

Summary
Despite extensive analyses, there remains an urgent need to delineate immune cell states that contribute to mortality in people critically ill with COVID-19. Here, we present high-dimensional profiling of blood and respiratory samples from people with severe COVID-19 to examine the association between cell-linked molecular features and mortality outcomes. Peripheral transcriptional profiles by single-cell RNA sequencing (RNA-seq)-based deconvolution of immune states are associated with COVID-19 mortality. Further, persistently high levels of an interferon signaling module in monocytes over time lead to subsequent concerted upregulation of inflammatory cytokines. SARS-CoV-2-infected myeloid cells in the lower respiratory tract upregulate CXCL10, leading to a higher risk of death. Our analysis suggests a pivotal role for viral-infected myeloid cells and protracted interferon signaling in severe COVID-19.
Keywords: COVID-19, severe COVID-19, COVID-19 outcome, inflammatory monocytes, type I interferon, inflammatory cytokines, machine learning, single-cell RNA-seq, gene modules
Graphical abstract
Highlights
-
•
Transcription-factor-driven gene modules define immune states in blood
-
•
Gene modules are associated with mortality in people critically ill with COVID-19
-
•
Inflammatory monocyte states precede upregulation of inflammatory cytokines
-
•
SARS-CoV-2-infected macrophages in lung express high levels of CXCL10
Cillo et al. identify transcriptional profiles in peripheral blood that are associated with mortality in people critically ill with COVID-19. Inflammatory monocyte signatures are correlated with CXCL10 in plasma and precede upregulation of inflammatory cytokines in blood. SARS-CoV-2-infected macrophages in the respiratory tract expressed CXCL10, linking peripheral and lung immune profiles.
Introduction
The emergence of SARS-CoV-2 led to a worldwide pandemic1 and subsequent clinical, translational, and basic research efforts to combat this global health crisis. Despite the rapid development and deployment of vaccines,2,3 COVID-19 is projected to be a major cause of critical illness and hospitalizations throughout 2021 and beyond before worldwide vaccine campaigns can control the pandemic. Furthermore, the emergence of highly transmissible viral variants (e.g., alpha, beta, delta, and gamma variants) is contributing to infections and hospitalizations4 and poses a threat to effective immunity following vaccination. Clinical trials examining pharmacologic interventions in severe COVID-19 have highlighted the efficacy of non-specific immunosuppression with corticosteroids5, 6, 7 and targeted interleukin (IL)-6 blockade in people treated soon after ICU admission.8 Despite these therapeutics, severe COVID-19 continues to carry a high mortality risk. Thus, in-depth analyses of cellular and molecular states, particularly of the immune system, that are associated with survival or death in severe COVID-19 disease are urgently needed.
SARS-CoV-2 infection can cause acute lung injury, leading to acute respiratory distress syndrome (ARDS),9 dysregulated inflammation,10 and hyper-coagulability.11 Studies dissecting the immunopathology of COVID-19 have evaluated people across the spectrum of disease severity (mild to critical) and noted lymphopenia9 with high levels of multiple inflammatory cytokines, C-reative protein (CRP),12 and D-dimer11 in the context of severe disease. Elevated levels of the cytokines IL-6, tumor necrosis factor (TNF)-α, IP-10 (CXCL10), IL-8 (CXCL8), and IL-10 have been reported in severe COVID-19 disease.13,14 Notably, the dysregulated levels of IL-6 in people with COVID-19 are lower compared with those in non-COVID ARDS, cytokine release syndrome, or sepsis, suggesting differences in the underlying etiology.15 Type I interferon (IFN) signaling is essential in moderating COVID-19 disease, as individuals with either IFN auto-antibodies16 or inborn errors of type I IFN17 production have a much higher risk of severe COVID-19. Paradoxically, a delayed and excessive type I IFN response is implicated in severe disease and leads to mortality in a mouse model of SARS-CoV-2 infection.18, 19, 20 Among the immune lineages altered in COVID-19, monocytes have been described to play an important role in the periphery and at the site of infection.21, 22, 23, 24, 25 We undertook high-dimensional cellular and molecular analyses of the immune system in critically ill ICU patients, utilizing flow cytometry, single-cell RNA sequencing (scRNA-seq), and cytokine profiling coupled with machine learning to reveal cellular states and gene modules that predicted mortality and underlie adverse pathogenesis.
Results
Clinical cohorts, subject characteristics, and study design
Following informed consent, we enrolled 42 consecutive critically ill people with acute hypoxemic respiratory failure and symptoms suggestive of COVID-19 in a prospective, observational cohort study, with limited longitudinal sampling. Based on reference-standard nasopharyngeal swab SARS-CoV-2 qPCR, COVID-19 was diagnosed in 35 individuals (COVID-19 group), whereas a non-COVID etiology of acute respiratory illness was identified in 7 persons who had negative SARS-CoV-2 qPCR (non-COVID ARDS group). Clinical details of this cohort have been described in detail.26 As controls in the study, we also included 10 healthy blood donors (healthy donor group). The median ages of non-COVID ARDS and COVID-19-infected individuals were 62 and 65 years, and many people had pre-existing conditions (Data S1). Individuals entered the ICU a median of 6 (interquartile range [IQR]: 4 to 11) days after symptom onset, and mortality at 90 days was 29% and 40% in non-COVID ARDS-affected- and COVID-19-affected individuals, respectively (Data S1). Comparisons of clinical covariates by death or survival (day 90) in individuals with COVID-19 showed trends toward higher mortality in older individuals and people with lower BMI, and we note that persons who received glucocorticoids were more likely to survive (Data S1). We obtained peripheral blood as well as endotracheal aspirates from subjects with COVID-19 to perform high-dimensional profiling of cell frequencies, cellular transcriptional states, and cytokines (Figure 1A; Data S1).
Figure 1.

Expansion of intermediate monocytes and plasmablasts is associated with COVID-19
(A) Schema highlighting the three clinical groups along with biological specimens obtained and assays performed.
(B) Flow cytometric analysis of immune cell frequencies in peripheral blood of healthy donors (one time point), non-COVID ARDS-affected individuals (day 1 ICU), and COVID-19-affected individuals (day 1 ICU). Comparisions with p values <0.05 by Wilcoxon rank-sum test are shown.
(C) Principal-component analysis (PCA) of immune cell frequencies displayed in (B).
(D) Weightings of immune cell frequencies that contribute to the PC1 embeddings in (C) and distinguish critically ill individuals (non-COVID ARDS/COVID-19) from healthy donors.
(E) scRNA-seq analysis of 98,327 cells showing the canonical immune lineages from peripheral blood of healthy donors (14,271 cells), non-COVID ARDS-affected individuals (13,060 cells), and COVID-19-affected individuals (78,922 cells).
(F) PCA performed by using differentially expressed immune cell gene modules (delineated with Arboreto; see Methods details) as molecular features of study participants in the three clinical groups.
(G) Weightings of the immune cell gene modules that are dominant contributors to PC1 and PC2 embeddings in (F). Flow cytometry analysis, 36 individuals (20 COVID-19-affected individuals, 6 non-COVID ARDS-affected individuals, and 10 healthy donors); scRNA-seq analysis, 79 distinct samples.
Expansion of intermediate monocytes and plasmablasts in COVID-19
We utilized high-dimensional flow cytometry to evaluate peripheral blood mononuclear cells (PBMCs) of critically ill individuals (COVID-19 and non-COVID ARDS) at post-enrollment day 1 in the ICU (Figures 1B and S1A–S1C). In agreement with other reports, we found that individuals with COVID-19 tended to have higher frequencies of plasmablasts27,28 and inflammatory monocytes29 and lower frequencies of CD4+ T cells.27 We did not find significant associations between cell frequencies and death or survival (Figures S2A and S2B). Although cycling CD8+ T cells that co-express human leukocyte antigen DR isotype (HLA-DR) and CD38 are associated with viral infection in general and COVID-19 in particular,27 there were no differences in frequencies of this CD8+ T cell subset on day 1 between deceased and surviving individuals in our cohort (Figures S2C and S2D). We performed principal-component analysis (PCA) using the immune cell frequencies (from Figure 1B) to determine whether these multi-variate features could stratify individuals by clinical group. When visualized along the PC1 axis, distributions of samples demonstrated considerable heterogeneity (Figure 1C). However, COVID-19 as well as non-COVID ARDS samples were shifted rightward on PC1, while healthy donors were shifted leftward. Consistent with analysis in Figure 1B, higher frequencies of myeloid cells and plasmablasts contributed to the PC1 distribution of samples from critically ill individuals, while higher frequencies of T and B cells distinguished the healthy donors (Figure 1D). Concomitant expansion of intermediate monocytes and plasmablasts has been noted in other viral diseases beyond COVID-19, such as Dengue virus.30 In this context, the inflammatory monocytes promote the differentiation of naive B cells into plasmablasts, which can be the source of protective or pathogenic antibodies. We note that severe COVID-19 has been associated with various types of auto-antibodies.31, 32, 33 Although assessment of cell frequencies revealed shifts in the composition of immune cells in COVID-19-affected and non-COVID ARDS-affected individuals versus healthy donors, these frequencies yielded no statistically significant associations with outcome in critically ill individuals.
