

Abstract
The affinity of an antibody for its antigen is primarily determined by the specific sequence and structural arrangement of the complementarity-determining regions (CDRs). Recent evidence, however, points toward a nontrivial relation between the CDR and distal sites: variations in the binding strengths have been observed upon mutating residues separated from the paratope by several nanometers, thus suggesting the existence of a communication network within antibodies, whose extension and relevance might be deeper than insofar expected. In this work, we test this hypothesis by means of molecular dynamics (MD) simulations of the IgG4 monoclonal antibody pembrolizumab, an approved drug that targets the programmed cell death protein 1 (PD-1). The molecule is simulated in both the apo and holo states, totalling 4 μs of MD trajectory. The analysis of these simulations shows that the bound antibody explores a restricted range of conformations with respect to the apo one, and that the global conformation of the molecule correlates with that of the CDR. These results support the hypothesis that pembrolizumab featues a multi-scale hierarchy of intertwined global and local conformational changes. The analysis pipeline developed in this work is general, and it can help shed further light on the mechanistic aspects of antibody function.
Subject terms: Computational biophysics, Biological physics
Introduction
The number of monoclonal antibodies (mAbs) employed for therapeutic applications dramatically increased in the recent years: from 1997 to 2013, 34 mAbs-based pharmaceuticals were approved in US or EU, while from 2014 to 2020, in only 7 years, the number of approved mAbs was 611. mAbs have been developed to treat a large variety of conditions, including cancer, autoimmune diseases and, very recently, COVID-192,3.
Even in the case of naked immunoglobuline drugs, which do not involve conjugation with radionuclides or small molecules4,5, engineering of the antibody sequence is routinely performed to optimize its therapeutic efficacy for a given function, through successive steps of humanization, affinity maturation, and modifications aimed at overcoming challenges in stability and manifacturing6. On the one side, the selection of the isotype, and therefore those structural/dynamical features of the constant region that come with it, leads to different immune responses, and is thus performed on the basis of the planned application7,8; on the other side, modifications of single residues can determine a higher therapeutic efficacy, as in the case of those mutations introduced in the Fc domain to enhance effector function and recruitment of additional proteins9–11. A remarkable example is the single-residue mutation that, in the hinge of IgG4 antibodies, prevents Fab-arm exchange12–14.
Modifications of this type, which may be distributed throughout the whole antibody sequence, are usually introduced for reasons that are not directly linked to antigen affinity modulation. Engineering efforts intended to increase specificity and affinity are in fact mostly focused on the residues of the six loops comprising the complementarity-determining region (CDR), because of their preeminent role in antigen binding15. However, the CDR loops are not the only possible loci of intervention; it is experimentally shown that both mutations near and far from the antigen-binding site can affect affinity16. Such mutations act by modulating the interdomain conformational dynamics of the antigen-binding fragment, which eventually reflects on the paratope, namely the antigen binding site. On a similar note, NMR relaxation dispersion experiments allowed researchers to detect important fluctuating residues that are not located in the CDR, whose point mutation can nonetheless increase antigen-antibody affinity17.
Empirical experimental optimization of binding affinity can be laborious and costly, both in terms of time and resources18. Molecular dynamics (MD) simulations, on the other hand, offer a valuable tool for the investigation of the interplay between stabilizing interactions, fluctuation correlations, and conformational variability at different levels of resolution and experimental conditions19–22. In silico structural investigation of immunoglobulins and antigen-antibody complexes has been successfully employed with different objectives, which include the detailed description of the dynamics of CDR loops and the transitions between their conformational states23–25, as well as the comprehension of structural rearrangements and allosteric modifications following antigen binding26–28.
Here, we employ atomistic MD simulations to investigate the internal dynamics of full-length pembrolizumab, a humanized IgG4 antibody used in immunotherapy, whose full structure has been experimentally solved29 (Fig. 1). Pembrolizumab, whose commercial success rate is expected to profoundly impact pharmaceutical market in the next years30, is approved for the treatment of melanoma, lung cancer, head and neck cancer, Hodgkin’s lymphoma and stomach cancer31–33. Its mechanism of action consists in binding to the programmed cell death protein 1 (PD-1), a 288-residues long receptor located on the membrane of T cells, B cells, and natural killer cells34. PD-1 promotes apoptosis of the lymphocyte when activated by the programmed cell death receptor ligands PD-L1 and PD-L2, whose expression is upregulated in malignant cells35. The large contact area between pembrolizumab and PD-1 hinders the binding of PD-L1 and PD-L2, thus preventing down-regulation of the anti-tumor activity of T cells36–38.
Figure 1.
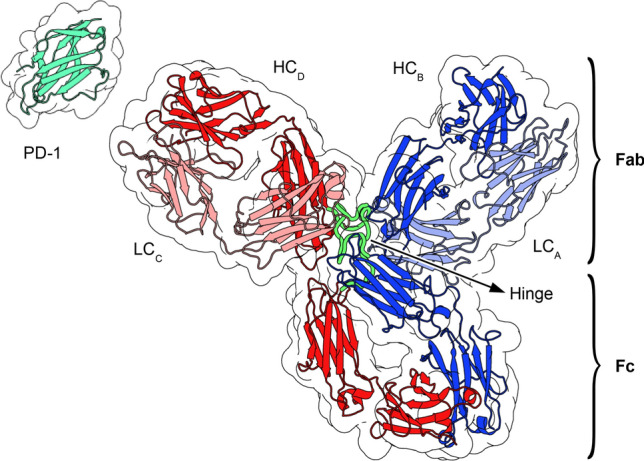
Graphical representation of the starting configuration of pembrolizumab employed in the MD simulations. LC stands for light chain, while HC stands for heavy chain. The antigen PD-1 is represented in the vicinity of the CDR.
We performed a total of 4 μs of dynamics of the deglycosylated antibody, both in presence and in absence of the antigen PD-1. The analysis pipeline, which combines structural analysis, investigation of chemical interactions, and information theory-based measures of correlation, allowed us to rationalize at a residue-level the observed conformational dynamics. Our simulations highlighted the particular role of key residues of the hinge, resulting in an asymmetric behavior of the two hinge segments in pembrolizumab; moreover, an interplay between large-scale conformation and binding state is observed, and the residues allowing such information routing from the paratope throughout the antibody structure are identified. We believe that the residue-level elucidation of the complex dynamics of potent and highly selective mAbs—of which pembrolizumab is a notable example—in relation to their binding state may assist in the process of antibody engineering, for the rational design of novel, optimized therapeutic agents.
