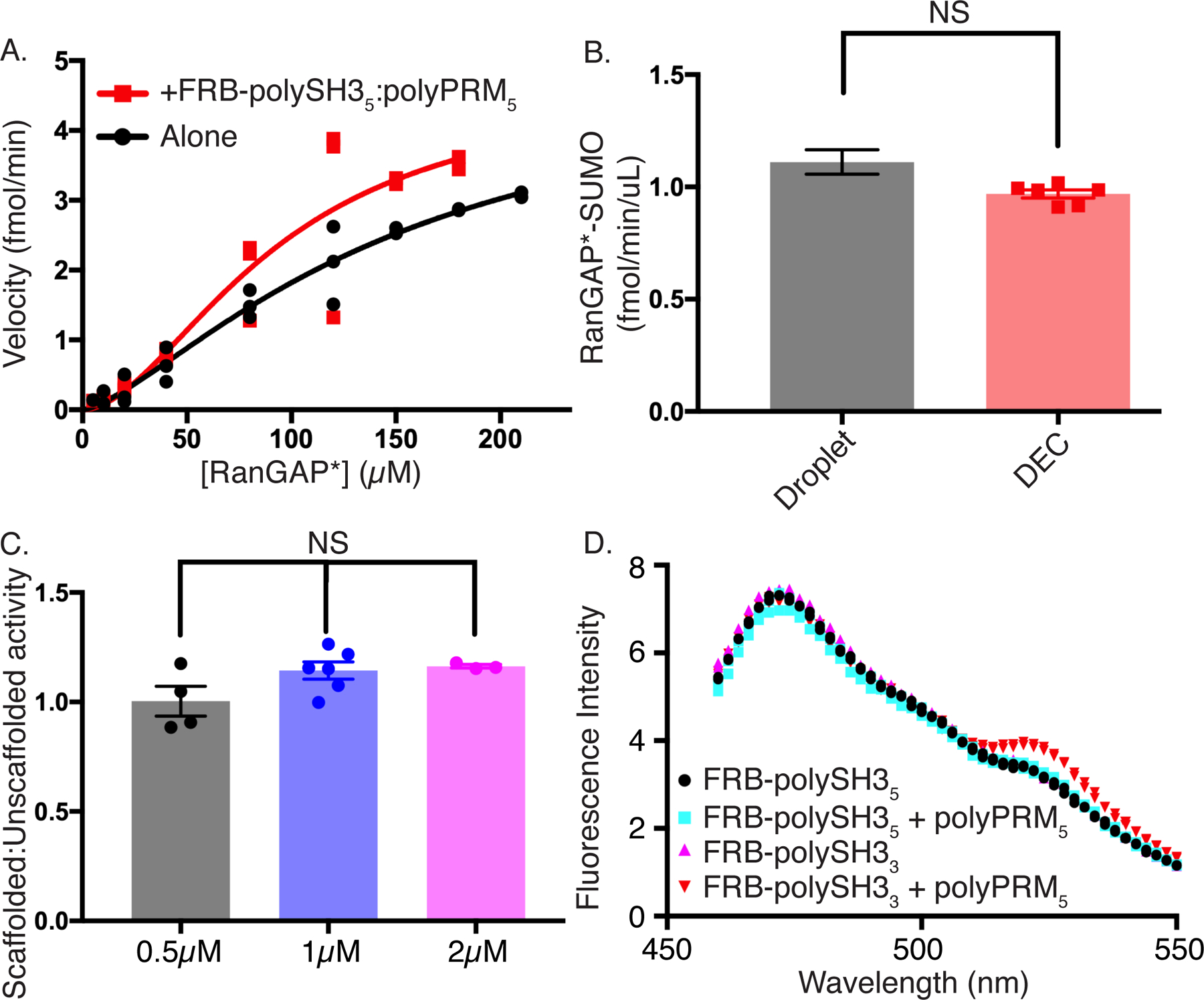
(A) Rate of production of SUMOylated RanGAP* as a function of RanGAP* concentration with (red) or without (black; same data as shown in Supplementary Figure 3D) subcritical concentrations of FRB-polySH35-polyPRM5. Each symbol represents the mean and standard deviation from n=3 (<150uM) and n=2 (≥150uM) independent experiments. Points without errors bars have standard deviations too small to show.
(B) Volume-normalized reaction rates of FRB-polySH35-polyPRM5 droplets (black) and droplet equivalent concentration (DEC, red). Error bars represent SEM from 6 independent experiments. Statistical significance was assessed by a two-tailed, unpaired Student’s t-test with a p-value cutoff of 0.05. Droplet rate was determined from the difference between the average total reaction rate and average bulk reaction rate, with errors propagated accordingly.
(C) Droplet:DEC rate ratio of FRB-polySH35-polyPRM5 droplets at total RanGAP* concentrations of 0.5 µM (black), 1 µM (blue), and 2 µM (green), corresponding to droplet concentrations of ~9 µM, ~32 µM, and ~40 µM, respectively. Error bars represent SEM from n=4, n=6, and n=3 independent experiments, respectively. Statistical significance was assessed by unpaired one way ANOVA with a p-value cutoff of 0.05.
(D) Fluorescence emission spectrum of FKBP-YPet-RanGAP* upon 445nm excitation of CyPet-FKBP-E2. Spectra recorded in the presence of FRB-polySH35 (black circles), FRB-polySH35 + polyPRM5 (cyan squares), FRB-polySH33 (magenta triangles) or FRB-polySH33 + polyPRM5 (inverted red triangles). Each point represents the mean and SD (error bars) of 2 independent experiments.
All figure panels have associated raw data.
We then examined the effects of LLPS by the pentameric scaffold system on the SUMOylation reaction. As with the trimeric scaffold, recruiting E2 and RANGAP* into the FRB-polySH35:polyPRM5 droplets substantially increased the SUMOylation rate. However, rates in droplets were identical within error to those in DEC conditions, showing no excess activity (Figure 6B). Moreover, doubling or halving the substrate concentration did not produce excess activity (Figure 6C). Thus, for the pentameric scaffold, which does not alter KM, the effects of phase separation on SUMOylation rates can be quantitatively described simply by mass action.
