

Abstract
Natural killer (NK) cells mediate the cytolysis of transformed cells and are currently used as an adoptive cellular therapy to treat cancer. Infection with human cytomegalovirus has been shown to expand a subset of “adaptive” NK cells expressing the activation receptor NKG2C that have preferred functional attributes distinct from conventional NK cells. Because NKG2C delivers a strong activating signal to NK cells, we hypothesized that NKG2C could specifically trigger NK-cell-mediated antitumor responses. To elicit a tumor-directed response from NKG2C+ NK cells, we created an anti-NKG2C/IL-15/anti-CD33 killer engager called NKG2C-KE that directs NKG2C+ cells to target CD33+ cells and tumor-associated antigen expressed by acute myelogenous leukemia cells. The NKG2C-KE induced specific degranulation, interferon-γ production, and proliferation of NKG2C-expressing NK cells from patients who reactivated cytomegalovirus after allogeneic transplantation. The NKG2C-KE was also tested in a more homogeneous system using induced pluripotent stem cell (iPSC)-derived NK (iNK) cells that have been engineered to express NKG2C at high levels. The NKG2C-KE triggered iNK-cell-mediated cytotoxicity against CD33+ cells and primary AML blasts. The NKG2C-KE-specific interaction with adaptive NK and NKG2C+ iNK cells represents a new immunotherapeutic paradigm that uniquely engages highly active NK cells to induce cytotoxicity against AML through redirected targeting.
Keywords: natural killer cell, killer engager, NKG2C, iNK, acute myeloid leukemia, adoptive transfer, immunotherapy, NK cells
Graphical abstract
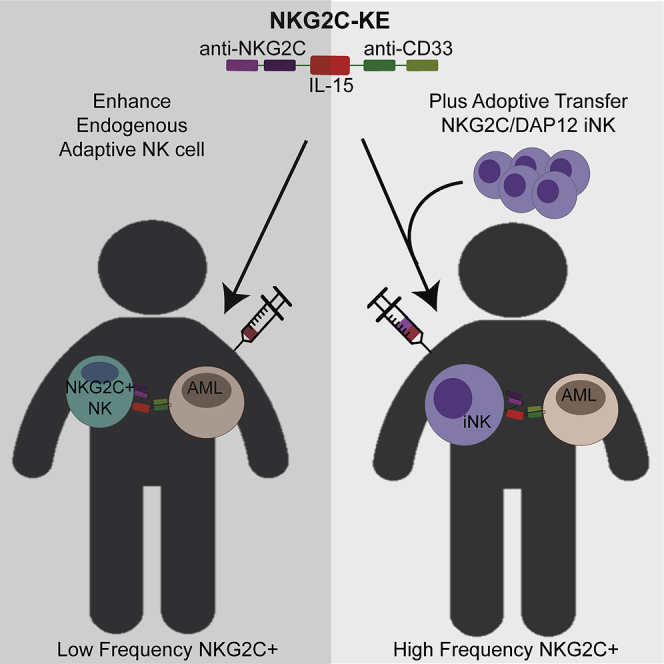
Chiu and colleagues design a strategy to make cytomegalovirus-induced-adaptive NK cells antigen specific by creating an anti-NKG2C/IL-15/anti-CD33 killer engager that directs NKG2C+ NK cells to CD33+ tumors. Combining this engager with iPSC-derived NK cells engineered to express NKG2C is the basis for an off-the-shelf therapeutic strategy.
Introduction
Natural killer (NK) cells are innate lymphocytes whose main function is to survey the body for virally infected and malignant cells. Upon recognition of a target cell, sufficient activation will trigger a functional response, resulting in the secretion of cytokines and cytolytic molecules that initiate apoptosis of the target. NK cell-mediated antitumor responses are triggered by the binding of germline-encoded activation receptors on NK cells to stress-induced ligands on tumor cells. The initiation of NK cell functional responses is highly regulated through the balance between inhibitory and activating signals. Inhibitory receptors such as the killer cell immunoglobulin-like receptors (KIRs) recognize major histocompatibility complex (MHC) class I molecules, which are generally found at higher frequencies on healthy tissues.1 Thus, NK cells are particularly active against cells that lack or downregulate MHC class I. Lack of inhibitory signals alone is insufficient to activate NK cells. Activating receptors, such as NKG2D, recognize inducible stress ligands such as MICA/B and ULBPs, while natural cytotoxicity receptors, such as NKp30, NKp44, and NKp46 recognize a variety of host- and pathogen-encoded ligands.2, 3, 4, 5 Once activated, NK cells degranulate, releasing perforin and granzyme in a directed manner. They also produce inflammatory cytokines such as interferon-γ (IFN-γ). However, many tumors maintain relatively normal levels of MHC class I and evade NK cell surveillance and elimination.
Despite the evasion of NK cell responses by tumor cells, NK cells have been shown to exhibit effective control of acute myeloid leukemia (AML) and other hematopoietic cancers. In addition to their ability to mediate natural cytotoxicity, NK cells also perform antibody-dependent cellular cytotoxicity (ADCC).6 ADCC is mediated by the low-affinity Fc receptor FcγRIIIa (CD16a). This Fc receptor is expressed at high levels on peripheral blood NK cells and, unlike most other activating receptors, triggers functional responses without the need for simultaneous engagement of a co-receptor.6,7 Our group previously described the production and function of a tri-specific molecule composed of anti-CD16 and anti-CD33 single-chain variable fragments (scFvs) linked by an interleukin-15 (IL-15) moiety (161533). This tri-specific killer engager (TriKE) effectively directs NK cell killing toward CD33-expressing target cells while also providing a proliferation and survival signal through IL-15.8 However, a variant of CD16 (CD16b) is highly expressed by neutrophils and binds the Fc portion of ADCC-mediating antibodies and the anti-CD16 component of TriKEs. Neutrophils, comprising a larger percentage of cells in peripheral blood, make them a potential sink for CD16 targeting agents that have a broad specificity for both CD16a and CD16b.7 Furthermore, a subset of monocytes expresses CD16a, creating a potential for off-target effects even when using engagers that specifically target CD16a. While the anti-CD16 component in the 161533 TriKE is potent, the potential neutrophil and monocyte sink could lead to unwanted side effects and a need for higher dosing. Thus, the use of NK cell-specific activating receptors other than CD16a for engager-mediated redirection may have importance for immunotherapy to treat cancer.
