

SUMMARY
The anorexigenic effect of serotonergic compounds has largely been attributed to activation of serotonin 2C receptors (Htr2cs). Using mouse genetic models in which Htr2c can be selectively deleted or restored (in Htr2c-null mice), we investigate the role of Htr2c in forebrain Sim1 neurons. Unexpectedly, we find that Htr2c acts in these neurons to promote food intake and counteract the anorectic effect of serotonergic appetite suppressants. Furthermore, Htr2c marks a subset of Sim1 neurons in the paraventricular nucleus of the hypothalamus (PVH). Chemogenetic activation of these neurons in adult mice suppresses hunger, whereas their silencing promotes feeding. In support of an orexigenic role of PVH Htr2c, whole-cell patch-clamp experiments demonstrate that activation of Htr2c inhibits PVH neurons. Intriguingly, this inhibition is due to Gαi/o-dependent activation of ATP-sensitive K+ conductance, a mechanism of action not identified previously in the mammalian nervous system.
Graphical Abstract
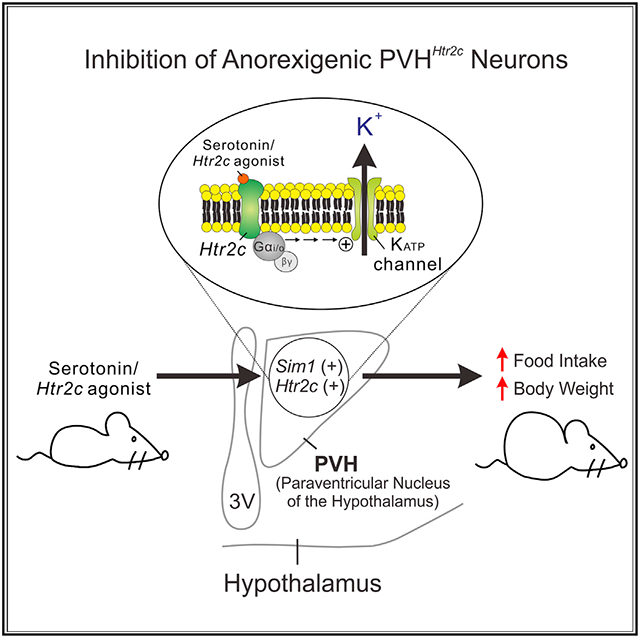
In brief
Yoo et al. show that Htr2c, the target of a former weight loss drug, can inhibit and promote food intake by coupling with distinct intracellular signaling events in different hypothalamic neurons. These findings may help explain the rather modest anti-obesity effects of Htr2c agonists.
INTRODUCTION
To combat the obesity epidemic as well as eating disorders such as anorexia nervosa, it is essential that we gain a better understanding of the complex regulation of feeding behaviors. Recent studies have identified several “feeding” neurons along the neuraxis where hunger and satiety cues are integrated with cognitive, emotional, and reward inputs (Kenny, 2011; Schwartz and Zeltser, 2013). These neuron populations may adopt different strategies to integrate neuronal and humoral signals and exert a positive or negative influence on food intake. For example, it has been shown that agouti-related protein (AgRP) neurons evoke voracious feeding behaviors through parallel and redundant forebrain feeding circuits, each of which is sufficient to drive food intake (Betley et al., 2013). On the other hand, multiple anorexigenic signals converge onto protein kinase C (PKC)-δ+ neurons in the amygdala, supporting a model where diverse anorectic information is processed in one central node (Cai et al., 2014).
The brain serotonin (5-hydroxytryptamine or 5-HT) system is a key target for weight loss therapies. Pharmacological agents that increase extracellular 5-HT content suppress food intake and reduce body weight (Wyler et al., 2017). The therapeutic potential of this pathway was highlighted by the demonstration that the anorexigenic actions of serotonergic agents such as d-fenfluramine (dFen, a key ingredient of the once-popular diet pill Fen/Phen) were mediated in part through the serotonin 2c receptor (Htr2c) (Tecott et al., 1995; Vickers et al., 1999). Subsequently, lorcaserin, an Htr2c-specific agonist, became one of the first US Food and Drug Administration (FDA)-approved weight loss drugs (Colman et al., 2012).
Htr2c is widely distributed in the brain, including in regions that are known to regulate food intake (Hoffman and Mezey, 1989). However, it is not clear whether the anorectic effects of 5-HT agents require coordinated actions of Htr2c at multiple feeding nodes or whether activation of Htr2c in discrete neuronal populations suppresses food intake in a parallel or convergent manner. We previously demonstrated that selective restoration of Htr2c only in pro-opiomelanocortin (POMC) neurons of Htr2c-null mice was sufficient to normalize body weight, food intake, and the anorectic responses to dFen (Xu et al., 2008). Conversely, deletion of Htr2c specifically in these neurons during development or in adulthood resulted in hyperphagia and a blunted anorectic response to dFen (Berglund et al., 2013; D’Agostino et al., 2018). These findings identified POMC neurons as one of the sites where Htr2c regulates food intake.
In addition to POMC neurons, Htr2c is enriched in neurons of the paraventricular nucleus of the hypothalamus (PVH) that express the transcription factor single-minded 1 (Sim1) (Heisler et al., 2007). PVH Sim1 neurons are physiologically relevant to regulation of feeding behaviors because haploinsufficiency of Sim1 results in reduced numbers of PVH neurons and causes early-onset hyperphagia and obesity in rodents and humans (Holder et al., 2004; Michaud et al., 2001). Furthermore, deletion of methyl CpG binding protein 2 (Fyffe et al., 2008) or melanocortin 4 receptor (MC4R) specifically in Sim1 neurons leads to hyperphagia and obesity in rodents (Li et al., 2021). Importantly, we demonstrated that the anorectic responses to 5-HT agents were blunted in mice heterozygous for Sim1, suggesting that Sim1 neurons represent another important node in the brain to regulate such responses (Xu et al., 2010). In the current study, we investigated the role of Htr2c in feeding regulation in Sim1 neurons using a combination of mouse genetic, chemogenetic, and electrophysiological analyses. Our studies led to the unexpected finding that Htr2c acts in Sim1 neurons to promote food intake and counteract the anorexigenic actions of 5-HT agents. Moreover, although activation of Htr2c depolarizes anorexigenic POMC neurons in a phospholipase C (PLC)-dependent manner (Sohn et al., 2011), we find that its actions on PVH neurons are via the Gαi/o pathway, resulting in hyperpolarization of PVH neurons.
RESULTS
Selective restoration of Htr2c only in Sim1 neurons promotes food intake
We previously generated and characterized a re-activatable Htr2c-null mouse (Htr2cnulll) in which transcription of endogenous Htr2c was impeded by a transcriptional blocker cassette. Specific to this model, physiological levels of Htr2c can be restored selectively in distinct cell groups upon Cre recombinase activity. Consistent with previous reports (Nonogaki et al., 1998; Xu et al., 2008), chow-fed Htr2cnull mice were hyperphagic in a fasting-refeeding test but had body weight gains comparable with their wild-type littermates until 20 weeks of age because of a concomitant increase in energy expenditure (Figure S1A). However, hyperphagia was exacerbated when these mice were fed a high-fat diet (HFD; 42% kcal from fat, 0.2% cholesterol), tipping the scale of energy balance. As a result, despite the sustained higher energy expenditure, male Htr2cnull mice rapidly gained more weight and became significantly heavier than their littermate controls after an average of 8–10 weeks of HFD feeding (Figure 1A).
Figure 1. Restoration of Htr2c only in Sim1 neurons increases food intake and body weight in male mice.

(A) Body weight curves; F(28, 294) = 9.802, p < 0.001.
