

Abstract
Traumatic brain injury (TBI), particularly of greater severity (i.e., moderate-to-severe), has been identified as a risk factor for all-cause dementia and Parkinson’s disease (PD), with risk for specific dementia sub-types being more variable. Among the limited studies involving neuropathological (post-mortem) confirmation, the association between TBI and risk for neurodegenerative disease increases in complexity, with polypathology often reported on examination. The heterogeneous clinical and neuropathological outcomes associated with TBI are likely reflective of the multifaceted post-injury acute and chronic processes that may contribute to neurodegeneration. Acutely in TBI, axonal injury and disrupted transport influences molecular mechanisms fundamental to the formation of pathological proteins, such as amyloid (A) β peptide and hyperphosphorylated tau (pTau). These protein deposits may develop into Aβ plaques, pTau positive neurofibrillary tangles and dystrophic neurites. These and other characteristic neurodegenerative diseases pathologies may then spread across brain regions. The acute immune and neuroinflammatory response involves alteration of microglia, astrocytes, oligodendrocytes, and endothelial cells, release of downstream pro- and anti-inflammatory cytokines and chemokines, and recruitment of peripheral immune cells. Though thought to be neuroprotective and reparative initially, prolongation of these processes may promote neurodegeneration. We review the evidence for TBI as a risk factor for neurodegenerative disorders, including Alzheimer’s dementia and PD, in clinical and neuropathological studies. Further, we describe the dynamic interactions between acute response to injury and chronic processes that may be involved in TBI-related pathogenesis and progression of neurodegeneration.
Keywords: Traumatic brain injury, Neurodegeneration, Dementia, Neuropathology, Neuroinflammation
Introduction
Systematic reviews have found that TBI of any severity is associated with a 63%-96% increased risk of all-cause dementia(1-3). However, the specific epidemiological factors and pathophysiological mechanisms that underlie the association between TBI and specific neurodegenerative pathologies is poorly understood. This is a challenging area of research for many reasons, including methodological variability across studies (e.g., differing diagnostic criteria or injury characterization, heterogeneity of clinical samples, self-report vs. clinician determined diagnosis), a lack of specific and sensitive biomarkers to detect and monitor post-TBI neurodegeneration, and a dearth of cohorts with combined reliable, detailed data that includes TBI exposure, structured clinical assessments with biofluid banking, and neuropathological evaluation. Acutely following TBI, there is a complex pathophysiological response to injury that involves neuroprotective and reparative processes, but prolonged activation of these processes may promote pathogenesis and progression of neurodegenerative processes. Here, we extend prior broad reviews of associations between TBI and neurodegeneration by summarizing evidence for risk of dementia and specific neurodegenerative diseases following TBI across all severities, as well as leading candidate mechanisms linking TBI and neurodegenerative outcomes.
TBI and neurodegeneration: large-scale studies of clinical diagnoses
All-cause dementia in moderate/severe or all severity TBI with loss of consciousness (LOC)
The majority of administrative cohort clinical research involving TBI of all severities report an association between TBI-exposure and increased risk of dementia(2, 4-12). For clarity, administrative cohort studies typically involve a large dataset, that was conducted using data derived from administrative system operations, such as patient registration, health record keeping, billing/payment transactions, etc. Of the four largest studies to date examining moderate/severe TBI exclusively and risk of all-cause dementia, hazard ratios [(HRs)(95% confidence interval)] for development of dementia range from 1.35 [(1.26-1.45)(4, 6, 7)] to 3.77 [3.63-3.91 (11)]. A retrospective population cohort study of ~40,000 working-age adults identified an association between moderate/severe TBI exposure and increased risk of developing all-cause dementia, as compared to those who sustained a mild TBI (mTBI), but TBI was not a significant risk factor for any dementia sub-type when specific dementia sub-types were examined (7).
Dementia sub-types and movement disorders in moderate/severe or all severity TBI with loss of consciousness (LOC)
Research focused on moderate/severe TBI exposure and distinct clinically diagnosed neurodegenerative diseases (e.g., Alzheimer’s disease [AD]) is limited, with most reports in the literature binning TBIs of all severity, or differentiating between the presence of LOC. In one study of World War II veterans using expert consensus clinical diagnostic criteria, those who sustained a moderate or severe TBI in early adulthood (approximately 40 years prior) were 2.3 and 4.5 times more likely to develop AD later in life, respectively, compared to a similar group of veterans with non-TBI histories(13). Other investigations aggregating TBI of all severities have yielded conflicting results, presumably due to the large degree of heterogeneity that can exist across samples (e.g., mild to severe TBI range, time since injury, methodology of investigation [case-control vs. cohort], and rigor of dementia sub-type diagnosis [ICD code vs. expert consensus vs. biomarker])(12, 14-22).
Two of the largest administrative and population-based cohort studies to date reported the risk of clinical Parkinson’s disease (PD) to be at least 1.8 times greater among those with a history of moderate/severe TBI, as compared to those without TBI, over an average follow-up duration of 4.6 years, adjusting for multiple demographic, medical, and psychiatric history factors(7, 23). The elevated risk of PD among those with a history of TBI is supported by meta-analyses involving TBI of all severities(5, 24). Research on multiple prospective cohorts reported elevated risk of incident PD among individuals with remote TBI history (moderate-to-severe), which was later consistent with post-mortem neuropathological examination at follow-up (20). A meta-analysis of 16 studies demonstrated a greater risk of amyotrophic lateral sclerosis (ALS; OR=1.5) among individuals with a history of TBI (aggregate of all severities), but secondary analyses raised concern for reverse-causation when taking into account injury time lags of greater than 1-year(25). Limited studies have reported a positive relationship between history of TBI and frontotemporal dementia (FTD) and more research in this area is needed(26-28).
All-cause dementia in mTBI
Administrative cohort and population-based studies have identified mTBI as a risk factor for all cause dementia. A recent nationwide Veterans Health Administration (VHA) cohort study (N> 350,000) with a mean age of 49.5 yrs., and average follow-up duration of 4.2 years, reported a 2.4-fold increased risk for dementia in veterans with mTBI without LOC, and a 3.2-fold risk in those who experienced TBI with LOC(11). Over periods from 5-25 years, population-based studies have robustly reported mTBI to increase risk of dementia by factors of 1.2 to 3.3(4, 6, 29, 30). Within these studies, limitations such as reliance on diagnostic codes lacking specificity or exclusions of subjects with mTBI who did not seek care may confound estimation of outcome risk. Methodological heterogeneity (e.g., exposure misclassification, different diagnostic criteria, dementia diagnosed clinically vs. via informant, etc.) across studies may also contribute to the range of exposure-outcome risk estimates across investigations(31).