Gene modules derived from scRNA-seq stratify subjects by clinical group
To query the transcriptional states of immune cells and generate high-resolution predictive features, we performed scRNA-seq analysis of PBMCs from COVID-19-affected and non-COVID ARDS-affected individuals and healthy donors (Methods details). Samples were obtained from critically ill individuals on days 1, 5, and 10 post-enrollment in the ICU, whereas those from healthy donors were obtained at one time point (Data S1). In total, we profiled 99,618 PBMCs by scRNA-seq (Figure 1E). We recovered a median of 1071 (IQR: 662 to 1544) PBMCs from 80 samples across disease states and time points. Canonical immune cell types were identified as previously described (Methods details)34 and were visualized across clinical groups by uniform manifold approximation and projection (UMAP).35
We next sought to determine whether the clinical groups (i.e., healthy donor, non-COVID ARDS, and COVID-19) could be better stratified by transcriptional states rather than by cell frequencies. To characterize transcriptional states across immune cell types, we used Arboreto within the pySCENIC framework (Methods details).36 First, we bioinformatically isolated all canonical immune cell types across all samples and time points (Figure S3A) and then utilized tree-based regression analysis in Arboreto to identify modules of co-expressed genes presumptively co-regulated by given transcription factors within each cell type (Figures S3B and S3C). Individual genes within a module were assigned a weight derived from the tree-based regression analysis, with higher-weighted genes being more significant contributors to a given gene module (Data S1). Finally, for each individual, we generated a single score for each cell-type-specific gene module by taking the median module score for each module within that subject and time point (Figures S3D–S3G). This approach resulted in each individual being described by a unique combination of gene module scores reflecting the underlying transcriptional state. We then asked whether we could use the resultant gene module scores to distinguish between the study groups. To do this, we first identified the top 10 most significantly different gene modules for each immune cell type among the subject groups (Data S1). We then performed PCA with these selected gene modules and visualized the top two PCs. This analysis revealed that PC1 stratified individuals by critical illness and that PC2 partially stratified COVID-19-affected and non-COVID ARDS-affected individuals (Figure 1F). Interestingly, three of the top five gene modules that enabled separation of COVID-19-affected and non-COVID ARDS-affected individuals from healthy control subjects were linked to monocytes (Figure 1G), consistent with the notion that monocytic activation states play an important role in COVID-19 disease pathogenesis. These results demonstrate that immune cell transcriptional states could better stratify subject groups compared to cell frequencies and prompted us to interrogate whether transcriptional states in PBMCs were associated with outcome in COVID-19-affected individual.
PBMC transcriptional signatures are predictive of outcome in COVID-19
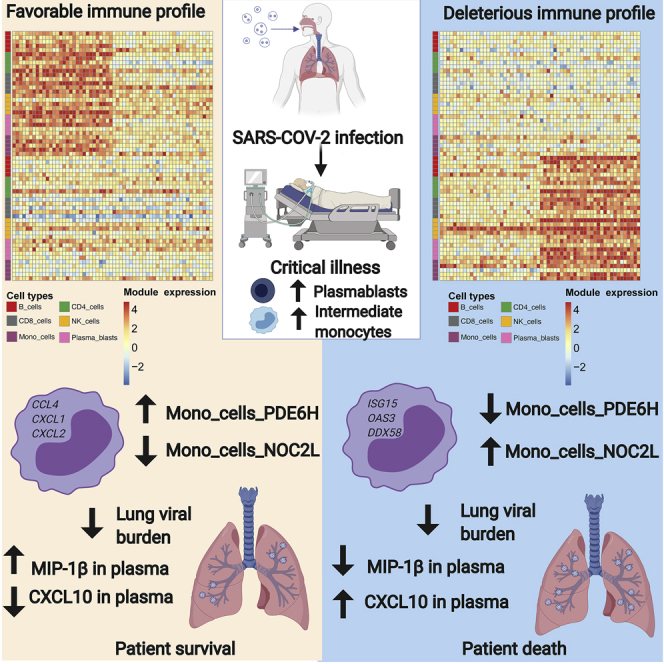
After using day 1 gene module scores to stratify individuals by clinical group, we next queried whether these transcriptional states were associated with COVID-19 outcomes (i.e., death versus survival). To address this question, we first performed a Wilcoxon rank sum test to identify gene module scores significantly associated with mortality. We found a total of 87 nominally statistically significant gene modules (Data S1), and when we visualized these gene modules across all individuals and performed co-clustering analysis, we observed a demarcation between individuals who survived and those who did not (Figure 2A). Notably, we identified several monocyte gene modules that were associated either with survival (e.g., Mono_cells_PDE6H) or with death (e.g., Mono_cells_NOC2L).
Figure 2.

Discovery and validation of a predictive model for COVID-19 mortality based on peripheral immune signatures
(A) Heatmap displaying hierarchical clustering of Arboreto-delineated gene modules that were statistically significantly associated with COVID-19 outcome. Samples (day 1 ICU) are arrayed in columns and gene modules in rows. Color bar on the left indicates the immune cell type from which each gene module was derived.
(B) First two PCs based on statistically significant gene modules from day 1 (A) reveals separation between individuals by outcome.
(C) Receiver operating curve (ROC) for LOOCV of individuals with COVID-19 from the University of Pittsburgh shows area under the curve (AUC) scores from 0.81 to 0.86 for three different models.
(D) UMAP of all PBMCs from Ren et al.37 used as an external validation cohort (top) and ROC for mortality prediction based on the models derived from our cohort (bottom).
(E) UMAP of all PBMCs from Liu et al.38 used as an external validation cohort (top) and ROC for mortality prediction based on the models derived from our cohort.
(F) UMAP of all PBMCs from Stephenson et al.39 used as an external validation cohort (top) and ROC for mortality prediction based on the models derived from our cohort.
Pitt cohort, 24 affected individuals; Ren et al., 33 affected individuals; Liu et al., 32 affected individuals; Stephenson et al., 54 affected individuals.
To evaluate whether there is a robust multivariate gene module signature associated with 90-day mortality, we first utilized the 87 significant gene module scores on day 1 to perform PCA. The resulting PC1 and PC2 embeddings stratified individuals who died from those that survived (Figure 2B). The separation between outcome evident in the first two PCs suggested that gene modules could be predictive of mortality. We next performed leave-one-out cross-validation (LOOCV) to evaluate the predictive power of this approach within our cohort. In a LOOCV framework, using either a support vector machine (SVM) classifier with a linear kernel, a random forest (RF), or a neural network (NNET) on the significant gene modules (with assessment of significance done separately within each fold to avoid signal leakage) yielded accurate stratification of the individuals who survived versus those that died (Methods details). Area under the curve (AUC) scores from the LOOCV ranged from 0.81 to 0.86 (Figure 2C), demonstrating that peripheral blood immune transcriptional signatures were predictive of outcome in our cohort. We also performed permutation testing (Methods details), comparing the accuracy across LOO folds versus permutated labels, and found that the LOO statistically significantly outperformed the permutated labels, as expected (Figure S4). Model accuracy and associated metrics are presented in Data S1.
After finding strong predictive power by LOOCV analysis, we next sought to determine whether the peripheral immune signatures derived from our discovery cohort could be validation in external cohorts. We identified three relevant external cohorts of scRNA-seq datasets of peripheral blood immune cells from individuals with COVID-1938, 37, 39 (Key resources table) and analyzed the major immune lineages from these cohorts (Methods details; Figures 2D–2F, top panels). We then generated cell-type-specific gene module scores for each individual in the three external cohorts, using the 87 significant gene modules identified in our discovery cohort. AUC scores across these studies were generally consistent with those from the LOOCV analysis, with ranges from 0.73 to 0.81 in Ren et al.,37 0.87 to 0.88 in Liu et al.,38 and 0.58 to 0.59 from Stephenson et al.39 (Figures 2D–2F, bottom panels). These data demonstrate that peripheral immune cell transcriptional signatures derived from our discovery cohort are generally predictive of death versus survival in COVID-19-affected individuals across multiple independent cohorts. Model accuracy and other associated model metrics across validation cohorts are present in Data S1.
Myeloid cell states are associated with plasma cytokine levels and drive pathogenesis
Given that our flow cytometry data showed elevated inflammatory monocytes in individuals with COVID-19 (Figure 1B), that higher frequencies of classical and intermediate monocytes were associated with ARDS/COVID-19 (Figure 1D), and that our predictive modeling identified monocyte cell states as associated with outcome (Figure 2A), we next interrogated the transcriptional signatures of monocytes in detail. To achieve this, we re-clustered all PBMC monocytes from all samples and visualized these cells using either canonical monocyte markers (Figure 3A) or unsupervised clustering (Figure 3B). Cluster 4 represented intermediate monocytes; clusters 1, 2, 3, 7, and 8 were classical monocytes; and cluster 6 represented non-classical monocytes.
Figure 3.

Peripheral cytokine levels are linked to monocyte cell states and are associated with mortality
(A) UMAPs showing canonical myeloid clusters in peripheral blood of individuals who died versus those who survived.
(B) Same UMAPs as (A), but myeloid cells are delineated using graph-based clustering.
(C) Frequency and magnitude of expression of genes reflective of bifurcated inflammatory monocyte states. Shown are the frequency of cells expressing the indicated gene (size of the dot) and its mean expression level in that cluster (color of the dot).
(D) Levels of MIP-1B (CCL4) in plasma trend toward a correlation with the monocyte gene module Mono_cells_PDE6H. p value derived from Spearman's correlation.
(E) Levels of CXCL10 in plasma are correlated with the monocyte gene module Mono_cells_NOC2L. p value derived from Spearman's correlation.
(F) Patients with higher levels of MIP-1B on day 1 in plasma have better survival versus those with lower MIP-1B. p value derived from Cox proportional hazards logrank test.
(G) Patients with higher levels of CXCL10 on day 1 in plasma have worse survival versus those with lower CXCL10. p value derived from Cox proportional hazards logrank test.
(H) Mono_cells_NOC2L gene module levels are strongly correlated between days 1 and 5 in the ICU. p value derived from Spearman's correlation.
(I) Mono_cells_NOC2L on day 5 is inversely correlated with Mono_cells_PDE6H on day 1. p value derived from Spearman's correlation.
(J) On day 5, Mono_cells_NOC2L is correlated with numerous inflammatory cytokines including TNF-α, IL6, and IL8.
scRNaseq data, 34,160 cells from 55 distinct samples; survival analysis based on 27 affected individuals; cytokine correlation analysis based on 20 affected individuals.
Next, we analyzed the association between the monocyte clusters and differentially expressed genes associated with outcome. Based on the aforementioned gene modules, we focused on key cytokine and IFN response genes. This analysis revealed the expression of CCL3, CCL4, CXCL1, CXCL2, and CXCL3 to be associated with the classical monocyte clusters 7 and 8, while the IFN response genes such as IFI44L, MX1, MX2, OAS3, and others associated with intermediate monocyte cluster 4 (Figure 3C). These findings suggest that a distinctive chemokine programmed state of classical monocytes in peripheral blood is associated with survival, while high levels of IFN signaling and viral sensing in intermediate monocytes are associated with mortality. To distinguish the activation states of canonical monocyte subsets, we performed gene set enrichment analysis34 (Data S1). Gene sets for IFN signaling were highly enriched in intermediate monocytes, consistent with viral sensing. We also utilized flow cytometry to analyze the expression levels of the SARS-CoV-2 entry receptor ACE2 (which has recently been shown to be an IFN-stimulated gene40) and cofactor neuropilin-1 (NRP1).41,42 ACE2 expression was significantly higher in both classical and intermediate monocytes in COVID-19-affected individuals as well as in non-COVID-19 ARDS-affected individuals, compared to healthy donors (Figures S5A and S5B). Notably, only intermediate monocytes from individuals with COVID-19 expressed NRP1 at significantly higher levels versus healthy controls (Figures S5C and S5D). Both intermediate and classical monocytes expressed higher levels of the inflammation-associated chemokine receptor CCR5 (Figures S5E and S5F). Consistent with our transcriptional analysis, flow cytometry revealed increased expression of activated and antiviral states in monocyte populations from individuals with COVID-19.
