

Abstract
Repeated cocaine use induces coordinated changes in gene expression that drive plasticity in the nucleus accumbens (NAc), an important component of the brain’s reward circuitry, and promote the development of maladaptive, addiction-like behaviors. Studies on the molecular basis of cocaine action identify transcription factors, a class of proteins that bind to specific DNA sequences and regulate transcription, as critical mediators of this cocaine-induced plasticity. Early methods to identify and study transcription factors involved in addiction pathophysiology primarily relied on quantifying the expression of candidate genes in bulk brain tissue after chronic cocaine treatment, as well as conventional overexpression and knockdown techniques. More recently, advances in next generation sequencing, bioinformatics, cell-type-specific targeting, and locus-specific neuroepigenomic editing offer a more powerful, unbiased toolbox to identify the most important transcription factors that drive drug-induced plasticity and to causally define their downstream molecular mechanisms. Here, we synthesize the literature on transcription factors mediating cocaine action in the NAc, discuss the advancements and remaining limitations of current experimental approaches, and emphasize recent work leveraging bioinformatic tools and neuroepigenomic editing to study transcription factors involved in cocaine addiction.
Introduction: Cocaine-Induced Plasticity in the Nucleus Accumbens
Neurocircuitry analyses in humans and rodents identify the nucleus accumbens (NAc) as a critical brain region involved in mediating the reinforcing and addicting properties of drugs of abuse [1]. The NAc receives convergent glutamatergic inputs from several forebrain regions involved in regulating addictive behaviors, including the prefrontal cortex (PFC), basolateral amygdala, and ventral hippocampus, among other regions, in concert with mostly dopaminergic inputs from the ventral tegmental area (VTA). Medium spiny projection neurons (MSNs) comprise ~95% of the neuronal population in the NAc, with the remaining neurons consisting of several types of local interneurons. MSNs are divided into two main subpopulations depending on their predominant expression of dopamine (DA) receptor subtypes. DA receptor 1-expressing MSNs (D1 MSNs) and DA receptor 2-expressing MSNs (D2 MSNs) exhibit distinct transcriptional profiles [2,3], unique afferent inputs and projection patterns [4,5], as well as different and often opposing contributions to addiction-related behaviors [6–12]. Several studies suggest that a small (<10%) subset of NAc MSNs express both receptor types, but their functioning and regulation is less well understood [3,13,14].
There is now a large literature that defines the effects of repeated cocaine exposure, including drug self-administration and prolonged withdrawal, on the functioning of the NAc and its constituent D1 and D2 MSNs [15–17]. Briefly, numerous adaptations have been characterized at the level of cell excitability, synaptic plasticity (function and morphology), and responsiveness to glutamatergic inputs [4,18–22]. While these neuroadaptations are likely mediated via a wide range of mechanisms, including protein modifications, trafficking, and local translation, increasing evidence supports the view that persistent changes in gene expression are also important contributors, especially to long-lasting drug-induced neuroplasticity that heightens the risk of relapse after prolonged abstinence [23–25].
Transcriptional Mechanisms Underlying Cocaine-Induced Plasticity
There are numerous mechanisms through which repeated exposure to drugs of abuse such as cocaine could induce changes in gene expression in NAc MSNs. The most straightforward scheme is that cocaine, by increasing levels of DA at the synapse, would increase activation of D1 and D2 receptors on MSN subpopulations, which through distinct intracellular signaling pathways would activate or suppress the activity of numerous transcription factors through changes in post-translational modifications or their total expression levels. Transcription factors are proteins that bind to specific sequences of DNA (known as response elements or motifs), usually 6-12 base pairs in length, within the regulatory regions (i.e., promoters and enhancers) of genes and then recruit numerous secondary regulatory proteins to the genes, with the result being the activation or suppression of transcription (Figure 1) [26]. Importantly, transcription factor binding may not affect the steady-state level of a gene’s expression but instead alter the transcriptional response of the gene to a future stimulus through gene priming or desensitization [24]. Numerous studies have demonstrated such patterns of cocaine regulation of gene expression. For example, a recent study from our laboratory found that cocaine self-administration alters the expression of many hundred genes in the NAc, based on RNA-sequencing (RNA-seq) analysis of bulk tissue dissections, many of which are sustained after extended withdrawal or even become apparent only after extended withdrawal [27]. Moreover, a cocaine challenge after extended withdrawal results in the induction or suppression of hundreds of genes that are not affected by initial cocaine exposure—a clear demonstration of the prominence of gene priming and desensitization in this brain region in response to a distant history of cocaine self-administration (Figure 2) [27,28].
Figure 1: Theoretical mechanisms of gene activation or suppression by transcription factors.
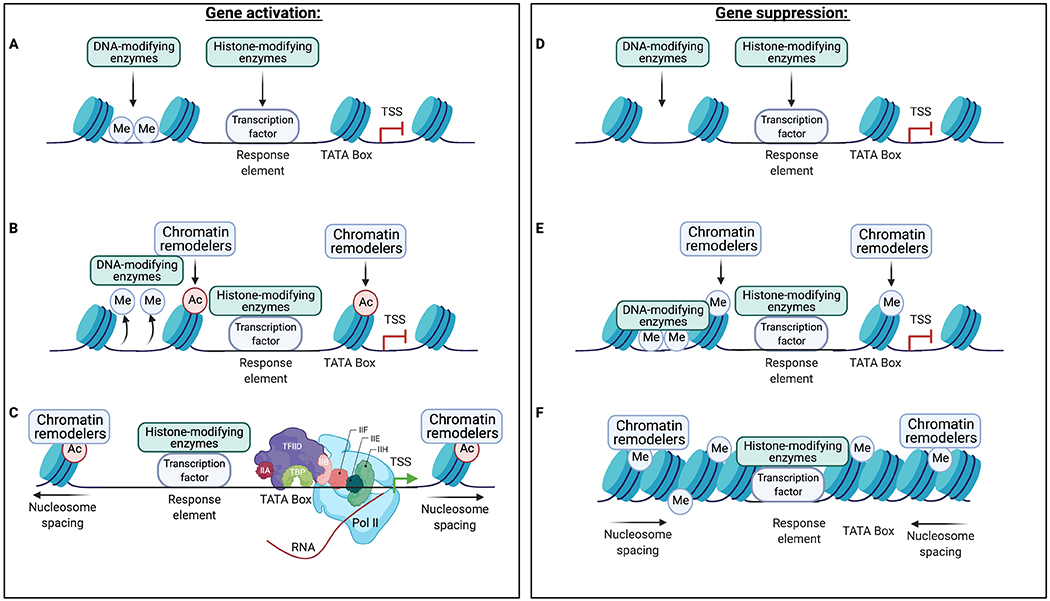
Transcription factors bind to specific DNA sequences, termed response elements, at their target genes and recruit secondary epigenetic enzymes and transcriptional machinery to regulate gene transcription. While many transcription factors either activate or suppress gene transcription at their target genes, some transcription factors may have opposite effects on transcription at different target genes via distinct mechanisms downstream of transcription factor binding. In this scheme, a transcription factor binds to its response element at a given target gene and recruits various chromatin-modifying enzymes (A). These enzymes remove repressive DNA methylation (Me) and deposit permissive modifications (e.g., acetylation [Ac]) on local histones (B), which are subsequently recognized by chromatin remodeling proteins that increase nucleosome spacing and enable recruitment of the transcriptional machinery (C). Conversely, a transcription factor bound to its response element (D) may recruit chromatin-modifying enzymes that deposit repressive modifications on local histones and DNA (E). These repressive modifications are then recognized by chromatin remodeling proteins that reduce nucleosome spacing and prevent the binding of transcriptional machinery (F). It is important to note that many additional proteins, chromatin modifications, and non-coding RNAs also regulate gene transcription, but these mechanisms remain poorly characterized in the context of drug-induced adaptations in NAc MSNs. TATA box denotes promoter region of the gene just 5’ of the transcription start site (TSS). The purple and light blue shapes in (C) show subunits of the basal transcription machinery and RNA polymerase II (Pol II).
Figure 2: Priming and desensitization of gene expression in NAc after prolonged withdrawal from cocaine self-administration.
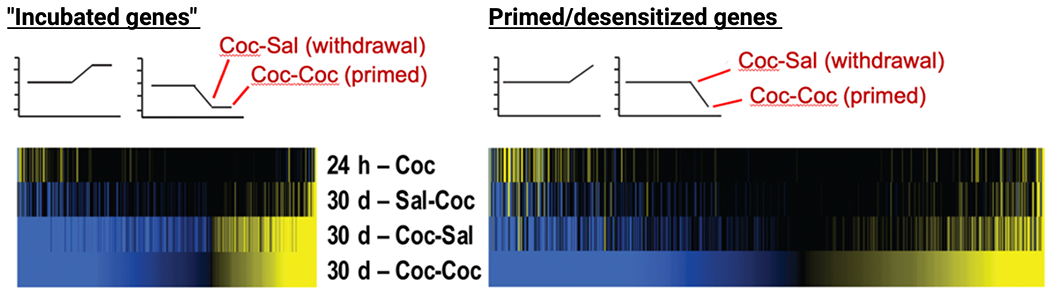
Mice self-administered saline (Sal) or cocaine (Coc) for 10 d. One cohort was analyzed 24 h after the last self-administration session (24 h - Coc). The other cohorts remained in their home cages for 30 d and then returned to the self-administration chambers and given an intraperitoneal injection of saline or cocaine yielding three groups: Sal-Coc (acute cocaine); Coc-Sal (30 d withdrawal, saline challenge: “Incubated genes”); Coc-Coc (30 d withdrawal, cocaine challenge: primed/desensitized genes). Heatmaps show RNAs whose expression levels exhibit statistically significant differential expression (yellow: upregulated; blue, downregulated) from all other conditions at both Coc-Sal and Coc-Coc conditions (left) or after the Coc-Coc condition (right). From Walker et al., 2018.
Studies over the past several decades—beginning in the early 1990s—have shown that rodents exposed chronically to cocaine—either investigator-delivered or self-administered—show differential expression or activity levels of numerous transcription factors in the NAc [25]. Those most studied to date include cAMP response element-binding protein (CREB), ΔFOSB, nuclear factor κB (NFκB), and early growth response protein 3 (EGR3) (Table 1A). Cocaine-induced regulation of these transcription factors alters the expression of their target genes, many of which are involved in long-term synaptic plasticity and modulating addiction-like behavior in rodents. As will be seen, technical advances introduced over the past few years have allowed a more unbiased and therefore comprehensive analysis of transcription factors involved in cocaine action, which is dramatically expanding the range of transcriptional mechanisms implicated in cocaine addiction [27].
Table 1:
Summary of studies investigating transcription factor gene expression or activity in bulk NAc tissue (A) or in a cell-type-specific manner (B).
| Transcription factor | Treatment duration | Cocaine dose | Withdrawal duration | Effect on Transcription factor expression | Reference |
|---|---|---|---|---|---|
| ΔFosB | 1 day | 0.5 mg/kg/inf | 0 hours 24 hours |
Increased ΔFOSB mRNA No effect on ΔFOSB mRNA |
Larson et al. 2010 |
| 1 day | 10 mg/kg | 24 hours | No effect on ΔFOSB protein | Robison et al. 2013 | |
| 7 days | 10 mg/kg | 24 hours | Increased ΔFOSB protein | Robison et al. 2013 | |
| 7 days | 10 mg/kg | 14 days | Increased ΔFOSB protein | Robison et al. 2013 | |
| 14 days | 15 mg/kg x 2 | 18-24 hours | Increased ΔFOSB protein | Perrotti et al. 2008 | |
| 14 days | 0.5 mg/kg/inf | 18-24 hours | Increased ΔFOSB protein | Perrotti et al. 2008 | |
| 18 days | 0.5 mg/kg/inf | 0 hours 24 hours 3 weeks |
Increased ΔFOSB protein; no effect on ΔFOSB mRNA Increased ΔFOSB protein; no effect on ΔFOSB mRNA No effect on ΔFOSB protein |
Larson et al. 2010 | |
|
| |||||
| CREB | 1 day | 30 mg/kg | 0 – 1.5 hours | Increased pCREB | Nazarian et al. 2009 |
| 1 day | 10 mg/kg | 20 minutes | Increased pCREB | Walters et al. 2003 | |
| 7 days | 15 mg/kg | 0 – 1 hour | Increased pCREB | Mattson et al. 2005 | |
| 14 days | 15 mg/kg x 2 | 0 – 1.5 hours | No effect on pCREB | Nazarian et al. 2009 | |
|
| |||||
| E2F3a | 7 days | 20 mg/kg | 24 hours | Increased E2F3a mRNA Increased E2F3a protein |
Cates et al. 2017 |
|
| |||||
| E2F3b | 7 days | 20 mg/kg | 24 hours | No effect on E2F3b mRNA | Cates et al. 2017 |
|
| |||||
| EGR3 | 7 days | 20 mg/kg | 24 hours | Decreased EGR3 mRNA; Decreased EGR3 protein | Chandra et al. 2015 |
| 10 days | 1 mg/kg/inf | 24 hours | Decreased EGR3 mRNA; Decreased EGR3 protein | Chandra et al. 2015 | |
| 10 days | 1 mg/kg/inf | 20 days | Decreased EGR3 protein in females No effect on EGR3 protein in males |
Engeln et al. 2019 | |
|
| |||||
| FOXO3a | 7 days | 20 mg/kg | 24 hours | Increased FOXO3a protein | Ferguson et al. 2015 |
|
| |||||
| MEF2 | 1 day | 20 mg/kg | 4 hours | No effect on MEF2 P-S404/4441 | Pulipparacharuvil et al. 2008 |
| 24 hours | Increased MEF2 P-S408/4441 | ||||
| 48 hours | No effect on MEF2 P-S404/4441 | ||||
| 7 days | 20 mg/kg | 4 hours | Increased MEF2 P-S408/4441 | Pulipparacharuvil et al. 2008 | |
| 24 hours | Increased MEF2 P-S408/4441 | ||||
| 48 hours | No effect on MEF2 P-S408/4441 | ||||
|
| |||||
| NFκB | 1 day | 20 mg/kg | 4-6 hours 24 hours |
Increased NFκB-dependent transcription No effect on NFκB-dependent transcription |
Russo et al. 2009 |
| 1 day | 20 mg/kg | 4 hours | No effect on protein expression of p105, IκB-γ p70, or p65 | Ang et al. 2001 | |
| 5 days | 20 mg/kg | 4-6 hours 24 hours |
Increased NFκB-dependent gene transcription Increased NFκB-dependent gene transcription |
Russo et al. 2009 | |
| 5 days | 20 mg/kg | 4-6 hours | Increased protein expression of p105 and p65 subunits No effect on protein expression of IκBβ and IκBα |
Russo et al. 2009 | |
| 14 days | 20 mg/kg | 4 hours | Increased protein expression of p105 Trending increase in protein expression of IκB-γ p70 and p65 |
Ang et al. 2001 | |
|
| |||||
| NR4A1 | 21 days | 0.7 mg/kg/inf | 24 hours | Increased NR4A1 mRNA | Carpenter et al. 2020 |
| 21 days | 0.7 mg/kg/inf | 28 days | No effect on NR4A1 mRNA | Carpenter et al. 2020 | |
| Transcription factor | Cell type | Treatment duration | Cocaine dose | Withdrawal duration | Effect on Transcription factor expression | Reference |
|---|---|---|---|---|---|---|
| ΔFOSB | D1 MSNs | 21 days | 0.5 mg/kg/inf | 24 hours | Increased ΔFOSB protein | Lobo et al. 2013 |
| 7 days | 20 mg/kg | 24 hours | Increased ΔFOSB protein | Lobo et al. 2013 | ||
| D2 MSNs | 21 days | 0.5 mg/kg/inf | 24 hours | No effect on ΔFOSB protein | Lobo et al. 2013 | |
| 7 days | 20 mg/kg | 24 hours | No effect on ΔFOSB protein | Lobo et al. 2013 | ||
|
| ||||||
| CREB | D1 MSNs | 7 days | 20 mg/kg | 24 hours | No effect on CREB protein | Lobo et al. 2013 |
| D2 MSNs | 7 days | 20 mg/kg | 24 hours | No effect on CREB protein | Lobo et al. 2013 | |
|
| ||||||
| E2F3a | D1 MSNs | 7 days | 20 mg/kg | 24 hours | Increased E2F3a mRNA | Cates et al. 2019 |
| D2 MSNs | 7 days | 20 mg/kg | 24 hours | No effect on E2F3a mRNA | Cates et al. 2019 | |
|
| ||||||
| E2F3b | D1 MSNs | 7 days | 20 mg/kg | 24 hours | No effect on E2F3b mRNA | Cates et al. 2019 |
| D2 MSNs | 7 days | 20 mg/kg | 24 hours | No effect on E2F3b mRNA | Cates et al. 2019 | |
|
| ||||||
| EGR3 | D1 MSNs | 7 days | 20 mg/kg | 24 hours | Increased ribosome-associated EGR3 mRNA | Chandra et al. 2015 |
| D2 MSNs | 7 days | 20 mg/kg | 24 hours | Decreased ribosome-associated EGR3 mRNA | Chandra et al. 2015 | |
MEF2 activity was measured by levels of inhibitory phosphorylation.