An alternative activating receptor of interest is the c-lectin heterodimer NKG2C/CD94. NKG2C is predominantly expressed on NK cells, although some subsets of T cells also express this receptor at low levels. NKG2C is an activating receptor that forms a heterodimer with CD94 and signals through the adaptor protein DAP12.9 Similar to CD16, the ligation of NKG2C triggers strong responses in resting NK cells in the absence of co-activation.10 NKG2C is predominantly expressed on resting NK cells that lack NKG2A, an inhibitory receptor that also forms a heterodimer with CD94.11, 12, 13 Individuals who have had prior exposure to human cytomegalovirus (CMV) have an increased proportion of NKG2C+ NK cells compared to CMV-naive individuals. Despite the positive association with CMV serostatus, CMV-seropositive individuals have highly variable frequencies of NKG2C+ NK cells.14 The NKG2C+ population following CMV exposure represents a distinct subset of NK cells, called adaptive NK cells, that exhibit enhanced ADCC, a genome-wide methylation signature similar to that of effector memory CD8+ T cells, and properties of immune memory.9,15, 16, 17, 18 Our group has established a clinical link between the frequency of adaptive NK cells post-hematopoietic cell transplantation (HCT) and protection from cancer relapse.19 The enhanced functionality of NKG2C+ adaptive NK cells against AML, coupled with its expression being largely limited to the NK cells, makes NKG2C a promising target for immune engagement. In this study, we describe the development of a target engager platform in which we created a novel anti-NKG2C/IL-15/anti-CD33 KE (NKG2C-KE) and combined it with a uniquely engineered NKG2C+ induced pluripotent stem cell (iPSC)-derived NK (iNK) cell to overcome the low and variable availability of NKG2C found on primary NK cells. We tested the efficacy of this platform to efficiently and specifically target and kill AML.
Results
Functional validation of a novel NKG2C-KE
To determine whether NKG2C engagement can be leveraged to specifically activate NK cells, an NKG2C-KE was generated using the sequence encoding an anti-NKG2C antibody previously described by our group.20 The variable heavy- and light-chain sequences of the anti-NKG2C antibody were cloned into a construct containing sequences encoding an anti-CD33 single-chain variable fragment (scFv) component to target AML and a wild-type IL-15 component sequence to link the two scFvs. Peripheral blood from healthy CMV-seropositive donors were screened by flow cytometry for the presence of NKG2C+ NK cells and then placed into two groups based on whether greater than (NKG2Chigh) or less than (NKG2Clow) 10% of NK cells were NKG2C+. Peripheral blood mononuclear cells (PBMCs) were isolated from each donor and co-cultured with the CD33+ AML cell line THP-1 with no treatment, recombinant human IL-15 (rhIL-15), or the NKG2C-KE in a 5-h flow cytometric functional assay. When compared to NKG2Chigh cells cultured with rhIL-15, NKG2Chigh cells cultured with NKG2C-KE exhibited increased degranulation at 0.3 nM (18.5% versus 13.3%, p = 0.05), 3 nM (21.9% versus 13.9%, p = 0.0001), and 30 nM concentrations (22.6% versus 13.6%, p < 0.0001) (Figure 1A). When comparing NKG2Clow and NKG2Chigh NK cells, the NKG2C-KE triggered more robust degranulation for NKG2Chigh NK cells at the 3 nM (10.7% versus 21.9%, p = 0.009) and 30 nM (12.2% versus 22.6%, p = 0.02) concentrations (Figure 1A). Similar to the degranulation response, NKG2Chigh NK cells produced more IFN-γ in response to the NKG2C-KE compared to rhIL-15. Compared to rhIL-15, IFN-γ was increased with the NKG2C-KE at 3 nM (8.1% versus 16.1%, p = 0.0001) and 30 nM concentrations (8.0% versus 15.9%, p = 0.0002). When comparing NKG2Clow to NKG2Chigh frequency donors, the NKG2Chigh NK cells produced more IFN-γ with the NKG2C-KE at 3 nM (4.4% versus 16.1%, p = 0.01) and 30 nM (5.5% versus 15.9%, p = 0.04). No statistically significant differences in degranulation or IFN-γ production were observed for NKG2Clow NK cells treated with NKG2C-KE compared to rhIL-15 (Figure 1B). To evaluate whether NK cell function correlated with the frequency of NKG2C expression, we performed a linear regression analysis. While there was no association between degranulation and the frequency of NKG2C expression on NK cells in rhIL-15 cultures, NK cells stimulated with the NKG2C-KE exhibited higher frequencies of degranulation that correlated with the percentages of NKG2C expression at all concentrations (0.3 nM [R2 = 0.7, p = 0.0002], 3 nM [R2 = 0.7, p = 0.0003], and 30 nM [R2 = 0.6, p = 0.0009]) (Figure 1C). Frequencies of IFN-γ production were also associated with NKG2C frequencies when NK cells were stimulated with various concentrations of the NKG2C-KE. These data indicate that the NKG2C-KE specifically activates NK cells based on NKG2C expression.
Figure 1.