Our findings that monocyte inflammatory states may play an important role in COVID-19 disease outcome prompted us to evaluate whether the Mono_cells_NOC2L and Mono_cells_PDE6H gene module scores were correlated with levels of inflammatory cytokines in plasma. Briefly, we measured levels of 44 cytokines on days 1, 5, and 10 from individuals with COVID-19 using the Meso Scale Discovery platform (Methods details). The Mono_cells_PDE6H module trended toward a significant correlation with macrophage inflammatory protein 1 β (MIP-1β) in plasma (Figure 3D), consistent with the inclusion of the gene for MIP-1β (CCL4) in this module. Conversely, the Mono_cells_NOC2L module correlated with CXCL10 in plasma on day 1 (Figure 3E), consistent with CXCL10 being strongly associated with response to IFN in monocytes and suggesting that the Mono_cells_NOC2L module is reflective of an IFN response in monocytes. We next evaluated the extent to which this chemokine was associated with survival. Using Cox proportional hazards regression (Methods details), we found that higher levels of plasma MIP-1β on day 1 were associated with improved survival (Figure 3E). We also found that higher levels of CXCL10 in plasma on day 1 trended toward an association with greater risk of death (Figure 3G). The outcome trends associated with these cytokines and their molecular linkages with the monocyte gene modules strengthened the overall interpretation of divergent monocytic states that could underlie disease trajectories. Importantly, these measures of monocyte states and plasma cytokines were not correlated with age (Figures S6A–S6D) or associated with whether individuals received glucocorticoids (Figures S6E–S6H). Therefore, the monocyte gene modules capture key aspects of divergent systemic inflammatory states in severe COVID-19 disease that are predictive of mortality.
We next used longitudinal data from COVID-19 samples to investigate the stability of the initial monocytic cell states (day 1 post-enrollment) and their temporal relationships with inflammatory cytokines. We first used a rank sum test to determine if monocyte gene modules on days 5 and 10 were associated with subsequent mortality and found that there were 60 monocyte modules nominally associated with outcome on day 5 but none on day 10 (using a two-sided alpha of 10% as a cutoff; Data S1). We next evaluated which monocyte modules on day 5 were correlated with Mono_cells_NOC2L or Mono_cells_PDE6H monocyte modules on day 1 (Data S1). This analysis revealed the Mono_cells_NOC2L module to be strongly correlated with itself at days 1 and 5 (Figure 3H). There was also a strong negative correlation between Mono_cells_PDE6H on day 1 and Mono_cells_NOC2L on day 5 (Figure 3I). These data demonstrate that monocyte states on day 1, as measured by NOC2L or PDE6H gene module scores, are dominated by one signature or the other and remain reflective of monocyte states on day 5.
Finally, we sought to evaluate the significance of protracted elevation of the Mono_cells_NOC2L gene module. To this end, we evaluated the relationships between Mono_cells_NOC2L levels and inflammatory cytokine levels in plasma on day 5 and found that Mono_cells_NOC2L was strongly correlated with inflammatory cytokines such as CXCL10, IL-6, TNF-α, and IL-8 on day 5 (Figure 3J). Taken together, these data suggest that sustained viral sensing and IFN signaling by monocytes (inferred by high Mono_cells_NOC2L levels) promote an inflammatory immune state involving a network of cytokines, leading to immunopathogenesis and organ damage. Importantly, high levels of Mono_cells_PDE6H on day 1 appear to dictate a disease course in which inflammatory cytokines do not become correlated at day 5.
SARS-CoV-2-infected respiratory tract myeloid cells have distinct transcriptional profiles
We next aimed to characterize immune transcriptional states by scRNA-seq in endotracheal aspirate (ETA) samples from mechanically ventilated individuals. Like our PBMC analysis, we visualized cells using UMAP and identified cell types by canonical gene expression profiles (Figure 4A). Compared to PBMCs, we identified cells with unique expression profiles consistent with epithelia and CD14+CD16+ macrophages in addition to populations found in PBMCs. Graph-based clustering revealed five distinct clusters of myeloid cells (clusters 1, 2, 4, 5, and 6), with the CD14+CD16+ macrophages having two distinct cell states (clusters 1 and 4; Figure 4B). Next, we evaluated expression of SARS-CoV-2 transcripts by scRNA-seq (Methods details). Briefly, inclusion of the SARS-CoV-2 genome in the GRCh38 reference sequence permitted mapping of viral sequences to individual cells.37,43,44 We found that CD14+CD16+ macrophages had the highest levels of SARS-CoV-2 transcript expression (as measured by the viral N gene; Figure 4C). When evaluated by graph-based clustering, we found that the CD14+CD16+ macrophage cluster 4 had the highest levels of viral transcripts, compared with a paucity of transcripts from CD14+CD16+ macrophage cluster 1 (Figure 4D). This suggested that infected myeloid cells may have a unique expression profile.
Figure 4.

CXCL10 is consistently expressed by infected myeloid cells in the lower respiratory tract of critically ill individuals with COVID-19
(A) UMAP showing the inferred cell types of all 6,328 cells from ETA samples from the University of Pittsburgh cohort.
(B) Same UMAP as (A), but showing clusters derived by graph-based clustering. Two distinct states of macrophages are present (clusters 1 and 4), and three other myeloid clusters were similar to expression profiles in peripheral blood.
(C) Macrophages show the highest recovery of viral transcripts across cell types.
(D) Of the macrophage clusters (B), cluster 4 shows the highest recovery of viral transcripts. Surprisingly, few transcripts were recovered from the other myeloid cell clusters.
(E) Differentially expressed genes associated with each myeloid cluster reveal a distinct profile of infected cells including expression of CXCL10.
(F–I) UMAPs of lower respiratory tract cells from individuals with COVID-19 from four external datasets.
(J) CXCL10 is one of the top genes associated with infected myeloid cells in the lower respiratory tract in a meta-analysis of four different external cohorts. The top 200 up- and downregulated genes are ordered by the weighted average derived from the meta-analysis, where the weight is based on the number of affected individuals in each study.
We next evaluated differentially expressed genes between myeloid cell clusters. Consistent with peripheral blood immune signatures, we found that intermediate monocytes had a strong IFN response signature (Figure 4E). Interestingly, infected CD14+CD16+ macrophages had high expression levels of specific cytokines, including CXCL10 (Figure 2G). This finding was suggestive of a role of infected myeloid cells in COVID-19 immunopathogenesis and motivated further interrogation in external cohorts.
We, therefore, performed a meta-analysis across datasets to determine if CXCL10 was consistently upregulated in infected myeloid cells in the lower respiratory tract (Methods details). We identified four studies in which scRNA-seq was performed on lower respiratory tract cells37,43,44 or nuclei45 (Figures 4F–4I) from individuals with COVID-19. Each of these studies also quantified levels of SARS-CoV-2 transcripts in cells/nuclei as described above. We next identified clusters of myeloid cells in each study and identified the myeloid cluster with the highest frequency of cells positive for SARS-CoV-2 transcripts (Figures S7A–S7D). Next, we determined the log fold-change for all genes between the infected myeloid cluster and all clusters for each individual and created a mean and SD of log fold-change for each study (Methods details; Data S1). A meta-analysis was then performed across all four studies, using a weighted average of the mean log fold-change from each individual study where the weight was dictated by the number of affected individuals (Methods details). The resulting heatmap of the top 200 up- and downregulated genes by meta-analysis was ordered by the weighted average and demonstrated that CXCL10 was among the top upregulated gene expressed by infected myeloid cells (Figure 4J). This meta-analysis strongly supports the notion that SARS-CoV-2-infected myeloid cells contribute to immunopathogenesis through upregulation of CXCL10.
Discussion
In this study, we sought to evaluate cellular and molecular features associated with the immunopathogenesis of severe COVID-19 illness and that were predictive of mortality. Our study highlights the importance of analyzing the initial immune state of individuals with COVID-19 early in their ICU course by deconvolution of transcriptional states of peripheral immune cells based on gene modules. The utilization of our cohort as a discovery cohort, in conjunction with results confirmed in three external cohorts, demonstrates the ability of a multidimensional peripheral immune signature to predict outcome in COVID-19. Earlier work in non-COVID ARDS has defined monocyte signatures in PBMCs that are indicative of disease progression46; our work demonstrates distinctive monocyte gene modules as part of a multivariate peripheral immune state that is predictive of COVID-19 disease mortality. Among the gene modules on day 1 that are predictive of outcome, we uncovered a bifurcation of monocyte inflammatory modules. Viral sensing and IFN response genes were reflective of a monocytic state (NOC2L module) that was sustained over 5 days in the ICU and correlated with serum cytokines CXCL10, IL-6, TNF-α, and IL-8. CXCL10 has been associated with severe COVID-19,13 and IL-6, TNF-α, and IL-8 have been associated with increased risk of death from COVID-19.14 In contrast, a divergent inflammatory monocytic state (PDE6H module) involving expression of the chemokine genes CCL3 (MIP-1α), CCL4 (MIP-1β), CXCL1, and CXCL2 was associated with survival.
