

Abstract
Cloud-hosted environments offer known benefits when computational needs outstrip affordable local workstations, enabling high-performance computation without a physical cluster. What has been less apparent, especially to novice users, is the transformative potential for cloud-hosted environments to bridge the digital divide that exists between poorly funded and well-resourced laboratories, and to empower modern research groups with remote personnel and trainees. Using cloud-based proteomic bioinformatic pipelines is not predicated on analyzing thousands of files, but instead can be used to improve accessibility during remote work, extreme weather, or working with under-resourced remote trainees. The general benefits of cloud-hosted environments also allow for scalability and encourage reproducibility. Since one possible hurdle to adoption is awareness, this paper is written with the nonexpert in mind. The benefits and possibilities of using a cloud-hosted environment are emphasized by describing how to setup an example workflow to analyze a previously published label-free data-dependent acquisition mass spectrometry data set of mammalian urine. Cost and time of analysis are compared using different computational tiers, and important practical considerations are described. Overall, cloud-hosted environments offer the potential to solve large computational problems, but more importantly can enable and accelerate research in smaller research groups with inadequate infrastructure and suboptimal local computational resources.
Keywords: cloud computing, remote access, high-performance computing, proteomic workflow
Graphical Abstract
INTRODUCTION
Remote hosted computational environments, often referred to as the cloud, allow for potentially easier setup, faster processing, and lower cost to build and manage than local workstations. Recently, with increased remote work due to pandemic driven lockdowns, globalization of scientific research (e.g., globally distributed research consortia), and the growth of public data repositories and reanalysis of public data sets, using cloud-hosted environments as the computational backbone of research offers increasing advantages. Specific to proteomics there are opportunities for increased use of cloud-hosted environments, leading to improved accessibility, scalability, and reproducibility. Currently, there is a plethora of cloud-based options and different benefits, with real and perceived bottlenecks to implementation. These benefits are not limited to utilizing cloud-hosted environments for high-throughput large-scale proteomics data, but can also prove essential for remote work, remote training, or disaster resilience. To help the reader, a glossary of terms is provided.
CLOUD COMPUTING
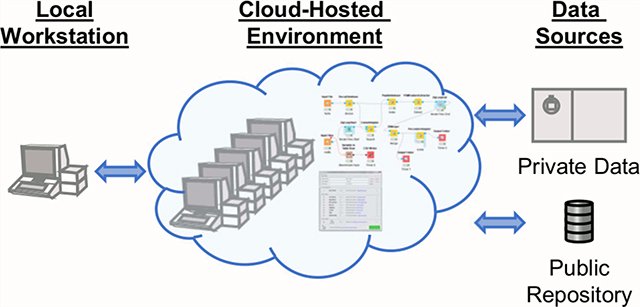
Depending on the available resources of both investigators and their institution/company, there exists a spectrum of local computational capacity. These resources may be available via remote access, but many cases exist when this is not possible often due to institutional security concerns. By utilizing cloud-hosted environments (Figure 1), computational work may continue even when local resources are inaccessible (due to lockdowns, extreme weather, inconsistent power, etc.) and provide access to remote colleagues and trainees (including those in other countries) that otherwise could be using inadequate local resources. Furthermore, using cloud-hosted environments allows users to quickly scale resources to accomplish larger tasks at appropriate times, instead of purchasing or upgrading local hardware that runs far below its potential most of the time and requires periodic system hardware and software maintenance. Depending on the cloud environment, adding computational resources can be as simple as “building” a new virtual cluster with the click of a button, using an instance with more cores or memory with an existing image, utilizing workflows that automatically scale across a virtual cluster depending on workload, or executing a function in a serverless framework. In theory, this means that every researcher can have the same computational capacity, regardless of location.
Figure 1.