Results
Pembrolizumab is a 1324-residues long therapeutic antibody, belonging to the IgG4 class. On both hinge segments, it presents the typical S228P mutation, introduced to prevent fab-arm exchange in IgG4 antibodies12–14. A schematic representation of the protein is given in Fig. S3, where an index is associated to each structural domain. The four chains forming the antibody are labelled A, B, C and D, and correspond to chains A, B, F and G of the original PDB file, respectively29. We label Fab1 the Fab domain formed by chains A and B, and Fab2 the one including chains C and D; similarly, Fc1 includes domains CH2 and CH3 from chain B, while Fc2 includes those from chain D. Throughout the manuscript, all references to residue indices follow the EU numbering scheme39.
We focused on the analysis of conformations and the rationale behind domain motion in dependence of the binding state; to this aim, the results presented here are based on a comparison between the deglycosylated antibody alone (apo form) and bound to its antigen, the ectodomain of the protein PD-1 (holo form). The latter, comprising 114 residues, corresponds to the region of PD-1 whose structure has been experimentally solved in complex with pembrolizumab Fab40. To allow for an unbiased comparison, the starting structure of the holo system was built on the same initial antibody conformation of the apo case; specifically, Fab2 of pembrolizumab (formed by chains C and D) was replaced with the Fab-PD1 complex (PDB ID: 5GGS), after structural alignment on the antibody domain.
Both apo and holo systems were simulated in four independent replicas, with a duration of 500 ns each. Relaxation of the starting structure to an equilibrated conformation was assessed by computing the root-mean-square deviation (RMSD) of C atoms of the full antibody (Figs. S4 and S5); in addition, the RMSD was computed for the of C atoms of Fab1, Fab2 and Fc independently, to verify the relaxation of each structural domain from the crystallographic structure (Figs. S6 and S7).
The presence of the bound antigen restricts the range of conformations of the full-length pembrolizumab
To facilitate the analysis of antibody flexibility, the conformations sampled from MD simulations are collected in clusters on the basis of their structural similarity (Fig. S8), as measured by the RMSD matrix of C atoms. The cluster analysis is performed separately and independently on the two systems, in order to highlight possible differences; in both cases, an RMSD threshold of 1.2 nm is used. In the simulations of pembrolizumab alone, 6 conformational clusters are identified, while in the case of the holo system the same clustering protocol leads to the identification of only 4 clusters; in the presence of the antigen, a limited conformational variability is indeed observed, as apparent from the narrow range of radii of gyration spanned by these clusters when compared to the apo case (Fig. 2). This is consistent with the visual inspection of the sampled conformations, whose representative structures are reported in Fig. 2. A similar population shift towards a more uniform conformational distribution upon antigen binding was observed through MD simulations of an IgG1 antibody27.
Figure 2.

(a) Mean and standard deviation of the antibody radii of gyration in the apo forms (left) and holo forms (right), averaged within each conformational cluster. In the holo case, the radius is computed for the antibody alone. The error bars correspond to the standard deviation within each cluster. The shaded areas correspond to the range of variability, which is greatly reduced in the holo case. (b) Representative structures of pembrolizumab in the apo (A) and holo (H) forms, for each conformational cluster. Chains AB are in blue, chains CD in red, and the antigen PD-1 in green.
For each system (apo or holo), clusters are indexed according to increasing radii of gyration; in this way, clusters and are the ones grouping the most compact conformations of the apo and holo systems, respectively. At a large scale, conformational clusters differ mainly in the relative arrangement of the Fab and Fc domains, reflecting the extent of packing. Clusters and include the equilibrated experimental structures of the apo and holo systems, respectively. These clusters are intermediate, in terms of radius of gyration, between clusters // and / for apo, and / and for holo; this means that the simulations captured both tendencies of the antibody to shrink and to expand. Interestingly enough, the conformations that are not sampled by the holo system correspond to particularly low radii of gyration, namely those found in clusters , , and . Compact conformations, particularly clusters , and , present a higher structural stability, as measured in terms of RMSD with respect to the representative structure of the cluster (Fig. S9).
The relative cluster population (quantified by the fraction of frames relative to the total number, Fig. S10), shows a large unbalance among conformational states. Despite the different population, each cluster nonetheless includes a sufficient sampling of the conformational basin, as proved by the convergence of the root-mean-square fluctuations (RMSF) of the antibody C atoms (Figs. S11 and S12). Comparison of the cluster sizes reveals that the unbound antibody evolves towards a compact conformation for most of the simulation time, while the holo has a more symmetric distribution of configurations, associated to larger and smaller radii of gyration compared to the initial structure. Also in the holo case, however, the time spent in open conformations is a small fraction of the full simulation time. This result is in agreement with the experimental observation that the compact conformation is the most populated for IgG4 molecules in solution, as observed in N TROSY NMR and SAXS experiments29,41. This conformational preference has been explained on the basis of the short hinge of IgG4 antibodies with respect to other IgG subclasses; IgG4 hinge has indeed a three amino acid deletion, compared to the one of IgG142. Another factor that can favour the compact arrangement is the tilted conformation of the CH2 domain in the Fc29, which displays a rigid-body rotation of relative to the position observed in the crystal structure of a truncated IgG4 Fc (PDB ID: 4C54). In the most compact conformations, the superposition of the pembrolizumab Fc and the one of 4C54 would lead to an overlap of CH2 and CL in Fab1 (Fig. S13). The peculiar CH2 conformation of pembrolizumab, which is present in the crystal structure, persists during the course of the simulations. The hypothesis that such CH2 domain rotation is found in solution was supported by a recent experimental observation of the reduced cleavage of pembrolizumab, mediated by the immunoglobulin-degrading enzyme from Streptococcus pyogenes, which binds the CH2–CH3 interface43. The asymmetric orientation of the two CH2 domains can also explain the contact area between Fabs and Fc, reported in Fig. S14. Here, the range of variability of the Fab1-Fc contact area greatly exceeds that of the Fab2-Fc pair, which remains low in the vast majority of the clusters; moreover, a significantly larger contact surface between Fab1 and Fc is observed in clusters and , with respect to the other cases. The rotated CH2 forms indeed a cavity that gives Fab1 a greater freedom to move, minimizing steric clashes with Fc. This is reflected also in clusters , and , where the symmetric arrangement of Fabs with respect to Fc leads to a slightly larger Fab2-Fc contact area than Fab1-Fc; and in cluster , and , where, although the Fc is clearly tilted toward the Fab1, the Fab1-Fc contact area is only slightly larger than the Fab2-Fc case. The large possibility of rearrangements of Fab1 with respect to the Fc is therefore one of the main determinants of the overall shape of pembrolizumab. We do not rule out the possibility of a conformational change of the CH2 domain in chain B, leading either to a symmetric FC conformation, or to a switching of the conformational properties of chains B and D; however, given the short hinge of pembrolizumab, we expect that prior breaking of the disulfide bonds in the hinge is necessary for the CH2 rotation to take place. Such an event, in turn, is unlikely to happen even in vitro or in vivo, given the stabilization of the disulfide bonds brought by the S228P mutation13.