Together, these data further support our model in which excess activity induced by the polySH33:polyPRM5 scaffolds results from tethering-dependent changes in KM. Further, because the KM effects are scaffold-specific, they show that LLPS driven by different scaffolds can have different effects on activity, with some systems acting purely through mass action and others acting additionally to change the kinetic parameters of the reaction.
Finally, we asked whether the FRB-polySH3:polyPRM oligomers could bring E2 and substrate into spatial proximity, which could decrease the apparent KM of the SUMOylation reaction40. We tagged FKBP-E2 and FKBP-RanGAP* with CyPet and YPet, respectively, fluorescent proteins that undergo fluorescence resonance energy transfer (FRET) at distances < 100 Å41. We incubated the labeled enzyme and substrate (plus rapamycin) with either the trimeric or pentameric FRB-polySH3 scaffold below the LLPS threshold concentration in the presence or absence of polyPRM5 and measured CyPet-YPet FRET. With the pentameric FRB-polySH35 scaffold, no FRET is observed either with or without polyPRM5. In contrast, while the trimeric FRB-polySH3 scaffold does not produce FRET on its own, further addition of polyPRM5 produces a reproducible FRET signal (Figures 6D, Supplementary Figure 6A). These data suggest that the trimeric scaffold brings E2 and substrate closer together on average than the pentameric scaffold, and, thus, has a greater effect on the SUMOylation reaction.
Condensate effects decrease at high substrate concentration
To better understand the interplay between partition coefficient, scaffolding effects on KM, and substrate concentration, we modeled the change in total reaction rate upon recruitment of enzyme and substrate into condensates. Initially, we assumed an unscaffolded KM/scaffolded KM (KM,US/KM,S) ratio of 3, as observed here (Figure 5C) and equal substrate and enzyme partition coefficients. Extended Data Figure 1A illustrates how the reaction rate ratio (= total scaffolded reaction / total unscaffolded reaction) varies with substrate concentration (expressed as [S]/KM,US) and partition coefficient. For low substrate concentrations, increased partition coefficient produces higher total reaction rate, as enzyme and substrate are concentrated together in the droplets. However, the effect of partition coefficient decreases as substrate concentration grows, with a pronounced shift in behavior near [S] ~ KM,US (Extended Data Figure 1A, inset), such that when [S] >> KM,US, recruitment of the system into droplets has no effect on the total reaction rate. If only substrate has a large partition coefficient, saturation of enzyme within the droplets can cause the total rate to decrease, due to depletion of substrate in the bulk (Extended Data Figure 2).
We also plotted the reaction rate ratio as a function of partition coefficient and KM,US/KM,S at low and high substrate concentrations (Extended Data Figures 1B and C, respectively). At low substrate concentration, increasing KM,US/KM,S substantially increases the rate ratio, up to a plateau value. This increase is synergistic with that due to partition coefficient. At high substrate concentration, the rate ratio is dampened; partition coefficient has no effect, and the KM ratio has a very minor effect that quickly saturates.
Finally, we examined the ratio of droplet to bulk reaction rate as a function of partition coefficient and substrate concentration. This ratio increases strongly with partition coefficient due co-enrichment of enzyme and substrate in the droplets and co-depletion in the bulk. As with the total rate, the ratio is damped at high substrate concentrations as the enzyme becomes saturated (Extended Data Figure 3A). The fraction of total activity in the droplets parallels these trends as well (Extended Data Figure 3B).
Discussion
The engineered system we developed here captures important aspects of natural biomolecular condensates. It is composed of a small number of multivalent scaffold molecules that produce the condensate through LLPS, and a larger number of client molecules that are recruited through binding to these scaffolds13,42. Recruitment of clients into the condensate can be triggered through environmental factors (here, rapamycin). Partition coefficients are in the 2 ~ 100 range9–12. Finally, the droplet volume fraction in our system, ~1 %, is similar to that observed for natural condensates9.
Using this system, we found that recruitment to phase separated scaffolds can enhance enzymatic reactions by up to 36-fold within the condensate compared to the surrounding bulk, and ~7-fold overall in the total reaction volume. Enhancement in the droplets is achieved through two different mechanisms, mass action and a scaffold-dependent decrease in KM of the reaction, which results from scaffold-induced molecular organization. The latter effect also accounts for the scaffold-dependent increase in reaction rate of the dilute phase, which contributes to the increased total rate.
These analyses allow us to quantitatively understand the effects of recruiting the SUMOylation cascade into phase separated droplets under the conditions used here with RanGAP* as substrate. In the absence of rapamycin, the solution has a very low rate (0.01 fmol/min/µL), which is evenly distributed in solution. Upon addition of rapamycin, E2 and RanGAP* are concentrated in the droplets and depleted from the bulk. Compared to the rate without rapamycin, the rate in the bulk increases due to the reduced KM,S to 0.05 fmol/min/µL. Within the droplets, the rate is 36-fold higher than in the bulk, at 1.8 fmol/min/µL. A droplet volume of 1.1%, thus yields a total reaction rate of 0.07 fmol/min/µL (0.989*0.05 fmol/min/µL + 0.011*1.8 fmol/min/µL; Figure 4B). Rapamycin increases the rate in both phases, producing a total increase of 7.3-fold. In the absence of rapamycin, the bulk phase contributes 98.9% of the total activity, while the droplet phase contributes 1.1%. In the presence of rapamycin, the bulk phase contributes 70% of total activity, while the droplet phase contributes 30%.