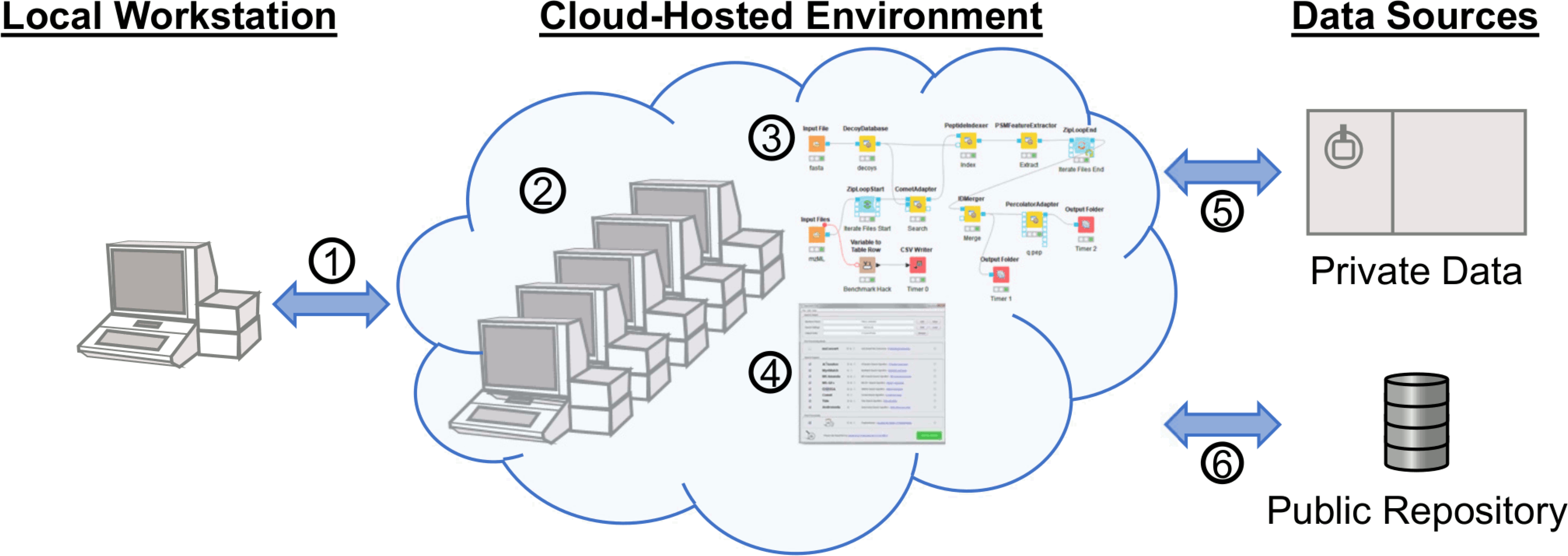
Generalized concept of proteomics in a cloud-hosted environment. The environment is accessed via any client with Internet access (l). Data for analysis can be retrieved directly to the environment from private data sources (5) and public data repositories (6). Computational resources can be assigned to the environment prior to analysis (2), at which point programs (4) or workflows (3) may be used to complete the analysis.
Cloud-based computing can trace a line from the time-sharing of the 1950s1 through the packet radio van of 19772 into the modern “cloud” available from public and commercial providers. In many regions across the globe, there are federally subsidized resources available to researchers through varied application processes. For instance, Jetstream (a distributed collaboration of the University of Indiana, University of Texas at Austin, and University of Arizona) on the National Science Foundation-funded Extreme Science and Engineering Discovery Environment (XSEDE) allows users to start instances using a catalog of Linux-based images, many already preconfigured for common research applications, and create single multi-CPU and high memory instances or multiple-node virtual clusters, and access these by web-based console or remote desktop. Setting up seemingly complicated virtual clusters can be accomplished with ease when a tutorial exists, exemplified by MAKER on Jetstream,3 Cactus on Amazon Web Services (AWS),4 and Bioconductor on AWS.5 The ease of using a service like Jetstream for a cloud novice cannot be understated. Similar to XSEDE, other publicly subsidized resources including ACI-REF (Advanced Cyberinfrastructure Research and Education Facilitators) Network, ELIXIR (the European life-sciences Infrastructure for biological Information), NCI (National Computational Infrastructure) Australia, and PRACE (Partnership for Advanced Computing in Europe) make free hosted computation time available to researchers. Although the free or low-cost nature of these services is preferred, commercial resources are also available and include AWS, Microsoft Azure, and Google Cloud, which also have free tiers or trials available. These services, both public and commercial, are the backbone of most large computational efforts from particle physics to population genomics.
PROTEOMICS IN THE CLOUD
Mass spectrometry-based proteomics is a broad term encompassing many applications6 used across different biological systems,7 and accordingly has an abundance of software tools available.8 Protein inference is made possible by peptide identification following database searching of tandem mass spectrometry data.9 Computation in many modern protein identification algorithms is performed in RAM with high-speed CPUs and varied I/O requirements. These computational requirements mean that high-performance local machines are perceived as better suited to database searching, as opposed to cloud-hosted environments, though there have been notable cost benefit analyses over the past decade showing the benefits of proteomics analysis in cloud-hosted environments.10,11 In contrast, nucleic acid sequencing computation has historically relied on massive parallelization on modestly appointed motherboards, and therefore was very amenable to cloud applications. Despite this historic precedent, with decreasing cloud computing costs there is an ever increasing list of cloud-based proteomic solutions including Bolt12 and ionbot.13 More importantly, there are software platforms specifically tuned to run proteomic data processing and analysis in the cloud, such as Galaxy-P.14,15 Even without being specifically tailored to a cloud environment, any software can be used in a cloud-hosted environment, though there may be concerns for licensing. Software that can run in a Linux environment is best for services like Jetstream or containers like Singularity, while software that can run in Windows may be used for services like AWS or containers like Docker. Examples of free or open-source proteomic software that can run in a Linux environment include Crux,16 EncyclopeDIA,17 ProteoWizard,18 SearchGUI,19 The OpenMS Proteomics Pipeline (referred to as OpenMS or TOPP),20,21 Trans-Proteomic Pipeline (TPP),22,23 X!Tan-dem,24 and FragPipe.25,26 Other tools that run in Windows but can also be run via command line in a Linux environment include MaxQuant,27,28 MetaMorpheus,29 and Spritz.30 In order to truly take advantage of the scalability of cloud environments, software that can work in a clustered environment is preferred. This relies on distributing tasks across nodes, often integrated with workflow engines including the Konstanz Information Miner (KNIME),31 Makeflow,32 Nextflow,33 Snakemake,34 Swift,35 and Toil,36 which frequently provide tutorials specific to scaling in cloud-hosted environments. A recent review of proteomic software, containerization, and workflow engines highlights the benefits related to scalability.37 Regardless, most proteomic applications have not made the transition to being capable of fully utilizing modern clustering options, though database searching has been shown to benefit greatly from parallelization.38 Notable noncommercial exceptions include MS-PyCloud,39 SEQUEST-PVM,40 UltraQuant, which uses Snakemake to run a containerized MaxQuant,41 and OpenMS-based tools, which can be run on a cluster using KNIME42 and Nextflow.43,44 Beyond workflows and virtual clusters, in the coming years computational steps will be offloaded onto serverless frameworks (i.e., function as a service; FaaS),45 blurring the line between local and cloud-hosted environments. Given the potential of cloud-hosted environments, it seems that we are on the cusp of seeing a shift to cloud-based solutions in proteomics.
