

Abstract
The Polb gene encodes DNA polymerase beta (Pol β), a DNA polymerase that functions in base excision repair (BER) and microhomology-mediated end-joining. The Pol β-Y265C protein exhibits low catalytic activity and fidelity, and is also deficient in microhomology-mediated end-joining. We have previously shown that the PolbY265C/+ and PolbY265C/C mice develop lupus. These mice exhibit high levels of antinuclear antibodies and severe glomerulonephritis. We also demonstrated that the low catalytic activity of the Pol β-Y265C protein resulted in accumulation of BER intermediates that lead to cell death. Debris released from dying cells in our mice could drive development of lupus. We hypothesized that deletion of the Neil1 and Ogg1 DNA glycosylases that act upstream of Pol β during BER would result in accumulation of fewer BER intermediates, resulting in less severe lupus. We found that high levels of antinuclear antibodies are present in the sera of PolbY265C/+ mice deleted of Ogg1 and Neil1 DNA glycosylases. However, these mice develop significantly less severe renal disease, most likely due to high levels of IgM in their sera.
Keywords: Systemic lupus erythematosus, Base excision repair, DNA polymerase beta, DNA glycosylase, Oxidative DNA Damage
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with an incidence of 5 to 20 per 100,000 in the US population (for our recent review see (1)). The disease presentation is heterogeneous, women are nine times more likely to develop SLE than men (for reviews see (2,3)), and lupus is 2 to 3 times more prevalent in people of Asian, Hispanic, Native American, and African ancestry than people of European ancestry. Monozygotic twin concordance is found to be as low as 25% and familial aggregation studies suggest that lupus results at least in part from genetic predisposition (4–8).
The majority of common alleles associated with a genetic predisposition to lupus map to genes that encode proteins that function in innate and adaptive immunity. Additionally, genetic variants of DNA repair genes are also linked to the development of lupus (for reviews see (1) (9)). DNA polymerase β (Pol β is a DNA polymerase that functions in base excision repair (BER) and also in V(D)J recombination and somatic hypermutation (1,10,11). The Pol β Y265C protein has a significantly decreased catalytic activity compared to the wild-type (WT) enzyme and exhibits strong mutator activity both in vitro and in vivo (12–14). In our previous work, we showed that expression of the Pol β-Y265C protein, encoded by a genetic variant of the Polb gene, leads to development of lupus in mice (15). Both the homozygous and heterozygous Polb mice, PolbY265C/C and PolbY265C/+, respectively, developed increased levels of antinuclear antibodies (ANA) compared to PolbY265+/+ (wild-type) mice and also significantly more severe renal disease, with immune complexes forming in kidneys. The complementarity determining region 3 (CDR3) of the immunoglobulin heavy (IgH) chain is significantly shorter in the PolbY265C/C versus Polb+/+, likely as a result of aberrant VDJ recombination (10,15). In addition, somatic hypermutation levels are significantly increased in the PolbY265C/C versus Polb+/+ mice.
During BER, a variety of DNA glycosylases, some with overlapping substrate specificities, recognize and remove damaged bases (for a review see (16)). Bifunctional DNA glycosylases with associated lyase activities also cleave the DNA backbone, followed by end remodeling that generates a 3’-OH and 5’-P. Pol β then fills in the resulting single nucleotide gap and the X-ray cross-complimenting 1 (XRCC1)-ligase IIIα complex seals the nick.
Antinuclear antibodies are thought to arise as a result of the accumulation of antigens, many of which are generated in response to dying cells. We previously showed that mouse embryo fibroblasts (MEFs) derived from the PolbY265C/C mice exhibited decreased survival and increased levels of apoptosis compared to Polb+/+ mice when grown at 20% oxygen (17). This is likely a result of the compromised ability of the Pol β Y265C DNA polymerase to fill single nucleotide gaps after removal of modified bases from the DNA of the MEFs or from its inability to fill gaps during microhomology-mediated end-joining (10). Double-strand breaks are then generated when replication forks encounter gaps in DNA, and if not rescued, lead to cell death. Therefore, we reasoned that deficiencies in DNA glycosylases that function upstream from Pol β gap-filling activity may lead to fewer dying cells, lower levels of ANA, and less severe kidney disease in the PolbY265C/C and PolbY265C/+ mice.
The 8-oxoguanine DNA glycosylase Ogg1 recognizes and removes 8-oxoguanine (8-oxoG) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) (18–24). Neil1 recognizes and removes predominantly 4,6-diamino-5-formamidopyrimidine (FapyA) and FapyG and some oxidized pyrimidines (25) (for a comprehensive review see (16). In the study described here, we show that PolbY265C/+ Ogg1−/−Neil1−/− mice produce higher levels of ANA compared to the Polb+/+ Ogg1−/− Neil1−/− mice. Strikingly, we observe less severe renal disease in the PolbY265C/+ Ogg1−/−Neil1−/− mice compared to PolbY265C/+ Ogg1+/+Neil1+/+ mice, likely as a result of high levels of IgM.
2. Materials and Methods
2.1. Anti-nuclear antibodies (ANA)
ANA was measured by coating human Hep-2 cells fixed slides (Antibodies Inc.) with mouse serum and incubating with an Alexa 488 goat-anti-mouse IgG secondary antibody (Invitrogen) as per manufacturer’s instructions. Coated slides were mounted with ProLong Gold anti-fade reagent (Invitrogen) and digitally photographed with a Nikon EVOS fluorescence microscope. Pixel Fluorescent intensity was measured by ImageJ® software.
2.2. Scoring of kidney pathology
The severity of kidney lesions was scored as previously published (Senejani et al., 2014) by characterization of mesangial thickening, mesangial cellularity, glomerular enlargement, segmental tuft atrophy, appearance of glomerular crescents, Bowman’s capsular fibrosis, glomerulosclerosis, perivascular round cell infiltration, tubular epithelial atrophy or proliferation, tubular dilation or casts. Each of these characteristics receive a score of 1, 2 or 3 for normal, mild, moderate, or severe pathology and were added to generate a final score. Scoring was performed by a board-certified veterinary pathologist who was blinded to the genotype of the mice. Characterization of IgG deposition was performed as described (15). The slides were scored blindly by two observers. Characterization of IgG deposition was performed as described (15).
2.3. Enzyme-linked immunosorbent assay (ELISA)
ELISAs were performed to determine the serum levels of total IgM and IgG in mice. Briefly, 96-well plates were coated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgM (1 :1000 dilution) overnight or IgG (1: 2000 dilution) for 2 hr. Plates were blocked with 1% bovine serum albumin (BSA) for 1 h to 2 h at room temperature and washed twice with wash solution (PBS, 1%BSA). This was followed by the addition of standards or serum samples to respective wells and incubated for 1 h to 2 h at 4°C. All the wells were washed four times with wash buffer for 10 min each and TMB ELISA substrate (ab171523, Abcam) was added to facilitate the detection as per manufacturer’s recommendations. The reaction was stopped using Stop solution (ab171529, Abcam) and data was acquired using a Spectra Max 340 ELISA plate reader (Molecular Devices) at the optical density of 450 nm (OD450).
2.4. Germinal center (GC) architecture
Cryosections were fixed in prechilled acetone for 20 min. Slides were then washed twice with 1x PBS for 10 min followed by blocking (3% w/v bovine serum albumin (BSA) and 0.1% v/v Tween 20 in PBS) for 20 min. This was followed by addition of primary antibodies that were diluted in washing solution (1% w/v BSA and 0.1% v/v Tween 20 in PBS). Biotinylated peanut agglutinin (Vector Labs, B-1075) was diluted 1:500, FITC rabbit anti-mouse IgD (eBioscience, 11-5993-82) was diluted 1:200, and anti-mouse CD4 (eBioscience, 15-0041-81) was diluted 1:300. Samples were incubated in a humidified chamber at room temperature for 2 h and then washed twice with washing solution for 5 min each wash. Next, a secondary antibody, streptavidin Alexa Fluor 555 (Thermo Fischer Scientific, S32355), diluted 1:500 in washing solution, was added and incubated in a humidified chamber at room temperature for 1 hour. Samples were washed three times with washing solution for 10 min for each wash. Finally, 5 uL of prolong gold anti-fade mountant (P36930) was used to mount each sample. Slides were stored at −20 °C until images were captured.
2.5. Confocal microscopy
Confocal images were captured using a Nikon Ti-E inverted spinning disk confocal microscope (Yale west campus imaging core facility, CT). Images were captured using three different lasers; 488 nm, 561 nm, and 647 nm lasers with the laser powers of 55%, 25% and 40%, respectively. Images were saved in a tagged image file format (TIF) and were further processed in ImageJ® to reduce background and assign a color to each channel.
2.6. TUNEL assay
DNA fragmentation ApopTag Fluorescein In Situ Apoptosis Detection Kit (S7110) was used to detect TUNEL-positive cells in paraffin-embedded kidneys as per manufacturer’s recommendations. Briefly, paraffin-embedded kidney sections were subjected to deparaffinization with xylene (5 min) followed by gradually lowering concentrations of ethanol (absolute, 95% and 70% for 5 min each) and distilled water in coplin jars. Tissue sections were then treated with proteinkinase-K for 15 min at room temperature for antigen retrieval and washed with PBS. This was followed by blocking sections for 1hr with blocking solution followed by adding TdT, antidigoxigeni-fluorescein and were mounted with ProLong Gold anti-fade reagent. Images were captured with Nikon EVOS fluorescence microscope. Number of TUNEL-positive cells were counted throughout the tissue per each field and the values were expressed as a percentage of total number of cells in the field from DAPI staining. TUNEL-positive cells in spleens were identified as previously described (15).