In addition to analysis of immune cell states in peripheral blood and plasma cytokine levels, our analysis of lower respiratory tract myeloid cells revealed that CXCL10 may play an important role in driving pathogenesis. Studies of Middle East respiratory syndrome (MERS) coronavirus have demonstrated infection of monocyte-derived macrophages with subsequent secretion of inflammatory cytokines such as CXCL10 and CXCL8 (IL-8).47 Consistent with our findings, SARS-CoV abortively infects human macrophages but triggers production of CXCL10 and CCL2.48,49 Furthermore, recent work demonstrated that infected myeloid cells in the lower respiratory tract of individuals with COVID-19 participate in a positive-feedback loop in which infected myeloid cells produce T cell chemoattractants, recruiting T cells into the lung.44 These T cells then secrete IFN-γ, contributing to release of inflammatory cytokines from macrophages50 and thereby promoting further T cell activation.44 In the context of our findings that elevated CXCL10 levels in plasma are predictive of death, we propose that higher levels of CXCL10 in peripheral blood are reflective of a higher degree of viral burden (and, correspondingly, greater immunopathogenesis) in the lung.
Our combined analysis of peripheral blood immune cell states, lower respiratory tract cells, and plasma cytokines provides important insight into the pathogenesis of severe COVID-19. A specific peripheral blood monocyte state characterized by the Mono_cells_NOC2L gene module was correlated with plasma levels of CXLC10, suggesting that peripheral blood immune signatures are reflective of the degree of infection in the lower respiratory tract (given that infected myeloid cells appear to be a dominant source of CXCL10). Interestingly, the Mono_cells_NOC2L was governed by genes associated with viral sensing, and type I IFN response and activated monocyte states were associated with elevated expression of ACE2 and NRP1. Plasma viremia is also associated with increased risk of mortality in individuals with COVID-19,51 suggesting that peripheral monocytes may become activated by inflammatory cytokines, express entry receptors such as ACE2, and become further activated by viral entry or opsonization, triggering innate viral sensors such as retinoic acid inducible gene I (RIG-I) in monocytes. This pathogenic process would not require productive viral infection of monocytes but could, nevertheless, contribute to viral pathogenesis by driving increasing immune-related inflammation.
The finding that CCL4 (MIP-1β) was associated with survival is consistent with its positive prognostic role in Dengue infection.52 In hepatitis C infection, higher MIP-1β was associated with viral control following treatment with antiviral therapy.53 Interestingly, MIP-1β is a type-I-IFN-dependent gene but does not directly inhibit viral replication. Instead, it promotes the recruitment of monocytes to infected tissue to prevent viral spread throughout the tissue.54 In this context, our findings suggest a model in which preferential expression of MIP-1β, in relation to other IFN response genes by monocytes, protects infected tissue in the lung during severe COVID-19.
High levels of inflammatory cytokines are associated with increased risk of death in individuals with COVID-19,14 but the immunologic drivers of these cytokine levels remain incompletely understood. Type I IFN signaling has been proposed to play both a protective and a pathogenic role in COVID-19 disease, depending on its kinetics.18, 19, 20 It is essential early in infection in moderating COVID-19 disease, as individuals with either IFN auto-antibodies16 or inborn errors of type I IFN17 production have a much higher risk of severe COVID-19. However, dysregulated IFN signaling at later times during COVID-19 disease progression appears to be pathogenic.55,56 Our analysis revealed that monocyte gene modules on day 1 dictate distinct disease trajectories, with the high levels of the IFN-response-associated Mono_cells_NOC2L module on day 1 correlating with its levels on day 5 in individuals. Additionally, the Mono_cells_NOC2L module on day 5 is correlated with numerous cytokines associated with disease severity and death, including CXCL10, IL-6, TNF-α, and IL-8. In support of this association, a late immune juncture beginning at 17 days following symptom onset (and corresponding to day 10 in our cohort) was recently described as associated with death in individuals with COVID-19.38 Thus, we infer that protracted IFN signaling (as reflected by consistently high Mono_cells_NOC2L module scores, ICU days 1–5) promotes an immune state leading to immunopathogenesis, organ damage, and death.
Our study (which is supported by validation studies with external cohorts) adds important details of immunopathogenesis. The high-dimensional approaches we applied to our cohort allowed us not only to characterize peripheral biomarkers that were predictive of mortality, but also to elucidate deeper insights into the biology underlying critical illness in COVID-19. Nevertheless, our findings may facilitate discovery of new diagnostics and therapeutics to improve outcome in severe COVID-19.
Limitations of the study
A limitation of our study is the relatively small number of individuals with COVID-19 in our cohort, which precludes in-depth analysis of relationships among flow cytometry measurements, transcriptional states of immune cells, and cytokines with clinical covariates such as diabetes, age, and gender. These covariates are important contributors to outcome and need to be more fully integrated with the high-dimensional immune system analyses in larger studies. Another limitation is that our derivation of immune signatures was based on samples from critically ill individuals obtained soon after admission to the ICU. This complicates validation in external cohorts that have limited sample available from critically ill individuals in the same time interval. While our immune signatures perform well in several cohorts, the predictive power is variable, and more extensive validation across appropriate cohorts is required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human HLA-DR conjugated to BUV395 | BD Biosciences | Cat# 564040; RRID:AB_2738558 |
| Mouse anti-human CD8 conjugated to BUV496 | BD Biosciences | Cat# 612942; RRID:AB_2870223 |
| Mouse anti-human CD4 conjugated to BUV563 | BD Biosciences | Cat# 741353; RRID:AB_2870854 |
| Mouse anti-human CD103 conjugated to BUV615 | BD Biosciences | Cat# 751285; RRID:AB_2875299 |
| Mouse anti-human CD45 conjugated to BUV661 | BD Biosciences | Cat# 750178; RRID:AB_2874383 |
| Mouse anti-human CD14 conjugated to BUV737 | BD Biosciences | Cat# 612763; RRID:AB_2870094 |
| Mouse anti-human CD19 conjugated to BUV805 | BD Biosciences | Cat# 742007; RRID:AB_287130 |
| Mouse anti-human Ki-67 conjugated to BV412 | Biolegend | Cat# 350506; RRID:AB_2563860 |
| Rat anti-human FOXP3 conjugated to eFluor450 | Thermo Fisher Scientific | Cat# 48-4776-42; RRID:AB_1834364 |
| Mouse anti-human CD38 conjugated to BV480 | BD Biosciences | Cat# 566186; RRID:AB_2739582 |
| Mouse anti-CD1c conjugated to PE-Cy5 in house | Biolegend | Cat# 331502; RRID:AB_1088995 |
| Mouse anti-human CD45RA | Biolegend | Cat# 304108; RRID:AB_314412 |
| Mouse anti-human CD62L conjugated to BV605 | Biolegend | Cat# 304834; RRID:AB_2562130 |
| Mouse anti-human CD15 conjugated to BV650 | Biolegend | Cat# 323034; RRID:AB_2563840 |
| Mouse anti-human CD25 conjugated to BV711 | Biolegend | Cat# 302636; RRID:AB_2562910 |
| Mouse anti-human CD20 conjugated to BV750 | BD Biosciences | Cat# 747062; RRID:AB_2871819 |
| Mouse anti-human CD141 conjugated to BV785 | Biolegend | Cat# 344116; RRID:AB_2572195 |
| Mouse anti-human CD36 conjugated to CD36 | Biolegend | Cat# 336204; RRID:AB_1575025 |
| Mouse anti-human CD3 conjugated to SparkBlue 550 | Biolegend | Cat# 344852; RRID:AB_2819985 |
| Mouse anti-human CD11b conjugated to PerCP/Cyanine5.5 | Biolegend | Cat# 393106; RRID:AB_2734453 |
| Mouse anti-human CD56 conjugated to PerCP-eFluor710 | Thermo Fisher Scientific | Cat# 46-0567-42; RRID:AB_10548939 |
| Mouse anti-human ACE2 conjugated to PE | R&D Systems | Cat# FAB933P-100 |
| Mouse anti-human CD16 conjugated to PE/Texas-Red | Biolegend | Cat# 302016; RRID:AB_314216 |
| Mouse anti-human CD27 conjugated to BV510 | Biolegend | Cat# 302836; RRID:AB_2562086 |
| Mouse anti-human CD138 conjugated to APC | Biolegend | Cat# 142506; RRID:AB_10962911 |
| Mouse anti-human CD11c conjugated to AF700 | Biolegend | Cat# 301648; RRID:AB_2819923 |
| Mouse anti-human CD192 conjugated to APC/Cy7 | Biolegend | Cat# 357220; RRID:AB_2566750 |
| Mouse anti-human CD16 conjugated to BV421 | Biolegend | Cat# 302038; RRID:AB_2561578 |
| Mouse anti-human CD195 conjugated to BV510 | Biolegend | Cat# 313707; RRID:AB_345307 |
| Mouse anti-human CD3 conjugated to BV650 | Biolegend | Cat# 100229; RRID:AB_11204249 |
| Mouse anti-human CD19 conjugated to BV650 | Biolegend | Cat# 302238; RRID:AB_2562097 |
| Mouse anti-human CD20 conjugated to BV650 | Biolegend | Cat# 302336; RRID:AB_2563806 |
| Mouse anti-human CD56 conjugated to BV650 | Biolegend | Cat# 362532; RRID:AB_2565602 |
| Mouse anti-human CD163 conjugated to BV711 | Biolegend | Cat# 333630; RRID:AB_2650972 |
| Mouse anti-human CCR2 conjugated to BV785 | Biolegend | Cat# 357234; RRID:AB_2800972 |
| Mouse anti-human ACE2 conjugated to APC | R&D Systems | Cat# FAB933A-100 |
| Mouse anti-human NRP1 conjugated to PE | Biolegend | Cat# 354504; RRID:AB_11219194 |
| Mouse anti-human CD36 conjugated to PE/Cyanine7 | Biolegend | Cat# 336222; RRID:AB_2716142 |
| Total-SeqC Hashtag Barcode 0251 | Biolegend | Cat# 394661; RRID:AB_2801031 |
| Total-SeqC Hashtag Barcode 0252 | Biolegend | Cat# 394663; RRID:AB_2801032 |
| Total-SeqC Hashtag Barcode 0253 | Biolegend | Cat# 394665; RRID:AB_2801033 |