Dementia sub-types and movement disorders in mTBI
A cohort of 2,223 individuals, including those with clinically diagnosed probable or definite AD across three countries, were two times more likely to report a history of mTBI than a first-degree relative control group(14). Conversely, two large-scale investigations failed to detect an association between prior mTBI and incidence of AD over shorter durations of follow-up [(two years or less)(13, 32)]. There is stronger evidence for an association with PD, with evidence from several large cohorts indicating that a single mTBI may increase the risk of PD later in life(23, 30, 33, 34). Adjusting for confounds, the VHA study cohort described above with over 325,000 veterans (half with TBI) reported a significant increase of incident PD (HR=1.6) among those with a history of mTBI [average follow-up duration 4.6 years)(23)] An independent administrative cohort study over a 25-year period reported similar findings (a PD incidence rate ratio of 1.6) among those with a history of mTBI. While retrospective studies have shown associations of mTBI history with other non-AD dementia sub-types, such as FTD(28), there is insufficient evidence to draw definitive conclusions.
Dementia, dementia sub-types, and movement disorders in repetitive TBI and ‘subconcussive’ impacts
Repetitive TBI, including subconcussive impacts, are associated with chronic traumatic encephalopathy (CTE) neuropathologic change at autopsy, but risk for dementia and movement disorders is less clear. While population studies have shown increased risk for all-cause dementia with greater number of TBIs sustained(4, 30), the implications of repetitive TBI for risk of specific dementia/movement disorder sub-types have been less reliable. Not distinguishing between TBI severity, multiple large-scale studies have failed to observe a significant elevation in risk for AD(13, 32) and ALS(25) among those with a history of multiple TBIs, as compared to those with one TBI or no prior head injury. Some studies have focused on risk of dementia or specific dementia sub-types among contact sport athletes. In the largest study of former professional American football players to date (N=2,552), the authors reported a dose response relationship between number of prior mTBIs and risk of mild cognitive impairment (participant-reported physician diagnosed), but not AD(35).
Elevated risk of dementia/movement disorders may be attributable to cumulative subconcussive or repetitive head impacts (RHI) that fall below the relative magnitude of clinically diagnosed concussive injury (36-39). Combined neurodegenerative disease (AD, PD, ALS) mortality among former professional American football players is reportedly increased compared to either the general population or former non-contact sport athletes(40, 41). Results regarding specific disease subtypes in former professional American football players have been inconclusive, or contradictory in some cases, with one study observing higher rates of AD and ALS, but not PD(40), and the other study reporting higher rates of PD, but not AD or ALS(41). Research on former high school football players participating in sport between 1957-1970 has not observed elevated rates neurodegenerative diseases or increased cognitive impairment compared to well-matched control groups(42, 43).
Neuropathological outcomes of TBI
Large-scale studies exploring associations between TBI and neuropathologically-confirmed neurodegenerative disease have been inconsistent in regard to linking TBI exposure with a specific disease. While some cohort and retrospective case-control studies reported elevated risk of AD neuropathology among those with a self-reported history of TBI(44, 45), two of the largest scale studies to date involving longitudinal cohorts have not observed such an association(18, 20). Research using in vivo positron emission tomography (PET) have more reliably shown an association between remote TBI and tau tracer uptake, as compared to amyloid tracer uptake, which has been more variable(46-50). In a community-based cohort, TBI with LOC >1 hour was not significantly associated with AD pathologic burden, but those individuals were more likely to have Lewy body disease and there was a significant association between TBI with LOC >1 hour and incident PD, as well as progression rate of parkinsonism(20). The same group also reported greater microvascular ischemic changes associated with TBI history(20). The role of vascular pathology is further supported by an administrative cohort study of individuals with TBI presenting to the ED or hospitalized in the state of California between 2005 and 2009, which reported TBI to be an independent risk factor for subsequent stroke over a median follow-up duration of 28 months(51).
Detecting and interpreting relationships between TBI and risk of neurodegenerative disease is complicated by the high prevalence of polypathology among these samples(45, 52-59). The notion of polypathology and coexistence of multiple proteinopathies has been reported across the spectrum of TBI (mild to severe, repetitive mTBI, RHI) in post-mortem studies(45, 53, 55, 57, 58, 60). Aging is a major consideration for neurodegenerative disease polypathology, since the incidence for every major neurodegenerative disease increases with age, and the majority of neuropathological research samples have been from aged individuals(61). Further understanding of the relationship between TBI, neurodegenerative disease, aging, and polypathology, will require consideration of many other factors, such as other environmental exposures, genetic influences (e.g., apolipoprotein E, type ε4 allele), lifestyle (e.g., diet, exercise), and systemic disease (e.g., diabetes, hypertension), etc. which are also associated with elevated levels of polypathology(61).
Acute to chronic processes and mechanisms associating TBI and neurodegenerative disease
Many pathological processes have been proposed to contribute to neurodegenerative disease in general, and also in association with TBI exposure(8, 13, 62, 63). As relationships between TBI and neurodegenerative disease are clarified, it will be critical to understand the direct and indirect pathways in which pathophysiological processes initiated or influenced by TBI exposure may contribute to age-related neurodegenerative disease and dementia(20, 62, 64-70). It is important to consider the structural effects of TBI, acutely and over time, to understand how neurodegenerative disease processes may unfold post-injury, as well as the way in which these structural changes interact with chronic processes. Below we review processes that occur acutely in response to injury and their evolution to the chronic post-injury phase. These processes are considered to be among the leading candidate mechanisms that may confer increased risk for dementia post-TBI, but other pathways, such as glymphatic dysfunction or vascular damage and dysfunction, also likely contribute to TBI-related neurodegenerative disease (36).
Axonal injury, white matter changes, and potential degeneration
Axonal damage is highly common in TBI(71), with extensive axonal severance (i.e., axotomy), and secondary axonal swelling and disruption of axonal transport in more severe injuries (72-75). Over time this manifests as degeneration of white matter with severe loss of neurons and alterations in glial populations(76). Chronic, progressive changes of white matter following TBI are supported in multiple studies(77, 78). Graham et al. reported that alterations in white matter microstructure using diffusion tensor imaging (DTI) were predictive of white matter atrophy in patients in the chronic phase of moderate-to-severe TBI(73). Similar microstructural alterations occur in the absence of macroscopic pathology (approximately 1/3 of moderate-to-severe TBI cases) and inversely correlate with cognitive and functional outcomes. Progressive white matter changes (i.e., decline in white matter neurite density) among patients wither lower injury severity (mTBI) have also been reported in a prospective cohort (approximately 50% with TBI-related MRI findings) presenting to the ED(79).
In younger athletes who sustain an mTBI, where neuroradiological findings are significantly less common, decreased neurite density white matter alterations can persist beyond clinical recovery, but recover more completely compared to control-group levels(80, 81). The longitudinal significance of this is unclear; but at older ages, diffuse white matter tract abnormalities on DTI have been recorded in clinically unimpaired former professional American football players with a history of remote sport concussion(s)(82). A study showing a U-shaped trajectory of diffusion-related changes (i.e., initial improvement in recovery with later decline) at subacute, 1-year and 5-year time points following combat concussive blast trauma may help explain the discrepancy between initial recovery and long-term white matter changes(83). Together, this research supports the notion that TBI exposure can lead to progressive white matter degeneration, possibly through secondary effects of oxidative stress, cellular hyperexcitability, and vasogenic, cytotoxic edema, inflammation, and Wallerian degeneration (reviewed in-depth elsewhere).(84) Interestingly, changes on DTI are not predictive of grey matter atrophy in the same patients, which suggests other mechanisms beyond axonal white matter changes, such as the processes reviewed below, may be involved in alterations of cortex(78).