A comparison between the conformational states in apo/holo clusters is facilitated by the RMSD matrix in Fig. S15. As expected, cluster shares a low RMSD with the open conformations of clusters and , while clusters and are closer to the compact conformations in clusters , , . The extremely compact conformation of cluster and the peculiar cluster are the ones sharing the lowest similarity with all the other clusters. appears indeed as a singular conformation, where the major axis of Fab1 is perpendicular to the plane of Fab2 and Fc; once again, this movement is permitted by the rotated CH2 domain. Although such bent conformation of Fab1 is not directly observed in the apo case, visual inspection of the trajectory reveals that the variable region of Fab 1 in cluster has the tendency to bend in a direction perpendicular to the plane of Fc-Fab2. This is confirmed by a principal components analysis (PCA) of the dynamics, performed on the C atoms of the structures grouped in the conformational clusters (Section S2.1, Figs. S16–S35). The entity of the conformational change, however, is larger in than in any apo cluster.
Role of key residues in stabilizing the conformational states
The determinants of the observed conformations are identified, at a finer level of detail, through the calculation of correlations between residues, as quantified by the mutual information (MI) of C atom fluctuations. MI, which captures both linear and non-linear contributions to amino acid displacements from a reference position, is used to build inter-residue networks of information pathways, which can be interpreted in the light of the inter-domain non-bonded interactions established in the course of the simulations. Tables S3 and S4 list the residues involved in inter-domain contacts, including hydrogen bonds, salt bridges, and hydrophobic interactions. These contacts are highlighted as important channels for information transfer by the results of the network analysis, and in particular by the calculation of edge betweenness (Figs. S36 and S37), which measures the centrality of a graph edge as the number of shortest paths crossing it44.
The most open conformations (clusters and ) are stabilized by a persistent electrostatic interaction between side chains of ASP, located in the CL domain of Fab2, and LYS, located between CH1 of Fab1 and the hinge, which results in a highly central edge of the interaction network; we can expect that mutation of one of these residues would destabilize the open state. Despite this common feature, stable contacts between Fab1 and Fc are absent in cluster , while in the two domains interact through electrostatic interactions. Moreover, the comparison of the non-bonded interactions between Fab1/Fab2 and Fc confirms the asymmetry of the contact distributions in the extended conformations: even though the molecule adopts an overall symmetric Y-shape, the larger number of interactions between Fc and Fab2 (with respect to Fab1) in clusters , and supports the hypothesis that further approaching of the two domains would be impaired by steric clashes.
In the most compact conformations (clusters , and ), a key stabilizing role is played by ARG in the CH1 domain of Fab1, which emerges as a central network node and is involved in electrostatic interactions with several Fab2 residues (ASP and LYS in particular, located in the CL domain). The network representations of closed conformations reveal also the large number of edges crossing the Fab1-Fc contact surface in the compact arrangements. While the residues involved in these key interactions belong mainly to the CH2 domains of chain B, in the case of cluster the surface of interaction is extended to distant regions of the Fc domain, namely the CH3 domain of chain B (through the backbone of MET and the side-chain of HIS) and the CH2 domain of chain D (through the side-chains of THR and LYS). The peculiar conformations in cluster are stabilized by a number of high betweenness contacts taking place outside of the hinge; fundamental for the stability of this compact and highly interconnected state are the electrostatic interactions between side chains of LYS-GLU, in the CL of Fab2 and in the CH2 of chain B, respectively, and between LYS/ARG in the AB loop of chain B CH245 and light-chain residues of Fab1 (specifically, hydrogen bonds LYS-ASP, ARG-GLU, and ARG-PRO).
These observations are strengthened by the analysis of communities in the MI-based network. The latter is divided into substructures (communities) with dense internal correlations, but sparse inter-community connections (see Section S1). Although the optimal community distribution closely reflects the natural subdivision of the antibody in structural domains, residues previously identified as promoting inter-domain connections fall within the same community (Figs. S38 and S39), thus supporting results from the investigations of non-bonded interactions and edge centrality. In addition, community analysis serves to detect, at a first level, connections between the hinge and the neighbouring domains; in this regard, a significant consistency is observed in the intermediate and fully open conformations of both apo and holo cases, where the hinge belongs to the same community as the CH1 domain of Fab2. Building on the high level of connection between the hinge and the rest of the molecule, a more detailed investigation of the role of the hinge in the overall confomational variability of the antibody is explored in the next section.
Role of the hinge in the observed conformational variability
As noted above, the hinge displays significant correlations with other domains, both in the apo and holo systems, as apparent from the MI matrices (Figs. S40 and S41); such correlations appear particularly strong in pembrolizumab when compared to those reported in a previous study of an IgG1 antibody46. In pembrolizumab, in fact, the hinge retains a complex network of interactions with nearby domains, thus functioning as more than just a flexible linker; a detailed account on the interactions involving the hinge residues, as emerging from network analysis and inspection of non-bonded contacts, is reported in Section S2.2. Given its role as interaction hub, in addition to allowing large-scale rearrangements47,48, the hinge region deserves therefore particular attention.