| Total-SeqC Hashtag Barcode 0254 | Biolegend | Cat# 394667; RRID:AB_2801034 |
| Total-SeqC Hashtag Barcode 0255 | Biolegend | Cat# 394669; RRID:AB_2801035 |
| Total-SeqC Hashtag Barcode 0256 | Biolegend | Cat# 394671; RRID:AB_2820042 |
| Biological samples | ||
| Whole blood from healthy donors | University of Pittsburgh | NA |
| Whole blood from non-COVID ARDS-affected individuals | University of Pittsburgh | NA |
| Whole blood from COVID-19-affected individuals | University of Pittsburgh | NA |
| Endotracheal aspirates from COVID-19-affected individuals | University of Pittsburgh | NA |
| Chemicals, peptides, and recombinant proteins | ||
| Zombie near-IR viability dye | Biolegend | Cat# 423105 |
| eFluor780 fixable viability dye | Thermo Fisher Scientific | Cat# 65-0865-18 |
| Critical commercial assays | ||
| Chromium Single Cell 5′ Library & Gel Bead Kit, 16 rxns | 10X Genomics | Cat# PN-1000006 |
| Chromium Single Cell 5′ Library Construction Kit, 16 rxns | 10X Genomics | Cat# PN-1000020 |
| Chromium Single Cell A Chip Kit, 48 rxns | 10X Genomics | Cat# PN-1000152 |
| Chromium i7 Multiplex Kit, 96 rxns | 10X Genomics | Cat# PN-120262 |
| V-PLEX Human Cytokine 44-plex Kit | Meso Scale Discovery | Cat# K15249D-2 |
| Deposited data | ||
| Single-cell RNaseq dataset | This study | Accession number: GEO: GSE180578 |
| Single-cell RNaseq dataset | Liao et al.43 | Accession number: GEO: GSE145926 |
| Single-cell RNaseq dataset | Grant et al.44 | Accession number: GEO: GSE155249 |
| Single-cell RNaseq dataset | Liu et al.37 | Accession number: GEO: GSE161918 |
| Single-cell RNaseq dataset | Ren et al.38 | Accession number: GEO: GSE158055 |
| Single-cell RNaseq dataset | Stephenson et al.39 | https://www.covid19cellatlas.org/ |
| Single-cell RNaseq dataset | Delorey et al.45 | Accession number: GEO: GSE171668 |
| Additional Data | This study | Mendeley Data: http://doi.org/10.17632/r83csstphc.2 |
| Software and algorithms | ||
| Red Hat Enterprise Linux Server (v7.7) | Red Hat | https://access.redhat.com/documentation/en-us/red_hat_enterprise_linux/7/html/7.7_release_notes/index |
| CellRanger (v3.1.0) | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| CiteSeqCount (v1.4.3) | Roelli57 | https://github.com/Hoohm/CITE-seq-Count |
| R (v3.6.0) | NA58 | https://www.r-project.org/ |
| Tidyverse (v1.3.0) | Wickham et al.59 | https://www.tidyverse.org/ |
| ggplot2 (v3.3.2) | Wickham et al.60 | https://ggplot2.tidyverse.org/ |
| patchwork (v1.0.0) | Pederson et al.61 | https://patchwork.data-imaginist.com/ |
| Irlba (v2.3.3) | Baglama et al.62 | https://cran.r-project.org/web/packages/irlba/index.html |
| ggplotify (v.0.0.5) | Yu et al.63 | https://cran.r-project.org/web/packages/ggplotify/index.html |
| pheatmap (v1.0.12) | Kolde et al.64 | https://CRAN.R-project.org/web/packages/pheatmap/index.html |
| RColorBrewer (v1.1-2) | Neuwirth et al.65 | https://CRAN.R-project.org/web/packages/RColorBrewer/index.html |
| survival (v3.2-7) | Therneau66 | https://CRAN.R-project.org/web/packages/survival/index.html |
| survminer (v0.4.8) | Kassambara et al. 67 | https://CRAN.R-project.org/web/packages/survminer/index.html |
| lubridate (v1.7.4) | Grolemund and Wickham68 | https://www.jstatsoft.org/v40/i03/ |
| pROC (v1.16.1) | Robin et al.69 | https://www.biomedcentral.com/1471-2105/12/77/ |
| singleseqgset (v0.1.2.9000) | Cillo et al.34 | https://github.com/arc85/singleseqgset |
| future (v1.16.0) | Bengtsson70 | https://CRAN.R-project.org/web/packages/future/index.html |
| future.apply (v1.4.0) | Bengtsson71 | https://CRAN.R-project.org/web/packages/future.apply/index.html |
| SCENIC (v.1.1.2-2) | Aibar et al.72 | https://github.com/aertslab/SCENIC |
| Reticulate (v1.14) | Ushey et al.73 | https://CRAN.R-project.org/web/packages/reticulate/index.html |
| python (bioconda v.3.7-2019.03) | Björn et al.74 | https://bioconda.github.io/ |
| FlowJo (v9.9.6) | BD Biosciences | https://www.flowjo.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dario A.A. Vignali (dvignali@pitt.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Following acquisition of written informed consent from patients or their legally authorized representatives, we enrolled 42 consecutive critically ill patients with acute hypoxemic respiratory failure and symptoms/signs suggestive of COVID-19 in a prospective, observational cohort study (University of Pittsburgh Institutional Review Board study number 20040036). Patients were hospitalized in ICUs at two hospitals (Presbyterian and Shadyside) within the University of Pittsburgh Medical Center system. All patients underwent at least one nasopharyngeal swab testing for SARS-CoV-2 qPCR (reference standard diagnosis at our Institution), which could be repeated at the discretion of the treating physicians when the first test was negative and significant clinical suspicion for COVID-19 remained. Based on SARS-CoV-2 qPCR results, 35 patients with at least one positive test were diagnosed with COVID-19 (COVID-19 group), whereas 7 patients had at least one negative SARS-CoV-2 qPCR test and were diagnosed with a non-COVID etiology of acute respiratory illness (non-COVID group). For comparisons against healthy controls, we also included a single blood biospecimen from 10 healthy donors.
From enrolled critically ill patients, we collected blood specimens upon enrollment (day 1), and then if the patients remained in the ICU, we collected follow-up blood samples on days 5 and 10 post-enrollment. From intubated patients, we also collected endotracheal aspirate (ETA) samples for profiling of the lower respiratory tract.
Demographics information include age, gender, and race, history of chronic diseases such as diabetes and chronic obstructive pulmonary disease (COPD), timing of disease onset and whether patients received mechanical ventilation, and outcome data are all presented in Table S1.
Methods details
Sample processing
Whole blood was drawn by venipuncture into EDTA tubes. Plasma was separated from whole blood by centrifugation at 400xg for 5 min with the brake off. Following removal of plasma, blood was diluted with Hank’s Buffered Saline Solution (HBSS), and diluted blood was layered of Ficoll-Hypaque. Density gradient centrifugation was performed by spinning at 400xg for 20 min with the brake turned off, and the PBMC layer was removed. PBMC were then washed twice with HBSS, and carry-over red blood cells were lysed using BD Pharm Lyse per the manufacturer’s instructions. Viable cells were counted using a Nexcelom Cellometer with acridine orange and propidium iodide. Endotracheal aspirates (ETA) were collected from intubated patients in the ICU based on the condition of the patient; samples were not collected if the patient’s status was poor. ETA were processed by diluting 1.5 mL of samples up to 15 mL with HBSS, pipetting vigorously to break up aggregated sputum. Cells from ETA were then pelleted by centrifugation at 400xg for 5 min, and red blood cells were lysed using BD Pharm Lyse per the manufacturer’s instructions. Viable cells were counted using a Nexcelom Cellometer as described above. PBMC and ETA cells were cryopreserved in 90% FBS and 10% DMSO. Plasma was frozen at −80°C. All experiments with COVID-19 patient samples were performed in a Biosafety Level 2+ facility (with appropriate precautions) at the University of Pittsburgh’s Center for Vaccine research.
Flow cytometry analyses
PBMC were stained for flow cytometry as previously described.34 Briefly, cryopreserved PBMC were thawed in a water bath at 37°C, then diluted to 15 mL with warm RPMI with 10% FBS. 1-2x105 cells were placed in 96 well plates and centrifuged at 400xg for 5 min. Supernatant was then removed, and cells were resuspended in antibody cocktails consisting of phosphate buffered saline with 10% FBS (PBS/FBS) and appropriately diluted antibodies. All antibodies were used at a 1:100 final dilution. Samples were stained for 15 min a 4°C, and were then washed by adding PBS/FBS and centrifuging for 5 min a 400xg. Viability dye in PBS (1:4000 dilution) was then added, and samples were once again incubated for 15 min at 4°C, followed by a subsequent wash step in PBS. Next, samples were fixed using Becton Dickinson (BD) Fix/Perm solution as per the manufacturer’s instructions. Following fixation and permeabilization, intracellular antibodies were added in BD Perm/Wash solution at appropriate concentrations; samples were once again incubated at 4°C and washed. Samples were then resuspended in PBS/FBS and acquired on the appropriate flow cytometer. Samples were stained for subsequent assessment on either a Cytek Aurora 5-laser spectral flow cytometer or a Becton Dickinson 5 laser Fortessa II for standard flow cytometry. The 28 antibodies were used for the Cytek Aurora panel are listed in the Key Resources Table. Briefly, they are HLA-DR, CD8, CD4, CD103, CD45, CD14, CD19, Ki67, FoxP3, CD38, CD1c, CD45RA, CD62L, CD15, CD25, CD20, CD141, CD36, CD3, CD11b, CD56, ACE2, CD16, CD27, CD138, CD11c, and CCR2. We also used Zombie NIR fixable viability dye (Biolegend) at a 1:1000 dilution for this panel. The 14 antibodies used for our monocyte specific Fortessa II panel are listed in the Key Resources Table. Briefly, they are HLA-DR, CD14, CD16, CCR5, CD3, CD19, CD20, CD56, CD163::BV711, CCR2, ACE2, CD11c, NRP1, and CD36. We also used eFluor 780 Fixable Viability Dye (ThermoFisher) for this panel at a 1:4000 dilution. Flow cytometry was performed in the Hillman Cancer Center Flow Cytometry Facility.
We used principal component analysis (PCA) to visualize the distribution of cell frequencies across patient cohorts. PCA was performed using the R package irlba. Centered and scaled values of all immune cell frequencies were used as input. Sample embeddings and variable loadings were extracted from resulting PCA and used for visualization.
