

Abstract
Background and Purpose:
In addition to binding to the classical high-affinity extracellular benzodiazepine binding site of the GABAA receptor, some benzodiazepines occupy transmembrane inter-subunit anaesthetic sites that bind etomidate (β+/α− sites) or the barbiturate derivative R-mTFD-MPAB (α+/β− and γ+/β− sites). We aimed to define the functional effects of these interactions on GABAA receptor activity and animal behaviour.
Experimental Approach:
With flumazenil blocking classical high-affinity extracellular benzodiazepine site effects, modulation of GABA-activated currents by diazepam, midazolam and flurazepam was measured electrophysiologically in wildtype and M2-15′ mutant α1β3γ2L GABAA receptors. Zebrafish locomotive activity was also assessed in the presence of each benzodiazepine plus flumazenil.
Key Results:
In the presence of flumazenil, micromolar concentrations of diazepam and midazolam both potentiated and inhibited wildtype GABAA receptor currents. β3N265M (M2-15′ in the β+/α− sites) and α1S270I (M2-15′ in the α+/β− site) mutations reduced or abolished potentiation by these drugs. In contrast, the γ2S280W mutation (M2-15′ in the γ+/β− site) abolished inhibition. Flurazepam plus flumazenil only inhibited wildtype receptor currents, an effect unaltered by M2-15′ mutations. In the presence of flumazenil, zebrafish locomotion was enhanced by diazepam at concentrations up to 30 μM and suppressed at 100 μM, suppressed by midazolam and enhanced by flurazepam.
Conclusions and Implications:
Benzodiazepine binding to transmembrane anaesthetic binding sites of the GABAA receptor can produce positive or negative modulation manifesting as decreases or increases in locomotion, respectively. Selectivity for these sites may contribute to the distinct GABAA receptor and behavioural actions of different benzodiazepines, particularly at high (i.e. anaesthetic) concentrations.
1 ∣. INTRODUCTION
γ-Aminobutyric acid type A (GABAA) receptors mediate the majority of inhibitory neurotransmission in the brain (Olsen & Sieghart, 2009; Sigel & Steinmann, 2012). Typical synaptic GABAA receptors are heteropentamers containing 2 α, 2 β and 1 γ subunit arranged β-α-β-α-γ counterclockwise when viewed from an extracellular perspective (Figure 1). They are targets of many sedative-hypnotic drugs, including benzodiazepines and general anaesthetic agents such as propofol, etomidate and barbiturates (Franks, 2006; Olsen & Sieghart, 2009). Upon binding to the GABAA receptor, such drugs stabilize the receptor in the open channel state thus enhancing (i.e. potentiating) the agonist actions of GABA (Ruesch et al., 2012; Weir et al., 2017). In spite of their similar agonist potentiating actions, benzodiazepines and general anaesthetics are generally considered to act via distinct receptor sites (Scott & Aricescu, 2019). The classical high affinity binding site for benzodiazepines is located at the α+/γ− subunit interface in the extracellular domain of the GABAA receptor, whereas binding sites for intravenous general anaesthetics have been found at subunit interfaces within the hydrophobic transmembrane domain (Chiara et al., 2013; Scott & Aricescu, 2019). Photoaffinity labelling and cysteine modification and protection studies further resolve these anaesthetic binding sites into two homologous but distinct classes (Chiara et al., 2013; Nourmahnad et al., 2016). One class of sites selectively binds etomidate and is located at the two β+/α− subunit interfaces, whereas the other class of sites selectively binds the barbiturate R-5-allyl-1-methyl-5-(m-trifluoromethyl-diazirynylphenyl) barbituric acid (R-mTFD-MPAB) and is located at the α+/β− and γ+/β− subunit interfaces. At each binding site, plus-faced M2-15′ mutations (β3N265M at the two etomidate binding sites or γ2S280W and α1S270I at the R-mTFD-MPAB sites) significantly reduce or abolish GABAA receptor sensitivity to intravenous general anaesthetics and other allosteric modulators that act via that site (Jayakar et al., 2015; Nourmahnad et al., 2016; Siegwart et al., 2002; Walters et al., 2000).
FIGURE 1.

Cross-sectional diagram of the α1β3ɣ2L GABAA receptor visualized at the level of the transmembrane domain. Alpha subunits are highlighted in blue, beta subunits are highlighted in red and the gamma subunit is highlighted in green. Respective 15′ mutations are represented by appropriately coloured circles on the M2 domain of each subunit. Etomidate and 5-(m-trifluoromethyl-diazirynylphenyl) barbituric acid (R-mTFD-MPAB) binding site locations are highlighted at the appropriate interfaces with grey and black ovals, respectively
Recent cryo-electron microscopic imaging studies reveal that in addition to binding to the classical extracellular benzodiazepine binding site of the GABAA receptor, some benzodiazepines can also bind to the transmembrane anaesthetic binding sites. For example, Masiulis et al. (2019) found that diazepam (but not alprazolam or flumazenil) bound to the etomidate binding sites located at the two β+/α− subunit interfaces. Mutagenesis studies by Walters et al. (2000) that first suggested that benzodiazepines can positively modulate GABAA receptors via this site indicate that such binding—similar to binding to the classical benzodiazepine site—increases peak receptor currents evoked by low GABA concentrations by up to twofold to threefold. However, the apparent affinity of diazepam to the etomidate binding site is 2–3 orders of magnitude lower than to the classical extracellular site (Drexler et al., 2010; McGrath et al., 2020; Walters et al., 2000). More recently, Kim et al. (2020) visualized diazepam binding to the R-mTFD-MPAB binding site located at the γ+/β− subunit interface. The functional significance of such binding is unknown.
The aim of the current studies was to assess the GABAA receptor and behavioural consequences of benzodiazepine binding to individual anaesthetic binding sites located in the transmembrane receptor domain. In our experiments, we used the benzodiazepine competitive antagonist flumazenil to prevent potentially confounding benzodiazepine action via the classical high affinity site and utilized GABAA receptors harbouring M2-15′ mutations to resolve benzodiazepine actions at each anaesthetic binding site. Our results show that some benzodiazepines can potentiate and inhibit GABAA receptor function by binding to specific transmembrane anaesthetic binding sites and that these two actions respectively decrease and increase spontaneous locomotive activity in a zebrafish larvae behavioural model.
2 ∣. METHODS
2.1 ∣. Electrophysiology
Oocytes were harvested from adult female Xenopus laevis frogs using procedures approved by the Institutional Animal Care and Use Committee (IACUC protocol #2010N000002) of the Massachusetts General Hospital (Boston, Massachusetts) and in accordance with the principles outlined in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (Bethesda, Maryland) and the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). Oocytes were injected with 0.5 or 1.0 ng of messenger RNA (mRNA) mixtures encoding for wildtype or mutated α1, β3 and γ2L subunits of the GABAA receptor at a ratio of 1α:1β:3γ. Following injection, oocytes were incubated at 18°C for 18–48 h in ND-96 buffer (96-mM NaCl, 2-mM KCl, 1.8-mM CaCl2, 1-mM MgCl2, 5-mM HEPES, pH = 7.4) supplemented with 0.05 mg·mL−1 of gentamicin, 0.025 mg·mL−1 ciprofloxacin and 0.1 mg·mL−1 ampicillin. Whole cell two electrode voltage-clamp experiments were carried out at room temperature, a holding potential of −50 mV, and using an oocyte chamber volume of 50 μL. Currents were monitored with a GeneClamp 500B amplifier, digitized at a rate of 2000 Hz with a DigiData 1550B, low pass filtered at 0.01 kHz and recorded using Clampex v. 10.6 software (all from Molecular Devices, San Jose, CA). Current traces were analysed in Clampfit 10.6 (Molecular Devices, San Jose, CA). Solutions were perfused using a gravity fed system with an average flow rate of 2 mL·min−1 and controlled by a VC3 8 channel valve commander (ALA Scientific Instruments, Farmingdale, NY).
2.2 ∣. Molecular biology
Coding DNA for the α1, β3 and γ2L subunits of human GABAA receptors were expressed in pCDNA3.1 vectors. Mutations at the M2-15′ site of each subunit were made as previously described (Szabo et al., 2019). Briefly, QuikChange kits (Agilent Technologies, Santa Clara, CA) were used for oligonucleotide-directed mutagenesis in the wildtype subunit expression plasmids. The entire cDNA sequence of each mutant plasmid was sequenced to confirm the presence of the desired mutation and ensure the absence of stray mutations.