(B) Heat production during light (06:00–18:00) and dark (18:00–06:00) phases of a day; F(2, 24) = 1.961, p = 0.16.
(C and D) Food intake (C; F(2, 24) = 13, p < 0.001) and (D) respiratory exchange ratio (F(2, 24) = 1.712, p = 0.2) in HFD-fed male mice.
(E and F) Percentage of food intake (chow) after an i.p. dose of dFen (E, 6 mg/kg) or mCPP (F, 5 mg/kg), compared with vehicle-treated mice during the first 2 h of refeeding (F(2, 24) = 13.49 and F(2, 24) = 14.5 respectively; p < 0.001).
(G) Food intake (chow) after an i.p. dose of vehicle (saline) or lorcaserin during the first 2 h of refeeding; p = 0.05.
Values represent mean ± SEM; n = 8–11; two-way ANOVA with Tukey’s post hoc tests in (A)–(D); one-way ANOVA with Tukey’s post hoc tests in (E) and (F); paired t test in (G); *p < 0.05, **p < 0.01, ***p < 0.001.
To examine whether selective restoration of Htr2c in Sim1 neurons was sufficient to normalize the body weight deficits of Htr2cnull mice, we bred re-activatable Htr2cnulll mice with Sim1::Cre mice (Balthasar et al., 2005) to generate Htr2cnull, Sim1::Cre mice in which endogenous levels of Htr2c were restored only in Sim1 neurons (hereafter called Htr2cSim1RE mice). We found that selective re-expression of Htr2c in Sim1 neurons failed to normalize hyperphagia or higher energy expenditure in chow-fed mice (Figures S1B-S1D). In contrast, HFD-fed Htr2cSim1RE mice gained significantly more weight and became obese faster than their wild-type (Sim1::Cre +/−) and Htr2cnull littermates (Figure 1A). We then subjected another cohort of mice to a combined indirect calorimetry system (TSE metabolic chambers) where food intake, energy expenditure, and physical activity were monitored continuously. We found that, although the abnormal increase in energy expenditure persisted and remained comparable between HFD-fed Htr2cnull and Htr2cSim1RE mice (Figure 1B), hyperphagia was exaggerated in Htr2cSim1RE mice because they consumed significantly more calories than Htr2cnull mice during the dark phase, when mice are normally active (Figure 1C). The respiratory exchange ratio was not different among Htr2cSim1RE, Htr2cnull, and wild-type mice (Figure 1D).
Next we investigated whether Htr2c in Sim1 neurons regulated the anorexigenic effects of serotonergic compounds such as dFen, a 5-HT releaser and uptake inhibitor, and meta-chlorophe-nylpiperazine (mCPP), a non-selective Htr2c agonist. Refeeding was measured in chow-fed mice after an overnight fast followed by an intraperitoneal (i.p.) dose of drug or saline. Despite the sustained effect on cumulative intake, the anorexigenic actions of dFen (6 mg/kg) and mCPP (5 mg/kg) were most prominent within the first 2 h following drug administration, whereas refeeding during subsequent hours was not statistically different between mice treated with drug or saline (Figures S1E-S1H). It has been reported previously reported that the anorexigenic effects of dFen and mCPP are blunted in conventional Htr2cnull mice (Tecott et al., 1995; Vickers et al., 1999). Similarly, we found that Htr2cnull mice showed attenuated anorectic responses to these agents (Xu et al., 2008). However, we unexpectedly found that such responses were blunted further in Htr2cSim1RE mice (Figures 1E and 1F). We then tested the response to lorcaserin, a selective agonist for Htr2c (Thomsen et al., 2008). As expected, lorcaserin (10 mg/kg) inhibited food intake in wild-type littermates but not in Htr2cnull mice (Figure 1G). Surprisingly, rather than suppressing food intake, lorcaserin appeared to increase food intake in Htr2cSim1RE mice (Figure 1G).
Selective ablation of Htr2c in Sim1 neurons reduces food intake
Results from the re-expression studies raised the intriguing possibility that, despite its anorexigenic function in hypothalamic POMC neurons, Htr2c may act in Sim1 neurons to promote food intake. To test this hypothesis, we selectively deleted Htr2c in Sim1 neurons by breeding floxed Htr2c mice (Berglund et al., 2013) with Sim1::Cre mice to generate Htr2cfl/Y; Sim1::Cre mice (hereafter called as Htr2cSim1KO). Sim1::Cre activity is enriched in neurons in the PVH and amygdala (Balthasar et al., 2005). In situ hybridization analyses demonstrated that Htr2c mRNAs were downregulated significantly in the PVH of Htr2cSim1KO mice, whereas levels within the amygdala were comparable between the two genotypes (Figures 2A-2D). Compared with their littermate controls (Htr2cfl/Y), Htr2cSim1KO mice had normal body weight and body composition at 6 weeks of age (Figures 2E and 2F). Moreover, these mice demonstrated normal glucoregulatory responses in glucose and insulin tolerance tests (Figures S2A and S2B). However, Htr2cSim1KO mice gained less weight and became significantly leaner than their littermate controls when fed an HFD (Figure 2E). Nuclear magnetic resonance analyses revealed that the decreased body weight in Htr2cSim1KO mice was due to a reduction in fat and lean mass (Figure 2F). Furthermore, metabolic chamber analyses of a separate cohort of mice showed that food intake was reduced in HFD-fed Htr2cSim1KO mice (Figure 2G), whereas energy expenditure (Figure 2H) and physical activity (Figure 2I) remained comparable with their littermate controls. Interestingly, we found that chow-fed Htr2cSim1KO mice exhibited an anorectic response to a sub-anorectic dose of dFen (1 mg/kg) that otherwise did not suppress refeeding (during the first 2 h) in wild-type littermates (Figure 2J; Wurtman and Wurtman, 1977). Likewise, the anorectic response to mCPP (5 mg/kg) was also potentiated in these mice (Figure 2K). These findings suggest that Htr2c in Sim1 neurons modulates the anorectic responses to 5-HT compounds.
Figure 2. Selective deletion of Htr2cs in Sim1 neurons reduces food intake and body weight in male mice.

(A–D) In situ hybridization of Htr2c mRNA in the PVH (A and B) and amygdala (C and D) of Htr2cfl/Y and Htr2cSim1KO mice.
(E) Body weight curves; F(15, 360) = 4.78, p < 0.001.
(F) Body composition; F(3, 96) = 3.91, p = 0.01.
(G) Food intake during light and dark phases of a day; F(1, 20) = 13.44, p = 0.002.
(H and I) Heat production (H; F(1, 20) = 0.33, p = 0.57) and (I) physical activity (F(1, 20) = 0.16, p = 0.7) in HFD-fed mice.
(J and K) Responses to dFen (J, 1 mg/kg) and mCPP (K, 5 mg/kg) in chow-fed Htr2cfl/Y and Htr2cSim1KO mice.
Values represent mean ± SEM; n = 11–15, two-way ANOVA with Tukey’s post hoc tests in (E)–(I); unpaired t tests in (J) and (K); *p < 0.05, **p < 0.01, ***p < 0.001.
PVH neurons are critical regulators of stress responses (Xu et al., 2019). Therefore, we investigated whether the observed decrease in food intake in Htr2cSim1KO mice was due to distress, such as increased anxiogenic and/or depressive behaviors. We tested two independent cohorts of control and Htr2cSim1KO mice that were fed chow (data not shown) or an HFD in a series of behavioral paradigms including the elevated plus maze, dark-light exploration, open field tests for anxiety-related behaviors, as well as the forced swim test for depression-like behaviors (Figures S2C-S2F). However, we found no differences between the genotypes in any of these tests, indicating that the reduced food intake was unlikely to be the result of mood disturbances.