TESTING PERFORMANCE IN THE CLOUD
It has been said that the future is already here, it is just not evenly distributed yet (paraphrased from William Gibson), which is especially true of cloud computing. Research in numerous fields including materials science, astronomy, and genomics rely heavily on cloud-based computing, while it is largely absent in proteomic research. Aside from knowledge of these resources and tools, a common hindrance is understanding the ease of use and estimating time and cost.10,11 With respect to time, it is difficult to directly compare cloud-hosted environments to local-hardware given the diversity and dynamic nature of computational time and costs, and the fact that processing time is affected by everything from algorithm, general code, and settings optimization, to the processing pipeline's physical arhitecture used for the cloud-hosted environment. For this reason, tools like the TPP Amazon simulator11 or an exploratory analysis such as presented here can help estimate the scale of time and costs. Since different search algorithms will use resuores differntily and react diffently to search settings (e.g., mass tolerance, database size, variable modifications, and quantification), users should benchmark their preferred tools with their typical data sets. Broader efforts such as the ongoing proteomic data analysis pipeline comparison led by the ELIXR Proteomics Commun-ity46 will help clarify pipeline performance. For the discussion herein, a previously published study47 with a follow-up analysis48 of label-free data-dependent acquisition shotgun proteomic data from mammalian urine was chosen. For this example, the data was analyzed using AWS Elastic Compute Cloud (EC2) instances with an arbitrarily chosen OpenMS-based Comet-Percolator workflow constructed with KNIME (Figure 2). The goal was to demonstrate time and costs with different computational resources on a ubiquitous commercial platform using a typical label-free data-dependent shotgun proteomic experiment.
Figure 2.
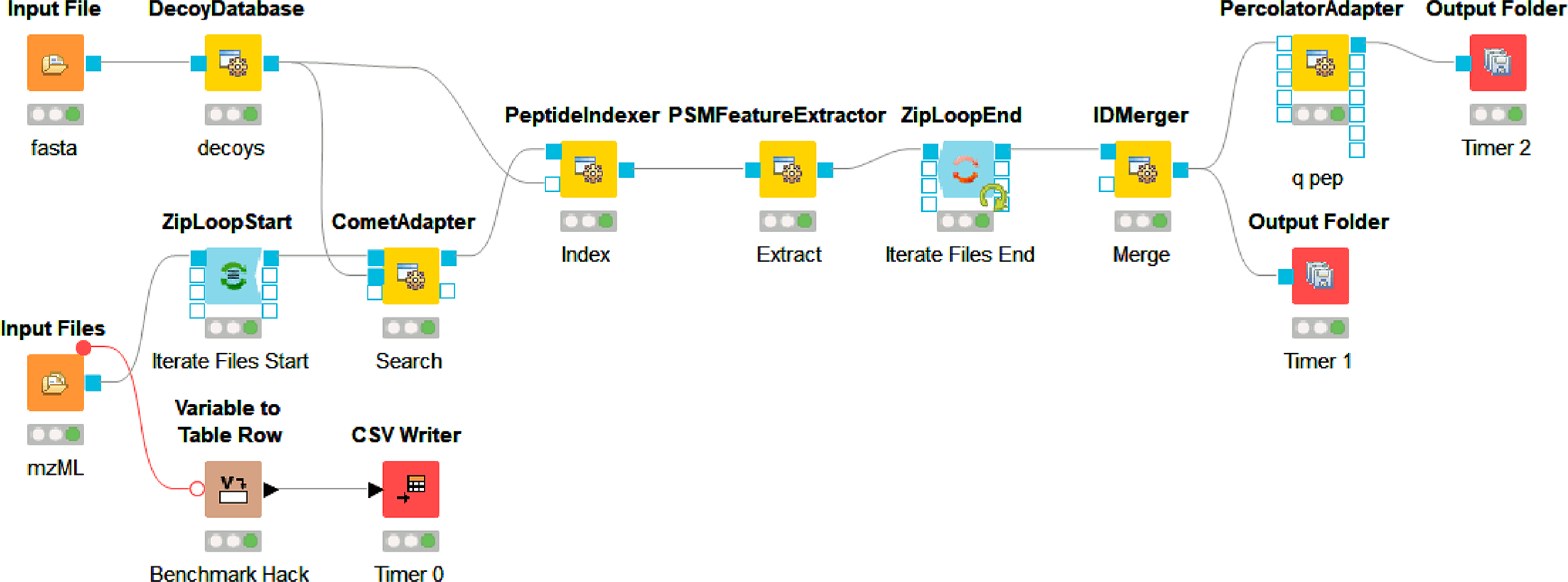
KNIME workflow using OpenMS adapters. OpenMS 2.6 KNIME nodes for a simple workflow loading spectra and protein sequence files, performing a search, and consolidating peptide and protein IDs found in the input spectra files. A simple approach to benchmarking was achieved through a standard KNIME node and file write timestamps.