2.3 ∣. Impact of benzodiazepine binding to nonclassical sites on currents mediated by wildtype and mutant GABAA receptors
Each oocyte was first assessed for adequate receptor expression with a 10-s application of 1-mM GABA prepared in ND-96 buffer. Adequate expression was defined as an evoked peak current amplitude of at least 0.3 μA, but less than 10 μA to avoid amplifier saturation. After a 5-min recovery (ND-96 wash) period, the oocyte was perfused with buffer containing a GABA concentration that our pilot experiments showed evoke a peak current amplitude that is approximately 5% of that evoked by 1-mM GABA (i.e. EC5 GABA) along with 200-μM flumazenil for 7 min (Table 1). One minute after beginning this 7-min GABA/flumazenil application, the desired concentration of benzodiazepine was added for 2 min and the impact of the benzodiazepine (in the presence of flumazenil) on GABA-evoked currents was recorded.
TABLE 1.
Summary of fitted parameters for activation of GABAA receptor currents by GABA for wildtype and mutant receptors
| GABAA receptor subtype | GABAa (μM) |
Normalized mean peak current amplitudeb (%) |
|---|---|---|
| α1β3γ2L | 3 | 4.9 (4.2–5.5) |
| α1β3(N265M)γ2L | 7 | 6.3 (5.7–7.0) |
| α1β3γ2L(S280W) | 3 | 6.0 (5.2–6.8) |
| α1(S270I)β3γ2L | 0.2 | 5.2 (4.6–5.7) |
| α1(S270I)β3γ2L(S280W) | 0.1 | 4.8 (4.2–5.4) |
| α1(S270I)β3(N265M)γ2L | 0.3 | 3.4 (3.0–3.9) |
| α1β3(N265M)γ2L(S280W) | 3.5 | 4.9 (4.4–5.5) |
| α1(S270I)β3(N265M)γ2L(S280W) | 0.15 | 6.7 (5.8–7.7) |
Note: Values given in parentheses are 95% CI.
For each receptor subtype, the GABA concentration used to evoke a peak current amplitude that is approximately 5% of that evoked by 1-mM GABA.
For all experiments, the actual peak current amplitude achieved as a percent of that evoked by 1-mM GABA.
We chose this protocol in order to allow us to (1), establish the baseline EC5 GABA response (in the presence of flumazenil) in the same oocyte immediately prior to the addition of the test benzodiazepine, (2) quantify the positive and/or negative modulation produced by the test benzodiazepine (in the presence of flumazenil) and (3), qualitatively assess whether that modulation was reversible in wildtype and mutant receptors. We did not quantitatively analyse the recovery after test benzodiazepine application as it was typically quite slow (presumably because it was rate-limited by benzodiazepine washout) and therefore heavily contaminated by the process of desensitization.
If the peak current amplitude evoked by the EC5 GABA concentration in an oocyte failed to fall within a range of 2%–8% of that produced by 1-mM GABA, the oocyte was discarded. To account for oocyte-to-oocyte variability in receptor expression, the benzodiazepine-inhibited or potentiated current amplitude was normalized to the baseline EC5 GABA response recorded immediately prior to benzodiazepine addition (Figure 2a,b). In experiments where two actions were observed (i.e. an inhibitory action followed by a potentiating action), the potentiated current amplitude was normalized to that of the immediately preceding inhibited current amplitude (Figure 2c).
FIGURE 2.

Representative traces illustrating (a–c) the approach to quantifying benzodiazepine actions on GABAA receptor currents activated by EC5 GABA in the presence of 200-μM flumazenil and (d) the impact of solution exchange on holding currents in oocytes expressing wildtype GABAA receptors. (e,f) Pilot studies to assess the effects of solution exchange and 200-μM flumazenil, respectively, on EC5 GABA-activated currents. Depending upon the identity of the benzodiazepine and its concentration, currents exhibited quantifiable (a) potentiation, (b) inhibition or (c) inhibition followed by potentiation. The boxes below each trace quantitatively define the amplitudes of the potentiated and/or inhibited currents. Where traces exhibited only potentiation (or inhibition), the potentiated (or inhibited) current amplitude was normalized to the current amplitude recorded immediately prior to benzodiazepine application. Where traces exhibited inhibition followed by potentiation, the inhibited current amplitude was normalized to the current amplitude recorded immediately prior to benzodiazepine application and the subsequent potentiated current was normalized to the immediately preceding inhibited current amplitude. (d) Switching between two syringes containing ND-96 buffer alone (top) or ND-96 buffer plus 200-μM flumazenil (bottom) did not produce artefacts resembling activated current. (e) Switching between two syringes containing EC5 GABA plus 200-μM flumazenil produced no change in the amplitude of currents mediated by wildtype or mutant GABAA receptors. Inset shows the protocol for switching between syringes and a typical resulting trace using wildtype GABAA receptors; (f) 200-μM flumazenil had small (<20%) mutation-dependent effects on the amplitude of currents evoked by EC5 GABA in some receptor constructs. Inset shows the protocol for applying flumazenil and a typical resulting trace using wildtype GABAA receptors. In all panels showing electrophysiological traces, the grey bars were used to highlight the time of (a–c) benzodiazepine application, (d) switching between syringes or (f) flumazenil application. Each trace or data point was derived using a different oocyte
Pilot studies confirmed that switching between two reservoir syringes in our perfusion system where both contained either buffer (Figure 2d, top) or 200-μM flumazenil (Figure 2d, bottom) produced no significant electrophysiological artefacts. Such studies also revealed that while switching between two reservoir syringes where both contained EC5 GABA + 200-μM flumazenil similarly had no effect (Figure 2e), switching from a reservoir syringe containing EC5 GABA alone to one that also contained 200-μM flumazenil had small (<20%) effects in some receptor constructs (Figure 2f). To correct for these flumazenil-induced effects on our assessments of nonclassical benzodiazepine actions, we included flumazenil not only during administration of the desired benzodiazepine but also when recording the baseline EC5 GABA response to which the benzodiazepine action is normalized as noted in the protocol described above.
2.4 ∣. Impact of diazepam binding to nonclassical GABAA receptor sites on the GABA concentration–response relationship
Each oocyte was first assessed for adequate receptor expression by perfusing it with 1-mM GABA. As in the above-described experiment, adequate expression was defined as an evoked peak current amplitude that was greater than 0.3 μA, but less than 10 μA. After a 5-min recovery period, the oocyte was perfused with 200-μM flumazenil for 10 s and then with the desired concentration of GABA (0.1–100 μM) along with 100-μM diazepam (or 0-μM diazepam for control studies) plus 200-μM flumazenil for 30 s. The resulting test peak GABA-activated current amplitude was normalized to the peak current amplitude evoked in the same oocyte by 1-mM GABA. The normalized peak current amplitudes were then plotted against the GABA concentration and fit to a Hill equation with the minimum and Hill slope constrained to 0% and 1, respectively.