During our simulations, the hinge shows relatively small variations in the radius of gyration (Fig. S42). Such result is expected on the basis of the presence of disulfide bonds between the two hinge segments, and of a number of transient intra- and inter-chain non-bonded interactions; among them, the most stable ones are the hydrogen bonds between the sidechain of SER in hinge 2 and the backbone of PRO in hinge 1, and between the sidechain of TYR in hinge 2 and the backbone of PRO in hinge 1 (Fig. 3).
Figure 3.

(a) Interaction stabilizing the hinge conformation; residues belonging to hinge 1 are labelled in red, while those belonging to hinge 2 are in blue. The disulfide bonds between the two CYS of chains B and D, and the CYS of chains B and D, are also shown. (b) Conformational ensembles of the hinge in its most compact and most extended conformations (clusters and respectively), after structural alignment on the cysteine residues. Chain B is represented in red, chain D in blue. The overall hinge shape is determined by the conformation of H2. (c) Per-residue flexibility of hinge backbone, as quantified by PAD parameter. The yellow-shaded areas correspond to the cysteine residues forming inter-chain disulfide bonds. Particularly high is the backbone plasticity of the second cysteine in the hinge 2 of the apo antibody, which might result from the torsional stress imposed by the high conformational variability of the Fc relative to the Fab in the apo simulations.
The two hinge segments from chain B and chain D are highly asymmetric (Fig. S43), with hinge 1 assuming significantly bent conformations (Fig. 3). Although they are mostly attributable to the numerous non-bonded Fab1-Fc interactions, which prevent an extended conformation of chain B (see previous section), they are also stabilized by a number of non-bonded interactions, such as the stacking interaction between the side-chains of TYR and PHE and the hydrogen bonds between PHE-GLY and GLY-CYS. Hinge 2 shows instead the largest degree of variability, and it appears to be the main determinant of the overall hinge shape.
Results from the previous sections highlighted a smaller range of conformations of the pembrolizumab molecule in presence of the antigen. In order to have a closer look at the residues responsible for the increased rigidity in the holo structures, we computed the PAD (protein angular dispersion) parameter of the angle , the latter being the sum of the backbone torsional angles and 49. PAD measures the per-residue backbone plasticity through the variance of its torsional angles (Fig. 3). It ranges from 0 to 180; a higher value corresponds to a higher backbone flexibility. PAD values of hinge 2 (connecting Fab2, where the antigen is bound, and Fc) are very similar in the apo and holo cases, and for a few residues are even slightly higher in the bound state (the average PAD is 60 for apo and 62 for bound). However, hinge 1 (connecting Fab1 and Fc) is, on average, more rigid in the holo than in the apo form (the average PAD is 61 for apo and 44 for holo). Since the flexibility of hinge 1 is responsible for the relative movements of Fab1 and Fc, the calculated PAD values are in line with the restricted conformational variability observed in the bound conformations. RMSF computed on the hinge C atoms confirms the reduction of flexibility in hinge 1 with respect to hinge 2 when going from the apo to the bound state (Fig. S44). Therefore, we suggest that the limited conformational variability of pembrolizumab in presence of the bound antigen is linked to a higher rigidity in the hinge region with respect to the apo case. Moreover, hinge 1 shares a higher MI with domains CH1 and CL of Fab2 than hinge 2 (Figs. S40 and S41), despite the latter being the direct extension of Fab2. Results from network analysis corroborate this observation: hinge 2 in the holo system does not include residues with high centrality, as opposed to the apo case (Tables S5 and S6).
The hinge is not the only region of the holo antibody where a decrease in backbone plasticity is observed, with respect to the apo case; from the PAD analysis, this is true also for the binding site of Fab2 (as expected for the presence of the antigen) and residues 242-252 and 338 in the CH2 domain of chain B (Fig. S45). Interestingly enough, this domain is indeed involved in high-betweenness contacts, as evidenced by the network analysis (Figs. S36 and S37), through highly central paths that put it in communication with Fab2. We notice here that a concurrent reduction in flexibility in the binding site, in hinge 1, and in the CH2 domain is not in contradiction with the observation that changes of conformational flexibility in antibodies follow Le Chatelier’s principle50,51: upon binding, counteracting changes in rigidity and flexibility occur at distant sites. Figure S45 reveals indeed that a reduction in the value of PAD in the paratope of Fab2 is associated to an increased backbone plasticity in distant regions of the holo system, especially in the chain D of the Fc domain (residues 252-255, 271, and 323-330).
Binding modes of PD-1 elicit different degrees of correlation within the antibody
The results from the previous section suggest the presence of a communication channel between the antigen-bound Fab2 and hinge 1, leading to a transfer of information that might modulate an interplay between binding and conformational state. In this regard, different conformational clusters correspond to different binding site conformations, as quantified by the distributions of the RMSD between C atoms in the paratope of the simulated system and the bound crystal structure40 (Fig. S46). At a first level of distinction, the presence of the antigen largely reduces the displacements in the binding site, as expected; moreover, in the apo case, a correspondence is observed between the entity of the displacements and the compactness of the conformation, with more compact clusters shifted toward larger values of RMSD. On the other hand, the displacements of the binding site in Fab1 are not affected by the presence of PD-1 on Fab2; their RMSD distributions are similar in the apo and holo case (Fig. S47).
In order to better understand the interplay between the binding site on Fab2 and the rest of the pembrolizumab structure, we performed an investigation of the stabilizing interactions between the antibody and the antigen in each holo cluster, followed by a study of the correlations elicited by the ligand within the antibody.
The complex formed by Fab2 and the protein PD-1 is shown in Fig. 4. The number of hydrogen bonds between the two molecules varies slightly among the distinct conformational clusters (Fig. S48); large deviations are instead observed in the persistence of the salt bridge formed between side-chains of ARG in the VH domain of chain D and ASP of PD-1 (Fig. 4), which has been identified as a key contact for the stabilization of the complex40. For all electrostatic interactions, the binding is strengthened when the antibody is in cluster , resulting in large and stable values of the contact surface area between pembrolizumab and PD-1 (Fig. S48). The RMSF of C atoms in the antigen molecule, after structural alignment of the antibody variable region, reflects the strength of the interactions, with fluctuations in that are approximately the half of those in the other clusters (Fig. S49). Furthermore, the distribution of the antigen RMSD in each cluster, calculated with respect to the experimental structure, reveals a lower dispersion in cluster (Fig. S49). This particular stability might be correlated to the rigidity of the antibody structure in this conformation (Fig. S9), and the corresponding absence of large-scale conformational changes perturbing the Fab/antigen complex. These observations are in agreement with the calculated values of binding enthalpies, obtained with the MM/PBSA approach52 and reported in Table S7. The values shown are relative to the simulation with the sole Fab in complex with the antigen, taken as the reference. In addition, for all of the conformational clusters, the simulation of the full-structure antibody results in a more stabilizing energy than in the simulation of the Fab alone, thus suggesting the importance of maintaining the full antibody structure in the studies of antibody/antigen binding free energies through MD simulation; a similar stabilizing effect was already observed upon inclusion in the simulation set-up of the constant region, with respect to the sole variable region53.