PVH Htr2c neurons bidirectionally regulate food intake
To investigate the neurochemical identity of PVH Htr2c neurons, we conducted Cre-mediated fate mapping using a Htr2c::Cre mouse we developed recently, in which expression of a Cre recombinase is driven by the endogenous regulatory sequences of Htr2c (Park et al., 2020). We found that expression of a Cre-activated tdTomato reporter (Ai14) marked a subset of adult PVH neurons. None of these neurons expressed PVH neuronal markers such as oxytocin (OXY; Figure S3), arginine vasopressin (AVP), or tyrosine hydroxylase (TH). PVH neurons expressing Mc4r mediate the anorectic effect of serotonin downstream of Htr2c/POMC neurons (Xu et al., 2010). However, none of the tomato-positive neurons in the PVH expressed Mc4r-GFP. Using double immunohistochemistry and in situ hybridization (Figure S3), we found that the majority of PVH Htr2c neurons expressed mRNAs for corticotropin-releasing hormone (Crh) or thyrotropin-releasing hormone (Trh). Furthermore, similar analyses showed that these neurons did not express pituitary adenylate cyclase-activating polypeptide (Pacap) or galanin (Gal).
To test whether PVH Htr2c neurons directly regulate food intake in vivo, we stereotaxically delivered adeno-associated viruses (AAVs) expressing Cre-dependent Designer Receptors Exclusively Activated by Designer Drugs (DREADD) constructs (Krashes et al., 2011) into the PVH of Htr2c::Cre mice (Figures 3A and 3B). Following an i.p. dose of clozapine-N-oxide (CNO; 1 mg/kg), we found elevated levels of Fos protein, a marker for enhanced neuronal activity, on the ipsilateral side of the PVH that received the stimulatory DREADD (hM3Dq) (Figure 3C; 95.89 ± 6.18 positive cells/section compared with 14.56 ± 4.02 positive cells/section on the contralateral side). Fos expression was found in the majority (86.84% ± 4.25% of 469 cells from three mice) of Htr2c neurons that had been targeted by the GFP-labeled hM3Dq (Figure 3D). Consistent with this, electrophysiological recordings of GFP-labeled neurons showed that bath application of CNO (5 μM) elicited rapid depolarization of membrane potentials in PVH Htr2c neurons (Figures 3E and 3F). In Htr2c::Cre mice that received unilateral or bilateral hM3Dq injections in the PVH, a dose of CNO (i.p., 1 mg/kg) suppressed fasting-induced refeeding (Figure 3G). Importantly, CNO had no effect on food intake in mice in which the stereotaxic injections were placed outside of the PVH (misinjection; Figure 3G), nor did it alter feeding in Htr2c::Cre mice that received injections of a control virus (AAV-DIO-mCherry; Figure S4). Therefore, these findings demonstrate that chemogenetic activation of PVH Htr2c neurons inhibits feeding.
Figure 3. PVH Htr2c neurons directly regulate food intake in male mice.

(A) Schematic for stereotaxic delivery of AAVs that contain stimulatory (hM3Dq) or inhibitory (hM4Di) DREADDs.
(B) Representative image showing unilateral targeting of GFP-labeled DREADD constructs in the PVH.
(C) Immunohistochemistry for Fos proteins.
(D) Double immunostaining for Fos and GFP. Arrowheads indicate double-labeled neurons.
(E) Bath application of CNO (5 μM) increased the firing rates of GFP-labeled PVH neurons.
(F) Change in membrane potentials.
(G) Stimulation of PVH Htr2c neurons suppressed fasting-induced refeeding; F(2, 36) = 3.511, p = 0.04.
(H) Food intake during the 1-h satiation period after fasting.
(I) Silencing of PVH Htr2c neurons promoted feeding; F(2, 28) = 4.832, p = 0.002.
Values represent mean ± SEM; *p < 0.05, **p < 0.01; two-way ANOVA with Sidak’s post hoc tests in (F), (G), and (I). Scale bars, 200 μm (B and C) and 20 μm (D).
To evaluate the effect of silencing PVH Htr2c neurons on food intake, we conducted bilateral stereotaxic injections of AAVs that contained the inhibitory DREADD (hM4Di). Overnight-fasted mice were initially satiated for 1 h before receiving either CNO or saline. We found that individual mice consumed similar amounts of food during the satiation period (Figure 3H). However, compared with saline-treated mice, silencing PVH Htr2c neurons increased food intake after CNO treatment (Figure 3I). These findings demonstrated that PVH Htr2c neurons can bidirectionally regulate food intake in vivo. The effects of chemogenetic manipulations (stimulation or silencing) on food intake were transient because there were no differences in cumulative food intake between saline- and CNO-treated mice after 24 h (Figure S4).
Htr2c agonists hyperpolarize PVH neurons via postsynaptic KATP channels
Our chemogenetic experiments demonstrated that activation of PVH Htr2c neurons produced an anorectic effect. In contrast, our genetic studies appeared to suggest that Htr2c acts in PVH neurons to promote feeding. Htr2c has been reported to couple to the Gαq/PLC pathway and, upon activation, to depolarize membrane potentials (Sohn et al., 2011). How could activation of Htr2c in anorexigenic PVH Htr2c neurons ultimately lead to an orexigenic effect? To investigate this apparent discrepancy, we studied the acute effects of Htr2c agonists on membrane potentials of genetically labeled Htr2c neurons in the arcuate nucleus of the hypothalamus (ARH) and the PVH.
We have demonstrated that activation of Htr2c depolarized a subset of ARH POMC neurons (Sohn et al., 2011). Consistent with this finding, mCPP (4 μM) depolarized a subpopulation of tomato-positive Htr2c neurons within the ARH (Figure S5). However, to our surprise, mCPP hyperpolarized the membrane potential of Htr2c neurons in the PVH by −9.9 ± 0.8 mV (28 of 76 cells, 36.8%; Figure 4A). The mCPP-induced hyperpolarization was not blunted by the presence of tetrodotoxin (TTX; 0.5 μM) and a cocktail of fast synaptic blockers (10 μM CNQX + 50 μM AP5 + 50 μM picrotoxin), supporting a postsynaptic event (−7.8 ± 0.9 mV, 6 of 11 cells, 54.5%; Figure 4B). We further tested the acute responses to the Htr2c-specific agonist lorcaserin and found that lorcaserin hyperpolarized PVH Htr2c neurons by −10.2 ± 2.2 mV (5 of 11 cells, 45.5%; Figure 4C). No depolarizing responses were recorded in the presence of mCPP (n = 76) or lorcaserin (n = 11). Moreover, lorcaserin-induced hyperpolarization of PVH Htr2c neurons was accompanied by an 18.6% ± 4.6% decrease in input resistance (from 1,074 ± 109 MΩ to 875 ± 103 MΩ, n = 5), which was comparable with that induced by mCPP treatment (decreased 20.5% ± 1.7% from 1,335 ± 53 MΩ to 1,056 ± 43 MΩ, n = 28). In both cases, the reversal potential (Erev; −94.7 ± 7.1 mV for lorcaserin, −97.0 ± 2.4 mV for mCPP) was indicative of activated potassium conductance (Figure 4D). Consistent with these findings, we found that the mCPP-induced hyperpolarization in PVH Htr2c neurons was completely reversed by the ATP-sensitive K+ (KATP) channel inhibitors tolbutamide (200 μM; Figures 4A and 4E) and glibenclamide (100 μM; Figure 4F). Similarly, 5-HT (30 μM) hyperpolarized PVH Htr2c neurons (−9.2 ± 1.4 mV, 6 of 12 cells, 50.0%; Figures 4G and 4H), presumably by increasing potassium conductance (22.8% ± 1.4% decrease in input resistance from 1,571 ± 74 MΩ to 1,216 ± 71 MΩ, Erev = −86.4 ± 4.5 mV, n = 6). Interestingly, we found that inhibition of Gαq proteins/PLC signaling with anti-Gαq antibodies or U73122 (a PLC inhibitor, 5 μM) failed to prevent mCPP-induced inhibition (Figures 4G and 4H, 11 of 26 cells [42.3%] pretreated with anti-Gαq antibodies hyperpolarized by −7.7 ± 1.4 mV; 4 of 13 cells [30.8%] pretreated with U73122 hyperpolarized by −6.0 ± 1.1 mV, respectively). It has been reported that KATP channels can couple to the Gαi/o pathway to hyperpolarize membrane potentials (Kurachi et al., 1992). In support of this, we found that application of pertussis toxin (PTX; 1 ng/μL), which inhibits Gαi/o proteins, prevented mCPP-induced hyperpolarization of PVH Htr2c neurons (Figures 4G and 4H). These findings suggest that Htr2c couples to Gαi/o-KATP pathway in PVH neurons to inhibit their activity and promote feeding.