2.7. Measurement of DNA base lesions in kidneys
Genomic DNA was isolated from kidneys using the Qiagen Blood and Cell Culture DNA Maxi kit without the use of phenol as per the manufacturers’ instructions. The eluted genomic DNA was ethanol-precipitated then washed 3 times with ethanol. Ethanol was removed, and the DNA was dissolved in water for 18 h at 4 °C, and then quantified using an absorption spectrophotometer (absorption of 1 = 50 μg DNA). Aliquots (50 μg) of the DNA samples were dried in a SpeedVac under vacuum. DNA samples were dissolved in 50 μL of an incubation buffer consisting of 50 mmol/L phosphate buffer (pH 7.4), 100 mmol/L KCl, 1 mmol/L EDTA and 0.1 mmol/L dithiothreitol. Aliquots of FapyA-13C,15N2, FapyG-13C,15N2, 8-OH-A-13C,15N2, 8-oxoG-15N5, and 5-OH-C-13C,15N2 were added as internal standards, which are a part of the NIST (National Institute of Standards and Technology) Standard Reference Material 2396 Oxidative DNA Damage Mass Spectrometry Standards (NIST SRM 2396) (for details see http://www.nist.gov/srm/index.cfm and https://www-s.nist.gov/srmors/view_detail.cfm?srm=2396). Subsequently, they were incubated with 1 μg of E.coli Fpg protein and 1 μg of E. coli Nth protein at 37 °C for 1 h to release the modified bases from DNA. For all measurements, 3 independently prepared DNA samples were used. An aliquot of 200 μL ethanol was added to precipitate DNA. After centrifugation, the supernatant fractions were separated, lyophilized and trimethylsilylated. Derivatized samples were analyzed by GC-MS/MS using multiple reaction monitoring (MRM) as described previously (26–28).
2.8. Isolation and culture of splenic B cells
Single-cell suspensions of murine splenocytes were obtained by mechanically digesting tissue between two frosted glass slides and passing it through 70 μm nylon mesh filters. Splenocyte suspensions were then washed with PBS, centrifuged and red blood cells (RBC) were lysed using 1X RBC lysis buffer made from 10X (TNB-4300-L100, TONBO Biosciences, CA). To obtain untouched B cells, CD43-expressing B cells and non-B cells were magnetically labeled and removed using mouse B cell isolation kit (MACS, Miltenyi, 130-090-862) according to manufacturer’s protocol. Briefly, single-cell suspensions were incubated with a cocktail of biotin-conjugated antibodies (CD43, CD4 and Ter-119) and anti-biotin MicroBeads. Cells were then washed with MACS buffer on LS separation columns. Untouched B cells were collected in the flow through of the columns. Purified B cells were cultured in RPMI 1640 medium (SH3002701, HyClone) supplemented with 2 mM L-glutamine, 5 × 10−5 M 2-mercaptoethanol, 1 mM sodium pyruvate and 10% fetal bovine serum (100–500, GeminiBio). B cells were labeled with 0.5 μM carboxyfluorescein diacetate-succinimyl ester (CFSE) (43801, Biolegend) before culture to observe cell division. Cells were cultured at a density of 0.5 × 106 cells per ml in the presence of 25 ng per ml of lipopolysaccharide (LPS) and 10 ng per ml of purified anti-mouse interleukin-4 (IL4) (RUO, BD Pharmingen) for 64 hr.
2.9. Flow Cytometry
Stimulated B cells were harvested at 64 hr and stained for surface expression of live/dead fixable aqua dead cell stain (Invitrogen, L34957), pacific blue anti-mouse CD45R (RUO, 558108, BD Pharmingen), PE-F(ab’)2 anti-mouse IgG1 (1072-09, SouthernBiotech) in the presence of TruStain FcX blocking antibody (anti-mouse CD16/32, BioLegend) and 7-AAD staining solution (420403, BioLegend). Samples were run on a STD-13 benchtop flow cytometer (Yale flow cytometry core facility, Yale School of Medicine, CT). A total of one million events were collected for each sample. Data were analyzed using FlowJo 10.5.3.
2.10. Comet assay
Equal numbers of cells (4 × 105) from early passages of mouse embryo fibroblasts (P0 or P1) were plated onto a 6-cm plate in the presence of DMEM containing 10% v/v FBS and 1x penicillin-streptomycin. After 1 day, the cells were incubated with media containing 25 μM menadione for 1 hr and subsequently recovered in fresh media for 0, 1, or 4 hr. After treatment and recovery, the cells were prepared and analyzed immediately using Cometslides (Trevigen, 4250–200-03). Image analysis of 100 to 125 cells from two to three littermates of each genotype was performed using CometScore software (TriTek).
2.11. Survival Assay
Approximately 100,000 MEFs were seeded into 6-well plates. The next day, media was removed and replaced with different concentrations of menadione (SelleckChem cat. # S1949). After 1 hr of treatment, the menadione was removed and replaced with growth media. The MEFs were counted 5 days after treatment.
2.12. Western Blot Analysis
Approximately 125,000 cells were seeded into 6-well plates. The next day, media was removed and replaced with 0, 20, 40 uM of menadione for 1 hr. The cells were then recovered for 3 hrs in media without menadione and lysed with Laemmli buffer. The lysates were resolved on a 10% SDS polyacrylamide gel and blotted onto a 0.2 uM pore nitrocellulose membrane. The blots were then probed with the following antibodies: histone H3 (Abcam, Ab18521), γ-H2AX (CST, 9718S), anti-rabbit (Cytiva, NA9340).
2.13. Statistical analysis
All statistical analyses were performed using GraphPad Prism software. Specific information about the analysis can be found in the Figure legends.
3. Results
3.1. Overview
The PolbY265C/C and PolbY265C/+ mice develop lupus as characterized by high levels of ANA and renal disease, including immune complex formation in the kidneys (15). The development of lupus could result from the aberrant immune repertoire of these mice. Alternatively, the mice may develop lupus as a result of the presence of high levels of antigens released by dying cells. Both mechanisms could underlie disease development and are not mutually exclusive. We surmised that the catalytically compromised Pol β Y265C protein would be significantly reduced in its ability to fill single nucleotide gaps arising from the removal of oxidative DNA damage by DNA glycosylases, leading to the accumulation of BER intermediates and cell death in mouse tissues. Therefore, we crossed the PolbY265C/+ mice with Ogg1−/− Neil1−/− mice, reasoning that deletion of these DNA glycosylases would ultimately result in the accumulation of fewer BER intermediates and perhaps ameliorate lupus that results from expression of the Pol β Y265C protein.
3.2. The PolbY265C/C Ogg1−/− Neil1−/− mice exhibit perinatal mortality
We crossed PolbY265C/+ Ogg1−/− Neil1−/− mice with each other to generate PolbY265C/C Ogg1−/− Neil1−/−, PolbY265C/+ Ogg1−/− Neil1−/−, and Polb+/+ Ogg1−/− Neil1−/− mice. As shown in Table 1, the expected numbers of mice of each genotype were born, but after two days the majority of PolbY265C/C Ogg1−/− Neil1−/− had died, with only one surviving mouse instead of the expected number of 17. These results demonstrate that the absence of the Ogg1 and Neil1 DNA glycosylases combined with a catalytically compromised Pol β protein results in perinatal lethality.
Table 1.
The PolBc/c, Ogg1−/−, Neil1−/− mice die shortly after birth.
| Number of mice | ||||||
|---|---|---|---|---|---|---|
| Polb +/+ , Ogg1 −/− , Neil1 −/− | PolB +/c , Ogg1 −/− , Neil1 −/− | PolB c/c , Ogg1 −/− , Neil1 −/− | ||||
| Expected | Observed | Expected | Observed | Expected | Observed | |
| Born (n=75) | 19 | 18 | 38 | 40 | 19 | 17 |
| Day two | 19 | 18 | 38 | 40 | 19 | 1* |
Number of expected and observed mice from 12 crosses of PolB+/c, Ogg1−/−, Neil1−/− x PolB+/c, Ogg1−/−, Neil1−/− parents. All pups were born at expected Mendelian ratios. However, within the first 24 hours almost all homozyogus mice died.
One PolBc/c, Ogg1−/−, Neil1−/− mouse survived.
3.3. The PolbY265C/+ Ogg1−/− Neil1−/− exhibit sensitivity to oxidative DNA damage.
MEFs isolated from the mice were treated with various concentrations of menadione for 1 h. The media was then replaced with fresh media and the cells were counted five days later. As shown in Figure 1, MEFs isolated from the PolbY265C/+ Ogg1−/− Neil1−/− mice exhibited greater sensitivity to menadione, an agent that induces oxidative damage, compared to MEFs isolated from the Polb+/+ Ogg1−/− Neil1−/−, Polb+/+ Ogg1+/+ Neil1+/+, and PolbC/+ Ogg1+/+ Neil1+/+ mice. This result suggests that the inability to initiate BER with the OGG1 and NEIL1 DNA glycosylases combined with the slow gap-filling of Pol β-Y265C polymerase results in increased cell death, perhaps as a result of a defective BER process.
Figure 1. MEFs isolated from the PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) mice are sensitive to menadione.