2.5 ∣. Zebrafish activity assay
Zebrafish (Danio rerio, Tubingen AB strain) were bred and used in accordance with established protocols approved by the Massachusetts General Hospital IACUC (protocol #2016N000586) and ARRIVE guidelines. Embryos were collected from mating of adult male/female pairs as needed. Larvae were maintained in 140-mm diameter Petri dishes containing E3 medium (5-mM NaCl, 0.17-mM KCl, 0.33-mM CaCl, 0.33-mM MgSO4, 2-mM HEPES, pH 7.4) and incubated at 28.5°C with a 14/10 h light/dark cycle. Larvae density did not exceed 100 fish per dish. Six or 7 days postfertilization, larval zebrafish (sex undetermined) were transferred in 100 μL of E3 buffer from the Petri dish to a standard 96-well plate using a cut and fire-polished 1000-μL pipette tip; 100 μL of E3 buffer solutions containing flumazenil and either diazepam, midazolam or flurazepam, prepared to twice the desired final concentration in E3 buffer, were then added to the wells to bringing the total volume to 200 μL. Diazepam was diluted from a DMSO stock solution. Final DMSO concentrations never exceeded 0.5%. Midazolam and flurazepam were prepared in deionized water. Immediately following the addition of benzodiazepines (0–100 μM) and 200-μM flumazenil (final concentrations), the 96-well plate was placed inside a Zebrabox (Viewpoint Behavioral Systems, Canada) and incubated in the dark chamber at 28°C for 15 min prior to recording. Spontaneous larvae activity was then recorded with an infrared video camera and analysed using Zebralab v3.2 software (Viewpoint Behavioral Systems, Canada), which quantifies each animal's motor activity by assessing changes in pixel intensity and summing the absolute value of intensity change for all pixels in a single pre-defined circular well over a 10-min recording period. Test larvae activity levels recorded in the presence of benzodiazepine (plus 200-μM flumazenil) were normalized to the control larvae activity level recorded on the same plate in the absence of benzodiazepine (i.e. 0-μM benzodiazepine plus 200-μM flumazenil) in E3 buffer to correct for any flumazenil-dependent effects and cohort variability. At each benzodiazepine concentration, the normalized activity levels of the eight larvae present on each plate were averaged. Five independent plates were run and the results of the five plates averaged. Thus, each data point on benzodiazepine concentration–response curves represent the average activity level derived from 40 larvae (8 larvae/plate × 5 plates). Each plate also contained eight larvae in wells containing buffer alone (no flumazenil). These larvae allowed us to confirm that flumazenil itself had no statistically significant effect on spontaneous motor activity (data not shown). After study, zebrafish larvae were euthanized in 0.5% tricaine followed by the addition of bleach (1:20 V:V).
2.6 ∣. Computational modelling
Benzodiazepine structures were constructed in Chemdraw Professional version 19.0.1.32 (PerkinElmer, Akron, OH) and imported into Spartan 14 version 1.2.0 (Wavefunction, Irvine, CA). The structures were then geometry optimized using ab initio quantum mechanics (Hartree–Fock 3-21 basis set) in vacuo and their molecular volumes defined using the built-in Molecular Properties function.
2.7 ∣. Data and statistical analysis
Data derived from electrophysiological experiments are reported as the mean of five individual experiments (each using a different oocyte) ± SD. The assumption of normality around reported mean values was confirmed using the Shapiro–Wilk test with an alpha value of 0.05. Data derived from zebrafish larvae experiment are reported as the mean value ± SEM obtained using five normalized activity values from separate plates that each held eight zebrafish larvae per benzodiazepine concentration. A one-way analysis of variance (ANOVA) with the Geisser–Greenhouse correction and a Dunnett multiple comparisons test was used to assess whether the inhibitory action of flumazenil was affected by the M2-15′ mutations with significance assumed for a P value <0.05. One-tailed Student's t-tests were used to compare sets of normalized peak currents to 100%. The false discovery rate for these t-tests was controlled using the Benjamini–Hochberg approach and reported P values were Benjamini–Hochberg adjusted with significance assumed for adjusted P values <0.05. Statistical comparisons between GABA concentration–response relationships used the extra sum-of-squares F test. All sample sizes were based on our previous experience with these experimental designs (Ma et al., 2017; McGrath et al., 2020). All fitting and statistical tests were performed with Graphpad Prism 8.0 for MacOS (San Diego, CA) or Microsoft Excel for the MAC version 16.45 (Redmond, WA).
2.8 ∣. Materials
All reagents used in the preparation of buffers for electrophysiology and zebrafish studies were purchased from either Sigma Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Waltham, MA). Flumazenil was purchased from Thermo Fisher Scientific. DMSO was purchased from VWR International (Radnor, PA). All benzodiazepines and GABA were purchased from Sigma-Aldrich.
2.9 ∣. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3 ∣. RESULTS
3.1 ∣. Diazepam both inhibits and potentiates GABA-activated wildtype GABAA receptor currents
Figure 3 shows the actions of the prototypical benzodiazepine diazepam (in the presence of 200-μM flumazenil) on currents evoked by EC5 GABA and mediated by wildtype α1β3γ2L GABAA receptors. The representative traces shown in Figure 3a reveal that diazepam had two distinct, opposing and concentration-dependent actions on GABAA receptor currents, even when flumazenil was present to prevent modulation via the classical benzodiazepine binding site. At lower diazepam concentrations (≤10 μM), the only discernible action of diazepam was to inhibit GABA-activated currents. The magnitude of this inhibition increased with diazepam concentration and reversed upon diazepam washout. However, at higher diazepam concentrations, a potentiating action became evident in current traces that also increased in magnitude with diazepam concentration and reversed upon diazepam washout. This produced traces in which the current amplitude initially decreased before increasing upon diazepam application. At the highest diazepam concentration studied (100 μM), the magnitude of the potentiating action was greater than the inhibitory one thus largely obscuring it. Figure 3b plots the mean ± SD amplitudes (n = 5) of the diazepam-inhibited and diazepam-potentiated currents observed in current traces as a function of diazepam concentration. At all diazepam concentrations ranging from 0.3 to 100 μM, the normalized amplitude of the inhibited current was statistically less than 100%, whereas that of the potentiated current was statistically greater than 100% only at diazepam concentrations ≥60 μM.
FIGURE 3.
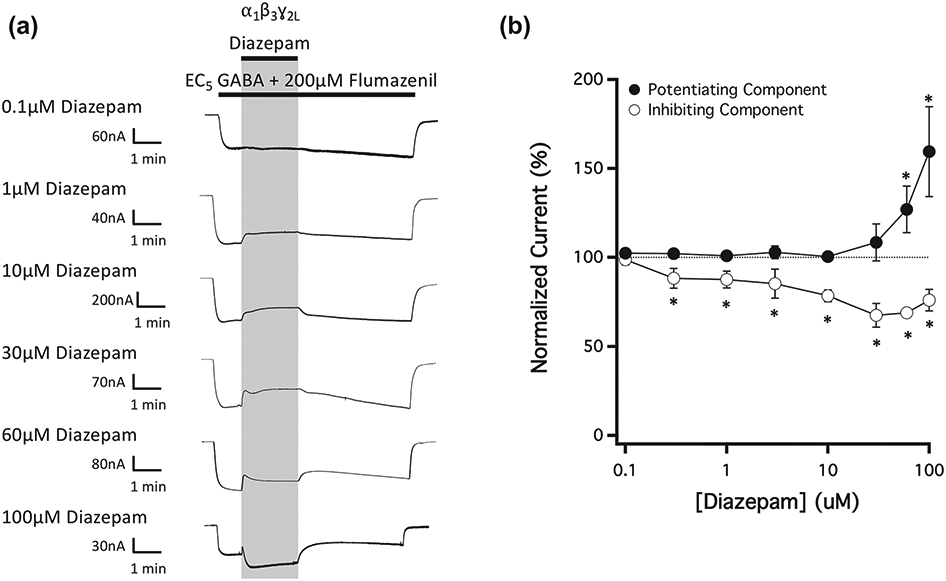
The concentration-dependence of diazepam action on currents mediated by wildtype GABAA receptors and activated by EC5 GABA in the presence of 200-μM flumazenil. (a) Representative traces showing the impact of diazepam on GABA-activated currents at the indicated concentrations. Each trace was derived using a different oocyte. (b) Diazepam concentration–response relationship for GABA-activated currents in the presence of 200-μM flumazenil. Each point represents the mean ± SD of normalized currents from five different oocytes. Closed circles represent the potentiated current amplitude and open circles represent the inhibited current amplitude. Each point was evaluated using a one-way t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. *P < 0.05
3.2 ∣. Mutations at transmembrane anaesthetic binding sites selectively eliminate the potentiating and inhibiting actions of diazepam
To assess the potential roles of the different transmembrane anaesthetic binding sites in mediating the GABAA receptor action described above, we introduced single subunit M2-15′ amino acid mutations into the receptor that have previously been shown to selectively reduce or abolish etomidate or R-mTFD-MPAB modulation via these sites (Jayakar et al., 2015; Nourmahnad et al., 2016; Siegwart et al., 2002). We then evaluated the impact of each mutation on diazepam action in the presence of flumazenil. Figure 4a shows that a β3N265M mutation at each of the two etomidate binding sites completely abolished the potentiating action seen in wildtype receptor-mediated currents, thus fully revealing the inhibitory action. Conversely, Figure 4b shows that the γ2S280W mutation at one of the two R-mTFD-MPAB binding sites (i.e., the one located at the γ+/β− subunit interface) completely abolished the inhibitory action seen in currents, thus fully revealing the potentiating action. Such unopposed potentiation in γ2S280W mutated receptors also appeared to accelerate the apparent rate of desensitization. Finally, Figure 4c shows that the α1S270I mutation at the other R-mTFD-MPAB binding site (i.e. the one located at the α+/β− subunit interface) had effects on diazepam action that were generally similar to that of the β3N265M mutation as it greatly reduced the potentiating action of diazepam, while largely preserving the inhibitory action. Figure 4d plots the mean ± SD normalized amplitude (n = 5) of the diazepam-inhibited and diazepam-potentiated currents observed in current traces mediated by mutant α1β3(N265M)γ2L, α1β3γ2L(S280W) and α1(S270I)β3γ2L GABAA receptors as a function of diazepam concentration and in the presence of flumazenil. It shows that inhibition of α1β3(N265M)γ2L GABAA receptor currents and potentiation of α1β3γ2L(S280W) GABAA receptor currents were statistically significant at all diazepam concentrations studied. At the highest concentration studied (100 μM), diazepam reduced the mean (± SD) amplitude of current traces mediated by α1β3(N265M)γ2L GABAA receptors and potentiated those mediated by α1β3γ2L(S280W) GABAA receptors to 15 ± 7% and 310 ± 40%, respectively, of that measured immediately prior to diazepam application. The plot also shows that while diazepam inhibition of α1(S270I)β3γ2L GABAA receptor currents was statistically significant at nearly all diazepam concentrations studied, potentiation did not reach statistical significance at any concentration.