In parallel, the field of epigenetics is providing a far more complete understanding of how the binding of a transcription factor to the regulatory region of a target gene influences that gene’s steady-state transcription or its potential to be induced or suppressed (i.e., primed or desensitized) in response to a subsequent stimulus. Transcription factors bound to their response elements may recruit hundreds of enzymes that control a host of post-translational modifications of histones or methylation of the DNA itself, as well as numerous other proteins that control the spacing of nucleosomes (composed of histone octomers) along the linear span of DNA (Figure 1). In general, mechanisms that increase nucleosome spacing increase gene transcription, while mechanisms that reduce nucleosome spacing exert the opposite effect. Transcription factors also influence the three-dimensional (3D) structure of chromatin, which controls the activity of a gene in part by bringing enhancers and promoters in close proximity via chromatin looping [29,30]. Knowledge of these epigenetic mechanisms is refining our understanding of how drug-regulated transcription factors actually alter the steady-state or potential expression levels of a given target gene, although the precise changes to chromatin that occur when an active transcription factor binds to a given gene in response to cocaine exposure remain mostly unknown and a major focus of current research. Knowledge of epigenetic mechanisms has also suggested alternative pathways—independent of transcription factors—through which cocaine might alter gene expression, that is, through direct effects on chromatin regulatory proteins. Indeed, experimental manipulations of numerous epigenetic mechanisms, including histone acetyltransferases or deacetylases, histone or DNA methyltransferases or demethylases, and microRNAs (miRNAs) in NAc neurons has been shown to control cocaine-induced changes in gene expression and associated behavioral abnormalities. We refer the reader to several recent reviews of such noncanonical mechanisms of drug-induced regulation of gene expression [23,31–35].
Technical Advancements Improve Insights Into Transcription Factor Function
Historically, studies characterizing the functions of transcription factors in animal models of cocaine addiction have been hindered by the lack of unbiased experimental approaches to identify transcription factors driving cocaine-induced neuroplasticity, quantify genome-wide gene expression and transcription factor binding, and causally define the mechanisms downstream of transcription factor regulation.
First, earlier methods to quantify gene expression—from PCR to gene expression microarrays—are limited by the number of targets they could measure, thus introducing experimental bias by quantifying a small subset of candidate genes. This approach hindered the ability to identify the diversity of transcription factors and related proteins that underlie the lasting actions of cocaine and other drugs of abuse in the NAc and other brain reward regions. More recently, next-generation sequencing has become commonplace in neuroscience laboratories and enabled the first unbiased, comprehensive (i.e., genome-wide) quantification of gene expression, protein-DNA interactions, epigenetic modifications on histones or DNA, and 3D chromatin architecture, among other properties of chromatin. These technologies are especially relevant for studies of transcription factors. For example, recent studies have employed RNA-seq to measure the effects of transcription factor manipulations on the NAc transcriptome [36,37]. Increasingly, RNA-seq is being applied to isolated subpopulations of neurons and even to individual cells [2,3]. In addition, chromatin immunoprecipitation paired with DNA sequencing (ChIP-seq) enables quantification of the binding of a single transcription factor across the genome. While early ChIP-seq protocols were limited by the requirement for large tissue quantities and high background signal, improved methods such as Cleavage Under Targets and Release Using Nuclease (CUT&RUN) [38] are amenable to quantifying transcription factor binding genome-wide in discrete regions of the mouse brain, even in selected cell types. Assay for transposase-accessible chromatin plus DNA sequencing (ATAC-seq) is a method to quantify open regions of chromatin (euchromatin), in which genes are accessible to transcription factors, chromatin-modifying enzymes, and transcriptional machinery for dynamic control of gene expression [39]. Interestingly, short DNA sequences occupied by transcription factors are depleted in ATAC-seq libraries, allowing researchers to deduce genome-wide transcription factor occupancy based on known transcription factor response elements [40]. Finally, genome-wide chromosome capture (HiC) is a technique that reveals the 3D arrangement of chromatin in the nucleus [41], which contributes to the regulation of gene expression through a variety of complex mechanisms including the formation of chromatin loops and topographically associated domains (TADs), regulating chromosome compartments, and localization within the nucleus [42]. Another technique, called chromatin interaction analysis using paired-end tag sequencing (ChIA-PET), combines genome-wide chromosome capture and ChIP technologies to identify gene segments in close proximity to chromatin-binding proteins, such as transcription factors [43]. Work in other tissues demonstrates that transcription factors regulate gene expression in part by altering chromatin topology [29,30,44], but this phenomenon remains unstudied in NAc MSNs.
Second, work from several laboratories has applied advanced computational approaches to derive more comprehensive biological insight from RNA-seq and other genome-wide datasets. For example, we recently analyzed RNA-seq data across numerous regions of the brain’s reward circuitry in animals self-administering cocaine or saline at several time points, namely, early vs. late withdrawal ± relapse [27]. This study revealed numerous transcription factors that are predicted to drive maladaptive behaviors during cocaine self-administration, many of which have not been studied in brain let alone in addiction-related phenomena. It is reassuring that most of the transcription factors identified in early studies using a candidate gene approach (e.g., CREB, ΔFOSB, NFκB, and EGR3) were also deduced as being important by unbiased bioinformatic analyses, suggesting that the field succeeded historically in identifying a subset of the transcription factors that are important for cocaine-induced plasticity, but neglected many others [27]. This work highlights the importance of leveraging unbiased, hypothesis-generating computational approaches to study the molecular basis of drug addiction.
Third, studies to characterize the causal relationship between an individual transcription factor binding to a single genomic locus and addiction-related molecular, neurophysiological, and behavioral endpoints have been hampered by the experimental techniques available to study gene function. Traditionally, such studies relied on overexpressing the transcription factor or a dominant-negative mutant, RNA interference (RNAi)-mediated suppression of RNA translation, or Cre recombinase-mediated knockout of the gene to study its downstream effects. However, these techniques—even when induced in the adult brain in a cell-type-specific fashion—alter total cellular levels of the transcription factor and therefore its binding at hundreds or thousands of genomic locations, which limits mechanistic insight for any individual target gene [45]. Previous studies have also shown significant off-target effects of RNAi, which remains a commonly used approach to study transcription factors in the brain, that further hinder insight into transcription factor function [46]. Additionally, these techniques typically induce changes in the expression levels of transcription factors that are far greater in magnitude than the changes observed in response to drugs of abuse, which may have distinct effects on downstream experimental measures than more physiologically relevant changes in gene expression [47]. The evolving field of locus-specific neuroepigenomic editing offers a powerful set of tools to vastly improve the quality of proof in studies of transcription factor function by selectively recruiting a transcription factor to its response element at a single target gene within a single type of neuron in a given brain region of interest (Box 1) [47–54]. Although still in early stages of development, this approach far more accurately establishes causal relationships between transcription factor binding at single gene and outcome measures at the transcriptional, molecular, cellular, circuit, and behavioral levels.
Box 1: Locus-Specific Neuroepigenomic Editing.
Current genetic and pharmacological approaches to regulate gene function have several limitations that hinder experimental insight. First, overexpression or knockout techniques often induce changes in gene expression that fall outside of the range observed under physiological or pathophysiological conditions. This is crucially important because empirical data demonstrate that gene regulation outside of a physiologically-relevant range can have different, even opposite, experimental outcomes [47,141,168]. Second, these techniques alter transcription factor binding at hundreds or thousands of genomic regions, making it difficult to determine the role of individual target genes. Lastly, gene knockdown strategies, such as RNA interference (RNAi) and dominant negative approaches, are prone to significant off-target effects [46].
Locus-specific neuroepigenomic editing utilizes a highly specific DNA binding domain tethered to a chromatin-modifying protein, allowing researchers to model a specific epigenetic change at a single genomic locus in a discrete cell population in the brains of awake, behaving animals. Current neuroepigenomic editing platforms include zinc finger proteins (ZFPs), transcriptional activator-like effectors (TALEs), and clustered regularly interspaced short palindromic repeats (CRISPR). These tools are modified to lack functional nuclease domains and are instead tethered to a transcriptional effector domain. ZFPs and TALEs bind directly to their target DNA sequence via protein-DNA interactions, while CRISPR uses a guide RNA (gRNA) to direct nuclease-dead Cas9 (dCas9) binding.
A wide range of effector domains have been tested in vivo including histone acetyltransferases (HATs), histone methyltransferases (HMTs), DNA methyltransferases (DNMTs), and a transcription factor [47–54]. For example, work from our laboratory used the CRISPR platform to guide a phosphomimetic (active) form of CREB (CREBS133D) to its response element at a single CREB target gene implicated in mediating resilience to chronic stress, zinc finger protein 189 (Zfp189) [48]. Unlike CREB overexpression experiments, this method causally links CREB binding at a single target gene to downstream experimental measures. A similar approach was used recently to target CREBS133D to the FosB gene [37].
In a related strategy, neuroepigenome editing tools have been used to control bidirectionally the expression of an endogenous transcription factor. For example, coupling a FosB-targeted ZFP fused to a histone acetyltransferase or methyltransferase increases or decreases, respectively, levels of expression of endogenous FOSB and ΔFOSB in the NAc [102,169,170]. While the induced FOSB/ΔFOSB can still influence all of its downstream targets, this approach avoids confounds associated with conventional overexpression and knockdown methods.
An important focus of future research is expanding the neuroepigenomic editing toolbox to study a wide range of transcription factors and better establish the molecular mechanisms downstream of transcription factor activation.
Integrating genome-wide next-generation sequencing approaches, cell-type-specific analyses, bioinformatic approaches, and neuroepigenomic editing represents an important frontier in elucidating the molecular basis of drug addiction. In the following sections, we synthesize the current literature on transcription factors driving cocaine-induced plasticity in the NAc, discuss new insights into the molecular mechanisms downstream of transcription factor regulation, and delve into the emerging literature using advanced computational methods and neuroepigenomic editing to delineate the function of novel transcription factors important in drug action.
Candidate Transcription Factors Mediating Cocaine-Induced Plasticity
CREB
CREB is a ubiquitously-expressed transcription factor implicated in diverse functions in the nervous system, including neurodevelopment, learning and memory, and neuronal plasticity [55,56]. CREB regulates the expression of several thousand target genes by forming homodimers and binding to cAMP response elements (CREs). It is generally thought that CREB is bound to target genes in its inactive state and, in response to a stimulus, phosphorylation of CREB at serine 133 enables the recruitment of CREB-binding protein (CBP), a histone acetyltransferase, and of numerous other regulatory proteins that ultimately facilitate CREB-mediated transcription. Notably, a recent study challenges this notion by demonstrating that transgenic mice lacking the serine 133 phosphorylation site retain certain CREB functions in the hippocampus [57]. These data suggest that the function of CREB phosphorylation at serine 133 may be context-dependent. Nonetheless, several protein kinases phosphorylate CREB at serine 133, including protein kinase A (PKA), Ca2+/calmodulin-dependent kinase IV (CaMKIV), and ribosomal S6 kinase (RSK), the latter being downstream of extracellular signal-regulated kinase (ERK) signaling [56]. Conversely, CaMKII phosphorylates CREB at serine 143, which inhibits CREB function by dissociating the CREB homodimer and precluding CBP recruitment [58]. An interesting and as yet unanswered question is the upstream signaling pathway (i.e., cAMP, Ca2+/calmodulin, or BDNF-TrkB-ERK-RSK) that mediates cocaine or other drug-induced phosphorylation and activation of CREB in the NAc (Figure 3).
Figure 3: Intracellular signaling pathways in D1 MSNs after chronic cocaine administration.