Adaptive NK cells from CMV seropositive healthy donors with higher frequencies of NKG2C respond to the NKG2C-KE
CMV seropositive donors were divided into groups based on expression of <10% or >10% NKG2C+ NK cells. These populations were then incubated for 5 h with THP1 tumor cells and stained for degranulation by CD107a (A) and IFN-γ production (B). Correlations between NKG2C frequency and degranulation or IFN-γ production are shown in (C). Graphs indicate the means ± SEMs analyzed using a 2-way ANOVA for (A) and (B). The correlation in (C) was analyzed using a linear regression analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
NKG2C-KE activates and expands NKG2C+ NK cells from HCT patients who experienced CMV reactivation
Our lab has previously identified a correlation between the reconstitution of “adaptive” NK cells (defined as CD57+NKG2C+) post-allogeneic HCT and protection from relapse.19 Furthermore, reconstitution of this unique NK cell subset is found predominantly in transplant recipients who were CMV seropositive and experienced CMV reactivation post-transplant. This association suggests enhanced antitumor function by adaptive NKG2C+ NK cells compared to canonical NK cells lacking NKG2C expression. Thus, we hypothesized that the NKG2C-KE would preferentially activate NK cell function in transplant recipients who reactivated CMV compared to patients who were CMV seronegative. PBMCs collected from patients 6 months post-transplant were co-cultured with THP-1 AML cells, which express high levels of CD33, in the presence or absence of the NKG2C-KE. Compared to the control no-treatment condition, CMV-reactivated patient NK cells degranulated >2-fold greater in the presence of the NKG2C-KE (16% versus 48.2%, p < 0.0001) (Figure 2A) and produced significantly more IFN-γ (4.3% versus 29.8%, p ≤ 0.0001) (Figure 2B). However, this effect was not observed when testing NK cells from post-transplant CMV-seronegative patients who reconstitute with low frequencies of NKG2C+ NK cells (Figures 2A and 2B).
Figure 2.

NKG2C-KE exhibit higher function in PBMCs from 6 months after transplantation in those who reactivate CMV
PBMCs from patients after hematopoietic transplant were stratified as to whether they were CMV seronegative and did not reactivate CMV or whether they reactivated CMV in the first 100 days after transplant. Respective samples were incubated with THP-1 tumor targets in a 5-h assay and stained for CD107a degranulation (A) and IFN-γ production (B). Correlation of CD107a degranulation (C) and IFN-γ production (D) with NKG2C expression on NK cells is shown. Representative samples of CMV-reactivated and CMV-seronegative staining following 7-day incubation with indicated treatment after CellTrace labeling (E). Compiled data of proliferated NK cells in CMV-reactivated (n = 9) and CMV-seronegative (n = 4) patients 6 months post-transplantation showing percentage of NKG2C (F) and ratio of NKG2C+:NKG2C− (G). Graphs indicate the means ± SEMs analyzed using a 2-way ANOVA and mixed-effects analysis for (A) and (B), linear regression for (C) and (D), and paired t tests for (F) and (G). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Due to the differences in frequencies and quality of NKG2C+ NK cells between CMV-seronegative patients and patients who reactivated CMV, we sought to determine whether the responsiveness to the NKG2C-KE correlated with the frequencies of NKG2C. We found a high correlation between NKG2C expression and degranulation (R2 = 0.6, p < 0.0001), as well as IFN-γ production (R2 = 0.9, p < 0.0001), further confirming the specificity of the NKG2C-KE (Figures 2C and 2D). We then used the post-transplant PBMCs to test whether the NKG2C-KE could induce selective proliferation of adaptive NK cells. We observed specific proliferation of NKG2C+ NK cells from CMV-reactivated patients with the NKG2C-KE when compared to rhIL-15, suggesting a targeted delivery of the IL-15 by NKG2C-KE (Figure 2E). While rhIL-15 resulted in the broad proliferation of all NK cells, the IL-15 in the context of the NKG2C-KE was selective for the NKG2C+ population. When evaluating the proportion of NK cells that proliferate in response to the NKG2C-KE, NKG2C+ cells constituted a larger proportion of proliferating cells than those treated with rhIL-15 (Figure 2F: 65.2% versus 34.8%, p < 0.0001), resulting in a higher NKG2C+:NKG2C− ratio with the NKG2C-KE (Figure 2G). In samples in which NK cells contained no or low frequencies of NKG2C, rhIL-15 and the NKG2C-KE triggered similar proportions of NK cell proliferation.
NKG2C-KE controls CD33+ AML and results in enhanced persistence of NKG2C+ adaptive NK cells in vivo
Based on our in vitro data showing the specificity of NKG2C+ NK cells and the NKG2C-KE, we wanted to validate our findings in vivo. We chose a well-established myeloid leukemia model in which we inject NOD scid gamma (NSG) mice that lack mouse lymphocytes (T, B, and NK cells) with a luciferase-labeled CD33+ HL-60 myeloid tumor.8, 21, 22 AML was established, and 3 days later, animals were treated with expanded PB NK cells from a CMV+ normal donor comprising 13.5% NKG2C+ and 91.3% CD16+ NK cells (Figures 3A and 3B). NKG2C-KE or a CD161533 TriKE8, 21 was administered 5 times weekly for 3 weeks with bioluminescence imaging (BLI) performed weekly. Blood was sampled on days 14 and 28 to evaluate for NK cell persistence and expansion. In comparison to control animals (average radiance: 4.8e−9 ± 2.1e−9), both the 161533 TriKE or NKG2C-KE in combination with NK cells mediated potent tumor control (1.2e−8 ± 2.3e−8 versus 1.3e−8 ± 1.8e−8, p = 0.99) (Figures 3C and 3D), despite the 7-fold difference in CD16 versus NKG2C target for the immune engager on expanded PB NK cells (Figure 3B). In vivo NK cells 28 days after adoptive transfer showed greater persistence of NKG2C+ NK cells when stimulated with the NKG2C-KE treated compared to 161533 TriKE showing preferential persistence/expansion of adaptive NK cells in vivo. At day 14, the decreased NKG2C on NK cells with NKG2C-KE treatment was a result of receptor occupancy as the mice undergoing active NKG2C-KE treatment blocked NKG2C detection by flow cytometry (Figure 3E).
Figure 3.