FIGURE 4.

The concentration-dependence of diazepam action on currents mediated by 15′ mutated GABAA receptors and activated by EC5 GABA in the presence of 200-μM flumazenil. (a) Representative traces showing the impact of diazepam on GABA-activated currents at the indicated concentrations in GABAA receptors containing a (a) β3N265M mutation, (b) ɣ2LS280W mutation, or (c) α1S270I mutation. Each trace was derived using a different oocyte. (d) Diazepam concentration–response relationship for GABA-activated currents in the presence of 200-μM flumazenil. Each point represents the mean ± SD of normalized currents from five different oocytes. Receptors containing a β3N265M, ɣ2LS280W, or α1S270I mutation are represented with red, green and blue circles, respectively. For receptors containing an α1S270I mutation where both potentiation and inhibition was observed during diazepam application, closed circles represent the potentiated current amplitudes and open circles represent the inhibited current amplitudes. Each point was evaluated using a one-way t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. *P < 0.05
We then characterized the impact of diazepam (in the presence of flumazenil) on doubly mutated GABAA receptors containing M2-15′ mutations at two (of the three) different transmembrane anaesthetic binding sites. Our goal was to isolate the action(s) of the drug at each transmembrane anaesthetic binding site by incorporating M2-15′ mutations that reduce/abolish modulation at the other two sites (Figure 5a-c). Thus, we utilized (a) α1(S270I)β3γ2L(S280W) GABAA receptors to isolate the actions of diazepam mediated via the two etomidate binding sites, (b) α1(S270I)β3(N265M)γ2L GABAA receptors to isolate the actions of diazepam mediated via the R-mTFD-MPAB binding site located at the γ+/β− subunit interface and (c) α1β3(N265M)γ2L(S280W) GABAA receptors to isolate the actions of diazepam mediated via the R-mTFD-MPAB binding site located at the α+/β− subunit interface. We observed that diazepam substantially potentiated receptors in which the etomidate binding sites were left as wildtype (i.e. α1(S270I)β3γ2L(S280W) GABAA receptors) and inhibited receptors in which the γ+/β− subunit interfacial R-mTFD-MPAB binding site was left as wildtype (i.e., α1(S270I)β3(N265M)γ2L GABAA receptors). Along with the single mutation studies, these results provide strong evidence that the etomidate and γ+/β− subunit interfacial R-mTFD-MPAB binding sites mediate diazepam potentiation and inhibition respectively. At the highest concentration studied (100 μM), diazepam potentiated α1(S270I)β3γ2L(S280W) GABAA receptors and inhibited α1(S270I)β3(N265M)γ2L GABAA receptors to 257 ± 40% and 17 ± 18%, respectively, of that measured immediately prior to diazepam application. Diazepam also potentiated α1β3(N265M)γ2L(S280W) GABAA receptors (Figure 5c). However, at every diazepam concentration studied, the magnitude of this potentiation was less than that observed in studies using α1(S270I)β3γ2L(S280W) GABAA receptors, reaching only 112 ± 5% of that measured immediately prior to diazepam application even at 100-μM diazepam.
FIGURE 5.

The concentration-dependence of diazepam action on currents mediated by 15′ doubly and triply mutated GABAA receptors and activated by EC5 GABA in the presence of 200-μM flumazenil. (a) Representative traces showing the impact of diazepam in the presence of 200-μM flumazenil at the indicated concentrations on GABA-activated currents mediated by (a) α1(S270I) β3ɣ2L(S280W) GABAA receptors, (b) α1(S270I)β3(N265M)ɣ2L GABAA receptors, (c) α1β3(N265M)ɣ2L(S280W) GABAA receptors and (d) α1(S270I)β3(N265M)ɣ2L(S280W) GABAA receptors. Each trace was derived using a different oocyte. (e) Diazepam concentration–response relationship for GABA-activated currents. Each point represents the mean ± SD of normalized currents from five different oocytes. Each point was evaluated using a one-way t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. *P < 0.05
Using triply mutated α1(S270I)β3(N265M)γ2L(S280W) GABAA receptors, we then tested whether sensitivity to both potentiation and inhibition by diazepam could be abolished by incorporating all three M2-15′ mutations into a single receptor construct. Figure 5d,e demonstrates that such mutant receptors are, indeed, insensitive to both of these modulatory actions.
3.3 ∣. Diazepam differentially affects the GABA concentration–response curve for activation of wildtype and mutant GABAA receptors
We also characterized the impact of 100-μM diazepam (in the presence of 200-μM flumazenil) on the GABA concentration–response relationship for activation and found that diazepam differentially affected wildtype and mutated GABAA receptors (Figure 6, Table 2). Specifically, diazepam had no significant effect on the GABA EC50 of wildtype receptors but significantly increased the EC50 of α1β3(N265M)γ2L and α1(S270I)β3γ2L GABAA receptors and decreased that of α1β3γ2L(S280W) GABAA receptors. Diazepam also significantly reduced the maximal current response of wildtype and mutant GABAA receptors. The magnitude of this reduction was largest in α1β3(N265M)γ2L and α1(S270I)β3γ2L GABAA receptors, intermediate in wildtype receptors and smallest in α1β3γ2L(S280W) GABAA receptors.
FIGURE 6.

The impact of 100-μM diazepam on GABA concentration–response relationships in wildtype and 15′ mutated GABAA receptors in the presence of 200-μM flumazenil. Concentration-response relationships for (a) wildtype α1β3ɣ2L GABAA receptors, (b) α1β3(N265M)ɣ2L GABAA receptors, (c) α1β3ɣ2L(S280W) GABAA receptors, or (d) α1(S270I)β3ɣ2L GABAA receptors. Each point represents the mean ± SD of normalized currents from five different oocytes. Curves were fit to a Hill equation with the minimum and Hill slope constrained to 0% and 1, respectively. Fitted parameters are reported in Table 2
TABLE 2.
Summary of fitted parameters for activation of GABAA receptor currents by GABA in the absence (control) or presence of diazepam (100 μM)
| GABAA receptor subtype | GABA EC50 (μM) |
Maximum (%) |
||
|---|---|---|---|---|
| Control | + 100-μM diazepam | Control | + 100-μM diazepam | |
| α1β3γ2L | 45 (39–53) | 57 (44–72) | 106 (103–109) | 78* (74–82) |
| α1β3(N265M)γ2L | 100 (82–120) | 350* (230–540) | 112 (107–117) | 69* (60–78) |
| α1β3γ2L(S280W) | 46 (38–55) | 15* (12–19) | 103 (99–107) | 86* (83–90) |
| α1(S270I)β3γ2L | 3.2 (2.4–4.2) | 6.3* (4.0–9.9) | 96 (91–100) | 57* (53–62) |
Note: Values were defined by fitting drug concentration–response curves to a Hill equation with the minimum and the Hill slope constrained to 0% and 1, respectively. The fitted maximum was normalized to the peak response elicited by 1-mM GABA. Values given in parentheses are 95% CI.
versus control value.