Our data and modeling illustrate the ability of condensates to impart substrate specificity through several different mechanisms. First, by selectively recruiting some substrates over others, condensates can direct flux through one pathway over another (Figure 2F). Second, substrates at low concentrations relative to their KM will benefit more from condensate/scaffold recruitment than those at high concentrations (Extended Data Figures 1 and 2). For substrates that possess substrate inhibition, recruitment can be inhibitory (Figure 3) and might serve to sequester the enzyme against other substrates. Finally, we have shown that different scaffolds can impact rates through different modes, with one acting purely through mass action and another through both mass action and a decrease in the apparent KM, resulting in excess activity (Figures 5 and 6). This system contrasts with another model system where an increase in reaction rate in the droplet due to mass action was tempered by reaction inhibition due to slowed diffusion43. Thus, the nature of the scaffold, and the way individual enzymes and substrates are recruited to it, can impact its effects on reaction rates. As noted above, other kinetic effects, such as changes in cooperativity, may negatively or positively alter condensate reaction rates. Additional quantitative biochemical and cellular studies, as well as modeling, will be necessary to understand the full spectrum of behaviors enabled by recruitment of enzymatic machinery into condensates.
The overall ~7-fold reaction enhancement that we observed here is of similar magnitude to effects seen for ribozyme cleavage, RNA polymerization, formate dehydrogenation, and glucose oxidation in other engineered coacervates in vitro23–25. A separate study reported dramatic inhibition of ribozyme activity within a different condensate system44. Similarly, concentrating a ubiquitin ligase along with its substrate in condensates formed by the tumor suppressor, SPOP, did not enhance reaction rate, suggesting competion between enhancing and inhibitory factors45. This diversity suggests that different condensates with distinct physical properties and capacities for molecular interactions may differentially affect recruited enzymes. These effects are likely subject to evolutionary pressures to achieve beneficial functions. Further studies comparing natural and engineered condensates will be needed to understand the degree to which evolution has tuned the effects of condensates on enzymatic systems, and the mechanisms by which such tuning occurs.
The degree of reaction enhancement reported here is modest. However, as mentioned above, while E1, E2 and substrate were all enriched in the condensates, only the E2 reaction was enhanced, since we chose conditions that saturated the E1 reaction to simplify the analysis. In other systems where multiple elements of a pathway are enriched in a condensate and all are functioning at non-saturating conditions, the flux through the cascade should increase by the product of the individual step enhancements. Thus, one can envision quite substantial increases in flux, and also specificity, in multi-step processes. Consistent with this idea, preliminary data on condensate-mediated modification with SUMO2, which unlike SUMO1 can be multiply conjugated to produce polySUMO chains, suggest rate increases upon droplet recruitment much greater than those observed here (Supplementary Figure 7). We believe that such multi-step cascades are the most likely beneficiaries of recruitment to condensates in vivo, an idea that may explain why many condensates recruit multiple components of complex pathways46,47.
Our model system has provided evidence of sequestration, substrate specificity, and factors beyond mass action in dictating the consequences of enzyme recruitment into condensates. As genomics, proteomics, and imaging improve our understanding of condensate properties, it will be important to understand how factors such as composition and dynamics further modulate these mechanisms to control cellular biochemistry. The ability to quantitatively measure biochemical activities in natural condensates, both reconstituted in vitro and in cells, represents an exciting (and challenging) direction for future studies.
Methods
Constructs
PolyPRM5, polySH33 and polySH35 were described previously8. Ubc9, SAE1, and SAE2 are all full-length human proteins. For all experiments SUMO1 was N-terminally fused to the GFP variant, ShadowG, and C-terminally truncated after glycine 97 so that it is conjugation-competent48. The RanGAP protein used here corresponds to residues 398–587 in human RanGAP. PML peptide corresponds to residues 480–495 in human PML I. Ubc9, SAE1, SAE2, RanGAP CTD, and PML peptide substrate were each fused to FKBP12 and/or their respective fluorescent proteins using PCR. FKBP and fluorescent proteins linked together via the sequence (GGS)4. FRB (residues 2025 – 2115 of human mTOR) was synthesized by IDT and ligated to the N-termini of polySH33 and polySH35 (linked via (GGS)4). Exact amino acid sequence of each protein following proteolytic removal of tags (see below) is provided in Supplementary Table 3. Poly-PRM5, FRB-polySH33, FRB-polySH35, RanGAP CTD, and PML peptide constructs all contain an N-terminal MBP (maltose-binding protein) tag followed by a cleavage site for the TeV protease (ENLYFQG), followed by the insert, then another TeV cleavage site at the C-terminus, followed by a His6-tag. The ShadowG-SUMO1 construct contains an N-terminal His8-tag, followed by a TeV cleavage site. E2 constructs contain an N-terminal His10-tag, followed by a TeV cleavage site, then the insert, then another TeV cleavage site on the C-terminus, then a polybasic tag (RK)5. The SAE1 construct contains an N-terminal His6-tag followed by a TeV cleavage site. The SAE2 construct contains an N-terminal MBP-tag, followed by a TeV cleavage site.