Figure 4. Activation of Ht2c hyperpolarizes PVH Htr2c neurons.
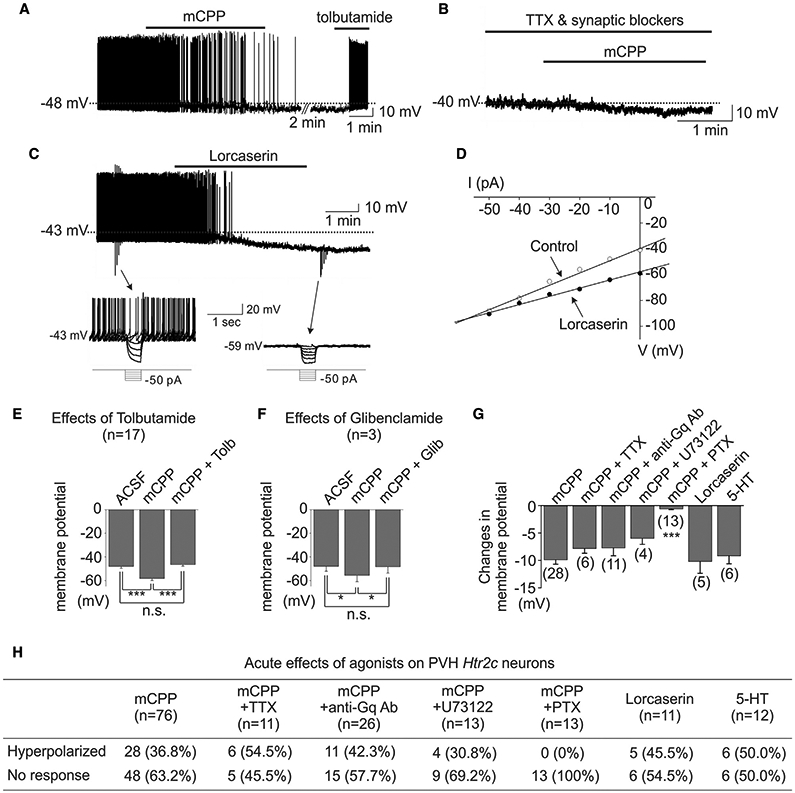
(A) Bath applications of mCPP caused prompt hyperpolarization of membrane potential, which was completely reversed by subsequent applications of tolbutamide.
(B) mCPP-induced hyperpolarization was not affected by addition of TTX and fast synaptic blockers in the bath solutions.
(C) Bath applications of lorcaserin, an Htr2c-specific agonist, caused prompt hyperpolarization of membrane potential. Downward deflections shown in the trace represent voltage responses to current steps, which are shown as insets below.
(D) Current-voltage (IV) relationship before (control) and after drug (lorcaserin) application. The Erev is close to the equilibrium potential for K+ (EK).
(E) Bar graphs summarizing the effects of tolbutamide on mCPP-induced hyperpolarization.
(F) Bar graphs summarizing the effects of glibenclamide.
(G) Bar graphs showing the average amplitude of mCPP-, lorcaserin-, or 5-HT-induced hyperpolarization.
(H) Table summarizing the numbers and percentages of the responses to mCPP treatment in recorded Htr2c neurons within the PVH.
Values represent mean ± SEM; *p < 0.05, ***p < 0.001; repeated-measures two-way ANOVA with Tukey’s or Bonferroni’s post hoc test in (E)–(G).
DISCUSSION
Despite its pharmacological effect on appetite suppression, serotonergic regulation of feeding is far more complex at the physiological level. For example, dietary depletion of tryptophan, a precursor of 5-HT biosynthesis, had little effect on food intake in healthy humans (Pagoto et al., 2009). Furthermore, mice completely lacking 5-HT in the brain or the whole body from early development are viable, fertile, and exhibit normal food intake and body weight (Mosienko et al., 2015). It has been shown that 5-HT can promote or inhibit food intake via different 5-HT receptors. For instance, its anorexigenic effect is largely attributed to activation of Htr2c and Htr1b (Lucas et al., 1998; Nonogaki et al., 1998). In contrast, treatment with 8-hydroxy-2-(di-n-pro-pylamino) tetralin (8-OH-DPAT), an Htr1a agonist, increases feeding in C57BL/6 mice (Ebenezer and Surujbally, 2007).
Employing mouse genetic models in which endogenous Htr2c is selectively ablated or restored in Sim1 neurons, our studies demonstrate that activation of Htr2c can produce orexigenic and anorexigenic effects. Similar to many other genes that are important for feeding regulation, Htr2c has broad distribution throughout the brain. Conventional wisdom has it that individual feeding regulators play coherent roles by promoting or inhibiting food intake at discrete central feeding nodes. Supporting this idea, the anorexigenic hormone leptin suppresses food intake by activating anorexigenic POMC neurons and inhibiting orexigenic AgRP neurons (Baver et al., 2014; Cowley et al., 2001; Hill et al., 2008). However, our study reveals that activation of Htr2c at different brain sites can have opposing effects on food intake. Contrary to its anorexigenic role in POMC neurons, Htr2c acts in Sim1 neurons to promote food intake. This is further supported by our findings that restoration of Htr2c only in Sim1 neurons exacerbates hyperphagia in Htr2cnull mice, whereas its selective deletion in the same neurons reduces food intake. Along this line, we show that the pharmacological effects of 5-HT agents can be differentially modulated by Htr2c in different brain locations. Opposite to its effect in POMC neurons, we find that Htr2c acts in Sim1 neurons to dampen anorectic responses. This is corroborated by the observation that lorcaserin, an Htr2c-specific agonist, suppressed feeding in wild-type animals but appeared to increase food intake in mice in which Htr2c was expressed exclusively in Sim1 neurons. We hypothesize that Htr2c in Sim1 neurons may provide important regulatory feedback that restricts the effectiveness and safety of serotonergic appetite suppressants. Moreover, these data may be a cellular correlate to the rather modest anti-obesity effects of lorcaserin that have been observed in humans (Tchang et al., 2020).
Sim1::Cre activity has been reported only in a handful of brain structures, including most prominently the PVH and medial amygdala (Balthasar et al., 2005). Our observation that Htr2c expression was largely preserved in the amygdala of Htr2cSim1KO mice suggests minimal overlap between Sim1::Cre and Htr2c expression in the amygdala and that the reduced food intake in these mice was not likely to be due to loss of amygdala Htr2c. These findings implicate the PVH as another critical brain site where Htr2c regulates food intake. Importantly, reactivation or deletion of Htr2c in Sim1 neurons had no effect on other physiological processes, such as energy expenditure, physical activity, or glucose homeostasis. Furthermore, mice lacking Htr2c in Sim1 neurons had normal responses in a battery of anxiety- and depression-related behavioral tests. Our findings suggest a dedicated role of Htr2c in Sim1 neurons for feeding regulation.