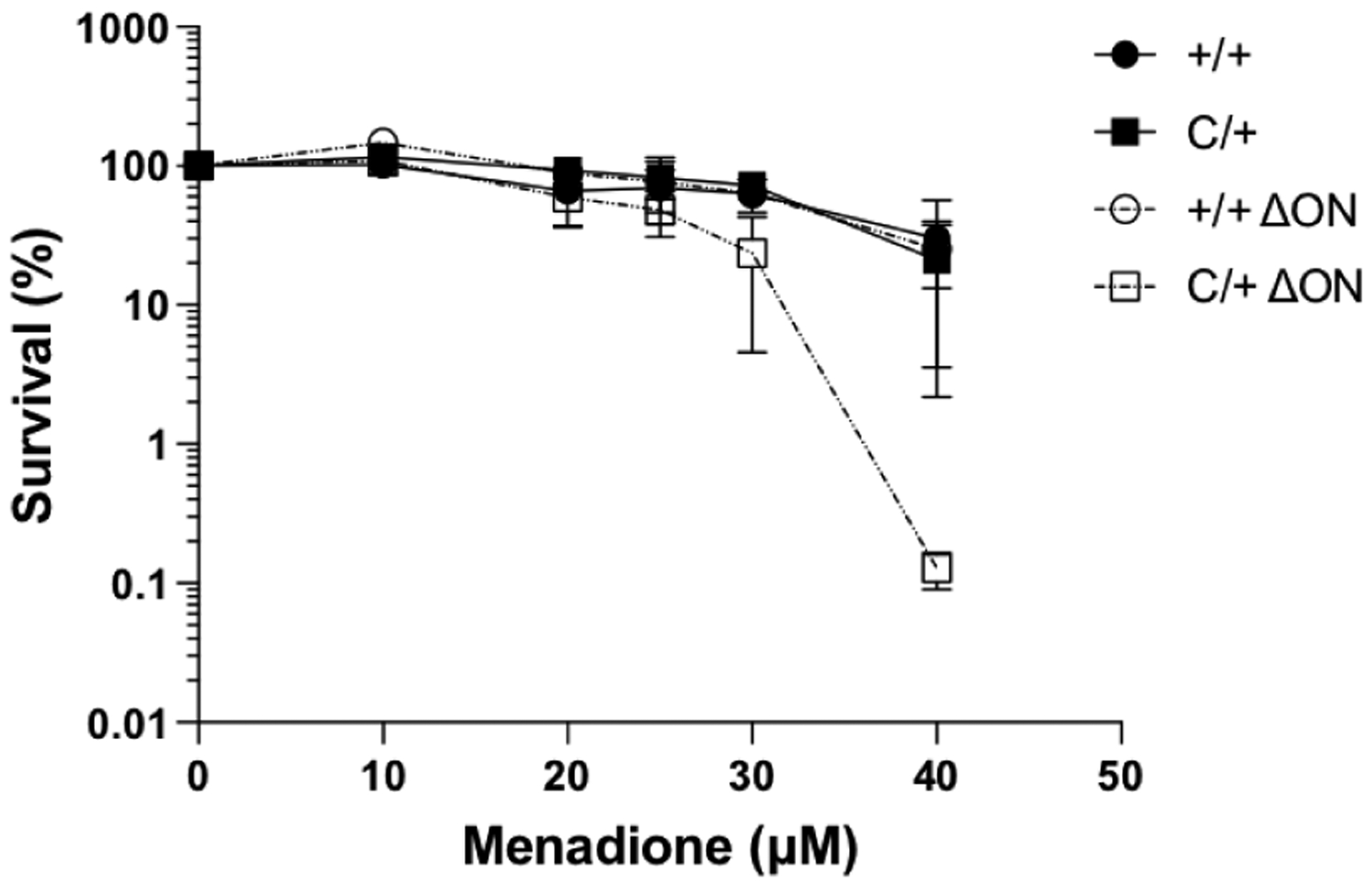
The cell survival assay was conducted as described in Methods.
3.4. Fewer Single-Strand Breaks in PolbY265C/+ Ogg1−/− Neil1−/− mouse embryo fibroblasts.
We isolated MEFs from the mice, treated them with menadione to generate oxidative damage, and monitored the levels of single-strand breaks using the alkaline comet assay. MEFs isolated from the PolbY265C/+ Ogg1−/− Neil1−/− and Polb+/+ Ogg1−/− Neil1−/− mice exhibited significantly lower alkaline comet Olive moments compared to Polb+/+ Ogg1+/+ Neil1+/+ and PolbY265C/+ Ogg1+/+ Neil1+/+ just after treatment with menadione (Figure 2). Although the Olive moments were similar in the Polb+/+ Ogg1−/− Neil1−/− and Polb+/+ Ogg1+/+ Neil1+/+ MEFs after four hours of recovery, the PolbY265C/+ Ogg1−/− Neil1−/− MEFs exhibited significantly lower Olive moments than the PolbY265C/+ Ogg1+/+ Neil1+/+ MEFs. This finding suggests that deletion of the Ogg1 and Neil1 DNA glycosylases results in fewer single-strand breaks in MEFs when the PolbY265C/+ cells are treated with an oxidizing agent. We also characterized levels of γH2AX, as an indication of the presence of double-strand breaks (DSBs), after treatment of cells with various concentrations of menadione followed by recovery. After treatment with 30 μM menadione followed by 3 hours of recovery, all of the MEFs cells exhibited elevated levels of γH2AX, with significantly elevated levels exhibited by the treated PolbY265C/+ Ogg1+/+ Neil1+/+ MEFs compared to untreated controls. MEFs isolated from the PolbY265C/+ Ogg1+/+ Neil1+/+ mice that were treated with 30 μM menadione and allowed to recover for 3 h exhibited significantly elevated levels of γH2AX compared to treated MEFs isolated from the Polb+/+ Ogg1+/+ Neil1+/+, the Polb+/+ Ogg1−/− Neil1−/−, and the PolbY265C/+ Ogg1−/− Neil1−/− mice (Figure 3 A, B). This indicates that fewer DSBs are present in the PolbY265C/+ MEFs on the Ogg1−/− Neil1−/− genetic background.
Figure 2. Alkaline DNA Olive moments for Polb+/+ Ogg1+/+ Neil1+/+ (+/+), Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON), PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+), and Polb+/+ Ogg1−/− Neil1−/− (C/+ ΔON) MEFs treated with menadione.

Single-strand breaks resulting from menadione treatment from were visualized with PicoGreen and quantified using CometScore (TriTeck) software. +/+ and C/+ MEFs accumulate significantly more single-strand breaks with compared to +/+ ΔON and C/+ ΔON. T-tests were used to determine if the means of each of the two groups were significantly different from each other.
Figure 3. MEFs isolated from the PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+) mice have high levels of double-strand breaks after three hours of recovery from menadione treatment.
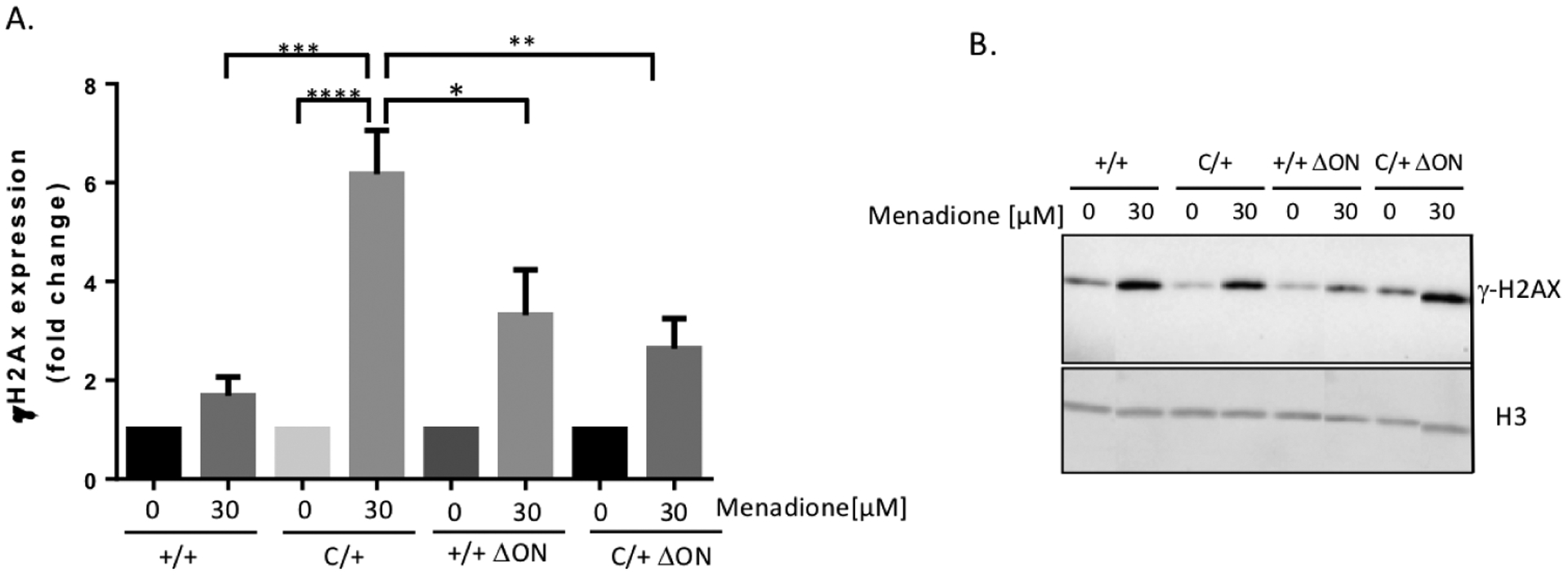
Cells were treated or not with 30 μM menadione for 1 h, followed by recovery in fresh media for 3 hours. Western blot analysis was then performed as described in Methods. Statistical analysis was performed in Graph-Pad Prism using 1-way ANOVA. A. Quantification of levels of γH2AX from 3 experiments. The fold-change is relative to untreated control. B. A representative western blot is shown.
3.5. The PolbY265C/+ Ogg1−/− Neil1−/− mice develop high levels of ANA
We aged the PolbY265C/+ Ogg1−/− Neil1−/− and the Polb+/+ Ogg1−/− Neil1−/− mice and monitored sera collected from the mice for ANA as described (15). Interestingly, the PolbY265C/+ Ogg1−/− Neil1−/− mice developed significantly higher levels of ANA than the Polb+/+ Ogg1−/− Neil1−/− controls at both 12 and 18 months of age (Figure 4A), similar to our previous findings with the PolbY265C/+ (15). Importantly, we observed increased numbers of germinal centers/field in the spleens of the PolbY265C/+ Ogg1−/− Neil1−/− versus Polb+/+ Ogg1−/− Neil1−/− mice, in line with increased levels of ANA (Figure 4B,C).
Figure 4. Anti-nuclear antibodies present in Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON) and PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) mouse serum.