Single-cell RNaseq library generation and sequencing
Single-cell Rnaseq (scRNaseq) was performed using 5′ v1 kit from 10X Genomics as per the manufacturer’s instructions. Libraries were created from either fresh PBMC or were prepared from batches of cryopreserved PBMC. For fresh processing, libraries were created immediately following isolation of cells. For cryopreserved samples, samples were thawed as described above for flow cytometry. Sample multiplexing was performed using CITEseq. To achieve this, cells were first stained with TotalSeq-C antibodies for 15 min at 4°C, followed by washing in PBS with 10% FBS and centrifugation for 5 min at 400xg. Two samples (each stained with unique TotalSeq-C antibodies) were then pooled and loaded per lane of the 10X chip to permit sample multiplexing. Libraries were prepared following the manufacturer’s instructions, with the additional preparation of cell hashing libraries using the manufacturer’s protocol for Feature Barcode libraries. Final libraries concentration and size distributions were quantified using a BioAnalyzer as per the manufacturer’s instructions. Samples were sequenced on a NovaSeq at the UPMC Genome Core using a read 1 length of 28 cycles, read 2 length of 91 cycles and an i7 read length of 8 cycles.
Generation of gene expression and feature barcode matrices
Following sequencing, NovaSeq runs were downloaded from the UPMC Genome Core to the University of Pittsburgh Center for Research Computing High Throughput Cluster. Samples were then demultiplex using bcl2fastq (Illumina), using a base mask of Y28, I8, Y91 and setting the stringency to allow no barcode mismatches. Following demultiplexing, gene expression reads were then aligned to the reference genome using CellRanger v3.1.0, and feature barcode matrices were created for each sample. Importantly, we added the reference sequence for SARS-CoV-2 (NC_045512.2) to the genome and the GTF to facilitate detection of SARS-CoV-2 transcripts. Cell hashing libraries were aligned to TotalSeq-C barcodes using CITE-seq-Count v1.4.3,75 using a sample-specific cell barcode whitelist (i.e., only cell barcodes from cells identified by CellRanger from each sample were included in the whitelist).
Identification of individual samples from cell hashing
After generation of gene expression and feature barcode matrices, downstream analysis was performed using Seurat v3.1.476 in R 3.6.0. Gene barcode matrices and associated feature barcode matrices containing cell hash expression values were read into R. To identify individual samples, feature barcode matrices were first log-normalized using a centered log ratio transformation (CLR) implemented in Seurat. Individual samples were identified by unique expression of the anticipated TotalSeq-C antibody and the absence of alternative TotalSeq-C antibodies. To permit automated identification of samples, k-means clustering was performed on CLR normalized expression values for each sample to identify cut-offs for negative and positive TotalSeq-C counts. Samples were visualized on bi-variate x/y plots to confirm adequate cutoffs. Cells with expression levels above the cut-offs for both TotalSeq-C antibodies were considered doublets and were excluded from downstream analysis.
Visualization of scRNaseq data and identification of cell types
We utilized the data integration workflow76 provided in Seurat to integrate between fresh and cryopreserved samples. Briefly, we independently identified highly variable features between fresh and cryopreserved samples and selected 2,000 integration features from this combined set of highly variable features. Selected integration features were then independently scaled across all cells in both fresh and cryopreserved samples, and PCA was performed using the scaled expression levels in each dataset. Next, integration anchors were identified using a reciprocal PCA across the first 30 PCs. Integrated data was then used for downstream PCA, visualization and clustering. Dimensionality reduction for visualization was next performed using Uniform Manifold Approximation Embedding (UMAP)35 implemented in Seurat. Clusters were identified using graph-based clustering in Seurat, and inspection of canonical lineage markers and their association with each cluster was used to identify cell types. One cluster of cells expressed high levels of genes involved in the cell cycle, but included multiple cell types. Cell types were resolved from this cluster by bioinformatically isolating these cells and inspecting lineage specific markers and then applying those labels to the cells in the overall UMAP.
Generation of gene module scores for immune lineages
To quantify gene co-express modules within each immune lineage, we first bioinformatically isolated each major immune lineage (i.e., CD4+ T cells, CD8+ T cells, B cells, plasmablasts, monocytes and NK cells) and verified there were no contaminating immune cells from other lineages present (e.g., there were no clusters expressing MS4A1 in the CD4+ T cell data, no clusters expressing CD14 in the CD8+ T cells, et cetera). Next, we utilized both the SCENIC72 R package (version 1.1.2-2) and the Arboreto package (version 0.1.0) implemented in PySCENIC36 in Python (Bioconda 3.7-2019.03). In R, genes were filtered based on a minimum count of 3 in 1% of cells, and expression in at least 1% of cells. Filtered expression matrices and a list of expressed transcription factors were then exported to be used in GRNBoost2 from the Aboreto package in Python. Gene modules identified by GRNBoost2 were filtered based on having greater than 20 genes and fewer than 200 genes, then scored across all cells in an expression dataset using the AddModuleScore function in Seurat. Briefly, the AddModuleScore function generates a score based on the aggregated expression level across a set of genes compared to a background set of genes with a similar express magnitude. Like to gene set enrichment, this allows a group of genes to be scored at the level of individual cells. Based on the modules scores, a median module scores were assigned to each patient for a given immune lineage. If less than 20 cells were present in a given patient sample, the median module score was imputed as the median across all patients.
Discovery of a predictive algorithm for mortality in COVID-19
We sought to leverage machine learning algorithms in conjunction with gene modules scores on day 1 post-enrollment in the ICU to predict outcome for critically ill patients with COVID-19. To do this, we utilized our cohort from the University of Pittsburgh as a discovery cohort to train a support vector machine (with a linear kernel), random forest, and neural net. To reduce the dimensionality prior to input into the machine learning algorithms, we first performed PCA using all scaled and centered gene modules scores from day 1 that were significantly associated with outcome (p < 0.05 by Wilcoxon rank sum test) as input. If a patient did not have a gene module score for a particular lineage due to too few cells present, the median gene module score from all samples was used as the interpolated value. We then used the embeddings for each patient in the first two principal components from the PCA as input for the machine learning algorithms. We implemented these algorithms through the R package caret.77
Cross-validation for the discovery cohort was performed using a leave-one-out analysis. To prevent data leakage between folds of cross validation, gene modules from day 1 samples that had a p < 0.05 for a Wilcoxon rank sum test between patients who survived versus those that died were selected within each fold. Then, modules were scaled and PCA was performed for dimensionality reduction in each fold, once again using PC1 and PC2 were used as predictive variables. Tuning parameters for each algorithm were selected internally within each round of cross-validation. Receiving operating curves and area under the curve were calculated using the R package pROC.69
Permutation testing for evaluating machine learning models
We performed permutation testing to determine if machine learning models outperformed the same models with permuted class labels. We determined the accuracy of each of the machine learning models when leaving one patient out for the Pitt cohort, and compare the resulting accuracy scores to models generated with the outcome labels for each patient permuted. We then compared the resulting accuracy using a t test.
Validation of the peripheral immune signature for mortality
To determine if the peripheral immune signatures derived from our discovery cohort were valid across cohorts, we identified and analysis 3 external datasets consisting of single-cell Rnaseq of PBMC from patients with COVID-19. Datasets were selected by having publicly available single-cell Rnaseq data from PBMC of COVID-19 patients and outcome data for each patient. For each of these external datasets, the processed data was downloaded from the Gene Expression Omnibus. Cell types annotations were used when provided by authors, and were characterized by canonical gene expression profiles otherwise. Patients were selected for inclusion based on availability of information pertaining to survival. Patients were retained in all instances where symptomatic disease was noted. Gene module scores for the statistically significant gene modules on day 1 in the discovery cohort were calculated for each immune lineage for each patient in the validation cohorts using the AddModuleScore function from Seurat. After gene module scores were calculated, the machine learning algorithms derived from the discovery cohort were applied to the validation cohorts. Receiver operating curves and area under the curve were calculated using the R package pROC69 as described above.
In-depth analysis of myeloid lineages
Further dissection of myeloid lineages was performed by bioinformatically isolating and re-clustering all myeloid cells. Datasets were once again integrated for downstream visualization and clustering based on fresh versus frozen status using the workflow described above. Differentially expressed genes were identified using a Wilcoxon rank sum test as implemented in Seurat. Gene set enrichment analysis was performed as previously described using the R package singleseqgset.34 Statistically significant gene sets were identified as those that had p values corrected for false discovery rate of less than 5%.
Soluble cytokine/chemokine quantification by Meso Scale Discovery
We utilized the V-PLEX Human Cytokine 44-plex Kit from Meso Scale Discovery (MSD) to quantify levels of chemokines and cytokines in plasma of patients with COVID-19. The following soluble markers were included: Eotaxin, Eotaxin-3, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-1RA, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-8 (HA), IL-9, IL-10, IL-12/IL-23p40, IL-13, IL-15, IL-16, IL-17A, IL-17A/F, IL-17B, IL-17C, IL-17D, IL-21, IL-22, IL-23, IL-27, IL-31, IP-10, MCP-1, MCP-4, MDC, MIP-1α, MIP-1β, MIP-3α, TARC, TNFα, TNFβ, TSLP, and VEGF-A. Assays were performed as per the manufacturer’s instructions. Briefly, plasma samples were thawed and on average 25 μL of plasma samples were diluted as recommended in a MSD 96-well assay plate. Calibrators were added to wells on each plate in parallel. Plates were then sealed and incubated overnight at 4°C. Next, the plates were washed 3 times with 150 μL per well of MSD wash buffer. Following washing, 25 μL of detection antibodies was added to each well, and plates were then sealed and incubated at room temperature for 2 h with shaking. After 2 h, the plates were once again washed 3 times with MSD wash buffer and 150 μL of MSD Read Buffer T was added to each well and the plates were analyzed on the MESO QuickPlex SQ 120MM. Sample concentrations for each marker were then calculated based on the respective standard curve. For analysis, the limit of detection was set at 50% of the lowest limit of detection across all analytes (i.e., 0.045 pg/mL).
Meta-analysis of infected myeloid cells in lower respiratory tract samples
We sought to evaluate whether there were consistent gene expression profiles of infected myeloid cells in the lower respiratory tract of patients with COVID-19. To do this, we first identified datasets in which single-cell Rnaseq was performed on either sputum, endotracheal aspirates, bronchoalveolar lavage (BAL) fluid, or lung tissues. We identified 4 studies that fit these criteria, from which we downloaded the processed gene expression datasets from the Gene Expression Omnibus. We note that 1 study used was a post-mortem autopsy study in which single nuclei were processed rather than single cells. We next visualized all cells from each study by UMAP, and either leveraged the cell type annotations noted by the authors when available or defined cells by canonical gene expression profiles otherwise. Likewise, we also used cluster annotations provide by the authors for downstream analysis or generated new graph-based clusters as previously described.