NKG2C-KE controls tumor as effectively as 161533 TriKE
(A) Schematic of mouse model. NSG mice were injected with 750,000 HL60-Luc cells, then 3 days later, BLI injected with 5 million thawed expanded NK cells. Mice received 5× weekly intraperitoneal (i.p.) injections of drug for 3 weeks; BLI at days 6, 13, 20, and 27; and bleeds at days 14 and 28. (B) Frozen expanded NK cells thawed and stained for CD16 and NKG2C. (C) Individual bioluminescence images separated into groups from days 6, 13, 20, 27, and 34. (D) Quantification of luminescence measured at days 0, 6, 13, 20, 27, and 34. (E) Blood from mice at days 14 and 28 stained for CD16 and NKG2C. Graphs indicate the means ± SDs analyzed using 2-way ANOVA and RM analysis used for (C) and (D). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Generation and functional attributes of iPSC-derived NK cells with transgenic expression of NKG2C
There is high variability in the frequency of NKG2C-expressing adaptive NK cells in the general population, and a minority of individuals harbor frequencies of these cells >10% in their PB.16 Therefore, to broaden the application of the NKG2C-KE, we used iPSC-derived NK cells to develop an ideal off-the-shelf NK cell to combine with NKG2C-KE. We and others have explored NK cell development from hematopoietic progenitors23,24 and more recently from renewable iPSCs.25 Production of iNK cells has been scaled up, and their use is being evaluated in the clinic (NCT03841110). Using this platform, two different iNK cell lines were created. One iNK cell line was engineered with NKG2C alone, and the other with both NKG2C and its signaling adaptor molecule, DAP12, for enhanced NKG2C expression and response. iPSCs were transduced, enriched for NKG2C expression, and banked to create a renewable starting material. Cells were then differentiated in a stepwise fashion through the CD34+ stage. CD34+ cells were then differentiated along the NK cell lineage using previously described methods.26 After expansion, the non-transduced parental iNK expressed low levels of NKG2C. In contrast, NKG2C-transduced iNK cells displayed significantly higher surface expression of NKG2C, which was further increased in iNK cells expressing both NKG2C and DAP12 (Figure 4A). This increase in NKG2C-expressing cells was reproducible, with the NKG2C and DAP12 iNK having a significantly higher NKG2C+ population compared to the non-transduced and NKG2C without DAP12 (65.32% versus 10.86 and 45.52, p > 0.01 and 0.001) (Figure 4B), and DAP12 was increased as well (Figure S1). The NKG2C and DAP12 iNK cells also expressed more NKG2C per cell, as shown by mean fluorescence intensity (MFI), compared to the NKG2C without DAP12 iNK (Figure 4C). While the NKG2C with DAP12 did not express intracellular markers of adaptive NK cells (PLZF, EAT2, or FcεRIγ), they did express slightly more DAP12-associated receptors NKp44 and KIRs at a higher density (Figure S2).
Figure 4.

iPSC-derived NK cells genetically modified to express NKG2C
(A) iNKs at the final stage of differentiation were stained for CD56 and NKG2C and analyzed by flow cytometry. (B) Quantification of %NKG2C for 5 batches of iNK at the final stage of differentiation for the indicated lines. (C) MFI of indicated iNKs showing NKG2C histograms. Graphs indicate the means ± SDs analyzed using 1-way ANOVA and RM analysis used for (B). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To test the combined therapeutic potential of the NKG2C-KE and NKG2C gene-edited iNK cells, two AML cell lines that highly express CD33 (HL-60 and THP-1) were used as targets in a series of functional assays. Non-transduced and gene-edited iNK cells were co-cultured with targets alone, with rhIL-15, or with the NKG2C-KE and evaluated for degranulation and cytokine production (Figures 5A–5D). In the transduced iNK, the NKG2C-KE induced significantly more degranulation against THP-1 targets compared to rhIL-15 (NKG2C: 16.1% versus 6.9%, p < 0.0001; NKG2C/DAP12: 17.2% versus 4.6%, p < 0.0001) and more IFN-γ production (NKG2C: 14.4% versus 6.9%, p = 0.003; NKG2C/DAP12: 23.0% versus 4.9%, p < 0.0001) (Figures 5A and 5B). Incubation with CD33− targets did not result in increased degranulation or IFN-γ (Figure S3). The functional responses of non-transduced cells were not affected by the NKG2C-KE, suggesting that low frequencies of NKG2C are insufficient to generate an enhanced response. A similar result was seen with HL-60 cells (Figures 5C and 5D). While the gene-edited iNK cells (NKG2C ± DAP12) exhibited similar levels of degranulation against both tumor cell lines, iNK cells co-transduced cells with both NKG2C and DAP12 produced significantly more IFN-γ than NKG2C-transduced cells without DAP12 against the THP-1 cells, but not the HL-60 cells. Stimulation with rhIL-15 induced a strong proliferative signal in both transduced and non-transduced iNK, but the NKG2C-KE mediated more proliferation for iNK cells co-transduced with both NKG2C and DAP12 (Figure 5E). The enhanced degranulation, IFN-γ production, and proliferative response indicate an important role for DAP12 in NKG2C-mediated functions in iNK cell products. Thus, we selected iNK cells engineered with both NKG2C and DAP12 for use in further experiments.
Figure 5.

iPSC transduced with NKG2C and DAP12 and differentiated into NK cells (iNK) exhibit CD33-specific function with the NKG2C-KE against AML targets
Non-transduced, NKG2C-transduced, and NKG2C/DAP12-transduced iNK were incubated with IL-15 or NKG2C-KE for 5 h with THP-1 (A and B) or HL60 (C and D). NK cells were stained for CD107a degranulation (A and C) and IFN-γ production (B and D) in 3 separate experiments pooled in the graphs. The indicated iNK lines were stained with CellTrace and incubated at 37°C for 7 days with indicated treatments as a measure of proliferation (E). Graphs indicate the means ± SDs analyzed using a 2-way ANOVA and RM analysis used for (A)–(D). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To directly evaluate tumor killing, a dynamic in vitro system was used in which the loss of fluorescently labeled target cells upon cell death can be quantified continuously over a 24-h period by live imaging. Non-transduced and NKG2C/DAP12-transduced iNK cells were co-cultured with THP-1 targets with and without rhIL-15 or NKG2C-KE and imaged every 30 min (Figures 6A and 6B). Non-transduced iNK cells exhibited modest natural cytotoxicity against THP-1 cells, which remained largely unchanged by the addition of rhIL-15, although statistically different, or the NKG2C-KE. In contrast, the addition of the NKG2C-KE to NKG2C/DAP12-transduced iNK cells resulted in substantially increased killing kinetics in the first 18 h of exposure, with statistical significance consistently at 6 and 12 h (Figure 6C).