Delineation of PVH feeding circuits is a challenge because the PVH consists of several functionally heterogeneous neuronal populations that differ in morphology, connectivity, and peptidergic identity (Biag et al., 2012; Swanson and Sawchenko, 1983). Our immunohistochemical analyses demonstrated that Htr2c marks a distinct population of PVH neurons that largely consist of subsets of parvocellular CRH and TRH neurons. These findings are in agreement with earlier histological data showing that 5-HT terminals in the PVH are enriched in the parvocellular division of the nucleus (Sawchenko et al., 1983). Likewise, numerous studies have established an inhibitory role of PVH neurons in food intake. With few exceptions (Krashes et al., 2014), activation of PVH neurons typically leads to suppression of food intake, whereas silencing or genetic ablation of these neurons causes hyperphagia (An et al., 2020; Atasoy et al., 2012; Li et al., 2019; Liu et al., 2017; Pei et al., 2014; Xi et al., 2013). Consistent with the anorexigenic effect of CRH and TRH (Krahn et al., 1988; Vijayan and McCann, 1977), we found that adult chemogenetic stimulation of PVH Htr2c neurons suppressed hunger-induced refeeding, whereas silencing of these neurons boosted consumption in satiated mice. These findings establish that PVH Htr2c neurons can bidirectionally regulate food intake in vivo. However, future studies are warranted to map the serotonergic inputs and downstream targets of these neurons. We have shown previously that PVH MC4Rs act downstream of the ARH Htr2c/POMC neurons to mediate the anorectic effect of 5-HT agents (Xu et al., 2010). Interestingly, PVH Htr2c neurons do not express MC4R-GFP. Furthermore, it has been shown that different hypothalamic nuclei are innervated by discrete serotonergic cell groups in the midbrain (Sawchenko et al., 1983). Therefore, we predict that Htr2c neurons in the ARH and PVH may receive inputs from discrete 5-HT neurons and activate under different physiological conditions.
Htr2c regulates food intake through homeostatic and non-homeostatic mechanisms. For example, dopamine (DA) neurons in the ventral tegmental area (VTA) express Htr2c and are activated by its specific agonist, lorcaserin (Xu et al., 2017). Given the importance of DA neurons in reward processing, Htr2c may regulate hedonic feeding in part through VTA DA neurons. In support of this notion, loss of Htr2c in these neurons attenuates lorcaserin’s inhibitory effect on binge-like eating but does not alter its anorectic effect on hunger-driven feeding (Xu et al., 2017). In the current study, we found that chow-fed Htr2cnulll and Htr2cSim1RE mice were hyperphagic but had comparable food intake in a fasting-refeeding paradigm. In contrast, Htr2cSim1RE mice consumed more food than Htr2cnulll mice when both were fed an HFD. Consistent with this finding, HFD-fed Htr2cSim1KO mice ate less than controls and were protected from HFD-induced weight gain. These findings support a role of Htr2c in Sim1 neurons in HFD-induced feeding adaptations. Many PVH Htr2c neurons express CRH, and a recent study has reported that reward consumption of sucrose solution inhibits PVH CRH neurons in freely behaving mice (Yuan et al., 2019). Although we showed that chemogenetic inhibition of PVH Htr2c neurons was sufficient to increase food intake in mice, future studies are warranted to determine whether HFD consumption has a similar inhibitory effect on PVH CRH neurons and whether it induces hyperphagia by inhibiting PVH CRH/Htr2c neurons.
Htr2c has been known to couple to the Gαq pathway and, upon activation, is expected to increase neuronal activity. In support of this, we demonstrated previously that Htr2c agonists depolarized ARH POMC neurons in a Gαq/PLC-dependent manner (Sohn et al., 2011). In the current study, we verified this finding and found that mCPP depolarized genetically labeled Htr2c/POMC neurons in the ARH of triple-transgenic Htr2c::Cre, R26R::tdTomato, Pomc::GFP mice. However, recordings in the same mice revealed that none of the PVH Htr2c neurons showed a depolarizing response to Htr2c agonist treatment. In contrast, half of them were hyperpolarized by mCPP or lorcaserin. These findings, along with the observation that adult inhibition of PVH Htr2c neurons increased food intake, corroborated the reactivation/deletion experiments and demonstrated that activation of PVH Htr2c promotes feeding. We further demonstrated that hyperpolarization of PVH neurons involved opening of postsynaptic KATP conductance. Most surprisingly, however, neither pretreatment with anti-Gαq antibodies nor a PLC inhibitor prevented Htr2c-mediated inhibition. Instead, it was blocked by the Gαi/o-sensitive PTX. Although functional coupling between Htr2c and Gαi/o has been described previously in Xenopus oocytes (Chen et al., 1994; Lucaites et al., 1996), we present evidence that this mechanism is important in the mammalian nervous system. These findings suggest that Htr2c can differentially regulate neuronal excitability and food intake in different neurons by coupling with distinct intracellular signaling events.
Limitations of the study
Our study utilizes sophisticated mouse genetic models in which Htr2c is selectively deleted or restored in Sim1::Cre neurons during early life. However, whether developmental manipulations of gene expression could have contributed to the observed phenotypes in adult mice remains unknown. We also cannot rule out the possibility that alterations in Htr2c expression in Sim1::Cre neurons may cause changes in gene transcription in these neurons or in other brain regions. Additionally, we use a Htr2c::Cre activated tdTomato reporter to mark Htr2c-expressing neurons in the PVH. Although we have validated this genetic tool in a previous study (Park et al., 2020), it may not capture the entire Htr2c neuron population. Finally, we use a chemogenetic approach to demonstrate that PVH Htr2c neurons are sufficient to drive feeding behaviors in vivo. Although this type of approach has been used in many studies, these manipulations may not recapitulate the normal physiological changes in the activity of PVH Htr2c neurons.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Chen Liu (chen.liu@utsouthwestern.edu).
Materials availability
No new unique reagents were generated in this study. Reagents will be made available upon completion of a Material Transfer Agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Southwestern Medical Center and Korea Advanced Institute of Science and Technology.
Mice
All mice were housed in a temperature-controlled room with a twelve-hour-light/dark cycle (lights on at 06:00 a.m., lights off at 6:00 p.m.) in the animal facility of the University of Texas Southwestern Medical Center at Dallas. Food and water were supplied ad libitum. Mice were fed with either regular chow (#2916, Harlan-Teklad, Madison, WI; 4.25% kcal from fat) or a high-fat diet (HFD, #88137, Harlan Teklad, 42% kcal from fat). Htr2cRN/Y; Sim1::Cre ± , Htr2cRN/Y, and Htr2c+/Y; Sim1::Cre ± mice were maintained on a C57BL/6 background, whereas Htr2cfl/Y; Sim1::Cre and Htr2cfl/Y mice were on a mixed C57BL/6 × 129 background. Htr2c::Cre mice were generated by conventional gene targeting in mouse embryonic stem cells.
METHOD DETAILS
Body weight and composition
Body weight was monitored weekly from weaning (four-week-old) to twenty-five weeks of age. In the HFD studies, mice were maintained on regular chow until six weeks old before being fed HFD. Body composition was assessed using the Bruker Minispec mq10 NMR analyzer at six and sixteen weeks of age.