Protein expression and purification
All proteins were purified similarly with slight variations. All proteins except SAE1 and SAE2 were grown in E. coli strain BL21TIR to an OD600 of ~0.8 and induced with 1 mM IPTG overnight at 18 °C. To produce the E1 heterodimer, plasmids encoding SAE1 (ampicillin resistant) and SAE2 (streptomycin resistant) were co-transformed into Rosetta (DE3) bacteria (Novagen-chloramphenicol resistant), grown to OD600 ~0.7, and induced with 1 mM IPTG overnight at 18 °C. For FRB-polySH3, RanGAP, PML peptide, and E1 proteins, overexpressing cells were lysed in buffer containing 50 mM Tris pH 8, 300 mM NaCl, 10 mM imidazole, 5 mM β-mercaptoethanol (BME). The lysate was cleared by centrifugation and the supernatant was applied to Ni NTA-agarose (Qiagen), which was washed with the same buffer. Proteins were eluted with buffer containing 50 mM Tris pH 8, 150 mM NaCl, 300 mM Imidazole, 5 mM BME. The eluate was loaded onto amylose resin (NEB), which was washed with 50 mM Tris pH 8, 50–150 mM NaCl, 1 mM DTT, and 1 mM EDTA (except E1). Proteins were eluted with 50 mM Tris pH 8, 50 mM NaCl, 1 mM DTT, 1 mM EDTA (except E1), and 50 mM maltose (Fisher). The amylose eluate was digested with TeV protease (~1:100) overnight at 4 °C, filtered (0.45 µm), and loaded onto anion exchange resin (Source15Q, GE Healthcare), and eluted with a linear gradient of NaCl (50–400 mM) in 50 mM Tris pH 8, 1 mM DTT, 1 mM EDTA (except E1) buffer. Protein containing fractions were collected, filtered, and polished using gel filtration (Superdex 75 or 200, GE Healthcare) in 50 mM Tris pH 8, 150 mM NaCl, 1 mM DTT buffer. E2 and polyPRM5 proteins were purified using Ni NTA-agarose (Qiagen) followed by cation exchange (Source15S, GE Healthcare) chromatography, TEV digestion, and cation exchange (Source15S) and gel filtration (Superdex 75 or 200) chromatographies. Lysis and Ni-NTA wash buffers were the same as above except with 500 mM NaCl. Ni-NTA elution buffer contained 300 mM imidazole pH 7, 150 mM NaCl, 5 mM BME. Cation exchange buffers were the same as anion exchange buffers above, except used 20 mM imidazole pH 7 instead of 50 mM Tris pH 8. Gel filtration buffers were the same as above. SUMO1 was purified using Ni NTA-agarose (Qiagen), dilution, digestion with TeV, and anion exchange (Source15Q) and gel filtration (Superdex 75) chromatographies. Lysis and Ni-NTA wash buffer was 50 mM Tris pH 8, 500 mM NaCl, 20 mM Imidazole, 5 mM BME. Ni-NTA elution buffer was 50 mM Tris pH 8, 50 mM NaCl, 400 mM Imidazole, 5 mM BME. Ni-NTA eluate was diluted ~5× into 25 mM Tris pH 8, 1 mM DTT buffer, then digested overnight with TeV (same as above). After filtration, the protein was separated by anion exchange chromatography, eluted using a 0–250 mM NaCl gradient. Gel filtration was same as above.
In vitro SUMOylation assay
All reactions were carried out in 100 µL (except bulk-see below) of 20 mM Tris, 110 mM potassium acetate, pH 7.5, 1 mM DTT at room temperature (~22 °C). For reactions with condensates, polyPRM5 was added last after all other components (including rapamycin) were added to delay condensate formation until all other components were thoroughly mixed. Reactions contained 90 nM E1, 100 nM E2, 1 µM RanGAP*, and 1 µM ShadowG-SUMO1, 15 µM FRB-polySH33 (or 9 µM FRB-polySH35) and 9 µM polyPRM5. Components were equilibrated for 1 hr, and then SUMOylation was initiated by addition of 1 mM ATP. Reactions included either 2 µM Rapamycin or 2% DMSO as indicated. For bulk samples, components were mixed as above, incubated at room temperature for 1 hour, centrifuged at 22 °C for 30 minutes at 21,000 g, and 50 µL was carefully transferred to a separate tube for assay. In all cases, samples were removed at the indicated timepoints (typically 0.5 – 10 minutes) and the reaction terminated by addition of an equal volume of 2X SDS PAGE loading buffer. Samples were not boiled, which allowed visualization of fluorescent proteins without stain. Samples were run on 10% SDS PAGE gels for 40 min at 240 V. Gels were either imaged directly (for fluorescent proteins) or stained with Coomassie blue and then imaged using a ChemiDoc gel imager (BioRad). Gel images were analyzed using Fiji, and band intensities (following background subtraction) were fit to extract kinetic parameters using Prism. Initial velocities were fit to either the standard Michaelis-Menten equation (V = (Vmax*[S]/KM+[S])), for PML peptide, or its variant with cooperativity (V = Vmax*[S]h/KMh+[S]h), for RanGAP* with or without scaffolds (more appropriate equation based on the F-statistic, with α > 0.05).
Microscopy
Corning 384-well clear bottom, untreated, assay plates were used for all microscopy experiments. Wells were treated with 5 M NaOH for 2 hours at room temperature, washed 15× with MilliQ (submerged to completely fill then emptied each time), blocked with 20% fatty acid free BSA (Fisher) for at least 2 hours at room temperature, washed 3–5 times with water and dried with argon. Samples were prepared identically to the SUMOylation reactions except without ATP. Fluorescence intensities were measured for pairs of proteins (E2 (mCherry) + substrate (EGFP), E2 (mCherry) + E1 (EGFP), and E2 (mCherry) + SUMO1 (ShadowG)) to avoid spectral overlap. We analogously measured fluorescence intensities in mixtures of FRB-polySH33 (0.5% FRB-polySH33-EGFP), polyPRM5 and rapamycin. Images were acquired on a spinning disk confocal microscope (Nikon) with an EMCCD camera (Andor) and a 20x objective. Six samples were imaged and five images per sample were taken to quantify fluorescence intensity within droplets. Separately, phase separated samples were centrifuged at 22,000 g for 30 minutes at 22 °C, and 50 µL were removed, transferred to adjacent wells, and imaged to give the fluorescence intensity of the bulk. A dilution series for each protein was used to generate a standard curve, which was imaged simultaneously with the droplet and bulk samples to avoid differences in microscope performance or settings. Bulk concentrations were independently measured using the same workflow but on a fluorimeter instead of a microscope; the two approaches returned the same result (within error). At each wavelength, 15 µm z-stacks (1 µm steps) were acquired for droplet images, or a single image was acquired for all other samples. Images were analyzed in MATLAB using a custom script as described previously13. Briefly, the component with largest partition coefficient was used to segment droplets and bulk in the background subtracted and flat-field corrected image. The average intensity and standard deviation were extracted for the droplets (restricted to 9 – 28 µm diameter) and bulk, and converted to concentrations based on standard curves for each fluorescent protein.