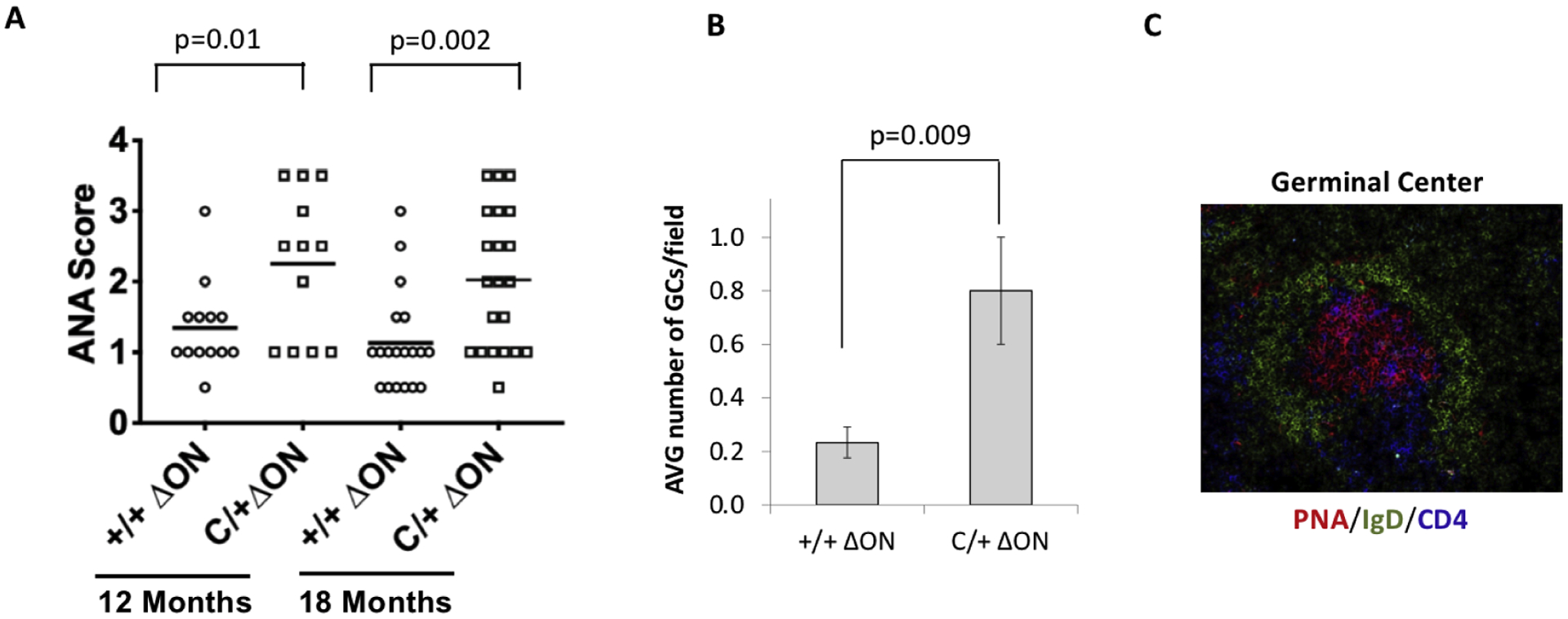
A. ANA was visualized with fluorescein (FITC)–conjugated goat anti–mouse IgG and scored from 1 (low) to 4 (high). C/+ ΔON serum contained higher levels of ANA with respect to +/+ ΔON. A t-test was used to determine if the means of the two groups were significantly different from each other. B. Quantification of germinal centers per field as described in methods. At least 50 fields were imaged. C. An example of the germinal center from the spleens of PolbY265C/+ Ogg1−/− Neil1−/− mice. PNA stains activated B cells, IgD labels immature B cells, and CD4 labels T cells.
Immunoglobulin M (IgM) is produced as a first response to an antigen. Class switch recombination (CSR) CSR converts IgM to IgG. To determine if CSR may be altered in our mice, we measured the levels of IgM and IgG in the sera of the mice. We discovered that the PolbY265C/+ Ogg1−/− Neil1−/− mice had significantly increased levels of IgM when compared to the Polb+/+ Ogg1−/− Neil1−/− (p=0.003), PolbY265C/+ Ogg1+/+ Neil1+/+ (p=0.001) and Polb+/+ Ogg1+/+ Neil1+/+ (p<0.0001) mice. (Figure 5A). Levels of IgG in each of the mouse strains were similar, but significantly lower than what is found in the sera of MRL/lpr mice, which is a lupus-prone mouse model (Figure 5B). Our results suggest that the presence of the Polb-Y265C allele is sufficient for the development of ANA and that deletion of the Ogg1 and Neil1 DNA glycosylases does not influence the levels of ANA in our mice. In addition, our results suggest that class switch recombination may be impacted by deletion of the Ogg1 and Neil1 DNA glycosylases in the presence of the Polb Y265C allele.
Figure 5. Serum from PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) mice has high levels of IgM.
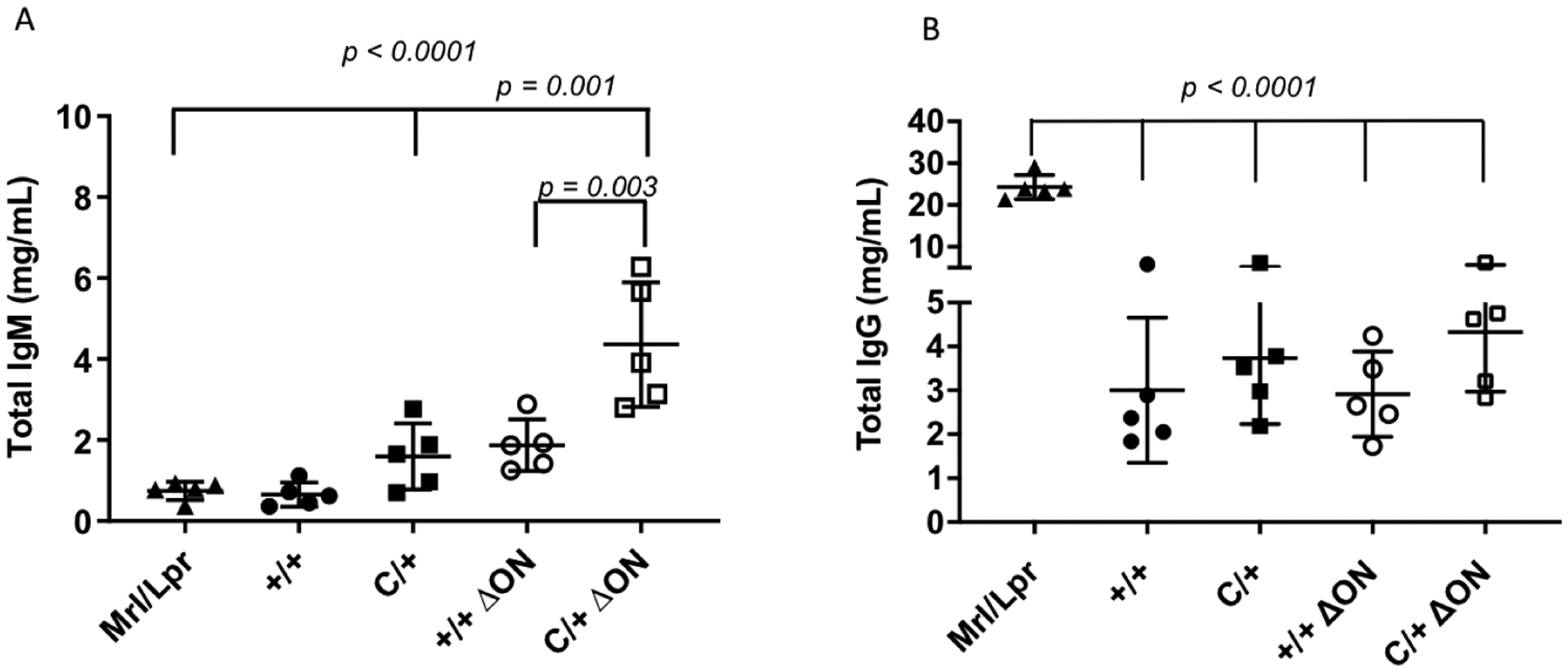
Elisa assays were used to quantify the levels of IgM and IgG in the sera. A. IgM. B. IgG. Serum from an MRL/lpr mouse is used as a control. A t-test was used to determine if the means of the two groups were significantly different from each other.
3.6. Less severe renal disease in the PolbY265C/+ Ogg1−/− Neil1−/− mice
High levels of IgM in sera are correlated with lower levels of organ damage and disease activity in patients with SLE and also in lupus-prone mouse models (for reviews see (29,30)). We previously found that the PolbY265C/+ mice developed severe renal disease that was consistent with the characteristics of lupus nephritis (15). Given the high levels of IgM in the PolbY265C/+ Ogg1−/− Neil1−/− mice, we scored hematoxylin- and eosin-stained sections of their kidneys using the scoring system described in methods. Remarkably, the PolbY265C/+ Ogg1−/− Neil1−/− mice exhibited significantly (p=0.02) less severe renal disease than the PolbY265C/+ Ogg1+/+ Neil1+/+ mice, whereas wild-type Polb mice on both backgrounds exhibited similarly low levels of renal disease severity (Figure 6). Immune complex formation is known to occur within the kidney in lupus-prone mice and in humans with lupus. We characterized immune complex formation in the kidneys of our mice and found that kidneys isolated from the PolbY265C/+ Ogg1−/− Neil1−/− exhibited significantly less immune complex deposition than kidneys from the PolbY265C/+ Ogg1+/+ Neil1+/+ mice, similar to what is observed in the kidneys of Polb+/+ Ogg1+/+ Neil1+/+ and Polb+/+ Ogg1−/− Neil1−/− mice (Figure 7). Next, we asked if the kidneys of our mice stained with TUNEL, which is a biomarker of apoptosis. Notably, the PolbY265C/+ Ogg1+/+ Neil1+/+ mice exhibited significantly higher percentages of TUNEL-positive cells than any of the other mouse genotypes (Figure 8). Statistically significant differences in the levels of 5-hydroxycytosine (5-OH-C), FapyG, FapyA, 8-oxoG, and 8-hydroxyadenine (8-OH-A) in the kidneys were not detected between the PolbY265C/+ Ogg1−/− Neil1−/− and PolbY265C/+ Ogg1+/+ Neil1+/+ (Figure 1S), suggesting that these modified bases are being removed by functionally redundant DNA glycosylases or do not occur at high levels in the kidneys of our mice. In combination, our results suggest that the deletion of Ogg1 and Neil1 in the presence of Polb Y265C protects against renal disease in this mouse model, perhaps as a result of high levels of IgM.
Figure 6. Renal disease severity is lower in the PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) mice.