Figure 4.

(a) Pembrolizumab Fab2 (cyan) in complex with PD-1 protein (red). The paratope is represented as a blue surface. (b) Residues involved in electrostatic interactions at the atigen/antibody interface; antigen residues are labelled in red. (c) Distance distributions between ARG99 in VH and ASP85 of PD-1. (d) Distributions of the intra- and inter- (e) domain correlation coefficients of antibody residues, for each conformational cluster of the holo state.
An ordering of conformational clusters similar to the one emerging from the strength of the binding interaction is reflected in the intensity of the residue-residue correlations within the antibody, as quantified by the generalized correlation coefficient (GCC)54; the latter corresponds to a normalized form of MI (Section S1), ranging from 0 (no correlation) to 1 (perfect correlation). Distributions of GCC values are shifted toward higher values in those clusters with the tightest binding (Fig. S50), particularly in cluster . For a more detailed inspection, we followed the example set in Palermo et al.55, and we computed the correlation score (CS) for each residue i as the sum of GCC values with all the other antibody residues. This allowed us to distinguish intra-domain scores (Fig. S51), where the summation extends to the residues belonging to the same structural domain, and inter-domain scores (Fig. S52), which take into account only residues belonging to the other structural domains, excluding the one that includes residue i. From the distributions of intra- and inter-CS values shown in Fig. 4, it is apparent that the change in correlations among the different clusters stems from an increased inter-correlation between the antibody domains in clusters and, to a smaller extent, in cluster . On the opposite, the intra-correlations do not show significant variations. In cluster , in particular, the inter-correlations are significantly higher in Fab2 and in the chain B of Fc, with respect to the other clusters (Fig. S52). This result can be explained on the basis of the tight binding between antigen and antibody in cluster , and of its largely distributed communication network, as highlighted above.
Mutual information shows the highest correlation between the antigen molecule and all the antibody domains in cluster (Fig. S53), especially in Fab1. To further investigate the interplay between paratope and such peculiar antibody conformation, simulations of pembrolizumab in the apo form were started from the representative structure of cluster , after removal of PD-1. Three 100ns-long replicas were performed to allow residues in the paratope to relax to new equilibrium conformations, while the antibody retains an overall conformation closely similar to the starting one. As shown in Fig. S54, the RMSD distribution of the residues in the binding site overlaps with the one obtained from the apo cluster with the highest structural similarities, namely cluster (Fig. S15); in addition to a large network of interactions, as observed in the case of the other compact apo clusters, the latter is characterized by particularly strong contacts between the Fab domains (Table S3). A similar transition of the binding site conformation is not obtained in the case of the other holo clusters, thus suggesting once again a complex interplay between the large-scale conformation of the antibody and the antigen binding site, as elicited by the highly extended correlation network of pembrolizumab in conformational states compatible with cluster .
Discussion and conclusions
While impressive progress has been made in the experimental characterisation and manipulation of antibodies, a detailed, atomistic investigation of their properties is still incomplete. This is particularly true for the interplay between the molecule structure and its dynamics, which is extremely rich and varied, as several studies have recently shown27,56–58.
If, on the one hand, the specific sequence of the CDR plays the most prominent role in the selectivity and binding affinity of the antibody, on the other hand the observation that a modulation of the binding strength can be effected though mutations in distal sites shows that the internal mechanics of these molecules can be extremely complex16,17. This is especially the case for IgG4 antibodies such as pembrolizumab, whose short hinge reduces the degree of flexibility and tightens the interactions among the various domains.
In this work, we have made use of atomistic MD simulations and information-theoretical analysis methods to elucidate the relation between the large-scale arrangement of pembrolizumab and the stability of the antigen binding. The analysis pipeline employed has allowed us to highlight a substantial conformational variability, a quality rather different from that one might observe in the case of loosely-connected rigid bodies. On the contrary, a complex pattern of structural arrangements, intramolecular communication pathways, and binding strength has emerged.
In accordance with experimental results, we observed that the antibody is prevalently found in a compact, asymmetric shape. This particular arrangement can be explained on the basis of the short hinge, which limits the conformational space accessible to Fab arms, as well as the rotated CH2 domain of chain B. Furthermore, the spectrum of structures sampled by the molecule is modulated by the binding state: the large conformational variability of the apo case is substantially restricted when the antibody is complexed with the antigen. Similarly, the flexibility of the hinge (hinge 1 in particular) is reduced in this latter case, with a high degree of correlation emerging between the binding site on Fab2 and hinge 1. The binding site was shown to bear strict ties with the rest of the molecule in general. The analysis of the distributions of the apo binding site RMSD, computed taking the experimentally resolved holo structure as a reference, highlighted a correlation between the binding site and the large-scale arrangement of the molecule as a whole.
The binding of the PD1 antigen to Fab2 is, in general, rather stable in all holo state simulations; however, the strength of the binding is different across the various conformational clusters. The trend follows the intensity of the inter-domain correlations not only within the antibody, but also between the antigen and the antibody itself; a particularly strong affinity is observed in a specific configuration in which Fab1 is bent toward Fab2 and Fc. The tight binding and high correlations in this cluster suggest an interplay between the binding mode and the intensity of correlations, which manifest themselves in an extended network of interactions.
The results reported in this work return a picture of antibodies as extremely complex molecules, with a rich pattern of structural and dynamical features. The analysis protocol here applied to pembrolizumab is completely general, which enables its widespread application to other antibodies, with the objective of acquiring an ever deeper understanding of their inner life and, in turn, providing sharper tools for the manipulation of these molecules for medical and technological purposes.