Droplet volume measurements
For direct measurements of droplet volume, we rapidly imaged a 200 µm z-stack through a droplet-containing sample using a spinning disk confocal fluorescence microscope. The images were subdivided into two 100 µm stacks. We joined each stack into a three-dimensional volume, identified droplets (3D objects counter-Fiji) and summed their volumes. Initially, small droplets were excluded by setting an intensity threshold based on the average intensity of all droplets with diameter > 2 µm. This threshold was then coupled with either a 1.3 µm or 1.0 µm diameter size threshold to identify smaller droplets. All measurements produced the same total droplet volume within error (0.9 ± 0.1 %), indicating that smaller droplets do not contribute appreciably to the volume measurement. Nevertheless, the volume determined with this approach may be somewhat lower than the true value due to undercounting of small/dim droplets. The lower stack encompassed the large droplets that had settled to the bottom of the well, while the top stack contained the small droplets that were still suspended in solution. Based on the dimensions of the well, the total solution height was approximately 5 mm, which equates to fifty 100 µm stacks. Since the second 100 µm stack represents the unsettled droplets, all the stacks except the first should be similar, so the droplet volume of the second stack was multiplied by 49 and added to the droplet volume of the first stack to give the total droplet volume in the stack. This was then divided by total volume of the stack to give the droplet volume fraction.
According to conservation of mass, the droplet volume fraction was determined from the total, droplet and bulk concentrations determined above by:
This numerator is depletion, and the denominator is the partition coefficient. By measuring the concentrations in the droplet and bulk, by knowing the total concentration, and by assuming no change in total volume upon droplet formation, we can calculate the droplet volume (as long as enough droplets are within the appropriate size range to accurately measure intensity).
FRET
All data were collected using a PTI fluorimeter with appropriate filters. Subcritical concentrations of the indicated scaffold mixtures were incubated with CyPet-FKBP-E2, FKBP-YPet-RanGAP* and rapamycin for 1 hr at 22 °C, centrifuged for 30 min at 21,000 g, 22 °C to remove droplets, and the supernatant was transferred to a fresh tube and imaged. Samples were excited at 445 nm and emissions collected at 460 – 550 nm at 2 nm intervals. Each curve was the average of two experiments.
Modeling
Modeling is described in the Supplementary Information.
Extended Data
Extended Data Figure 1. Sensitivity of enhanced condensate activity to KM, substrate concentration, and partition coefficient.
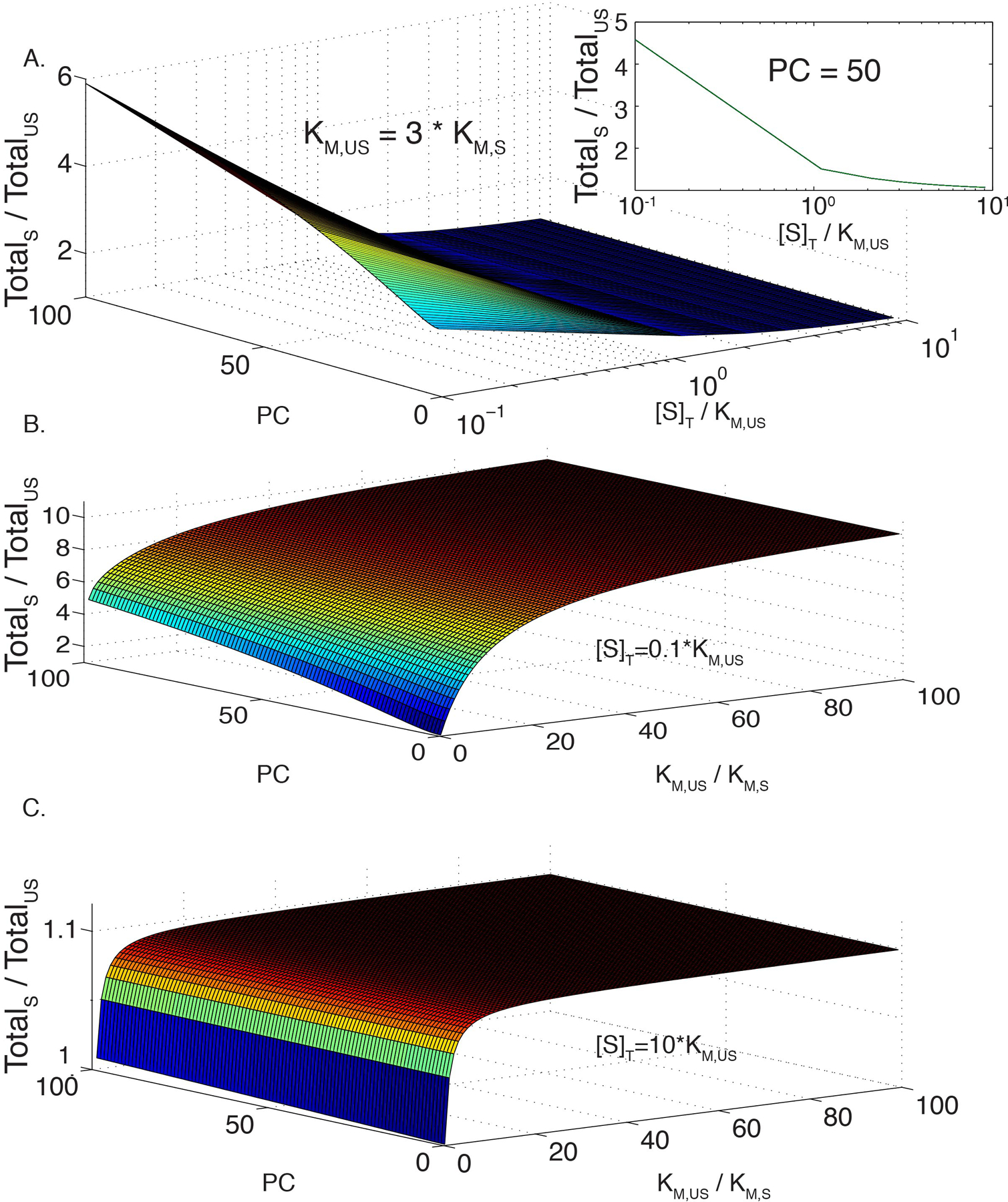
(A) Modeled ratio of total reaction rate in a phase separated solution, with and without recruitment of enzyme and substrate to the scaffold (TotalS and TotalUS, respectively, as a function of substrate concentration (plotted as [S]/KM,US) and partition coefficient (PC). Modeled for KM,US = 70 and KM,S = 17, as measured for FRB-polySH33+polyPRM5, with identical PC values for enzyme and substrate. Modeling assumes simple, hyperbolic Michaelis-Menten kinetics (see Methods). Color scale is a relative representation of the z-axis values and goes from low (blue) to high (red). Inset is a plot of TotalS:TotalUS rate as a function of substrate concentration at a fixed partition coefficient of 50.