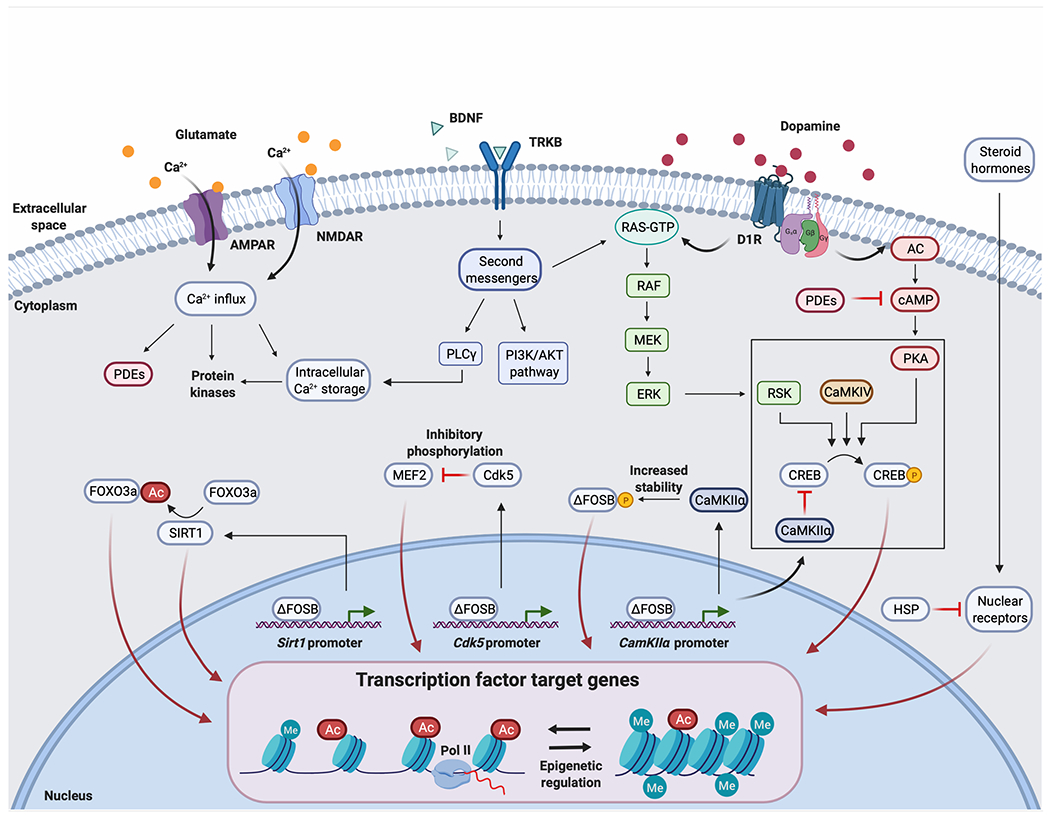
Chronic cocaine potentiates dopaminergic, glutamatergic, and BDNF-TRKB signaling in D1 MSNs in the NAc. Here, we represent several known intracellular signaling pathways that contribute to cocaine-induced regulation of transcription factor activity, but many additional pathways are involved in regulating these and other transcription factors, mechanisms which remain less well understood. Post-translational modifications of transcription factors are important regulators of their activity, and may have an inhibitory or activating effect depending on the type of modification (phosphorylation, acetylation, etc.) and site of modification. Each regulated transcription factor then controls the expression levels of a unique set of target genes. This is illustrated for ΔFOSB, the accumulation of which after chronic cocaine exposure activates or represses the expression of many intracellular signaling molecules, including CDK5, CaMKIIα, and SIRT1, among many others. Each of these targets in turn influences the activity of many additional transcription factors. Delineating the complex web of interactions among intracellular messengers and transcription factors remains an important frontier in understanding the molecular basis of cocaine addiction. The box around CREB regulation denotes the fact that these phosphorylation events occur in the cell nucleus, whereas it remains uncertain whether post-translational modification of the other transcription factors shown occurs in the nucleus or cytoplasm. Note that Ca2+ entry is shown for both AMPARs and NMDARs, although only AMPARs of particular subunit compositions flux Ca2+. Ca2+ can also enter cells through voltage-gated Ca2+ channels (not shown).
CREB was first implicated in drug addiction based on its known role as an effector for cAMP signaling and on evolving evidence that the cAMP pathway is upregulated in several brain regions in response to psychostimulant or opioid drugs of abuse [59]. Chronic cocaine administration was demonstrated subsequently to increase CREB activity within the NAc (Table 1A), with this induction mediating tolerance- and dependence-like effects to cocaine’s behavioral actions (Table 2A). Specifically, transgenic- or viral-mediated overexpression of wildtype or constitutively active CREB (i.e., CREBS133D) in NAc neurons was found to reduce rewarding responses to cocaine and elicit a negative emotional state, whereas overexpression of a dominant negative mutant form of CREB (i.e., CREBS133A) exerted the opposite effect [45,55,60]. CREB induction in NAc neurons was also shown to drive increased cocaine self-administration and relapse presumably through a negative reinforcement mechanism [61], while antisense-mediated knockdown of CREB decreased cocaine self-administration [62]. One study using transgenic mice expressing CREBS133A under the control of the D1 receptor promoter suggests that the tolerance-like effects induced by CREB activation are driven at least in part by D1-expressing neurons [63]. Effects of CREB in D2 MSNs, where it is also induced by drug exposure, have not yet been examined. CREB activation has been shown to play a similar negative reinforcement role with respect to opioid addiction [55,64], although its role for other drugs of abuse is not yet established with certainty.
Table 2:
Summary of studies analyzing behavioral and neuronal plasticity after transcription factor manipulation in NAc neurons (A) or in a cell-type-specific manner (B).
| Transcription factor | Manipulation | Locomotor sensitization | Conditioned place preference | Self-administration | Dendritic spines | Electrophysiology | Reference |
|---|---|---|---|---|---|---|---|
| ΔFOSB | Overexpression | Increased | Increased | Increased FR1 self-admin Left-shift dose response Increased breakpoint |
Increased thin and stubby spines No effect on mushroom spines |
Colby 2003, Kelz 1999, Robison 2013, McClung and Nestler 2003 | |
|
| |||||||
| ΔFOSB | Knockdown | No effect | Decreased | Peakman 2003 | |||
|
| |||||||
| CREB | Overexpression | Decreased | No effect on FR5 self-admin Left-shift dose response Increased breakpoint Increased reinstatement |
Increased silent synapses | Increased excitability, upstate duration, and firing frequency | Brown 2011, Carlezon 1998, Dong 2006, Huang 2008, Larson 2011, McClung and Nestler 2003 | |
|
| |||||||
| CREB | Knockdown | Increased | Increased | No effect on FR5 self-admin Right-shift dose response Decreased breakpoint No effect on reinstatement |
Blocks cocaine-induced silent synapses | Decreased excitability and action potentials per upstate, no effect on duration upstate | Vialou 2012, Larson 2011, Huang 2008, Dong 2006, Choi 2006, Carlezon 1998, Brown 2011 |
|
| |||||||
| NFκB | IKK overexpression | Increased | Increased spine density | Russo et al. 2009 | |||
|
| |||||||
| NFκB | IKK knockdown | Decreased | Decreased spine density | Russo et al. 2009 | |||
|
| |||||||
| MEF2 | MEF2-VP161 | Increased | Increased | Blocks cocaine-induced increase in spine density | Pulipparacharuvil et al. 2008 | ||
|
| |||||||
| MEF2 | MEF2A/2D shRNA | Decreased | Increased spine density | Pulipparacharuvil et al. 2008 | |||
|
| |||||||
| FOXO3a | Overexpression | Increased | Ferguson et al. 2015 | ||||
|
| |||||||
| FOXO3a | Knockdown | No effect | Ferguson et al. 2015 | ||||
|
| |||||||
| E2F3a | Overexpression | Increased | Increased | Cates et al. 2017 | |||
|
| |||||||
| E2F3a | Knockdown | Decreased | Decreased | Cates et al. 2017 | |||
|
| |||||||
| E2F3b | Overexpression | No effect | No effect | Cates et al. 2017 | |||
|
| |||||||
| E2F3b | Knockdown | No effect | No effect | Cates et al. 2017 | |||
|
| |||||||
| NR4A1 | dCas9-VP64 | Decreased | No effect on FR1 self-admin No effect on reinstatement (24 hr WD) Decreased reinstatement (28 d WD) |
Carpenter et al. 2020 | |||
|
| |||||||
| NR4A1 | dCas9-KRAB | Increased | Carpenter et al. 2020 | ||||
|
| |||||||
| ΔFOSB | Overexpression in D1 MSNs | Increased | Increased | Increased spine density | Decreased AMPAR:NMDAR | Gueter et al. 2013 | |
| Overexpression in D2 MSNs | No effect | No effect | No effect on spine density | Increased AMPAR:NMDAR | Gueter et al. 2013 | ||
|
| |||||||
| CREB | D1-A-CREB2 | Increased | Increased | No effect on FR1 self-admin | Bilbao et al. 2014 | ||
|
| |||||||
| EGR3 | Overexpression in D1 MSNs | Increased | Increased | Chandra et al. 2015 | |||
| Overexpression in D2 MSNs | Decreased | Decreased | Increased extinction and decreased reinstatement in females Decreased extinction and increased reinstatement in males |
Chandra et al. 2015, Engeln et al. 2019 |
|||
| Knockdown in D1 MSNs | Decreased | Decreased | Chandra et al. 2015 | ||||
| Knockdown in D2 MSNs | Increased | Increased | Chandra et al. 2015 | ||||
MEF2-VP16 is a constitutively active form of MEF2.
D1-A-CREB represents the expression of a dominant negative mutant of CREB (A-CREB) under the control of the Drd1 promoter.
CREB regulation in other brain regions.
Cocaine-induced regulation of CREB activity is observed in many additional brain regions. Specifically, previous work shows that chronic cocaine regulates the phosphorylation state of CREB in the dorsal striatum, basolateral amygdala, and central amygdala [65–67]. CREB phosphorylation in the CA1 and CA3 regions of the hippocampus remained unaffected. Another study demonstrated that chronic amphetamine administration regulates CREB activity in the olfactory bulb, thalamus, hypothalamus, motor cortex, somatosensory cortex, hippocampus, and amygdala, among other regions [68,69]. Although the functions of CREB in these brain regions have not been investigated in addiction models, one study investigated the effect of manipulating CREB activity in the VTA on cocaine reward [70]. CREB overexpression in the rostral VTA increases the rewarding effects of cocaine, while overexpression of mCREB induces an aversive response. Surprisingly, overexpression of CREB or mCREB in the caudal VTA has the opposite effect on cocaine reward. These data suggest that CREB oppositely regulates drug reward within subregions of the VTA. One possible explanation for this phenomenon is that transduced neurons in the caudal VTA were predominantly dopaminergic, which project to the NAc and other forebrain structures, whereas in the rostral VTA the transduced cells were mostly GABAergic interneurons.
CREB target genes.
Earlier work relied on the use of microarrays to identify genes in NAc that are induced or suppressed upon overexpression of wildtype or dominant-negative mutant CREB [45], or gene promoters that bind endogenous CREB after chronic cocaine administration [71]. Many of the implicated genes suggested an important role for CREB in modulating glutamatergic neurotransmission. CREB facilitates the induction of the NMDA receptor (NMDAR) subunit 2B (NR2B) after chronic cocaine administration, which is necessary for the formation of silent synapses [72,73]. In addition, CREB increases brain-derived neurotrophic factor (BDNF) expression, a neurotrophin that binds to tropomyosin receptor kinase B (TRKB) and regulates intracellular signaling [74]. BDNF-TRKB signaling in NAc promotes cocaine intake [75] and increases expression of the AMPA receptor (AMPAR) subunit, GLUA1, leading to increased expression of Ca2+-permeable AMPARs and to LTP induction at silent synapses [76]. As discussed previously, these molecular adaptations are essential for the development of incubation of craving during drug withdrawal. CREB was also shown to increase the intrinsic excitability of NAc MSNs, which was directly linked to decreased locomotor sensitization to cocaine and which is consistent with the hypothesis that CREB activation promotes drug tolerance and dependence [55,77]. Together, these studies suggest important molecular mechanisms by which CREB regulates behavioral responses to cocaine.
Another CREB target gene is prodynorphin (Pdyn) [60], which encodes an endogenous opioid peptide associated with the dysphoria experienced during withdrawal from drugs of abuse [78]. Pdyn was first considered as a CREB target in addiction models based on its known regulation by CREB in other systems and by the presence of a well-characterized CRE site in its promoter. Chronic cocaine was shown to induce dynorphin expression in NAc [60], an effect selective for D1 MSNs where Pdyn is expressed selectively [79]. Dynorphin is thought to exert its dysphoric effects by engaging in a negative feedback loop between D1 MSNs in the NAc and dopaminergic projections from the VTA [80]. According to this scheme, cocaine increases CREB-mediated dynorphin expression; dynorphin is then released onto DA neurons in the VTA where it activates Gi/o-coupled κ opioid receptors (KORs) and attenuates DA neuron firing. This mechanism is supported by data showing that intra-cerebroventricular administration of a KOR antagonist is sufficient to block the reduction in cocaine reward caused by CREB overexpression [60].
These earlier studies of CREB function and its target genes relied on the use of CREB overexpression and knockdown techniques, which fall short in defining causal relationships between CREB binding at a single target gene and downstream experimental measures. This is because these approaches alter CREB binding at hundreds or thousands of genes, as noted earlier [45]. Recent work from our laboratory developed a clustered regularly interspaced short palindromic repeats (CRISPR)-based neuroepigenomic editing tool that enables targeted recruitment of the constitutively active mutant of CREB (CREBS133D) to a CRE site at a single target gene in a defined cell population in awake, behaving animals (Box 1) [37,47,48]. Targeting CREBS133D to any of several target genes that contain a CRE site induces that gene selectively, an effect not seen using a CREB mutant lacking the serine 133 phosphorylation site (CREBS133A), highlighting the specificity of this approach. Similar tools could be developed for any other transcription factor and used to study its downstream molecular mechanisms, but this has not yet been achieved. The earlier studies of CREB also fall short of defining the cellular specificity of CREB regulation and action, relying on overexpression or knockdown approaches that target all NAc neurons and on analysis of target genes in homogenized NAc tissue. A major need moving forward is greater attention to cell-type-specific analyses to define, for example, CREB targets selectively in D1 and D2 MSNs (Box 2).
Box 2: Cell-Type-Specificity of Transcription Factor Actions.