Acutely upregulated amyloid precursor protein evolving to beta-amyloid (Aβ) plaques
The upregulation of amyloid precursor protein (APP) acutely after TBI is well-established in experimental and human neuropathological studies (66, 67). Since amyloid precursor protein contains the peptide sequence for Aβ(85), the potential link between TBI and AD is clear. From days to months post-injury, initial accumulation of APP evolves with elevated co-accumulation of Aβ peptide and enzymes, including β-secretase 1 (BACE1), presenilin-1 (PS-1), and caspase-3(68), that contribute to Aβ generation in axons of white and grey matter(86). Aβ peptide accumulation following axonal injury may occur during transport disruption via decreased kinesin-1, a motor protein that transports cargo [(in this case, APP, BACE1, and PS-1)(87-89)]. Accumulating Aβ may be released into the extracellular space as axons and their cell bodies degenerate, either as consequence of Aβ accumulation itself, or directly due to trauma. It is not known whether this can lead to AD neuropathologic change, but studies of severe TBI ranging from 4 hours to 2.5 years post-injury have reported a prevalence of 30-38% of post-mortem and surgical tissues containing the presence of Aβ plaques(62, 65, 69). Similar rates of Aβ plaque accumulation were reported in a study of longer-term survivors of severe TBI ranging from 1-47 years (45), suggesting there may be a threshold at which the Aβ cascade is initiated, and potentially that Aβ pathology in TBI may not be inherently progressive. Prospective cohort studies with sensitive and specific Aβ biomarker (neuroimaging and/or biofluid) monitoring are required to understand relationships between TBI and development and progression of Aβ pathology.
Acute hyperphosphorylation of tau evolving to neurofibrillary tangles (NFTs)
Hyperphosphorylation of microtubule-associated protein (MAP) tau has been observed acutely (24 hours) and up to 7-days post-TBI in experimental models and post-mortem samples 4 hours to 5 weeks post-injury(64, 65, 90, 91). Neurofibrillary tangles (NFTs), an advanced form of hyperphosphorylated tau and a hallmark pathological lesion in AD and other tauopathies, are rarely observed in the acute/subacute post-injury period. This might suggest that post-injury pathological tau (pTau) later progresses to NFT pathology, or that acute TBI-related pTau and long-term NFT pathology are unrelated(36, 65, 90-92).
Liberation of microtubule-dissociated tau, from axonal shearing injury and potentially other TBI-related mechanisms, may promote hyperphosphorylation and accumulation of pathogenic tau proteoforms(36, 91, 93). Multiple protein kinases and phosphatases regulate tau hyperphosphorylation(94); experimental studies have recorded elevated levels of delta-secretase, glycogen synthase kinase-3β, and c-Jun N-terminal kinase following TBI in regions of tau hyperphosphorylation(95, 96). Reduction or elimination of the above enzymes can reduce hyperphosphorylation and accumulation of tau hyperphosphorylation post-TBI(95, 96). Delta-secretase results in tau cleavage at N368, producing tau fragments that are more amenable to hyperphosphorylation(96). Diverse lifestyle, genetic, environmental, and health factors (sleep, stress, viral/bacterial infection, bridging integrator 1 gene status) can influence these and other processes associated with pathological tau production [(reviewed elsewhere)(94)].
There is mounting evidence supporting pathways of transcellular tau progression, which could underlie extensive pTau burden in some neurodegenerative settings, and which may be important in post-TBI neurodegeneration. In a recent study, wild type mice inoculated with brain homogenates from mice who underwent experimental severe TBI (closed cortical impact - CCI) exhibited diffuse, progressive tau pathology along with behavioral and synaptic dysfunction suggesting that a self-sustained neurodegenerative process could occur following TBI-related accumulation of hyperphosphorylated tau (92). Transcellular spreading may occur through neuronal internalization of tau released from injured cells, or potentially through trans-synaptic, exosomal, secretory (interstitial fluid, CSF, etc.), or glymphatic pathways(97-101).
Inflammatory and Immune response post-TBI (acute to chronic)
A neuroinflammatory response occurs following acute injury, which includes release of pro- and anti-inflammatory cytokines, neurotrophic factor modulation, cell migration and phagocytosis and other processes, to clear debris and heal the damaged cells and tissues(102). Neuroinflammatory responses are tightly regulated and evolve rapidly, ideally with resolution soon after the injury, when homeostasis is restored and major structural damage is resolved. However, the human brain is limited in its capacity to heal significant structural injury, and some post-TBI neuroinflammatory processes can persist(103-108). This pro-inflammatory state is mediated primarily by microglia/macrophages and astrocytes, and their communication through cytokine and cell-surface markers(109).
Microglia are important for synaptic pruning, immune surveillance, post-injury immune response coordination, debris clearance, and regeneration (particularly synaptic). Chronic microglia changes can be widespread long after TBI and are correlated with structural brain changes over time(103, 105, 110, 111). Astrocytes could function to preserve neurons post-TBI through processes such as repair of the blood-brain-barrier (BBB)/recalibration of neurovascular coupling, neuronal recovery including synaptogenesis, and neurotransmitter metabolism(112, 113). While the above acute glia activation may have beneficial effects on attenuating injury effects and initial repair, prolonged and chronic activation of these processes contribute to deleterious effects of aging and neurodegeneration(114, 115). For example, following demarcation and “sealing off” of injury, reactive astrocytes could contribute to adverse long-term problems, as they hinder a number of growth-related functions including inhibition of synaptic formation, integration of new neurons, and axonal growth(116, 117).
Persistent neuroinflammation (characterized by elevated MHCII, CR3/43, CD68, NADPH oxidase [NOX2], and GFAP) has been observed for up to 1-year post-TBI in animal studies (rats and mice) using different experimental injury models(103, 118). Reactive microglia post-mortem (CR3/43+ & CD68+) and in vivo (higher PK-11195 binding detected by PET scans in TBI patients) have been reported up to 18 years after TBI(106, 119). Recent RNA profiling studies show a robust pro-inflammatory response with increased expression of Nfκb driven cytokines and chemokines occurring acutely. The subacute phase of inflammation was dominated by interferon (IFN) type I signaling. “Primed” microglia with associated alterations in Iba-1 morphology have also been detected chronically post-TBI(120, 121). The transition to persistent neuroinflammation noted above correlates with extensive brain injury including hippocampal cell death and neurodegeneration, lesion site expansion, cortical atrophy, and myelin loss(103, 118, 121-123). Microglia depletion at 1-month post-CCI decreased hippocampal neurodegeneration, cortical lesion expansion, and neurobehavioral deficits (107, 124).