3.4 ∣. Distinct actions of midazolam and flurazepam on GABA-activated wildtype and mutant GABAA receptor currents
We then studied the actions of two additional benzodiazepines—midazolam and flurazepam—on currents evoked by EC5 GABA and mediated by wildtype α1β3γ2L GABAA receptors in the presence of flumazenil (Figure 7). The representative traces shown in Figure 7a,b reveal that midazolam and flurazepam differentially affected GABA-evoked currents. Similar to diazepam, midazolam exhibited both inhibitory and potentiating receptor actions with the inhibitory one most apparent at lower midazolam concentrations and the potentiating one most apparent at higher ones. At the highest midazolam concentration studied (100 μM), the inhibitory action was almost completely masked by the potentiating one with its only evidence being a surge current visible in current traces upon terminating midazolam application. We did not quantitatively analyse these surge currents, which occur upon drug washout when inhibition is removed while activation continues (Adodra & Hales, 1995). In contrast, the only discernible action of flurazepam at any concentration was to inhibit currents. Figure 7c plots the mean ± SD normalized amplitudes (n = 5) of the inhibited and potentiated currents observed in traces as a function of midazolam concentration and those of the inhibited current as a function of flurazepam concentration. It shows that the amplitudes of the inhibitory components produced by flurazepam and midazolam were statistically significant at concentrations as low as 100 and 300 nM, respectively.
FIGURE 7.
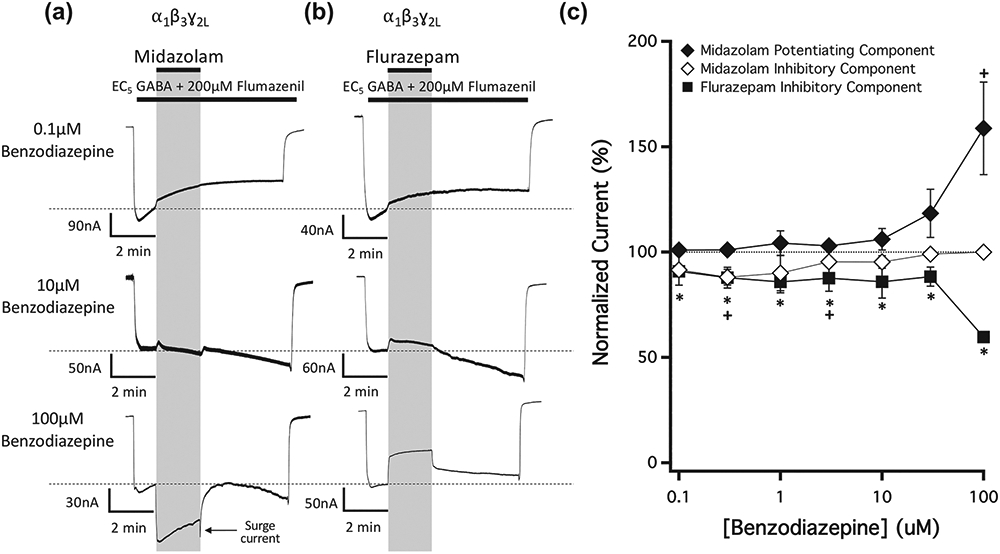
The concentration-dependence of midazolam and flurazepam action on currents mediated by wildtype GABAA receptors and activated by EC5 GABA in the presence of 200-μM flumazenil. Representative traces showing the impact of (a) midazolam and (b) flurazepam on GABA-activated currents at the indicated concentrations. Each trace was derived using a different oocyte. (c) Benzodiazepine concentration–response relationships for GABA-activated currents in the presence of 200-μM flumazenil. Each point represents the mean ± SD of normalized currents from five different oocytes. Midazolam is represented by diamonds and flurazepam by squares. For midazolam, in which both potentiated and inhibited currents were commonly present, closed diamonds represent the potentiated currents and open diamonds represent the inhibited current. Each data point was evaluated using a one-way Student's t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. +P < 0.05, (flurazepam)
Figure 8 shows the impact of the three M2-15′ mutations on the actions of midazolam in the presence of 200-μM flumazenil on currents evoked by EC5 GABA. Similar to their effects on the potentiating and inhibitory actions of diazepam, the β3N265M mutation abolished the potentiating action of midazolam and the γ2S280W mutation abolished the inhibitory action of midazolam (Figure 8a,b). Whereas the α1S270I mutation only reduced the potentiating action of diazepam (Figure 4c), it completely abolished that of midazolam (Figure 8c). Figure 8d plots the mean ± SD normalized amplitudes (n = 5) of the inhibited and potentiated currents observed in traces evoked by EC5 GABA and mediated by GABA receptors harboring each of these three mutations as a function of midazolam concentration.
FIGURE 8.

The concentration-dependence of midazolam action on currents mediated by 15′ mutated GABAA receptors in the presence of 200-μM flumazenil. Representative traces showing the impact of midazolam on GABA-activated currents at the indicated concentrations in GABAA receptors containing a (a) β3N265M mutation, (b) ɣ2LS280W mutation, or (c) α1S270I mutation. Each trace was derived using a different oocyte. (d) Midazolam concentration–response relationship for GABA-activated currents in the presence of 200-μM flumazenil. Each point represents the mean ± SD of normalized currents from five different oocytes. Receptors containing a β3N265M, ɣ2LS280W, or α1S270I mutation are represented with red, green and blue diamonds, respectively. Each data point was compared using a one-way t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. *P < 0.05
We also tested whether any of the three M2-15′ mutations abolished the inhibitory action of flurazepam. The representative traces shown in Figure 9a-c reveal that mutant α1β3(N265M)γ2L, α1β3γ2L(S280W) and α1(S270I)β3γ2L GABAA receptors were all similarly inhibited by flurazepam. Figure 9d plots the mean ± SD normalized amplitudes (n = 5) of the inhibited currents observed in traces mediated by these receptor mutants as a function of flurazepam concentration along with those obtained using wildtype receptors for comparison. A one-way ANOVA confirmed that there were no significant differences between the normalized amplitudes of flurazepam-inhibited currents when mediated by mutant receptors versus wildtype receptors.
FIGURE 9.

The concentration-dependence of flurazepam action on currents mediated by 15′ mutated GABAA receptors and activated by EC5 GABA in the presence of 200-μM flumazenil. Representative traces showing the impact of midazolam on GABA-activated currents at the indicated concentrations in GABAA receptors containing a (a) β3N265M mutation, (b) ɣ2LS280W mutation, or (c) α1S270I mutation. Each trace was derived using a different oocyte. (d) Flurazepam concentration–response relationship for GABA-activated currents in the presence of 200-μM flumazenil. Each point represents the mean ± SD of normalized currents from five different oocytes. Data from wildtype receptors or receptors containing a β3N265M, ɣ2LS280W, or α1S270I mutation are represented with black, red, green and blue squares, respectively. Each data point was evaluated using a one-way t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. A one-way analysis of variance (ANOVA) with the Geisser–Greenhouse correction and a Dunnett multiple comparisons test confirmed that the inhibitory action of flumazenil was unaffected by any of the M2-15′ mutations. *P < 0.05
3.5 ∣. The impact of the S280W mutation on GABAA receptor inhibition: A benzodiazepine survey
The above results indicate that the potentiating actions of diazepam and midazolam are reduced or eliminated by M2-15′ mutations in the two β+/α− etomidate binding sites and the α+/β− R-mTFD-MPAB binding site, while their inhibitory actions are eliminated by a M2-15′ mutation in the γ+/β− R-mTFD-MPAB binding site. Flurazepam is different: It evinces no potentiation in the presence of flumazenil and its inhibitory effects are unaltered by the γ2S280W mutation. We surveyed eight additional benzodiazepines to test for inhibition sensitive to the γ2S280W mutation. To isolate γ2S280W mutation effects on benzodiazepine inhibition, we utilized a strategy that first eliminated benzodiazepine potentiation by incorporating both β3N265M and α1S270I mutations into receptors. Comparisons of inhibitory effects in the α1(S270I)β3(N265M)γ2L background with α1(S270I)β3(N265M)γ2L(S280W) are shown in Figure 10 for 11 benzodiazepines combined with flumazenil on EC5 GABA currents. Figure 10a shows the effects of diazepam, midazolam and flurazepam (all at 100 μM) plus flumazenil in the double mutant versus triple mutant GABAA receptors. In the presence of flumazenil, α1(S270I)β3(N265M)γ2L GABAA receptors were inhibited by all three benzodiazepines, without evidence of potentiation (Figure 10a, blue traces). Consistent with the mutagenesis studies described above, incorporation of the γ2LS280W mutation to form α1(S270I)β3(N265M)γ2L(S280W) GABAA receptors abolished the inhibitory actions of diazepam and midazolam, but not flurazepam (Figure 10a, grey traces). For these three benzodiazepines and the eight additional ones, Figure 10b compares the normalized current amplitude recorded in the presence of benzodiazepine (mean ± SD; n = 5) when mediated by α1(S270I)β3(N265M)γ2L versus α1(S270I)β3(N265M)γ2L(S280W) GABAA receptors in the presence of flumazenil. Incorporation of the γ2LS280W mutation significantly reduced the inhibitory actions of diazepam, midazolam, nordiazepam, estazolam, alprazolam, clobazam, nitrazepam and fludiazepam but had no significant effect on the inhibitory actions of flurazepam, prazepam and imidazenil.