To date, the majority of studies on transcription factors in the context of cocaine-induced neural plasticity have involved analysis of bulk tissue and viral gene transfer techniques paired with neuron-specific promoters or neurotropic viruses. While these early studies have yielded a great deal of information on transcription factor function, they are inherently limited due to the rich heterogeneity of cell types in the brain and their unique contributions to brain function and pathophysiology. In the NAc, D1 and D2 MSNs differ in their transcriptional profiles [2,3], synaptic inputs and projection targets [4,5], cellular response to drugs of abuse [16], and effects on addiction-like behaviors [6–12]. In addition, local interneuron populations as well as glial and endothelial cells modulate the microcircuitry within the NAc to influence behavior. As a result, studies on homogenized brain tissue or with pan-neuronal gene manipulations are limited with respect to the insight they can provide into the cell-type-specific actions of transcription factors and their target genes.
For example, previous work demonstrated that ΔFOSB overexpression in NAc neurons increases the expression of NFκB—leading researchers to overexpress NFκB in NAc neurons and study the downstream effects on cocaine-induced plasticity and behavioral phenotypes [113,133]. However, NFκB is expressed predominantly in microglia and endothelial cells. There is no doubt that NFκB signaling in neurons influences neuronal plasticity, but it remains unclear if the cocaine-mediated induction of NFκB occurs in a subset of NAc neurons or rather in microglia or endothelial cells, suggesting that NFκB overexpression in NAc neurons may not accurately portray the functions of drug-induced NFκB.
Notably, there are some advantages to conducting initial analyses on bulk tissue rather than isolating specific cell types. A study from our laboratory showed that overexpression of zinc finger protein 189 (ZFP189), a CREB-regulated putative transcription factor, in PFC neurons dramatically alters the expression of genes in other cell types, especially endothelial cells [48]. By focusing on a single cell population, this finding would likely have been overlooked. Therefore, bulk analyses and cell-type-specific studies are often complementary, but to date the majority of studies on transcription factors in cocaine-induced plasticity in the NAc have not included cell-type-specific analyses.
Fortunately, there is now a growing toolbox for studying transcription factors in a cell-type-specific manner within a defined brain region of awake, behaving rodents. Several methods are available to isolate specific cell types from brain tissue for downstream analyses, including fluorescence-activated cell sorting (FACS), fluorescence-activated nuclei sorting (FANS), magnetic-activated cell sorting (MACS), and RiboTag affinity purification. Work from our laboratory has applied some of these techniques to study D1 vs. D2 MSN subtypes in the NAc [2,171]. Coupling these methods with RNA-seq, ChIP-seq, ATAC-seq, and other approaches can be leveraged to study the cell-type-specific effects of transcription factor manipulations on the transcriptome, genome-wide transcription factor binding, and chromatin accessibility, respectively. A major technical challenge for the field is improving experimental protocols to lower the amount of required input tissue for these protocols, which is very limited for samples of rodent brain tissue after microdissection and cell sorting. As mentioned in the main text, CUT&RUN is an alternative method to ChIP-seq that is amenable to lower input samples [38]. In addition, viral-mediated gene transfer methods are available to manipulate transcription factors in an inducible manner and selectively in many types of neurons or glia [172]. An important avenue for the field moving forward is to leverage these technologies to study transcription factors in a cell-type-specific manner, which will ultimately lead to an improved understanding of the molecular changes in the brain that drive maladaptive, addiction-like behaviors and elucidate potential therapeutic targets for cocaine use disorder.
ΔFOSB
ΔFOSB is a truncated variant of the full-length FOSB protein, generated by alternative splicing, which has been shown to be an important component of the transcriptional and behavioral responses to drugs of abuse [31,81]. The FOS family of transcription factors (cFOS, FOSB, FRA1, and FRA2) form heterodimers with members of the JUN family (cJUN, JUNB, and JUND) to form activator protein 1 (AP1) complexes that regulate transcription at their respective target genes. ΔFOSB was first identified as being the only FOS or JUN family transcription factor that is induced in NAc in response to chronic cocaine exposure [82]. Since this initial demonstration, ΔFOSB has been demonstrated to be induced in NAc by several classes of drugs of abuse (Table 1A) [83], and this induction is selective for D1 MSNs except for opioids which induce the protein equally in D2 MSNs as well (Table 1B) [84]. Elevated ΔFOSB protein levels are also observed in the NAc of human patients with cocaine or opioid addiction [85,86]. Drug induction of ΔFOSB is highly complex, with four transcription factors implicated: CREB, EGR3, serum response factor (SRF), and E2 factor 3a (E2F3a) (Figure 4) [87–89]. All four factors bind to response elements within a small upstream promoter region and there is evidence that they work cooperatively to induce the FosB gene. Additionally, several chromatin mechanisms have been identified to occur in concert with FosB gene induction, including dynamic regulation of histone acetylation and methylation [28,90–93] and DNA methylation [94]. In fact, the FosB gene is one of the most robustly primed genes after prolonged withdrawal from cocaine; identifying the “chromatin scars” that persist at FosB during withdrawal to mediate this priming is a high priority of current research. There is no evidence for cocaine regulation of FosB splicing. Rather, the progressive accumulation of ΔFOSB protein, in the absence of accumulation of full-length FOSB, observed during chronic drug administration is attributed to its unique stability. ΔFOSB lacks the degron domains present in full-length FOSB and all other FOS family proteins, which reduce its targeting for proteasomal and other proteolytic degradation [95]. The half-life of ΔFOSB is increased further by its phosphorylation at serine 27, and perhaps other residues, by casein kinase 2 and CaMKII (Figure 3) [85,96].
Figure 4: Postulated mechanisms regulating FosB gene expression after cocaine administration.
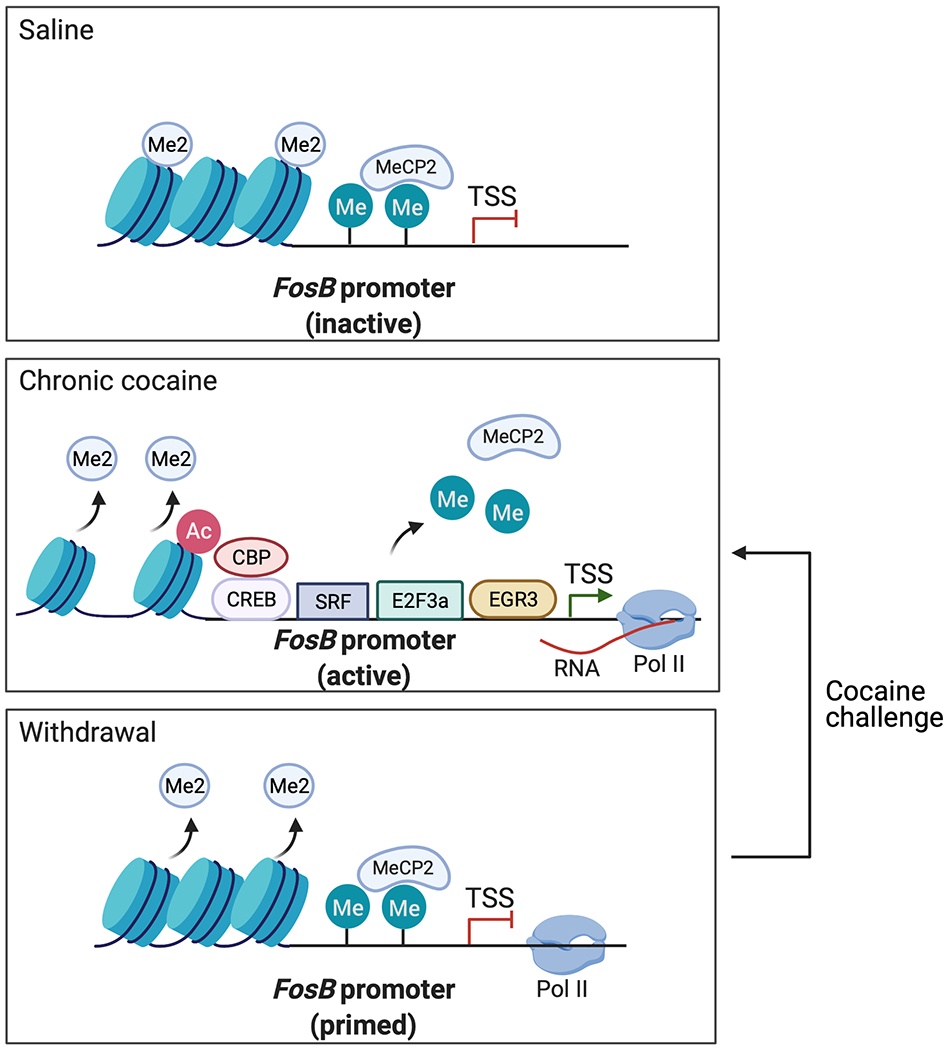
Chronic cocaine exposure promotes the recruitment of four known transcription factors, permissive epigenetic modifications, and RNA Pol II to the FosB locus, in concert with the removal of repressive histone dimethylation (Me2) and repressive DNA methylation and MeCP2 binding. After prolonged withdrawal, the FosB locus remains in a “primed” state, as indicated by persisting, long-lived removal of repressive histone methylation marks and stalling of RNA Pol II near the gene’s transcription start site (TSS). Cocaine re-exposure after prolonged withdrawal is thought to revert the FosB locus to the activated state, contributing further to the accumulation of ΔFOSB protein in NAc D1 MSNs.
Experiments that overexpress ΔFOSB, or a dominant negative mutant of a JUN protein (ΔcJUN or ΔJUND) that antagonizes AP1-mediated transcription, in NAc neurons—by use of inducible genetic mutant mice or viral-mediated gene transfer—indicate that drug-induced accumulation of ΔFOSB in D1 MSNs increases an animal’s sensitivity to the rewarding effects of cocaine and other drugs of abuse, increases cocaine self-administration, and promotes relapse, presumably through positive reinforcement (Table 2B) [31,85,97–101]. The functional consequences of ΔFOSB induction in D2 MSNs, which occurs in response to opioid drugs alone or to chronic consumption of natural rewards [84], remains a subject of active investigation. More recent work, utilizing neuroepigenome editing tools to control the expression levels of endogenous ΔFOSB and thereby avoid the confounds of conventional overexpression approaches (Box 2), have validated some of these findings [102]. In any event, the unique temporal features of ΔFOSB—as a stable product of an immediate early gene, along with its role in promoting drug reward and intake, support the notion that it serves as a “molecular switch” towards an addiction-like state [103].
ΔFOSB regulation in other brain regions.
While the majority of studies on ΔFOSB in addiction models have focused on the NAc, drugs of abuse induce ΔFOSB expression in many other brain regions [83]. For example, chronic cocaine elevates ΔFOSB expression in the PFC, orbitofrontal cortex (OFC), dorsal striatum, hippocampus, hypothalamus, amygdala, and VTA [83,104,105]. Among these brain regions, the functions of drug-induced ΔFOSB are most well-characterized in the OFC and hippocampus.
Cocaine self-administration increases ΔFOSB expression in the OFC [106], which is associated with increased impulsivity, cocaine intake, and locomotor response to cocaine [107,108]. An interesting difference between cocaine induction of ΔFOSB in OFC vs. NAc is that drug self-administration causes much greater ΔFOSB induction in OFC compared to forced-drug administration, whereas in NAc equivalent induction is seen with volitional and non-volitional intake. In any event, these results show that ΔFOSB activation in the NAc and OFC increases addiction-like behaviors [99,107,108], suggesting that pharmacological inhibition of ΔFOSB may be an effective treatment for cocaine use disorder (see Transcription Factors as Therapeutic Targets for Cocaine Use Disorder). Microarray analyses point toward many putative ΔFOSB target genes in the OFC that could mediate these behavioral effects, including those encoding mGLUR5, GABAA receptor subunits, and substance P [106].
In the hippocampus, ΔFOSB is induced by cocaine and involved in learning and memory [109]. By contrast, human patients with cocaine addiction show decreased levels of FOSB and ΔFOSB, as well as several ΔFOSB target genes, in the hippocampus relative to matched control subjects [110]. Knockdown of ΔFOSB-mediated transcription using ΔJUND in this brain region is associated with decreased cocaine CPP, possibly by impairing the paired context association rather than reducing the reward salience of cocaine [105].
ΔFOSB target genes.
As with CREB, earlier work used microarray techniques, in combination with ΔFOSB or mutant cJUN/JUND overexpression in NAc neurons, to identify ΔFOSB target genes [45,71]. These studies suggest that ΔFOSB facilitates the cocaine-mediated induction of the GLUA2 AMPAR subunit in NAc MSNs [111]. Increased GLUA2 expression would be expected to reduce the number of Ca2+-permeable AMPARs and thus decrease MSN excitability, which is observed after ΔFOSB overexpression in D1 MSNs [100]. This finding is consistent with evidence that ΔFOSB induction is necessary and sufficient for the formation of immature spines in D1 MSNs in response to chronic cocaine exposure [28,85,100], which as stated above are a postulated required substrate for longer-term drug-induced behavioral plasticity. However, the range of target genes through which ΔFOSB controls spine growth and morphology remains mostly unknown.
In parallel, microarray data pointed to hundreds of genes potentially being influenced by ΔFOSB [45,71]. Some interesting examples include cyclin-dependent kinase 5 (CDK5), NFκB, CaMKII, G9a (a repressive histone methyltransferase), and sirtuin 1 (SIRT1), all subsequently validated as bona fide targets that control neural and behavioral responses to cocaine [28,85,112–114]. ΔFOSB was also shown to suppress the induction of cFos in the NAc by recruiting histone deacetylase 1 (HDAC1) to the cFos promoter [115], which could contribute to the molecular switch—selective induction of ΔFOSB over other FOS family proteins—in the chronic drug-treated state. HDACs catalyze the removal of acetyl groups from histones and other proteins.
Recent work from our laboratory seeks to capture more physiologically-relevant target genes for ΔFOSB in a more comprehensive fashion by utilizing CRISPR-mediated, locus-specific neuroepigenomic editing to induce endogenous ΔFOSB selectively in D1 MSNs, D2 MSNs, or both cell types and identify all regulated transcripts using RNA-seq [37]. This approach takes advantage of the tool, outlined above, that targets CREBS133D to a single target—in this case FosB—and characterizes the downstream consequences. This study demonstrated partly distinct gene targets for ΔFOSB in D1 vs. D2 MSNs, with large differences in gene targets seen in each cell for male vs. female mice as well. A major need for the field is to now overlap this RNA-seq approach with direct measures of endogenous ΔFOSB binding to its genomic targets genome-wide. Such measures have proven challenging technically using ChIP-seq but appear more feasible with CUT&RUN given the latter method’s greater sensitivity and selectivity [116].