Persistent glial inflammation or glia priming following TBI may adversely affect the ability of microglia to appropriately respond to future immune challenges such as infection (producing higher levels of inflammatory cytokines). In the injured mouse brain (30 days post-injury), primed MHCII+ microglia are hyper-reactive to acute challenge with LPS(121). Specifically, peripheral LPS administration resulted in exaggerated expression of IL-1β, impaired cognitive performance, and protracted sickness and depressive-like behavior(121, 125). Following TBI in humans, secondary insults across the lifespan can include a repeat head injury, peripheral immune challenge, or even aging-related changes and pre-clinical neurodegenerative disease(126, 127).
Independent of TBI, chronic glial activation has been implicated in the pathogenesis and progression of diverse neurodegenerative diseases [(AD, PD, ALS)(128)]. For example, reactive astrocytes may elevate risk of various neurodegenerative diseases through pro-inflammatory processes, disruption of BBB integrity, and glymphatic system dysfunction (via changes in aquaporin-4 expression and activation of matrix metalloproteinases [MMPs-3 and −9]), all of which have also been observed following TBI(129-133). The neurodegenerative disease pathological peptides, and their associated toxicities, may also perpetuate the glial response to injury, exacerbating secondary injury, promoting pathological deposits, and perpetuating neurodegeneration(134). Together, this supports the notion that neuroinflammation can persist and progress diffusely as part of Wallerian degeneration, exacerbate neuropathological deposits, and contribute to risk of neurodegenerative disease.
Future directions
There are major gaps in knowledge with regard to the relationships between TBI and diverse neurodegenerative disease. Post-mortem assessments, by definition, represent a single point in time, and therefore inferences on sequence, rate, and magnitude of progression and/or regression of pathological processes are inherently speculative without sensitive and specific biomarkers. The influences of genetic, biological, and environmental contributors to the development and progression of neurodegenerative disease are at best poorly understood. Sensitive and specific biomarkers for TBI-related neurodegeneration are lacking, but will be required not just to monitor for progression but to understand potential interactions across comorbidities and polypathologies. Secondary injury resulting from prolonged activation of reactive processes, such as neuroinflammation, adds further uncertainty. Research in each of these important areas is ongoing and essential to fill critical gaps in knowledge.
Neuroimaging and biomarkers
Given the complex spatial and temporal interactions of injury-related processes and the role of diverse modifying factors (e.g., age, genetics, lifestyle activities), investigations into the mechanism and course of neurodegenerative disease following TBI require a range of multimodal neuroimaging and fluid biomarkers. Structural [(e.g., volumetric and diffusion tensor imaging)(73, 78, 135)], functional [(e.g., functional connectivity and cerebral blood flow)(136, 137)], and molecular [(PET)(46, 48-50, 106, 110)], imaging findings have been reported in acute and long-term TBI. These modalities will be essential to provide complimentary information regarding the longitudinal temporal course of changes that can occur post-TBI. For example, greater PET ligand signal of neuroinflammation has been observed as co-localizing in regions with the greatest disease progression (e.g., neuropathological deposition in primary progressive aphasia) in longitudinal non-TBI samples(138, 139).
As a non-invasive and cost-effective method of measurement, systemic biofluid biomarkers (cerebral spinal fluid or serum/plasma) provide a cost-effective approach for serial sampling and monitoring, as well as opportunity for correlation with neuroimaging studies, to learn about the dynamic course of TBI progression. In clinically normal and cognitive impaired non-TBI samples, serum levels of tau [(p-tau181)(140)], amyloid [(Aβ1-42)(141)], axonal damage [(i.e., neurofilament light chain)(142)], and inflammation [(C-reactive protein, interleukin-6)(143)], have demonstrated utility in predicting disease progression and decline of age-related neurodegenerative disease. Validation of these markers reflecting pathological processing occurring in the brain (i.e., corresponding tracer uptake on PET imaging) has also been demonstrated(140, 141). Combined with multimodal neuroimaging, functional/cognitive assessment, and neuropathological validation, continued studies of fluid biomarkers will be critical to detect and intervene in TBI-associated neurodegeneration.
Therapeutic targets and prospects
Development of effective interventions for neurodegenerative diseases has largely proven to be unsuccessful to this point, with hundreds of failed clinical trials for AD alone. Although clinical trials of anti-amyloid therapy have not been optimal for the treatment of AD (144), this approach may have potential utility in mitigating the effect of TBI given the consistent upregulation of amyloid precursor protein in acute injury and future risk for dementia. Anti-pathological tau therapies hold promise as another treatment for AD and other tauopathies and are under development [(Phase 1 and 2 trials)(145)]. Given the prevalence of polypathology described above, future treatment approaches will likely involve combination therapy(146).
Summary and conclusion
TBI is widely regarded as a risk factor for all-cause dementia, particularly when sustained at older ages. TBI-mediated risk for specific neurodegenerative disease sub-types is less clear. Some administrative and population-based cohort studies have reported higher rates of dementia among those with a history of mTBI, though studies that use structured psychometric assessments and standardized diagnostic criteria and include subjects with less severe injuries (i.e., those not requiring acute care), are needed. Well-controlled, prospective cohort studies of outcomes following TBI, with neuropathological verification are lacking but underway(147). Polypathology in the context of a remote TBI is extremely challenging and fraught with opportunities for over-interpretation. While neuropathology is the gold standard for diagnosis of neurodegeneration, sensitive and specific psychometric, biofluid and/or imaging biomarkers are desperately needed to finally enable detection and monitoring of neurodegenerative disease processes during life. This will improve understanding of genetic, biological, and environmental drivers behind TBI-related acute and chronic changes in the brain and the potential for neurodegeneration.
Figure 1.