EXAMPLE CLOUD SET-UP
Although AWS EC2 was chosen for this example, most services offer similar remote desktop access. This means that even for the cloud novice, making productive use of instances does not require command line work, but instead can look just like the computer they are already using by access via remote clients such as Windows Remote Desktop (RDP), Virtual Network Computing (VNC), Team Viewer or other available remote desktop options. For this example, a community Amazon Machine Image (AMI: Windows_Server-2019-English-Full-Base-2020.09.09) was used as a c5d.xlarge instance to set up the software and run analysis. The c5d instance types provide very fast local (to the instance’s motherboard) scratch disk space that is erased upon shut down of the on-demand instance. The 19 raw files from PRIDE PXD00901949 of approximately 1.3 gigabytes each were copied to the instance using FTP. It should be noted that transfer speeds tend to be faster between a public cloud-hosted environment and a public data repository since they tend to be network proximal; for example, there seem to be fewer hops and bigger pipes between MassIVE and XSEDE. Additional tools such as Globus or Aspera can make high transfer speed through proximity for data that are not in public repositories. For the analysis, CSL16 was omitted and the other 18 files were used. Once the raw files were loaded onto the instance, MSConvert 3.0.20280 was installed to derive MS2 mzML files. The remove_duplicates.py script50 was used to collapse duplicate fasta entries prior to workflow execution. The KNIME 4.2.2 scientific workflow platform was installed with OpenMS 2.6.0 nodes and a simple workflow was built using KNIME OpenMS tutorials as a guide.42,51 Broadly, a mix of OpenMS native nodes including DecoyDatabase, PeptideIndexer, PSMFeatureExtractor, and IDMerger, and adapter nodes for the Comet search engine52 and Percolator53 were used, and the specific KNIME workflow with required files and settings for replication locally or in cloud-hosted environments is publicly available,54 as well as the final AMI (ami-0dead6b478bd16281 on us-east-2 region). These different software were chosen to demonstrate the capabilities and possibilities of this approach. Following completion of the workflow with the c5d.xlarge instance type, outputs, and benchmark times were saved from the scratch drive to a long-term EBS (Elastic Block Store) volume and the instance was shut down. Two further iterations were completed in the same manner by rebooting and rerunning the same workflow using the c5d.2xlarge and c5d.12xlarge instance types. Only the threads parameter was changed and the resulting benchmarking information was saved after confirming the idXML outputs. For all three instance types tested, the number of parallel threads allowed to be used by the CometAdapter was set at one less than the number of cores available to the instance type. A representative completed search result was retained in long-term storage, and the results could be transferred elsewhere by various manners including browser-based file uplnoad from the desktop environment before shutting down.
TIME AND COST
In the case of a commercial provider, it is recommended to use a modest computational tier with low hourly cost for learning the system and setting up the workflow. It is also important to note that on-demand pricing of services like EC2 requires instances to be manually shut down when not in use. For high-end instances, if not shut down, the monthly bill can easily exceed many thousand dollars. For this specific example, three c5d instance types were compared and analysis time and cost were determined using the same data, workflow, and search parameters (Table 1). As stated before, the time and cost aspect of this comparison is extremely dynamic and will change depending on computational speeds of instances (which are periodically upgraded), software (and their updates), and search settings (e.g., number of variable modifications). This is also important to note when comparing to running on local resources or in other cloud-hosted environments. For comparison, there are benchmarks available for this specific data set analyzed on different hardware with different software, which is being updated here.55 Specific to the results described herein, unsurprisingly the search was quicker with the higher performance tiers, but this came at a monetary cost, similar to other studies.10,11 Though this trade-off is important to note, in situations where accessibility is the main concern, it demonstrates that a modestly powered instance can perform well at a low cost, especially since “set it and forget it” is a common approach when analyzing proteomic results locally. Still, any costs may prove prohibitive when resources are limited, though with federally subsidized resources like Jetstream, this is surmountable. Budgeting dynamic costs versus one-time hardware purchases is also difficult, but it is expected that this cost model will continue to be easier to cover as institutional views shift to preferring cloud-hosted environments versus local infrastructure. There are additional concerns beyond this discussion concerning privacy and security concerns of using cloud-hosted environments for certain types of data, and this may also affect costs. Whether speed or cost is a priority is up to each user and situation, but given the elastic nature of resource allocation, this decision can be made dynamically, further emphasizing the power of working in a cloud-hosted environment.