Metabolic cage studies
Data for food intake, meal patterns, energy expenditure, and physical activities were collected using a combined indirect calorimetry system (TSE Systems GmbH, Bad Homburg, Germany). Experimental animals were individually acclimated in the metabolic chambers for five days before data collection. During data collection, mice were initially fed the chow diet in the first two days of data collection and switched to the HFD diet for the next three days in the metabolic cages.
Behavioral analyses
All tests were carried out in the UTSW Rodent Behavior Core. Elevated plus maze, dark light exploration, open field, and forced swim tests were conducted as previously described (Liu et al., 2010). At least two independent cohorts of mice were tested in all of the behavioral paradigms. All tests were conducted during the light cycle of the day between 10:30 am and 6:00 pm. All equipment was cleaned thoroughly with 70% ethanol between individual tests to remove odor cues. Individual behavioral experiments were performed at least forty-eight hours apart from one another. The tester/scorer was blind to genotype identification.
Response to d-Fen, mCPP, and lorcaserin
We measured food intake in overnight-fasted mice hourly (up to six hours) after an intraperitoneal dose of D-Fen (6 mg/kg; Sigma-Aldrich), mCPP (3 mg/kg; Sigma-Aldrich), lorcaserin (10 mg/kg, a gift from Dr. Kathryn A. Cunningham) or sterile saline. Drugs and saline were administered in a counterbalanced manner as previously described (Xu et al., 2008; Xu et al., 2010). Food was given to individually housed mice thirty minutes after treatment.
Blood glucose, glucose, and insulin tolerance tests
For GTTs, chow-fed weight-matched animals were fasted for 18 hours (starting at 3 p.m.) with water provided ad libitum. At 9:00 a.m. of the next day, blood glucose levels were monitored at 15, 30, 60, 90, and 120 minutes after an i.p. dose of glucose (1.5 g/kg body weight). For ITTs, chow-fed weight-matched mice were fasted for 2 hours with water ad libitum on the experiment day. Mice were given an i.p. dose of insulin (1 U/kg, Eli Lilly and Company, HI-210), and blood glucose levels were monitored at 15, 30, 60, 90, and 120 minutes after insulin injection. Blood glucose was analyzed using an AlphaTrak (Abbott Laboratories, North Chicago, IL) meter designed for use in rodents.
Histology
Mice were anesthetized with an overdose of chloral hydrate (500mg/kg, i.p.) followed by transcardial perfusion with 10% formalin (in DEPC-treated saline). Brains were collected and then subjected to a two-hour postfix with 10% formalin (in DEPC-treated saline) before being cryoprotected in 30% sucrose (in 1XPBS) overnight at 4°C. On the next day, twenty-five mm coronal brain sections were prepared with a freezing microtome. The free-floating sections were stored in cryoprotectant at −20°C until needed for immunohistochemistry or RNA in situ hybridization. For immunostaining, sections were stained with primary antibodies against oxytocin (Cat# ab2078, abcam), arginine vasopressin (Cat# PA5-19819, Thermo Scientific), tyrosine hydroxylase (Cat# ab112, abcam), c-FOS (Cat# sc-52-G Santa Cruz Biotechnology), and GFP (Cat# A6455, Life Technologies) followed by secondary antibodies with Alexa Fluor® 488 (Life Technologies) or DAB chromogen (DAKO) according to manufacturers’ protocols. In situ hybridization was performed as described previously (Vianna et al., 2012). Riboprobes were made from amplified PCR fragments with T7 and T3 promoters incorporated into the primer sequences so that the PCR product could be directly used as a template for the synthesis of the sense and antisense riboprobes.
Electrophysiology
Whole-cell patch-clamp recordings from Htr2c neurons were conducted as previously described (Sohn et al., 2011). Briefly, 3- to 5-week-old male or female mice were deeply anesthetized with i.p. injection of 7% chloral hydrate or isoflurane inhalations and trans-cardially perfused with a modified ice-cold artificial CSF (ACSF) (described below), in which an equiosmolar amount of sucrose was substituted for NaCl. The mice were then decapitated, and the entire brain was removed, and immediately submerged in ice-cold, carbogen-saturated (95% O2 and 5% CO2) ACSF (126 mM NaCl, 2.8 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 5 mM glucose). A brain block containing the hypothalamus was made. Coronal sections (250 μm) were cut with a Leica VT1000S or VT1200S Vibratome and then incubated in oxygenated ACSF at room temperature for at least 1 h before recording. Slices were transferred to the recording chamber and allowed to equilibrate for 10–20 min before recording. The slices were bathed in oxygenated ACSF (32°C–34°C) at a flow rate of ~4 ml/min.The pipette solution for whole-cell recording was modified to include an intracellular dye (Alexa Fluor 488) for whole-cell recording: 120 mM K-gluconate, 10 mM KCl, 10 mM HEPES, 5 mM EGTA, 1 mM CaCl2, 1 mM MgCl2, and 2 mM MgATP, 0.03 mM Alexa Fluor 488 hydrazide dye, pH 7.3. Epifluorescence was briefly used to target fluorescent cells, at which time the light source was switched to infrared differential interference contrast imaging to obtain the whole-cell recording (Zeiss Axioskop FS2 Plus equipped with a fixed stage and a QuantEM:512SC electron-multiplying charge-coupled device camera or Nikon Eclipse FN1 equipped with a fixed stage and an optiMOS scientific CMOS camera). Electrophysiological signals were recorded using an Axopatch 700B amplifier (Molecular Devices), low-pass filtered at 2–5 kHz, and analyzed offline on a PC with pCLAMP programs (Molecular Devices). Recording electrodes had resistances of 2.5–5 MΩ when filled with the K-gluconate internal solution. Input resistance was assessed by measuring voltage deflection at the end of the response to a hyperpolarizing rectangular current pulse steps (500 ms of −10 to −50 pA or −5 to −25 pA). TTX, CNQX, AP5, picrotoxin, U73122, pertussis toxin, and 5-HT were obtained from Tocris. Anti-Gq antibody was obtained from Merck Millipore. Tolbutamide and glibenclamide were obtained from Sigma-Aldrich. All solutions were made according to the manufacturer’s specifications. Stock solutions of U73122 were made by dissolution in DMSO (Sigma-Aldrich). The concentration of DMSO in the external solution was maintained below 0.1%. Stock solutions of tolbutamide were made by dissolution in 100% ethanol, with the final ethanol concentration in ACSF less than 0.5%. Stock solutions of all other drugs were made by dissolution in distilled water. Membrane potential values were not compensated to account for junction potential (−8 mV).
Stereotaxic injections
Detailed experimental procedures were carried out according to a published protocol (Cetin et al., 2006). Briefly, six to eight-week-old Htr2c-Cre mice were deeply anesthetized by an i.p. dose of ketamine/xylazine. Mice were placed into a stereotaxic frame and a small opening was made in the skull directly over the injection site (bregma, AP, −0.7mm, DV, −4.5mm, ± 0.2mm from bregma). Coordinates for stereotaxic injections were obtained from the Paxinos mouse brain atlas. About 75-100 nL of AAV viruses that contain either stimulatory (hM3Dq) or inhibitory (hM4Di) DREADD constructs (AAV serotype 2 or 8, UNC viral core) were pressure injected into the PVH (unilateral or bilateral). After injection, all animals were allowed to recover for at least three weeks before being used for the feeding studies.