FIGURE 10.

Current inhibition produced by benzodiazepines (100 μM) in EC5 GABA-activated currents mediated by doubly mutated α1(S270I)β3(N265M)ɣ2L GABAA receptors versus triply mutated α1(S270I)β3(N265M)ɣ2L(S280W) GABAA receptors activated with EC5 GABA in the presence of 200-μM flumazenil. (a) Representative traces showing the impact of 100-μM diazepam, midazolam, or flurazepam on currents mediated by α1(S270I)β3(N265M)ɣ2L receptors (blue line) or α1(S270I)β3(N265M)ɣ2L(S280W) receptors (grey line). Each trace was derived using a different oocyte. (b) Bar graph comparing the actions of benzodiazepines on the normalized amplitudes of currents mediated by α1(S270I)β3(N265M)ɣ2L receptors (blue bars) versus α1(S270I)β3(N265M)ɣ2L(S280W) receptors (grey bars). Each bar represents the mean ± SD normalized amplitude from 5 different oocytes. Each benzodiazepine was compared across α1(S270I)β3(N265M)ɣ2L receptors and α1(S270I)β3(N265M)ɣ2L(S280W) receptors with a Student's t-test and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. *P < 0.05
3.6 ∣. The behavioural actions of benzodiazepines parallel their GABAA receptor actions
To assess the potential behavioural implications of benzodiazepine interactions with nonclassical GABAA receptor binding sites, we measured the spontaneous locomotive activity of individual zebrafish larvae in the presence of steady-state concentrations of diazepam, midazolam or flurazepam, each in the presence of flumazenil. We hypothesized that net inhibition of GABAA receptors would produce CNS excitation and increase locomotive activity whereas net potentiation of these receptors would sedate zebrafish larvae and reduce locomotive activity (Baraban et al., 2005). For the three benzodiazepines, Figure 11 plots the benzodiazepine concentration–response relationships for spontaneous zebrafish larva activity. To facilitate comparisons between the concentration-dependent behavioural and receptor actions of these drugs, GABAA receptor data for net effects of the three drugs were overlaid on these plots. The net receptor current amplitude was defined simply as the average of the potentiated and inhibited current amplitudes. Thus, net current amplitudes less than 100% signify net inhibition of GABAA receptor function while those greater than 100% signify net potentiation of receptor function.
FIGURE 11.

Comparison of GABAA receptor modulation and zebrafish spontaneous motor activity. Concentration–response relationships of (a) diazepam, (b) midazolam and (c) flurazepam on spontaneous activity in zebrafish (closed symbols) were plotted on the same horizonal axis as the net current amplitude generated in wildtype GABAA receptors (open symbols). Normalized activity levels above 100% represent increases in motor activity and those below 100% represent decreases in motor activity. Net current amplitudes below 100% represent net inhibition of GABAA receptor currents, whereas those above 100% represent net potentiation of GABAA receptor currents. For zebrafish activity, each data point represents the mean ± SEM of five experiments of eight fish each (total of 40 fish per data point). Each point was compared using a one-way Student's t-test against a hypothetical mean of 100% and an adjusted P value was derived using a Benjamini–Hochberg correction for multiple comparisons. *P < 0.05
Figure 11a shows that both spontaneous locomotive activity and net receptor current amplitude exhibited diazepam concentration–response curves that were biphasic. With increasing diazepam concentrations up to 30 μM, locomotive activity tended to increase relative to that of control larvae exposed to flumazenil alone while the net GABAA receptor current amplitude decreased. However, this trend reversed at 100-μM diazepam as locomotive activity decreased to a level that was below control while net GABAA receptor current amplitude increased. Figure 11b,c plots the analogous data for midazolam and flurazepam, respectively. For these drugs, spontaneous locomotive activity and net receptor current amplitude exhibited monophasic benzodiazepine concentration–response curves. In the case of midazolam, motor activity tended to decrease while net current amplitude increased. Conversely, in the case of flurazepam, motor activity tended to increase while net current amplitude decreased.
4 ∣. DISCUSSION
The goal of the current studies was to assess the functional consequences of benzodiazepine binding to individual transmembrane anaesthetic binding sites on the GABAA receptor and to define the impact of these interactions on animal behaviour. Our results reveal that diazepam and midazolam can both potentiate and inhibit wildtype GABAA receptors even when flumazenil is present to prevent action via the classical high affinity benzodiazepine site. They further show that these potentiating and inhibiting actions can be selectively abolished by introducing M2-15′ mutations into specific inter-subunit transmembrane anaesthetic binding sites. These are mutations that have previously been shown to selectively abolish the positive modulatory actions of anaesthetics and the negative modulatory actions of convulsants that act via each of these sites (Jayakar et al., 2015; Nourmahnad et al., 2016; Siegwart et al., 2002). Specifically, a β3N265M mutation at each of the two etomidate binding sites abolished the potentiating actions of diazepam and midazolam, whereas a γ2S280W mutation at the R-mTFD-MPAB binding site located at the γ+/β− subunit interface abolished their inhibitory actions. Taken together with previous cryo-electron microscopic studies documenting benzodiazepine binding to these anaesthetic binding sites (Kim et al., 2020; Masiulis et al., 2019), these results strongly suggest that benzodiazepine binding to the two etomidate binding sites produces allosteric GABAA receptor potentiation, whereas benzodiazepine binding to the R-mTFD-MPAB binding site located at the γ+/β− subunit interface conversely produces allosteric GABAA receptor inhibition. This interpretation was further supported by the results of our double mutant receptor studies as we observed significant diazepam-induced potentiation of receptors when the M2-15′ residue at the two etomidate binding sites was left as wildtype and significant inhibition in mutant receptors when the M2-15′ residue at the R-mTFD-MPAB binding site located at the γ+/β− subunit interface was left as wildtype.
We also found that an α1S270I mutation at the R-mTFD-MPAB binding site located at the α+/β− subunit interface had an effect that was similar to that of a β3N265M mutation as it significantly reduced (diazepam) or abolished (midazolam) the receptor potentiating actions of these benzodiazepines. However, we are more circumspect regarding whether this represents an actual benzodiazepine binding site as previous structural studies have not found evidence of benzodiazepines binding to this site. Thus, it is possible that this mutation acts allosterically to reduce benzodiazepine binding to (or positive modulation via) the etomidate sites, which are located on the opposite face of the α1 subunit (Figure 1). Alternatively, binding to this site may have been missed in those studies because its affinity is quite low; in our double mutation α1β3(N265M)γ2L(S280W) GABAA receptor studies to isolate diazepam binding to this site, statistically significant potentiation occurred only at our highest concentration (100 μM) and its magnitude was quite small.