Table 1.
Time and Cost of Running the Same 18 Injection Search-Only Workflow on Three Instance Typesa
| c5d.large | c5d.2xlarge | c5d.12xlarge | |
|---|---|---|---|
| processors available | 2 | 8 | 48 |
| processors used | 1 | 7 | 47 |
| run rate ($/h) | 0.22 | 0.84 | 5.08 |
| search time (hours:minutes) | 3:54 | 1:15 | 0:31 |
| total workflow time (hours:minutes) | 4:01 | 1:21 | 0:37 |
| cost ($) | 0.88 | 1.13 | 3.13 |
Each raw file is approximately 1.3 gigabytes and contains roughly 65 000 MS2 scans. The comparison of the large versus the 12× large c5 instance found that the higher performance is about three times more expensive but six times faster.
RESULT HANDLING AND REPRODUCIBILITY
Best practices for using a cloud-hosted environment will vary across fields, but in proteomic data analysis the primary computational bottlenecks are file conversion, processing spectra, peptide identification, protein inference, and relative quantification if applicable. Downstream steps such as differential analysis or enrichment analysis can be performed with fewer computational resources, meaning these steps are likely more appropriate on local systems. Following completion of the search steps, results files can be retrieved to local workstations. Typically, result files can be explored using the same software used to generate the data or software-specific viewers (e.g., PeptideShaker for SearchGUI output). Alternatively, flat file exports (e.g., csv) may be shared between users. It is also possible to stay completely within the cloud by using one of a growing number of cloud-based services for statistical analysis and result sharing (e.g., SimpliFi56).
One of the most important benefits of using cloud-hosted environments is the opportunities for reproducibility.57 Images can be shared privately or publicly between users, allowing others to reproduce the same operating system and software versions and, if desired, settings as the original analysis. Similar to previous cloud-based proteomic analyses that supplied AMIs to encourage reproducibility,10,11 the AMI used in this example has been shared publicly on EC2 (ami-0dead6b478bd16281 on us-east-2 region), and is ready for use following modification of KNIME memory allocation and CometAdapter threads to match the instance’s resources. An image may also be cloned and modified if a user wants to update or change software, thus allowing for comparison of results all while preserving the original environment. Another way to achieve reproducibility is by using containers. For example, software from GitHub can be packaged into a Singularity container58 that can be linked with other workflow steps using Nextflow. In this way, more complex sets of software with different dependencies can work together in a pipeline that can be used in a cloud-hosted environment. The reason this is preferred is that software is exactly preserved and shared via GitHub and the container can also be made available via repositories such as Singularity Container Registry,59 DockerHub60 or BioContainers,61 while optimized application-specific Nextflow workflows are available via nf-core.62 Together, this degree of portability and reproducibility enables replication by anyone on any system.
REAL AND PERCEIVED LIMITATIONS
With the concurrent advancements in proteomic data repositories and software along with pricing and performance of cloud-hosted environments, there are fewer limitations than ever to take proteomics to the cloud. In addition to the points addressed in previous sections, data transfer speed and storage costs present different limitations. Transferring data from repositories or from a private resource to cloud-hosted environments can be very fast depending on where the actual servers are located. Although it can be tedious and unintuitive, it is worthwhile to choose services and host locations with data sources in mind. In the future, there will be improved integration of data repositories with cloud-hosted resources (e.g., Google’s Cloud Life Sciences public data sets), which will increase usability and reduce data storage costs. Currently, the cost of storage used with a computational instance will vary from free to minimal depending on the service, but it is a fraction of the computational cost if managed properly. Finally, maybe the most crucial limitation to adoption of many of the resources and tools is the perceived diffculty. The proteomics community could address this by creating more prebuilt proteomic-centric images (similar to those available for the TPP11), while software and pipeline developers could provide detailed vignettes using real data on different cloud-hosted environments.
FUTURE OUTLOOK
When researchers look to the cloud it is often to accomplish tasks that are not possible with local workstations. Although proteomics researchers are adept at using local resources to accomplish large computational tasks, there is far-reaching potential in developing and utilizing cloud-hosted environments for proteomic needs. The resilience of using remote resources should not be understated in the current climate, and though not foolproof, they are lockdown proof. More importantly, as the global flow of people has slowed, utilizing the cloud to avoid lost time with trainees in other countries or to maintain research with distant colleagues is invaluable and can be facilitated using shared cloud resources. Moreover, these benefits are applicable beyond mass spectrometry-based proteomics since other similar mass spectrometry-based domains can benefit from cloud-hosted environments, including imaging, lipidomics, and metabolomics.37 As it becomes more common to work in cloud-hosted environments we will see benefits that will continue to drive the field forward.