Methods
System setup
The crystallographic structure of pembrolizumab in the apo form (Ab1, PDB ID: 5DK3)29 was used as a starting structure for the MD simulations. Missing residues (positions 230-232 of chain B and 230-235 of chain D, located in the hinge region) were modelled with Chimera59, using an IgG2a antibody (PDB ID: 1IGT60) as a template. Glycans were removed from the crystal structure, so as to isolate the role of structural features of the antibody itself in the emerging dynamics. The system in the holo form (Ab2) was obtained by replacing one Fab in Ab1 with the crystallized structure of Fab-PD1 complex (PDB ID: 5GGS)40, after structural alignment. The Fab-fragment alone bound to the PD-1 (Ab3) was also simulated, in order to assess possible differences in the interaction with the antigen with respect to the full-length antibody.
Simulation protocol
MD simulations were performed with the Gromacs 2018 software61,62. The amber99SB-ILDN force field63 was used to define the topology of the systems. The protein was solvated in a box of TIP3P water molecules64 and the charge was neutralized with Cl and Na ions at physiological concentration (150 mM). The final number of atoms was 571932 in Ab1, 618265 in Ab2, and 193009 in Ab3; of these, the number of protein atoms was 20218, 21976 and 8317, respectively. Energy minimization was performed until the maximum force reached the value of 500 kJ mol nm. NVT and NPT equilibrations were performed for 100 ps each using the Velocity-rescale thermostat65 and the Parrinello-Rahman barostat66. Temperature was set at 300 K and pressure at 1 bar, and the time constants were set to ps and ps. Constraints were applied to hydrogen-containing bonds through the LINCS algorithm67. A cutoff of 10 Å was used for Van der Waals and short-range Coulomb interactions; long range electrostatics was treated with Particle Mesh Ewald. Ab1 and Ab2 were simulated for a total of 2 μs each, in 4 independent replicas of 500 ns. Ab3 was simulated for 500 ns. Additional simulations of the apo state were started from the representative holo conformations, after removal of the antigen; the structures were solvated and equilibrated using the above-mentioned procedure, and three 100ns-long replicas were simulated for each system.
Analyses
Clustering of the simulation frames according to structural similarity was performed with an in-house script using hierarchical clustering (UPGMA)68. The distance used in the cluster definition is based on the root-mean-square deviation between C of all the structure pairs sampled in the simulations, following superimposition with the mdtraj algorithm that translates and rotates the structures for optimal alignment69. Calculation of atomic displacement, residue fluctuations, interatomic distances and contact surface areas, were performed using Gromacs 2018 utilities, while calculation of MM/PBSA energy was performed with the software g_mmpbsa52. Additional details on the MM/PBSA method and on the PAD calculation are reported in the SI. The number of hydrogen bonds was computed with VMD70, setting an angle cutoff of 25° and a maximum donor-acceptor distance of 3 Å.
Principal component analysis
Principal component analysis (PCA) was performed for each cluster using C atoms positions. Calculation, diagonalization and analysis of the covariance matrices were performed using Gromacs tools gmx covar and gmx anaeig62. In order to visualize the direction of movements captured by the eigenvectors, porcupine plots were generated using the extreme projections on the first three principal component, and visualized with VMD; here, the direction of the arrow on each C atom represents the direction of motion, while the length of the arrow characterizes the movement strength.
Correlations
Mutual information (MI) was calculated as a measure of inter-residue communications. MI is defined as follows:
| 1 |
where and are the displacements of the C atoms with respect to their average positions, and are the probability functions of finding the i-th or j-th atoms with a displacement equal to or , is the joint probability function. The calculation was performed with in-house scripts, and the displacement of each atom was divided in 100 discrete bins covering the range between 0 and the largest distance of that atom from its equilibrium position as sampled during the simulations. From the mutual information, the generalized correlation coefficient (GCC) is computed as:
| 2 |
GCC represents a measure of normalized MI, ranging from 0 (no correlation) to 1 (perfect correlation)54. Calculations were performed through in-house scripts.
GCC was in turn employed to calculate the correlation score (CS). For each residue i, is computed as the sum of the generalized correlation coefficient values between residue i and the other protein residues:
| 3 |
In the case of the intra-domain CS, the sum extends to the residues belonging to the same structural domain as residue i; in the case of the inter-domain CS, the summation takes into account only residues belonging to all the other structural domains, excluding that of residue i.
Network analysis
Networks were defined as sets of interconnected nodes centered on the C atoms. The total number of nodes of each system corresponds therefore to the number of residues. A couple of nodes is considered connected by an edge if any heavy atoms of the two residues is within a distance of 4.5 Å for at least 75% of the simulation time. These cutoffs were selected after a convergence study based on the Community Repartition Difference (see SI).
Each edge is weighted according to the generalized correlation coefficient measure; specifically, the weight of the edge between nodes i and j is defined as:
| 4 |
where is the generalized correlation coefficient (equation 2).
The network analysis was performed with the Python implementation found in Melo et al.71, which makes use of the NetworkX package72 and is optimized using Cython73 and Numba74. In the weighted networks, the sets of communities were identified using the Girvan-Newman algorithm75,76. The importance of the edge for communication within the network was measured in terms of edge betweenness, which is defined as the number of shortest pathways that cross the edge; this quantity was calculated with the commonly used Floyd–Warshall algorithm77,78. For a detailed description of the methods used for network analysis, the reader is referred to the Supplementary Information.
Supplementary Information
Acknowledgements
The authors thank Emiliano Biasini and Attilio Vittorio Vargiu for an insightful reading of the manuscript. This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant 758588).
Author contributions
R.P. designed the project; T.T. and M.R. performed the simulations and the analyses; all authors contributed to the interpretation of the results and to the writing of the manuscript.
Data availibility
The raw data produced and analysed in this work are freely available on the Zenodo repository https://doi.org/10.5281/zenodo.5018432.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Thomas Tarenzi and Marta Rigoli.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-02323-x.