Figure 6.

Rapid killing kinetics with NKG2C-KE and NKG2C/DAP12 iNK
Tumor targets stained with CellTrace for tracking were incubated in the Incucyte live imaging system with iNK and indicated treatments. (A) Images from a representative experiment at indicated times for NKG2C-KE-treated iNK. (B) Images taken every 30 min to track cell numbers over time from a representative experiment. (C) Cell numbers over time in 5 separate experiments at representative times. Graphs indicate the means ± SDs analyzed using a 2-way ANOVA and RM analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
NKG2C/DAP12-transduced iNK cells targeted with the NKG2C-KE mediate cytotoxicity against primary AML
The functional assays described above tested targeting of AML cell lines. To test the functional capacity of NKG2C/DAP12 iNK cells activated with the NKG2C-KE against a more physiologic target, we performed additional function experiments using primary AML blasts. Five AML patient samples, containing 47%–97% CD33+ blasts were used as targets in a flow cytometry-based functional assay and all expressing HLA-E (Figure S4). The non-transduced iNK cells exhibited low levels of degranulation against the blasts, whether treated with or without rhIL-15 or the NKG2C-KE (Figure 7A). Similar functional responses were observed for NKG2C/DAP12 iNK cells in the presence or absence of rhIL-15, even though rhIL-15 has a stronger IL-15 signal than NKG2C-KE. However, when combined with the NKG2C-KE, the NKG2C/DAP12 iNK cells exhibited 2-fold greater degranulation against primary AML blasts. While rhIL-15 potentiated IFN-γ production by both iNK lines, the greatest induction was observed with the NKG2C/DAP12-transduced cells targeted with the NKG2C-KE (Figure 7B). The killing of primary AML blasts was also evaluated by the determination of viable AML cell counts after 2 days of co-culture (Figure 7C). No difference in AML cell numbers was observed when comparing non-transduced iNK cell co-cultures and NKG2C/DAP12-transduced iNK cell co-cultures in the absence of rhIL-15 or the NKG2C-KE. rhIL-15 alone induced a strong natural cytotoxic response from both iNK cell lines, resulting in substantial AML killing. Similar to the THP-1 AML cell line, directing NKG2C/DAP12-transduced iNK cells with the NKG2C-KE to the CD33 antigen resulted in the robust and nearly complete elimination of primary AML targets (rhIL-15, 3298; NKG2C-KE, 486.7; p = 0.02).
Figure 7.

NKG2C-KE and NKG2C/DAP12 iNK kill primary AML targets
Primary AML cells from 5 patients were incubated with indicated iNK for 5 h and stained for CD107a degranulation (A) and IFN-γ production (B). Primary AML cells (n = 3) were stained with CellTrace and incubated for 48 h with iNK and counted by flow cytometry (C). Graphs indicate the means ± SEMs analyzed using 2-way ANOVA and RM analysis used for (A)–(D). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Discussion
We have previously described a TriKE platform that includes an anti-CD16 moiety to target and trigger NK cells to mediate ADCC.8 However, CD16 is broadly expressed by a variety of other lymphocyte subsets such as granulocytes, monocytes, and macrophages.7 CD16 expression on the surface of these cells could bind the anti-CD16 component of a TriKE and act as a sink, sequestering the TriKE away from NK cells and limiting its availability and efficacy or perhaps an undesired toxicity effect. Thus, we sought to investigate targeting of other activating receptors expressed on NK cells that could trigger potent functional responses and the expression of which are more cell type restricted. NKG2C, a strong activating receptor expressed exclusively by cytotoxic lymphocytes, meets both of those requirements.
A 161533 TriKE has been shown to elicit a potent NK response against CD33 tumor targets.8 This 161533 TriKE is undergoing clinical trials for testing in patients with high-risk hematological malignancies (NCT03214666). Confirmation of CD33 targeting with a TriKE allowed us to take an already-validated tumor targeting platform and create a new platform by swapping the anti-CD16 moiety with an anti-NKG2C. A WT IL-15 linker is included in the new NKG2C-KE construct, as we have previously shown that incorporation of IL-15 signaling yields both NK proliferation and activation to enhance antitumor efficacy.21
While NKG2C expression is largely limited to NK cells, the proportion of cells that expresses this activation receptor are generally small in healthy individuals.14 Larger populations of NK cells that express this receptor arise in allogeneic HCT patients who reactivate CMV.27 Here, we demonstrate an enhanced antitumor response triggered by the NKG2C-KE when endogenous PB NK cells from CMV-reactivated HCT recipients were exposed to CD33+ AML cell lines. Furthermore, the magnitude of the response directly correlated to the percentage of NK cells that expressed NKG2C. Interestingly, the NKG2C-targeting component also appears to direct the IL-15 element of the NKG2C-KE to those NK cells that express NKG2C, resulting in an increased proliferative response of that specific NK cell population. This suggests that the NKG2C-KE is mimicking the CMV signal that is normally seen through NKG2C ligation with human leukocyte antigen-E (HLA-E). This indicates that the NK cell targeting component is specific and will likely not have considerable off-target effects. We also demonstrated that the ability of the NKG2C-KE to control tumors in vivo is comparable to that of the 161533 TriKE, despite the difference in CD16 and NKG2C expression in the starting population. In this population, CD16 is expressed at almost 7-fold higher levels than NKG2C, indicating the effectiveness of the NKG2C-KE. The NKG2C-KE was also able to maintain a higher NKG2C+ population in vivo, which was not seen with the 161533 TriKE, further suggesting a directed targeting of the IL-15 element in the NKG2C-KE.