To perform a meta-analysis to robustly characterize expression profiles of infected myeloid cells, we next identified the most highly infected myeloid cell cluster in each individual study. This cluster was identified as the cluster with the highest frequency of infected cells as determined by SARS-CoV-2 transcript levels. Within each study, we then determined the log fold-change in gene expression for the infected myeloid cluster for each individual patient and created a mean and standard deviation in log fold-change for each study. Meta-analysis was then performed by taking a weighted average of the log fold-change in gene expression, where the weighting was determined by the number of patients in each study. Results of this meta-analysis were then visualized by heatmap to identify genes consistently up- and downregulated in infected myeloid cells.
Quantification and statistical analysis
Survival analysis was performed using Cox’s proportional hazard regression analysis as implement in the R package survival.78,66 Likelihood ratio tests were used to determine the statistical significance of the survival models. Wilcoxon rank sum tests were used to evaluate differences in immune cell frequencies and mean fluorescence intensity by flow cytometry, to calculate differentially expressed genes from the scRNaseq analysis, and identify gene modules that were statistically different between patients who survived and those that died. Spearman’s correlation was used to calculate the correlation between gene module scores and cytokine levels in plasma, between gene modules on day 1 and day 5, and between cytokines and gene modules on day 5. A two-sided alpha of 5% was considered significant unless otherwise noted.
Acknowledgments
We thank the affected individuals and their families for their willingness to participate in this study. We also thank the frontline clinical providers (nurses, physicians, respiratory therapists, and research coordinators) for their assistance in collecting biospecimens from individuals in the ICU. We wish to thank everyone in the Vignali (Vignali-lab.com) and Bruno labs for all their constructive comments and advice during this project. This research was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided. We thank the Hillman Cytometry Facility for assistance with flow cytometry and the Center for Vaccine Research for access to Biosafety Level 2+ Facilities. We thank the following institutions for research funding: Immune Transplant and Therapy Center at UPMC (D.A.A.V., T.C.B., and A.M.); NIH P01 AI108545 (D.A.A.V.); NIH P01 HL103455 (A.M.); NIH P01 HL114453 (P.R., J.S.L., and B.J.M.); the Hillman Post-doctoral Fellowship for Innovative Cancer Research (A.R.C.); and NIH T32 AI089443 and NIH F31 AI147638 (S.G.). The graphical abstract was created in part using Biorender.com.
Author contributions
D.A.A.V., T.C.B., and A.M. conceived and directed the project. D.A.A.V., T.C.B., and A.M. obtained funding for the project. G.D.K., B.M., J.S.L., Y.Z., and A.M. coordinated patient enrollment, clinical phenotyping and data collection, and acquisition of biospecimens. A.R.C., C.C., F.S., and C. Liu processed samples and/or performed scRNA-seq library preparation. A.R.C. and F.S. performed bioinformatics analysis. A.S., C.J.W., and G.D. performed sample processing and soluble cytokine assays. A.S., T.C.B., A.T.R., C. Lampenfeld, S.O., and S.G. performed flow cytometry staining and analysis. S.K., T.C.B., and A.T.R. performed flow cytometry. T.C.B., L.P.A., S.K., A.S., and A.T.R. analyzed flow cytometry data. P.R., A.T.R., P.V.B., J.D., H.S., J.S.L., A.M., and G.D.K. provided intellectual input on viral infections of the lung, computational genomics, and/or machine learning. A.R.C., J.D., H.S., A.M., T.C.B., and D.A.A.V. interpreted the results and wrote the manuscript. All authors edited and approved the manuscript.
Declaration of interests
D.A.A.V.: cofounder and stockholder for Novasenta, Tizona, Trishula, Potenza; stockholder for Oncorus, Werewolf, Apeximmune; patents licensed and royalties for Astellas, BMS, Novasenta; scientific advisory board member for Tizona, Werewolf, F-Star, Bicara, and Apeximmune; consultant for Astellas, BMS, Almirall, Incyte, G1 Therapeutics; research funding for BMS, Astellas, and Novasenta. G.D.K.: research funding for Karius, Inc. T.C.B: research funding for Alkermes and Pfizer; consultant for Walking Fish Therapeutics, iTeos Therapeutics, and BeSpoke Therapeutics. The remaining authors declare no competing interests.
Published: December 2, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100476.
Contributor Information
Anthony R. Cillo, Email: arc85@pitt.edu.
Alison Morris, Email: amorris@upmc.edu.
Tullia C. Bruno, Email: tbruno@pitt.edu.
Dario A.A. Vignali, Email: dvignali@pitt.edu.
Supplemental information
Data and code availability
-
•
Data: Both raw and processed transcriptomic are available through the Gene Expression Omnibus (GEO: GSE180578). Additional data are available via Mendeley Data (Mendeley Data: http://doi.org/10.17632/r83csstphc.2).
-
•
Code: The Seurat package was used for scRNaseq normalization, scaling, dimensionality reduction, UMAP visualization, clustering, and differential gene expression analysis. Code for these steps is available through Seurat’s website (https://satijalab.org/seurat/). Code for gene set enrichment analysis is available at https://www.github.com/arc85/singleseqgset. Code is provided at https://github.com/arc85/covid_analyses files to demonstrate gene module discovery, training of machine learning algorithms and leave one out cross validation analysis, and meta-analysis of infected myeloid cells.
-
•
General statement: Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
References
- 1.Hopkins J. 2020. Coronavirus Resource Center.https://coronavirus.jhu.edu/ [Google Scholar]
- 2.FDA . 2020. FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine. Action Follows Thorough Evaluation of Available Safety, Effectiveness, and Manufacturing Quality Information by FDA Career Scientists, Input from Independent Experts.https://www.fda.gov/news-events/press-announcements/fda-takes-key-action-fight-against-covid-19-issuing-emergency-use-authorization-first-covid-19 [Google Scholar]
- 3.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., et al. C4591001 Clinical Trial Group Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauring A.S., Hodcroft E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA. 2021;325:529–531. doi: 10.1001/jama.2020.27124. [DOI] [PubMed] [Google Scholar]
- 5.Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sterne J.A.C., Murthy S., Diaz J.V., Slutsky A.S., Villar J., Angus D.C., Annane D., Azevedo L.C.P., Berwanger O., Cavalcanti A.B., et al. WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA. 2020;324:1330–1341. doi: 10.1001/jama.2020.17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan H., Peto R., Henao-Restrepo A.M., Preziosi M.P., Sathiyamoorthy V., Abdool Karim Q., Alejandria M.M., Hernández García C., Kieny M.P., Malekzadeh R., et al. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021;384:497–511. doi: 10.1056/NEJMoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shankar-Hari M., Vale C.L., Godolphin P.J., Fisher D., Higgins J.P.T., Spiga F., Savovic J., Tierney J., Baron G., Benbenishty J.S., et al. WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group Association Between Administration of IL-6 Antagonists and Mortality Among Patients Hospitalized for COVID-19: A Meta-analysis. JAMA. 2021;326:499–518. doi: 10.1001/jama.2021.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., et al. China Medical Treatment Expert Group for Covid-19 Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cummings M.J., Baldwin M.R., Abrams D., Jacobson S.D., Meyer B.J., Balough E.M., Aaron J.G., Claassen J., Rabbani L.E., Hastie J., et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395:1763–1770. doi: 10.1016/S0140-6736(20)31189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao D., Zhou F., Luo L., Xu M., Wang H., Xia J., Gao Y., Cai L., Wang Z., Yin P., et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: a retrospective cohort study. Lancet Haematol. 2020;7:e671–e678. doi: 10.1016/S2352-3026(20)30217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q., Ding X., Xia G., Chen H.G., Chen F., Geng Z., Xu L., Lei S., Pan A., Wang L., Wang Z. Eosinopenia and elevated C-reactive protein facilitate triage of COVID-19 patients in fever clinic: A retrospective case-control study. EClinicalMedicine. 2020;23:100375. doi: 10.1016/j.eclinm.2020.100375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I., et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- 14.Del Valle D.M., Kim-Schulze S., Huang H.H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leisman D.E., Ronner L., Pinotti R., Taylor M.D., Sinha P., Calfee C.S., Hirayama A.V., Mastroiani F., Turtle C.J., Harhay M.O., et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020;8:1233–1244. doi: 10.1016/S2213-2600(20)30404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., et al. HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., et al. COVID-STORM Clinicians. COVID Clinicians. Imagine COVID Group. French COVID Cohort Study Group. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. NIAID-USUHS/TAGC COVID Immunity Group Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.S., et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5:eabd1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Major J., Crotta S., Llorian M., McCabe T.M., Gad H.H., Priestnall S.L., Hartmann R., Wack A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369:712–717. doi: 10.1126/science.abc2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Israelow B., Song E., Mao T., Lu P., Meir A., Liu F., Alfajaro M.M., Wei J., Dong H., Homer R.J., et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J. Exp. Med. 2020;217:e20201241. doi: 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y., Liu X., Le W., Xie L., Li H., Wen W., Wang S., Ma S., Huang Z., Ye J., et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell. 2020;11:740–770. doi: 10.1007/s13238-020-00762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang D., Guo R., Lei L., Liu H., Wang Y., Wang Y., Qian H., Dai T., Zhang T., Lai Y., et al. Frontline Science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J. Leukoc. Biol. 2021;109:13–22. doi: 10.1002/JLB.4HI0720-470R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kvedaraite E., Hertwig L., Sinha I., Ponzetta A., Hed Myrberg I., Lourda M., Dzidic M., Akber M., Klingström J., Folkesson E., et al. Karolinska KI/K COVID-19 Study Group Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2018587118. e2018587118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabo P.A., Dogra P., Gray J.I., Wells S.B., Connors T.J., Weisberg S.P., Krupska I., Matsumoto R., Poon M.M.L., Idzikowski E., et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity. 2021;54:797–814.e6. doi: 10.1016/j.immuni.