QUANTIFICATION AND STATISTICAL ANALYSIS
Replicate information is indicated in the figure legends. All results are given as mean ± SEM and analyzed by using statistical tools implemented in Prism (GraphPad version 9). Statistical analyses were performed using the Student’s t test and regular one-way or two-way analysis of variance (ANOVA). Differences with p < 0.05 were considered to be significant. p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti- oxytocin | Abcam | RRID: AB_302818, Cat# ab2078 |
| Anti- arginine vasopressin | Thermo Scientific | RRID: AB_11003086, Cat# PA5-19819 |
| Anti- tyrosine hydroxylase | Abcam | RRID: AB_297840, Cat# ab112 |
| Anti-c-Fos | Santa Cruz | RRID: AB_2629503, Cat# sc-52-G |
| Anti- GFP | Life Technologies | RRID: AB_221570, Cat# A6455 |
| Anti-Gq/11α | Merck Millipore | RRID: AB_310221, Cat# 06-709 |
| Bacterial and virus strains | ||
| hSyn-DIO-mCherry | Addgene plasmid | RRID:Addgene_50459, Cat# 50459 |
| hSyn-DIO-hM3D(Gq)-mCherry | Addgene plasmid | RRID:Addgene_44361, Cat# 44361 |
| hSyn-DIO-hM4D(Gi)-mCherry | Addgene plasmid | RRID:Addgene_44362, Cat# 44362 |
| Chemicals, peptides, and recombinant proteins | ||
| Insulin (Humulin) | Lilly | Cat# HI-210 |
| Dextrose 50% Solution | Hospira | Cat# 00409-6648-02 |
| D-Fen | Sigma | Cat #F112 |
| mCPP | Sigma | Cat #C-089 |
| Lorcaserin | A gift from Dr. Kathryn A. Cunningham | N/A |
| Chloral hydrate | Sigma | Cat #C8383 |
| Sucrose | Sigma | Cat #S0389 |
| CNO | Sigma | Cat #SML2304 |
| CNQX disodium salt | Tocris | Cat #1045 |
| DL-AP5 | Tocris | Cat #0105 |
| Picrotoxin | Tocris | Cat #1128 |
| TTX citrate | Tocris | Cat #1069 |
| U73122 | Tocris | Cat #1268 |
| Pertussis toxin | Tocris | Cat #3097 |
| 5-HT | Tocris | Cat #3547 |
| Tolbutamide | Sigma | Cat #T0891 |
| Glibenclamide | Sigma | Cat #G0639 |
| Rodent Diet With 42 kcal% Fat | Harlan Teklad | Cat# 88137 |
| Standard chow | Harlan Teklad | Cat# 2916 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: JAX:000664 |
| Mouse: Sim1::Cre | Bradford B Lowell | RRID: MGI:3692526 |
| Mouse: Htr2c::Cre | Chen Liu | Park et al., 2020 |
| Mouse: Htr2cnull | Joel K Elmquist | RRID: MGI:3841486 |
| Mouse: Htr2cflox | Joel K Elmquist | RRID: MGI:5569357 |
| Mouse: Gt(ROSA)26Sor(RCL-tdT)-D (Ai14) | The Jackson Laboratory | RRID: JAX: 007914 |
| Oligonucleotides | ||
| See Table S1 for oligo nucleotide information | N/A | |
| Other | ||
| Indirect calorimetry system – TSE PhenoMaster | TSE Systems Germany | N/A |
| EchoMRI | EchoMRI Corporation | N/A |
| Vibratome | Leica | N/A |
| Glucometer | AlphaTrak | N/A |
| GraphPad version 9 | GraphPad Software Inc | https://www.graphpad.com |
Highlights.
Selective restoration of Htr2c in Sim1 neurons promotes high-fat diet (HFD) feeding
Selective deletion of Htr2c in Sim1 neurons reduces HFD feeding
PVH Htr2c neurons bidirectionally regulate food intake
Htr2c is coupled to the Gαi/o-KATP pathway in PVH neurons to inhibit their activity
ACKNOWLEDGMENTS
The authors would like to thank the staff of the UTSW Transgenic, Behavioral Phenotyping, and Metabolic Phenotyping Cores. This work was supported by the NIH (R01 DK088423 and P50 DA033935 to K.A.C and R01 DK114036 to C.L.) and the National Research Foundation of Korea (NRF-2019R1A2C 2005161 to J.-W.S.). The authors thank Dr. Joel K. Elmquist for helpful comments that improved the manuscript as well as Efferent Manuscript Services for editing services.
Footnotes
DECLARATION OF INTERESTS
K.A.C. has current research funding from VidaLibreBio, Inc. for research unrelated to this study.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109997.
REFERENCES
- An JJ, Kinney CE, Tan JW, Liao GY, Kremer EJ, and Xu B (2020). TrkB-expressing paraventricular hypothalamic neurons suppress appetite through multiple neurocircuits. Nat. Commun 11, 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Betley JN, Su HH, and Sternson SM (2012). Deconstruction of a neural circuit for hunger. Nature 488, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. (2005). Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505. [DOI] [PubMed] [Google Scholar]
- Baver SB, Hope K, Guyot S, Bjørbaek C, Kaczorowski C, and O’Connell KM (2014). Leptin modulates the intrinsic excitability of AgRP/NPY neurons in the arcuate nucleus of the hypothalamus. J. Neurosci 34, 5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund ED, Liu C, Sohn JW, Liu T, Kim MH, Lee CE, Vianna CR, Williams KW, Xu Y, and Elmquist JK (2013). Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J. Clin. Invest 123, 5061–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Cao ZF, Ritola KD, and Sternson SM (2013). Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell 155, 1337–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biag J, Huang Y, Gou L, Hintiryan H, Askarinam A, Hahn JD, Toga AW, and Dong HW (2012). Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J. Comp. Neurol 520, 6–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Haubensak W, Anthony TE, and Anderson DJ (2014). Central amygdala PKC-δ(+) neurons mediate the influence of multiple anorexigenic signals. Nat. Neurosci 17, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin A, Komai S, Eliava M, Seeburg PH, and Osten P (2006). Stereotaxic gene delivery in the rodent brain. Nat. Protoc 1, 3166–3173. [DOI] [PubMed] [Google Scholar]
- Chen Y, Baez M, and Yu L (1994). Functional coupling of the 5-HT2C serotonin receptor to G proteins in Xenopus oocytes. Neurosci. Lett 179, 100–102. [DOI] [PubMed] [Google Scholar]
- Colman E, Golden J, Roberts M, Egan A, Weaver J, and Rosebraugh C (2012). The FDA’s assessment of two drugs for chronic weight management. N. Engl. J. Med 367, 1577–1579. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, and Low MJ (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. [DOI] [PubMed] [Google Scholar]
- D’Agostino G, Lyons D, Cristiano C, Lettieri M, Olarte-Sanchez C, Burke LK, Greenwald-Yarnell M, Cansell C, Doslikova B, Georgescu T, et al. (2018). Nucleus of the Solitary Tract Serotonin 5-HT2C Receptors Modulate Food Intake. Cell Metab. 28, 619–630.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenezer IS, and Surujbally A (2007). The effects of 8-hydroxy-2-(di-n-propylamino)-tetralin (8-OH-DPAT) on food intake in non-deprived C57BL6 mice. Eur. J. Pharmacol 559, 184–188. [DOI] [PubMed] [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, and Zoghbi HY (2008). Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59, 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler LK, Pronchuk N, Nonogaki K, Zhou L, Raber J, Tung L, Yeo GS, O’Rahilly S, Colmers WF, Elmquist JK, and Tecott LH (2007). Serotonin activates the hypothalamic-pituitary-adrenal axis via serotonin 2C receptor stimulation. J. Neurosci 27, 6956–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, and Elmquist JK (2008). Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Invest 118, 1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BJ,and Mezey E (1989). Distribution of serotonin 5-HT1C receptor mRNA in adult rat brain. FEBS Lett. 247, 453–62. [DOI] [PubMed] [Google Scholar]
- Holder JL Jr., Zhang L, Kublaoui BM, DiLeone RJ, Oz OK, Bair CH, Lee YH, and Zinn AR (2004). Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am. J. Physiol. Endocrinol. Metab 287, E105–E113. [DOI] [PubMed] [Google Scholar]