ACKNOWLEDGMENTS
The author wishes to thank Devin Drew (Thermo Fisher Scientific) for his assistance in implementing searches on AWS and for his expertise in using proteomic software in cloud environments. The author also wishes to thank Phillip Wilmarth (Oregon Health and Science University), Brian Beck (University of Texas at Austin), and the anonymous peer-reviewers for critical insight and feedback. The identification of certain commercial equipment, instruments, software, or materials does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
GLOSSARY
- Cloud computing
Computing performed with remote resources. Arguably, the first “cloud” drawn as an information system visual was for the first transatlantic demonstration of connectivity among ARPANET, SATNET, and PRNET in 1977.2
- Container
An encapsulated environment typically containing only one program and associated dependencies, not to be confused with a virtual appliance, which is more akin to an image. A container can be run with a set of input and output arguments via a command or in a workflow.
- Image
A complete snapshot (“template”) of a computational environment including operating system.
- Instance
The instantiation of an instance type (putting a machine image onto an instance type and “spinning it up” in a server farm somewhere).
- Instance type
A set of attributes describing number of processor cores, memory, and I/O resources having a cost per unit time.
- Pipeline
A generic term referring to linked steps of analysis. Workflow engines generate workflows, often referred to as pipelines.
- Running instance
An instance that is accumulating cost attributable to its instance type and associated data storage and I/O.
- Stopped (or not running) instance
An instance accumulating cost attributable only to the storage of it is image.
- Time-sharing (computing term)
A concept dating from the 1950s relating to a computer system handling a number of problems (for different users) concurrently.1
- Workflow engines
Typically a visual tool to link steps that are wrappers or containers of other programs. May be used in different environments including clustering if capable.
Footnotes
The author declares no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00920
REFERENCES
- (1).Lebo H 100 Days: How Four Events in 1969 Shaped America; Rowman and Littlefield Publishers, 2019. [Google Scholar]
- (2).Cerf VG The day the Internet age began. Nature 2009, 461 (7268), 1202–3. [DOI] [PubMed] [Google Scholar]
- (3).Devisetty UK; Tuteja R MAKER 2.31.9 with CCTOOLS Jetstream Tutorial. https://cyverse.atlassian.net/wiki/spaces/TUT/pages/258736333/MAKER+2.31.9+with+CCTOOLS+Jetstream+Tutorial (accessed 26 October 2020). [Google Scholar]
- (4).Running Cactus on AWS. https://github.com/ComparativeGenomicsToolkit/cactus/blob/master/doc/running-in-aws.md (accessed 26 October 2020).
- (5).Bioconductor in the cloud. https://www.bioconductor.org/help/bioconductor-cloud-ami/ (accessed 16 November 2020).
- (6).Pino LK; Rose J; O’Broin A; Shah S; Schilling B Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochem. Soc. Trans. 2020, 48 (5), 1953–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Heck M; Neely BA Proteomics in Non-model Organisms: A New Analytical Frontier. J. Proteome Res. 2020, 19 (9), 3595–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tsiamis V; Ienasescu HI; Gabrielaitis D; Palmblad M; Schwämmle V; Ison J One Thousand and One Software for Proteomics: Tales of the Toolmakers of Science. J. Proteome Res. 2019, 18 (10), 3580–3585. [DOI] [PubMed] [Google Scholar]
- (9).Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–89. [DOI] [PubMed] [Google Scholar]
- (10).Halligan BD; Geiger JF; Vallejos AK; Greene AS; Twigger SN Low cost, scalable proteomics data analysis using Amazon’s cloud computing services and open source search algorithms. J. Proteome Res. 2009, 8 (6), 3148–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Slagel J; Mendoza L; Shteynberg D; Deutsch EW; Moritz RL Processing shotgun proteomics data on the Amazon cloud with the trans-proteomic pipeline. Mol. Cell Proteomics 2015, 14 (2), 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prakash A; Ahmad S; Majumder S; Jenkins C; Orsburn B Bolt: a New Age Peptide Search Engine for Comprehensive MS/MS Sequencing Through Vast Protein Databases in Minutes. J. Am. Soc. Mass Spectrom. 2019, 30 (11), 2408–2418. [DOI] [PubMed] [Google Scholar]
- (13).ionbot. https://ionbot.cloud (accessed 26 October 2020).
- (14).Stewart PA; Kuenzi BM; Mehta S; Kumar P; Johnson JE; Jagtap P; Griffin TJ; Haura EB The Galaxy Platform for Reproducible Affinity Proteomic Mass Spectrometry Data Analysis. Methods Mol. Biol. (N. Y., NY, U. S.) 2019, 1977, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).GalaxyP - Access to software. http://galaxyp.org/access-galaxyp/ (accessed 26 October 2020).