(B) Modeled ratio of TotalS to TotalUS as a function of PC and the change in KM upon recruitment of enzyme and substrate to the scaffold, KM,US/KM,S. Total substrate concentration, [S]T, set to 0.1 * KM,US.
(C) Same as (B), except [S]T set to 10 * KM,US.
Extended Data Figure 2. Total scaffold rate can be less than total unscaffolded activity in certain regimes if enzyme partitioning is much less than substrate partitioning.
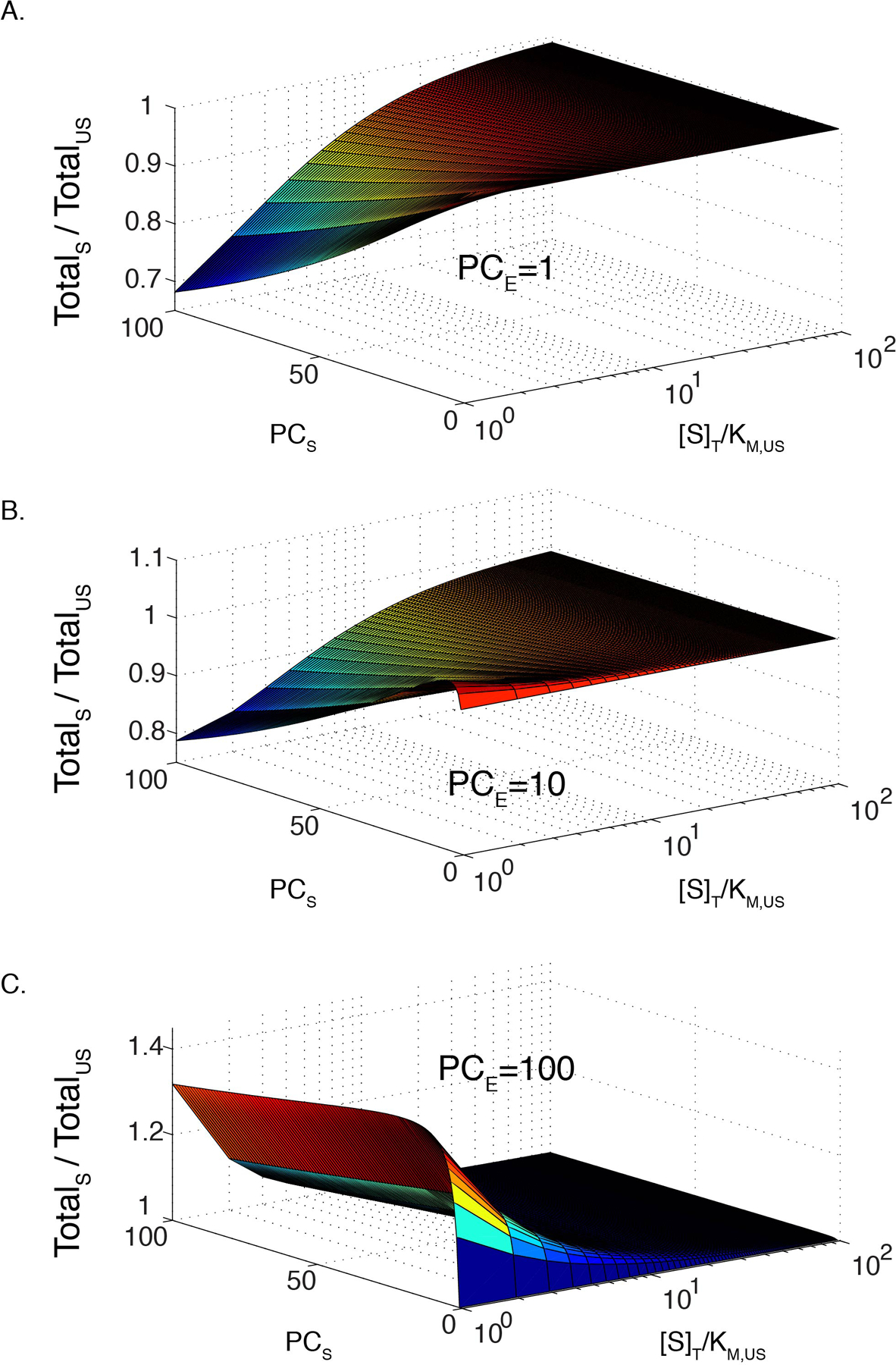
(A)-(C) Modeled ratio of total reaction rate in a phase separated solution, with and without recruitment of enzyme and substrate to the scaffold as a function of substrate concentration and substrate partition coefficient (PCS). Both reactions have KM = 70. Enzyme partitioning (PCE) is 1 (A), 10 (B), and 100 (C); enzyme concentration, [E] = 0.1[S]. Model based on 0.01 droplet volume fraction.
Extended Data Figure 3. Droplet rate increases rapidly relative to bulk as a function of partition coefficient.