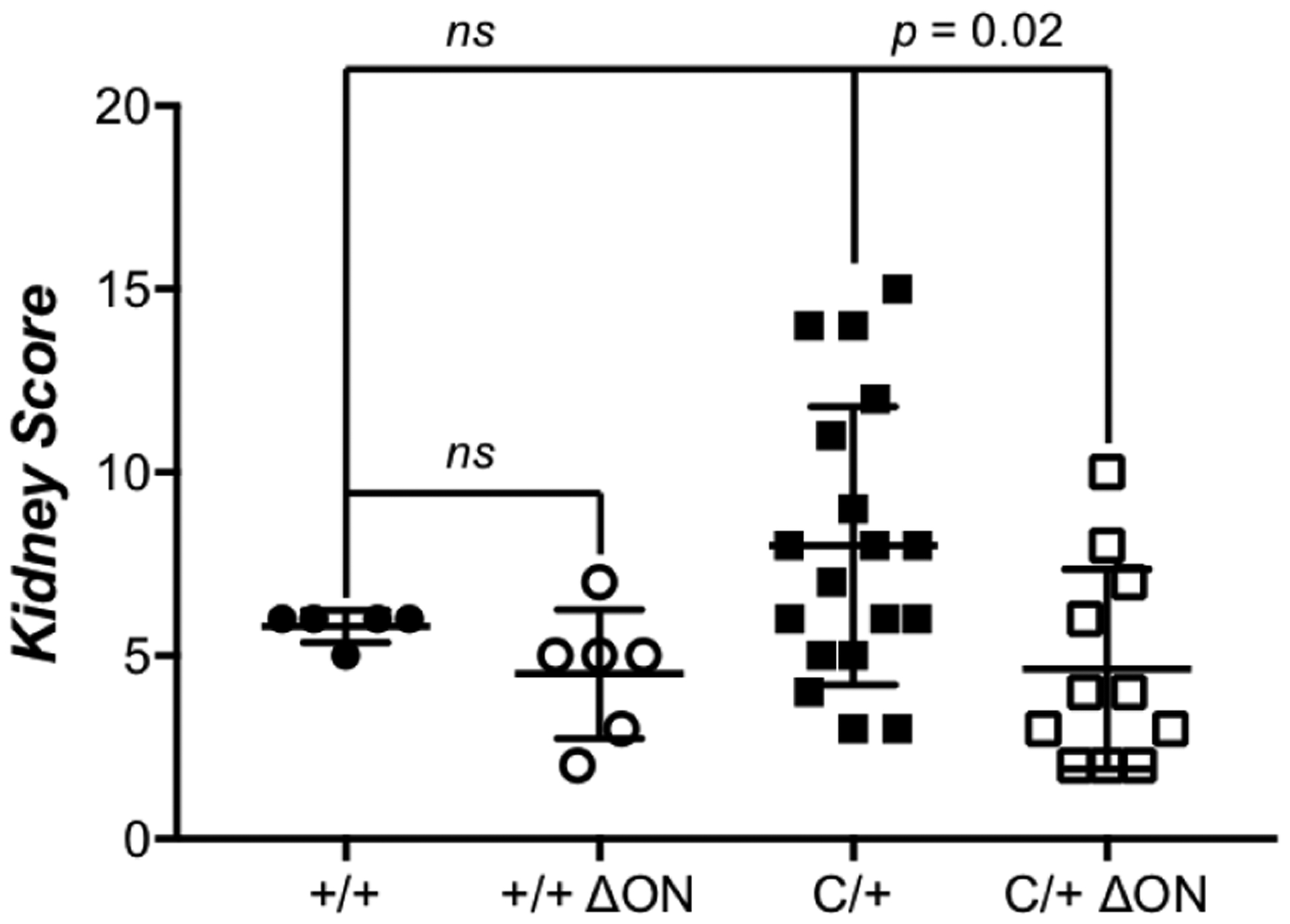
Kidney tissue from mice was fixed in histological formalin solution fixative, embedded in paraffin and sectioned. H&E staining was subsequently performed with the tissue sections. The severity of kidney lesions was scored on a scale from 1 (least severe) to 20 (most severe) as described in methods. Scores of kidneys from Polb+/+ Ogg1+/+ Neil1+/+ (+/+), Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON), PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+), and PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) are shown. Significant renal disease, as determined by a t-test, was observed with sections from C/+ mice, whereas kidney scores from +/+, +/+ ΔON, and C/+ ΔON mice were comparable.
Figure 7. The PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) have less immune complex deposition.
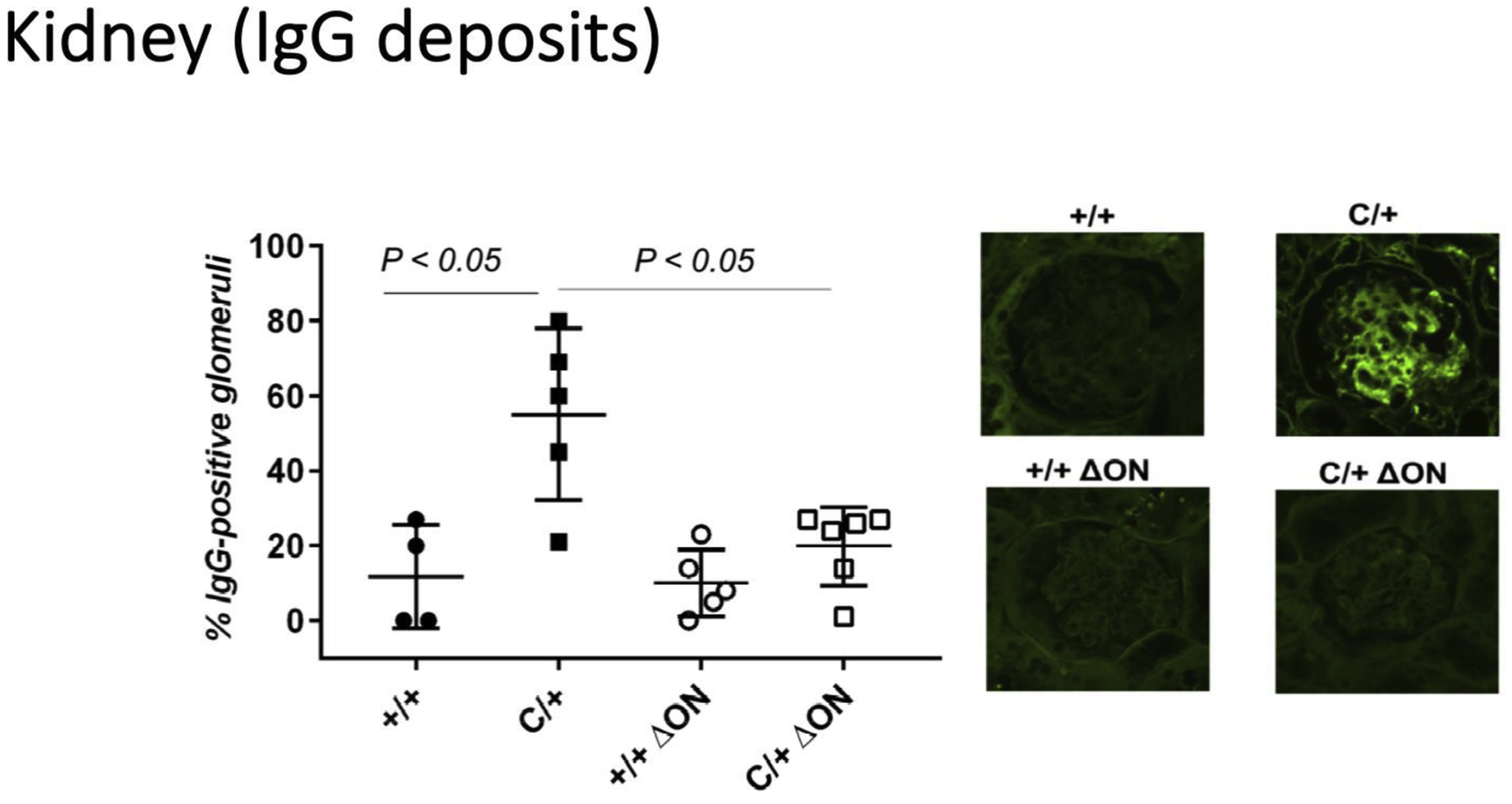
Formalin-fixed paraffin embedded kidney sections from Polb+/+ Ogg1+/+ Neil1+/+ (+/+), PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+), Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON), PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) were stained with FITC–conjugated goat anti–mouse IgG. The number of IgG-positive glomeruli was then normalized to the total number of glomeruli present in the tissue section. Significantly higher levels of IgG-positive cells were present in C/+ kidney sections.
Figure 8. The PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) have fewer TUNEL positive cells.
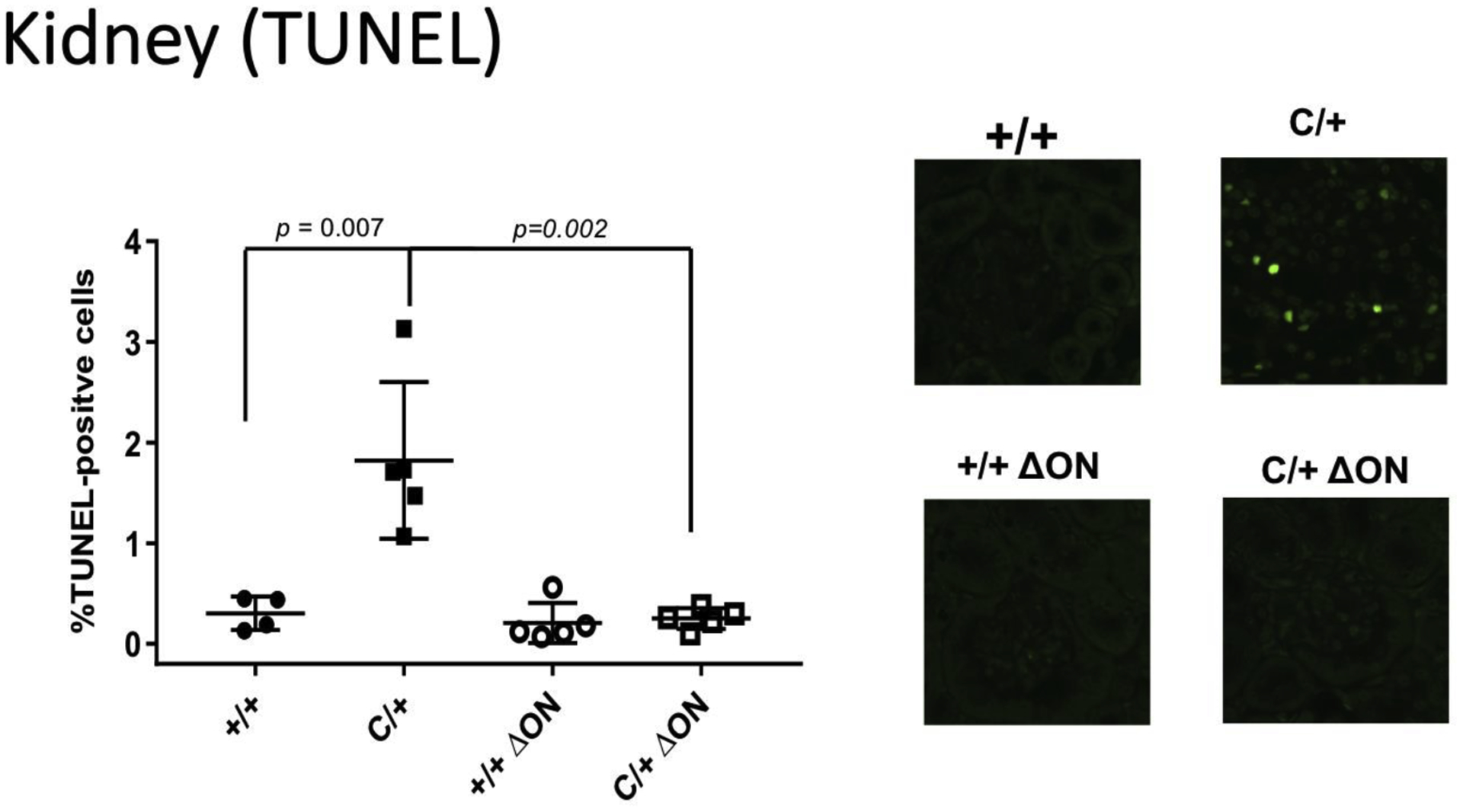
Paraffin embedded kidney sections from Polb+/+ Ogg1+/+ Neil1+/+ (+/+), PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+), Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON), PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) were stained with TUNEL. The total number of TUNEL-positive cells was then normalized to the total cells in each field. Significantly higher levels of TUNEL positive cells were present in C/+ kidney sections.