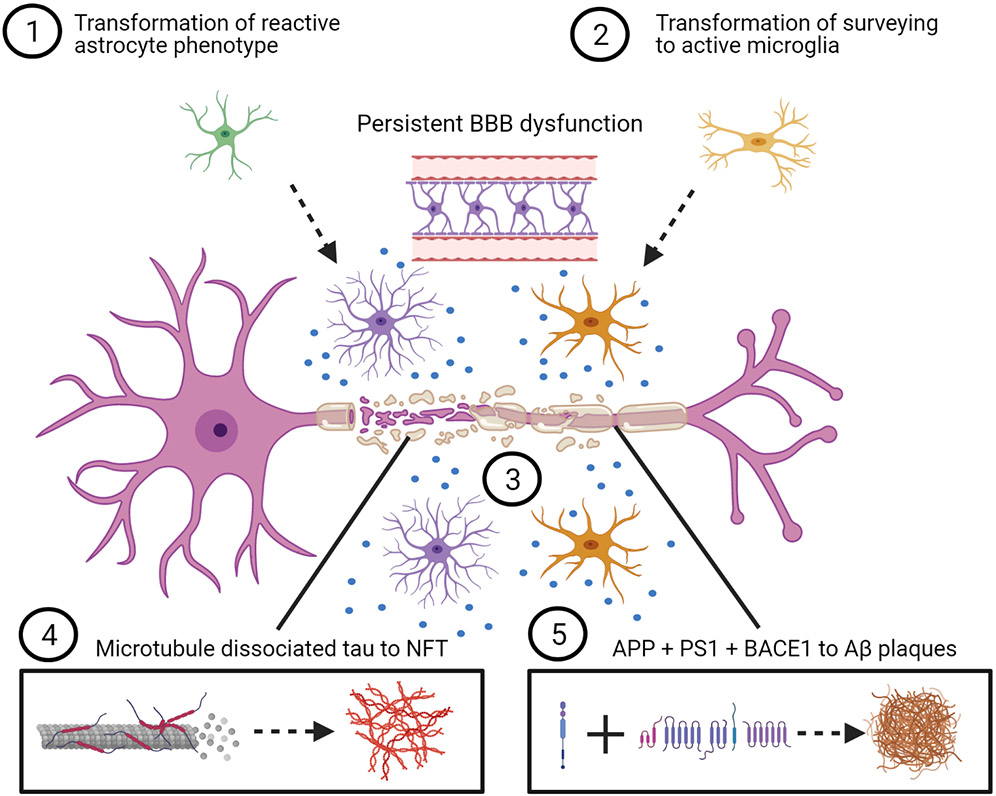
Conceptual representation of the post–traumatic brain injury evolution of acute to chronic response, potentially including neurodegenerative disease pathways. 1) Initial protective activation of astrocytes persists toward chronic morphology, contributing to deleterious effects of advanced aging and neurodegeneration, including prolonged proinflammatory cytokine release, disruption of BBB integrity, and glymphatic system disturbance. 2) Resident microglia react, potentially exacerbating neurotoxicity through prolonged activation or primed morphology, resulting in local and diffuse proinflammatory cytokine release (represented by blue dots), contributing to longer-term secondary injury. 3) Insult because of injury results in structural and myelin damage to the axon. Progressive Wallerian degeneration occurs through axonal transport disruption and secondary disconnection. 4) Sheering forces result in microtubule-dissociated tau and aberrant phosphorylation, possibly through coaccumulation of multiple protein kinases and phosphatases that regulate tau hyperphosphorylation, such as delta-secretase, glycogen synthase kinase-3β, and c-Jun N-terminal kinase. Progressive accumulation of phosphorylated tau exacerbates mitochondrial transport disruption and cell apoptosis. 5) APP is acutely increased, potentially leading to coaccumulation of Aβ peptide, BACE1, and PS1 in axons of white and gray matter over time after injury. Aβ, amyloid-β; APP, amyloid precursor protein; BACE1, β-secretase 1; BBB, blood-brain barrier; NFT, neurofibrillary tangle; PS1, presenilin-1. Created with BioRender.com.
Acknowledgements
BLB acknowledges support from the National Institute of Neurological Disorders and Stroke (NINDS) under the National Institutes of Health (NIH) under the award L30 NS113158. KDOC acknowledges support from NINDS under the awards RF1NS115268, U54 NS115322 and U54 NS115266, from the National Institute of Aging under the NIH through award R01 AG061028, from the National Institute on Disability, Independent Living, and Rehabilitation Research under award 90DPTB0009, and also from the Department of Defense under awards W81XWH-17-1-0330 and W81XWH-18-1-0580. CDK acknowledges support from NINDS under awards RF1NS115268, R01 AG061028 and U54 NS115322 and from the NIA under awards R01 AG061028, P50 AG066509, from the Department of Defense under award W81XWH-17-1-0330, and from the Nancy and Buster Alvord Endowment.
Footnotes
Competing Interests:
The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li Y, Li Y, Li X, Zhang S, Zhao J, Zhu X, et al. (2017): Head Injury as a Risk Factor for Dementia and Alzheimer's Disease: A Systematic Review and Meta-Analysis of 32 Observational Studies. PLoS One. 12:e0169650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang CH, Lin CW, Lee YC, Huang CY, Huang RY, Tai YC, et al. (2018): Is traumatic brain injury a risk factor for neurodegeneration? A meta-analysis of population-based studies. BMC Neurol. 18:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snowden TM, Hinde AK, Reid HMO, Christie BR (2020): Does Mild Traumatic Brain Injury Increase the Risk for Dementia? A Systematic Review and Meta-Analysis. J Alzheimers Dis. 78:757–775. [DOI] [PubMed] [Google Scholar]
- 4.Fann JR, Ribe AR, Pedersen HS, Fenger-Gron M, Christensen J, Benros ME, et al. (2018): Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. 5:424–431. [DOI] [PubMed] [Google Scholar]
- 5.Perry DC, Sturm VE, Peterson MJ, Pieper CF, Bullock T, Boeve BF, et al. (2016): Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J Neurosurg. 124:511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordstrom A, Nordstrom P (2018): Traumatic brain injury and the risk of dementia diagnosis: A nationwide cohort study. PLoS Med. 15:e1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raj R, Kaprio J, Korja M, Mikkonen ED, Jousilahti P, Siironen J (2017): Risk of hospitalization with neurodegenerative disease after moderate-to-severe traumatic brain injury in the working-age population: A retrospective cohort study using the Finnish national health registries. PLoS Med. 14:e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K (2014): Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 71:1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang HK, Lin SH, Sung PS, Wu MH, Hung KW, Wang LC, et al. (2012): Population based study on patients with traumatic brain injury suggests increased risk of dementia. J Neurol Neurosurg Psychiatry. 83:1080–1085. [DOI] [PubMed] [Google Scholar]
- 10.Kornblith E, Peltz CB, Xia F, Plassman B, Novakovic-Apopain T, Yaffe K (2020): Sex, race, and risk of dementia diagnosis after traumatic brain injury among older veterans. Neurology. 95:e1768–e1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K (2018): Association of Mild Traumatic Brain Injury With and Without Loss of Consciousness With Dementia in US Military Veterans. JAMA Neurol. 75:1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K (2014): Traumatic brain injury and risk of dementia in older veterans. Neurology. 83:312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. (2000): Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 55:1158–1166. [DOI] [PubMed] [Google Scholar]
- 14.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, et al. (2000): Head injury and the risk of AD in the MIRAGE study. Neurology. 54:1316–1323. [DOI] [PubMed] [Google Scholar]
- 15.Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, et al. (1990): The association between head trauma and Alzheimer's disease. Am J Epidemiol. 131:491–501. [DOI] [PubMed] [Google Scholar]
- 16.Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, et al. (1997): Alzheimer's disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 62:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suhanov AV, Pilipenko PI, Korczyn AD, Hofman A, Voevoda MI, Shishkin SV, et al. (2006): Risk factors for Alzheimer's disease in Russia: a case-control study. Eur J Neurol. 13:990–995. [DOI] [PubMed] [Google Scholar]
- 18.Sugarman MA, McKee AC, Stein TD, Tripodis Y, Besser LM, Martin B, et al. (2019): Failure to detect an association between self-reported traumatic brain injury and Alzheimer's disease neuropathology and dementia. Alzheimers Dement. 15:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tripodis Y, Alosco ML, Zirogiannis N, Gavett BE, Chaisson C, Martin B, et al. (2017): The Effect of Traumatic Brain Injury History with Loss of Consciousness on Rate of Cognitive Decline Among Older Adults with Normal Cognition and Alzheimer's Disease Dementia. J Alzheimers Dis. 59:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crane PK, Gibbons LE, Dams-O'Connor K, Trittschuh E, Leverenz JB, Keene CD, et al. (2016): Association of Traumatic Brain Injury With Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurol. 73:1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dams-O'Connor K, Gibbons LE, Bowen JD, McCurry SM, Larson EB, Crane PK (2013): Risk for late-life re-injury, dementia and death among individuals with traumatic brain injury: a population-based study. J Neurol Neurosurg Psychiatry. 84:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmes E, Ostbye T, Steenhuis RE (2011): Incremental contribution of reported previous head injury to the prediction of diagnosis and cognitive functioning in older adults. Brain Inj. 25:338–347. [DOI] [PubMed] [Google Scholar]
- 23.Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K (2018): Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology. 90:e1771–e1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jafari S, Etminan M, Aminzadeh F, Samii A (2013): Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov Disord. 28:1222–1229. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe Y, Watanabe T (2017): Meta-analytic evaluation of the association between head injury and risk of amyotrophic lateral sclerosis. Eur J Epidemiol. 32:867–879. [DOI] [PubMed] [Google Scholar]