Considering how small the NKG2C-expressing NK populations are in CMV-seronegative individuals and that the strength of the antitumor response correlated to the quantity of NKG2C on the NK cell surface, we generated iNK cells that express NKG2C and its signaling adaptor, DAP12. We tested the killing capacity of these cells in combination with NKG2C-KE as a rationally designed platform to specifically target these potent effector cells toward cancer cells. NKG2C-KE in combination with the NKG2C/DAP12-transduced iNK cells elicited a stronger degranulation and cytokine response than NKG2C/DAP12-transduced iNK cells alone or the NKG2C-KE combined with non-transduced iNK cells. While iNK cells express endogenous DAP12, increased DAP12 expression enhanced iNK cell proliferation in response to NKG2C-KE engagement. These cells also expressed slightly higher DAP12-associated proteins, but lacked intracellular adaptive NK cell markers showing that the mechanism of developing an adaptive NK cell response to CMV goes well beyond the forced expression of NKG2C and DAP12. The enhanced effector function of NKG2C/DAP12-transduced iNK cells is reflected in the robust killing of both CD33+ AML cell lines and nearly complete elimination of primary AML blasts. In addition to combining NKG2C+ iNK cells with NKG2C-KE to target AML, we are also exploring the innate function of NKG2C+ iNK cells to target HLA-E+ cancer cells. Many cancers use HLA-E to suppress and evade effector cell function, and the application of NKG2C+ iNK cells to target these cancer populations may serve as a ubiquitous cancer therapeutic strategy.
The data presented here illustrate the potential for a bi- or tri-specific KE to mediate tumor-targeted NK cell responses through activation receptors other than CD16. Furthermore, NK cell targeting through NKG2C resulted in the specific proliferation of NKG2C+ NK cells, demonstrating a unique ability to target the IL-15 component of the NKG2C-KE to a specific NK cell subset. The use of an NKG2C engager in combination with a tailor-made, off-the-shelf iNK cell product through adoptive transfer has the potential to generate strong antitumor response even in patients who have an exhausted immune cell compartment and who naturally lack NKG2C+ NK cells. Furthermore, the use of NKG2C, as opposed to CD16, as the target for immune engagement will allow for enhanced NK cell responses while limiting the possibility of sequestration of the engager by other cell types. The study described here demonstrates the therapeutic utility and versatility of combining a multipurpose and specific NK cell engager, which can redirect and activate endogenous NK cells, and an off-the-shelf NK cell product, which is fine-tuned through uniform engineering, to elicit a strong and directed elimination of targeted cancer cells.
Materials and Methods
Protein production
The final construct of the NKG2C-KE was spliced into the Minicircle plasmid (SBI: MN502A-1) with a CMV promoter into the multicloning site. The NKG2C-KE portion contains a start codon, export sequence, an anti-NKG2C scFv, an 18-amino acid sequence flanking WT IL-15, an anti-CD33 scFv, and a 10xHis-Tag. The NKG2C-KE plasmid was transfected into Expi293F (Thermo Fisher Scientific, Waltham, MA: A14527) cells using Expifectimine (Thermo Fisher Scientific: A14524) following the manufacturer’s instructions. Supernatant was harvested at days 4–5 post-transfection when cell viability dropped below 80%. The supernatant was then incubated with HisPur cobalt resin (Thermo Fisher Scientific: 89965) for 1 h. Resin was washed 3 times, then the NKG2C-KE was eluted from HisPur resin through a column using 250 mM imidazole. Protein was desalted using a PD-10 column (GE Healthcare, Chicago, IL) per the manufacturer’s instructions. Protein was run on a Tris-based gel, and purity was assessed using GelCode Blue (Thermo Fisher Scientific; 24592) stain. The 161533 TriKE was made as previously described.8 Briefly, plasmid was transformed into Escherichia coli strain BL21 (DE3) (EMD), and then harvested 2 h later by pelleting. Then, bacteria were resuspended and inclusion bodies harvested, and washed to remove endotoxin. Protein was then refolded and purified using fast protein liquid chromatography (FPLC) ion exchange chromatography.
Healthy donors and patient samples
Healthy donor blood was obtained from Memorial Blood Bank (Minneapolis, MN) and processed to isolate PBMCs using density gradient Ficoll-Paque (GE Healthcare). PBMCs were either cryopreserved in liquid nitrogen or used fresh. AML blasts were obtained and cryopreserved from an apheresis of a de novo (no prior chemotherapy) AML patient with normal cytogenetics and 45%–93% AML blast frequencies. The samples were managed by the Heme Malignancy Tissue Bank at the University of Minnesota and the Translational Therapy Shared Resource. All of the human samples, including AML patient blasts, were used with approval from the institutional review board (IRB) at the University of Minnesota. Informed consent was given in compliance with guidelines by the Committee on the Use of Human Subjects in Research and in accordance with the Declaration of Helsinki. All cells were cultured in RPMI-1640 (Thermo Fisher Scientific) with 10% heat inactivated fetal bovine serum (FBS) and PenStrep supplementation at 37°C and 5% CO2. PBMCs and AML samples were thawed and rested overnight before use.
Generation of expanded NK cells
Human PBMCs obtained from the above protocol were enriched for NK cells (STEMCELL). Then cells were cultured for 14 days with feeder cells of irradiated K562 with 41BBL and membrane-bound IL-21 in RPMI with 10% heat-inactivated FBS, PenStrep, and 50 IU/mL IL-2. The media were changed every 2–3 days. Feeder cells were added twice at days 0 and 7.
Generation of NKG2C+ NK cells from iPSCs
Human iPSC culture and differentiation to iCD34+ and iNK cells were performed as previously described.26 At the beginning of the iNK cell differentiation culture, iCD34 cells were plated on stroma cells in B0 media supplemented with cytokines that support NK cell differentiation from hematopoietic progenitors.26 After iNK cell specification, iNK cells were harvested and co-cultured with modified K562 in supplemented B0 media for expansion. K562 cells were propagated in RPMI 1640 media (Thermo Fisher Scientific) containing 10% FBS (HyClone).