3.7. Cell death after CSR in the PolbY265C/+ Ogg1−/− Neil1−/− mice
CSR is a deletional recombination process in which B cells switch from expressing IgM or IgD to expressing IgG, IgA, or IgE on their cell surface (for a review see (31)). The result is improvement in the antibody response to eliminate pathogens. Given that the PolbY265C/+ Ogg1−/− Neil1−/− mice have high levels of IgM in their sera, we hypothesized that CSR was aberrant. To test our hypothesis, we isolated splenic B cells from our mice and performed in vitro CSR assays followed by flow analysis (see Figure 2S for gating strategy), as described in methods. Compared to the Polb+/+ Ogg1+/+ Neil1+/+ control (Figure 9A,E), B cells isolated from the PolbY265C/+ Ogg1+/+ Neil1+/+ (Figure 9B,E), and PolbY265C/+ Ogg1−/− Neil1−/− (Figure 9D,E) mice produced lower percentages of IgG1+ B220 cells, but this was not statistically significant. Although cell division, as monitored by dilution of carboxyfluorescein succinimidyl ester (CFSE), was similar in each of the B cell cultures (Figure 9F), significantly higher percentages of switched IgG1 B cells from the PolbY265C/+ Ogg1−/− Neil1−/− (Figure 10 D,E) versus cells isolated from PolbY265C/+ Ogg1+/+ Neil1+/+ (Figure 10B,E) and Polb+/+ Ogg1+/+ Neil1+/+ (Figure 10A,E) mice were marked with 7AAD (Figure 10, Figure 2S). This suggests that once PolbY265C/+ Ogg1−/− Neil1−/− cells switch from IgM to IgG1, they have greater potential to undergo cell death. There was no difference in the levels of TUNEL staining in the spleens of these mice (Figure 10F).
Figure 9. Class switch recombination is not altered in the PolbY265C/+ Ogg1−/− Neil1−/− mice.
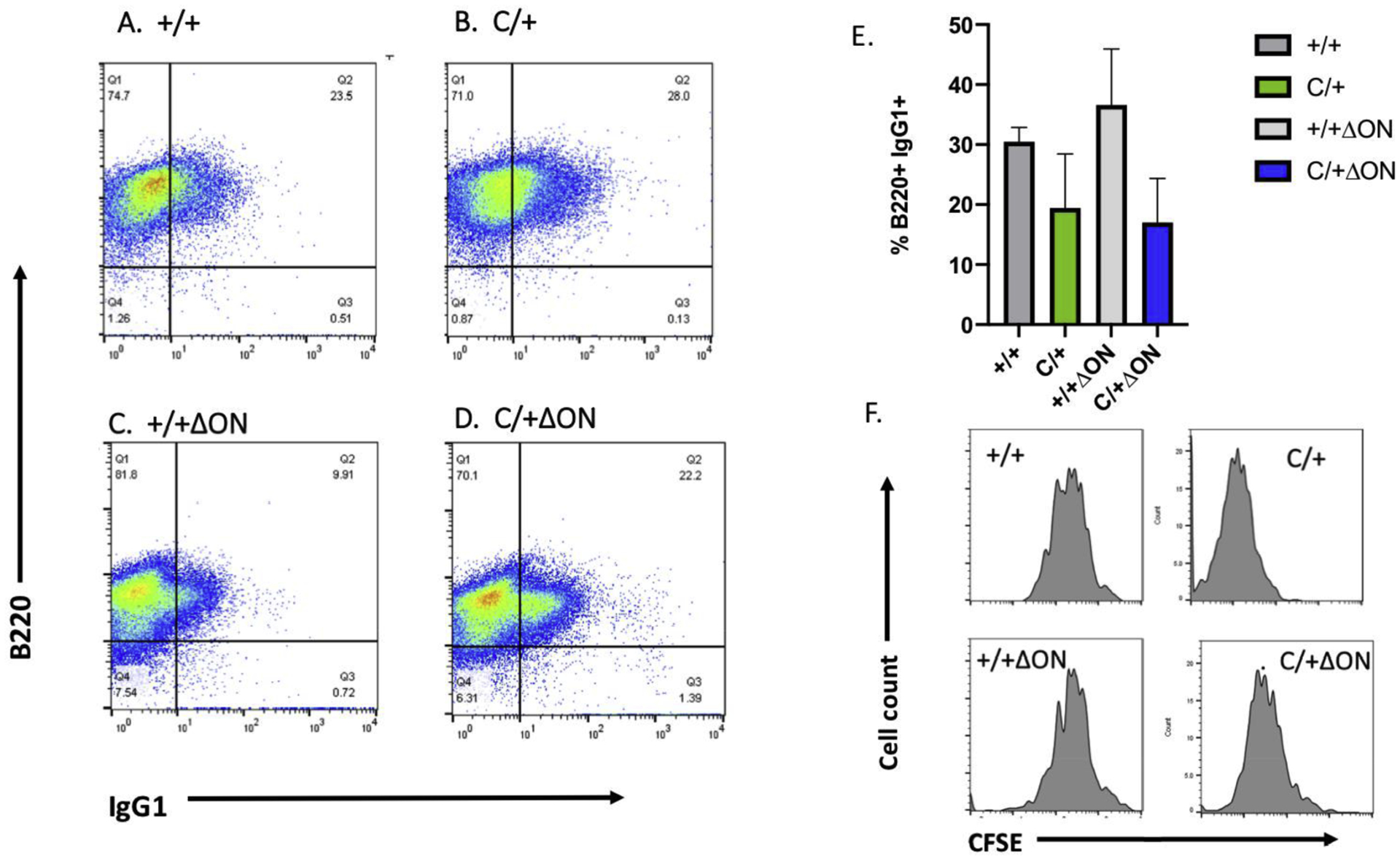
Representative flow cytometric analysis of LPS/IL-4-stimulated mouse splenic B cells surface stained with anti-CD45R/B220-PB, anti-IgG1-PE and live/dead fixability dye from: (A) Polb+/+ Ogg1+/+ Neil1+/+ (+/+), (B) PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+), (C) Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON), (D) PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON). The percentage of IgG1+B220+ cells represent CSR to IgG1 at 64 hr. (E) Statistical analyses of three independent flow cytometric analysis experiments of LPS/IL-4-stimulated splenic B cells switching to IgG1 at 64 hr from +/+, C/+, +/+ ΔON and C/+ ΔON mice. A Student’s t test was used to calculate statistical significance. (F) Representative cell proliferation curves of LPS/IL-4-stimulated splenic B cells from +/+, C/+, +/+ ΔON and C/+ ΔON mice. B cells were stained with CFSE, harvested at 64 h and analyzed by flow cytometry.
Figure 10: The PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON) mice have increased percentage of 7-AAD+ IgG1+ cells.