- 26.Kalkonde YV, Jawaid A, Qureshi SU, Shirani P, Wheaton M, Pinto-Patarroyo GP, et al. (2012): Medical and environmental risk factors associated with frontotemporal dementia: a case-control study in a veteran population. Alzheimers Dement. 8:204–210. [DOI] [PubMed] [Google Scholar]
- 27.Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC (2003): Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosurg Psychiatry. 74:1574–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deutsch MB, Mendez MF, Teng E (2015): Interactions between traumatic brain injury and frontotemporal degeneration. Dement Geriatr Cogn Disord. 39:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC (2013): Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 8:e62422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morissette MP, Prior HJ, Tate RB, Wade J, Leiter JRS (2020): Associations between concussion and risk of diagnosis of psychological and neurological disorders: a retrospective population-based cohort study. Fam Med Community Health. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godbolt AK, Cancelliere C, Hincapie CA, Marras C, Boyle E, Kristman VL, et al. (2014): Systematic review of the risk of dementia and chronic cognitive impairment after mild traumatic brain injury: results of the International Collaboration on Mild Traumatic Brain Injury Prognosis. Arch Phys Med Rehabil. 95:S245–256. [DOI] [PubMed] [Google Scholar]
- 32.Mehta KM, Ott A, Kalmijn S, Slooter AJ, van Duijn CM, Hofman A, et al. (1999): Head trauma and risk of dementia and Alzheimer's disease: The Rotterdam Study. Neurology. 53:1959–1962. [DOI] [PubMed] [Google Scholar]
- 33.Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K (2015): Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol. 77:987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldman SM, Tanner CM, Oakes D, Bhudhikanok GS, Gupta A, Langston JW (2006): Head injury and Parkinson's disease risk in twins. Ann Neurol. 60:65–72. [DOI] [PubMed] [Google Scholar]
- 35.Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, et al. (2005): Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery. 57:719–726; discussion 719-726. [DOI] [PubMed] [Google Scholar]
- 36.Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang XL, et al. (2018): Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain. 141:422–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKee AC, Alosco ML, Huber BR (2016): Repetitive Head Impacts and Chronic Traumatic Encephalopathy. Neurosurg Clin N Am. 27:529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alosco ML, Koerte IK, Tripodis Y, Mariani M, Chua AS, Jarnagin J, et al. (2018): White matter signal abnormalities in former National Football League players. Alzheimers Dement (Amst). 10:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein TD, Alvarez VE, McKee AC (2015): Concussion in Chronic Traumatic Encephalopathy. Curr Pain Headache Rep. 19:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehman EJ, Hein MJ, Baron SL, Gersic CM (2012): Neurodegenerative causes of death among retired National Football League players. Neurology. 79:1970–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen VT, Zafonte RD, Chen JT, Kponee-Shovein KZ, Paganoni S, Pascual-Leone A, et al. (2019): Mortality Among Professional American-Style Football Players and Professional American Baseball Players. JAMA Netw Open. 2:e194223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janssen PH, Mandrekar J, Mielke MM, Ahlskog JE, Boeve BF, Josephs K, et al. (2017): High School Football and Late-Life Risk of Neurodegenerative Syndromes, 1956–1970. Mayo Clin Proc. 92:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deshpande SK, Hasegawa RB, Rabinowitz AR, Whyte J, Roan CL, Tabatabaei A, et al. (2017): Association of Playing High School Football With Cognition and Mental Health Later in Life. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abner EL, Nelson PT, Schmitt FA, Browning SR, Fardo DW, Wan L, et al. (2014): Self-reported head injury and risk of late-life impairment and AD pathology in an AD center cohort. Dement Geriatr Cogn Disord. 37:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson VE, Stewart W, Smith DH (2012): Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiner MW, Harvey D, Hayes J, Landau SM, Aisen PS, Petersen RC, et al. (2017): Effects of traumatic brain injury and posttraumatic stress disorder on development of Alzheimer's disease in Vietnam Veterans using the Alzheimer's Disease Neuroimaging Initiative: Preliminary Report. Alzheimers Dement (N Y). 3:177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asken BM, Mantyh WG, La Joie R, Strom A, Casaletto KB, Staffaroni AM, et al. (2021): Association of remote mild traumatic brain injury with cortical amyloid burden in clinically normal older adults. Brain Imaging Behav. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohamed AZ, Cumming P, Gotz J, Nasrallah F, Department of Defense Alzheimer's Disease Neuroimaging I (2019): Tauopathy in veterans with long-term posttraumatic stress disorder and traumatic brain injury. Eur J Nucl Med Mol Imaging. 46:1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stern RA, Adler CH, Chen K, Navitsky M, Luo J, Dodick DW, et al. (2019): Tau Positron-Emission Tomography in Former National Football League Players. N Engl J Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahata K, Kimura Y, Sahara N, Koga S, Shimada H, Ichise M, et al. (2019): PET-detectable tau pathology correlates with long-term neuropsychiatric outcomes in patients with traumatic brain injury. Brain. 142:3265–3279. [DOI] [PubMed] [Google Scholar]
- 51.Burke JF, Stulc JL, Skolarus LE, Sears ED, Zahuranec DB, Morgenstern LB (2013): Traumatic brain injury may be an independent risk factor for stroke. Neurology. 81:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. (2013): The spectrum of disease in chronic traumatic encephalopathy. Brain. 136:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. (2017): Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA. 318:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ling H, Morris HR, Neal JW, Lees AJ, Hardy J, Holton JL, et al. (2017): Mixed pathologies including chronic traumatic encephalopathy account for dementia in retired association football (soccer) players. Acta Neuropathol. 133:337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W (2019): Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol. 138:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walt GS, Burris HM, Brady CB, Spencer KR, Alvarez VE, Huber BR, et al. (2018): Chronic Traumatic Encephalopathy Within an Amyotrophic Lateral Sclerosis Brain Bank Cohort. J Neuropathol Exp Neurol. 77:1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kenney K, Iacono D, Edlow BL, Katz DI, Diaz-Arrastia R, Dams-O'Connor K, et al. (2018): Dementia After Moderate-Severe Traumatic Brain Injury: Coexistence of Multiple Proteinopathies. J Neuropathol Exp Neurol. 77:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson VE, Stewart W, Trojanowski JQ, Smith DH (2011): Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 122:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iacono D, Lee P, Edlow BL, Gray N, Fischl B, Kenney K, et al. (2020): Early-Onset Dementia in War Veterans: Brain Polypathology and Clinicopathologic Complexity. J Neuropathol Exp Neurol. 79:144–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, et al. (2015): Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 130:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. (2018): Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 141:2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI (1994): Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 57:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Early AN, Gorman AA, Van Eldik LJ, Bachstetter AD, Morganti JM (2020): Effects of advanced age upon astrocyte-specific responses to acute traumatic brain injury in mice. J Neuroinflammation. 17:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, et al. (2007): Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 208:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, et al. (2004): Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 190:192–203. [DOI] [PubMed] [Google Scholar]