References
- 1.Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13:1860476. doi: 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu R-M, et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scourfield DO, et al. The role and uses of antibodies in covid-19 infections: A living review. Oxford Open Immunol. 2021;2:iqab003. doi: 10.1093/oxfimm/iqab003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017;16:315–337. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 5.Larson SM, Carrasquillo JA, Cheung N-KV, Press OW. Radioimmunotherapy of human tumours. Nat. Rev. Cancer. 2015;15:347–360. doi: 10.1038/nrc3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumet C, Pottier J, Gouilleux-Gruart V, Watier H. Insights into the igg heavy chain engineering patent landscape as applied to igg4 antibody development. MAbs. 2019;11:1341–1350. doi: 10.1080/19420862.2019.1664365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood. 2016;127:1097–1101. doi: 10.1182/blood-2015-09-625343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samsudin F, Yeo JY, Gan SK-E, Bond PJ. Not all therapeutic antibody isotypes are equal: The case of igm versus igg in pertuzumab and trastuzumab. Chem. Sci. 2020;11:2843–2854. doi: 10.1039/c9sc04722k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Mathieu M, Brezski RJ. Igg fc engineering to modulate antibody effector functions. Protein Cell. 2018;9:63–73. doi: 10.1007/s13238-017-0473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front. Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, et al. Asymmetrical fc engineering greatly enhances antibody-dependent cellular cytotoxicity (adcc) effector function and stability of the modified antibodies. J. Biol. Chem. 2014;289:3571–3590. doi: 10.1074/jbc.M113.513366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handlogten MW, et al. Prevention of fab-arm exchange and antibody reduction via stabilization of the igg4 hinge region. MAbs. 2020;12:1779974. doi: 10.1080/19420862.2020.1779974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva J-P, Vetterlein O, Jose J, Peters S, Kirby H. The s228p mutation prevents in vivo and in vitro igg4 fab-arm exchange as demonstrated using a combination of novel quantitative immunoassays and physiological matrix preparation. J. Biol. Chem. 2015;290:5462–5469. doi: 10.1074/jbc.M114.600973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbener P, et al. Functional relevance of in vivo half antibody exchange of an igg4 therapeutic antibody-drug conjugate. PLoS ONE. 2018;13:e0195823. doi: 10.1371/journal.pone.0195823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu ML, Gilliland GL. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016;38:163–173. doi: 10.1016/j.sbi.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Koenig P, et al. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc. Natl. Acad. Sci. 2017;114:E486–E495. doi: 10.1073/pnas.1613231114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanaka S, Moriwaki Y, Tsumoto K, Sugase K. Elucidation of potential sites for antibody engineering by fluctuation editing. Sci. Rep. 2017;7:1–10. doi: 10.1038/s41598-017-10246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cannon DA, et al. Experimentally guided computational antibody affinity maturation with de novo docking, modelling and rational design. PLoS Comput. Biol. 2019;15:e1006980. doi: 10.1371/journal.pcbi.1006980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel JS, Ytreberg FM. Fast calculation of protein–protein binding free energies using umbrella sampling with a coarse-grained model. J. Chem. Theory Comput. 2018;14:991–997. doi: 10.1021/acs.jctc.7b00660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lees WD, Stejskal L, Moss DS, Shepherd AJ. Investigating substitutions in antibody-antigen complexes using molecular dynamics: A case study with broad-spectrum, influenza a antibodies. Fronti. Immunol. 2017;8:143. doi: 10.3389/fimmu.2017.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Codina N, et al. An expanded conformation of an antibody fab region by X-ray scattering, molecular dynamics, and smfret identifies an aggregation mechanism. J. Mol. Biol. 2019;431:1409–1425. doi: 10.1016/j.jmb.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida K, et al. Exploring designability of electrostatic complementarity at an antigen-antibody interface directed by mutagenesis, biophysical analysis, and molecular dynamics simulations. Sci. Rep. 2019;9:4482. doi: 10.1038/s41598-019-40461-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández-Quintero ML, Heiss MC, Pomarici ND, Math BA, Liedl KR. Antibody cdr loops as ensembles in solution vs. canonical clusters from X-ray structures. MAbs. 2020;12:1744328. doi: 10.1080/19420862.2020.1744328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernández-Quintero ML, Kraml J, Georges G, Liedl KR. Cdr-h3 loop ensemble in solution-conformational selection upon antibody binding. MAbs. 2019;11:1077–1088. doi: 10.1080/19420862.2019.1618676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernández-Quintero ML, Kroell KB, Hofer F, Riccabona JR, Liedl KR. Mutation of residue 71 h results in different paratope states in solution. Front. Immunol. 2021;12:243. doi: 10.3389/fimmu.2021.630034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al Qaraghuli MM, Kubiak-Ossowska K, Ferro VA, Mulheran PA. Antibody-protein binding and conformational changes: Identifying allosteric signalling pathways to engineer a better effector response. Sci. Rep. 2020;10:1–10. doi: 10.1038/s41598-020-70680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Nussinov R, Ma B. Antigen binding allosterically promotes fc receptor recognition. MAbs. 2019;11:58–74. doi: 10.1080/19420862.2018.1522178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J, Nussinov R, Ma B. Mechanisms of recognition of amyloid- (a) monomer, oligomer, and fibril by homologous antibodies. J. Biol. Chem. 2017;292:18325–18343. doi: 10.1074/jbc.M117.801514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scapin G, et al. Structure of full-length human anti-pd1 therapeutic igg4 antibody pembrolizumab. Nat. Struct. Mol. Biol. 2015;22:953–958. doi: 10.1038/nsmb.3129. [DOI] [PubMed] [Google Scholar]
- 30.Urquhart L. Top companies and drugs by sales in 2019. Nat. Rev. Drug Discov. 2020;19:228–229. doi: 10.1038/d41573-020-00047-7. [DOI] [PubMed] [Google Scholar]
- 31.Patnaik A, et al. Phase i study of pembrolizumab (mk-3475; anti-pd-1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 2015;21:4286–4293. doi: 10.1158/1078-0432.CCR-14-2607. [DOI] [PubMed] [Google Scholar]