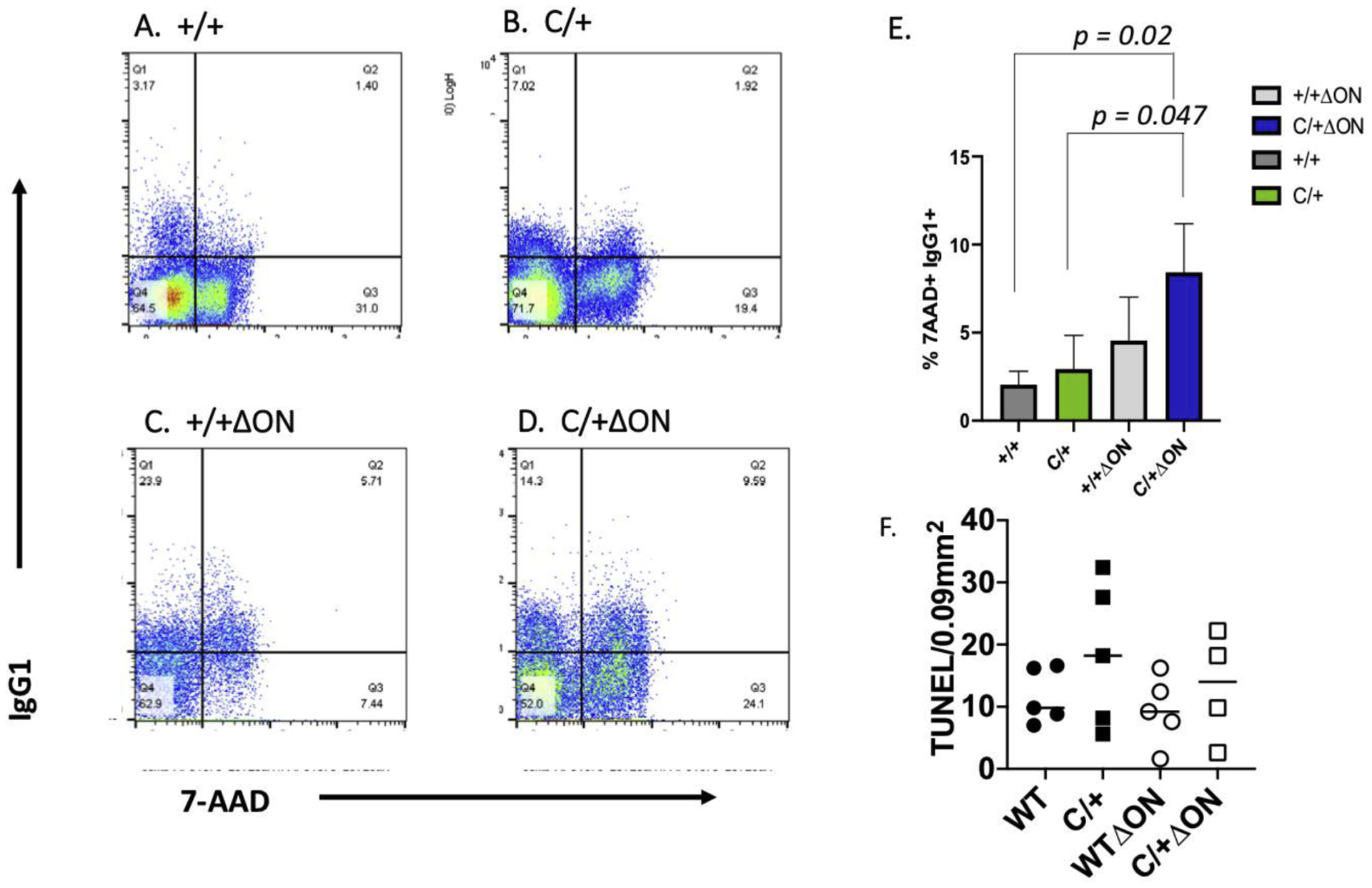
Representative flow cytometric analysis of LPS/IL-4-stimulated mouse splenic B cells stained with anti-CD45R/B220-PB, anti-IgG1-PE, 7-AAD and live/dead fixability dye from: (A) Polb+/+ Ogg1+/+ Neil1+/+ (+/+), (B) PolbY265C/+ Ogg1+/+ Neil1+/+ (C/+), (C) Polb+/+ Ogg1−/− Neil1−/− (+/+ ΔON), (D) PolbY265C/+ Ogg1−/− Neil1−/− (C/+ ΔON). The percentage of IgG1+7-AAD+ cells represent switched IgG1 B cells that were dying at 64 hr. (E) Statistical analysis of three independent flow cytometric analysis experiments of dying IgG1 cells from +/+, C/+, +/+ ΔON and C/+ ΔON mice at 64 hr. A Student’s t test was used to calculate statistical significance. Significantly higher levels of 7-AAD+ IgG1+ cells were observed in C/+ ΔON mice. (F) TUNEL-positive cells in spleens of the mouse genotypes shown. TUNEL staining in five sections of the spleens of each of the mice were quantified. Each mark represents the average number of TUNEL stained cells per 0.09 mm2 from one mouse.
4. Discussion
The goal of our study was to determine if deletion of DNA glycosylases, which function upstream of Pol β in the BER pathway of oxidatively-induced DNA damage, impact the development of lupus in the PolbY265C/+ lupus-prone mouse model. We found the ANA levels remain elevated upon deletion of Ogg1 and Neil1 in the PolbY265C/+ mice. However, deletion of these DNA glycosylases results in fewer BER intermediates in cells, high levels of IgM in sera and less severe renal disease in mice. Our results are consistent with the interpretation that high levels of ANA in our mouse model are not sufficient for the development of lupus nephritis. Our results also suggest that diminishing the levels of potential BER intermediates in the kidney or facilitation of the phagocytosis of dying cells by IgM leads to less severe renal disease.
4.1. Aberrant BER leads to the development of lupus nephritis
The PolbY265C/+ Ogg1+/+ Neil1+/+ mice develop severe renal disease compared to wild-type controls, as a function of age (17). Because deletion of two major DNA glycosylases, namely, Ogg1 and Neil1, in the PolbY265C/+ mice results in less severe renal disease, it is suggested that aberrant BER of oxidatively-damaged DNA plays a role in the development of renal disease. This aberrant BER in the presence of the Pol β Y265C protein is likely defined by the accumulation of single nucleotide gaps. We propose that accumulation of gaps in DNA eventually leads to cell death in the kidney, as observed in the PolbY265C/+ Ogg1+/+ Neil1+/+ mice. Dying cells release cellular debris that acts as a “planted antigen” (reviewed in (32,33)), leading to the deposition of IgG complexes in this kidney, as we observe in the PolbY265C/+ Ogg1+/+ Neil1+/+ mice. We propose that deletion of Ogg1 and Neil1 in the PolbY265C/+ mice leads to less initiation of BER and also lower accumulation of BER intermediates in the presence of catalytically compromised Pol β Y265C. A caveat to this proposal is that we do not observe increased levels of specific modified bases in the kidneys of the PolbY265C/+ Ogg1−/− Neil1−/− versus PolbY265C/+ Ogg1+/+ Neil1+/+ mice as we would have expected. This indicates that other types of modified bases that we did not profile accumulate in the kidneys of the PolbY265C/+ Ogg1+/+ Neil1+/+ and that removal of these lesions could be responsible, at least in part, for the increased percentages of TUNEL-labeled cells in these mice. An alternative explanation is that high levels of IgM in the PolbY265C/+ Ogg1−/− Neil1−/− mice bind to the self-antigens on the dying cells in the kidneys of the PolbY265C/+ Ogg1−/− Neil1−/− mice and enhance their clearance by phagocytes as described (29,34–36). If this were happening in the PolbY265C/+ Ogg1−/− Neil1−/− mice, we might not be able to detect dying cells in the kidney using TUNEL. Importantly, antigen would not be available to facilitate immune complex deposition in the kidney, consistent with our observation of less severe renal disease in our mice.
4.2. Cell death after class switch in B cells from the PolbY265C/+ Ogg1−/− Neil1−/− mice
High levels of IgM are present in the PolbY265C/+ Ogg1−/− Neil1−/−mice. We hypothesized that this may be a result of defective CSR, but showed that CSR from IgM to IgG1 appears to be normal. However, significantly increased percentages of PolbY265C/+ Ogg1−/− Neil1−/− IgG1+ cells are marked with 7-AAD after switching from IgM to IgG1, suggesting that they are dying. This result indicates that the combination of deficient Ogg1 and Neil1 activities with a catalytically slow Pol β leads to cell death after class switch recombination.
Upon stimulation of B cells to undergo CSR, activation-induced cytidine deaminase (AID) and subsequent processing of the resulting uracils by the BER pathway results in the formation of DSBs in G-rich switch regions. Processing of DSBs by the non-homologous end-joining machinery results in deletional recombination and isotype switching. Pol β, Ogg1, and Neil1 are all expressed in stimulated germinal center B cells (37–39). B cells deleted of Pol β undergo CSR at slightly higher levels than controls but have significantly increased numbers of double-strand breaks compared to wild-type cells in switch regions (39). This suggests that Pol β functions in the repair of DSBs in switch regions, likely by microhomology-mediated end-joining (10). Ogg1-deficient B cells are not defective in class switch recombination (38). However, in both of these studies the levels of 7-AAD switched cells were not characterized. To the best of our knowledge, CSR has not been characterized in Neil1Δ mice. However, upon immunization, Neil1Δ mice exhibited decreased numbers of germinal center B cells, reduced somatic hypermutation, and lower levels of antibody production, likely as a result of inefficient repair of modified DNA bases in activated B cells (37). The levels of IgM in the sera of the mice were not measured in any of these studies.
The underlying mechanism of cell death in IgG1 switched cells from the PolbY265C/+ Ogg1−/− Neil1−/− may be related to defective repair of DSBs in switch regions combined with defective removal of oxidative damage during proliferation of B cells. Pol β Y265C has significantly compromised catalytic activity, so its ability to function in the repair of breaks in switch regions may also be compromised. Defective removal of 8-oxoG or FapyG by Ogg1 could lead to incorporation of adenine opposite these lesions in highly proliferative cells. Upon removal by MUTYH DNA glycosylase, accumulation of single nucleotide gaps would occur as a result of the low catalytic activity of Pol β Y265C. These gaps could be converted to DSBs leading to replication fork collapse. Alternatively, given that 8-oxoG is more susceptible to oxidation than guanine, it could be oxidized to spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh), which are replication and transcription blocking lesions (40–43). Repair of replication blocking oxidatively-induced DNA lesions is suggested to occur by a Neil1 replication-coupled pathway (44), which, in the absence of Neil1, is less efficient and leads to replication fork collapse. We suggest that replication fork collapse combined with the presence of lingering breaks in switch regions could result in B cell death. Our results showing the MEFs isolated from the PolbY265C/+ Ogg1−/− Neil1−/− mice are sensitive to menadione support this suggestion. Expression of polymerase-defective Pol β Y265C in B cells is important for development of ANA, but alone is not sufficient for high levels of cell death following CSR. Increased percentages of B cells from the PolbY265C/+ Ogg1+/+ Neil1−/− mice are marked with 7-AAD after CSR, but this was not statistically significant.
4.3. High levels of IgM lead to less severe renal disease
As pointed out earlier, high levels of IgM in sera are correlated with less severe autoimmune disease in humans and in mice. We suggest that the high levels of IgM in the PolbY265C/+ Ogg1−/− Neil1−/− mice lead to less severe renal disease than what is observed in the PolbY265C/+ Ogg1+/+ Neil1+/+ mice, perhaps because IgM can facilitate phagocytosis of dying cells (for a review see (29)). We have shown that defective CSR is not the underlying explanation for the high levels of IgM in the PolbY265C/+ Ogg1−/− Neil1−/− mice. Circulating IgM is thought to be derived from B-1 cells that reside in the bone marrow and spleen and that are selected in the presence of self-antigen. This implies that the levels of B-1 cells in the PolbY265C/+ Ogg1−/− Neil1−/− may be higher than what is present in the other strains of mice characterized in our study. How the combined defects in Polb and DNA glycosylases would lead to increased levels of B-1 cells is not readily apparent. One speculation is that proliferative cells in the PolbY265C/+ Ogg1−/− Neil1−/− mice, including B cells, may undergo cell death at levels exceeding what is present in the other genotypes of mice in our study. This could lead to higher levels of self-antigen that would play a role in the selection of B1 cells in our mice, and this will be a topic of future studies.