- 66.Masumura M, Hata R, Uramoto H, Murayama N, Ohno T, Sawada T (2000): Altered expression of amyloid precursors proteins after traumatic brain injury in rats: in situ hybridization and immunohistochemical study. J Neurotrauma. 17:123–134. [DOI] [PubMed] [Google Scholar]
- 67.Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH (2002): Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 61:1056–1068. [DOI] [PubMed] [Google Scholar]
- 68.Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT (2002): Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J Neurotrauma. 19:601–614. [DOI] [PubMed] [Google Scholar]
- 69.Roberts GW, Gentleman SM, Lynch A, Graham DI (1991): beta A4 amyloid protein deposition in brain after head trauma. Lancet. 338:1422–1423. [DOI] [PubMed] [Google Scholar]
- 70.Tan XL, Sun M, Brady RD, Liu S, Llanos R, Cheung S, et al. (2018): Transactive Response DNA-Binding Protein 43 Abnormalities after Traumatic Brain Injury. J Neurotrauma. [DOI] [PubMed] [Google Scholar]
- 71.Johnson VE, Stewart W, Smith DH (2013): Axonal pathology in traumatic brain injury. Exp Neurol. 246:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Povlishock JT, Katz DI (2005): Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 20:76–94. [DOI] [PubMed] [Google Scholar]
- 73.Jolly AE, Balaet M, Azor A, Friedland D, Sandrone S, Graham NSN, et al. (2020): Detecting axonal injury in individual patients after traumatic brain injury. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR (1989): Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 15:49–59. [DOI] [PubMed] [Google Scholar]
- 75.Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH (2012): Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol. 233:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raghupathi R, Graham DI, McIntosh TK (2000): Apoptosis after traumatic brain injury. J Neurotrauma. 17:927–938. [DOI] [PubMed] [Google Scholar]
- 77.Tang-Schomer MD, Patel AR, Baas PW, Smith DH (2010): Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 24:1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Graham NSN, Jolly A, Zimmerman K, Bourke NJ, Scott G, Cole JH, et al. (2020): Diffuse axonal injury predicts neurodegeneration after moderate-severe traumatic brain injury. Brain. 143:3685–3698. [DOI] [PubMed] [Google Scholar]
- 79.Palacios EM, Owen JP, Yuh EL, Wang MB, Vassar MJ, Ferguson AR, et al. (2020): The evolution of white matter microstructural changes after mild traumatic brain injury: A longitudinal DTI and NODDI study. Sci Adv. 6:eaaz6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu YC, Harezlak J, Elsaid NMH, Lin Z, Wen Q, Mustafi SM, et al. (2020): Longitudinal white-matter abnormalities in sports-related concussion: A diffusion MRI study. Neurology. 95:e781–e792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Churchill NW, Caverzasi E, Graham SJ, Hutchison MG, Schweizer TA (2019): White matter during concussion recovery: Comparing diffusion tensor imaging (DTI) and neurite orientation dispersion and density imaging (NODDI). Hum Brain Mapp. 40:1908–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tremblay S, Henry LC, Bedetti C, Larson-Dupuis C, Gagnon JF, Evans AC, et al. (2014): Diffuse white matter tract abnormalities in clinically normal ageing retired athletes with a history of sports-related concussions. Brain. 137:2997–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mac Donald CL, Barber J, Andre J, Panks C, Zalewski K, Temkin N (2019): Longitudinal neuroimaging following combat concussion: sub-acute, 1 year and 5 years post-injury. Brain Commun. 1:fcz031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar-Queralt A, Shah SB, et al. (2017): Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell Mol Neurobiol. 37:571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chambers S (2008): Gerard Manley Hopkins and the kinesthetics of conviction. Vic Stud. 51:7–35. [DOI] [PubMed] [Google Scholar]
- 86.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH (2004): Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 165:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. (2005): Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 307:1282–1288. [DOI] [PubMed] [Google Scholar]
- 88.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001): Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 414:643–648. [DOI] [PubMed] [Google Scholar]
- 89.Terwel D, Dewachter I, Van Leuven F (2002): Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. Neuromolecular Med. 2:151–165. [DOI] [PubMed] [Google Scholar]
- 90.Smith C, Graham DI, Murray LS, Nicoll JA (2003): Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. 29:496–502. [DOI] [PubMed] [Google Scholar]
- 91.Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011): Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci. 31:9513–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, et al. (2018): Induction of a transmissible tau pathology by traumatic brain injury. Brain. 141:2685–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, et al. (2017): Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. 8:1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y (2020): Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener. 15:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tran HT, Sanchez L, Brody DL (2012): Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol. 71:116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu Z, Wang ZH, Liu X, Zhang Z, Gu X, Yu SP, et al. (2020): Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer's disease pathology. Prog Neurobiol. 185:101730. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, et al. (2017): The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. 12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP (2013): Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, et al. (2016): Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 19:1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, et al. (2014): Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 34:16180–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. (2011): In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 31:13110–13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Corps KN, Roth TL, McGavern DB (2015): Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 72:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014): Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 73:14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nagamoto-Combs K, McNeal DW, Morecraft RJ, Combs CK (2007): Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J Neurotrauma. 24:1719–1742. [DOI] [PubMed] [Google Scholar]
- 105.Scott G, Hellyer PJ, Ramlackhansingh AF, Brooks DJ, Matthews PM, Sharp DJ (2015): Thalamic inflammation after brain trauma is associated with thalamo-cortical white matter damage. J Neuroinflammation. 12:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. (2011): Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 70:374–383. [DOI] [PubMed] [Google Scholar]