- 32.Nghiem PT, et al. Pd-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N. Engl. J. Med. 2016;374:2542–2552. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldberg SB, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: Early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–983. doi: 10.1016/S1470-2045(16)30053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharpe AH, Pauken KE. The diverse functions of the pd1 inhibitory pathway. Nat. Rev. Immunol. 2018;18:153. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 35.Ahmadzadeh M, et al. Tumor antigen-specific cd8 t cells infiltrating the tumor express high levels of pd-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivashko IN, Kolesar JM. Pembrolizumab and nivolumab: PD-1 inhibitors for advanced melanoma. Am. J. Health Syst. Pharm. 2016;73:193–201. doi: 10.2146/ajhp140768. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, et al. Looking for the optimal pd-1/pd-l1 inhibitor in cancer treatment: A comparison in basic structure, function, and clinical practice. Front. Immunol. 2020;11:1–10. doi: 10.3389/fimmu.2020.01088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edelman GM, et al. The covalent structure of an entire g immunoglobulin molecule. Proc. Natl. Acad. Sci. 1969;63:78–85. doi: 10.1073/pnas.63.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JY, et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 2016;7:13354. doi: 10.1038/ncomms13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian X, et al. In-depth analysis of subclass-specific conformational preferences of igg antibodies. IUCrJ. 2015;2:9–18. doi: 10.1107/S205225251402209X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aalberse RC, Schuurman J. Igg4 breaking the rules. Immunology. 2002;105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deveuve Q, Lajoie L, Barrault B, Thibault G. The proteolytic cleavage of therapeutic monoclonal antibody hinge region: More than a matter of subclass. Front. Immunol. 2020;11:168. doi: 10.3389/fimmu.2020.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freeman LC. A set of measures of centrality based on betweenness. Sociometry. 1977;1:35–41. [Google Scholar]
- 45.Lefranc M-P, Lefranc G. Imgt and 30 years of immunoinformatics insight in antibody v and c domain structure and function. Antibodies. 2019;8:29. doi: 10.3390/antib8020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kortkhonjia E, et al. Probing antibody internal dynamics with fluorescence anisotropy and molecular dynamics simulations. MAbs. 2013;5:306–322. doi: 10.4161/mabs.23651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roux KH, Strelets L, Michaelsen TE. Flexibility of human igg subclasses. J. Immunol. 1997;159:3372–3382. [PubMed] [Google Scholar]
- 48.Dangl JL, et al. Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. EMBO J. 1988;7:1989–1994. doi: 10.1002/j.1460-2075.1988.tb03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caliandro R, Rossetti G, Carloni P. Local fluctuations and conformational transitions in proteins. J. Chem. Theory Comput. 2012;8:4775–4785. doi: 10.1021/ct300610y. [DOI] [PubMed] [Google Scholar]
- 50.Li T, et al. Redistribution of flexibility in stabilizing antibody fragment mutants follows Le Chatelier’s principle. PLoS ONE. 2014;9:e92870. doi: 10.1371/journal.pone.0092870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li T, et al. Rigidity emerges during antibody evolution in three distinct antibody systems: Evidence from qsfr analysis of fab fragments. PLoS Comput. Biol. 2015;11:e1004327. doi: 10.1371/journal.pcbi.1004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumari R, Kumar R, Consortium OSDD, Lynn A. G\_mmpbsa: A gromacs tool for high-throughput mm-pbsa calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 53.Knapp B, Dunbar J, Alcala M, Deane CM. Variable regions of antibodies and t-cell receptors may not be sufficient in molecular simulations investigating binding. J. Chem. Theory Comput. 2017;13:3097–3105. doi: 10.1021/acs.jctc.7b00080. [DOI] [PubMed] [Google Scholar]
- 54.Lange OF, Grubmüller H. Generalized correlation for biomolecular dynamics. Proteins Struct. Funct. Bioinform. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 55.Palermo G, et al. Protospacer adjacent motif-induced allostery activates crispr-cas9. J. Am. Chem. Soc. 2017;139:16028–16031. doi: 10.1021/jacs.7b05313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adhikary R, et al. Adaptive mutations alter antibody structure and dynamics during affinity maturation. Biochemistry. 2015;54:2085–2093. doi: 10.1021/bi501417q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janda A, Bowen A, Greenspan NS, Casadevall A. Ig constant region effects on variable region structure and function. Front. Microbiol. 2016;7:22. doi: 10.3389/fmicb.2016.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yanaka S, et al. Dynamic views of the fc region of immunoglobulin g provided by experimental and computational observations. Antibodies. 2019;8:1–10. doi: 10.3390/antib8030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF, et al. Ucsf chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 60.Harris LJ, Larson SB, Hasel KW, McPherson A. Refined structure of an intact igg2a monoclonal antibody. Biochemistry. 1997;36:1581–1597. doi: 10.1021/bi962514+. [DOI] [PubMed] [Google Scholar]
- 61.Bekker H, et al. Gromacs: A parallel computer for molecular dynamics simulations. Phys. Comput. 1993;92:252–256. [Google Scholar]
- 62.Abraham MJ, et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 63.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins Struct. Funct. Bioinform. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 65.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126(1):1. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 66.Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 67.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 68.Sokal RR, Michener CD. A statistical method for evaluating systematic relationships. Univ. Kans. Sci. Bull. 1958;28:1409–1438. [Google Scholar]
- 69.McGibbon RT, et al. Mdtraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015;109:1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 71.Melo MC, Bernardi RC, De La Fuente-nunez C, Luthey-Schulten Z. Generalized correlation-based dynamical network analysis: A new high-performance approach for identifying allosteric communications in molecular dynamics trajectories. J. Chem. Phys. 2020;153:134104. doi: 10.1063/5.0018980. [DOI] [PubMed] [Google Scholar]
- 72.Hagberg, A., Swart, P. & Chult, D. Exploring network structure, dynamics, and function using networkx. Tech. Rep. (Los Alamos National Lab. (LANL), 2008).
- 73.Behnel S, et al. Cython: The best of both worlds. Comput. Sci. Eng. 2011;13:31–39. [Google Scholar]
- 74.Lam, S. K., Pitrou, A. & Seibert, S. Numba: A llvm-based python jit compiler. In Proceedings of the Second Workshop on the LLVM Compiler Infrastructure in HPC, 1–6 (2015).
- 75.Girvan M, Newman ME. Community structure in social and biological networks. Proc. Natl. Acad. Sci. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008;2008:P10008. [Google Scholar]
- 77.Floyd RW. Algorithm 97: Shortest path. Commun. ACM. 1962;5:345. doi: 10.1145/367766.368168. [DOI] [Google Scholar]
- 78.Warshall S. A theorem on Boolean matrices. J. ACM. 1962;9:11–12. doi: 10.1145/321105.321107. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data produced and analysed in this work are freely available on the Zenodo repository https://doi.org/10.5281/zenodo.5018432.