Supplementary Material
Highlights.
Lupus results from aberrant base excision repair
Deficiency in the removal of oxidative damage leads to less severe renal disease
High levels of IgM correlate with less severe renal disease in our mouse model
Acknowledgement
Certain commercial equipment or materials are identified in this paper in order to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Author Statement Funding
This research was supported by R01ES019179 and 1R35ES031708 from the National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
There are no competing interests.
Refrences
- 1.Meas R, Burak MJ and Sweasy JB (2017) DNA repair and systemic lupus erythematosus. DNA Repair (Amst). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petri M (2008) Sex hormones and systemic lupus erythematosus. Lupus, 17, 412–415. [DOI] [PubMed] [Google Scholar]
- 3.Tedeschi SK, Bermas B and Costenbader KH (2013) Sexual disparities in the incidence and course of SLE and RA. Clin Immunol, 149, 211–218. [DOI] [PubMed] [Google Scholar]
- 4.Alarcon-Segovia D, Alarcon-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, Pons-Estel BA and Grupo Latinoamericano de Estudio del Lupus, E. (2005) Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum, 52, 1138–1147. [DOI] [PubMed] [Google Scholar]
- 5.Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A and Mack TM (1992) A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum, 35, 311–318. [DOI] [PubMed] [Google Scholar]
- 6.Block SR, Winfield JB, Lockshin MD, D’Angelo WA and Christian CL (1975) Studies of twins with systemic lupus erythematosus. A review of the literature and presentation of 12 additional sets. The American journal of medicine, 59, 533–552. [DOI] [PubMed] [Google Scholar]
- 7.Gulati G and Brunner HI (2018) Environmental triggers in systemic lupus erythematosus. Semin Arthritis Rheum, 47, 710–717. [DOI] [PubMed] [Google Scholar]
- 8.Kuo CF, Grainge MJ, Valdes AM, See LC, Luo SF, Yu KH, Zhang W and Doherty M (2015) Familial Risk of Sjogren’s Syndrome and Co-aggregation of Autoimmune Diseases in Affected Families: A Nationwide Population Study. Arthritis Rheumatol, 67, 1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo MS and Tsokos GC (2018) Recent developments in systemic lupus erythematosus pathogenesis and applications for therapy. Curr Opin Rheumatol, 30, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray S, Breuer G, DeVeaux M, Zelterman D, Bindra R and Sweasy JB (2018) DNA polymerase beta participates in DNA End-joining. Nucleic Acids Res, 46, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K and Wilson SH (1996) Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature, 379, 183–186. [DOI] [PubMed] [Google Scholar]
- 12.Washington SL, Yoon MS, Chagovetz AM, Li SX, Clairmont CA, Preston BD, Eckert KA and Sweasy JB (1997) A genetic system to identify DNA polymerase beta mutator mutants. Proc Natl Acad Sci U S A, 94, 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opresko PL, Sweasy JB and Eckert KA (1998) The mutator form of polymerase beta with amino acid substitution at tyrosine 265 in the hinge region displays an increase in both base substitution and frame shift errors. Biochemistry, 37, 2111–2119. [DOI] [PubMed] [Google Scholar]
- 14.Clairmont CA, Narayanan L, Sun KW, Glazer PM and Sweasy JB (1999) The Tyr-265-to-Cys mutator mutant of DNA polymerase beta induces a mutator phenotype in mouse LN12 cells. Proc Natl Acad Sci U S A, 96, 9580–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senejani AG, Liu Y, Kidane D, Maher SE, Zeiss CJ, Park HJ, Kashgarian M, McNiff JM, Zelterman D, Bothwell AL et al. (2014) Mutation of Polb causes lupus in mice. Cell Rep, 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace SS, Murphy DL and Sweasy JB (2012) Base excision repair and cancer. Cancer Lett, 327, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senejani AG, Dalal S, Liu Y, Nottoli TP, McGrath JM, Clairmont CS and Sweasy JB (2012) Y265C DNA polymerase beta knockin mice survive past birth and accumulate base excision repair intermediate substrates. Proc Natl Acad Sci U S A, 109, 6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu R, Nash HM and Verdine GL (1997) A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr Biol, 7, 397–407. [DOI] [PubMed] [Google Scholar]
- 19.Arai K, Morishita K, Shinmura K, Kohno T, Kim SR, Nohmi T, Taniwaki M, Ohwada S and Yokota J (1997) Cloning of a human homolog of the yeast Ogg1 gene that is involved in the repair of oxidative DNA damage. Oncogene, 14, 2857–2861. [DOI] [PubMed] [Google Scholar]
- 20.Aburatani H, Hippo Y, Ishida T, Takashima R, Matsuba C, Kodama T, Takao M, Yasui A, Yamamoto K and Asano M (1997) Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res, 57, 2151–2156. [PubMed] [Google Scholar]
- 21.Roldan-Arjona T, Wei YF, Carter KC, Klungland A, Anselmino C, Wang RP, Augustus M and Lindahl T (1997) Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc Natl Acad Sci U S A, 94, 8016–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjoras M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T and Seeberg E (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J, 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asagoshi K, Yamada T, Okada Y, Terato H, Ohyama Y, Seki S and Ide H (2000) Recognition of formamidopyrimidine by Escherichia coli and mammalian thymine glycol glycosylases. Distinctive paired base effects and biological and mechanistic implications. J Biol Chem, 275, 24781–24786. [DOI] [PubMed] [Google Scholar]
- 24.Dherin C, Radicella JP, Dizdaroglu M and Boiteux S (1999) Excision of oxidatively damaged DNA bases by the human alpha-hOgg1 protein and the polymorphic alpha-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res, 27, 4001–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M and Mitra S (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A, 99, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dizdaroglu M, Coskun E and Jaruga P (2015) Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic Res, 49, 525–548. [DOI] [PubMed] [Google Scholar]
- 27.Reddy PT, Jaruga P, Kirkali G, Tuna G, Nelson BC and Dizdaroglu M (2013) Identification and quantification of human DNA repair protein Neil1 by liquid chromatography/isotope-dilution tandem mass spectrometry. J Proteome Res, 12, 1049–1061. [DOI] [PubMed] [Google Scholar]
- 28.Jaruga P, Coskun E, Kimbrough K, Jacob A, Johnson WE and Dizdaroglu M (2017) Biomarkers of oxidatively induced DNA damage in dreissenid mussels: A genotoxicity assessment tool for the Laurentian Great Lakes. Environ Toxicol, 32, 2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen TT and Baumgarth N (2016) Natural IgM and the Development of B Cell-Mediated Autoimmune Diseases. Crit Rev Immunol, 36, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gronwall C and Silverman GJ (2014) Natural IgM: beneficial autoantibodies for the control of inflammatory and autoimmune disease. J Clin Immunol, 34 Suppl 1, S12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stavnezer J and Schrader CE (2014) IgH chain class switch recombination: mechanism and regulation. J Immunol, 193, 5370–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shlomchik MJ and Madaio MP (2003) The role of antibodies and B cells in the pathogenesis of lupus nephritis. Springer Semin Immunopathol, 24, 363–375. [DOI] [PubMed] [Google Scholar]
- 33.Kramers K, Stemmer C, Monestier M, van Bruggen MC, Rijke-Schilder TP, Hylkema MN, Smeenk RJ, Muller S and Berden JH (1996) Specificity of monoclonal anti-nucleosome auto-antibodies derived from lupus mice. J Autoimmun, 9, 723–729. [DOI] [PubMed] [Google Scholar]
- 34.Ogden CA, Kowalewski R, Peng Y, Montenegro V and Elkon KB (2005) IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity, 38, 259–264. [DOI] [PubMed] [Google Scholar]
- 35.Quartier P, Potter PK, Ehrenstein MR, Walport MJ and Botto M (2005) Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. Eur J Immunol, 35, 252–260. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y, Park YB, Patel E and Silverman GJ (2009) IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J Immunol, 182, 6031–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori H, Ouchida R, Hijikata A, Kitamura H, Ohara O, Li Y, Gao X, Yasui A, Lloyd RS and Wang JY (2009) Deficiency of the oxidative damage-specific DNA glycosylase Neil1 leads to reduced germinal center B cell expansion. DNA Repair (Amst), 8, 1328–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ucher AJ, Linehan EK, Teebor GW, Schrader CE and Stavnezer J (2012) The DNA glycosylases Ogg1 and Nth1 do not contribute to Ig class switching in activated mouse splenic B cells. PLoS One, 7, e36061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X and Stavnezer J (2007) DNA polymerase beta is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med, 204, 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munk BH, Burrows CJ and Schlegel HB (2008) An exploration of mechanisms for the transformation of 8-oxoguanine to guanidinohydantoin and spiroiminodihydantoin by density functional theory. J Am Chem Soc, 130, 5245–5256. [DOI] [PubMed] [Google Scholar]
- 41.Alenko A, Fleming AM and Burrows CJ (2017) Reverse Transcription Past Products of Guanine Oxidation in RNA Leads to Insertion of A and C opposite 8-Oxo-7,8-dihydroguanine and A and G opposite 5-Guanidinohydantoin and Spiroiminodihydantoin Diastereomers. Biochemistry, 56, 5053–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aller P, Ye Y, Wallace SS, Burrows CJ and Doublie S (2010) Crystal structure of a replicative DNA polymerase bound to the oxidized guanine lesion guanidinohydantoin. Biochemistry, 49, 2502–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh J, Fleming AM, Xu J, Chong J, Burrows CJ and Wang D (2020) RNA polymerase II stalls on oxidative DNA damage via a torsion-latch mechanism involving lone pair-pi and CH-pi interactions. Proc Natl Acad Sci U S A, 117, 9338–9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hegde ML, Hegde PM, Bellot LJ, Mandal SM, Hazra TK, Li GM, Boldogh I, Tomkinson AE and Mitra S (2013) Prereplicative repair of oxidized bases in the human genome is mediated by Neil1 DNA glycosylase together with replication proteins. Proc Natl Acad Sci U S A, 110, E3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.