The impact of diazepam (in the presence of flumazenil) on the GABA concentration–response curve for wildtype and mutated GABAA receptor activation is generally consistent with the above interpretation (Figure 6, Table 2). In wildtype receptors, 100-μM diazepam had significant potentiating and inhibiting receptor actions. These two opposing actions tended to offset one another, leaving the GABA EC50 essentially unchanged and the maximum peak current response only modestly (but statistically significantly) reduced. In receptors containing β3N265M or α1S270I mutations, potentiation is reduced (or eliminated) and thus, the predominant action of 100-μM diazepam (plus flumazenil) was to inhibit receptors. Consequently, it acts as a negative allosteric modulator, shifting the GABA concentration–response curve rightward (significantly increasing the GABA EC50) and further reducing the maximum peak current. Conversely, in receptors containing the γ2S280W mutation, inhibition is eliminated, and thus 100-μM diazepam (plus flumazenil) only had potentiating receptor actions. Consequently, it acts as a positive allosteric modulator, shifting the GABA concentration–response curve leftward (thus significantly reducing the GABA EC50) and lessening the reduction in the maximum peak current response compared to that observed using wildtype receptors.
In contrast to diazepam and midazolam, flurazepam exhibited only inhibitory GABAA receptor actions in the presence of flumazenil. Furthermore, these actions were unaffected by any of the M2-15′ mutations, suggesting that—unlike inhibition by diazepam and midazolam—they are not mediated by any of the inter-subunit transmembrane anaesthetic binding sites. A survey of eight additional benzodiazepines in the presence of flumazenil revealed differing impacts of the γ2S280W mutation on their inhibitory activities. Thus, our studies define two distinct mechanisms for benzodiazepine-induced inhibition of GABAA receptors. One mechanism is abolished by the γ2S280W mutation and reflects binding to the R-mTFD-MPAB binding site located at the γ+/β− subunit interface, whereas the other mechanism is unaffected by the γ2S280W mutation and likely reflects binding elsewhere on the receptor. Diazepam and midazolam are examples of benzodiazepines that inhibit via the former γ2S280W mutation-sensitive mechanism, whereas flumazenil is an example of one that inhibits via the latter γ2S280W mutation-insensitive mechanism. While the location of the binding site responsible for this latter mechanism is undefined by our studies, Baur et al. (2008) have suggested that, at least in the case of flurazepam such inhibition is mediated by binding to a subunit interfacial site located in the extracellular domain. Flurazepam binding sites in the extracellular domain were similarly suggested by ligand-bound crystal structures of the Erwinia ligand-gated ion channel (ELIC), a bacterial homologue of the vertebrate GABAA receptor (Spurny et al., 2012). Figure 12 displays the chemical structures of all of the benzodiazepines evaluated in our studies. The three γ2S280W mutation-insensitive benzodiazepines were determined from computational modelling to have the largest molecular volumes due to the presence of substituent groups on the diazepine ring. This suggests that the binding of these drugs to the R-mTFD-MPAB binding site located at the γ+/β− subunit interface is prevented by steric hindrance.
FIGURE 12.

Molecular structures of the benzodiazepines and their molecular volumes. The structures of benzodiazepines whose inhibitory activities are insensitive to the ɣ2S280W GABAA receptor mutation are highlighted by the blue boxes
We used a zebrafish larvae activity assay to assess the potential behavioural implications of benzodiazepine binding to these transmembrane anaesthetic binding sites. This aquatic animal model offers several important advantages over analogous assays used in mammals (Basnet et al., 2019). First, it is performed at clearly defined, steady-state effect site drugs concentrations. These conditions are necessary for evaluating the concentration-dependent actions of drugs but are difficult to achieve with intravenous agents such as benzodiazepines in mammals because they are strongly impacted by pharmacokinetic processes such as tissue distribution and metabolism (Riss et al., 2008). Second, the effect of protein binding on free aqueous drug concentrations is minimized by the large reservoir of drugs present in the water within each well. In the case of benzodiazepines, this is especially important as they are highly protein-bound in blood (Dubey et al., 1989; Jones et al., 1988). Third, a large number of animals can be efficiently studied to more precisely define drug potency and efficacy. In our studies, each data point was derived using 40 zebrafish larvae. Finally, video analysis of individual larval movement is completely automated, eliminating any potential observer bias. The results of our zebrafish studies support the hypothesis that the GABAA receptor potentiating and inhibiting actions that we observed have behavioural consequences. Under conditions where benzodiazepines produced net potentiation of GABAA receptor function, we recorded a reduction in zebrafish locomotive activity consistent with sedation. In contrast, under conditions where these benzodiazepines produced net inhibition of GABAA receptor function, we recorded an increase in zebrafish locomotive activity consistent with CNS excitation.
The benzodiazepine concentrations that we found modulate GABAA receptor function via transmembrane anaesthetic binding sites exceed those needed to produce anxiolysis or sedation (Bond et al., 1977; Steiner et al., 2016). However, brain slice experiment along with in vivo animal and human studies suggest that such concentrations are not only achieved when these drugs are used at the higher doses needed to induce anaesthesia but may be required to produce the behavioural hallmarks of the anaesthetic state: hypnosis and lack of responsiveness to painful stimuli (Cao et al., 2019; Downes & Courogen, 1996; Little & Bichard, 1984). For example, Drexler et al. (2010) estimated that a plasma diazepam concentration of approximately 40 μM was needed to produce hypnosis in rodents. Studies by Gamble et al. (1981) similarly suggest that in the absence of premedication, anaesthesia is induced by midazolam in humans only when plasma midazolam concentrations are in the micromolar range.
A potential limitation of this study was the use of mutations to resolve benzodiazepine actions at individual transmembrane anaesthetic binding sites as it is conceivable that they themselves may have introduced novel actions absent in wildtype receptors. However, we believe that such a limitation was unlikely to have significantly impacted our studies as the results of our single and double mutation studies—which utilized different mutations at each binding site—led us to identical conclusions.
In conclusion, our studies show that nonclassical benzodiazepine binding to specific anaesthetic binding sites on the GABAA receptor produces either potentiation or inhibition. Specifically, benzodiazepine binding to the etomidate binding sites located at the two β+/α− subunit interfaces produces allosteric receptor potentiation whereas binding to the R-mTFD-MPAB binding site located at the γ+/β− subunit interface produces allosteric receptor inhibition. Some benzodiazepines (e.g. flurazepam) can also inhibit GABAA receptor by binding to another site(s) whose location is undefined by our studies. Our data suggest that these GABAA receptor potentiating and inhibiting actions can manifest in vivo as reductions or increases, respectively, in locomotive activity.
What is already known
Etomidate positively modulates GABAA receptors by binding to a set of inter-subunit transmembrane sites.
Benzodiazepines binding to these sites similarly produces positive modulation.
What does this study add
By occupying other anaesthetic binding sites, benzodiazepines can also negatively modulate GABAA receptor function.
Positive and negative GABAA receptor modulation via anaesthetic binding sites has significant behavioural effects.
What is the clinical significance
Binding to anaesthetic sites may contribute to benzodiazepine action, particularly at high benzodiazepine doses.
ACKNOWLEDGEMENTS
This work was funded by grants GM122806 (to DER) and GM128989 (to SAF) from the National Institutes of Health, Bethesda, MD and the Department of Anesthesia, Critical Care, and Pain Medicine, Massachusetts General Hospital, Boston, Massachusetts.