- (16).Park CY; Klammer AA; Käll L; MacCoss MJ; Noble WS Rapid and accurate peptide identification from tandem mass spectra. J. Proteome Res. 2008, 7 (7), 3022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; MacLean BX; Villén J; MacCoss MJ Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9 (1), 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak MY; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30 (10), 918–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vaudel M; Barsnes H; Berven FS; Sickmann A; Martens L SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11 (5), 996–9. [DOI] [PubMed] [Google Scholar]
- (20).Kohlbacher O; Reinert K; Gröpl C; Lange E; Pfeifer N; Schulz-Trieglaff O; Sturm M TOPP–the OpenMS proteomics pipeline. Bioinformatics 2007, 23 (2), e191–7. [DOI] [PubMed] [Google Scholar]
- (21).Röst HL; Sachsenberg T; Aiche S; Bielow C; Weisser H; Aicheler F; Andreotti S; Ehrlich HC; Gutenbrunner P; Kenar E; Liang X; Nahnsen S; Nilse L; Pfeuffer J; Rosenberger G; Rurik M; Schmitt U; Veit J; Walzer M; Wojnar D; Wolski WE; Schilling O; Choudhary JS; Malmström L; Aebersold R; Reinert K; Kohlbacher O OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13 (9), 741–8. [DOI] [PubMed] [Google Scholar]
- (22).Keller A; Eng J; Zhang N; Li XJ; Aebersold R A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005, 1, 2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Deutsch EW; Mendoza L; Shteynberg D; Farrah T; Lam H; Tasman N; Sun Z; Nilsson E; Pratt B; Prazen B; Eng JK; Martin DB; Nesvizhskii AI; Aebersold R A guided tour of the Trans-Proteomic Pipeline. Proteomics 2010, 10 (6), 1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fenyö D; Beavis RC A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 2003, 75 (4), 768–74. [DOI] [PubMed] [Google Scholar]
- (25).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).FragPipe. https://github.com/Nesvilab/FragPipe (accessed 26 October 2020).
- (27).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26 (12), 1367–72. [DOI] [PubMed] [Google Scholar]
- (28).Sinitcyn P; Tiwary S; Rudolph J; Gutenbrunner P; Wichmann C; Yilmaz Ş; Hamzeiy H; Salinas F; Cox J MaxQuant goes Linux. Nat. Methods 2018, 15 (6), 401. [DOI] [PubMed] [Google Scholar]
- (29).Solntsev SK; Shortreed MR; Frey BL; Smith LM Enhanced Global Post-translational Modification Discovery with MetaMorpheus. J. Proteome Res. 2018, 17 (5), 1844–1851. [DOI] [PubMed] [Google Scholar]
- (30).Cesnik AJ; Miller RM; Ibrahim K; Lu L; Millikin RJ; Shortreed MR; Frey BL; Smith LM Spritz: A Proteogenomic Database Engine. J. Proteome Res. 2020, 10.1021/acs.jproteo-me.0c00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Berthold MR; Cebron N; Dill F; Gabriel TR; Kötter T; Meinl T; Ohl P; Thiel K; Wiswedel B KNIME - the Konstanz information miner: version 2.0 and beyond. SIGKDD Explor. Newsl. 2009, 11 (1), 26–31. [Google Scholar]
- (32).Albrecht M; Donnelly P; Bui P; Thain D Makeflow: A portable abstraction for data intensive computing on clusters, clouds, and grids. In Proceedings of the 1st ACM SIGMOD Workshop on Scalable Workflow Execution Engines and Technologies; 2012; pp 1–13. [Google Scholar]
- (33).Di Tommaso P; Chatzou M; Floden EW; Barja PP; Palumbo E; Notredame C Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35 (4), 316–319. [DOI] [PubMed] [Google Scholar]
- (34).Köster J; Rahmann S Snakemake–a scalable bioinformatics workflow engine. Bioinformatics 2012, 28 (19), 2520–2. [DOI] [PubMed] [Google Scholar]
- (35).Wilde M; Hategan M; Wozniak JM; Clifford B; Katz DS; Foster I Swift: A language for distributed parallel scripting. Parallel Computing 2011, 37 (9), 633–652. [Google Scholar]
- (36).Vivian J; Rao AA; Nothaft FA; Ketchum C; Armstrong J; Novak A; Pfeil J; Narkizian J; Deran AD; Musselman-Brown A; Schmidt H; Amstutz P; Craft B; Goldman M; Rosenbloom K; Cline M; O’Connor B; Hanna M; Birger C; Kent WJ; Patterson DA; Joseph AD; Zhu J; Zaranek S; Getz G; Haussler D; Paten B Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35 (4), 314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Perez-Riverol Y; Moreno P Scalable Data Analysis in Proteomics and Metabolomics Using BioContainers and Workflows Engines. Proteomics 2020, 20 (9), e1900147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mohammed Y; Mostovenko E; Henneman AA; Marissen RJ; Deelder AM; Palmblad M Cloud parallel processing of tandem mass spectrometry based proteomics data. J. Proteome Res. 2012, 11 (10), 5101–8. [DOI] [PubMed] [Google Scholar]
- (39).Chen L; Zhang B; Schnaubelt M; Shah P; Aiyetan P; Chan D; Zhang H; Zhang Z MS-PyCloud: An open-source, cloud computing-based pipeline for LC-MS/MS data analysis. bioRxiv, May 13, 2018, 320887. 10.1101/320887. [DOI] [Google Scholar]
- (40).Sadygov RG; Eng J; Durr E; Saraf A; McDonald H; MacCoss MJ; Yates JR 3rd Code developments to improve the efficiency of automated MS/MS spectra interpretation. J. Proteome Res. 2002, 1 (3), 211–5. [DOI] [PubMed] [Google Scholar]
- (41).UltraQuant. https://github.com/kentsisresearchgroup/UltraQuant (accessed 26 October 2020).