Among the aforementioned gene targets for ΔFOSB are several that themselves control gene expression (e.g., those encoding cFOS, NFκB, G9a, and SIRT1), which underscores the complexity of transcriptional regulation (Figure 3). For example, cocaine and morphine increase ΔFOSB binding at the Sirt1 gene promoter, a class III HDAC, in the NAc, which is associated with increased Sirt1 gene expression in this brain region [112]. Pharmacological activation or overexpression of SIRT1 in NAc neurons increases cocaine-induced reward, locomotor sensitization, and dendritic spine density [71,112,117]. The target genes of SIRT1 were identified using ChIP-seq. This work revealed the forkhead transcription factor (FOXO) family as important SIRT1 target genes [117], which we will describe later in this review.
While this review focuses on NAc MSNs, it is worth highlighting that local interneuron populations also regulate the behavioral response to cocaine and influence neuronal plasticity at NAc MSNs, but the cocaine-induced transcriptional mechanisms within these cell populations remain poorly understood [118–121]. One study investigated such mechanisms in the somatostatin-expressing interneurons within the NAc by RNA-seq and deduced AP1-mediated transcription as a major upstream regulator of cocaine’s effects on gene expression in this cell type. In particular, repeated cocaine selectively induced JUND, without induction of any other FOS or JUN family member [121]. Viral-mediated overexpression of a dominant negative mutant of JUND selectively in this cell type of NAc was shown subsequently to reduce locomotor responses to cocaine. This finding underscores a major conclusion of this review, which is the need for more cell-type-specific investigations of transcription factor action in drug abuse models (Box 2).
EGR3
The EGR family of transcription factors (EGR1-4) are products of immediate early genes that are implicated in synaptic and behavioral plasticity. This involvement prompted their study in addiction models where EGR3 was found to be induced in the NAc by chronic cocaine administration [122]. In this brain region, EGR3 is responsive to intracellular pathways downstream of DA and BDNF signaling, both of which are elevated by cocaine [89]. Subsequent investigation demonstrated that chronic cocaine increases EGR3 expression in D1 MSNs but reduces it in D2 MSNs (Table 1B) [89]. The regulation observed in D1 MSNs may be mediated by ΔFOSB, which preferentially accumulates in D1 MSNs and exhibits increased binding at the Egr3 promoter following chronic cocaine treatment [71], although direct functional validation of EGR3 regulation by ΔFOSB has not yet been demonstrated. EGR3 overexpression in D1 MSNs in the NAc increases cocaine reward and locomotor sensitization, while overexpression in D2 MSNs has the opposite effect (Table 2B) [89]. Conversely, miRNA-mediated knockdown of EGR3 in D1 or in D2 MSNs reverses these behavioral phenotypes. Interestingly, EGR3 overexpression in D2 MSNs in the NAc delays extinction and promotes relapse-like behavior in male mice, whereas this manipulation increases extinction and attenuates relapse-like behavior in female mice [123]. This latter finding highlights a major need in the field, namely, to examine sex differences in transcriptional regulation in drug abuse models, something that has received relatively little attention to date. The data also indicate cell-type-specific actions of EGR3 in the NAc, further emphasizing the importance of greater focus on this variable in future studies of transcription factors (Box 2).
EGR3 target genes.
Multiple synaptic plasticity-related proteins, transcription factors, and chromatin-modifying enzymes implicated in cocaine-induced plasticity exhibit altered EGR3 binding in the NAc after chronic cocaine administration [89,124]. Specifically, chronic cocaine increases EGR3 binding at the promoters of genes encoding CaMKIIα, CREB, FOSB/ΔFOSB, SIRT1, nuclear receptor subfamily 4 group A member 2 (NR4A2), and peroxisome proliferator-activated receptor gamma coactivator-1α (PGC1α). These same genes show increased expression in D1 MSNs following repeated cocaine administration. The promoter region of the gene encoding PGC1α, a transcriptional coactivator, contains binding sites for multiple transcription factors discussed in this review, including CREB, ΔFOSB, NFκB, EGR3, and MEF2, which underscores the complexity and cooperative nature of transcriptional regulation [124]. PGC1α overexpression in D1 MSNs in the NAc elevates cocaine reward and locomotor sensitization, while overexpression in D2 MSNs has the opposite effect [124].
By contrast, cocaine administration decreases EGR3 binding at the promoters of G9a and DNA methyltransferase 3a (DNMT3a), which is associated with reduced expression of these target genes in D1 MSNs [89]. G9a is also a target for ΔFOSB which, as noted earlier, has been shown to robustly control cocaine-elicited plasticity [28,125,126]. Likewise, DNMT3a expression in the NAc is linked to neuronal and behavioral plasticity after chronic treatment with cocaine. DNMT3a expression is reduced in this region during the early stages of withdrawal from chronic cocaine, possibly due to reduced EGR3 binding, but then becomes elevated after prolonged withdrawal from the drug [89,127]. DNMT3a overexpression in NAc neurons decreases the rewarding effects of cocaine, but increases dendritic spine density [127]. Interestingly, one study suggests that a specific DNMT3a isoform, termed DNMT3a2, is elevated after cocaine self-administration and regulates cue-induced reinstatement [128].
Another line of evidence suggests that EGR3 regulates physiological responses to chronic cocaine administration by regulating energy balance in NAc MSNs [129]. Chronic cocaine alters EGR3 binding and gene expression of multiple genes involved in mitochondrial function including those encoding dynamin-related protein 1 (Drp1), nuclear respiratory factor 2 (Nrf2), and DNA polymerase gamma subunit (Polg). Further, miRNA-mediated knockdown of EGR3 in the NAc blocks the cocaine-induced increase in small mitochondria in D1 MSNs. However, the consequences of mitochondrial regulation by chronic cocaine exposure on NAc MSN function remain poorly understood.
NFκB
NFκB is a transcription factor classically studied in the context of the immune system and inflammation, but more recent studies point to a role for NFκB in drug-induced synaptic plasticity [113,130–132]. NFκB transcription factors are composed of dimerized subunits (RELA/p65, RELB, cREL, p50, and p52) and bind to specific response elements called κB sites. The first indication that NFκB might be implicated in cocaine action came from a microarray study which demonstrated induction of an NFκB subunit in NAc upon overexpression of ΔFOSB [133]. Chronic cocaine administration was subsequently shown to upregulate the expression of the p65 and p50 subunits in the NAc, which is associated with increased permissive histone marks (Table 1) [71,113,133]. Expression of a constitutively active inhibitory-κB kinase (IKKca), which activates NFκB, in NAc neurons increases cocaine reward and dendritic spine density of NAc MSNs (Table 2A) [113].
NFκB target genes.
Several target genes for NFκB have been identified in the context of addiction models. One such target is Bdnf, which is discussed earlier in this review [74]. Other putative targets revealed by motif analysis are genes encoding the JUN family of transcription factors [134]. As discussed previously, JUN proteins heterodimerize with ΔFOSB or other FOS family proteins to generate AP1 transcription factors, which suggests that NFκB may indirectly regulate ΔFOSB-mediated transcription via interactions with JUN proteins. In addition, several NFκB target genes converge on the endogenous opioid system. NFκB regulates genes that encode the δ opioid receptor (DOR), μ opioid receptor (MOR), and dynorphin, the primary ligand for KORs. The role of each of these receptor subtypes in addiction pathophysiology has been reviewed in detail elsewhere [78].
Studies of NFκB in addiction models underscore a major weakness of the earlier literature: most NFκB subunits in brain are highly enriched in microglia and endothelial cells [135], while efforts to date to manipulate their expression in adult brain in addiction models used neurotropic viruses. NFκB signaling no doubt functions in neurons, but these observations emphasize the importance of now better defining the cell-type-specificity of transcription factor regulation by a drug of abuse within the NAc or another brain region, and to then study the causal consequences of this regulation by manipulating the factor selectively within that one cell type. Such studies are becoming increasingly feasible with advances in cell-targeting approaches (Box 2).
MEF2
The myocyte-enhancing factor 2 family of transcription factors (MEF2A-D) have been shown to play a role in regulating neuronal survival, actin remodeling, and synaptic plasticity [136–138]. Genome-wide analysis of putative MEF2 target genes point to multiple genes implicated in structural plasticity, including N-WASP, WAVE3, Homer1a, ARC, and protocadherins [136]. Experiments in cell culture suggest that MEF2 also targets several transcription factors discussed elsewhere in this review, such as FOSB, EGR3, and nuclear receptor 77 (NUR77; also known as NR4A1) [139]. Based on this knowledge, work from our laboratory interrogated whether MEF2 transcription factors might be regulated in NAc by cocaine (Table 1A). We found that acute and chronic cocaine administration increases inhibitory phosphorylation of MEF2A/2D in the NAc and, by use of viral manipulations of MEF2 levels in NAc neurons, that this suppression of MEF2A/2D activity is required for cocaine’s induction of dendritic spine density of NAc MSNs. Expressing a constitutively active form of MEF2 (MEF2-VP16) was shown to elevate cocaine-induced locomotor sensitization and reward (Table 2A) [136].
CDK5 is an important regulator of MEF2 transcription factors. As noted earlier, after cocaine administration, ΔFOSB promotes transcription of CDK5 in NAc, which catalyzes inhibitory phosphorylation of MEF2 proteins [136]. This once again underscores the complex web of interactions among transcription factors and their regulatory proteins. Blocking CDK5 activity in NAc is associated with reduced spinogenesis, but with increased cocaine place conditioning and locomotor sensitization [114,140–142]. Therefore, reduced CDK5 activity, which releases the brake on MEF2-dependent transcription, elicits similar phenotypes as MEF2 overexpression. Together, these studies suggest that cocaine potentiates CDK5 activity via induction by ΔFOSB, which then attenuates MEF2 activity to further facilitate cellular and behavioral plasticity to cocaine (Figure 3).
Additional Transcription Factors Driving Cocaine-Induced Plasticity From Unbiased Sequencing Datasets
Bioinformatic-centric approaches to analyze sequencing datasets
The ever-increasing use of RNA-seq and other next-generation sequencing approaches in neuroscience laboratories has driven novel bioinformatic efforts to optimally utilize these vast datasets to derive meaningful biological insight.
Integrating next-generation sequencing and bioinformatics is particularly relevant for elucidating transcription factor function, as many transcription factors have known motifs (response elements) within regulatory regions of their target genes [26]. One such bioinformatic approach, known as hypergeometric optimization of motif enrichment (HOMER), scans the promoter and other regions of differentially expressed genes (DEGs) for the enrichment of known transcription factor motifs [143]. Recent work from our laboratory used HOMER to identify transcription factors in NAc and several other brain reward regions associated with addiction-like behavior after prolonged withdrawal ± relapse from cocaine self-administration, many of which had not been previously linked to cocaine addiction [27].
Another bioinformatic approach involves transcriptomic network analysis, such as weighted gene co-expression network analysis (WGCNA) and multiscale embedded gene expression network analysis (MEGENA) [144,145]. Here, expressed RNAs are subdivided into distinct modules, a collection of genes exhibiting coordinated co-expression across conditions within a given dataset, with each module containing hub genes, or predicted drivers of other genes in the same module. By searching for modules enriched in DEGs after cocaine self-administration, or genes whose expression is correlated with addiction-like behaviors, researchers can deduce those genes that are most important in driving the molecular pathology of cocaine addiction. These methods have revealed novel molecular mechanisms underlying brain pathology in Parkinson’s disease [146], Alzheimer’s disease [147,148], and depression [48,149,150], and are now being applied in addiction research [151].
Below, we discuss recent work that has employed unbiased bioinformatic tools to identify previously unexplored transcription factors driving addiction-like behaviors. These efforts are providing compelling evidence that bioinformatic-centric approaches to analyzing large sequencing datasets can reveal novel biological insight into addiction pathophysiology. At the same time, we should note that the semantic distinction between candidate factors and deduced unbiased factors becomes arbitrary since the initial focus on NFκB in addiction models was based on an open-ended microarray analysis of ΔFOSB targets. It is likewise reassuring that the more recent unbiased analyses of upstream transcriptional regulators of cocaine-induced gene expression alterations implicate CREB, AP1 (FOS and JUN family proteins), and EGR family transcription factors [27], indicating that the earlier hunches of their involvement were indeed well placed.
FOXO3a
Our laboratory paired ChIP-seq with de novo motif analysis and demonstrated that the FOXO transcription factor family motif is enriched in genes bound by SIRT1 in the NAc after chronic cocaine administration [117]. Based on this insight, we went on to demonstrate that cocaine increases the expression of FOXO3a, the FOXO family member most ubiquitously expressed in the brain, as well as known FOXO3a target genes in the NAc [117]. However, other FOXO family members are expressed in the NAc and their role in cocaine-induced plasticity remains unexplored [152]. Viral-mediated FOXO3a overexpression in NAc neurons, like SIRT1 overexpression, potentiates the rewarding effects of cocaine [112,117]. Based on data demonstrating that cocaine induction of SIRT1 in NAc is mediated by ΔFOSB [71,112], these results suggest that FOXO3a may be part of a larger ΔFOSB-mediated network of transcriptional regulation (Figure 3).
SIRT1 overexpression in the NAc reduces acetylation of FOXO3a, possibly through direct deacetylation by SIRT1, which increases the transcriptional activity of FOXO3a (Figure 3). Decreased genomic binding of SIRT1 was also associated with elevated histone 4 lysine 15 acetylation (H4K15ac) in the NAc after chronic cocaine administration. Notably, SIRT1 itself does not bind DNA in a sequence-specific manner, but rather is recruited to specific genes through interactions with DNA-binding proteins, such as transcription factors. These data suggest that SIRT1 influences cocaine-induced plasticity in the NAc through two distinct mechanisms: activation of FOXO3a-dependent transcriptional programs through deacetylation of FOXO3a and regulation of H4K15ac levels. Further studies are needed to identify key target genes of FOXO3a in the context of cocaine exposure, which represents an interesting focus of future research given the implication of FOXO-mediated transcription in a wide range of nervous system functions.
E2F3a
Transcriptome analysis across the brain’s reward circuitry after cocaine self-administration predicted the E2F family of transcription factors as key upstream regulators of genes whose altered expression by chronic cocaine or cocaine withdrawal is associated with addiction-like behavior in the NAc [27]. The E2F family was similarly implicated in a study that integrated ChIP-seq data from NAc for several prominent histone modifications after chronic cocaine administration, which suggested a role for E2F proteins not only in overall transcriptional regulation by cocaine but in cocaine regulation of alternative splicing as well [153]. These are striking findings in that E2F proteins had not previously been implicated in addiction-related phenomena and barely studied at all in neuronal function, yet the proteins are highly expressed in neurons.