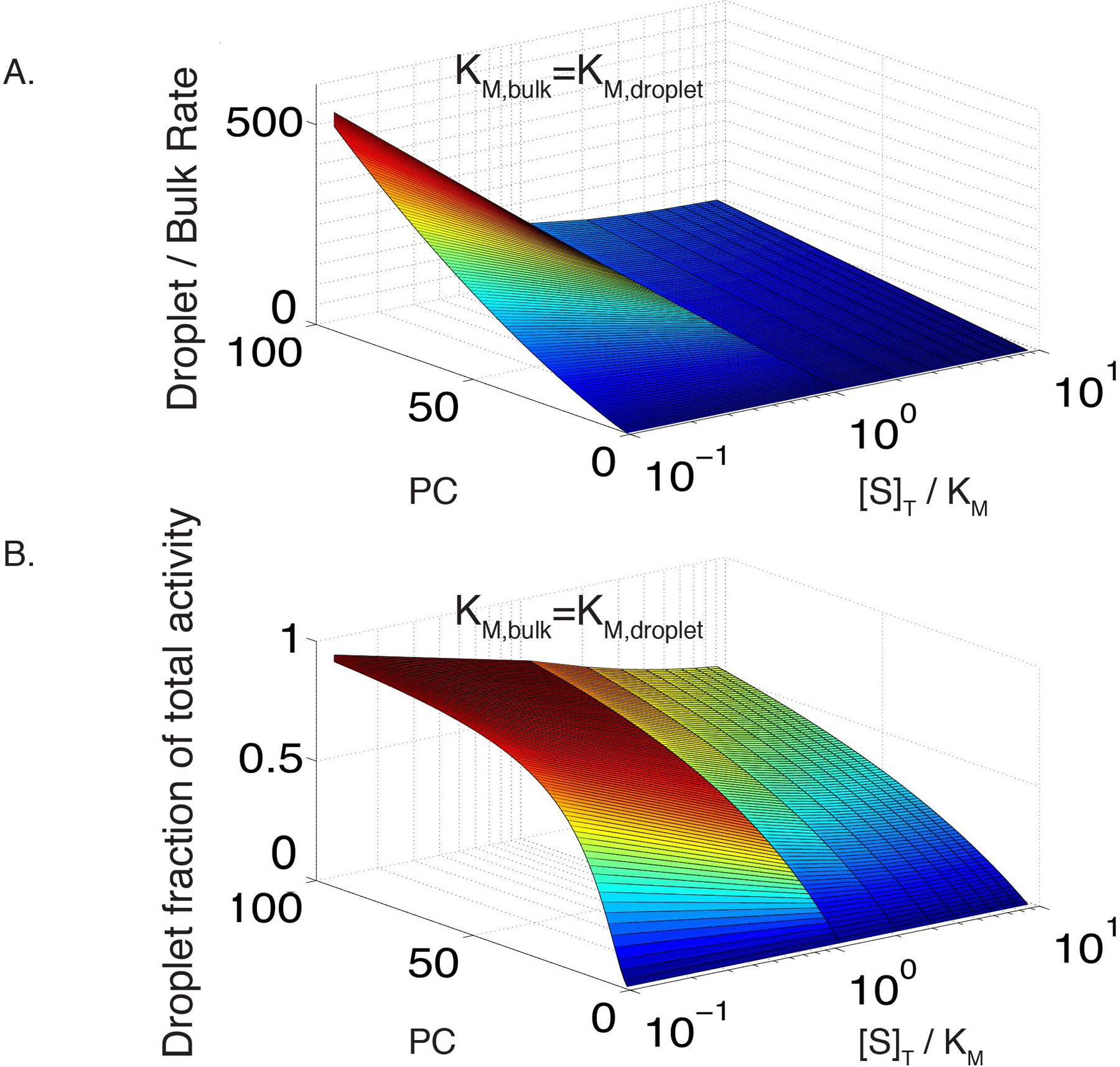
(A) Modeled ratio of droplet and bulk reaction rates as a function of substrate concentration and partition coefficient (PC). Both reactions are scaffolded and have KM = 17. Enzyme partitioning is identical to substrate partitioning, and [E] = 0.1[S].
(B) Modeled fractional activity contributed by the droplet phase as a function of substrate concentration and partition coefficient (PC). Conditions same as in (A), with a 0.01 droplet volume fraction.
Supplementary Material
Acknowledgements
We thank Salman Banani and Allyson Rice for constructs, and all members of the Rosen Lab, past and present, for helpful advice and discussions. Research was supported by the Howard Hughes Medical Institute, a Paul G. Allen Frontiers Group Distinguished Investigator Award and a grant from the Welch Foundation (I-1544 to M.K.R.).
Footnotes
Declaration of Interests
M.K.R. is a founder of Faze Medicines.
Code Availability
The Matlab code used to generate Extended Data Figure 1, 2 and 3 is provided in the Supplementary Information.
Reagent Availability
All reagents are available upon request from the authors.
Data Availability
All raw data have been deposited in the Dryad Database, with accession number doi:10.5061/dryad.sj3tx9636.