Abbreviations:
- EC5
GABA concentration that evokes a peak current amplitude that is 5% of that evoked by 1-mM GABA
- R-mTFD-MPAB
R-5-allyl-1-methyl-5-(m-trifluoromethyl-diazirynylphenyl) barbituric acid
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Adodra S, & Hales TG (1995). Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1-7 neurones by propofol. British Journal of Pharmacology, 115(6), 953–960. 10.1111/j.1476-5381.1995.tb15903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA, Veale EL, & CGTP collaborators. (2019). The concise guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176(Suppl 1), S142–S228. 10.1111/bph.14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Taylor MR, Castro PA, & Baier H (2005). Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience, 131(3), 759–768. 10.1016/j.neuroscience.2004.11.031 [DOI] [PubMed] [Google Scholar]
- Basnet RM, Zizioli D, Taweedet S, Finazzi D, & Memo M (2019). Zebrafish larvae as a behavioral model in neuropharmacology. Biomedicine, 7(1), 23. 10.3390/biomedicines7010023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur R, Tan KR, Luscher BP, Gonthier A, Goeldner M, & Sigel E (2008). Covalent modification of GABAA receptor isoforms by a diazepam analogue provides evidence for a novel benzodiazepine binding site that prevents modulation by these drugs. Journal of Neurochemistry, 106(6), 2353–2363. 10.1111/j.1471-4159.2008.05574 [DOI] [PubMed] [Google Scholar]
- Bond AJ, Hailey DM, & Lader MH (1977). Plasma concentrations of benzodiazepines. British Journal of Clinical Pharmacology, 4(1), 51–56. 10.1111/j.1365-2125.1977.tb00666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Yan H, Yu G, & Su R (2019). Flumazenil-insensitive benzodiazepine binding sites in GABAA receptors contribute to benzodiazepine-induced immobility in zebrafish larvae. Life Sciences, 239, 117033. 10.1016/j.lfs.2019.117033 [DOI] [PubMed] [Google Scholar]
- Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, & Cohen JB (2013). Specificity of inter-subunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. The Journal of Biological Chemistry, 288(27), 19343–19357. 10.1074/jbc.M113.479725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes H, & Courogen PM (1996). Contrasting effects of anesthetics in tadpole bioassays. The Journal of Pharmacology and Experimental Therapeutics, 278(1), 284–296. [PubMed] [Google Scholar]
- Drexler B, Zinser S, Hentschke H, & Antkowiak B (2010). Diazepam decreases action potential firing of neocortical neurons via two distinct mechanisms. Anesthesia and Analgesia, 111(6), 1394–1399. 10.1213/ANE.0b013e3181f9c035 [DOI] [PubMed] [Google Scholar]
- Dubey RK, McAllister CB, Inoue M, & Wilkinson GR (1989). Plasma binding and transport of diazepam across the blood–brain barrier. No evidence for in vivo enhanced dissociation. The Journal of Clinical Investigation, 84(4), 1155–1159. 10.1172/JCI114279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP (2006). Molecular targets underlying general anaesthesia. British Journal of Pharmacology, 147(Suppl 1), S72–S81. 10.1038/sj.bjp.0706441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble JA, Kawar P, Dundee JW, Moore J, & Briggs LP (1981). Evaluation of midazolam as an intravenous induction agent. Anaesthesia, 36(9), 868–873. 10.1111/j.1365-2044.1981.tb08859 [DOI] [PubMed] [Google Scholar]
- Jayakar SS, Zhou X, Savechenkov PY, Chiara DC, Desai R, Bruzik KS, Miller KW, & Cohen JB (2015). Positive and negative allosteric modulation of an α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor by binding to a site in the transmembrane domain at the γ+-β− interface. The Journal of Biological Chemistry, 290(38), 23432–23446. 10.1074/jbc.M115.672006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DR, Hall SD, Jackson EK, Branch RA, & Wilkinson GR (1988). Brain uptake of benzodiazepines: Effects of lipophilicity and plasma protein binding. The Journal of Pharmacology and Experimental Therapeutics, 245(3), 816–822. [PubMed] [Google Scholar]
- Kim JJ, Gharpure A, Teng J, Zhuang Y, Howard RJ, Zhu S, Noviello CM, Walsh RM Jr., Lindahl E, & Hibbs RE (2020). Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature, 585(7824), 303–308. 10.1038/s41586-020-2654-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley E, Stanford SC, Kendall DE, Alexander SP, Cirino G, Docherty JR, George CH, Insel PA, Izzo AA, Ji Y, Panettieri RA, Sobey CG, Stefanska B, Stephens G, Teixeira M, & Ahluwalia A (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177, 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little HJ, & Bichard AR (1984). Differential effects of the benzodiazepine antagonist Ro 15-1788 after “general anaesthetic” doses of benzodiazepines in mice. British Journal of Anaesthesia, 56(10), 1153–1160. 10.1093/bja/56.10.1153 [DOI] [PubMed] [Google Scholar]
- Ma C, Pejo E, McGrath M, Jayakar SS, Zhou X, Miller KW, Cohen JB, & Raines DE (2017). Competitive antagonism of anesthetic action at the γ-aminobutyric acid type a receptor by a novel etomidate analog with low intrinsic efficacy. Anesthesiology, 127(5), 824–837. 10.1097/ALN.0000000000001840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiulis S, Desai R, Uchanski T, Serna Martin I, Laverty D, Karia D, Malinauskas T, Zivanov J, Pardon E, Kotecha A, & Steyaert J (2019). GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature, 565(7740), 454–459. 10.1038/s41586-018-0832-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath M, Hoyt H, Pence A, Jayakar SS, Zhou X, Forman SA, Cohen JB, Miller KW, & Raines DE (2020). Competitive antagonism of etomidate action by diazepam: In vitro GABAA receptor and in vivo zebrafish studies. Anesthesiology, 133(3), 583–594. 10.1097/ALN.0000000000003403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourmahnad A, Stern AT, Hotta M, Stewart DS, Ziemba AM, Szabo A, & Forman SA (2016). Tryptophan and cysteine mutations in M1 helices of α1β3γ2L γ-aminobutyric acid type a receptors indicate distinct intersubunit sites for four intravenous anesthetics and one orphan site. Anesthesiology, 125(6), 1144–1158. 10.1097/ALN.0000000000001390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, & Sieghart W (2009). GABA a receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology, 56(1), 141–148. 10.1016/j.neuropharm.2008.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, … Würbel H (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riss J, Cloyd J, Gates J, & Collins S (2008). Benzodiazepines in epilepsy: Pharmacology and pharmacokinetics. Acta Neurologica Scandinavica, 118(2), 69–86. 10.1111/j.1600-0404.2008.01004.x [DOI] [PubMed] [Google Scholar]
- Ruesch D, Neumann E, Wulf H, & Forman SA (2012). An allosteric coagonist model for propofol effects on α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology, 116(1), 47–55. 10.1097/ALN.0b013e31823d0c36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S, & Aricescu AR (2019). A structural perspective on GABAA receptor pharmacology. Current Opinion in Structural Biology, 54, 189–197. 10.1016/j.sbi.2019.03.023 [DOI] [PubMed] [Google Scholar]
- Siegwart R, Jurd R, & Rudolph U (2002). Molecular determinants for the action of general anesthetics at recombinant α2β3γ2γ-aminobutyric acidA receptors. Journal of Neurochemistry, 80(1), 140–148. 10.1046/j.0022-3042.2001.00682.x [DOI] [PubMed] [Google Scholar]
- Sigel E, & Steinmann ME (2012). Structure, function, and modulation of GABAA receptors. The Journal of Biological Chemistry, 287(48), 40224–40231. 10.1074/jbc.R112.386664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurny R, Ramerstorfer J, Price K, Brams M, Ernst M, Nury H, Verheij M, Legrand P, Bertrand D, Bertrand S, Dougherty DA, de Esch IJP, Corringer PJ, Sieghart W, Lummis SCR, & Ulens C (2012). Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proceedings of the National Academy of Sciences of the United States of America, 109(44), E3028–E3034. 10.1073/pnas.1208208109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner C, Steurer MP, Mueller D, Zueger M, & Dullenkopf A (2016). Midazolam plasma concentration after anesthesia premedication in clinical routine—An observational study: Midazolam plasma concentration after anesthesia premedication. BMC Anesthesiology, 16(1), 105. 10.1186/s12871-016-0262-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Nourmahnad A, Halpin E, & Forman SA (2019). Monod-Wyman-Changeux allosteric shift analysis in mutant α1β3γ2L GABAA receptors indicates selectivity and crosstalk among intersubunit transmembrane anesthetic sites. Molecular Pharmacology, 95(4), 408–417. 10.1124/mol.118.115048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RJ, Hadley SH, Morris KD, & Amin J (2000). Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nature Neuroscience, 3(12), 1274–1281. 10.1038/81800 [DOI] [PubMed] [Google Scholar]
- Weir CJ, Mitchell SJ, & Lambert JJ (2017). Role of GABAA receptor subtypes in the behavioural effects of intravenous general anaesthetics. British Journal of Anaesthesia, 119, i167–i175. 10.1093/bja/aex369 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.