The highest expressed E2F family member in NAc is E2F3, which undergoes alternative splicing to produce E2F3a and E2F3b isoforms that, based on work in other tissues, serve as transcriptional activators or repressors [154]. Animals treated with chronic cocaine show elevated expression of E2F3a exclusively in D1 MSNs, while E2F3b remains unchanged (Table 1) [36,88]. Viral-mediated overexpression of E2F3a in NAc neurons increased cocaine-induced locomotor sensitization and reward, while E2F3a knockdown had the opposite effect (Table 2A), with manipulation of E2F3b having no effect on cocaine responses [36].
At the transcriptional level, E2F3a overexpression in NAc neurons was sufficient to recapitulate a large subset of the changes in overall gene expression observed by RNA-seq after chronic cocaine administration, as well as changes in alternative splicing seen in response to the drug [36]. Fgfr1, Tle2, and Ptbp1 are examples of genes regulated similarly by cocaine and E2F3a overexpression in this brain region. Together, these findings support an important role for E2F3a in mediating behavioral and transcriptional plasticity induced by chronic cocaine administration.
One mechanism by which E2F3a facilitates cocaine-elicited plasticity might be through its induction of ΔFOSB. The FosB gene contains an E2F binding site ~500 base pairs upstream of the transcription start site (TSS) and the binding of E2F3, but not of other E2F family members, is increased at this site of the FosB gene in NAc after chronic cocaine. Further, E2F3a overexpression in this brain region increases FosB and ΔFosB mRNA expression, while E2F3a knockdown partially blocks the induction of ΔFOSB by cocaine [88], consistent with the involvement of several other transcription factors as stated earlier. These data support the scheme that E2F3a is an upstream regulator of ΔFOSB in NAc, a further example of the inter-connected web of cocaine-elicited transcriptional mechanisms (Figure 3). Of note, cocaine regulates E2F3b, not E2F3a, in the PFC, where E2F3b exerts very different transcriptional and behavioral effects as seen for E2F3a in NAc [155]. These observations highlight the importance of characterizing transcriptional mechanisms of drug addiction in many other brain reward regions beyond the NAc.
Nuclear receptors
In the same transcriptomic analysis from animals self-administering cocaine and examined at different withdrawal time points ± relapse, numerous nuclear receptors were identified as upstream regulators of genes associated with addiction-like behaviors [27]. Most of these nuclear receptors have not previously been linked to addiction-related phenomena. One such nuclear receptor is retinoic X receptor alpha (RXRα, also known as NR2B1), an isotype of the RXR family of nuclear receptors. RXRα is activated by retinoic acid and regulates the expression of its target genes as a homo- or heterodimer. RXRα provides a particularly powerful test of bioinformatic predictions because, unlike the other transcription factors discussed in this review, its own transcription in NAc is not affected by cocaine when measured at the bulk tissue level. Nonetheless, based on its deduction as an important upstream regulator, we proceeded to study RXRα in addiction models. We showed that RXRα is a potent, positive regulator of cocaine reward in NAc of both female and male rodents, and that overexpressing RXRα in neurons of this brain region modulates many of the same genes that are altered by cocaine self-administration [156]. While RXRα is expressed predominantly in microglia [135], it is also expressed in NAc interneurons [3]. Ongoing work on this nuclear receptor seeks to define whether its regulation of cocaine responses occurs via microglia or this neuronal cell type, or both, and to then identify the target genes mediating cocaine-induced neuroplasticity.
A recent study from Carpenter and colleagues identified NR4A1 (NUR77), as a predicted mediator of cocaine-induced neuroplasticity using RNA-seq following cocaine self-administration plus early or late withdrawal [157]. Chronic cocaine administration transiently increases NR4A1 expression, but the expression returns to baseline after extended withdrawal. However, there is an enrichment of permissive histone marks (H3K27ac and H3K4me3) at the gene after extended withdrawal, suggesting that Nr4a1 remains primed for reactivation by cocaine. This study employed CRISPR activation and interference (CRISPRa/i) tools to bidirectionally regulate endogenous NR4A1 expression in the NAc; these tools are discussed in greater detail elsewhere in this review (Box 1). CRISPR-mediated activation of NR4A1 in the NAc attenuates cocaine reward and reinstatement after prolonged withdrawal, while CRISPR-mediated knockdown increases cocaine reward (Table 2A).
In addition, pharmacological activation of NR4A1, using peripheral administration of cytosporone-B (Csn-B), decreases cocaine reward, thus replicating the effect of CRISPR-mediated activation of NR4A1 in the NAc on cocaine reward. This study provides promising preliminary evidence that NR4A1 warrants further investigation as a possible treatment for cocaine use disorder. These data are further evidence that unbiased next-generation sequencing approaches are useful for understanding the molecular basis of drug addiction and that this work can be leveraged to develop novel treatments for drug addiction.
Transcription Factors as Therapeutic Targets for Cocaine Use Disorder
Research over the last decade suggests that transcription factors are viable targets for therapeutic intervention in human disease. Multiple approved medications are agonists or antagonists of glucocorticoid, mineralocorticoid, or gonadal steroid hormone receptors, all of which are transcription factors. Another class of compounds, called thiazolidinediones, are used to treat type II diabetes by modulating the peroxisome proliferators activated receptor (PPAR) nuclear receptor family [158]. Additionally, a selective inhibitor of AP1 transcription factors (T-5224) is under investigation for the treatment of rheumatoid arthritis [159], cancer [160], and intravertebral disc degeneration [161], among others. This compound selectively disrupts DNA binding of AP1 complexes without affecting the activity of other transcription factors. Applying a similar approach to disrupt dimerization or the DNA binding properties of additional transcription factors may present a novel treatment avenue for drug addiction.
Identifying small molecule regulators of transcription factors for the treatment of cocaine use disorder represents a major challenge for the field. These compounds must be able to cross the blood-brain barrier and achieve sustained bioavailability in target brain structures to regulate transcription factor function without inducing peripheral toxicity or detrimental off-target interactions. In addition, many transcription factors, such as CREB, are ubiquitously expressed throughout the brain and peripheral organs, suggesting that small molecule regulators of these transcription factors would disrupt normal physiological function. By contrast, growing evidence suggests that small molecule inhibitors of ΔFOSB may be effective treatments for cocaine use disorder because ΔFOSB is highly enriched in striatal regions, ΔFOSB overexpression in the NAc or OFC increases addiction-like behavior in preclinical models of drug addiction [99,107,108], elevated ΔFOSB expression is observed in the NAc of human patients with cocaine addiction [85], and the long-term stability of ΔFOSB is thought to contribute to heightened vulnerability to relapse after prolonged withdrawal from drugs of abuse [103]. However, evaluating the therapeutic efficacy of ΔFOSB inhibitors for cocaine addiction will require careful investigation because ΔFOSB is also involved in important physiological functions such as learning and memory [109], resilience to chronic stress [162], motivation for natural rewards [163] and, in the periphery, osteoblast and adipocyte function [164]. To date, several compounds have been identified that disrupt the DNA binding of ΔFOSB [165,166], but studies investigating the efficacy of optimized ΔFOSB inhibitors in preclinical models of drug addiction are still needed. The ongoing efforts to develop small molecule regulators of transcription factors demonstrate that, beyond identifying innumerable targets for these transcriptional regulators that could serve as the basis for new addiction treatments, transcription factors themselves should be seen as a potential focus for therapeutic discovery for drug addiction.
Concluding Remarks
Cocaine and other psychostimulants increase neurotransmission at dopaminergic synapses, leading to altered intracellular signaling on postsynaptic neurons, such as NAc MSNs. Transcription factors respond to these intracellular messengers—being activated or inactivated in the cytoplasm and transported into the nucleus or having upstream protein kinases transported into the nucleus where they activate resident transcription factors. In the nucleus, transcription factors bind to specific response elements across the genome, within genic regions and at distant enhancers, to regulate the expression of hundreds or thousands of genes by recruitment of RNA polymerase II and its associated transcriptional machinery or, conversely, proteins that suppress gene expression (Figure 1). Drug-induced regulation of transcription factors, including alterations in their activation by upstream protein kinases or in their own transcription, mediates lasting changes in the steady-state expression, as well as in the expression potential (priming/desensitization), of a host of target genes which then underlie many of the maladaptations observed in the brain’s reward circuitry that drive drug addiction.
The new tools of locus-specific neuroepigenomic editing are improving the level of proof that can now be obtained to directly link a transcription factor to its downstream functional—transcriptional, cellular, synaptic, and ultimately circuit—consequences (Box 1). This work is most advanced for cocaine, with similar approaches now required to better understand transcriptional responses to opioids and several other classes of abused drugs. We focused in this review on NAc, which is by far the most studied brain region, but parallel studies are needed for many other reward regions also implicated in drug addiction.
Despite the progress made to date in studying a small handful of cocaine-regulated transcription factors, advanced bioinformatic analyses of genome-wide datasets are identifying large numbers of additional transcription factors that are predicted to be key upstream regulators of differentially regulated genes after cocaine self-administration and withdrawal that remain unexplored in addiction models [27,157]. Further, the detailed molecular mechanisms that link receptor actions of drugs of abuse to downstream regulation of transcription factors remain in general poorly understood (Figure 3). Mechanisms mediating the induction of ΔFOSB in NAc by cocaine illustrate that such upstream pathways are highly complex and therefore especially difficult to delineate in vivo (Figure 4). Yet a focus on in vivo models is essential because it is well known that regulatory events that occur in vivo are often not captured in cultured neurons.
Likewise, a great deal more needs to be learned about the detailed changes in chromatin architecture at downstream target genes that work in concert with transcription factors to mediate gene regulation. For example, most transcription factors activate some target genes, but suppress others, and little is known about the mechanisms that determine such actions. There remains a paucity of studies examining transcription factor binding in discrete regions of brain, let alone in specific cell types in those regions, with ChIP-seq lacking the sensitivity to produce high quality datasets for most transcription factors in microdissected brain regions like the NAc. By contrast, alternative approaches, such as CUT&RUN, offer far greater sensitivity and are showing potential for providing such crucial data, as found recently for mapping cocaine-induced binding of endogenous ΔFosB genome-wide [116]. Combinatorial analysis of such data with a host of other chromatin measures, such as ChIP-seq for histone modifications, whole genome bisulfite sequencing to quantify DNA methylation, or HiC to map the 3D structure of chromatin, will help address these outstanding questions. Combinatorial approaches are required as well to understand how cocaine regulation of multiple transcription factors, occurring simultaneously within the same cell type (e.g., D1 MSNs), is integrated to achieve the ultimate time-dependent effects of cocaine on gene expression. Moreover, it is increasingly essential to carry out such studies in a cell-type-specific manner since it is known from other tissues, and from early work in brain as outlined in this review, that the target genes of a given transcription factor differ dramatically from one cell type to another (Box 2).
Another challenge is to better understand the stimulus specificity that has been observed for some transcription factor actions. It was stated earlier that ΔFOSB mediates the ability of chronic cocaine and of chronic morphine to induce Sirt1 in NAc [112]. By contrast, Sirt2, another sirtuin isoform, is also induced in NAc by cocaine via ΔFOSB, whereas it is not induced by morphine even though both drugs induce ΔFOSB to the same extent in D1 MSNs. What mediates this stimulus specificity? Do the drugs induce ΔFOSB in distinct subsets of D1 MSNs? Are there other transcriptional changes induced by cocaine in D1 MSNs that are permissive for Sirt2 induction, or by morphine that preclude Sirt2 induction?
These are just some of the outstanding questions that need to be answered in delineating the precise transcriptional steps through which cocaine or other drugs of abuse change brain reward mechanisms to drive an addicted state, particularly in the context of improved animal models capturing specific behavioral characteristics of substance use disorders [167]. Elaborating the likely scores of transcription factors that contribute to this transcriptional regulation, and the hundreds of target genes through which each factor acts to produce its downstream molecular, cellular, circuit, and behavioral plasticity, will fundamentally improve our understanding of the molecular basis of drug addiction and provide a template to guide efforts at developing improved treatment and prevention measures for substance use disorders.
Acknowledgements:
Several figures in this review were created using Biorender.com
Footnotes
Conflicts of interest:
None.