Cell lines, antibodies, and reagents
THP-1 and HL-60 cells were cultured in RPMI 1640 (GIBCO) with 10% heat-inactivated FBS and PenStrep supplementation. THP-1 and HL-60 cells were cultured to a density of between 0.2 and 2 million cells per milliliter. Cell lines were purchased from ATCC in 2016. Fluorochrome-conjugated antibodies were purchased from BioLegend (San Diego, CA): anti-CD56 (clone HCD56), anti-IFNγ (clone XMG1.2), anti-CD45 (clone HI30), anti-CD34 (clone 561), anti-NKp44 (clone P44-8), anti-KIRs (clone HP-MA4, DX27, DX9), anti-NKG2D (clone 1D11), and anti-HLA-E (clone 3D12); BD Biosciences (San Jose, CA): anti-CD3 (clone UCHT1); Thermo Fisher Scientific: Live/Dead Aqua (product # L34966), CellTrace Violet (C34557), Live/Dead Near IR (L34976), CellTrace FarRed (C34564); R&D Systems (Minneapolis, MN): anti-NKG2C (clone 134591), anti-PLZF (clone 6318100), anti-DAP12 (clone 406288); Millipore (Burlington, MA): anti-FcεRIγ (polyclonal); Proteintech (Rosemont, IL): anti-EAT2 (SH2D1B) (polyclonal); Beckman Coulter (Indianapolis, IN): anti-NKG2A (clone z199); Sartorius (France): caspase 3/7 apoptosis assay reagent FITC (Sartorius 4440).
NK cell function assays
Effectors were incubated with targets with or without NKG2C-KE at a 2:1 effector:target (E:T) ratio. Anti-CD107a antibody was added at the beginning of co-culture. One hour into the incubation, GolgiStop and GolgiPlug were added to each well and incubated for an additional 4 h. At the end of the 5-h incubation, cells were stained with Live/Dead Aqua per the manufacturer’s instructions before being surface stained for CD3 and CD56. Cells were then fixed with 2% paraformaldehyde in PBS for 20 min. and permeabilized with 0.1% Triton X for 5 min, followed by intracellular staining for IFN-γ. Samples were analyzed on an LSRII Flow Cytometer (BD, Ashland, OR) and analyzed with FlowJo software (BD). For the analysis of proliferation, cells were stained with CellTrace Violet and then incubated with NKG2C-KE or rhIL-15 (R&D Systems) for 7 days at 37°C, 5% CO2. At the end of 7 days, cells were washed and stained with Live/Dead Near IR and then surface stained with antibodies against CD3, CD56, and NKG2C. For live imaging, THP-1 cells were stained with CellTrace Far Red before plating. IncuCyte caspase 3/7 apoptosis assay reagent was added to each well. iNK cells were added at a 5:1 E:T ratio. Plates were then placed in Incucyte S3 (Sartorius) for 24 h. Images were taken every 30 min. Graphs were created using values for live THP-1 cell counts normalized to no effector control values and to initial plated cell counts. All of the conditions were run in triplicate. Primary AML cells were stained with CellTrace Violet and then co-incubated with iNK cells and NKG2C-KE for 2 days. Cells were stained with antibodies against CD45 and CD34 and analyzed by flow cytometry. The remaining AML cells with a CellTrace+ CD45intermediate CD34+ cells were counted.
In vivo mouse study
The in vivo study was conducted in accordance with the Institutional Animal Care and Use Committee at the University of Minnesota. The HL60-Luc model was previously described.8, 21, 22 Briefly, female NSG mice were injected with 750,000 HL60-Luc intravenously. Then, 3 days later, BLI was performed on mice and mice were divided into groups with an equal tumor load. Mice were injected with 5 million expanded NK cells intravenously and drug treatments were started at 5 times weekly for 3 weeks in accordance with our previous models. Treatments were injected intraperitoneally. BLI was then measured at days 6, 13, 27, and 35, and facial vein bleeding was done at days 14 and 28. Red blood cells were lysed and then stained for hCD45, mCD45, CD3, CD56, NKG2C, and CD16.
Statistics
GraphPad Prism (GraphPad Prism Software, La Jolla, CA) was used to plot graphs with error bars showing means ± SEMs or ±SD where appropriate and correlation curves with 95% confidence interval (CI). GraphPad was also used to calculate linear regression t tests, one-way ANOVA, and two-way ANOVA with and without repeated-measurement (RM) analysis where appropriate and to determine statistical significance as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Specific statistical analysis used for each graph is indicated in the figure legends.
Acknowledgments
We would like to acknowledge the Flow Cytometry Core and Translational Therapy Laboratory. This work was supported by NIH grants NCI F30-CA232481-01A1 (to E.C.) NCI P01 CA111412 (to J.S.M., D.A.V., and M.F.), NCI P01 65493 (to J.S.M.), R35 CA197292 (to J.S.M.), P30 CA077598 (to J.S.M. and M.F.), US Department of Defense grant CA150085 (to M.F.), the Minnesota Masonic Charities, and the Killebrew-Thompson Memorial Fund. T.L., R.B., and B.V. are employees of Fate Therapeutics and directly contributed to this manuscript. F.C., K.J.M., and J.S.M. earn income from and hold stock in Fate Therapeutics, who partially funded the iPSC-derived NK cell component of this work. These conflicts have been reviewed and managed by the University of Minnesota (and Oslo University, for K.J.M.) in accordance with their conflict of interest policies.
Author contributions
Conceptualization, E.C., M.F., F.C., and J.S.M.; methodology, M.F., F.C., R.B., B.V., and J.S.M.; investigation, E.C.; resources, Z.D., H.W., K.T., T.L., and R.B.; writing – original draft, E.C. and Z.D.; writing – review & editing, M.F., F.C., Z.D., D.A.V., and J.S.M.; visualization, E.C.; supervision, M.F., F.C., D.A.V., K.J.M., and J.S.M.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.018.