- 107.Witcher KG, Bray CE, Chunchai T, Zhao F, O'Neil SM, Gordillo AJ, et al. (2021): Traumatic brain injury causes chronic cortical inflammation and neuronal dysfunction mediated by microglia. J Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barrett JP, Henry RJ, Shirey KA, Doran SJ, Makarevich OD, Ritzel RM, et al. (2020): Interferon-beta Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration. J Neurosci. 40:2357–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. (2017): Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Coughlin JM, Wang Y, Minn I, Bienko N, Ambinder EB, Xu X, et al. (2017): Imaging of Glial Cell Activation and White Matter Integrity in Brains of Active and Recently Retired National Football League Players. JAMA Neurol. 74:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loane DJ, Kumar A (2016): Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol. 275 Pt 3:316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV (2006): Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 129:2761–2772. [DOI] [PubMed] [Google Scholar]
- 113.Burda JE, Bernstein AM, Sofroniew MV (2016): Astrocyte roles in traumatic brain injury. Exp Neurol. 275 Pt 3:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001): Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 21:7153–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J (2018): Microglia in neurodegeneration. Nat Neurosci. 21:1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, et al. (2004): Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 24:5016–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang X, Hasan O, Arzeno A, Benowitz LI, Cafferty WB, Strittmatter SM (2012): Axonal regeneration induced by blockade of glial inhibitors coupled with activation of intrinsic neuronal growth pathways. Exp Neurol. 237:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen H, Desai A, Kim HY (2017): Repetitive Closed-Head Impact Model of Engineered Rotational Acceleration Induces Long-Term Cognitive Impairments with Persistent Astrogliosis and Microgliosis in Mice. J Neurotrauma. 34:2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013): Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 136:28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Witcher KG, Bray CE, Dziabis JE, McKim DB, Benner BN, Rowe RK, et al. (2018): Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia. 66:2719–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP (2014): Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry. 76:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, et al. (1997): Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 14:715–727. [DOI] [PubMed] [Google Scholar]
- 123.Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J (2012): Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation. 9:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, et al. (2020): Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. J Neurosci. 40:2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Palin K, Cunningham C, Forse P, Perry VH, Platt N (2008): Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol Dis. 30:19–29. [DOI] [PubMed] [Google Scholar]
- 126.Witcher KG, Eiferman DS, Godbout JP (2015): Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 38:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Henry CJ, Huang Y, Wynne AM, Godbout JP (2009): Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain BehavImmun. 23:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li K, Li J, Zheng J, Qin S (2019): Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 10:664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Candelario-Jalil E, Yang Y, Rosenberg GA (2009): Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 158:983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, et al. (2011): The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 114:92–101. [DOI] [PubMed] [Google Scholar]
- 131.Abrahamson EE, Ikonomovic MD (2020): Brain injury-induced dysfunction of the blood brain barrier as a risk for dementia. Exp Neurol. 328:113257. [DOI] [PubMed] [Google Scholar]
- 132.Bolte AC, Dutta AB, Hurt ME, Smirnov I, Kovacs MA, McKee CA, et al. (2020): Meningeal lymphatic dysfunction exacerbates traumatic brain injury pathogenesis. Nat Commun. 11:4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang Z, Wong LW, Su Y, Huang X, Wang N, Chen H, et al. (2020): Blood-brain barrier integrity in the pathogenesis of Alzheimer's disease. Front Neuroendocrinol. 59:100857. [DOI] [PubMed] [Google Scholar]
- 134.Smith JA, Das A, Ray SK, Banik NL (2012): Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 87:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ding K, Marquez de la Plata C, Wang JY, Mumphrey M, Moore C, Harper C, et al. (2008): Cerebral atrophy after traumatic white matter injury: correlation with acute neuroimaging and outcome. J Neurotrauma. 25:1433–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim J, Whyte J, Patel S, Avants B, Europa E, Wang J, et al. (2010): Resting cerebral blood flow alterations in chronic traumatic brain injury: an arterial spin labeling perfusion FMRI study. J Neurotrauma. 27:1399–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Venkatesan UM, Dennis NA, Hillary FG (2015): Chronology and chronicity of altered resting-state functional connectivity after traumatic brain injury. J Neurotrauma. 32:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pascual B, Funk Q, Zanotti-Fregonara P, Cykowski MD, Veronese M, Rockers E, et al. (2021): Neuroinflammation is highest in areas of disease progression in semantic dementia. Brain. [DOI] [PubMed] [Google Scholar]
- 139.Datta G, Colasanti A, Rabiner EA, Gunn RN, Malik O, Ciccarelli O, et al. (2017): Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain. 140:2927–2938. [DOI] [PubMed] [Google Scholar]
- 140.Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Rodriguez JL, Snellman A, et al. (2021): Time course of phosphorylated-tau181 in blood across the Alzheimer's disease spectrum. Brain. 144:325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. (2018): High performance plasma amyloid-beta biomarkers for Alzheimer's disease. Nature. 554:249–254. [DOI] [PubMed] [Google Scholar]
- 142.Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Lantero Rodriguez J, Snellman A, et al. (2021): Longitudinal Associations of Blood Phosphorylated Tau181 and Neurofilament Light Chain With Neurodegeneration in Alzheimer Disease. JAMA Neurol. 78:396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dik MG, Jonker C, Hack CE, Smit JH, Comijs HC, Eikelenboom P (2005): Serum inflammatory proteins and cognitive decline in older persons. Neurology. 64:1371–1377. [DOI] [PubMed] [Google Scholar]
- 144.Kozin SA, Barykin EP, Mitkevich VA, Makarov AA (2018): Anti-amyloid Therapy of Alzheimer's Disease: Current State and Prospects. Biochemistry (Mosc). 83:1057–1067. [DOI] [PubMed] [Google Scholar]
- 145.Cummings J, Blennow K, Johnson K, Keeley M, Bateman RJ, Molinuevo JL, et al. (2019): Anti-Tau Trials for Alzheimer's Disease: A Report from the EU/US/CTAD Task Force. J Prev Alzheimers Dis. 6:157–163. [DOI] [PubMed] [Google Scholar]
- 146.Cummings JL, Tong G, Ballard C (2019): Treatment Combinations for Alzheimer's Disease: Current and Future Pharmacotherapy Options. J Alzheimers Dis. 67:779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Edlow BL, Keene CD, Perl DP, Iacono D, Folkerth RD, Stewart W, et al. (2018): Multimodal Characterization of the Late Effects of Traumatic Brain Injury: A Methodological Overview of the Late Effects of Traumatic Brain Injury Project. J Neurotrauma. 35:1604–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]