References:
- 1.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. The Lancet Psychiatry. 2016;3(8):760–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kronman H, Richter F, Labonté B, Chandra R, Zhao S, Hoffman G, et al. Biology and Bias in Cell Type-Specific RNAseq of Nucleus Accumbens Medium Spiny Neurons. Sci Rep. 2019;9(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savell KE, Tuscher JJ, Zipperly ME, Duke CG, Phillips RA, Bauman AJ, et al. A dopamine-induced gene expression signature regulates neuronal function and cocaine response. Sci Adv. 2020;6(26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrientos C, Knowland D, Wu MMJ, Lilascharoen V, Huang KW, Malenka RC, et al. Cocaine-Induced Structural Plasticity in Input Regions to Distinct Cell Types in Nucleus Accumbens. Biol Psychiatry. 2018;84(12):893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Chen Z, Fan G, Li A, Yuan J, Xu T. Cell-type-specific afferent innervation of the nucleus accumbens core and shell. Front Neuroanat. 2018;12:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15(6):816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lobo MK, Covington HE, Chaudhury D, Friedman AK, Sun HS, Damez-Werno D, et al. Cell type–specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science (80- ). 2010;330(6002):385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soares-Cunha C, de Vasconcelos NAP, Coimbra B, Domingues AV, Silva JM, Loureiro-Campos E, et al. Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol Psychiatry. 2019;25(12):3241–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole SL, Robinson MJF, Berridge KC. Optogenetic self-stimulation in the nucleus accumbens: D1 reward versus D2 ambivalence. PLoS One. 2018;13(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardo-Garcia TR, Garcia-Keller C, Penaloza T, Richie CT, Pickel J, Hope BT, et al. Ventral pallidum is the primary target for accumbens D1 projections driving cocaine seeking. J Neurosci. 2019;39(11):2041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Peña CJ, et al. In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci U S A. 2016;113(10):2726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soares-Cunha C, Coimbra B, David-Pereira A, Borges S, Pinto L, Costa P, et al. Activation of D2 dopamine receptor-expressing neurons in the nucleus accumbens increases motivation. Nat Commun. 2016;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, Kalivas PW. Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat Neurosci. 2015;18(9):1230–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thibault D, Loustalot F, Fortin GM, Bourque MJ, Trudeau LÉ. Evaluation of D1 and D2 Dopamine Receptor Segregation in the Developing Striatum Using BAC Transgenic Mice. PLoS One. 2013;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lüscher C The Emergence of a Circuit Model for Addiction. Annu Rev Neurosci. 2016;39:257–76. [DOI] [PubMed] [Google Scholar]
- 16.Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith ACW, et al. The nucleus accumbens: Mechanisms of addiction across drug classes reflect the importance of glutamate homeostasis. Pharmacol Rev. 2016;68(3):816–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright WJ, Dong Y. Psychostimulant-Induced Adaptations in Nucleus Accumbens Glutamatergic Transmission. Cold Spring Harb Perspect Med. 2020;10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: Mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33(6):267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang YH, Schlüter OM, Dong Y. Silent Synapses Speak Up: Updates of the Neural Rejuvenation Hypothesis of Drug Addiction. Neuroscientist. 2015;21(5):451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Y, Taylor JR, Wolf ME, Shaham Y. Circuit and synaptic plasticity mechanisms of drug relapse. J Neurosci. 2017;37(45):10867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf ME. Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci. 2016;17(6):351–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gipson CD, Kupchik YM, Kalivas PW. Rapid, transient synaptic plasticity in addiction. Neuropharmacology. 2014;76:276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nestler EJ, Lüscher C. The Molecular Basis of Drug Addiction: Linking Epigenetic to Synaptic and Circuit Mechanisms. Neuron. 2019;102(1):48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mews P, Walker DM, Nestler EJ. Epigenetic Priming in Drug Addiction. Cold Spring Harb Symp Quant Biol. 2019;83:131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2(2):119–28. [DOI] [PubMed] [Google Scholar]
- 26.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell. 2018;172(4):650–65. [DOI] [PubMed] [Google Scholar]
- 27.Walker DM, Cates HM, Loh YHE, Purushothaman I, Ramakrishnan A, Cahill KM, et al. Cocaine Self-administration Alters Transcriptome-wide Responses in the Brain’s Reward Circuitry. Biol Psychiatry. 2018;84(12):867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maze I, Covingtoni HE, Dietz DM, Laplant Q, Renthal W, Russo SJ, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science (80- ). 2010;327(5962):213–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Shendure J. Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol Cell. 2019;76(2):306–19. [DOI] [PubMed] [Google Scholar]
- 30.Stadhouders R, Filion GJ, Graf T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 2019;569(7756):345–54. [DOI] [PubMed] [Google Scholar]
- 31.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12(11):623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mews P, Calipari ES, Day J, Lobo MK, Bredy T, Abel T. From Circuits to Chromatin: The Emerging Role of Epigenetics in Mental Health. J Neurosci. 2021;41(5):873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenny PJ. Epigenetics, microRNA, and addiction. Dialogues Clin Neurosci. 2014;16(3):335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker DM, Nestler EJ. Neuroepigenetics and addiction. In: Handbook of Clinical Neurology. 2018. p. 747–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pierce RC, Fant B, Swinford-Jackson SE, Heller EA, Berrettini WH, Wimmer ME. Environmental, genetic and epigenetic contributions to cocaine addiction. Neuropsychopharmacology. 2018;43(7):1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cates HM, Heller EA, Lardner CK, Purushothaman I, Peña CJ, Walker DM, et al. Transcription Factor E2F3a in Nucleus Accumbens Affects Cocaine Action via Transcription and Alternative Splicing. Biol Psychiatry. 2018;84(3):167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lardner CK, van der Zee Y, Estill MS, Salery M, Kronman HG, Cunningham AM, et al. Gene-targeted, CREB-mediated induction of ΔFosB controls distinct downstream transcriptional patterns within D1 and D2 medium spiny neurons. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meers MP, Bryson TD, Henikoff JG, Henikoff S. Improved CUT&RUN chromatin profiling tools. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109(1):21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Z, Schulz MH, Look T, Begemann M, Zenke M, Costa IG. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019;20(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-). 2009;326(5950):289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17(11):661. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Poh HM, Peh SQ, Sia YY, Li G, Mulawadi FH, et al. ChIA-PET analysis of transcriptional chromatin interactions. Methods. 2012;58(3):289–99. [DOI] [PubMed] [Google Scholar]
- 44.Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet. 2018;50(2):238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nat Neurosci. 2003;6(11):1208–15. [DOI] [PubMed] [Google Scholar]
- 46.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21(6):635–7. [DOI] [PubMed] [Google Scholar]
- 47.Yim YY, Teague CD, Nestler EJ. In vivo locus-specific editing of the neuroepigenome. Nat Rev Neurosci. 2020;21(9):471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorsch ZS, Hamilton PJ, Ramakrishnan A, Parise EM, Salery M, Wright WJ, et al. Stress resilience is promoted by a Zfp189-driven transcriptional network in prefrontal cortex. Nat Neurosci. 2019;22(9):1413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamilton PJ, Lim CJ, Nestler EJ, Heller EA. Neuroepigenetic editing. In: Methods in Molecular Biology. 2018. p. 113–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Day JJ. New approaches to manipulating the epigenome. Dialogues Clin Neurosci. 2014;16(3):345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu SJ, Heller EA. Recent advances in neuroepigenetic editing. Curr Opin Neurobiol. 2019;59:26–33. [DOI] [PubMed] [Google Scholar]
- 52.Lu Z, Liu Z, Mao W, Wang X, Zheng X, Chen S, et al. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis. 2020;11(2):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen LF, Lin YT, Gallegos DA, Hazlett MF, Gómez-Schiavon M, Yang MG, et al. Enhancer Histone Acetylation Modulates Transcriptional Bursting Dynamics of Neuronal Activity-Inducible Genes. Cell Rep. 2019;26(5):1174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167(1):233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlezon WA, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28(8):436–45. [DOI] [PubMed] [Google Scholar]
- 56.Shaywitz AJ, Greenberg ME. CREB: A Stimulus-Induced Transcription Factor Activated by A Diverse Array of Extracellular Signals. Annu Rev Biochem. 2002;68(1):821–61. [DOI] [PubMed] [Google Scholar]
- 57.Briand LA, Lee BG, Lelay J, Kaestner KH, Blendy JA. Serine 133 phosphorylation is not required for hippocampal CREB-mediated transcription and behavior. Learn Mem. 2015;22(2):109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu X, McMurray CT. Calmodulin kinase II attenuation of gene transcription by preventing cAMP response element-binding protein (CREB) dimerization and binding of the CREB-binding protein. J Biol Chem. 2001;276(3):1735–41. [DOI] [PubMed] [Google Scholar]
- 59.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science (80- ). 1997;278(5335):58–63. [DOI] [PubMed] [Google Scholar]
- 60.Carlezon WA, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, et al. Regulation of cocaine reward by CREB. Science (80- ). 1998;282(5397):2272–5. [DOI] [PubMed] [Google Scholar]
- 61.Larson EB, Graham DL, Arzaga RR, Buzin N, Webb J, Green TA, et al. Overexpression of CREB in the Nucleus Accumbens Shell Increases Cocaine Reinforcement in Self-Administering Rats. J Neurosci. 2011;31(45):16447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi KH, Whisler K, Graham DL, Self DW. Antisense-induced reduction in nucleus accumbens cyclic AMP response element binding protein attenuates cocaine reinforcement. Neuroscience. 2006;137(2):372–83. [DOI] [PubMed] [Google Scholar]
- 63.Bilbao A, Rieker C, Cannella N, Parlato R, Golda S, Piechota M, et al. CREB activity in dopamine D1 receptor expressing neurons regulates cocaine-induced behavioral effects. Front Behav Neurosci. 2014;8:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barrot M, Olivier JDA, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99(17):11435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edwards S, Graham DL, Bachtell RK, Self DW. Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci. 2007;25:2201–13. [DOI] [PubMed] [Google Scholar]
- 66.Benedetto M, D’Addario C, Candeletti S, Romualdi P. Alterations of CREB and DARPP-32 phosphorylation following cocaine and monoaminergic uptake inhibitors. Brain Res. 2007;1128:33–9. [DOI] [PubMed] [Google Scholar]
- 67.Lee D, Bian S, Rahman A, Shim Y-B, Shim I, Choe E. Repeated cocaine administration increases N-methyl-D-aspartate NR1 subunit, extracellular signal-regulated kinase and cyclic AMP response element-binding protein phosphorylation and glutamate release in the rat dorsal striatum. Eur J Pharmacol. 2008;590:157–62. [DOI] [PubMed] [Google Scholar]
- 68.Yin H-S, Chen K, Kalpana S, Shih JC. Differential Effects of Chronic Amphetamine and Baclofen Administration on cAMP Levels and Phosphorylation of CREB in Distinct Brain Regions of Wild Type and Monoamine Oxidase B-Deficient Mice. Synapse. 2006;60:573–84. [DOI] [PubMed] [Google Scholar]
- 69.Shaw-Lutchman TZ, Impey S, Storm D, Nestler EJ. Regulation of CRE-mediated transcription in mouse brain by amphetamine. Synapse. 2003;48(1):10–7. [DOI] [PubMed] [Google Scholar]
- 70.Olson VG, Zabetian CP, Bolanos CA, Edwards S, Barrot M, Eisch AJ, et al. Regulation of Drug Reward by cAMP Response Element-Binding Protein: Evidence for Two Functionally Distinct Subregions of the Ventral Tegmental Area. J Neurosci. 2005;25(23):5553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, Maze I, et al. Genome-wide Analysis of Chromatin Regulation by Cocaine Reveals a Role for Sirtuins. Neuron. 2009;62(3):335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown TE, Lee BR, Mu P, Ferguson D, Dietz D, Ohnishi YN, et al. A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J Neurosci. 2011;31(22):8163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang YH, Lin Y, Brown TE, Han MH, Saal DB, Neve RL, et al. CREB modulates the functional output of nucleus accumbens neurons: A critical role of N-methyl-D-aspartate glutamate receptor (NMDAR) receptors. J Biol Chem. 2008;283(5):2751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X, Wolf ME. Multiple faces of BDNF in cocaine addiction. Behav Brain Res. 2015;279:240–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10(8):1029–37. [DOI] [PubMed] [Google Scholar]
- 76.Itami C, Kimura F, Kohno T, Matsuoka M, Ichikawa M, Tsumoto T, et al. Brain-derived neurotrophic factor-dependent unmasking of “silent” synapses in the developing mouse barrel cortex. Proc Natl Acad Sci U S A. 2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, et al. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9(4):475–7. [DOI] [PubMed] [Google Scholar]
- 78.Darcq E, Kieffer BL. Opioid receptors: Drivers to addiction? Nat Rev Neurosci. 2018;19(8):499–514. [DOI] [PubMed] [Google Scholar]
- 79.Steiner H, Gerfen CR. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J Neurosci. 1993;13(12):5066–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47:24–32. [DOI] [PubMed] [Google Scholar]
- 81.Nestler EJ. Transcriptional mechanisms of addiction: Role of ΔFosB. Philos Trans R Soc B Biol Sci. 2008;363(1507):3245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, et al. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13(5):1235–44. [DOI] [PubMed] [Google Scholar]
- 83.Perrotti LI, Weaver RR, Robison B, Renthal W, Maze I, Yazdani S, et al. Distinct patterns of ΔFosB induction in brain by drugs of abuse. Synapse. 2008;62(5):358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, et al. ΔFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci. 2013;33(47):18381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robison AJ, Vialou V, Mazei-Robison M, Feng J, Kourrich S, Collins M, et al. Behavioral and structural responses to chronic cocaine require a feedforward loop involving ΔFosB and calcium/calmodulin-dependent protein kinase II in the nucleus accumbens shell. J Neurosci. 2013;33(10):4295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jabbour L, Cartee F, Caisee M, Haynes K. ΔFosB expression in human postmortem brain tissue from opioid overdoses. In: 47th Annual Meeting of the Society for Neurosciences. 2017. [Google Scholar]
- 87.Vialou V, Feng J, Robison AJ, Ku SM, Ferguson D, Scobie KN, et al. Serum response factor and cAMP response element binding protein are both required for cocaine induction of ΔFosB. J Neurosci. 2012;32(22):7577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cates HM, Lardner CK, Bagot RC, Neve RL, Nestler EJ. Fosb induction in nucleus accumbens by cocaine is regulated by E2F3a. eNeuro. 2019;6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chandra R, Francis TC, Konkalmatt P, Amgalan A, Gancarz a. M, Dietz DM, et al. Opposing Role for Egr3 in Nucleus Accumbens Cell Subtypes in Cocaine Action. J Neurosci. 2015;35(20):7927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Damez-Werno D, LaPlant Q, Sun HS, Scobie KN, Dietz DM, Walker IM, et al. Drug experience epigenetically primes Fosb gene inducibility in rat nucleus accumbens. J Neurosci. 2012;32(30):10267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DEH, Truong HT, et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48(2):303–14. [DOI] [PubMed] [Google Scholar]