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bain W., Yang H., Shah F.A., Suber T., Drohan C., Al-Yousif N., DeSensi R.S., Bensen N., Schaefer C., Rosborough B.R., et al. COVID-19 versus Non-COVID-19 Acute Respiratory Distress Syndrome: Comparison of Demographics, Physiologic Parameters, Inflammatory Biomarkers, and Clinical Outcomes. Ann. Am. Thorac. Soc. 2021;18:1202–1210. doi: 10.1513/AnnalsATS.202008-1026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A., et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y., Fu B., Zheng X., Wang D., Zhao C., Qi Y., Sun R., Tian Z., Xu X., Wei H. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl. Sci. Rev. 2020;7:998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwissa M., Nakaya H.I., Onlamoon N., Wrammert J., Villinger F., Perng G.C., Yoksan S., Pattanapanyasat K., Chokephaibulkit K., Ahmed R., Pulendran B. Dengue virus infection induces expansion of a CD14(+)CD16(+) monocyte population that stimulates plasmablast differentiation. Cell Host Microbe. 2014;16:115–127. doi: 10.1016/j.chom.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuo Y., Estes S.K., Ali R.A., Gandhi A.A., Yalavarthi S., Shi H., Sule G., Gockman K., Madison J.A., Zuo M., et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020;12:eabd3876. doi: 10.1126/scitranslmed.abd3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang E.Y., Mao T., Klein J., Dai Y., Huck J.D., Liu F., Zheng N.S., Zhou T., Israelow B., Wong P., et al. Diverse Functional Autoantibodies in Patients with COVID-19. medRxiv. 2020 doi: 10.1101/2020.12.10.20247205. [DOI] [PubMed] [Google Scholar]
- 33.Chang S.E., Feng A., Meng W., Apostolidis S.A., Mack E., Artandi M., Barman L., Bennett K., Chakraborty S., Chang I., et al. New-Onset IgG Autoantibodies in Hospitalized Patients with COVID-19. medRxiv. 2021 doi: 10.1101/2021.01.27.21250559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cillo A.R., Kürten C.H.L., Tabib T., Qi Z., Onkar S., Wang T., Liu A., Duvvuri U., Kim S., Soose R.J., et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity. 2020;52:183–199.e9. doi: 10.1016/j.immuni.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018 doi: 10.1038/nbt.4314. Published online December 3, 2018. [DOI] [PubMed] [Google Scholar]
- 36.Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- 37.Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184:1895–1913.e1819. doi: 10.1016/j.cell.2021.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C., Martins A.J., Lau W.W., Rachmaninoff N., Chen J., Imberti L., Mostaghimi D., Fink D.L., Burbelo P.D., Dobbs K., et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell. 2021;184:1836–1857.e1822. doi: 10.1016/j.cell.2021.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Cambridge Institute of Therapeutic Immunology and Infectious Disease-National Institute of Health Research (CITIID-NIHR) COVID-19 BioResource Collaboration Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/s41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziegler C.G.K., Allon S.J., Nyquist S.K., Mbano I.M., Miao V.N., Tzouanas C.N., Cao Y., Yousif A.S., Bals J., Hauser B.M., et al. HCA Lung Biological Network. Electronic address: lung-network@humancellatlas.org. HCA Lung Biological Network SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. 2020;181:1016–1035.e19. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., van der Meer F., Kallio K., Kaya T., Anastasina M., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daly J.L., Simonetti B., Klein K., Chen K.E., Williamson M.K., Antón-Plágaro C., Shoemark D.K., Simón-Gracia L., Bauer M., Hollandi R., et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370:861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 44.Grant R.A., Morales-Nebreda L., Markov N.S., Swaminathan S., Querrey M., Guzman E.R., Abbott D.A., Donnelly H.K., Donayre A., Goldberg I.A., et al. NU SCRIPT Study Investigators Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delorey T.M., Ziegler C.G.K., Heimberg G., Normand R., Yang Y., Segerstolpe Å., Abbondanza D., Fleming S.J., Subramanian A., Montoro D.T., et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Y., Rosborough B.R., Chen J., Das S., Kitsios G.D., McVerry B.J., Mallampalli R.K., Lee J.S., Ray A., Chen W., Ray P. Single cell RNA sequencing identifies an early monocyte gene signature in acute respiratory distress syndrome. JCI Insight. 2020;5:135678. doi: 10.1172/jci.insight.135678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou J., Chu H., Li C., Wong B.H., Cheng Z.S., Poon V.K., Sun T., Lau C.C., Wong K.K., Chan J.Y., et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J. Infect. Dis. 2014;209:1331–1342. doi: 10.1093/infdis/jit504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tseng C.T., Perrone L.A., Zhu H., Makino S., Peters C.J. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J. Immunol. 2005;174:7977–7985. doi: 10.4049/jimmunol.174.12.7977. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J., Chu H., Chan J.F., Yuen K.Y. Middle East respiratory syndrome coronavirus infection: virus-host cell interactions and implications on pathogenesis. Virol. J. 2015;12:218. doi: 10.1186/s12985-015-0446-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bain W.G., Peñaloza H.F., van der Geest R., Sullivan M., Ross M., Kitsios G.D., Methe B., McVerry B.J., Morris A., Watson A.M., et al. Lower Respiratory Tract Myeloid Cells Harbor SARS-Cov-2 and Display an Inflammatory Phenotype. Chest. 2021;159:963–966. doi: 10.1016/j.chest.2020.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fajnzylber J., Regan J., Coxen K., Corry H., Wong C., Rosenthal A., Worrall D., Giguel F., Piechocka-Trocha A., Atyeo C., et al. Massachusetts Consortium for Pathogen Readiness SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 2020;11:5493. doi: 10.1038/s41467-020-19057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bozza F.A., Cruz O.G., Zagne S.M., Azeredo E.L., Nogueira R.M., Assis E.F., Bozza P.T., Kubelka C.F. Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma as predictive factors for severity. BMC Infect. Dis. 2008;8:86. doi: 10.1186/1471-2334-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carlin A.F., Aristizabal P., Song Q., Wang H., Paulson M.S., Stamm L.M., Schooley R.T., Wyles D.L. Temporal dynamics of inflammatory cytokines/chemokines during sofosbuvir and ribavirin therapy for genotype 2 and 3 hepatitis C infection. Hepatology. 2015;62:1047–1058. doi: 10.1002/hep.27971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parekh N.J., Krouse T.E., Reider I.E., Hobbs R.P., Ward B.M., Norbury C.C. Type I interferon-dependent CCL4 is induced by a cGAS/STING pathway that bypasses viral inhibition and protects infected tissue, independent of viral burden. PLoS Pathog. 2019;15:e1007778. doi: 10.1371/journal.ppat.1007778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roelli Patrick, bbimber, Flynn Bill, santiagorevale, Gui Gege. Patrick Roelli, bbimber, Bill Flynn, santiagorevale, & Gege Gui. Zenodo. 2019 doi: 10.5281/zenodo.2590196. [DOI] [Google Scholar]
- 58.R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2019 [Google Scholar]
- 59.Wickham Hadley, Averick Mara, Bryan Jennifer, Chang Winston, McGowan Lucy, François Roman, et al. Journal of Open Source Software. 2019;4(43) doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 60.Wickham Hadley. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 2016. [Google Scholar]
- 61.Pedersen Thomas. patchwork: The Composer of Plots version 1.0.0. R package. 2019 [Google Scholar]
- 62.Baglama Jim, Reichel Lothar, Lewis B.W. Jim Baglama, Lothar Reichel and B. W. Lewis (2019). irlba: Fast Truncated Singular Value Decomposition and Principal Components Analysis for Large Dense and Sparse Matrices version 2.3.3. R package. 2019 [Google Scholar]
- 63.Yu Guangchuang. ggplotify: Convert Plot to ‘grob’ or ‘ggplot’ Object version 0.0.5. R package. 2020 [Google Scholar]
- 64.Kolde Raivo. pheatmap: Pretty Heatmaps version 1.0.12. R package. 2019 [Google Scholar]
- 65.Neuwirth Erich. RColorBrewer: ColorBrewer Palettes version 1.1-2. R package. 2014 [Google Scholar]
- 66.Therneau T.M. 2020. Using Time Dependent Covariates and Time Dependent Coefficients in the Cox Model.https://cran.pau.edu.tr/web/packages/survival/vignettes/timedep.pdf [Google Scholar]
- 67.Kassambara Alboukadel, Losinski Marcin, Biecek Przemyslaw. survminer: Drawing Survival Curves using ‘ggplot2’ version 0.4.8. R package. 2020 [Google Scholar]
- 68.Grolemund Garrett, Wickham Hadley. Dates and Times Made Easy with lubridate. Journal of Statistical Software. 2011;40(3) [Google Scholar]
- 69.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bengtsson Henrik. future: Unified Parallel and Distributed Processing in R for Everyone version 1.15.1. R package. 2019 [Google Scholar]
- 71.Bengtsson Henrik. future.apply: Apply Function to Elements in Parallel using Futures version 1.3.0. R package. 2019 [Google Scholar]
- 72.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ushey Kevin, Allaire J.J., Tang Yuan. reticulate: Interface to ‘Python’ version 1.14. R package. 2019 [Google Scholar]
- 74.Grüning Björn, Dale Ryan, Sjödin Andreas, Chapman Brad, Rowe Jillian, Tomkins-Tinch Christopher, et al. the Bioconda Team Bioconda: Sustainable and Comprehensive Software Distribution for the Life Sciences. Nature Methods. 2018 doi: 10.1038/s41592-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., 3rd, Smibert P., Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuhn M. Building predictive models in R using the caret Package. J. Stat. Softw. 2008:28. [Google Scholar]
- 78.Therneau T.M.G., Patricia M. Springer; 2000. Modeling Survival Data: Extending the Cox Model. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: Both raw and processed transcriptomic are available through the Gene Expression Omnibus (GEO: GSE180578). Additional data are available via Mendeley Data (Mendeley Data: http://doi.org/10.17632/r83csstphc.2).
-
•
Code: The Seurat package was used for scRNaseq normalization, scaling, dimensionality reduction, UMAP visualization, clustering, and differential gene expression analysis. Code for these steps is available through Seurat’s website (https://satijalab.org/seurat/). Code for gene set enrichment analysis is available at https://www.github.com/arc85/singleseqgset. Code is provided at https://github.com/arc85/covid_analyses files to demonstrate gene module discovery, training of machine learning algorithms and leave one out cross validation analysis, and meta-analysis of infected myeloid cells.
-
•
General statement: Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.