Supplemental information
References
- 1.Kumar S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology. 2018;154:383–393. doi: 10.1111/imm.12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raulet D.H., Gasser S., Gowen B.G., Deng W., Jung H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013;31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantoni C., Bottino C., Vitale M., Pessino A., Augugliaro R., Malaspina A., Parolini S., Moretta L., Moretta A., Biassoni R. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med. 1999;189:787–796. doi: 10.1084/jem.189.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pessino A., Sivori S., Bottino C., Malaspina A., Morelli L., Moretta L., Biassoni R., Moretta A. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998;188:953–960. doi: 10.1084/jem.188.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jelenčić V., Lenartić M., Wensveen F.M., Polić B. NKG2D: a versatile player in the immune system. Immunol. Lett. 2017;189:48–53. doi: 10.1016/j.imlet.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Lanier L.L., Ruitenberg J.J., Phillips J.H. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol. 1988;141:3478–3485. [PubMed] [Google Scholar]
- 7.Unkeless J.C., Shen Z., Lin C.-W., DeBeus E. Function of human Fc γ RIIA and Fc γ RIIIB. Semin. Immunol. 1995;7:37–44. doi: 10.1016/1044-5323(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 8.Vallera D.A., Felices M., McElmurry R., McCullar V., Zhou X., Schmohl J.U., Zhang B., Lenvik A.J., Panoskaltsis-Mortari A., Verneris M.R., et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016;22:3440–3450. doi: 10.1158/1078-0432.CCR-15-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Della Chiesa M., Falco M., Muccio L., Bertaina A., Locatelli F., Moretta A. Impact of HCMV Infection on NK Cell Development and Function after HSCT. Front. Immunol. 2013;4:458. doi: 10.3389/fimmu.2013.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Béziat V., Sleiman M., Goodridge J.P., Kaarbø M., Liu L.L., Rollag H., Ljunggren H.G., Zimmer J., Malmberg K.J. Polyclonal expansion of NKG2C+ NK cells in TAP-deficient patients. Front. Immunol. 2015;6:507. doi: 10.3389/fimmu.2015.00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaiser B.K., Pizarro J.C., Kerns J., Strong R.K. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc. Natl. Acad. Sci. USA. 2008;105:6696–6701. doi: 10.1073/pnas.0802736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauterbach N., Wieten L., Popeijus H.E., Voorter C.E.M., Tilanus M.G.J. HLA-E regulates NKG2C+ natural killer cell function through presentation of a restricted peptide repertoire. Hum. Immunol. 2015;76:578–586. doi: 10.1016/j.humimm.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Picardi A., Mengarelli A., Marino M., Gallo E., Benevolo M., Pescarmona E., Cocco R., Fraioli R., Tremante E., Petti M.C., et al. Up-regulation of activating and inhibitory NKG2 receptors in allogeneic and autologous hematopoietic stem cell grafts. J. Exp. Clin. Cancer Res. 2015;34:98. doi: 10.1186/s13046-015-0213-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heath J., Newhook N., Comeau E., Gallant M., Fudge N., Grant M. NKG2C(+)CD57(+) Natural Killer Cell Expansion Parallels Cytomegalovirus-Specific CD8(+) T Cell Evolution towards Senescence. J. Immunol. Res. 2016;2016:7470124. doi: 10.1155/2016/7470124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luetke-Eversloh M., Hammer Q., Durek P., Nordström K., Gasparoni G., Pink M., et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog. 2014;10:e1004441. doi: 10.1371/journal.ppat.1004441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlums H., Cichocki F., Tesi B., Theorell J., Beziat V., Holmes T.D., Han H., Chiang S.C., Foley B., Mattsson K., et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. 2015;42:443–456. doi: 10.1016/j.immuni.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foley B., Cooley S., Verneris M.R., Curtsinger J., Luo X., Waller E.K., Anasetti C., Weisdorf D., Miller J.S. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol. 2012;189:5082–5088. doi: 10.4049/jimmunol.1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tesi B., Schlums H., Cichocki F., Bryceson Y.T. Epigenetic Regulation of Adaptive NK Cell Diversification. Trends Immunol. 2016;37:451–461. doi: 10.1016/j.it.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Cichocki F., Taras E., Chiuppesi F., Wagner J.E., Blazar B.R., Brunstein C., Luo X., Diamond D.J., Cooley S., Weisdorf D.J., Miller J.S. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight. 2019;4:e125553. doi: 10.1172/jci.insight.125553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merino A., Zhang B., Dougherty P., Luo X., Wang J., Blazar B.R., Miller J.S., Cichocki F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Invest. 2019;129:3770–3785. doi: 10.1172/JCI125916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Felices M., Lenvik T.R., Kodal B., Lenvik A.J., Hinderlie P., Bendzick L.E., Schirm D.K., Kaminski M.F., McElmurry R.T., Geller M.A., et al. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol. Res. 2020;8:1139–1149. doi: 10.1158/2326-6066.CIR-19-0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arvindam U.S., van Hauten P.M.M., Schirm D., Schaap N., Hobo W., Blazar B.R., Vallera D.A., Dolstra H., Felices M., Miller J.S. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia. 2021;35:1586–1596. doi: 10.1038/s41375-020-01065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dezell S.A., Ahn Y.O., Spanholtz J., Wang H., Weeres M., Jackson S., Cooley S., Dolstra H., Miller J.S., Verneris M.R. Natural killer cell differentiation from hematopoietic stem cells: a comparative analysis of heparin- and stromal cell-supported methods. Biol. Blood Marrow Transplant. 2012;18:536–545. doi: 10.1016/j.bbmt.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller J.S., Alley K.A., McGlave P. Differentiation of natural killer (NK) cells from human primitive marrow progenitors in a stroma-based long-term culture system: identification of a CD34+7+ NK progenitor. Blood. 1994;83:2594–2601. [PubMed] [Google Scholar]
- 25.Hermanson D.L., Bendzick L., Pribyl L., McCullar V., Vogel R.I., Miller J.S., Geller M.A., Kaufman D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells. 2016;34:93–101. doi: 10.1002/stem.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cichocki F., Bjordahl R., Gaidarova S., Mahmood S., Abujarour R., Wang H., Tuininga K., Felices M., Davis Z.B., Bendzick L., et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy. Sci. Transl. Med. 2020;12:eaaz5618. doi: 10.1126/scitranslmed.aaz5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cichocki F., Cooley S., Davis Z., DeFor T.E., Schlums H., Zhang B., Brunstein C.G., Blazar B.R., Wagner J., Diamond D.J., et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia. 2016;30:456–463. doi: 10.1038/leu.2015.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.