- 92.Levine a. a., Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci. 2005;102(52):19186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, et al. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIα in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35(4):913–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology. 2010;35(12):2450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ulery-Reynolds PG, Castillo MA, Vialou V, Russo SJ, Nestler EJ. Phosphorylation of ΔFosB mediates its stability in vivo. Neuroscience. 2009;158(2):369–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carle TL, Ohnishi YN, Ohnishi YH, Alibhai IN, Wilkinson MB, Kumar A, et al. Proteasome-dependent and -independent mechanisms for FosB destabilization: Identification of FosB degron domains and implications for ΔFosB stability. Eur J Neurosci. 2007;25(10):3009–19. [DOI] [PubMed] [Google Scholar]
- 97.Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, et al. An essential role for ΔFosB in the nucleus accumbens in morphine action. Nat Neurosci. 2006;9(2):205–11. [DOI] [PubMed] [Google Scholar]
- 98.Kelz MB, Chen J, Carlezon WA, Whisler K, Gilden L, Beckmann AM, et al. Expression of the transcription factor ΔFosB in the brain controls sensitivity to cocaine. Nature. 1999;401(6750):272–6. [DOI] [PubMed] [Google Scholar]
- 99.Colby CR, Whisler K, Steffen C, Nestler EJ, Self DW. Striatal cell type-specific overexpression of ΔFosB enhances incentive for cocaine. J Neurosci. 2003;23(6):2488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grueter BA, Robison AJ, Neve RL, Nestler EJ, Malenka RC. FosB differentially modulates nucleus accumbens direct and indirect pathway function. Proc Natl Acad Sci. 2013;110(5):1923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peakman MC, Colby C, Perrotti LI, Tekumalla P, Carle T, Ulery P, et al. Inducible, brain region-specific expression of a dominant negative mutant of c-Jun in transgenic mice decreases sensitivity to cocaine. Brain Res. 2003;970(1–2):73–86. [DOI] [PubMed] [Google Scholar]
- 102.Heller EA, Cates HM, Peña CJ, Sun H, Shao N, Feng J, et al. Locus-specific epigenetic remodeling controls addiction-and depression-related behaviors. Nat Neurosci. 2014;17(12):1720–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nestler EJ, Barrot M, Self DW. ΔFosB: A sustained molecular switch for addiction. Proc Natl Acad Sci U S A. 2001;98(20):11042–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chocyk A, Czyrak A, Wedzony K. Acute and repeated cocaine induces alterations in FosB/ΔFosB expression in the paraventricular nucleus of the hypothalamus. Brain Res. 2006;1090(1). [DOI] [PubMed] [Google Scholar]
- 105.Gajewski PA, Eagle AL, Williams ES, Manning CE, Lynch H, McCornack C, et al. Epigenetic regulation of hippocampal fosb expression controls behavioral responses to cocaine. J Neurosci. 2019;39(42). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Winstanley CA, LaPlant Q, Theobald DEH, Green TA, Bachtell RK, Perrotti LI, et al. ΔFosB induction in orbitofrontal cortex mediates tolerance to cocaine-induced cognitive dysfunction. J Neurosci. 2007;27(39). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Winstanley CA, Bachtell RK, Theobald DEH, Laali S, Green TA, Kumar A, et al. Increased impulsivity during withdrawal from cocaine self-administration: Role for ΔFosB in the orbitofrontal cortex. Cereb Cortex. 2009;19(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Winstanley CA, Green TA, Theobald DEH, Renthal W, LaPlant Q, DiLeone RJ, et al. ΔFosB induction in orbitofrontal cortex potentiates locomotor sensitization despite attenuating the cognitive dysfunction caused by cocaine. Pharmacol Biochem Behav. 2009;93(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eagle AL, Gajewski PA, Yang M, Kechner ME, Al Masraf BS, Kennedy PJ, et al. Experience-dependent induction of hippocampal ΔFosB controls learning. J Neurosci. 2015;35(40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gajewski PA, Turecki G, Robison AJ. Differential expression of FosB proteins and potential target genes in select brain regions of addiction and depression patients. PLoS One. 2016;11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kelz MB, Chen J, Carlezon WA, Whisler K, Gilden L, Beckmann AM, et al. Expression of the transcription factor ΔFosB in the brain controls sensitivity to cocaine. Nature. 1999;401(6750):272–6. [DOI] [PubMed] [Google Scholar]
- 112.Ferguson D, Koo JW, Feng J, Heller E, Rabkin J, Heshmati M, et al. Essential Role of SIRT1 Signaling in the Nucleus Accumbens in Cocaine and Morphine Action. J Neurosci. 2013;33(41):16088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, et al. Nuclear Factor kappa B Signaling Regulates Neuronal Morphology and Cocaine Reward. J Neurosci. 2009;29(11):3529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, et al. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410(6826):376–80. [DOI] [PubMed] [Google Scholar]
- 115.Renthal W, Carle TL, Maze I, Covington HE, Truong H- T, Alibhai I, et al. ΔFosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28(29):7344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yeh S-Y, Estill MS, Lardner CK, Shen L, Nestler EJ. The Role of ΔFosB in the Development of Drug Addiction: Identifying ΔFosB Transcriptional Targets in Nucleus Accumbens. SfN Glob Connect. 2021; [Google Scholar]
- 117.Ferguson D, Shao N, Heller E, Feng J, Neve R, Kim H-D, et al. SIRT1-FOXO3a Regulate Cocaine Actions in the Nucleus Accumbens. J Neurosci. 2015;35(7):3100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schall TA, Wright WJ, Dong Y. Nucleus accumbens fast-spiking interneurons in motivational and addictive behaviors. Mol Psychiatry. 2021;26:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee JH, Ribeiro EA, Kim J, Ko B, Kronman H, Jeong YH, et al. Dopaminergic Regulation of Nucleus Accumbens Cholinergic Interneurons Demarcates Susceptibility to Cocaine Addiction. Biol Psychiatry. 2020;88(10):746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee J, Finkelstein J, Choi JYY, Witten IBB. Linking Cholinergic Interneurons, Synaptic Plasticity, and Behavior during the Extinction of a Cocaine-Context Association. Neuron. 2016;90(5):1071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ribeiro EA, Salery M, Scarpa JR, Calipari ES, Hamilton PJ, Ku SM, et al. Transcriptional and physiological adaptations in nucleus accumbens somatostatin interneurons that regulate behavioral responses to cocaine. Nat Commun. 2018;9(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Donovan KJ, Tourtellotte WG, Milbrandt J, Baraban JM. The EGR family of transcription-regulatory factors: Progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22(4):167–73. [DOI] [PubMed] [Google Scholar]
- 123.Engeln M, Mitra S, Chandra R, Gyawali U, Fox ME, Dietz DM, et al. Sex-Specific Role for Egr3 in Nucleus Accumbens D2-Medium Spiny Neurons Following Long-Term Abstinence From Cocaine Self-administration. Biol Psychiatry. 2019;87(11):992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chandra R, Engeln M, Francis TC, Konkalmatt P, Patel D, Lobo MK. A Role for Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1α in Nucleus Accumbens Neuron Subtypes in Cocaine Action. Biol Psychiatry. 2017;81(7):564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Anderson EM, Larson EB, Guzman D, Wissman AM, Neve RL, Nestler EJ, et al. Overexpression of the histone dimethyltransferase G9a in nucleus accumbens shell increases cocaine self-administration, stress-induced reinstatement, and anxiety. J Neurosci. 2018;38(4):803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anderson EM, Sun H, Guzman D, Taniguchi M, Cowan CW, Maze I, et al. Knockdown of the histone di-methyltransferase G9a in nucleus accumbens shell decreases cocaine self-administration, stress-induced reinstatement, and anxiety. Neuropsychopharmacology. 2019;44(8):1370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Laplant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren BL, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13(9):1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cannella N, Oliveira AMM, Hemstedt T, Lissek T, Buechler E, Bading H, et al. Dnmt3a2 in the nucleus accumbens shell is required for reinstatement of cocaine seeking. J Neurosci. 2018;38(34):7516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cole S, Chandra R, Harris M, Patel I, Wang T, Kim H, et al. Cocaine-induced neuron subtype mitochondrial dynamics through Egr3 transcriptional regulation. bioRxiv. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Boersma MCH, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK. A Requirement for Nuclear Factor- B in Developmental and Plasticity-Associated Synaptogenesis. J Neurosci. 2011;31(14):5414–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-κB. Trends Neurosci. 2011;34(5):316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ang E, Chen J, Zagouras P, Magna H, Holland J, Schaeffer E, et al. Induction of nuclear factor-κB in nucleus accumbens by chronic cocaine administration. J Neurochem. 2001;79(1):221–4. [DOI] [PubMed] [Google Scholar]
- 134.Wang Y, Teng H, Sapozhnikov DM, Du Q, Zhao M. Transcriptome sequencing reveals candidate NF-κB target genes involved in repeated cocaine administration. Int J Neuropsychopharmacol. 2018;21(7):697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron. 2019;101(2):207–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, et al. Cocaine Regulates MEF2 to Control Synaptic and Behavioral Plasticity. Neuron. 2008;59(4):621–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science (80- ). 1999;286(5440):785–90. [DOI] [PubMed] [Google Scholar]
- 138.Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science (80- ). 2006;311(5763):1008–12. [DOI] [PubMed] [Google Scholar]
- 139.Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, et al. Genome-Wide Analysis of MEF2 Transcriptional Program Reveals Synaptic Target Genes and Neuronal Activity-Dependent Polyadenylation Site Selection. Neuron. 2008;60(6):1022–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Norrholm SD, Bibb JA, Nestler EJ, Ouimet CC, Taylor JR, Greengard P. Cocaine-induced proliferation of dendritic spines in nucleus accumbens is dependent on the activity of cyclin-dependent kinase-5. Neuroscience. 2003;116(1):19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Benavides DR, Quinn JJ, Zhong P, Hawasli AH, DiLeone RJ, Kansy JW, et al. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J Neurosci. 2007;27(47):12967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Taylor JR, Lynch WJ, Sanchez H, Olausson P, Nestler EJ, Bibb JA. Inhibition of Cdk5 in the nucleus accumbens enhances the locomotor-activating and incentive-motivational effects of cocaine. Proc Natl Acad Sci U S A. 2007;104(10):4147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Song WM, Zhang B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput Biol. 2015;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang Q, Zhang Y, Wang M, Song WM, Shen Q, McKenzie A, et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat Commun. 2019;10(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Neff RA, Wang M, Vatansever S, Guo L, Ming C, Wang Q, et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv. 2021;7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang M, Li A, Sekiya M, Beckmann ND, Quan X, Schrode N, et al. Transformative Network Modeling of Multi-omics Data Reveals Detailed Circuits, Key Regulators, and Potential Therapeutics for Alzheimer’s Disease. Neuron. 2021;109(2):257–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bagot RCC, Cates HMM, Purushothaman I, Lorsch ZSS, Walker DMM, Wang J, et al. Circuit-wide Transcriptional Profiling Reveals Brain Region-Specific Gene Networks Regulating Depression Susceptibility. Neuron. 2016;90(5):969–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Labonté B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23(9):1102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Teague CD, Zhou X, Walker D, Holt L, Zhang B, Nestler E. Phosphodiesterase 1b is an Upstream Regulator of a Key Gene Network in the Nucleus Accumbens Driving Addiction-Like Behaviors. In: Virtual NIDA Genetics and Epigenetics Consortium Meeting. 2021. [Google Scholar]
- 152.Hoekman MFM, Jacobs FMJ, Smidt MP, Burbach JPH. Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expr Patterns. 2006;6(2):134–40. [DOI] [PubMed] [Google Scholar]
- 153.Feng J, Wilkinson M, Liu X, Purushothaman I, Ferguson D, Vialou V, et al. Chronic cocaine-regulated epigenomic changes in mouse nucleus accumbens. Genome Biol. 2014;15(4):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Julian LM, Vandenbosch R, Pakenham CA, Andrusiak MG, Nguyen AP, McClellan KA, et al. Opposing regulation of Sox2 by cell-cycle effectors E2f3a and E2f3b in neural stem cells. Cell Stem Cell. 2013;12(4):440–52. [DOI] [PubMed] [Google Scholar]
- 155.Cates HM, Bagot RC, Heller EA, Purushothaman I, Lardner CK, Walker DM, et al. A novel role for E2F3b in regulating cocaine action in the prefrontal cortex. Neuropsychopharmacology. 2019;44(4):776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Godino A, Salery M, Futamura R, Browne C, Martinez-Rivera F, Hamilton P, et al. Retinoic x receptor alpha signaling in the nucleus accumbens contributes to cocaine- and opioid-induced transcriptional and behavioral alterations. Society for Neuroscience 2019. 2019. [Google Scholar]
- 157.Carpenter MD, Hu Q, Bond AM, Lombroso SI, Czarnecki KS, Lim CJ, et al. Nr4a1 suppresses cocaine-induced behavior via epigenetic regulation of homeostatic target genes. Nat Commun. 2020;11(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nanjan MJ, Mohammed M, Prashantha Kumar BR, Chandrasekar MJN. Thiazolidinediones as antidiabetic agents: A critical review. Vol. 77, Bioorganic Chemistry. 2018. [DOI] [PubMed] [Google Scholar]
- 159.Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, et al. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol. 2008;26(7). [DOI] [PubMed] [Google Scholar]
- 160.Kamide D, Yamashita T, Araki K, Tomifuji M, Tanaka Y, Tanaka S, et al. Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Sci. 2016;107(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Makino H, Seki S, Yahara Y, Shiozawa S, Aikawa Y, Motomura H, et al. A selective inhibition of c-Fos/activator protein-1 as a potential therapeutic target for intervertebral disc degeneration and associated pain. Sci Rep. 2017;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nestler EJ. δfosB: A transcriptional regulator of stress and antidepressant responses. Eur J Pharmacol. 2015;753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wallace DL, Vialou V, Rios L, Carle-Florence TL, Chakravarty S, Kumar A, et al. The influence of ΔFosB in the nucleus accumbens on natural reward-related behavior. J Neurosci. 2008;28(41):10272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sabatakos G, Sims NA, Chen J, Aoki K, Kelz MB, Amling M, et al. Overexpression of ΔFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat Med. 2000;6(9). [DOI] [PubMed] [Google Scholar]
- 165.Li Y, Liu Z, Aglyamova G, Chen J, Chen H, Bhandari M, et al. Discovery of phenanthridine analogues as novel chemical probes disrupting the binding of DNA to ΔFosB homodimers and ΔFosB/JunD heterodimers. Bioorganic Med Chem Lett. 2020;30(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang Y, Cesena TI, Ohnishi Y, Burger-Caplan R, Lam V, Kirchhoff PD, et al. Small molecule screening identifies regulators of the transcription factor ΔfosB. ACS Chem Neurosci. 2012;3(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Venniro M, Banks ML, Heilig M, Epstein DH, Shaham Y. Improving translation of animal models of addiction and relapse by reverse translation. Nat Rev Neurosci. 2020;21(11):625–43. [DOI] [PubMed] [Google Scholar]
- 168.Heller EA, Hamilton PJ, Burek DD, Lombroso SI, Peña CJ, Neve RL, et al. Targeted epigenetic remodeling of the cdk5 gene in nucleus accumbens regulates cocaine- and stress-evoked behavior. J Neurosci. 2016;36(17):4690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hamilton PJ, Burek DJ, Lombroso SI, Neve RL, Robison AJ, Nestler EJ, et al. Cell-Type-Specific Epigenetic Editing at the Fosb Gene Controls Susceptibility to Social Defeat Stress. Neuropsychopharmacology. 2018;32(2):272–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aleyasin H, Flanigan ME, Golden SA, Takahashi A, Menard C, Pfau ML, et al. Cell-type-specific role of Δfosb in nucleus accumbens in modulating intermale aggression. J Neurosci. 2018;38(26):5913–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kronman H, Torres-Berrio A, Sidoli S, Issler O, Godino A, Ramakrishnan A, et al. Long-term behavioral and cell type-specific molecular effects of early life stress are mediated by H3K79me2 dynamics in medium spiny neurons. Nat Neurosci. 2021;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nectow AR, Nestler EJ. Viral tools for neuroscience. Nat Rev Neurosci. 2020;21(12):669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]