- Kenny PJ (2011). Reward mechanisms in obesity: new insights and future directions. Neuron 69, 664–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn DD, Gosnell BA, Levine AS, and Morley JE (1988). Behavioral effects of corticotropin-releasing factor: localization and characterization of central effects. Brain Res. 443, 63–69. [DOI] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, Vong L, Pei H, Watabe-Uchida M, Uchida N, et al. (2014). An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi Y, Tung RT, Ito H, and Nakajima T (1992). G protein activation of cardiac muscarinic K+ channels. Prog. Neurobiol 39, 229–246. [DOI] [PubMed] [Google Scholar]
- Li MM, Madara JC, Steger JS, Krashes MJ, Balthasar N, Campbell JN, Resch JM, Conley NJ, Garfield AS, and Lowell BB (2019). The Paraventricular Hypothalamus Regulates Satiety and Prevents Obesity via Two Genetically Distinct Circuits. Neuron 102, 653–667.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Yoo ES, Li X, Wyler SC, Chen X, Wan R, Arnold AG, Birnbaum SG, Jia L, Sohn JW, and Liu C (2021). The atypical antipsychotic risperidone targets hypothalamic melanocortin 4 receptors to cause weight gain. J. Exp. Med 218, e20202484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Maejima T, Wyler SC, Casadesus G, Herlitze S, and Deneris ES (2010). Pet-1 is required across different stages of life to regulate serotonergic function. Nat. Neurosci 13, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Conde K, Zhang P, Lilascharoen V, Xu Z, Lim BK, Seeley RJ, Zhu JJ, Scott MM, and Pang ZP (2017). Enhanced AMPA Receptor Trafficking Mediates the Anorexigenic Effect of Endogenous Glucagon-like Peptide-1 in the Paraventricular Hypothalamus. Neuron 96, 897–909.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucaites VL, Nelson DL, Wainscott DB, and Baez M (1996). Receptor subtype and density determine the coupling repertoire of the 5-HT2 receptor subfamily. Life Sci. 59, 1081–1095. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Yamamoto A, Scearce-Levie K, Saudou F, and Hen R (1998). Absence of fenfluramine-induced anorexia and reduced c-Fos induction in the hypothalamus and central amygdaloid complex of serotonin 1B receptor knock-out mice. J. Neurosci 18, 5537–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Lévy E, Mitchell GA, Himms-Hagen J, and Fan CM (2001). Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet 10, 1465–1473. [DOI] [PubMed] [Google Scholar]
- Mosienko V, Beis D, Pasqualetti M, Waider J, Matthes S, Qadri F, Bader M, and Alenina N (2015). Life without brain serotonin: reevaluation of serotonin function with mice deficient in brain serotonin synthesis. Behav. Brain Res 277, 78–88. [DOI] [PubMed] [Google Scholar]
- Nonogaki K, Strack AM, Dallman MF, and Tecott LH (1998). Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nat. Med 4, 1152–1156. [DOI] [PubMed] [Google Scholar]
- Pagoto SL, Spring B, McChargue D, Hitsman B, Smith M, Appelhans B, and Hedeker D (2009). Acute tryptophan depletion and sweet food consumption by overweight adults. Eat. Behav 10, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Williams KW, Liu C, and Sohn JW (2020). A neural basis for tonic suppression of sodium appetite. Nat. Neurosci 23, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H, Sutton AK, Burnett KH, Fuller PM, and Olson DP (2014). AVP neurons in the paraventricular nucleus of the hypothalamus regulate feeding. Mol. Metab 3, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW, Steinbusch HW, and Verhofstad AA (1983). The distribution and cells of origin of serotonergic inputs to the paraventricular and supraoptic nuclei of the rat. Brain Res. 277, 355–360. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, and Zeltser LM (2013). Functional organization of neuronal and humoral signals regulating feeding behavior. Annu. Rev. Nutr 33, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Xu Y, Jones JE, Wickman K, Williams KW, and Elmquist JK (2011). Serotonin 2C receptor activates a distinct population of arcuate pro-opiomelanocortin neurons via TRPC channels. Neuron 71, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, and Sawchenko PE (1983). Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu. Rev. Neurosci 6, 269–324. [DOI] [PubMed] [Google Scholar]
- Tchang BG, Abel B, Zecca C, Saunders KH, and Shukla AP (2020). An up-to-date evaluation of lorcaserin hydrochloride for the treatment of obesity. Expert Opin. Pharmacother 21, 21–28. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, and Julius D (1995). Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature 374, 542–546. [DOI] [PubMed] [Google Scholar]
- Thomsen WJ, Grottick AJ, Menzaghi F, Reyes-Saldana H, Espitia S, Yuskin D, Whelan K, Martin M, Morgan M, Chen W, et al. (2008). Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J. Pharmacol. Exp. Ther 325, 577–587. [DOI] [PubMed] [Google Scholar]
- Vianna CR, Donato J Jr., Rossi J, Scott M, Economides K, Gautron L, Pierpont S, Elias CF, and Elmquist JK (2012). Cannabinoid receptor 1 in the vagus nerve is dispensable for body weight homeostasis but required for normal gastrointestinal motility. J. Neurosci 32, 10331–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers SP, Clifton PG, Dourish CT, and Tecott LH (1999). Reduced satiating effect of d-fenfluramine in serotonin 5-HT(2C) receptor mutant mice. Psychopharmacology (Berl.) 143, 309–314. [DOI] [PubMed] [Google Scholar]
- Vijayan E, and McCann SM (1977). Suppression of feeding and drinking activity in rats following intraventricular injection of thyrotropin releasing hormone (TRH). Endocrinology 100, 1727–1730. [DOI] [PubMed] [Google Scholar]
- Wurtman JJ, and Wurtman RJ (1977). Fenfluramine and fluoxetine spare protein consumption while suppressing caloric intake by rats. Science 198, 1178–1180. [DOI] [PubMed] [Google Scholar]
- Wyler SC, Lord CC, Lee S, Elmquist JK, and Liu C (2017). Serotonergic Control of Metabolic Homeostasis. Front. Cell. Neurosci 11, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi D, Roizen J, Lai M, Gandhi N, and Kublaoui B (2013). Paraventricular nucleus Sim1 neuron ablation mediated obesity is resistant to high fat diet. PLoS ONE 8, e81087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Jones JE, Kohno D, Williams KW, Lee CE, Choi MJ, Anderson JG, Heisler LK, Zigman JM, Lowell BB, and Elmquist JK (2008). 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron 60, 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Jones JE, Lauzon DA, Anderson JG, Balthasar N, Heisler LK, Zinn AR, Lowell BB, and Elmquist JK (2010).Aserotonin and melanocortin circuit mediates D-fenfluramine anorexia. J. Neurosci 30, 14630–14634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, He Y, Cao X, Valencia-Torres L, Yan X, Saito K, Wang C, Yang Y, Hinton A Jr., Zhu L, et al. (2017). Activation of Serotonin 2C Receptors in Dopamine Neurons Inhibits Binge-like Eating in Mice. Biol. Psychiatry 81, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lu Y, Cassidy RM, Mangieri LR, Zhu C, Huang X, Jiang Z, Justice NJ, Xu Y, Arenkiel BR, and Tong Q (2019). Identification of a neurocircuit underlying regulation of feeding by stress-related emotional responses. Nat. Commun 10, 3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Wu W, Chen M, Cai F, Fan C, Shen W, Sun W, and Hu J (2019). Reward Inhibits Paraventricular CRH Neurons to Relieve Stress. Curr. Biol 29, 1243–1251.e4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.