References
- 1.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298, doi: 10.1038/nrm.2017.7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin Y & Brangwynne CP Liquid phase condensation in cell physiology and disease. Science 357, doi: 10.1126/science.aaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Sheth U & Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805–808, doi: 10.1126/science.1082320 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheu-Gruttadauria J & MacRae IJ Phase Transitions in the Assembly and Function of Human miRISC. Cell 173, 946–957 e916, doi: 10.1016/j.cell.2018.02.051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su X et al. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science, doi: 10.1126/science.aad9964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ditlev JA et al. A composition-dependent molecular clutch between T cell signaling condensates and actin. eLife 8, doi: 10.7554/eLife.42695 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lafontaine DLJ, Riback JA, Bascetin R & Brangwynne CP The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol 22, 165–182, doi: 10.1038/s41580-020-0272-6 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Li P et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340, doi: 10.1038/nature10879 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing W, Muhlrad D, Parker R & Rosen MK A quantitative inventory of yeast P body proteins reveals principles of composition and specificity. eLife 9, doi: 10.7554/eLife.56525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guillen-Boixet J et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 181, 346–361 e317, doi: 10.1016/j.cell.2020.03.049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang P et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 181, 325–345 e328, doi: 10.1016/j.cell.2020.03.046 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders DW et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 181, 306–324 e328, doi: 10.1016/j.cell.2020.03.050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banani SF et al. Compositional Control of Phase-Separated Cellular Bodies. Cell 166, 651–663, doi: 10.1016/j.cell.2016.06.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyon AS, Peeples WB & Rosen MK A framework for understanding the functions of biomolecular condensates across scales. Nat Rev Mol Cell Biol 22, 215–235, doi: 10.1038/s41580-020-00303-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banjade S & Rosen MK in eLife Vol. 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Case LB, Zhang X, Ditlev JA & Rosen MK Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science 363, 1093–1097, doi: 10.1126/science.aau6313 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodruff JB et al. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell 169, 1066–1077 e1010, doi: 10.1016/j.cell.2017.05.028 (2017). [DOI] [PubMed] [Google Scholar]
- 18.King MR & Petry S. Phase separation of TPX2 enhances and spatially coordinates microtubule nucleation. Nat Commun 11, 270, doi: 10.1038/s41467-019-14087-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dewey DC, Strulson CA, Cacace DN, Bevilacqua PC & Keating CD Bioreactor droplets from liposome-stabilized all-aqueous emulsions. Nat Commun 5, 4670, doi: 10.1038/ncomms5670 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Du M & Chen ZJ DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709, doi: 10.1126/science.aat1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freeman Rosenzweig ES et al. The Eukaryotic CO2-Concentrating Organelle Is Liquid-like and Exhibits Dynamic Reorganization. Cell 171, 148–162 e119, doi: 10.1016/j.cell.2017.08.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang WYC et al. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 363, 1098–1103, doi: 10.1126/science.aau5721 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poudyal RR et al. Template-directed RNA polymerization and enhanced ribozyme catalysis inside membraneless compartments formed by coacervates. Nat Commun 10, 490, doi: 10.1038/s41467-019-08353-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love C et al. Reversible pH-Responsive Coacervate Formation in Lipid Vesicles Activates Dormant Enzymatic Reactions. Angewandte Chemie 59, 5950–5957, doi: 10.1002/anie.201914893 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kojima T & Takayama S. Membraneless Compartmentalization Facilitates Enzymatic Cascade Reactions and Reduces Substrate Inhibition. ACS Appl Mater Interfaces 10, 32782–32791, doi: 10.1021/acsami.8b07573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castellana M et al. in Nat Biotechnol Vol. 32 1011–1018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langdon EM et al. mRNA structure determines specificity of a polyQ-driven phase separation. Science 360, 922–927, doi: 10.1126/science.aar7432 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brady JP et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc Natl Acad Sci U S A 114, E8194–E8203, doi: 10.1073/pnas.1706197114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stroberg W & Schnell S. Do Cellular Condensates Accelerate Biochemical Reactions? Lessons from Microdroplet Chemistry. Biophys J 115, 3–8, doi: 10.1016/j.bpj.2018.05.023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi J, Chen J, Schreiber SL & Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242, doi: 10.1126/science.273.5272.239 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Schlatter S, Senn C & Fussenegger M. Modulation of translation-initiation in CHO-K1 cells by rapamycin-induced heterodimerization of engineered eIF4G fusion proteins. Biotechnol Bioeng 83, 210–225, doi: 10.1002/bit.10662 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Muller S, Hoege C, Pyrowolakis G & Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol 2, 202–210, doi: 10.1038/35056591 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Varejao N, Lascorz J, Li Y & Reverter D. Molecular mechanisms in SUMO conjugation. Biochemical Society transactions 48, 123–135, doi: 10.1042/BST20190357 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Lallemand-Breitenbach V & de The H. PML nuclear bodies: from architecture to function. Current opinion in cell biology 52, 154–161, doi: 10.1016/j.ceb.2018.03.011 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Melchior F. SUMO--nonclassical ubiquitin. Annual review of cell and developmental biology 16, 591–626, doi: 10.1146/annurev.cellbio.16.1.591 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Bernier-Villamor V, Sampson DA, Matunis MJ & Lima CD Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 108, 345–356, doi: 10.1016/s0092-8674(02)00630-x (2002). [DOI] [PubMed] [Google Scholar]
- 37.Coey CT et al. E2-mediated small ubiquitin-like modifier (SUMO) modification of thymine DNA glycosylase is efficient but not selective for the enzyme-product complex. J Biol Chem 289, 15810–15819, doi: 10.1074/jbc.M114.572081 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balcells C et al. Macromolecular crowding effect upon in vitro enzyme kinetics: mixed activation-diffusion control of the oxidation of NADH by pyruvate catalyzed by lactate dehydrogenase. J Phys Chem B 118, 4062–4068, doi: 10.1021/jp4118858 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Park SH, Zarrinpar A & Lim WA Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science 299, 1061–1064, doi: 10.1126/science.1076979 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Gao Y, Roberts CC, Toop A, Chang CE & Wheeldon I. Mechanisms of Enhanced Catalysis in Enzyme-DNA Nanostructures Revealed through Molecular Simulations and Experimental Analysis. Chembiochem 17, 1430–1436, doi: 10.1002/cbic.201600224 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Nguyen AW & Daugherty PS Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol 23, 355–360, doi: 10.1038/nbt1066 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Ditlev JA, Case LB & Rosen MK Who’s In and Who’s Out-Compositional Control of Biomolecular Condensates. Journal of molecular biology 430, 4666–4684, doi: 10.1016/j.jmb.2018.08.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Küffner AM et al. Acceleration of an Enzymatic Reaction in Liquid Phase Separated Compartments Based on Intrinsically Disordered Protein Domains. ChemSystemsChem 2, doi: 10.1002/syst.202000001 (2020). [DOI] [Google Scholar]
- 44.Drobot B et al. Compartmentalised RNA catalysis in membrane-free coacervate protocells. Nat Commun 9, 3643, doi: 10.1038/s41467-018-06072-w (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouchard JJ et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol Cell 72, 19–36 e18, doi: 10.1016/j.molcel.2018.08.027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Correll CC, Bartek J & Dundr M. The Nucleolus: A Multiphase Condensate Balancing Ribosome Synthesis and Translational Capacity in Health, Aging and Ribosomopathies. Cells 8, doi: 10.3390/cells8080869 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woodruff JB et al. in Science Vol. 348 808–812 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamada K et al. Characterization of the C-terminal diglycine motif of SUMO-1/3. Biosci Biotechnol Biochem 76, 1035–1037, doi: 10.1271/bbb.120019 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data have been deposited in the Dryad Database, with accession number doi:10.5061/dryad.sj3tx9636.