- (42).OpenMS Nodes for KNIME. https://www.knime.com/community/bioinf/openms (accessed 26 October 2020).
- (43).Perez-Riverol Y; Heumos L; Gabernet G; Garci M nf-core/ proteomicslfq, v. 1.0.0 - Lovely Logan; Zenodo, 2020. [Google Scholar]
- (44).Bichmann L; Gupta S; Rosenberger G; Kuchenbecker L; Sachsenberg T; Alka O; Pfeuffer J; Kohlbacher O; Röst H DIAproteomics: A multi-functional data analysis pipeline for data-independent-acquisition proteomics and peptidomics. bioRxiv, December 9, 2020, 2020.12.08.415844. 10.1101/2020.12.08.415844. [DOI] [PubMed] [Google Scholar]
- (45).Chard R; Skluzacek TJ; Li Z; Babuji Y; Woodard A; Blaiszik B; Tuecke S; Foster I; Chard KJ Serverless supercomputing: High performance function as a service for science. arXiv, August 14, 2019, arXiv:1908.04907. [Google Scholar]
- (46).Comparison, benchmarking and dissemination of proteomics data analysis pipelines. https://elixir-europe.org/about-us/commissioned-services/proteomics-pipelines (accessed 26 October 2020).
- (47).Neely BA; Prager KC; Bland AM; Fontaine C; Gulland FM; Janech MG Proteomic Analysis of Urine from California Sea Lions (Zalophus californianus): A Resource for Urinary Biomarker Discovery. J. Proteome Res. 2018, 17 (9), 3281–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wilmarth P Sea_lion_urine_SpC. https://github.com/pwilmart/Sea_lion_urine_SpC (accessed 26 October 2020).
- (49).PRIDE Project PXD009019. ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2018/06/PXD009019 (accessed 24 September 2020).
- (50).fasta_utilities. https://github.com/pwilmart/fasta_utilities/ (accessed 26 October 2020).
- (51).OpenMS Tutorial. https://sourceforge.net/p/open-ms/code/HEAD/tree/Tutorials/UM_2014/Handout/handout.pdf?format=raw (accessed 26 October 2020).
- (52).Eng JK; Jahan TA; Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (53).The M; MacCoss MJ; Noble WS; Käll L Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J. Am. Soc. Mass Spectrom. 2016, 27 (11), 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Neely BA An example KNIME-OpenMS workflow for benchmarking cloud-hosted environments. Zenodo, 2020. [Google Scholar]
- (55).Cloud_desktop_benchmarks. https://github.com/pwilmart/Cloud_desktop_benchmarks (accessed 26 October 2020).
- (56).SimpliFi. https://simplifi.protifi.com/ (accessed 26 October 2020).
- (57).Madduri R; Chard K; D’Arcy M; Jung SC; Rodriguez A; Sulakhe D; Deutsch E; Funk C; Heavner B; Richards M; Shannon P; Glusman G; Price N; Kesselman C; Foster I Reproducible big data science: A case study in continuous FAIRness. PLoS One 2019, 14 (4), e0213013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).The Singularity Recipe. https://singularity.lbl.gov/docs-recipes (accessed 26 October 2020).
- (59).Singularity Container Registry. https://singularity-hub.org/ (accessed 26 October 2020).
- (60).Docker Hub. https://hub.docker.com/ (accessed 26 October 2020).
- (61).Bai J; Bandla C; Guo J; Alvarez RV; Vizcaíno JA; Bai M; Moreno P; Grüning BA; Sallou O; Perez-Riverol Y BioContainers Registry: searching for bioinformatics tools, packages and containers. bioRxiv, July 22, 2020, 2020.07.21.187609. 10.1101/2020.07.21.187609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ewels PA; Peltzer A; Fillinger S; Patel H; Alneberg J; Wilm A; Garcia MU; Di Tommaso P; Nahnsen S The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38 (3), 276–278. [DOI] [PubMed] [Google Scholar]